
Submitted:
04 May 2023
Posted:
05 May 2023
You are already at the latest version
Abstract
Fibroblast activation protein (FAP) is a membrane-tethered serine protease overexpressed in the reactive stromal fibroblasts of > 90% human carcinomas, which makes it a promising target for developing radiopharmaceuticals for imaging and therapy of carcinomas. Here, we synthesized two novel (R)-pyrrolidin-2-yl-boronic acid-based FAP-targeted ligands; SB02055 (DOTA-conjugated (R)-(1-((6-(3-(piperazin-1-yl)propoxy)quinoline-4-carbonyl)glycyl)pyrrolidin-2-yl)boronic acid) and SB04028 (DOTA-conjugated ((R)-1-((6-(3-(piperazin-1-yl)propoxy)quinoline-4-carbonyl)-D-alanyl)pyrrolidin-2-yl)boronic acid). natGa- and 68Ga-complexes of both ligands were evaluated in preclinical studies and compared to previously reported natGa/68Ga-complexed PNT6555. Enzymatic assays showed that FAP binding affinities (IC50) of natGa-SB02055, natGa-SB04028 and natGa-PNT6555 were 0.41±0.06, 13.9±1.29 and 78.1±4.59 nM, respectively. PET imaging and biodistribution studies in HEK293T:hFAP tumor-bearing mice showed that while [68Ga]Ga-SB02055 presented with a nominal tumor uptake (1.08±0.37 %ID/g), [68Ga]Ga-SB04028 demonstrated clear tumor visualization with ~1.5-fold higher tumor uptake (10.1±0.42 %ID/g) compared to [68Ga]Ga-PNT6555 (6.38±0.45 %ID/g). High accumulation in the bladder indicated renal excretion of all three tracers. [68Ga]Ga-SB04028 displayed low background level uptake in most normal organs, and comparable to [68Ga]Ga-PNT6555. However, since its tumor uptake was considerably higher than [68Ga]Ga-PNT6555, the corresponding tumor-to-organ uptake ratios for [68Ga]Ga-SB04028 were also significantly greater than [68Ga]Ga-PNT6555. Our data demonstrate that (R)-(((quinoline-4-carbonyl)-D-alanyl)pyrrolidin-2-yl)boronic acid is a promising pharmacophore for the design of FAP-targeted radiopharmaceuticals for cancer imaging and radioligand therapy.
Keywords:
fibroblast activation protein α (FAP-α)
; cancer-associated fibroblasts (CAFs)
; FAP inhibitors (FAPIs)
; PET imaging
; gallium-68
; (R)-pyrrolidin-2-yl-boronic acid-based radiopharmaceuticals
1. Introduction
Cancer associated fibroblasts (CAFs) found in the tumor microenvironment are functionally and phenotypically distinct from normal fibroblasts found in non-cancerous tissues [1]. Immuno-histochemical studies performed by Chesa et al. revealed that CAFs in several different primary and metastatic carcinomas, including colorectal, breast, ovarian, bladder, and lung carcinomas, overexpress a transmembrane glycoprotein called fibroblast activation protein (FAP) [2]. FAP is a cell surface serine hydrolase shown to have dipeptidyl exopeptidase and endopeptidase activities [3]. FAP has a restricted normal tissue distribution including normal fibroblasts, but is overexpressed in the reactive stromal fibroblasts of > 90% of human epithelial carcinomas (breast, lung, colorectal etc.) [2,3,4,5]. Moreover, its enzymatic activity is considered crucial for the promotion of tumor growth [6]. FAP’s constrained expression combined with its role in invasion and tumor metastasis, makes it a promising target for developing FAP-targeted radiopharmaceuticals for imaging and therapy of carcinomas. In this regard, several different strategies including use of antibodies [7,8], CAR T-cells [9,10] and FAP vaccines [11] and most significantly small-molecule inhibitors [6,12,13,14,15], have been explored for FAP-targeting.
The two most noteworthy groups exploited for the development of small-molecule based FAP inhibitors are either 2-cyanopyrrolidine derivatives or 2-pyrrolidinylboronic acid derivatives (boroPro). Additionally, most of the research so far has been directed towards identifying inhibitors that are selective for FAP over dipeptidyl peptidases (DPPs), with which it shares exopeptidase specificity, and over cytosolic prolyl endopeptidase (PREP), with which it shares endopeptidase specificity [16].
Interestingly, FAP has been reported to exhibit an absolute requirement for glycine at P2 position as an endopeptidase [17,18]. This may explain why most small-molecule FAP inhibitors are based upon an X-Gly-Pro sequence. Jansen et al. presented N-(4-quinolinoyl)-Gly-(2-cyanopyrrolidine) as an extremely potent FAP inhibitor (IC50 = 10.3±0.4 nM), with high selectivity indices (SI) over both DPPs (> 103) and PREP (> 83-fold) [19]. Remarkably, substitution of the nitrile on the P1 pyrrolidine ring with a more electrophilic boronic acid warhead yielded N-(4-quinolinoyl)-Gly-boroPro that demonstrated 2.8-fold higher FAP binding affinity (IC50 = 3.7±0.2 nM) than the former. Moreover, it retained selectivity over DPPs (> 103) with a ~ 3-fold selectivity over PREP [14].
