
Submitted:
03 May 2023
Posted:
04 May 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The COVID-19 pandemic has underscored the importance of swift responses and the necessity of dependable technologies for vaccine development. Our team previously developed a fast cloning system for the modified vaccinia virus Ankara (MVA) vaccine platform. In this study, we report the construction and preclinical testing of a recombinant MVA vaccine obtained using this system. We obtained recombinant MVA expressing the unmodified full-length SARS-CoV-2 spike (S) protein containing the D614G amino acid substitution (MVA-Sdg) and a version expressing a modified S protein containing amino acid substitutions designed to stabilize the protein a in a pre-fusion conformation (MVA-Spf). S protein expressed by MVA-Sdg was found to be expressed and correctly processed and transported to the cell surface, where it efficiently produced cell-cell fusion. Version Spf, however, was not proteolytically processed and despite being transported to the plasma membrane, it failed to induce cell-cell fusion. We assessed both vaccine candidates in prime-boost regimens in the susceptible transgenic K18-human angiotensin converting enzyme 2 (K18-hACE2) mice and in golden Syrian hamsters. Robust immunity and protection from disease was induced with either vaccine in both animal models. Remarkably, the MVA-Spf vaccine candidate produced higher levels of antibodies, a stronger T cell response, and a higher degree of protection from challenge. In addition, the levels of SARS-CoV-2 in the brain of MVA-Spf inoculated mice was decreased to undetectable levels. Those results add to our current experience and range of vaccine vectors and technologies for developing a safe and effective COVID-19 vaccine
Keywords:
Poxvirus
; Modified Vaccinia virus Ankara
; vaccine
; SARS-CoV-2
; COVID-19
; recombinant viral vectors
; immunization
1. Introduction
Measures for transmission control and safe and efficient vaccines have emerged as prime factors in dealing with the Coronavirus disease 2019 (COVID-19) pandemic caused by the Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Since the spread of the disease, unprecedented efforts have been applied to develop COVID-19 vaccine candidates, resulting in numerous SARS-CoV-2-specific vaccines being evaluated in clinical trials in addition to the approved vaccines [1].
Multiple studies have identified the SARS-CoV-2 spike (S) protein as the most critical target antigen for COVID-19 vaccine development, similarly to other coronaviruses [2,3]. The trimeric S protein is a prominent immunological component of the virion surface and plays a crucial role in SARS-CoV-2 binding and entry to the host cell, mediating the interaction of the virus with the cellular receptor angiotensin-converting enzyme 2 (ACE2) and the subsequent fusion of the virus envelope with the host cell membrane. It has been clearly established that the production of S-specific antibodies that can neutralize the virus is crucial to confer protection against SARS-CoV-2 infection [4,5,6].
Diverse vaccine technologies have been explored in the race to generate suitable vaccines for COVID-19, including protein, attenuated live vectors and mRNA vaccines (for review see [7]). However, the current vaccines have limitations in providing long-term protection and may have varying levels of efficiency in preventing virus transmission [8,9,10]. One of the live attenuated vaccine vectors that can provide, alone or in combination, improved immune responses and protection is based on the Poxvirus Modified Vaccinia Ankara (MVA) [11,12,13]. MVA constitutes an efficient vaccine for Orthopoxvirus infections such as Smallpox and Mpox [14,15]. In addition to its use as a vaccine, and due to its proven safety profile, MVA virus recombinants have been explored as potential vaccines for diverse diseases. Since the development of MVA recombinant technology, recombinants expressing influenza A virus genes were shown to induce serum antibody levels and increased T-cell responses [16,17]. Since then, various immunization approaches using recombinant MVA have been evaluated in preclinical and clinical studies with successful results. For instance, recombinant MVA vaccines expressing HIV proteins have been tested alone or in combination with DNA-based or adenoviral vector vaccines to activate protective immune responses against AIDS [18,19]. MVA vector vaccines have also been successful in vaccine development against other infectious diseases with a global impact such as tuberculosis [20] and malaria [21]. In addition, recombinant MVA vaccines have been tested for emerging pathogens such as West Nile fever [22] and Ebola virus [23,24]. Notably, recombinant MVA expressing Ebola virus glycoprotein is authorized in Europe as an Ebola vaccine in a combined immunization regimen [25,26,27] (https://www.ema.europa.eu/en/medicines/human/EPAR/mvabea.
Since 2003, new beta coronaviruses causing atypical acute necrotizing pneumonia, the severe acute respiratory syndrome coronavirus (SARS-CoV), the Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-Cov2, have emerged in human populations [28]. In response to the emergence of human diseases caused by beta coronaviruses such as SARS-CoV and MERS-CoV, modified vaccinia virus Ankara (MVA) has been investigated as a viral vector vaccine, with the envelope spike (S) protein being a key target for neutralizing antibodies and protection [29,30]. A recombinant MVA expressing the SARS-CoV S antigen was assessed in various animal models, and was found to stimulate SARS-CoV-specific immune responses, including SARS-CoV-neutralizing antibodies and S-antigen-specific T cells, in mice, rabbits, and rhesus macaques [31,32]. Similarly, a recombinant MVA expressing the S protein of MERS-CoV was also developed and tested [33,34,35,36,37,38].
Not surprisingly, after the upsurge of the COVID-19 pandemic MVA was chosen by several groups as a possible vaccine platform for the prevention of the disease [39,40,41,42,43,44,45,46,47,48,49,50,51]. Since then, a number of different MVA-based vectors have been used to generate S-expressing MVA recombinants that have been tested in animals. So far, several studies have shown that such MVA-based vaccine candidates expressing SARS-CoV-2 S are able to confer protection against SARS-CoV-2 disease, in a way similar to that observed in coronavirus diseases caused by SARS-CoV and MERS. As a whole, those studies illustrate a variety of situations regarding the gene sequence, construction of the recombinants, inoculation protocols and characterization of the ensuing protection in different animal models.
In this work, we have generated vaccine candidates using our MVA-cloning system, and show that the resulting MVA recombinants produce a glycosylated, full-length SARS-CoV-2 S protein that is correctly transported to the cell membrane. Inoculation of mice and hamsters with those recombinants induced a strong immune response that included both humoral and cell-mediated responses and were able to protect animals from lethal SARS-CoV-2 infection. We further show that an MVA recombinant expressing a modified, prefusion-stabilized SARS-CoV-2 S has superior immunizing and protecting capacity, as tested in K18-hACE2 mice and golden Syrian hamsters.
2. Materials and Methods
2.1. Ethics Statement
All mouse experiments were approved by the Ethical Review Committee at the INIA-CISA and Comunidad de Madrid (Permit number: PROEX 037/15), Spain, in strict accordance with EU guidelines 2010/63/UE about protection of animals used for experimentation and other scientific purposes, and Spanish Animal Welfare Act 32/2007, and the Institutional Animal Care and Use Committees (IACUC) of Institutional Biosafety Committee of Yale University (IBSCYU), US. All the animals were housed under specific pathogen-free conditions and allowed to acclimatize in the facilities provided to the biosafety level 3 (BSL3) animal facilities at the Animal Health Research Center (INIA-CISA) of Madrid and at the Yale animal facilities (BSL2 and 3), supported by Yale Animal Resources Center (YARC). All standard operating procedures and protocols for IVIS imaging of SARS-CoV-2 infected animals under ABSL-3 conditions were approved by IACUC, IBSCYU and YARC. All mouse in vivo imaging, blood draw and virus inoculation experiments were done under anesthesia using regulated flow of isoflurane: oxygen mix to minimize pain and discomfort to the animals. BALB/c, C57BL/6 (B6) wild-type and heterozygous C57BL/6 (B6), hACE2-expressing transgenic B6 mice were obtained from Jackson Laboratory. Male and female 8-10 week-old mice were used for all the experiments. Heterozygous mice were crossed and genotyped to select heterozygous mice for experiments and confirmed by Automated Genotyping (TransnetYX).
Experiments with infectious SARS-CoV-2 using golden Syrian hamsters were conducted under appropriate ABSL-3 laboratories at Texas Biomedical Research Institute, San Antonio, Texas, US. Experiments were approved by the Texas Biomedical Research Institutional Biosafety (BSC) and Institutional Animal Care and Use (IACUC) committees, under protocols BSC20-004, BSC21-026, RDC21-026 and IACUC 1722 MA, respectively.
2.2. Cells and viruses.
Baby hamster kidney BHK-21 cells (ATCC CCL10) were grown in BHK-21 Glasgow minimal essential medium (Gibco BRL) containing 5% FBS, 3 g/ml tryptose phosphate broth, 0.01 M HEPES, antibiotics, and glutamine. Virus infections were performed with 2% FBS for all cell types. African green monkey kidney cell line BS-C-1 cells (ATCC CCL-26) were grown in Eagle’s Minimal Essential Medium (EMEM) supplemented with 0.1 mg/ml penicillin, 0.1 mg/ml streptomycin, 2 mM L-glutamine (Lonza) and 5% fetal bovine serum (FBS). African green monkey kidney epithelial Vero E6 cells (ATCC, CRL-1586) were grown in DMEM supplemented with 5% FBS, 0.1 mg/ml penicillin, 0.1 mg/ml streptomcin, 2 mM L-glutamine (Lonza). All cells were maintained in a 5% CO2 incubator at 37oC. Human Expi293F cells (Thermo Fisher Scientific) were grown in suspension in 293F expression medium (Thermo Fisher Scientific).
MVA -ΔF13L is an MVA virus that has replaced the F13L coding sequence with a red fluorescent protein (dsRed) gene [52]. All Vaccinia virus infections were performed with 2% FBS for all cell types.
SARS-CoV-2, USA-WA1/2020 strain, was obtained from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources, NR-52281) and the 2019 n-CoV/USA_WA1/2019 isolate of SARS-CoV-2 expressing nanoluc (nLuc) luciferase was obtained from Craig B Wilen, Yale University and generously provided by K. Plante and Pei-Yong Shi, World Reference Center for Emerging Viruses and Arboviruses, University of Texas Medical Branch. The viruses were amplified in African green monkey kidney epithelial Vero E6 cells. Briefly, Vero E6 cells were infected at low multiplicity of infection (MOI, 0.01) for 72 h and cell culture supernatants were collected, clarified, aliquoted and stored at -80ºC until use. Virus stocks were titrated in Vero E6 cells by plaque assay and immunostaining. SARS-CoV-2 stocks were sequenced by using next-generation sequencing [53].
2.3. Design and generation of MVAs.
Isolation of recombinant MVA viruses was carried out by inserting genes into the viral genome by homologous recombination occurring between the viral genome and plasmid DNA transfected. Selection of recombinant MVA was performed by plaque formation following restoration of the F13L gene [54,55]. The inserted sequence includes a combined early/late Poxviral promoter and open reading frames encoding different versions of the protein S gene. The complete SARS-CoV-2 S gene codon optimized to vaccinia virus codon usage (M.L. and R.B., unpublished), was synthesized gy Genscript Inc. and cloned into the transfer vector pMVA, to produce pMVA-S. The pMVA transfer plasmid contains the F13L gene of MVA, recombination flanks and a vaccinia virus early/late promoter to direct the transcription of the foreign gene [52,54].
pMVA-S was subsequently used to generate different S gene versions by site-directed mutagenesis. Mutations were introduced using the Q5 Site Directed Mutagenesis kit (New England Biolabs) following manufacturer´s instructions. Mutations K986P and V987P were introduced into pMVA-S by using oligonucleotides S4800-Q5-F TAGATTAGATccacctGAAGCTGAAGTACAAATAGATAGATTAATAAC and S4800-Q5-R GATAATATATCATTTAATACAGAAGATATAG, to generate plasmid pMVA-S-PP. Mutations R682S and R685S were introduced into plasmid pMVA-S-PP by using oligonucleotides S3910-Q5-F gcctctTCTGTAGCTAGCCAATCTATAATAG and S3910-Q5-R tctagaTGGAGAATTAGTTTGAGTTTGATAAG generating plasmids pMVA-S-SS-PP. Mutation D614G was introduced into plasmid pMVA-S and pMVA-S-SS-PP by using oligonucleotides S3690-Q5-F TTATATCAAGgaGTAAATTGTACTGAAGTAC and S3690-Q5-R TACAGCTACTTGATTAGAAG to generate pMVA-SDG and pMVA-S-DG-SS-PP (also termed pMVA-Spf).
Recombinant viruses MVA-S, MVA-Sdg and MVA-Spf were generated by infecting BHK-21 cells with MVAΔF13L at MOI of 0.05 and transfecting them with the corresponding transfer plasmids. Cell cultures were harvested at 48 h.p.i. and MVAs were selected and cloned by plaque isolation assay by using standard procedures [56].
2.4. Western blot.
BHK-21 cells were seeded in 6-well plates and the whole cell lysates (WCL) were prepared in denaturant buffer (80 mM Tris-HCl, pH 6.8, 2 % sodium dodecyl sulfate [SDS], 10 % glycerol, 0.01 % bromophenol blue solution and 0.71 M 2-mercaptoethanol). After SDS-polyacrylamide gel electrophoresis (PAGE), proteins were transferred to PVDF membranes and incubated 1 h at room temperature (RT) with primary antibody diluted in phosphate-buffered saline (PBS) containing 0.05 % Tween-20 and 1 % nonfat dry milk. Primary antibodies were Rabbit polyclonal anti-Receptor Binding Domain (RBD) antibody (GeneTex, ref GTX135709) diluted 1:500; Mouse monoclonal anti-S antibody (GeneTex, ref GTX632604) diluted 1:10000; Rat monoclonal anti-F13 diluted 1:100. After extensive washing with PBS-0.05 % Tween-20, membranes were incubated with HRP-conjugated secondary antibodies: polyclonal goat anti-rabbit IgG (Dako P0448) diluted 1:3000, polyclonal goat anti-mouse (GeneTex GTX213111) diluted 1:2000 and polyclonal rabbit anti-rat (Dako P0450) diluted 1:3000. After removal of unbound secondary antibodies by extensive washing using PBS-0.05 % Tween-20, membranes were incubated for 1 min with a 1:1 mix of solution A (2.5 mM luminol [Sigma], 0.4 mM p-coumaric acid [Sigma], 100 mM Tris-HCl, pH 8.5) and solution B (0.018 % H2O2, 100 mM Tris-HCl, pH 8.5). Luminescence was recorded using a Molecular Imager Chemi Doc-XRS (Bio-Rad).
2.5. Immunofluorescence.
BHK-21 cells were seeded in coverslips, washed with PBS and fixed by ice-cold 4 % paraformaldehyde for 12 min. Those samples that were subsequently permeabilized were treated with PBS containing 0.1 % Triton X-100 for 15 min at RT. Subsequently, all preparations were treated with PBS containing 0.1 M glycine for 5 min and incubated with primary antibodies in PBS-20 % FBS for 30 min, followed by incubation with secondary antibodies diluted 1:300 in PBS-20 % FBS. Primary antibodies used included rabbit polyclonal anti-RBD (GeneTex, ref GTX135709) diluted 1:500, rat monoclonal anti-B5 diluted 1:100, an anti-mouse IgG-Alexa Fluor 488, and an anti-Rat IgG-Alexa Fluor 594 (Invitrogen) both diluted 1:300.
2.6. Protein expression and purification.
Expi293F cells were grown to a density of 1x106 cells/mL at 37°C with 8% CO2 with regular agitation (150 rpm). Cells were transfected with a plasmid encoding SARS-CoV-2 RBD [57] using ExpiFectamine 293 transfection reagent, following manufacturer instructions (Thermo Fisher Scientific). At day 4 post-transfection, cell culture supernatants were clarified and the recombinant RBD was purified by Ni-NTA agarose resin (Qiagen), desalted and concentrated using Amicon columns (Millipore) Purified SARS-CoV-2 S RBD was snap-frozen at liquid nitrogen and stored in aliquots at 80°C until further use. Protein purity was confirmed as one single-band on SDS-PAGE. Proteins were quantified by Pierce protein BCA assay kit (Thermo Scientific).