Contrary to the notion that FAP exhibits an absolute requirement for glycine at the P2 position, two different groups identified small-molecule FAP inhibitors bearing a P2 D-alanine. Tran et al. were the first to show FAP’s tolerability for a P2 D-alanine [20]. They reported N-acetyl-D-Ala-boroPro to exhibit a modest potency with a Ki(FAP) of 350 nM, compared to N-acetyl-Gly-boroPro that possessed a Ki(FAP) of 23 nM. In the study published by Bachovchin and co-workers, N-acetyl-D-Ala-boroPro was also shown to have a modest FAP binding potency (IC50 = 2900±600 nM) [21]. Importantly, in the same study, N-(pyridine-4-carbonyl)-D-Ala-boroPro (ARI-3099) was identified as a highly potent FAP inhibitor with an IC50 of 36±4.8 nM. Its P2 glycine congener was reported to have ~75-fold higher FAP binding affinity. Notably, compared to N-(benzoyl)-D-Ala-boroPro (IC50(FAP) = 54±2.9 nM), N-(4-quinolinoyl)-D-Ala-boroPro (IC50(FAP) = 6.4±1.4 nM) performed superiorly with ~8-fold higher FAP binding affinity. The FAP/PREP selectivity values were 160 and 33 for the quinoline- and benzoyl-based inhibitors, respectively, indicating that the quinoline-based inhibitors are more selective for binding to FAP. Nevertheless, the findings from the two studies validated FAP’s ability to tolerate a P2 D-alanine in the context of an X-boroPro-based inhibitor.
The radiolabeled small-molecule FAP inhibitor called [125I]MIP-1232 (Figure 1) was reported in 2015 and was based on (R)-(1-(benzoylglycyl)pyrrolidin-2-yl)boronic acid framework [22]. Although the probe did not serve its primary purpose of atherosclerosis imaging, the authors presented its relevance as a radiotracer for imaging FAP-positive tumor tissues. [125I]MIP-1232 was reported to strongly accumulate in FAP-positive SK-Mel-187 melanoma xenograft in vitro.
Recently, a new SPECT tracer [99mTc]Tc-HYNIC-D-Ala-boroPro ([99mTc]Tc-iFAP, Figure 1) targeting FAP was reported to exhibit 7.05±1.13 %ID/g tumor uptake and 11.18±1.54 %ID/g kidney uptake in an induced Hep-G2 tumor model in mice at 30 min post injection (pi) [23]. The tumor and kidney uptake values reportedly dropped by ~ 27% (5.18±0.82 %ID/g) and ~ 33% (7.46±1.02 %ID/g), respectively, at 2 h pi with minimal uptake in most normal organs. In its clinical study, [99mTc]Tc-iFAP was seen to accumulate in the primary tumors and lymph node metastases of patients with different cancers [24]. The mean radiation equivalent dose for the kidney (5.2±0.8 mSv) was stated to be more than twice the effective dose (2.3±0.4 mSv) after administrating 740 MBq of the tracer. The authors concluded the study by declaring the need for additional clinical studies to further validate the performance of [99mTc]Tc-iFAP.
The development of targeted radiotracers by Haberkorn group is one of the most significant and clinically relevant applications of FAP inhibitors to date. By expanding on the framework previously outlined by Jansen et al. they generated a panel of specific and high affinity FAP inhibitors (FAPIs), undergoing almost complete internalization of the ligand-enzyme complex [25,26,27]. These derivatives were synthesized using different chemical modifications to attach various chelators via a linker to the quinolinoyl moiety in N-(4-quinolinoyl)-Gly-(2-cyanopyrrolidine). Of these, 68Ga-labeled FAPI-02 [28], FAPI-04 (Figure 1) and FAPI-46 [29], 99mTc-labeled FAPI-34 [30], and 18F-labeled FAPI-42 [31] and FAPI-74 [32] are the most advanced and validated in clinical studies for cancer imaging.
Most recently, [68Ga]Ga/[177Lu]Lu/[225Ac]Ac-PNT6555 were presented as highly potent FAP-targeted theranostic pairs in two different conference abstracts [33,34]. PNT6555 (Figure 1) is a DOTA-conjugated FAP-targeted ligand based on a (R)-(1-((4-(aminomethyl)benzoyl)-D-alanyl)pyrrolidin-2-yl)boronic acid scaffold. [177Lu]Lu-PNT6555 was reported to show little retention in normal tissues but a high level of tumor retention (>10 %ID/g) was observed up to 168 h pi. A single dose of [177Lu]Lu/[225Ac]Ac-PNT6555 was reported to exhibit a dose-dependent anti-tumor efficacy in a HEK-mFAP murine tumor model, and a phase I clinical trial of [177Lu]Lu-PNT6555 is currently underway [35]. No peer-reviewed publications on imaging or ex-vivo biodistribution analysis of [68Ga]Ga-, [177Lu]Lu- and/or [225Ac]Ac-PNT6555 are currently available.
There are varying preclinical and clinical reports on the association between uptake intensity of radiolabeled FAPIs and FAP expression. For example, while [68Ga]Ga-FAPI-04 is observed to have high uptake in the bone and muscle in mice, the former is not observed in humans [26,36,37,38,39,40]. However, non-tumor-specific uptake of [68Ga]Ga-FAPI-04 and [68Ga]Ga-FAPI-46 was recently reported in muscles and degenerative lesions mostly associated with joints and vertebral bones [39]. Although having additional clinical studies would help ascertain these ambiguous associations, efforts towards development of FAP-targeted inhibitors with improved pharmacokinetic properties and enhanced FAP specificity compared to the already existing FAPIs is warranted.
Compared to nitrile, the boronic acid warhead has been reported to have a higher FAP binding affinity [14,19], possibly due to its stronger electrophilic character, and could have a faster pharmacokinetics in vivo due to its highly hydrophilic nature. Furthermore, previous findings confirm that FAP can tolerate a P2 D-alanine apart from a P2 glycine [20,21]. Lastly, a P3 quinolinoyl moiety has been previously optimized for developing radioligands for FAP targeting by Haberkorn et al. [25,27]. With this aim in mind, we designed two novel boronic acid-based DOTA-conjugated FAP-targeted ligands; [68Ga]Ga-SB02055 and [68Ga]Ga-SB04028 (Figure 1), bearing a N-(4-quinolinoyl)-Gly-boroPro and N-(4-quinolinoyl)-D-Ala-boroPro pharmacophores, respectively. Addition of chelator DOTA via a piperazine-based linker (Figure 1) allowed formation of stable complexes with the PET isotope 68Ga. We posited that these novel boronic acid-based tracers may exhibit higher tumor uptake by virtue of their more potent FAP inhibition and demonstrate superior pharmacokinetics by undergoing quick clearance from all the non-target organs/tissues and serve as PET imaging agents with excellent imaging contrast. Both tracers were subjected to a thorough preclinical evaluation including in vitro FAP binding assays, in vivo stability testing and, PET/CT imaging and ex vivo biodistribution studies using HEK293T:hFAP tumor xenograft mouse model. The results were then compared with those obtained from [68Ga]Ga-PNT6555 which is currently been evaluated in the clinic [35].
2. Results
2.1. Synthesis of 68Ga- and natGa-Complexed DOTA-Conjugated FAP-Targeted Ligands
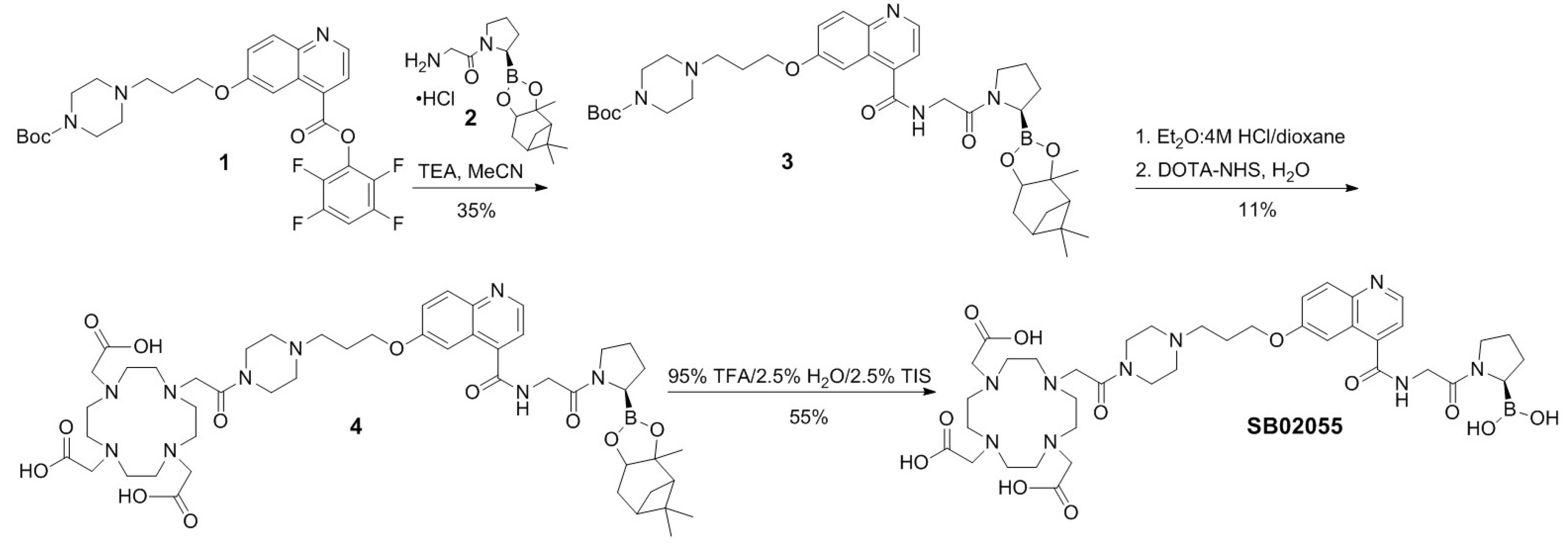
Detailed information on the synthesis, purification, and characterizations of all intermediates, precursors and natGa/68Ga-complexed ligands is provided in Supplementary Materials. For the synthesis of SB02055 (Scheme 1), compound 1 [41] was coupled overnight with compound 2 [42,20] to obtain compound 3 in a 35% yield. Compound 3 was Boc-deprotected by stirring it overnight with diethyl ether (Et₂O):4M HCl/dioxane (v/v) in a 1:1 ratio. The mixture was then evaporated before reacting it with DOTA-NHS to afford compound 4 as a pinanediol ester of precursor SB02055 in 11% yield over 2 steps. Compound 4 was treated with a cleavage cocktail consisting of 95% trifluoroacetic acid (TFA)/2.5% H2O/2.5% triisopropylsilane (TIS) for 4 h at room temperature (RT) to finally afford the desired precursor SB02055 in 55% yield.
For the synthesis of SB04028 (Scheme 2), compound 5 prepared following literature procedures [23] was coupled with compound 1 overnight to afford compound 6 in a 47% yield. Compound 6 was then treated overnight with Et₂O:4M HCl/dioxane (v/v) in a 1:1 ratio for removing the Boc-group. The resulting mixture was evaporated before reacting it with DOTA-NHS to obtain compound 7 as a pinanediol ester of SB04028 in 14% yield over 2 steps. Finally, compound 7 was treated with a cleavage cocktail consisting of 95% TFA/2.5% H2O/2.5% TIS for 4 h at RT to yield the desired precursor SB04028 in 63% yield.
natGa-complexed standards were prepared by incubating respective precursors with excess natGaCl3 in NaOAc buffer (0.1 N, pH 4.5), followed by HPLC purification [43]. natGa-SB02055, natGa-SB04028 and natGa-PNT6555 were obtained in 36-98% yields. 68Ga labeling was performed in HEPES buffer (2 M, pH 5.0) [43]. HPLC purification of the crude reaction yielded the desired 68Ga-labeled analogues in 19-58% decay-corrected radiochemical yield with > 92% radiochemical purity and > 9.1 GBq/µmol molar activity.