2.7. Mice immunization and SARS-CoV-2 challenge
Groups of 8-10 weeks old BALB/c mice (n=5) were intraperitoneally, intramuscularly or subcutaneously immunized at 0 and 21 days with 107 plaque forming units (PFU) of MVA or MVA recombinants MVA-Sdg, MVA-Spf or MVA. Intraperitoneally immunized mice were bled one and two weeks post-prime, and two weeks post-boost, to determine IgM and IgG against SARS-CoV-2 S protein and the presence of neutralizing SARS-CoV-2 antibodies. Intramuscular and subcutaneously immunized mice were bled two weeks post-boost to determine IgG titers against SARS-CoV-2 S protein. Neutralizing titers were determined against infectious SARS-CoV-2 in a plaque reduction assay .
Groups of 8-10 weeks old C57BL/6 (B6) mice (n=4) were intramuscularly immunized at 0 and 21 days with 107 PFU of MVA or MVA recombinants. All animals were euthanized at day 15 post-boost, and their blood and spleens were harvested to analyze specific cellular and humoral immune responses.
Groups of 8-10 week old male and female K18-hACE2 mice were intramuscularly immunized at 0 and 21 days with 107 PFU of MVA-Sdg, MVA-Spf[1] or MVA. Two weeks after the second immunization, all animals were bled to determine antibodies against SARS-CoV-2 S protein, and were intranasally challenged with 1 x 105 PFU of 2019n-CoV/USA_WA1/2019 isolate of SARS-CoV-2 expressing nLuc in 30 μl volume of sterile PBS, under anesthesia (0.5 - 5 % isoflurane delivered using precision Dräger vaporizer with oxygen flow rate of 1 L/min). Body weight was measured and recorded daily. The starting body weight was set to 100 %. For survival experiments, mice were monitored every 24 h. Lethargic and moribund mice or mice that had lost more than 20% of their initial body weight were humanely euthanized and considered to have succumbed to infection for Log rank survival plots.
2.8. Enzyme-linked immunosorbent assay (ELISA) to SARS-CoV-2 S.
Indirect ELISA were used to determine specific anti-S antibody levels in sera from immunized mice. 96-well MaxiSorp plates (Nunc, USA) were coated with 150 ng/well of purified recombinant RDB protein expressed in expi293 cells, overnight at 4°C. Coated wells were subsequently blocked with PBS containing 0.1% Tween20 (PBST) and 3% dried milk for 1 hour at RT. Wells were then washed three times with washing buffer PBST. Mice sera collected before challenge were diluted in PBST 1% dried milk and plates were incubated for 1h at RT. After three washes, wells were incubated for 1h at RT with an anti-mouse IgG-HRP secondary antibody (Dako cat. number P0447) diluted 1:2000 in PBST or anti-mouse IgG-HRP secondary antibody (cat. number AP128P, Sigma-Aldrich) diluted 1:1000 for 1h at RT. After washing three times with PBST, the reaction was developed with 50 μL of substrate solution TMB (Sigma) and stopped by adding the same volume of 3 NH2SO4. Results were expressed as optical density (OD) measured at 450 nm.
2.9. Ex vivo flow cytometric analysis
To evaluate the induced cellular immune response, four mice immunized with MVA-Sdg, MVA-Spf or MVA-wt were euthanized at day 15 post-immunization and their spleens were harvested for analysis by intracellular cytokine staining (ICS) assay. A total of 106 splenocytes were stimulated with 0.6 nmol (approximately 1 μg/peptide) of a pep-tide pool of the S protein (The PepTivator® Peptide Pools, Miltenyi Biotec) composed by peptides of 15 amino acid length with 11 amino acid overlapping previously selected for efficient in vitro stimulation of antigen-specific CD4+ and CD8+ Tcells. Cells were main-tained during 6 hours of stimulation in RPMI 1640 supplemented with 10% fetal calf se-rum and containing brefeldin A (5 mg/ml) to increase the accumulation of IFN-γ in the re-sponding cells. After stimulation, cells were washed, stained for the surface markers, fixed, permeabilized and stained intracellularly using appropriate fluorochromes. To an-alyze the adaptive immune responses, the following fluorochrome-conjugated antibodies were used: CD3-APC, CD4-FITC, CD8-PerCP and IFN-γ-PE. All antibodies were were purchased from BioLegend. Data were acquired by FACS analysis on a Cube 8 (Sysmex) and were analyzed with FlowJo softw
2.10. Plaque reduction neutralization (PRNT) assay.
A PRNT assay for SARS-CoV-2 serology was performed as previously described [58]. Experiments were conducted with 1200 PFU/well of a recombinant SARS-CoV-2 USA-WA1/2020 expressing nLuc. One day prior to infection, 2x104 Vero E6 cells were seeded per well in a 96 well flat bottom plate and incubated overnight at 37°C under 5% CO2 to permit cell adherence. Three-fold dilutions of the heat inactivated sera (1h 56C) were performed in a separate 96 well culture plate using DMEM supplemented with penicillin (100 U/mL), streptomycin (100 mg/mL), HEPES, L-Glutamine (0.3 mg/mL), 0.12% sodium bicarbonate, 10% FBS (all from Thermo Fisher Scientific), in a Biosafety Level 3 (BSL3) laboratory. The 1200 PFU SARS-CoV-2 USA-WA1/2020 live virus was prepared in DMEM containing 10% FBS and combined with an equivalent volume of respective mouse sera dilutions for 1h at RT. After incubation, all media was removed from the 96 well plates seeded with Vero E6 cells and virus:sera mixtures were added to each respective well at a volume corresponding to 600 PFU per well and incubated for 1h further at 37°C. Both virus only and media only conditions were included in this assay as controls. After 1h incubation, virus:sera mixtures were removed from wells without disrupting the cell monolayers. Each antibody concentration (100 μL) was added to its respective Vero E6-seeded well in addition to an equivalent volume of DMEM containing 10% FBS and was then incubated for 72 h. Media was then discarded and replaced with 10% formaldehyde for 24 h for fixation. After fixation, formaldehyde solution was removed from wells and cells were stained by addition of 50 μL of crystal violet solution for visualization. PRNT50 titers were calculated as the reciprocal (log10) of the highest dilution of serum that neutralized 50% of the control virus input.
Neutralization titers were alternatively determined by using a Vesicular Stomatitis Virus (VSV) pseudotyped with SARS-CoV-2 S glycoprotein lacking the C-terminal aminoacids located in the cytoplasmic domain [59], using a modified VSV pseudotyping protocol (M. Lorenzo and R. Blasco, manuscript in preparation).
2.11. Bioluminescence Imaging (BLI) of SARS-CoV-2 infection
All the imaging was carried out using IVIS Spectrum® 985 in XIC-3 animal isolation chamber (PerkinElmer) that provided biological isolation of anesthetized mice or individual organs during the imaging procedure. All mice were anesthetized via isoflurane inhalation (3 - 5 % isoflurane, oxygen flow rate of 1.5 L/min) prior and during BLI using the XGI-8 Gas Anesthesia System. Prior to imaging, 100 μL of nanoluciferase substrate, furimazine (NanoGloTM, Promega, Madison, WI) diluted 1:40 in endotoxin-free PBS was retroorbitally administered to mice under anesthesia. The mice were then placed into XIC-3 animal isolation chamber (PerkinElmer) pre-saturated with isoflurane and oxygen mix. The mice were imaged in both dorsal and ventral position. The animals were then imaged again after euthanasia and necropsy by spreading additional 200 μL of substrate on to exposed intact organs. Infected areas of interest identified by carrying out whole-body imaging after necropsy were isolated, washed in PBS to remove residual blood and placed onto a clear plastic plate. Additional droplets of furimazine in PBS (1:40) were added to organs and soaked in substrate for 1-2 min before BLI. Images were acquired and analyzed with the manufacturer’s Living Image v4.7.3 in vivo software package. Image acquisition exposures were set to auto, with imaging parameter preferences set in order of exposure time, binning, and f/stop, respectively. Images were acquired with luminescent f/stop of 2, photographic f/stop of 8. Binning was set to medium. Comparative images were compiled and batch-processed using the image browser with collective luminescent scales. Photon flux was measured as luminescent radiance (p/sec/cm2/sr). During luminescent threshold selection for image display, luminescent signals were regarded as background when minimum threshold levels resulted in displayed radiance above non-tissue-containing or known uninfected regions. To determine the pattern of virus spread, the image sequences were acquired during the indicated days following intranasal administration of SARS-CoV-2.
2.12. Measurement of viral burden and analysis of signature inflammatory cytokines mRNA
Organs analyzed (lung and brain, from infected or uninfected mice) were collected, weighted, and homogenized in 0.8 mL of complete RPMI media containing penicillin-streptomycin and homogenized in 2 mL tube containing 1.5 mm Zirconium beads with BeadBug 6 homogenizer (Benchmark Scientific, TEquipment Inc). Virus titers were measured by quantitative real-time PCR (qRT-PCR) and plaque assay. RNA transcripts for inflammatory cytokines were also measured by qRT-PCR. First, total RNA was extracted from 100 μL of the homogenized tissues using TRIzol reagent (Invitrogen) and purified using NucleoSpin RNA columns (Macherey-Nagel), reverse transcribed with iScript advanced cDNA kit (Bio-Rad Cat#1725036) and qRT-PCR analysis was performed using SYBR Green Real-time PCR assay for determining copies of SARS-CoV-2 N gene RNA, following manufacturer conditions. Second, serially diluted clarified tissue homogenates were used to infect Vero-E6 cell monolayers. Viral titers per gram of tissue were quantified using standard plaque assay. Briefly, the 4x105 Vero-E6 cells were seeded on 12-well plate. Cells were infected 24h after seeding with 100 μL of serially diluted clarified tissue homogenates. After 1 hour viral adsorption, cells were overlaid with 1mL of pre-warmed 0.6% Avicel (RC-5811059 FMC BioPolymer) in complete DMEM medium. Plaques were resolved at 72 h post infection by fixing in 10 % formaldehyde overnight at RT followed by staining for 20 min with 1% crystal violet solution (Sigma-Aldrich). Plates were rinsed in water to visualize virus plaques.
2.13. Experiments in golden Syrian hamsters
Four-weeks-old specific-pathogen-free (SPF) female golden Syrian hamsters were purchased from Charles River, USA, and maintained in micro-isolator cages at ABSL-2 before and during vaccination with the recombinant MVA. To conduct the challenge experiments with SARS-CoV-2, hamsters were transferred to ABSL-3. Protection experiments were conducted essentially as previously described [53,60,61]. To evaluate in vivo safety of the recombinant MVA, hamsters (n=4/group) were sedated in an isoflurane chamber and vaccinated intranasally with 1 x 107 PFU of MVA-Sdg, MVA-Spf, MVA, or unvaccinated with PBS, using a total volume of 100 μL. Hamsters were vaccinated using a prime and boost regimen, with animals receiving the boost vaccination 3 weeks after prime vaccination. After prime and boost vaccination, hamsters were monitored daily for morbidity (changes in body weight and clinical signs of infection) and mortality (survival) for 21 (prime) or 36 (booster) days. Sera were collected one day before prime vaccination , at 21 days after prime vaccination, and one day before challenge with SARS-CoV-2. For viral challenge, hamsters were sedated with isoflurane and intranasally infected with 2 x 105 PFU of SARS-CoV-2 in a volume of 100 μL. At days 2 and 4 post-challenge, hamsters were humanely sacrificed and nasal turbinates and lungs were collected for pathology and viral titration. Half of the organs were homogenized in PBS using a Precellys tissue homogenizer (Bertin Instruments). Tissue homogenates were centrifuged at 21,500 x g for 5 min and supernatants were used to calculate viral titers using plaque assay. The other half of the organs were kept in 10% neutral buffered formalin for pathology observation. Lung samples were photographed to show gross lesions. Images were used for macroscopic pathology scoring analysis by measuring the distributions of pathological lesions, including consolidation, congestion, and pneumonic lesions using NIH ImageJ software. The area of pathological lesions was converted into percent of the total lung surface area.
2.14. Statistical analysis
Data were analyzed using GraphPad Prism version 8.0.1 (GraphPad Software, San Diego, CA, USA). Survival curves for each immunized group were compared in search of statistical differences using log-rank test. Data from the IgG and IgM ELISA and ICS assays, as well as viraemia levels, cytokine expression levels and gross lesions in lungs were performed using an independent-samples Student's t test. Virus neutralization assays were analyzed using by two-way ANOVA with post hoc Tukey test for multiple comparisons. A P-value lower than 0.05 was considered significant in all cases.
3. Results
3.1. Isolation of MVA recombinants expressing SARS-CoV-2 S.
Recombinant vaccine candidates were generated by inserting SARS-CoV-2 S gene versions in the vaccinia (strain MVA) viral genome by using a fast insertion/selection procedure [52,54]. In our constructs, S gene was placed downstream of a strong synthetic Poxviral early/late promoter and inserted in an intergenic region between the viral F13L and F12L genes. The insertion does not result in the inactivation of any viral genes, thus preserving the genetic content of the original vector. Recombinant viruses expressing three different versions of the S gene were generated. MVA-S contains the complete gene for the original reference isolate (Wuhan-Hu-1, NCBI Reference Sequence: NC_045512.2). A modified version involved a sequence variant (Sdg) incorporating the D614G mutation that became prevalent at the beginning of the pandemic and is known to increase virus infectivity in humans [62]. Finally, we introduced a set of mutations in the Sdg backbone to make a version stabilized in the pre-fusion conformation. Those included two amino acid substitutions (K986P and V987P) that were first tested in the homologous MERS coronavirus and are known to prevent the conformational change of the protein keeping it in its prefusion state [63]. Also, to block the proteolytical maturation of the S protein, two additional amino acid substitutions (R682S and R685S) located around the proteolytic cleavage site [64] were introduced. The construct containing those four mutations was termed Spf (“pre-fusion”).
3.2. S protein expression by MVA recombinants.
After the generation of the different recombinant MVA viruses, expression of the different S proteins was assessed by Western blot and immunofluorescence in infected BHK-21 cells. Western blot with anti-RBD and anti-S2 antibodies revealed several protein products in MVA-S and MVA-Sdg infected cell extracts (Figure 1). The larger forms detected (180-200 kDa) were compatible with the size of the complete protein and different glycosylation states. In addition, we detected smaller forms with an apparent mass between 90-120 kDa, that were compatible with the products (S1 and S2) generated by proteolytic cleavage in the region of the fusion peptide. Notably, the smaller forms were not present in MVA-Spf infected cell extracts, confirming that the introduced mutations effectively prevented the proteolytic cleavage between the S1 and S2 domains in the S protein.
To further characterize the expression and subcellular location of the heterologous proteins, immunofluorescence assays were carried out. Indirect immunofluorescence on fixed and permeabilized cells produced an intracellular pattern compatible with endoplasmatic reticulum, Golgi Complex and plasma membrane staining (Figure 1C). Cell surface expression of the S protein was further confirmed by antibody staining on non-permeabilizing conditions both in MVA-Sdg and MVA-Spf infected cells (Figure 1D). Overall, these results indicated correct expression and transport of the S proteins expressed by MVA recombinants.
3.3. Fusion phenotype in cells infected by MVA recombinants.
Mature SARS-CoV-2 S protein mediates the entry of the virus in the target cells through fusion of the viral membrane with the plasma membrane. This activity can produce cell-cell fusion between cells expressing an active S protein and the SARS-CoV-2 virus receptor (ACE2). We have used this membrane fusion activity to check the integrity of the S proteins being expressed by the MVA recombinants and to confirm the predicted effect of the introduced mutations. BSC-1 cells were infected at a high multiplicity of infection with the different MVA recombinants and monitored for cell fusion. At 18 h post-infection, MVA-Sdg caused massive fusion of BSC-1 cells, producing large syncytia (Figure 2). In contrast, cell fusion activity could not be detected in cells infected with the unmodified MVA virus, indicating that cell fusion was specifically caused by the SARS-CoV-2 S protein. Interestingly, expression of the prefusion-stabilized S protein by the MVA-Spf virus did not induce detectable cellular fusion, indicating that the inserted mutations are not only impeding the proteolytic cleavage but also preventing the SARS-CoV-2 S protein-mediated fusion activity.