2.2. In Vitro Fluorescence-based Binding Assay
The enzymatic cleavage of a fluorogenic substrate (Suc-Gly-Pro-AMC) by the recombinant human FAP (rhFAP) was inhibited by natGa-SB02055, natGa-SB04028 and natGa-PNT6555 in a dose-dependent manner (Figure 2). IC50 values calculated for natGa-SB02055, natGa-SB04028 and natGa-PNT6555 were found to be 0.41±0.06, 13.9±1.29 and 78.1±4.59 nM, respectively (n = 3).
2.3. Ex Vivo Biodistribution and PET/CT Imaging Studies
The PET/CT image acquired at 1 h pi with [68Ga]Ga-SB04028 showed a clear visualization of the HEK293T:hFAP tumor xenograft [44], with the tumor uptake being comparatively higher than [68Ga]Ga-PNT6555 (Figure 3). Moreover, [68Ga]Ga-SB04028 displayed a faster clearance from most normal organs/tissues, with a relatively lower kidney uptake and slightly superior tumor/background contrast ratios compared to [68Ga]Ga-PNT6555. On the other hand, while the uptake of [68Ga]Ga-SB02055 in most normal organs/tissues was minimal, the tumor uptake of [68Ga]Ga-SB02055 was also negligible on the PET/CT image (Figure 3). High accumulation of all the three radiotracers in the urinary bladder indicated excretion via the renal pathway. Co-injection of [68Ga]Ga-SB04028 with FAPI-04 (0.5 mg/mouse) reduced the tumor uptake to the background level (Figure 3).
Data from biodistribution studies (Figure 4 and Table S1) for all three tracers conducted at 1 h pi in HEK293T:hFAP tumor-bearing mice were found to be concordant with their respective PET/CT images. The tumor uptake values were found to be 1.08±0.37, 10.1±0.42 and 6.38±0.45 %ID/g for [68Ga]Ga-SB02055, [68Ga]Ga-SB04028 and [68Ga]Ga-PNT6555, respectively. All tracers were excreted rapidly via the renal pathway with low but comparable kidney uptake (1.74±0.95 %ID/g for [68Ga]Ga-SB02055, 2.10±0.33 %ID/g for [68Ga]Ga-SB04028 and 2.29±0.43 %ID/g for [68Ga]Ga-PNT6555). [68Ga]Ga-SB02055 displayed retention slighter higher than background muscle uptake (0.63±0.20 %ID/g) in some normal organs, particularly the blood (3.13±1.25 %ID/g), pancreas (2.07±0.83 %ID/g), bone (1.14±0.37 %ID/g) and thyroid (1.49±0.47 %ID/g). Akin to the control tracer [68Ga]Ga-PNT6555, [68Ga]Ga-SB04028 exhibited minimal uptake in most normal organs/tissues, but demonstrated superior tumor/background contrast ratios than [68Ga]Ga-PNT6555.
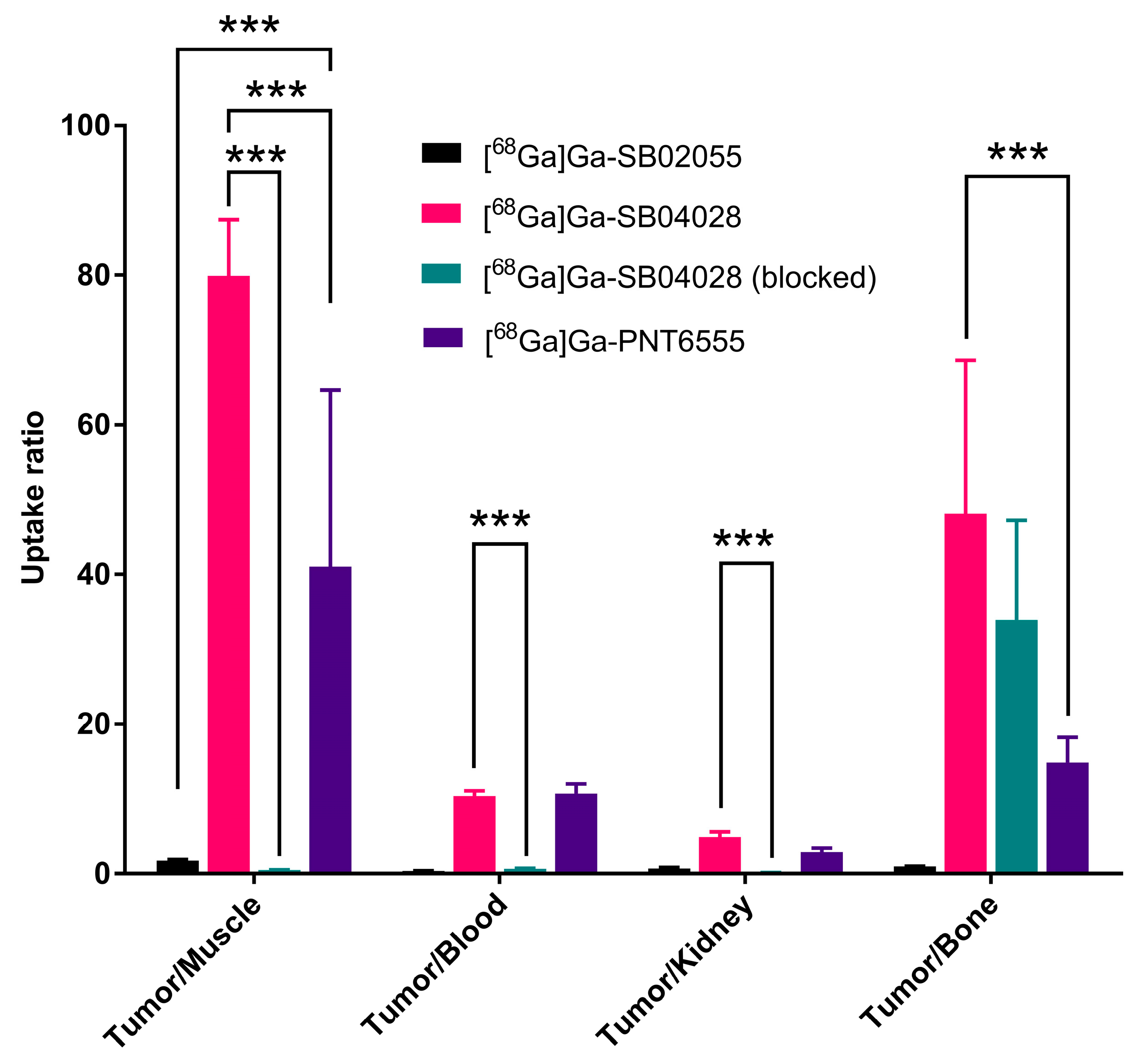
The tumor/muscle, tumor/kidney and tumor/bone ratios for [68Ga]Ga-SB04028 were considerably higher than those for [68Ga]Ga-PNT6555 (79.9±7.50 vs 52.6±5.86, 4.89±0.72 vs 2.87±0.59 and 48.1±20.5 vs 14.9±3.34, respectively) (Figure 5 and Table S1). These uptake ratios were significantly lower for [68Ga]Ga-SB02055 (tumor/muscle: 1.72±0.19; tumor/kidney: 0.67±0.15; tumor/bone: 0.95±0.06) compared to both control [68Ga]Ga-PNT6555 as well as [68Ga]Ga-SB04028. Additionally, tumor/blood ratios for [68Ga]Ga-SB04028 (10.4±0.70) and [68Ga]Ga-PNT6555 (10.7±1.29) were comparable and the tumor/blood ratio was much lower for [68Ga]Ga-SB02055 (0.35±0.03).
2.4. In Vivo Stability Studies
The in vivo stability studies revealed [68Ga]Ga-SB04028 to be considerably stable in blood plasma of male NRG mice (n = 3) at 15 min pi compared to [68Ga]Ga-SB02055. While no intact [68Ga]Ga-SB02055 was present in the mouse plasma at 15 min pi, 46.6±0.21% of radioactivity from [68Ga]Ga-SB04028 was found to be present as intact tracer. Fractions of tracers remaining intact in the urine at 15 min pi for [68Ga]Ga-SB02055 and [68Ga]Ga-SB04028 were 93.5±0.91% and 53.6±7.11%, respectively (Figures S1-S4).
3. Discussion
The FAP binding affinity of natGa-SB02055 (0.41±0.06 nM) was found to be ~ 190-fold higher than natGa-PNT6555 (78.1±4.59 nM), whereas that of natGa-SB04028 (13.9±1.29 nM) was ~ 5.6-fold higher than natGa-PNT6555 in the rhFAP enzymatic assays (Figure 2). This is consistent with previous reports demonstrating bicyclic heteroaromatic quinolinoyl-based FAP inhibitors to be more potent than their monocyclic homoaromatic benzoyl-based congeners [13,14,19,21,45]. Additionally, FAP seems to prefer P2 glycine (SB02055) over a P2 D-alanine (SB04028). It is to be noted that, P2 glycine bearing N-(4-quinolinoyl)-Gly-boroPro was observed to exhibit an IC50(FAP) of 3.7±0.2 nM by Jansen and co-workers [14]. Interestingly its P2 D-alanine analogue, N-(4-quinolinoyl)-D-Ala-boroPro, was shown to exhibit an IC50(FAP) of 6.4±1.4 nM only by Bachovchin et al. [21] in a separate study and hence cannot be pitted head to head for comparison.
Surprisingly, findings from the in vitro enzymatic assay for natGa-SB02055 were not in concordance with the data from PET/CT imaging and biodistribution analysis of its radioactive counterpart obtained from HEK293T:hFAP tumor-bearing mice at 1 h pi. [68Ga]Ga-SB02055 exhibited a nominal tumor uptake (1.08±0.37 %ID/g) close to the background levels that was ~ 6-fold lower than [68Ga]Ga-PNT6555 (6.38±0.45 %ID/g) and primarily underwent renal excretion (Figure 3 and Figure 4 and Table S1). However, uptake in normal organs, particularly the blood (3.13±1.25 %ID/g), pancreas (2.07±0.83 %ID/g), bone (1.14±0.37 %ID/g) and thyroid (1.49±0.47 %ID/g) was greater than the background muscle uptake (0.63±0.20 %ID/g).
We initially suspected lack of in vivo stability of [68Ga]Ga-SB02055 to be a reason for its nominal tumor uptake. Upon subjecting it to in vivo stability testing at 15 min pi, we found no intact [68Ga]Ga-SB02055 in the mouse plasma. Interestingly, the urine samples collected at the same time presented with > 93% intact [68Ga]Ga-SB02055 (Figures S1 and S2). The low tumor uptake at 1 h pi could possibly be a consequence of [68Ga]Ga-SB02055 undergoing quick clearance from the blood and rapid renal excretion into the urine in ≤ 15 min of injecting the tracer in vivo. Furthermore, an unidentified highly hydrophilic 68Ga-labeled species accounting for > 99% plasma fraction was observed at 15 min pi during the stability testing. The higher than background level uptake in the blood could supposedly be attributed to this uncharacterized 68Ga-labeled species. Further investigation of [68Ga]Ga-SB02055 in this regard is currently underway.