3.4. Induction of antibody responses in BALB/c mice.
Several works have shown that cloning of heterologous antigens in the F13L locus of the MVA genome is related with the induction of potent immune responses against the specific antigens in immunized animals. To assess the induction of a humoral immune response by the MVA vaccine candidates, groups of BALB/c mice (n=5) were intraperitoneally inoculated with two doses of 107 PFU of MVA, MVA-Sdg or MVA-Spf, in a three week-interval. Serum was harvested from whole blood of all mice 7- and 14-days post-prime (d.p.p.), and 14 days post-boost (d.p.b.), and total IgM and IgG titers were determined. Specific antibodies to the S RBD could be detected in all mice immunized with MVA-Sdg or MVA-Spf (Figure 3A,B). Significant enhancement in the titers of anti-RBD IgM and IgG in groups immunized with MVA-Spf was evident when compared with those immunized with MVA-Sdg (Figure 3B,C). Virus neutralizing antibodies were detected in all mice immunized with MVA-Spf, and in three out of five mice immunized with MVA-Sdg (Figure 3D). Importantly, SARS-CoV-2 neutralizing antibody titers in serum were significantly higher (p-value = 0.008) in animals immunized with MVA-Spf when compared to mice immunized with MVA-Sdg, indicating a superior ability of MVA expressing the pre-fusion stabilized SARS-CoV-2 S protein in eliciting a SARS-CoV-2 neutralizing humoral response.
Next, we wondered whether the inoculation route could influence the immuno-genicity of our recombinant MVA-based vaccine candidates. Intraperitoneal injection of mice is often used to evaluate potency/immunogenicity of MVA vaccine candidates. However, intramuscular (IM) or subcutaneous (SC) injection routes are preferred for human use and widely accepted for vaccinia MVA vaccine administration. To test whether these alternative routes were functional for our vaccine candidates, we evaluated the immunogenicity of MVA-Spf when administered by either subcutaneous or intramuscular inoculation routes. Significant anti-RBD IgG titers were detected in all MVA-Spf immunized mice, independently of the administration route (Figure 3E). Virus neutralizing antibodies were also induced, reaching slightly higher titers after intramuscular immunization compared to those obtained by the subcutaneous route (Figure 3F). Based on these results, intramuscular injection was chosen as the preferred route for administration in subsequent experiments.
3.5. Induction of Humoral and Cellular Responses in C57BL6 (B6) Mice.
The ability of the vaccine candidates to induce an S-specific cellular immune response was also investigated. To that end, groups of B6 mice (n=4) were immunized following a prime-boost strategy with 107 PFU of MVA-Sdg, MVA-Spf or MVA control, in a three-week interval. Serum from whole blood was harvested at 21 d.p.p., and at 14 d.p.b., for the analysis of the humoral immune response (Figure 4). Two weeks after the second inoculation, spleen and serum were harvested from mice and both humoral and cellular response were analyzed. As in immunized BALB/c mice, inoculation of our vaccine candidates resulted in significant virus neutralization antibody titers compared with MVA-inoculated B6 mice. Importantly, after the first dose of MVA-Spf, high neutralizing antibody titers were observed. which were higher in animals immunized with MVA-Spf compared to those immunized with MVA-Sdg. As in the previous experiments, boost inoculation induced enhanced antibody neutralization titers. As in the case of BALB/c mice, neutralizing antibody titers in serum were significantly higher in B6 mice immunized with MVA-Spf compared to mice immunized with MVA-Sdg, indicating a superior immunogenicity of MVA-Spf.
Splenic T-cell responses measured by intracellular cytokine staining (ICS) were detected against peptides spanning the full length of the S protein. After stimulation, splenocytes from animals vaccinated with MVA-Spf showed a moderate (non-significant) increase of CD8+IFN-γ+ and CD4+IFN-γ+ levels compared to the MVA (control) and MVA-Sdg immunization groups (Figure 4D), which indicates a more robust cell-mediated response after immunization with MVA-Spf.
3.6. Immunogenicity and protection efficacy of recombinant MVAs in K18-hACE2 Mice.
3.6.1. Humoral response in K18-hACE2 mice.
Prior to evaluating the ability of the vaccine candidates to protect against a lethat SAR-CoV-2 infection, we compare the immunogenicity of MVA-Spf or MVA-Sdg in an animal model suitable for testing SARS-CoV2 protection. To that end, we used transgenic mice expressing hACE2 receptor regulated by the cytokeratin 18 (K18) gene promoter (K18-hACE2). Groups of mice were intramuscularly inoculated with the MVA recombinants following a homologous prime-boost immunization strategy as stated above. Binding antibody titers were measured in sera samples obtained 2 weeks after boost from immunized animals by ELISA using the purified S-RBD. Both immunized groups had significant levels of specific anti S-RBD antibodies compared with the control group, being significantly higher in MVA-Spf immunized mice than in the MVA-Sdg immunized mice (Figure 5A). To test the neutralizing capacity of the anti-SARS-CoV-2 antibodies induced by the vaccine candidates, a PRNT assay was performed using the same serum samples. NT50 values revealed a significant increase of the neutralizing activity in the sera from immunized mice compared to the control group. A significantly increase in NT50 values was also observed in sera from MVA-Spf immunized mice compared with to MVA-Sdg immunized mice (p < 0.001) (Figure 5B).
3.6.2. Protection in K18-hACE2 mice
We next evaluated the ability of the recombinant MVAs to confer protection against SARS-CoV-2 challenge. To that end, K18-hACE2 mice (n=9) were immunized with the previously described MVA recombinants and subsequently challenged intranasally with 105 PFU of SARS-CoV-2 per mouse at two weeks post-boost. Presence of virus in brain and lung homogenates was determined in four out of nine immunized animals of each group. To that end, SARS-CoV-2 RNA was measured by qRT-PCR (Figure 5C) and SARS-CoV-2 titers were determined by plaque assay on Vero E6 cells (Figure 5D). High relative expression levels of viral RNA were detected in lungs and brains of control mice which had been inoculated with MVA. In contrast, infection in MVA-Spf immunized mice was abolished or under the level of detection by qRT-PCR in all four animals, and only one mouse in the MVA-Sdg immunization group showed viral replication. High levels of infective viral particles were observed in control mice by plaque assay, reaching 105 and 1010 PFU per mg of tissue in lung and brain, respectively. Conversely, an absence of infectious viral particles in these organs was confirmed in MVA-Spf immunized mice. Similarly, MVA-Sdg vaccinated mice did not present infective virus, except for one mouse that displayed a viral titer of 5x107 PFU/mg of tissue in brain (same animal that was positive by qRT-PCR), although this titer was 1000-fold lower than those in the control group.
Survival rates, percentage of body weight loss and clinical signs were also analyzed and monitored daily two weeks post-challenge in the five remaining mice of each group. By day 6-7 post-infection, control mice lost 20% or more of their initial body weight, showing severe clinical signs of infection. Those that did not succumb to infection were humanely euthanized (Figure 5E,F). Conversely, a partial protection (60% surviving mice) was observed in the group immunized with MVA-Sdg, with only one of the two animals showing signs of infection. Significantly, total protection was achieved in MVA-Spf immunized mice, as determined by the absence of body weight loss and clinical signs of infection (Figure S1).
In parallel, we also employed a BLI to monitor the impact of vaccination on SARS-CoV-2 replication and spread in K18-hACE2 mice using a reporter SARS-CoV-2 expressing nLuc luciferase (Figure 6A) [65]. In control mice, nLuc signal was first detected in the lung at 2 dpi where it continued to increase until 5-6 dpi with infection becoming systemic and reaching cervical lymph nodes and brain (imaging in ventral position) leading to death (Figure 6B and Figure S2). In contrast, luminescent signals were undetectable in the lungs or brains of MVA-Sdg or MVA-Spf immunized animals throughout the course of infection, with the exception of one MVA-Sdg immunized mouse that succumbed to infection at 8 dpi. Interestingly, we observed nLuc signal in the nares of control-immunized mice at 5-6 dpi, indicating presence of transmissible virus which was absent in any of the MVA-Sdg or MVA-Spf immunized animals (Figure 6B-D).
In addition, to observe viral spread with enhanced sensitivity and resolution, body and individual organs were imaged after necropsy. Most organs analyzed from control mice showed nLuc activity, with the highest signal in the brain followed by the lung and nasal cavity (Figure 6E and Figure S3). In contrast, mice immunized with MVA-Spf, that remained healthy throughout the experiment and displayed no signs of disease (including body weight loss phenotype), lacked detectable nLuc activity, lung infection, or neuroinvasion. Among MVA-Sdg immunized mice, 60% remained protected from infection with undetectable nLuc activity in their organs, and only one mice showed viral neuroinvasion at 7 dpi and died one day later.
3.6.3. MVA-Spf controls cytokine response in the brain of SARS-CoV-2 infected K18-hACE2 mice.
SARS-CoV-2 infection triggers a hyper-induction of proinflammatory cytokines, also known as cytokine storm or cytokine release syndrome (CRS), which is one of the key aspects that significantly contributes to the pathogenesis caused by this SARS-CoV-2 infection. To analyze if these MVA vaccine candidates can prevent this phenomenon, RNA transcripts of different proinflammatory cytokines (Il-1b, Il-6, TNF, IFN-γ) were measured in brain and lung at 6 d.p.i. by qRT-PCR. No significant differences were observed in lung among groups (Figure 7). Interestingly, a general decrease in the expression level of these cytokines was observed in the brain of immunized animals, especially in those immunized with MVA-Spf (Figure 7). Additionally, other inflammatory molecules related with the cytokine storm induced by SARS-CoV-2, as Ccl2 (also involved in neuroinflammatory processes [66]) and Cxcl10, were upregulated in control animals. Finally, adhesion molecules as VCAM1 and E-selectin were also upregulated in control mice (Figure 7), possibly indicating vascular injury in this tissue.
3.7. Protective Capacity of MVA vaccines in golden Syrian hamsters.
To further demonstrate the feasibility of using MVA expressing SARS-CoV-2 S as a COVID-19 vaccine, we evaluated the safety, immunogenicity, and protection efficacy in golden Syrian hamsters, which we and others have been previously shown to be susceptible and represent a good animal model for SARS-CoV-2 infection, including vaccine development [53,60,61]. To that end, golden Syrian hamsters were either mock-vaccinated with PBS or vaccinated with 107 PFU of MVA, MVA-Sdg, or MVA-Spf using a prime and boost approach (Figure 8A). After vaccination, clinical signs and body weight were measured daily for 21 (prime) and 36 (prime and boost) days. In both cases, hamsters vaccinated with MVA, MAV-Sdg or MVA-Spf did not develop any apparent clinical sign of infection, any changes in body weight, or mortality (data not shown), demonstrating that vaccination with 107 PFU of MVA, MVA-Sdg, or MVA-Spf is safe. At days -1, 21 (prime) and 35 (prime and boost) serum samples were collected and used to evaluate neutralizing antibody responses using a Vesicular Stomatitis Virus (VSV) pseudotyped-based assay or infectious SARS-CoV-2 (Figure 8B). We were able to detect the presence of neutralizing antibodies in the sera of hamsters vaccinated with MVA-Sdg after boost (day 35) while neutralizing antibodies were readily detected by day 21 (prime) in hamsters vaccinated with MVA-Spf. Notably, by day 35 (primer + boost) the levels of neutralizing antibodies were higher than those detected after prime vaccination and higher than those obtained by vaccination with MVA-Sdg (Figure 8B), demonstrating that vaccination with MVA-Spf is able to induce earlier and higher levels of neutralizing antibodies than the MVA-Sdg vaccine. As expected, we were not able to detect SARS-CoV-2 neutralizing antibodies in hamsters mock (PBS)-vaccinated or vaccinated with MVA, at any days post-vaccination.
To assess protection efficacy, vaccinated hamster were next challenged with 2x105 PFU of SARS-CoV-2 and nasal turbinate and lungs were collected at days 2 and 4 after challenge to assess the presence of virus, and to evaluate pathology in the lungs. Mock (PBS)-vaccinated hamsters challenged with SARS-CoV-2, and unvaccinated and mock-challenged hamsters were included as internal controls. As expected lungs from unvaccinated hamsters challenged with SARS-CoV-2 show pathology lesions at both days post-challenge but higher at day 4 than day 2 after SARS-CoV-2 challenge (Figure 9). Similar pathology lesions were observed in the lungs of hamsters vaccinated with MVA. Notably, pathology lesions were significantly lower in hamsters vaccinated with MVA-Sdg or MVA-Spf at days 2 and 4 post-challenge as compared to mock- or MVA-vaccinated hamsters. As expected, we did not observe any pathology lesions in the lungs of hamsters that were no challenged with SARS-CoV-2 at either day 2 or 4 post-challenge.
Notably, these pathology results correlated with the viral titers in the nasal turbinate and lungs (Figure 10). Higher levels of SARS-CoV-2 were detected at both days 2 and 4 and in the nasal turbinate and lungs of hamster mock (PBS)-vaccinated hamster. Similarly, high levels of SARS-CoV-2 replication were also observed in the nasal turbinate and lungs of hamsters vaccinated with MVA at days 2 and 4 after challenge with SARS-CoV-2. Levels of SARS-CoV-2 in the nasal turbinate and lungs of hamsters vaccinated with MVA-Sdg at both days after challenge with SARS-CoV-2 were significantly lower than those of PBS- or MVA-vaccinated hamsters. Likewise, significantly lower levels of SARS-CoV-2 were detected in the nasal turbinate and lungs of hamsters vaccinated with MVA-Spf at either day 2 or 4 post-infection. Notably, in the case of hamsters vaccinated with MVA-Spf we were not able to detect the presence of SARS-CoV-2 in the nasal turbinate or lungs at day 4 post-challenge. These results demonstrate that vaccination with MVA-Sdg and more notable with MVA-Spf can protect hamster from SARS-CoV-2 infection, with lower levels of viral replication in the upper (nasal turbinate) or lower (lung) respiratory tract as compared to unvaccinated animals.
4. Discussion
The recent COVID-19 pandemic has demonstrated the usefulness of vaccines to control the spread of infectious diseases and has reminded us of the importance of updating and improving vaccine technologies. Besides approved vaccines based on messenger RNA and non-replicating adenoviruses, additional technologies offer specific advantages that may expand our tool plethora for vaccination strategies. Among relevant aspects to consider in choosing vaccine platforms are the cost, thermal stability and ease of administration, and the optimal characteristics of vaccines in particular circumstances should be sought. In this respect, vaccine candidates that provide different advantages with respect to those already approved are of interest.
In this study, we have taken advantage of an efficient MVA cloning/selection system to isolate MVA recombinants expressing SARS-CoV-2 S glycoprotein to test them as COVID-19 vaccine candidates. In preclinical testing, we demonstrated that those candidates are suitable to achieve protection from SARS-CoV-2 induced pathogenicity in both mice and hamsters, as has been noted with MVA recombinants obtained with different procedures [39,40,41,46,67] . Even though unmodified S protein has been shown to induce protection, we show that a prefusion-stabilized version (Spf) is clearly superior in the induction of immunity, including neutralizing antibodies, and in protection from disease, in agreement with previous works [43,45,47].
Overall, the results obtained by vaccination with MVA recombinants expressing SARS-CoV-2 S protein render a positive vaccine profile with MVA-Spf providing effective protection against SARS-CoV-2 induced pathogenesis in both mice and hamsters. This includes reducing lesions in the lungs, a major site of virus replication and a crucial factor in COVID-19 pathogenesis. Besides the induction of immunity, a reduction in viral RNA and virus titers in the respiratory tract was detected. Also, we have shown a complete absence of virus replication in the brain of animals vaccinated with MVA-Spf. However, MVA-Sdg only partially decreased neuroinvasion. Thus, it seems likely that vaccine potency is crucial to prevent spread of the SARS-CoV-2 infection to the brain. In this respect, several vaccination strategies have resulted in varied levels of brain protection [51,68,69], which is a significant factor due to the likely relationship with COVID-19 neurological consequences.