Next, encouraged by earlier findings regarding FAP’s ability to tolerate a P2 D-alanine in the context of an X-boroPro-based inhibitor [20,21], we sought to evaluate the effect of substituting the P2 glycine in [68Ga]Ga-SB02055 with a D-alanine to yield [68Ga]Ga-SB04028. [68Ga]Ga-SB04028 differed from [68Ga]Ga-SB02055 in only the P2 amino acid residue, while rest of the molecular structure was kept alike.
The PET/CT imaging data was consistent with the data from the biodistribution analysis performed at 1 h pi in HEK293T:hFAP tumor-bearing mice. [68Ga]Ga-SB04028 demonstrated > 1.5 fold higher tumor uptake (10.1±0.42 %ID/g) than [68Ga]Ga-PNT6555 (6.38±0.45 %ID/g) (Figure 3 and Figure 4 and Table S1) and concordant with the findings from FAP inhibition assays where natGa-SB04028 was realized to be more potent than natGa-PNT6555. Additionally, high accumulation of both [68Ga]Ga-SB04028 and [68Ga]Ga-PNT6555 in the bladder indicated their renal excretion. The uptake of [68Ga]Ga-SB04028 in most normal organs/tissues was minimal and comparable to [68Ga]Ga-PNT6555, particularly in the muscle (0.13±0.01 vs 0.12±0.02 %ID/g), blood (0.97±0.03 vs 0.60±0.04 %ID/g), kidney (2.10±0.33 vs 2.29±0.43 %ID/g) and bone (0.24±0.09 vs 0.45±0.12 %ID/g) (Figure 4 and Table S1). However, since the tumor uptake of [68Ga]Ga-SB04028 was considerably greater than [68Ga]Ga-PNT6555, the corresponding tumor/organ uptake ratios for [68Ga]Ga-SB04028 were found to be significantly higher than [68Ga]Ga-PNT6555 (tumor/muscle: 79.9±7.50 vs 52.6±5.86; tumor/kidney: 4.89±0.72 vs 2.87±0.59; tumor/bone: 48.1±20.5 vs 14.9±3.34), except tumor/blood ratios (10.4±0.70 vs 10.7±1.29 %ID/g), which were equivalent for the two (Figure 5 and Table S1). In general, [68Ga]Ga-SB04028 demonstrated superior tumor/background contrast ratios compared to [68Ga]Ga-PNT6555 as evident from both PET/CT imaging (Figure 3) and biodistribution analysis (Figure 5 and Table S1).
Imaging and biodistribution analysis of mice co-injected [68Ga]Ga-SB04028 with FAPI-04 (0.5 mg/mouse) revealed a ~ 97% reduction in the tumor uptake (10.1±0.42 %ID/g vs 0.30±0.04 %ID/g) substantiating in vivo FAP specificity of our lead candidate [68Ga]Ga-SB04028 (Figure 3 and Figure 4, Table S1). Moreover, as anticipated since there was no significant reduction observed in the kidney uptake of [68Ga]Ga-SB04028 (2.10±0.33 %ID/g vs 2.53±0.57 %ID/g), we can conclusively state that retention in the kidneys is by virtue of the our tracer’s propensity to undergo renal elimination and is not FAP-mediated.
Unlike its P2 glycine analogue [68Ga]Ga-SB02055, [68Ga]Ga-SB04028 with a P2 D-alanine residue was found to be relatively more stable during in vivo stability testing. Strikingly, > 47% of [68Ga]Ga-SB04028 remained intact in the mouse plasma at 15 min pi (Figures S3-S4). The remaining fractions corresponded to currently uncharacterized but highly hydrophilic 68Ga-labeled species. The urine samples from the mouse presented with > 54% intact tracer, with the remaining portions co-eluting more or less with the uncharacterized 68Ga-labeled species found in the plasma samples for [68Ga]Ga-SB04028.
The exact nature and identity of the 68Ga-labeled species eluting prior to each of the intact tracer during in vivo stability testing has not been characterized. However, it has been shown that boronic acids under basic (physiological) conditions could be converted to other species including: (1) Degradation products formed under physiological pH conditions due to the inherent susceptibility of boronic acids to undergo oxidation [46,47]. The empty p orbital of boron in boronic acid is prone to attack by nucleophilic oxygen of reactive oxygen species (ROS), resulting in formation of labile boric esters and/or oxidative de-boronation products. Notably, it is the same attribute that allows boronic acid-based ligands to react with the nucleophilic hydroxyl of the catalytic serine residue of FAP and form reversible covalent adducts. (2) Anionic boronic acid/ hydroxyboronate anion formed under more basic conditions as a result of hydroxyl (OH-) attack on the highly electrophilic boron atom [48]. Naturally these ionic species will have a tendency to elute before the neutral boronic acid forms. (3) Boronate esters formed under aqueous conditions at a physiological pH of ~7.4 [48]. Efforts are underway to characterize these species.
The goal of the present work was to develop novel boroPro-based PET imaging agents with superior pharmacokinetics properties and excellent imaging contrast for easy and reliable diagnosis of FAP-overexpressing carcinomas. Although our lead candidate [68Ga]Ga-SB04028 demonstrated superior tumor/background contrast ratios compared to [68Ga]Ga-PNT6555, there is still some scope for further optimization: (1) A 4,4-difluoro substituent at the (2S)-2-cyanopyrrolidine ring was previously reported to enhance FAP inhibitory activity compared to that of unsubstituted analogue [12,13,14]. It would be interesting to generate a difluorinated derivative of [68Ga]Ga-SB04028 for preclinical investigation, although its synthesis might be a bit challenging; and (2) In terms of diagnostic radiopharmaceuticals, 99mTc is the most widely used with SPECT imaging accounting for > 70% of all the imaging procedures performed in the field of nuclear medicine. Replacing DOTA with a suitable chelator to allow radiolabeling with 99mTc is definitely worth exploiting.
4. Materials and Methods
4.1. Synthesis of natGa and 68Ga-Complexed DOTA-Conjugated FAP-Targeted Ligands
Detailed information on the synthesis, purification, and characterizations of the intermediates, precursors (SB02055 and SB04028), natGa-complexed analogues (natGa-SB02055, natGa-SB04028 and natGa-PNT6555) and their 68Ga-complexed analogues ([68Ga]Ga-SB02055, [68Ga]Ga-SB04028 and [68Ga]Ga-PNT6555) is provided in the Supplementary Materials.
4.2. Cell Culture
HEK293T cells were obtained from American Type Culture Collection (Manassas, VA, USA). The IMPACT Rodent Pathogen Test (IDEXX BioAnalytics, Columbia, MO, USA) confirmed that the cells were pathogen-free. Detailed information on the generation of HEK293T:hFAP cells has been previously reported by our group [44]. HEK293T:hFAP cells were cultured in DMEM GlutaMAX™ medium supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (100 µg/mL) at 37 °C in a Panasonic Healthcare (Tokyo, Japan) MCO-19AIC humidified incubator containing 5% CO2. Cells were grown until 80–90% confluence and washed with sterile PBS (pH 7.4) and collected.
4.3. In Vitro Fluorescence based Binding Assay
The half maximal inhibitory concentration (IC50) values of the FAP-targeted ligands were measured using in vitro enzymatic assay following previously reported procedures [41,44]. Briefly, rhFAP (Biolegend, San Diego, CA, USA; 0.2 µg/mL, 50 µL) was added into a Costar clear bottom 96-well plate. Varied concentrations (25 pM to 1 µM) of natGa-complexed ligands were added to each well (in duplicate) containing the rhFAP. PBS was added to the control well (in duplicate). After incubating for 30 min at 37 °C, fluorescence substrate Suc-Gly-Pro-AMC (Bachem, Bubendorf, Switzerland; 20 mM, 50 µL) was added to each well. The velocities of AMC release were measured kinetically at λex = 380 nm, λem = 460 nm at 60 min at 37 °C using a FlexStation 3 Multi-Mode Microplate Reader.
4.4. Ex Vivo Biodistribution and PET/CT Imaging Studies
Imaging and biodistribution studies were performed using immunodeficient male NRG (NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ) mice following previously reported procedures [41,44]. All experiments were conducted according to the guidelines established by the Canadian Council on Animal Care and approved by Animal Ethics Committee of the University of British Columbia. The mice were subcutaneously inoculated with 7.5×106 HEK293T:hFAP cells in the left dorsal flank. When the tumors grew to 6–8 mm in diameter, the mice were used for PET/CT imaging and biodistribution studies.
PET/CT imaging experiments were carried out using a Siemens Inveon micro PET/CT scanner (Knoxville, TN, USA). Tumor bearing mice were injected with ~4 to 6 MBq of 68Ga-labeled tracer through a lateral caudal tail vein under 2.5% isoflurane in oxygen anesthesia, followed by recovery and free roaming in its cage during the uptake period. A 10 min localization CT scan was acquired using 3 overlapping positions to cover the entire mouse. CT scan was used for attenuation and scatter correction, and anatomical localization. A list mode acquisition was then performed for 15 min at 1 h pi with the mouse under isoflurane sedation. The images were reconstructed using 3D OSEM/MAP iterative methods.
For biodistribution studies, the mice were injected with ~1-2 MBq of the 68Ga-labeled tracer, using the exact procedures as described above. At 1 h pi, the mice were euthanized by CO2 inhalation. Blood was withdrawn by cardiac puncture, and organs/tissues of interest were collected, weighed, and counted using a Perkin Elmer (Waltham, MA, USA) Wizard2 2480 automatic gamma counter. The uptake values in percent injected dose per gram of tissues (%ID/g) for different tracers were calculated.
For blocking studies, HEK293T:hFAP tumor-bearing mice were co-injected [68Ga]Ga-SB04028 with an excess of FAPI-04 (0.5 mg/mouse) as a competitor. Imaging and biodistribution studies were performed at 1 h pi similar to the unblocked mice.
4.5. In Vivo Stability Studies
In vivo stability studies were performed using healthy (non-tumor bearing) male NRG mice following previously reported procedures [43,49]. Briefly, [68Ga]Ga-SB02055 (6.32±0.16 MBq) and [68Ga]Ga-SB04028 (9.90±0.26 MBq) were injected via the lateral caudal vein into the mice (n = 3). At 15 min pi, the mice were sedated and euthanized, and urine and blood were collected. The blood was processed to obtain plasma by adding 0.5 mL of acetonitrile to 0.5-0.8 mL of whole blood, then vortexing and centrifuging the resulting mixture on a benchtop mini-centrifuge (Fisherbrand™ Mini-Centrifuge 100-240V, 50/60Hz Universal Plug) for 20 min. The supernatant was collected and filtered to give blood plasma. The plasma and urine samples were analyzed via radio-HPLC by using the same conditions as those used for quality control of these 68Ga-labeled radioligands.
4.6. Statistical Analysis
Data were reported as mean±standard deviation (SD) and analyzed with GraphPad Prism, version 9.5.1. Two way ANOVA and multiple t-tests were performed for all tumor and organ/tissue uptake values in the biodistribution studies of [68Ga]Ga-SB02055, [68Ga]Ga-SB04028, and [68Ga]Ga-PNT6555 in the HEK293T:hFAP tumor model. Statistical significance was defined at p < 0.05 using the Holm–Sidak method.