With the ongoing COVID-19 pandemic, numerous candidate vaccines are being investigated at an unprecedented pace. Among the hefty experience gained during the pandemic, the results presented in this study support further development of the MVA-Spf vaccine and hopefully add to our current understanding and knowledge base for improvement of poxvirus-based vaccines.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, M.M.L., A.N., J.O., L.M.S. A.Marín. and R.B.; methodology, M.M.L, K.C., A.Marín., P.D.U., J.M.S., J.P, and C.Y.; formal analysis, M.M.L, K.C., S.M., S.U., L.J., C.Y. and J.P., P.K.; investigation, M.M.L, K.C., A.Marín., L.J., I.U., S.M., S.U., E.C., G.L., C.Y., J.P., A.Matía. and J.M.S. A.N., ; resources, M.M.L. and J.M.S.; writing—original draft preparation, R.B., J.O. L.M.S, A.M. and R.B.; writing—review and editing, I.U., J.O. L.M.S, A.M. and R.B.; visualization, K.C., A.B., S.M., S.U., L.J., A.N.; supervision, J.O., L.M.S., A.Marín, E.F., and R.B.; project administration, R.B.; funding acquisition, J.O., L.M.S., E..F., and R.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Instituto de Salud Carlos III, Fondo COVID-19 de proyectos de investigación sobre SARS-CoV-2 y la enfermedad COVID-19 grant COV20/00901, and grant PID2021-128466OR-I00 funded by funded by MCIN/AEI/10.13039/501100011033 as part of Plan Estatal de Investigación Científica, Desarrollo e Innovación. This research work was funded by the European Commission – NextGenerationEU, through CSIC's Global Health Platform (PTI Salud Global) grant SGL2103059. All experiments using bioluminescent imaging approach were supported by NIH grant R01AI163395 to WM. Research on SARS-CoV-2 in L.M-S laboratory was partially supported by the San Antonio Partnership for Precision Therapeutics, the San Antonio Medical Foundation, and the Texas Biomedical Research Institute Forum Foundation.
Data Availability Statement
The data presented in this study are available on request from the corresponding authors.
Acknowledgments
We want to thank Dr. Thomas Moran at the Icahn School of Medicine at Mount Sinai for providing us with the SARS-CoV cross-reactive N protein monoclonal Ab 1C7. We also thank BEI Resources for providing the SARS-CoV-2 USA-WA1/2020 isolate (NR-52281).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Kyriakidis, N.C.; Lopez-Cortes, A.; Gonzalez, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines 2021, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV--a target for vaccine and therapeutic development. Nat Rev Microbiol 2009, 7, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Hillyer, C.; Du, L. Neutralizing Antibodies against SARS-CoV-2 and Other Human Coronaviruses. Trends Immunol 2020, 41, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, Z.A.; Al-Bashir, S.M.; Alzoubi, H.; Al-Salih, N.S.; Aldamen, A.A.; Abdulazeez, A.Z. The Role of Immunity in the Pathogenesis of SARS-CoV-2 Infection and in the Protection Generated by COVID-19 Vaccines in Different Age Groups. Pathogens 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, N.J.; Kibble, M.; Wong, A.; Silverwood, R.J.; Knuppel, A.; Williams, D.M.; Hamilton, O.K.L.; Lee, P.H.; Bridger Staatz, C.; Di Gessa, G.; et al. Antibody levels following vaccination against SARS-CoV-2: associations with post-vaccination infection and risk factors in two UK longitudinal studies. Elife 2023, 12. [Google Scholar] [CrossRef]
- Samrat, S.K.; Tharappel, A.M.; Li, Z.; Li, H. Prospect of SARS-CoV-2 spike protein: Potential role in vaccine and therapeutic development. Virus Res 2020, 288, 198141. [Google Scholar] [CrossRef]
- Cattel, L.; Giordano, S.; Traina, S.; Lupia, T.; Corcione, S.; Angelone, L.; La Valle, G.; De Rosa, F.G.; Cattel, F. Vaccine development and technology for SARS-CoV-2: Current insight. J Med Virol 2022, 94, 878–896. [Google Scholar] [CrossRef]
- Yang, H.; Xie, Y.; Li, C. Understanding the mechanisms for COVID-19 vaccine's protection against infection and severe disease. Expert Rev Vaccines 2023, 22, 186–192. [Google Scholar] [CrossRef]
- Wilder-Smith, A. What is the vaccine effect on reducing transmission in the context of the SARS-CoV-2 delta variant? Lancet Infect Dis 2022, 22, 152–153. [Google Scholar] [CrossRef]
- Feikin, D.R.; Higdon, M.M.; Abu-Raddad, L.J.; Andrews, N.; Araos, R.; Goldberg, Y.; Groome, M.J.; Huppert, A.; O'Brien, K.L.; Smith, P.G.; et al. Duration of effectiveness of vaccines against SARS-CoV-2 infection and COVID-19 disease: results of a systematic review and meta-regression. Lancet 2022, 399, 924–944. [Google Scholar] [CrossRef]
- Volz, A.; Sutter, G. Modified Vaccinia Virus Ankara: History, Value in Basic Research, and Current Perspectives for Vaccine Development. Adv Virus Res 2017, 97, 187–243. [Google Scholar] [CrossRef]
- Sutter, G.; Staib, C. Vaccinia vectors as candidate vaccines: the development of modified vaccinia virus Ankara for antigen delivery. Curr Drug Targets Infect Disord 2003, 3, 263–271. [Google Scholar] [CrossRef]
- Volkmann, A.; Williamson, A.L.; Weidenthaler, H.; Meyer, T.P.H.; Robertson, J.S.; Excler, J.L.; Condit, R.C.; Evans, E.; Smith, E.R.; Kim, D.; et al. The Brighton Collaboration standardized template for collection of key information for risk/benefit assessment of a Modified Vaccinia Ankara (MVA) vaccine platform. Vaccine 2020. [Google Scholar] [CrossRef]
- Stittelaar, K.J.; van Amerongen, G.; Kondova, I.; Kuiken, T.; van Lavieren, R.F.; Pistoor, F.H.; Niesters, H.G.; van Doornum, G.; van der Zeijst, B.A.; Mateo, L.; et al. Modified vaccinia virus Ankara protects macaques against respiratory challenge with monkeypox virus. J Virol 2005, 79, 7845–7851. [Google Scholar] [CrossRef]
- Zaeck, L.M.; Lamers, M.M.; Verstrepen, B.E.; Bestebroer, T.M.; van Royen, M.E.; Gotz, H.; Shamier, M.C.; van Leeuwen, L.P.M.; Schmitz, K.S.; Alblas, K.; et al. Low levels of monkeypox virus-neutralizing antibodies after MVA-BN vaccination in healthy individuals. Nat Med 2023, 29, 270–278. [Google Scholar] [CrossRef]
- Sutter, G.; Wyatt, L.S.; Foley, P.L.; Bennink, J.R.; Moss, B. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine 1994, 12, 1032–1040. [Google Scholar] [CrossRef]
- Altenburg, A.F.; Kreijtz, J.H.; de Vries, R.D.; Song, F.; Fux, R.; Rimmelzwaan, G.F.; Sutter, G.; Volz, A. Modified vaccinia virus ankara (MVA) as production platform for vaccines against influenza and other viral respiratory diseases. Viruses 2014, 6, 2735–2761. [Google Scholar] [CrossRef]
- Iyer, S.S.; Amara, R.R. DNA/MVA Vaccines for HIV/AIDS. Vaccines (Basel) 2014, 2, 160–178. [Google Scholar] [CrossRef]
- Ondondo, B.O.; Yang, H.; Dong, T.; di Gleria, K.; Suttill, A.; Conlon, C.; Brown, D.; Williams, P.; Rowland-Jones, S.L.; Hanke, T.; et al. Immunisation with recombinant modified vaccinia virus Ankara expressing HIV-1 gag in HIV-1-infected subjects stimulates broad functional CD4+ T cell responses. Eur J Immunol 2006, 36, 2585–2594. [Google Scholar] [CrossRef]
- McShane, H.; Behboudi, S.; Goonetilleke, N.; Brookes, R.; Hill, A.V. Protective immunity against Mycobacterium tuberculosis induced by dendritic cells pulsed with both CD8(+)- and CD4(+)-T-cell epitopes from antigen 85A. Infect Immun 2002, 70, 1623–1626. [Google Scholar] [CrossRef]
- Schneider, J.; Gilbert, S.C.; Blanchard, T.J.; Hanke, T.; Robson, K.J.; Hannan, C.M.; Becker, M.; Sinden, R.; Smith, G.L.; Hill, A.V. Enhanced immunogenicity for CD8+ T cell induction and complete protective efficacy of malaria DNA vaccination by boosting with modified vaccinia virus Ankara. Nat Med 1998, 4, 397–402. [Google Scholar] [CrossRef]
- Volz, A.; Lim, S.; Kaserer, M.; Lulf, A.; Marr, L.; Jany, S.; Deeg, C.A.; Pijlman, G.P.; Koraka, P.; Osterhaus, A.D.; et al. Immunogenicity and protective efficacy of recombinant Modified Vaccinia virus Ankara candidate vaccines delivering West Nile virus envelope antigens. Vaccine 2016, 34, 1915–1926. [Google Scholar] [CrossRef]
- Yoshikawa, T. Third-generation smallpox vaccine strain-based recombinant vaccines for viral hemorrhagic fevers. Vaccine 2021, 39, 6174–6181. [Google Scholar] [CrossRef]
- Tomori, O.; Kolawole, M.O. Ebola virus disease: current vaccine solutions. Curr Opin Immunol 2021, 71, 27–33. [Google Scholar] [CrossRef]
- Milligan, I.D.; Gibani, M.M.; Sewell, R.; Clutterbuck, E.A.; Campbell, D.; Plested, E.; Nuthall, E.; Voysey, M.; Silva-Reyes, L.; McElrath, M.J.; et al. Safety and Immunogenicity of Novel Adenovirus Type 26- and Modified Vaccinia Ankara-Vectored Ebola Vaccines: A Randomized Clinical Trial. JAMA 2016, 315, 1610–1623. [Google Scholar] [CrossRef]
- Ewer, K.; Rampling, T.; Venkatraman, N.; Bowyer, G.; Wright, D.; Lambe, T.; Imoukhuede, E.B.; Payne, R.; Fehling, S.K.; Strecker, T.; et al. A Monovalent Chimpanzee Adenovirus Ebola Vaccine Boosted with MVA. N Engl J Med 2016, 374, 1635–1646. [Google Scholar] [CrossRef]
- Tapia, M.D.; Sow, S.O.; Lyke, K.E.; Haidara, F.C.; Diallo, F.; Doumbia, M.; Traore, A.; Coulibaly, F.; Kodio, M.; Onwuchekwa, U.; et al. Use of ChAd3-EBO-Z Ebola virus vaccine in Malian and US adults, and boosting of Malian adults with MVA-BN-Filo: a phase 1, single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation trial, and a nested, randomised, double-blind, placebo-controlled trial. Lancet Infect Dis 2016, 16, 31–42. [Google Scholar] [CrossRef]
- Nova, N. Cross-Species Transmission of Coronaviruses in Humans and Domestic Mammals, What Are the Ecological Mechanisms Driving Transmission, Spillover, and Disease Emergence? Front Public Health 2021, 9, 717941. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Y.; Wu, H.; Luo, B.; Chen, J.; Li, W.; Jiang, S. Identification of immunodominant sites on the spike protein of severe acute respiratory syndrome (SARS) coronavirus: implication for developing SARS diagnostics and vaccines. J Immunol 2004, 173, 4050–4057. [Google Scholar] [CrossRef]
- Sui, J.; Li, W.; Murakami, A.; Tamin, A.; Matthews, L.J.; Wong, S.K.; Moore, M.J.; Tallarico, A.S.; Olurinde, M.; Choe, H.; et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc Natl Acad Sci U S A 2004, 101, 2536–2541. [Google Scholar] [CrossRef]
- Bisht, H.; Roberts, A.; Vogel, L.; Bukreyev, A.; Collins, P.L.; Murphy, B.R.; Subbarao, K.; Moss, B. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc Natl Acad Sci U S A 2004, 101, 6641–6646. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, L.; Qin, C.; Ba, L.; Yi, C.E.; Zhang, F.; Wei, Q.; He, T.; Yu, W.; Yu, J.; et al. Recombinant modified vaccinia virus Ankara expressing the spike glycoprotein of severe acute respiratory syndrome coronavirus induces protective neutralizing antibodies primarily targeting the receptor binding region. J Virol 2005, 79, 2678–2688. [Google Scholar] [CrossRef]
- Alharbi, N.K.; Aljamaan, F.; Aljami, H.A.; Alenazi, M.W.; Albalawi, H.; Almasoud, A.; Alharthi, F.J.; Azhar, E.I.; Barhoumi, T.; Bosaeed, M.; et al. Immunogenicity of High-Dose MVA-Based MERS Vaccine Candidate in Mice and Camels. Vaccines (Basel) 2022, 10. [Google Scholar] [CrossRef]
- Alharbi, N.K.; Padron-Regalado, E.; Thompson, C.P.; Kupke, A.; Wells, D.; Sloan, M.A.; Grehan, K.; Temperton, N.; Lambe, T.; Warimwe, G.; et al. ChAdOx1 and MVA based vaccine candidates against MERS-CoV elicit neutralising antibodies and cellular immune responses in mice. Vaccine 2017, 35, 3780–3788. [Google Scholar] [CrossRef]
- Haagmans, B.L.; van den Brand, J.M.; Raj, V.S.; Volz, A.; Wohlsein, P.; Smits, S.L.; Schipper, D.; Bestebroer, T.M.; Okba, N.; Fux, R.; et al. An orthopoxvirus-based vaccine reduces virus excretion after MERS-CoV infection in dromedary camels. Science 2016, 351, 77–81. [Google Scholar] [CrossRef]
- Koch, T.; Dahlke, C.; Fathi, A.; Kupke, A.; Krahling, V.; Okba, N.M.A.; Halwe, S.; Rohde, C.; Eickmann, M.; Volz, A.; et al. Safety and immunogenicity of a modified vaccinia virus Ankara vector vaccine candidate for Middle East respiratory syndrome: an open-label, phase 1 trial. Lancet Infect Dis 2020, 20, 827–838. [Google Scholar] [CrossRef]
- Song, F.; Fux, R.; Provacia, L.B.; Volz, A.; Eickmann, M.; Becker, S.; Osterhaus, A.D.; Haagmans, B.L.; Sutter, G. Middle East respiratory syndrome coronavirus spike protein delivered by modified vaccinia virus Ankara efficiently induces virus-neutralizing antibodies. J Virol 2013, 87, 11950–11954. [Google Scholar] [CrossRef]
- Volz, A.; Kupke, A.; Song, F.; Jany, S.; Fux, R.; Shams-Eldin, H.; Schmidt, J.; Becker, C.; Eickmann, M.; Becker, S.; et al. Protective Efficacy of Recombinant Modified Vaccinia Virus Ankara Delivering Middle East Respiratory Syndrome Coronavirus Spike Glycoprotein. J Virol 2015, 89, 8651–8656. [Google Scholar] [CrossRef]
- Garcia-Arriaza, J.; Garaigorta, U.; Perez, P.; Lazaro-Frias, A.; Zamora, C.; Gastaminza, P.; Del Fresno, C.; Casasnovas, J.M.; Sorzano, C.O.S.; Sancho, D.; et al. COVID-19 vaccine candidates based on modified vaccinia virus Ankara expressing the SARS-CoV-2 spike induce robust T- and B-cell immune responses and full efficacy in mice. J Virol 2021, 95. [Google Scholar] [CrossRef]
- Tscherne, A.; Schwarz, J.H.; Rohde, C.; Kupke, A.; Kalodimou, G.; Limpinsel, L.; Okba, N.M.A.; Bosnjak, B.; Sandrock, I.; Odak, I.; et al. Immunogenicity and efficacy of the COVID-19 candidate vector vaccine MVA-SARS-2-S in preclinical vaccination. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Lazaro-Frias, A.; Perez, P.; Zamora, C.; Sanchez-Cordon, P.J.; Guzman, M.; Luczkowiak, J.; Delgado, R.; Casasnovas, J.M.; Esteban, M.; Garcia-Arriaza, J. Full efficacy and long-term immunogenicity induced by the SARS-CoV-2 vaccine candidate MVA-CoV2-S in mice. NPJ Vaccines 2022, 7, 17. [Google Scholar] [CrossRef]
- Mooij, P.; Garcia-Arriaza, J.; Perez, P.; Lazaro-Frias, A.; Verstrepen, B.E.; Boszormenyi, K.P.; Mortier, D.; Fagrouch, Z.; Kiemenyi-Kayere, G.; Niphuis, H.; et al. Poxvirus MVA Expressing SARS-CoV-2 S Protein Induces Robust Immunity and Protects Rhesus Macaques From SARS-CoV-2. Front Immunol 2022, 13, 845887. [Google Scholar] [CrossRef]
- Perez, P.; Lazaro-Frias, A.; Zamora, C.; Sanchez-Cordon, P.J.; Astorgano, D.; Luczkowiak, J.; Delgado, R.; Casasnovas, J.M.; Esteban, M.; Garcia-Arriaza, J. A Single Dose of an MVA Vaccine Expressing a Prefusion-Stabilized SARS-CoV-2 Spike Protein Neutralizes Variants of Concern and Protects Mice From a Lethal SARS-CoV-2 Infection. Front Immunol 2021, 12, 824728. [Google Scholar] [CrossRef]
- Kulkarni, R.; Chen, W.C.; Lee, Y.; Kao, C.F.; Hu, S.L.; Ma, H.H.; Jan, J.T.; Liao, C.C.; Liang, J.J.; Ko, H.Y.; et al. Vaccinia virus-based vaccines confer protective immunity against SARS-CoV-2 virus in Syrian hamsters. PLoS One 2021, 16, e0257191. [Google Scholar] [CrossRef]
- Liu, R.; Americo, J.L.; Cotter, C.A.; Earl, P.L.; Erez, N.; Peng, C.; Moss, B. One or two injections of MVA-vectored vaccine shields hACE2 transgenic mice from SARS-CoV-2 upper and lower respiratory tract infection. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Bosnjak, B.; Odak, I.; Barros-Martins, J.; Sandrock, I.; Hammerschmidt, S.I.; Permanyer, M.; Patzer, G.E.; Greorgiev, H.; Gutierrez Jauregui, R.; Tscherne, A.; et al. Intranasal Delivery of MVA Vector Vaccine Induces Effective Pulmonary Immunity Against SARS-CoV-2 in Rodents. Front Immunol 2021, 12, 772240. [Google Scholar] [CrossRef]
- Meyer Zu Natrup, C.; Tscherne, A.; Dahlke, C.; Ciurkiewicz, M.; Shin, D.L.; Fathi, A.; Rohde, C.; Kalodimou, G.; Halwe, S.; Limpinsel, L.; et al. Stabilized recombinant SARS-CoV-2 spike antigen enhances vaccine immunogenicity and protective capacity. J Clin Invest 2022, 132. [Google Scholar] [CrossRef]
- Routhu, N.K.; Gangadhara, S.; Lai, L.; Davis-Gardner, M.E.; Floyd, K.; Shiferaw, A.; Bartsch, Y.C.; Fischinger, S.; Khoury, G.; Rahman, S.A.; et al. A modified vaccinia Ankara vaccine expressing spike and nucleocapsid protects rhesus macaques against SARS-CoV-2 Delta infection. Sci Immunol 2022, 7, eabo0226. [Google Scholar] [CrossRef]
- Americo, J.L.; Cotter, C.A.; Earl, P.L.; Liu, R.; Moss, B. Intranasal inoculation of an MVA-based vaccine induces IgA and protects the respiratory tract of hACE2 mice from SARS-CoV-2 infection. Proc Natl Acad Sci U S A 2022, 119, e2202069119. [Google Scholar] [CrossRef]
- Perez, P.; Astorgano, D.; Albericio, G.; Flores, S.; Sanchez-Cordon, P.J.; Luczkowiak, J.; Delgado, R.; Casasnovas, J.M.; Esteban, M.; Garcia-Arriaza, J. Intranasal administration of a single dose of MVA-based vaccine candidates against COVID-19 induced local and systemic immune responses and protects mice from a lethal SARS-CoV-2 infection. Front Immunol 2022, 13, 995235. [Google Scholar] [CrossRef]
- Villadiego, J.; Garcia-Arriaza, J.; Ramirez-Lorca, R.; Garcia-Swinburn, R.; Cabello-Rivera, D.; Rosales-Nieves, A.E.; Alvarez-Vergara, M.I.; Cala-Fernandez, F.; Garcia-Roldan, E.; Lopez-Ogayar, J.L.; et al. Full protection from SARS-CoV-2 brain infection and damage in susceptible transgenic mice conferred by MVA-CoV2-S vaccine candidate. Nat Neurosci 2023, 26, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Puig, J.M.; Blasco, R. Isolation of vaccinia MVA recombinants using the viral F13L gene as the selective marker. Biotechniques 2005, 39, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Chiem, K.; Park, J.G.; Oladunni, F.; Platt, R.N., 2nd; Anderson, T.; Almazan, F.; de la Torre, J.C.; Martinez-Sobrido, L. Rescue of SARS-CoV-2 from a Single Bacterial Artificial Chromosome. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Puig, J.M.; Lorenzo, M.M.; Blasco, R. Isolation of recombinant MVA using F13L selection. Methods Mol Biol 2012, 890, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Blasco, R.; Moss, B. Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene 1995, 158, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Earl, P.L.; Moss, B. Generation of Recombinant Vaccinia Viruses. Curr Protoc Mol Biol 2017, 117, 16–17. [Google Scholar] [CrossRef]
- Stadlbauer, D.; Amanat, F.; Chromikova, V.; Jiang, K.; Strohmeier, S.; Arunkumar, G.A.; Tan, J.; Bhavsar, D.; Capuano, C.; Kirkpatrick, E.; et al. SARS-CoV-2 Seroconversion in Humans: A Detailed Protocol for a Serological Assay, Antigen Production, and Test Setup. Curr Protoc Microbiol 2020, 57, e100. [Google Scholar] [CrossRef]
- Amanat, F.; Stadlbauer, D.; Strohmeier, S.; Nguyen, T.H.O.; Chromikova, V.; McMahon, M.; Jiang, K.; Arunkumar, G.A.; Jurczyszak, D.; Polanco, J.; et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat Med 2020, 26, 1033–1036. [Google Scholar] [CrossRef]
- Schmidt, F.; Weisblum, Y.; Muecksch, F.; Hoffmann, H.H.; Michailidis, E.; Lorenzi, J.C.C.; Mendoza, P.; Rutkowska, M.; Bednarski, E.; Gaebler, C.; et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J Exp Med 2020, 217. [Google Scholar] [CrossRef]
- Park, J.G.; Oladunni, F.S.; Rohaim, M.A.; Whittingham-Dowd, J.; Tollitt, J.; Hodges, M.D.J.; Fathallah, N.; Assas, M.B.; Alhazmi, W.; Almilaibary, A.; et al. Immunogenicity and protective efficacy of an intranasal live-attenuated vaccine against SARS-CoV-2. iScience 2021, 24, 102941. [Google Scholar] [CrossRef]
- Piepenbrink, M.S.; Park, J.G.; Oladunni, F.S.; Deshpande, A.; Basu, M.; Sarkar, S.; Loos, A.; Woo, J.; Lovalenti, P.; Sloan, D.; et al. Therapeutic activity of an inhaled potent SARS-CoV-2 neutralizing human monoclonal antibody in hamsters. Cell Rep Med 2021, 2, 100218. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc Natl Acad Sci U S A 2017, 114, E7348–E7357. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat Struct Mol Biol 2020, 27, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Prevost, J.; Ladinsky, M.S.; Stone, H.; Lu, M.; Anand, S.P.; Beaudoin-Bussieres, G.; Symmes, K.; Benlarbi, M.; Ding, S.; et al. Live imaging of SARS-CoV-2 infection in mice reveals that neutralizing antibodies require Fc function for optimal efficacy. Immunity 2021, 54, 2143–2158. [Google Scholar] [CrossRef]
- Howe, C.L.; LaFrance-Corey, R.G.; Goddery, E.N.; Johnson, R.K.; Mirchia, K. Neuronal CCL2 expression drives inflammatory monocyte infiltration into the brain during acute virus infection. J Neuroinflammation 2017, 14, 238. [Google Scholar] [CrossRef]
- Boudewijns, R.; Perez, P.; Lazaro-Frias, A.; Van Looveren, D.; Vercruysse, T.; Thibaut, H.J.; Weynand, B.; Coelmont, L.; Neyts, J.; Astorgano, D.; et al. MVA-CoV2-S Vaccine Candidate Neutralizes Distinct Variants of Concern and Protects Against SARS-CoV-2 Infection in Hamsters. Front Immunol 2022, 13, 845969. [Google Scholar] [CrossRef]
- Dangi, T.; Class, J.; Palacio, N.; Richner, J.M.; Penaloza MacMaster, P. Combining spike- and nucleocapsid-based vaccines improves distal control of SARS-CoV-2. Cell Rep 2021, 36, 109664. [Google Scholar] [CrossRef]
- Ku, M.W.; Authie, P.; Bourgine, M.; Anna, F.; Noirat, A.; Moncoq, F.; Vesin, B.; Nevo, F.; Lopez, J.; Souque, P.; et al. Brain cross-protection against SARS-CoV-2 variants by a lentiviral vaccine in new transgenic mice. EMBO Mol Med 2021, 13, e14459. [Google Scholar] [CrossRef]
Figure 1.
MVA recombinants expressing SARS-CoV-2 S protein. A) Schematic representation of the three expressed versions of the S protein. S corresponds to the original SARS-CoV-2 Wuhan-Hu-1 NC_045512.2 sequence. Sdg harbors a mutation for aminoacid change D614G in the S2 domain. Version Spf contains, in addition to the D614G mutations, four extra mutations (R682S, R685S, K986P and V987P) to prevent maturation and the conformational change of the protein. Domains of the S protein are indicated: NTD, N-terminal domain; RBD, receptor binding domain; F, fusion peptide; HH, heptad repeats; T, transmembrane domain; C, cytoplasmic tail. B) Western blot analysis of cells infected with recombinant MVAs. The unmodified MVA virus (MVA), and the recombinants MVA-S, MVA-Sdg and MVA-Spf were used to infect BHK-21 cells. Cell extracts were prepared at 24 hours post-infection and analyzed using antibodies directed to the RBD domain or to the S2 portion of the S protein. As an infection control, Vaccinia virus F13 protein was detected using a specific rat monoclonal antibody. C) and D) Subcellular location of S protein. BHK cells were infected for 18h and stained with specific antibodies under non-permeabilizing (C) or permeabilizing (D) conditions. A specific antibody against the RBD domain was used to label the extracellular domain of the S protein. An antibody against the B5 transmembrane protein of the vaccinia virus was used as control. Scale bar, 10μm.
Figure 1.
MVA recombinants expressing SARS-CoV-2 S protein. A) Schematic representation of the three expressed versions of the S protein. S corresponds to the original SARS-CoV-2 Wuhan-Hu-1 NC_045512.2 sequence. Sdg harbors a mutation for aminoacid change D614G in the S2 domain. Version Spf contains, in addition to the D614G mutations, four extra mutations (R682S, R685S, K986P and V987P) to prevent maturation and the conformational change of the protein. Domains of the S protein are indicated: NTD, N-terminal domain; RBD, receptor binding domain; F, fusion peptide; HH, heptad repeats; T, transmembrane domain; C, cytoplasmic tail. B) Western blot analysis of cells infected with recombinant MVAs. The unmodified MVA virus (MVA), and the recombinants MVA-S, MVA-Sdg and MVA-Spf were used to infect BHK-21 cells. Cell extracts were prepared at 24 hours post-infection and analyzed using antibodies directed to the RBD domain or to the S2 portion of the S protein. As an infection control, Vaccinia virus F13 protein was detected using a specific rat monoclonal antibody. C) and D) Subcellular location of S protein. BHK cells were infected for 18h and stained with specific antibodies under non-permeabilizing (C) or permeabilizing (D) conditions. A specific antibody against the RBD domain was used to label the extracellular domain of the S protein. An antibody against the B5 transmembrane protein of the vaccinia virus was used as control. Scale bar, 10μm.
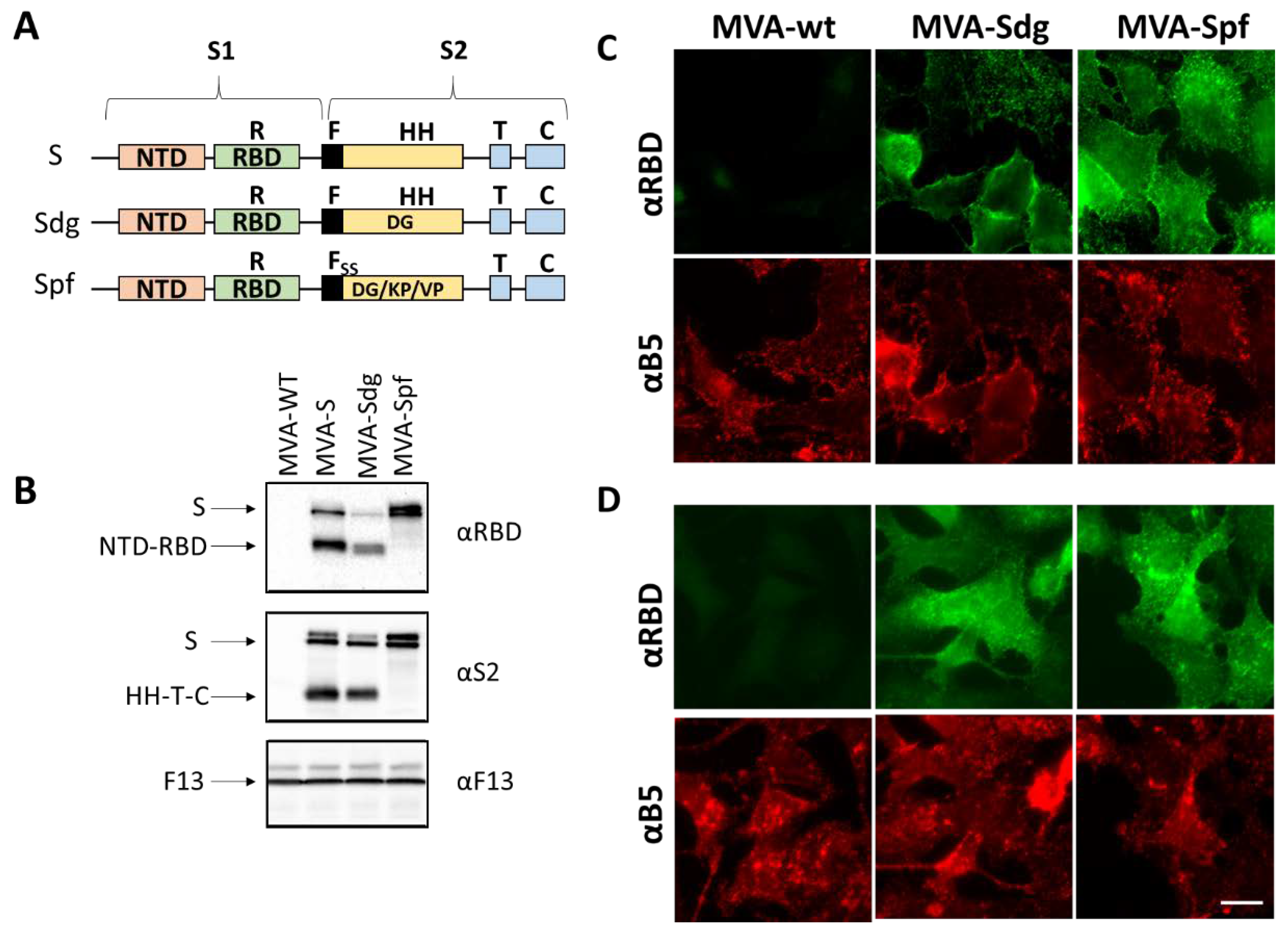
Figure 2.
Cell-cell fusion activity in BSC-1 cells infected with MVA virus recombinants. BSC-1 cells were infected with the indicated MVA recombinants at a MOI of 3 and incubated for 18h. Note the large syncytia (red arrow) caused by infection with MVA-Sdg and their absence in cells infected with MVA-Spf expressing the pre-fusion stabilized form of the S glycoprotein. Scale bar, 100 μm.
Figure 2.
Cell-cell fusion activity in BSC-1 cells infected with MVA virus recombinants. BSC-1 cells were infected with the indicated MVA recombinants at a MOI of 3 and incubated for 18h. Note the large syncytia (red arrow) caused by infection with MVA-Sdg and their absence in cells infected with MVA-Spf expressing the pre-fusion stabilized form of the S glycoprotein. Scale bar, 100 μm.
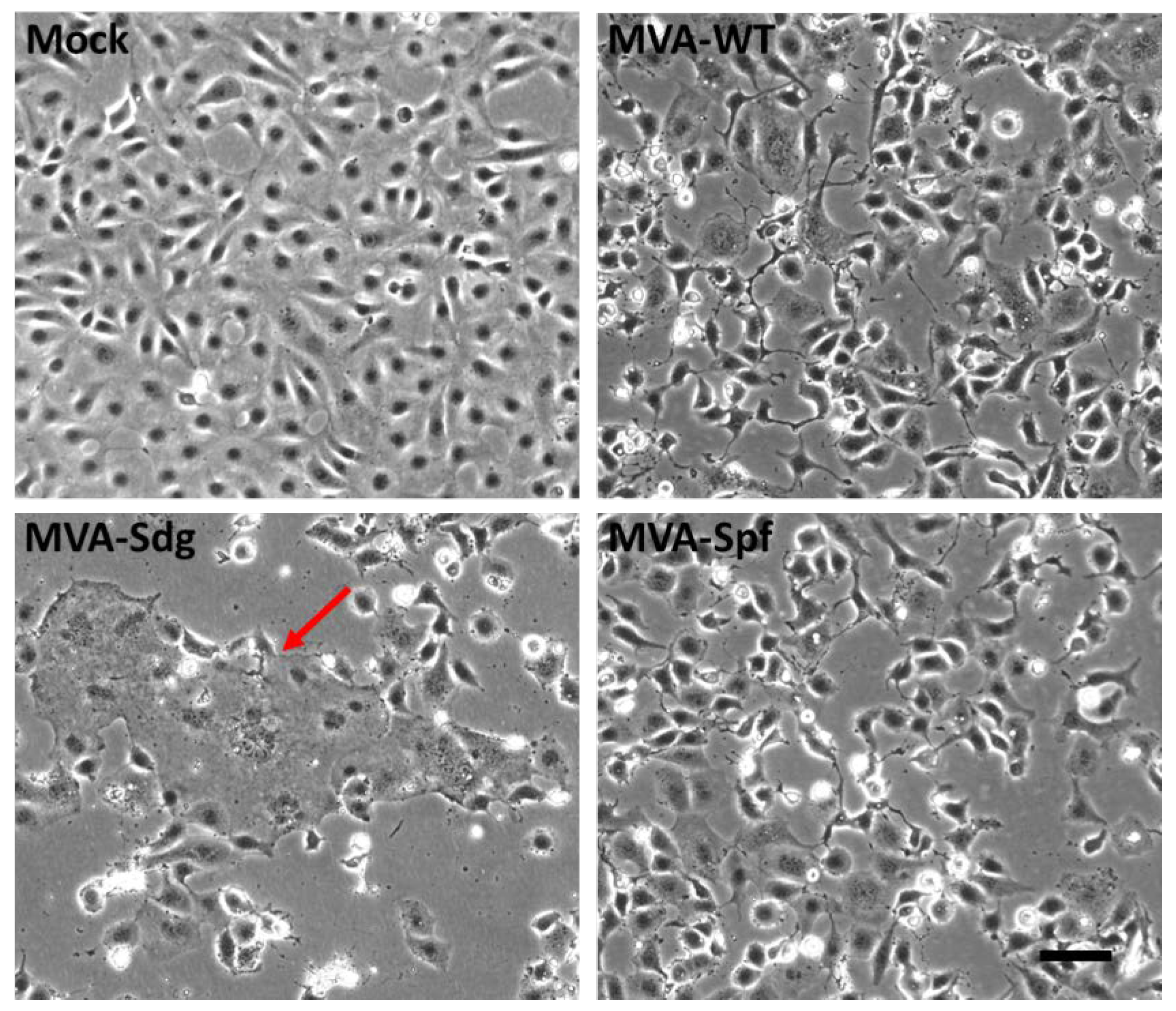
Figure 3.
Humoral immune responses in immunized BALB/c mice. A) immunization schedule. BALB/c mice were (B,C,D) intraperitoneal injected with two doses of 107 PFU of MVA-Sdg or MVA-Spf. A group immunized with MVA-wt was included as control. Mice were bled at the indicated time-points.. B) IgM titers obtained by ELISA to the S-RBD domain with the sera (dilution 1:200) of the vaccinated animals at 7 days post-inoculation. C) IgG levels specific to the RBD domain of the S glycoprotein at 14 days post-prime (d.p.p.) or boost (d.p.b) (sera dilution 1:200). Points represent individual values for each mouse, bars represent the mean values of each group and errors bars represent SD. Asterisks denote significant differences between groups (* P < 0.05; ** P < 0.033) (The Mann–Whitney U test). D) Serum neutralizing antibody titers 14 days after the second inoculation of the recombinant MVA. Points represent individual values for each mouse, bars represent the mean values of each group and errors bars represent SD. Asterisks denote significant differences between groups (** P < 0.033) (The Mann–Whitney U test). E, F) Comparison of the intramuscular (IM) and subcutaneous (SC) inoculation routes. BALB/c mice were intramuscular or subcutaneously immunized with two doses of 107 PFU of MVA-Spf or MVA-wt (control). Points represent individual values for each mouse, bars represent the mean values of each group and errors bars represent SD. Asterisks denote significant differences between groups (** P < 0.033) (The Mann–Whitney U test). E) Level of RBD-specific antibodies at 14 days post boost (d.p.b.) as determined by ELISA. Mouse sera were four-fold diluted starting from 1:50. Points represent mean values for each group and errors bars represent SD. Asterisks denote significant differences between group immunized by subcutaneous or intramuscular route (* P < 0.05). Both inoculation routes were significantly different from the control group (two-way ANOVA (post hoc Tukey test for multiple comparisons)). F) Level of neutralizing antibodies 14 d.p.b in PRNT assays using infectious SARS-CoV-2. Points represent individual values for each mouse, bars represent the mean values of each group and errors bars represent SD. Asterisks denote significant differences between groups (** P < 0.033) (two-way ANOVA (post hoc Tukey test for multiple comparisons)).
Figure 3.
Humoral immune responses in immunized BALB/c mice. A) immunization schedule. BALB/c mice were (B,C,D) intraperitoneal injected with two doses of 107 PFU of MVA-Sdg or MVA-Spf. A group immunized with MVA-wt was included as control. Mice were bled at the indicated time-points.. B) IgM titers obtained by ELISA to the S-RBD domain with the sera (dilution 1:200) of the vaccinated animals at 7 days post-inoculation. C) IgG levels specific to the RBD domain of the S glycoprotein at 14 days post-prime (d.p.p.) or boost (d.p.b) (sera dilution 1:200). Points represent individual values for each mouse, bars represent the mean values of each group and errors bars represent SD. Asterisks denote significant differences between groups (* P < 0.05; ** P < 0.033) (The Mann–Whitney U test). D) Serum neutralizing antibody titers 14 days after the second inoculation of the recombinant MVA. Points represent individual values for each mouse, bars represent the mean values of each group and errors bars represent SD. Asterisks denote significant differences between groups (** P < 0.033) (The Mann–Whitney U test). E, F) Comparison of the intramuscular (IM) and subcutaneous (SC) inoculation routes. BALB/c mice were intramuscular or subcutaneously immunized with two doses of 107 PFU of MVA-Spf or MVA-wt (control). Points represent individual values for each mouse, bars represent the mean values of each group and errors bars represent SD. Asterisks denote significant differences between groups (** P < 0.033) (The Mann–Whitney U test). E) Level of RBD-specific antibodies at 14 days post boost (d.p.b.) as determined by ELISA. Mouse sera were four-fold diluted starting from 1:50. Points represent mean values for each group and errors bars represent SD. Asterisks denote significant differences between group immunized by subcutaneous or intramuscular route (* P < 0.05). Both inoculation routes were significantly different from the control group (two-way ANOVA (post hoc Tukey test for multiple comparisons)). F) Level of neutralizing antibodies 14 d.p.b in PRNT assays using infectious SARS-CoV-2. Points represent individual values for each mouse, bars represent the mean values of each group and errors bars represent SD. Asterisks denote significant differences between groups (** P < 0.033) (two-way ANOVA (post hoc Tukey test for multiple comparisons)).
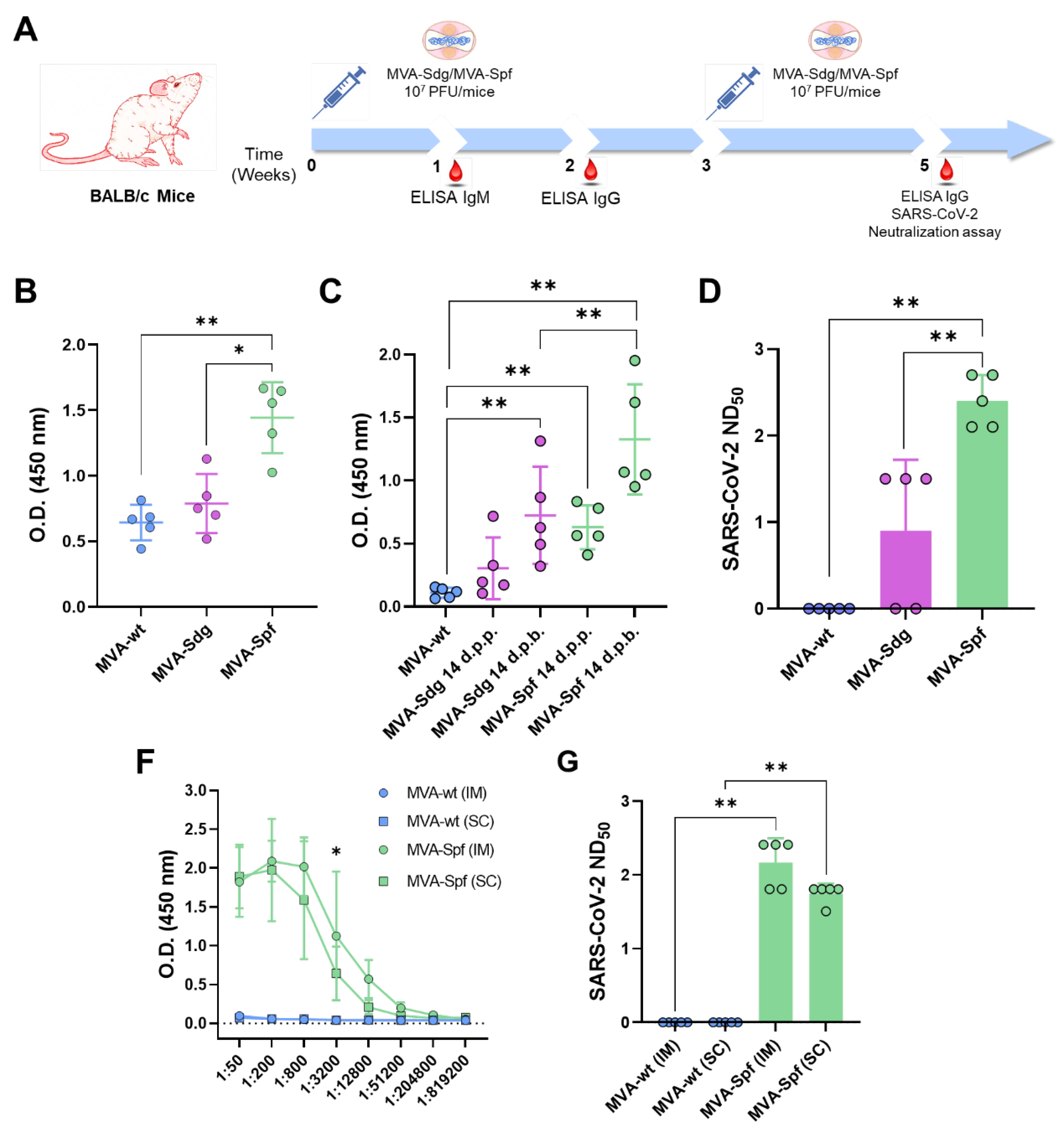
Figure 4.
Evaluation of immunogenicity of MVA-based vaccine candidates in B6 mice. A) Immunization schedule. Animals were immunized with two doses of 107 PFUs in a 3-week interval. Mice were bled 21 d.p.p. and 14 d.p.b to check antibody induction. Two weeks post-boost, animals were sacrificed and spleen cells isolated to analyze the cellular immune response. B) ) IgG levels specific to the RBD domain of the S glycoprotein at 14 d.p.b (sera dilution 1:200). Asterisks denote significant differences between groups (* P < 0.05) (The Mann–Whitney U test). C) Detection of SARS-CoV-2 neutralizing antibodies in mice sera at 21 d.p.p. and 15 d.p.b. Asterisks denote significant differences between groups by two-way ANOVA (post hoc Tukey test for multiple comparisons) (* P < 0.05). D) Cellular immune responses against the S protein of SARS-CoV-2 in immunized mice. Percentage of CD4+IFN-γ+ and CD8+IFN-γ+ T cells after stimulation with a panel of peptides spanning the S protein. Points represent individual values for each mouse, bars represent the mean values of each group and error bars represent SD. No significant differences between immunized and control mice were found (The Mann–Whitney U test).
Figure 4.
Evaluation of immunogenicity of MVA-based vaccine candidates in B6 mice. A) Immunization schedule. Animals were immunized with two doses of 107 PFUs in a 3-week interval. Mice were bled 21 d.p.p. and 14 d.p.b to check antibody induction. Two weeks post-boost, animals were sacrificed and spleen cells isolated to analyze the cellular immune response. B) ) IgG levels specific to the RBD domain of the S glycoprotein at 14 d.p.b (sera dilution 1:200). Asterisks denote significant differences between groups (* P < 0.05) (The Mann–Whitney U test). C) Detection of SARS-CoV-2 neutralizing antibodies in mice sera at 21 d.p.p. and 15 d.p.b. Asterisks denote significant differences between groups by two-way ANOVA (post hoc Tukey test for multiple comparisons) (* P < 0.05). D) Cellular immune responses against the S protein of SARS-CoV-2 in immunized mice. Percentage of CD4+IFN-γ+ and CD8+IFN-γ+ T cells after stimulation with a panel of peptides spanning the S protein. Points represent individual values for each mouse, bars represent the mean values of each group and error bars represent SD. No significant differences between immunized and control mice were found (The Mann–Whitney U test).
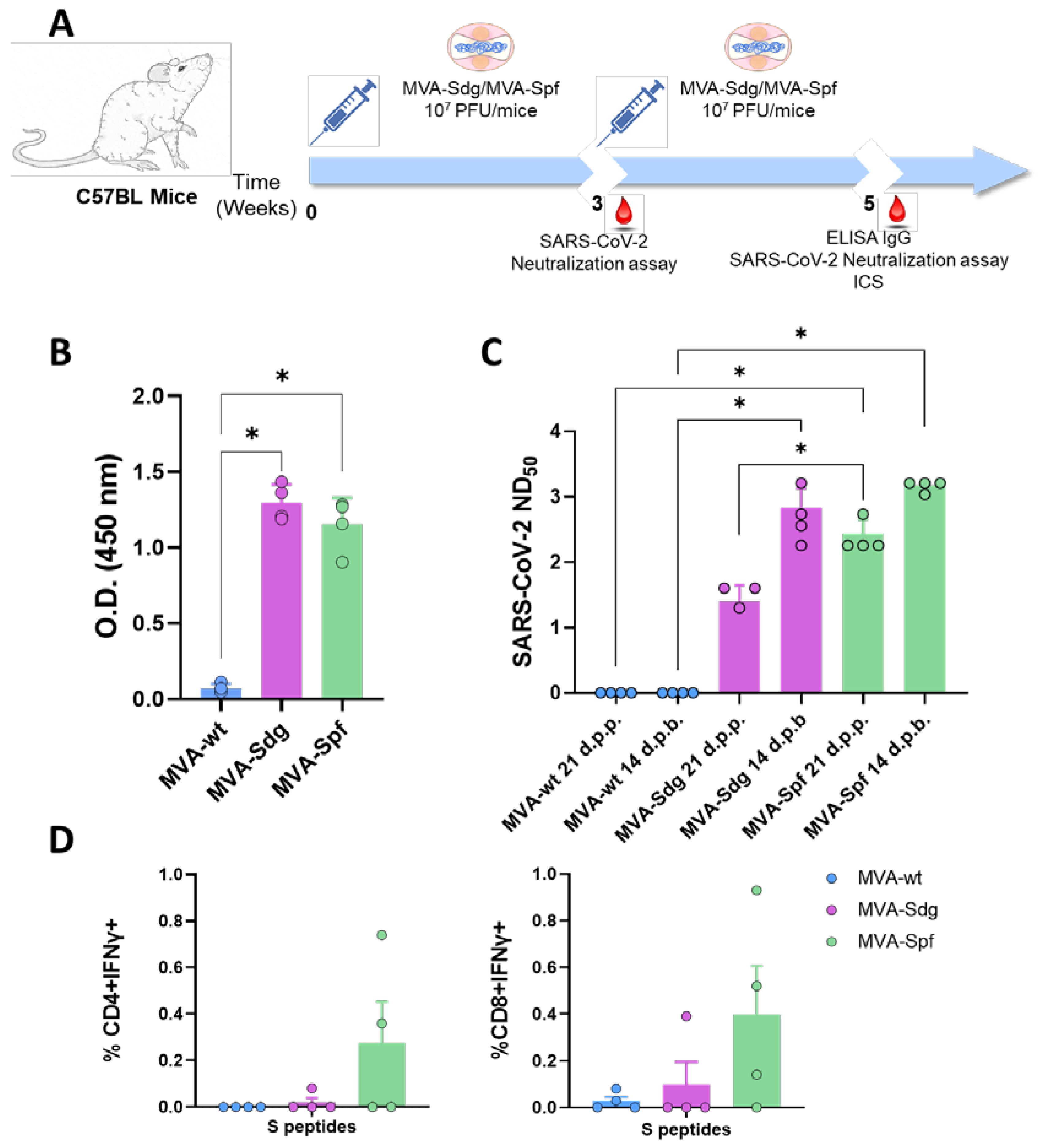
Figure 5.
Responses and protecion in K18-hACE2 mice immunized with the recombinant MVAs. A) Titration of anti-RBD antibodies prechallenge (left) and post challenge (right). Points represent mean values for each group and errors bars represent SD. Asterisks denote sig-nificant differences between group immunized with MVA-Sdg and MVA-Spf (* P < 0.05; ** P < 0.033; *** P < 0.0021). Both immunization groups were significantly different from the control group (two-way ANOVA (post hoc Tukey test for multiple comparisons). B) Neutralizing antibody titers in sera at 14 days post infection. Points represent individual values for each mouse, bars represent the mean values of each group and errors bars represent SD. Asterisks denote significant differences between groups by two-way ANOVA (post hoc Tukey test for multiple comparisons) (** P < 0.033; *** P < 0.0021; **** P < 0.0002). C) Viral RNA levels detected by quantitative RT-PCR in brain and lung. Points represent mean values for each group and errors bars represent SD. Asterisks denote significant differences between groups (* P < 0.05) (The Mann–Whitney U test). D) Viral infectious particlesin lungs and brain tissues after challenge. Points represent mean values for each group and errors bars represent SD. Asterisks denote significant differences between groups (* P < 0.05) (The Mann–Whitney U test). E) Survival after challenge with a lethal dose of SARS-CoV-2. Curves were found statistically significant compared with nonimmunized survival curve as calculated by Log-rank test (P value < 0.05). F) Percentage of body weight loss caused by SARS-CoV- 2 infection as average within groups (left) or individual animals (right).
Figure 5.
Responses and protecion in K18-hACE2 mice immunized with the recombinant MVAs. A) Titration of anti-RBD antibodies prechallenge (left) and post challenge (right). Points represent mean values for each group and errors bars represent SD. Asterisks denote sig-nificant differences between group immunized with MVA-Sdg and MVA-Spf (* P < 0.05; ** P < 0.033; *** P < 0.0021). Both immunization groups were significantly different from the control group (two-way ANOVA (post hoc Tukey test for multiple comparisons). B) Neutralizing antibody titers in sera at 14 days post infection. Points represent individual values for each mouse, bars represent the mean values of each group and errors bars represent SD. Asterisks denote significant differences between groups by two-way ANOVA (post hoc Tukey test for multiple comparisons) (** P < 0.033; *** P < 0.0021; **** P < 0.0002). C) Viral RNA levels detected by quantitative RT-PCR in brain and lung. Points represent mean values for each group and errors bars represent SD. Asterisks denote significant differences between groups (* P < 0.05) (The Mann–Whitney U test). D) Viral infectious particlesin lungs and brain tissues after challenge. Points represent mean values for each group and errors bars represent SD. Asterisks denote significant differences between groups (* P < 0.05) (The Mann–Whitney U test). E) Survival after challenge with a lethal dose of SARS-CoV-2. Curves were found statistically significant compared with nonimmunized survival curve as calculated by Log-rank test (P value < 0.05). F) Percentage of body weight loss caused by SARS-CoV- 2 infection as average within groups (left) or individual animals (right).
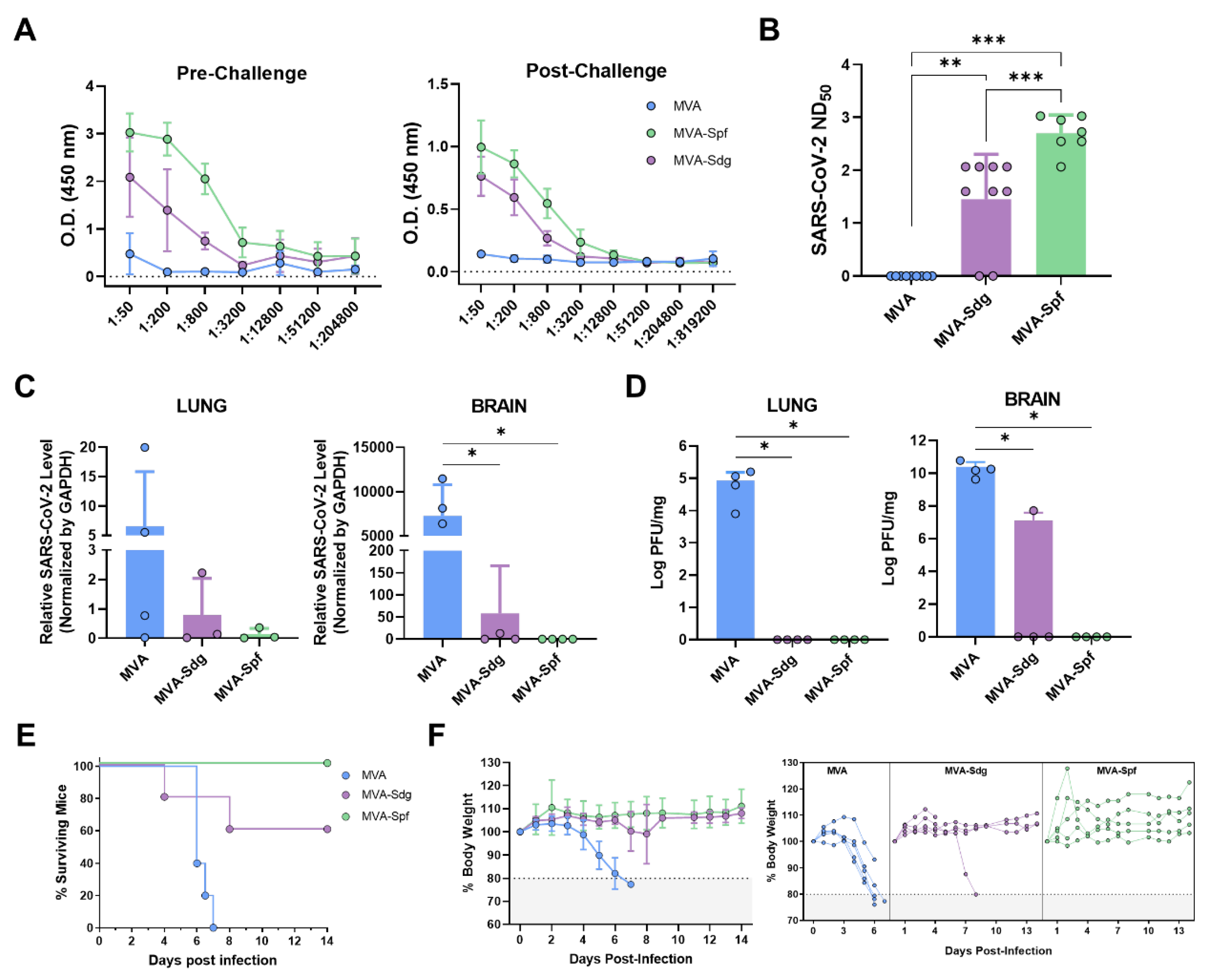
Figure 6.
Replication and spread of SARS-CoV-2-WA-nLuc in vaccinated K18-hACE2 mice. A) Experimental design to evaluate the in vivo efficacy of MVA vaccine. Animals were infected intranasally and infection monitored using bioluminescence imaging. B) Representative BLI images of SARS-CoV-2 WA-nLuc infected mice in ventral (v) and dorsal (d) position. Scale bars denote flux (photons/s) in log scale. C-D) Temporal quantification of nLuc signals as flux (photons/s) computed non-invasively. E-F) Ex vivo imaging of indicated organs and quantification of nLuc signal as flux(photons/s) in log scale after necropsy. Each curve in C-D and each data point in F represents an individual mouse. Grouped data in C-D and F were analyzed by 2-way ANOVA followed by Sidak’s multiple comparison tests. ∗, p < 0.05; ∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; Mean values ± SD are depicted.
Figure 6.
Replication and spread of SARS-CoV-2-WA-nLuc in vaccinated K18-hACE2 mice. A) Experimental design to evaluate the in vivo efficacy of MVA vaccine. Animals were infected intranasally and infection monitored using bioluminescence imaging. B) Representative BLI images of SARS-CoV-2 WA-nLuc infected mice in ventral (v) and dorsal (d) position. Scale bars denote flux (photons/s) in log scale. C-D) Temporal quantification of nLuc signals as flux (photons/s) computed non-invasively. E-F) Ex vivo imaging of indicated organs and quantification of nLuc signal as flux(photons/s) in log scale after necropsy. Each curve in C-D and each data point in F represents an individual mouse. Grouped data in C-D and F were analyzed by 2-way ANOVA followed by Sidak’s multiple comparison tests. ∗, p < 0.05; ∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; Mean values ± SD are depicted.
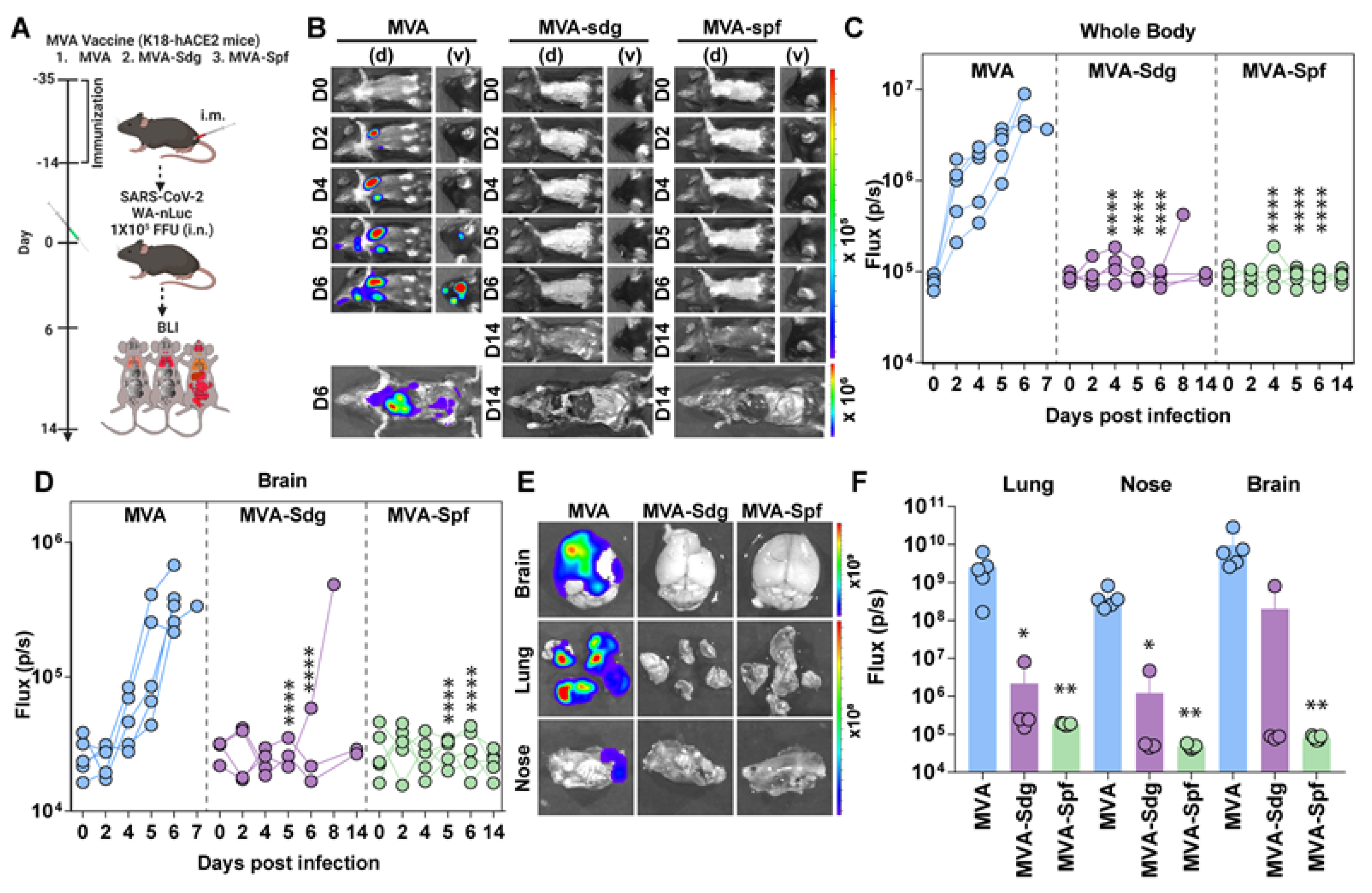
Figure 7.
Relative expression of cytokines and chemokines in lung and brain at 6 d.p.i. measured by qRT-PCR. Blue bars, MVA; purple bars, MVA-Sdg; green bars, MVA-Spf. Mouse GAPDH was used as a housekeeping gene.
Figure 7.
Relative expression of cytokines and chemokines in lung and brain at 6 d.p.i. measured by qRT-PCR. Blue bars, MVA; purple bars, MVA-Sdg; green bars, MVA-Spf. Mouse GAPDH was used as a housekeeping gene.
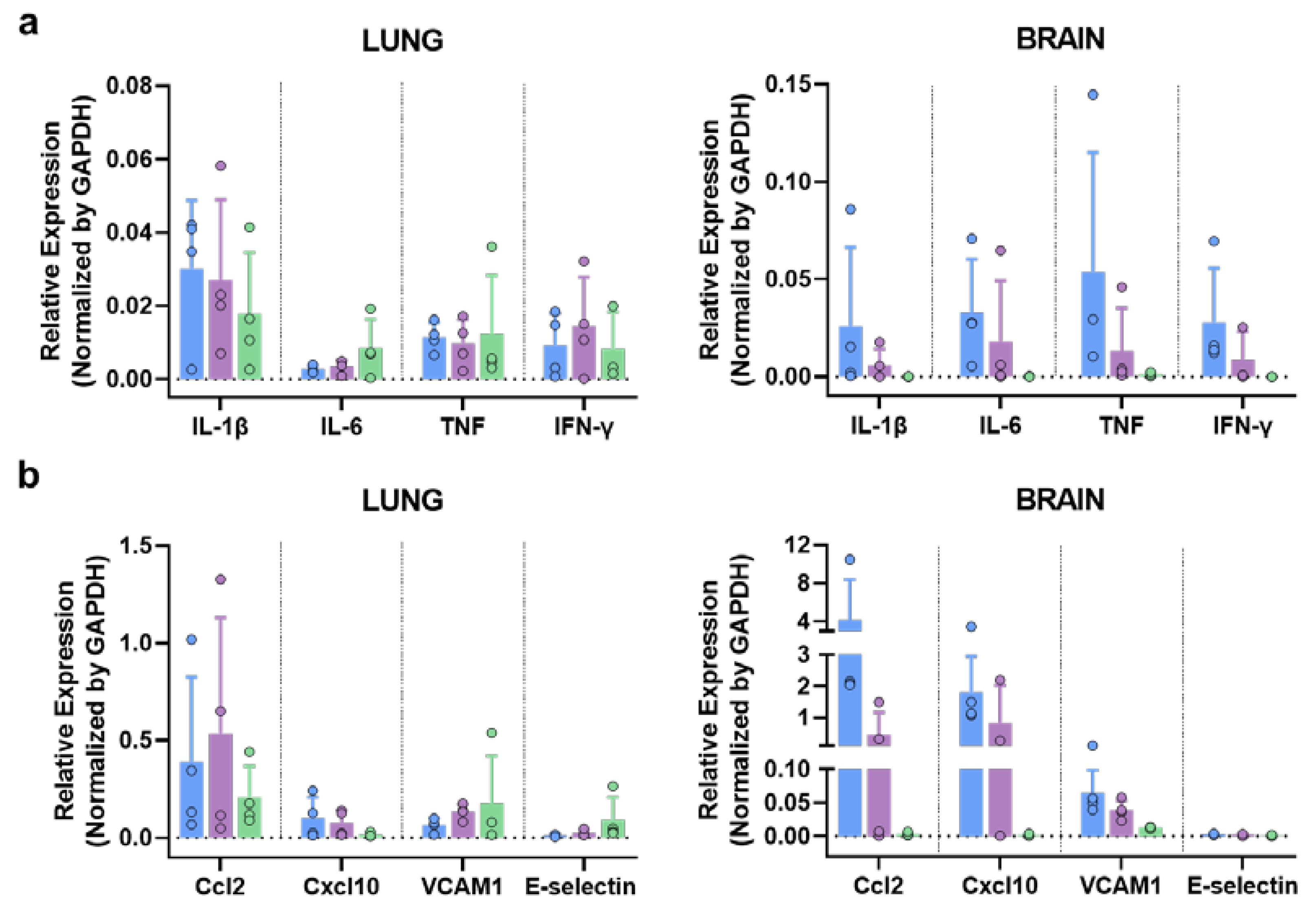
Figure 8.
SARS-CoV-2 neutralizing antibodies in sera of vaccinated golden Syrian hamsters. A) Timeline of the experiment (days). B) Neutralization titers in sera from hamsters. Neutralization titers were obtained using a VSV pseudotyped-based assay (left) or infectious SARS-CoV-2 (right). Titers in the left panel correspond to day 21 (one inoculation) and 35 (two inoculations) as indicated in the horizontal axis, and those in the right panel correspond to sera from day 35, immediately before challenge.
Figure 8.
SARS-CoV-2 neutralizing antibodies in sera of vaccinated golden Syrian hamsters. A) Timeline of the experiment (days). B) Neutralization titers in sera from hamsters. Neutralization titers were obtained using a VSV pseudotyped-based assay (left) or infectious SARS-CoV-2 (right). Titers in the left panel correspond to day 21 (one inoculation) and 35 (two inoculations) as indicated in the horizontal axis, and those in the right panel correspond to sera from day 35, immediately before challenge.
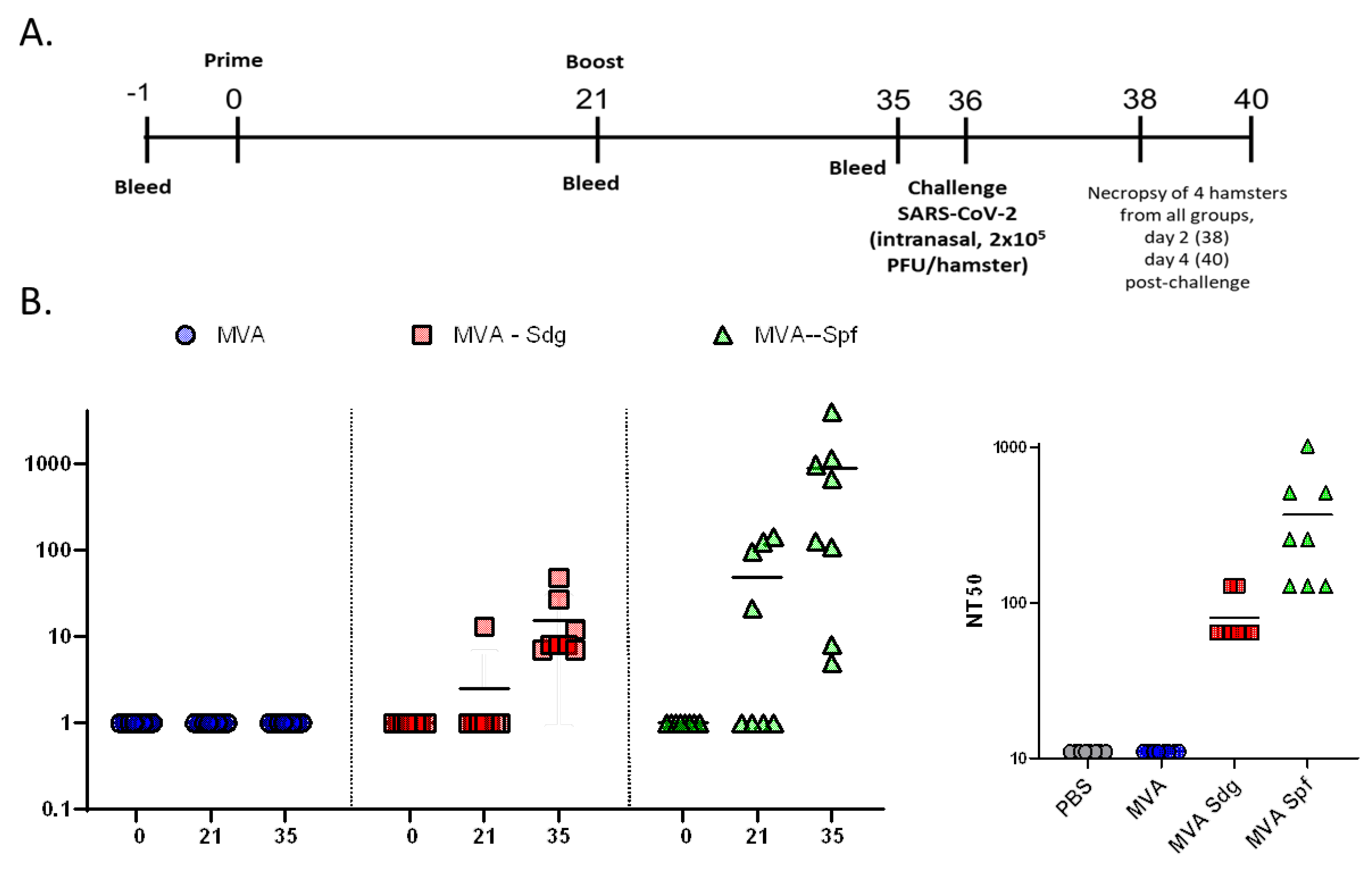
Figure 9.
Protection from lung pathology in the hamster model. Images of the lungs from hamsters at days 2 and 4 after challenge with SARS-CoV-2. Note the complete absence of red lesions in animals vaccinated with MVA-Spf. Lower panel shows the gross lung pathology score of the animals in each of the experimental groups.
Figure 9.
Protection from lung pathology in the hamster model. Images of the lungs from hamsters at days 2 and 4 after challenge with SARS-CoV-2. Note the complete absence of red lesions in animals vaccinated with MVA-Spf. Lower panel shows the gross lung pathology score of the animals in each of the experimental groups.
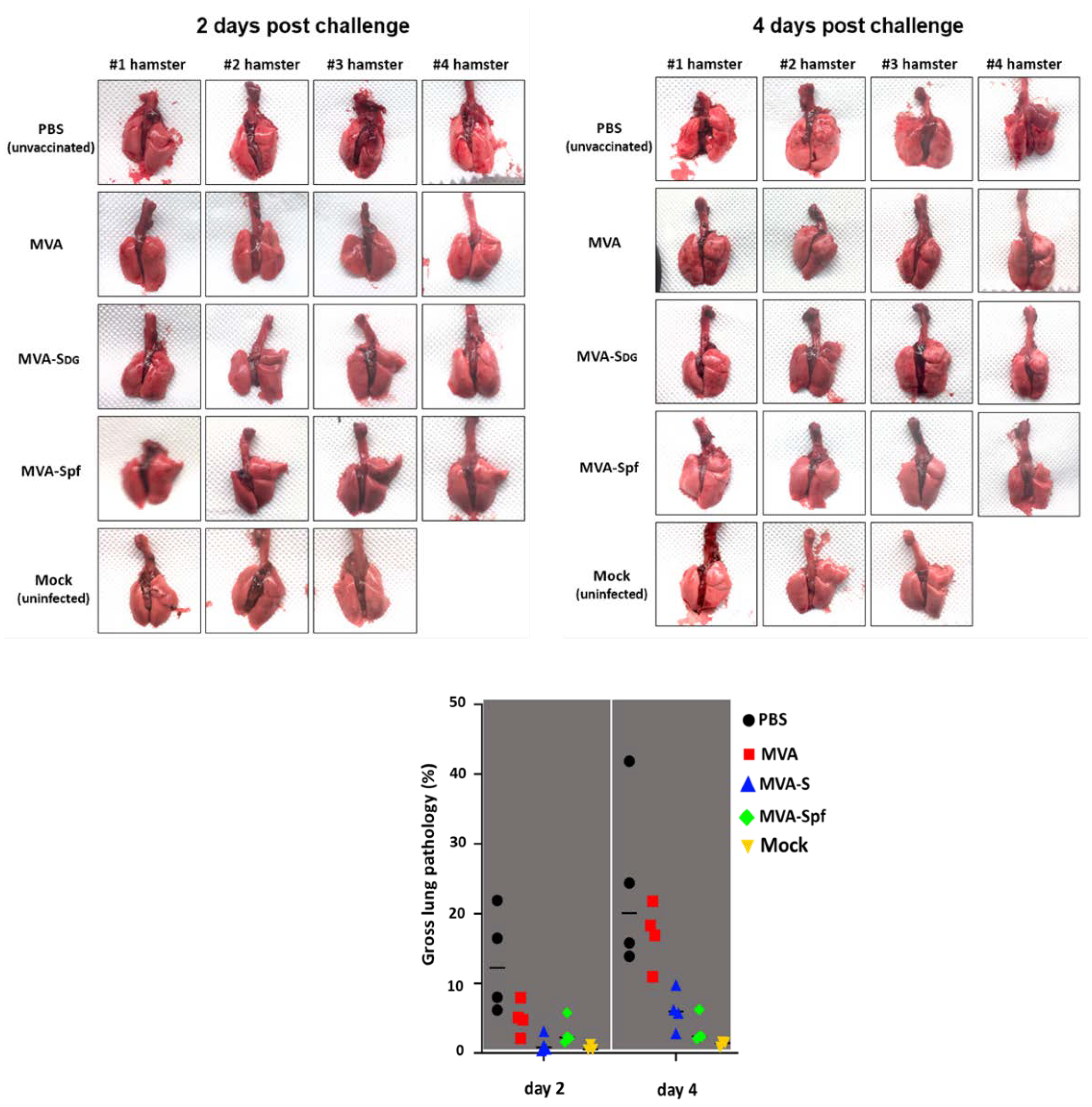
Figure 10.
Protection in the hamster infection model. SARS-CoV-2 viral titers at 2 and 4 days after challenge were determined by plaque assay. Dotted line indicates the detection limit in our assay. Values below the limit are plotted as 0. .
Figure 10.
Protection in the hamster infection model. SARS-CoV-2 viral titers at 2 and 4 days after challenge were determined by plaque assay. Dotted line indicates the detection limit in our assay. Values below the limit are plotted as 0. .
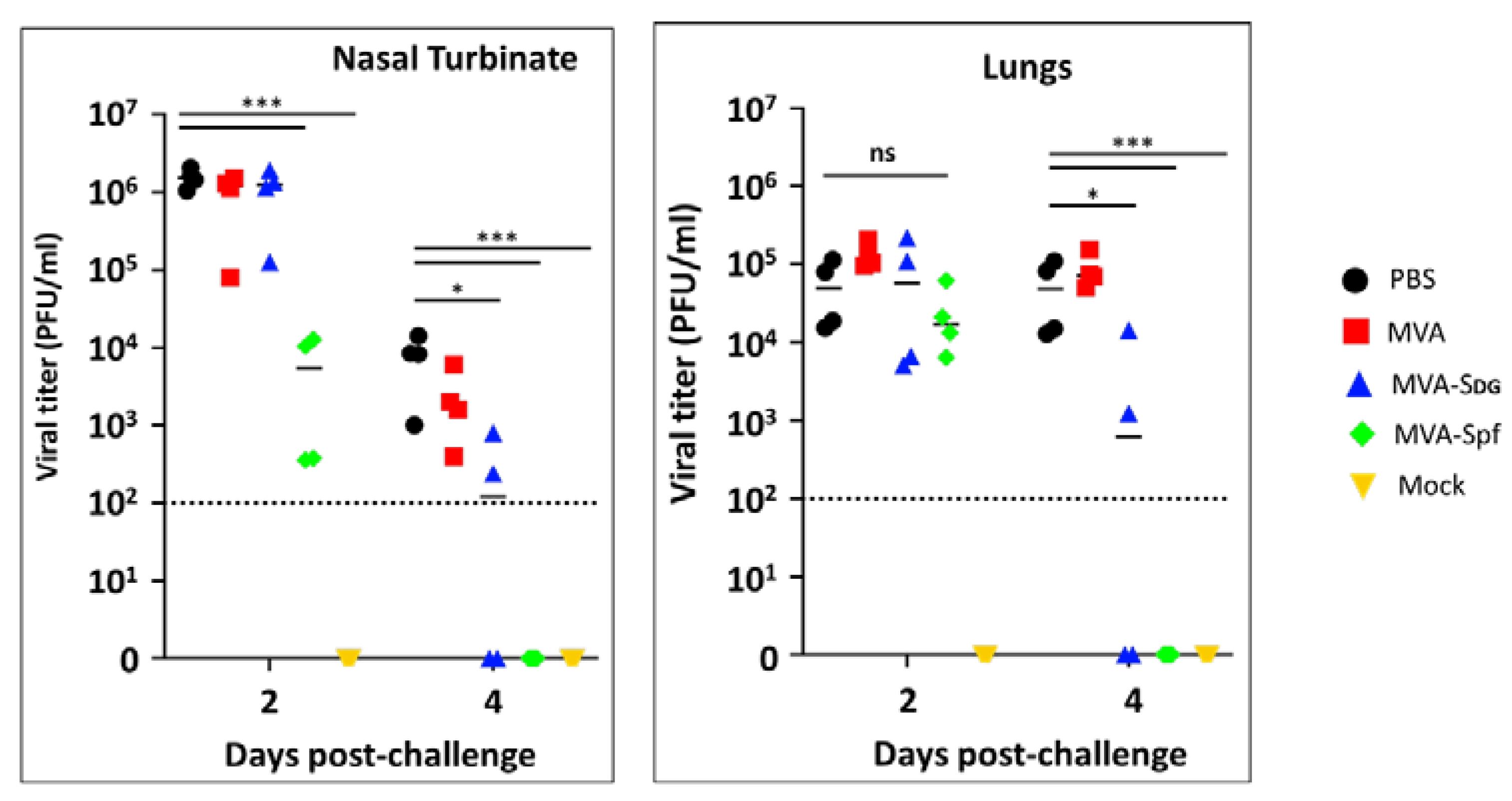
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