5. Conclusions
We successfully synthesized and evaluated two novel 68Ga-labeled (R)-pyrrolidin-2-yl-boronic acid-derived PET tracers for FAP-targeted cancer imaging. While the P2 glycine-containing [68Ga]Ga-SB02055 displayed minimal tumor uptake, its P2 D-alanine congener [68Ga]Ga-SB04028 demonstrated significantly higher tumor uptake and superior imaging contrast compared to [68Ga]Ga-PNT6555. (R)-(((quinoline-4-carbonyl)-D-alanyl)pyrrolidin-2-yl)boronic acid is a highly promising pharmacophore that can be used to develop PET imaging agents with superior pharmacokinetics properties and excellent imaging contrasts for visualization of FAP-overexpressing carcinomas. Replacement of DOTA with suitable chelators/prosthetic groups to allow radiolabeling with two of the most widely used imaging isotopes, 99mTc and 18F, is warranted in the future.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Detailed synthetic procedures and results for the preparation of FAP-targeted ligands and their natGa/68Ga-complexed analogues; Table S1: Biodistribution and tumor/organ uptake ratios of [68Ga]Ga-SB02055, [68Ga]Ga-SB04028 and [68Ga]Ga-PNT6555 in HEK239T:hFAP tumor-bearing mice; Figure S1: Representative radio-HPLC chromatograms from in vivo stability study performed to determine the percent intact fraction of [68Ga]Ga-SB02055 in mouse plasma and urine samples collected at 15 min post-injection; Figure S2: Radio-HPLC analysis of [68Ga]Ga-SB02055; Figure S3: Representative radio-HPLC chromatograms from in vivo stability study performed to determine the percent intact fraction of [68Ga]Ga-SB04028 in mouse plasma and urine samples collected at 15 min post-injection; Figure S4: Radio-HPLC analysis of [68Ga]Ga-SB04028.
Author Contributions
Conceptualization, K.-S.L. and S.B; methodology, S.B., H.-T.K., H.M., Z.Z. and A.A.W.L.W.; validation, S.B., H.-T.K., H.M., Z.Z. and A.A.W.L.W.; formal analysis, S.B., H.-T.K., Z.Z. and A.A.W.L.W; investigation, S.B. H.-T.K., H.M., Z.Z. and A.A.W.L.W.; resources, F.B. and K.-S.L.; data curation, S.B., H.-T.K., Z.Z. A.A.W.L.W. and K.-S.L.; writing—original draft preparation, S.B.; writing—review and editing, K.-S.L.; visualization, S.B.; supervision, K.-S.L. and F.B.; project administration, K.-S.L.; funding acquisition, K.-S.L. and F.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Canadian Institutes of Health Research (PJT-180300) and Pancreas Centre BC.
Institutional Review Board Statement
The animal study protocol (A20-0113) was approved by the Animal Ethics Committee of the University of British Columbia on September 30, 2020.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data generated from this study are available in the text and Supplementary Materials.
Acknowledgments
We thank Nadine Colpo for her technical assistance with PET/CT imaging.
Conflicts of Interest
F.B. and K.-S.L. are co-founders and consultants of Alpha-9 Theranostics Inc., and receive research funding from the company. H.-T.K. is a part time employee of Alpha-9. F.B., K.-S.L., H.-T.K. H.M. and Z.Z. hold shares and/or options in Alpha-9. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Xing, F. Cancer Associated Fibroblasts (CAFs) in Tumor Microenvironment. Front Biosci 2010, 15, 166. [Google Scholar] [CrossRef]
- Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Cell Surface Glycoprotein of Reactive Stromal Fibroblasts as a Potential Antibody Target in Human Epithelial Cancers. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 7235–7239. [Google Scholar] [CrossRef]
- Scanlan, M.J.; Raj, B.K.M.; Calvo, B.; RErrFIG, W.J. Molecular Cloning of Fibroblast Activation Protein a, a Member of the Serine Protease Family Selectively Expressed in Stromal Fibroblasts of Epithelial Cancers. Proc. Natl. Acad. Sci. USA 1994. [Google Scholar] [CrossRef]
- Tchou, J.; Zhang, P.J.; Bi, Y.; Satija, C.; Marjumdar, R.; Stephen, T.L.; Lo, A.; Chen, H.; Mies, C.; June, C.H.; et al. Fibroblast Activation Protein Expression by Stromal Cells and Tumor-Associated Macrophages in Human Breast Cancer. Human Pathology 2013, 44, 2549–2557. [Google Scholar] [CrossRef]
- Kesch, C.; Yirga, L.; Dendl, K.; Handke, A.; Darr, C.; Krafft, U.; Radtke, J.P.; Tschirdewahn, S.; Szarvas, T.; Fazli, L.; et al. High Fibroblast-Activation-Protein Expression in Castration-Resistant Prostate Cancer Supports the Use of FAPI-Molecular Theranostics. Eur J Nucl Med Mol Imaging 2021, 49, 385–389. [Google Scholar] [CrossRef]
- Cheng, J.D.; Valianou, M.; Canutescu, A.A.; Jaffe, E.K.; Lee, H.-O.; Wang, H.; Lai, J.H.; Bachovchin, W.W.; Weiner, L.M. Abrogation of Fibroblast Activation Protein Enzymatic Activity Attenuates Tumor Growth. Molecular Cancer Therapeutics 2005, 4, 351–360. [Google Scholar] [CrossRef]
- Scott, A.M.; Wiseman, G.; Adjei, A.; Lee, F.-T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; Larson, S.M.; et al. A Phase I Dose-Escalation Study of Sibrotuzumab in Patients with Advanced or Metastatic Fibroblast Activation Protein-Positive Cancer.
- Welt, S.; Divgi, C.R.; Scott, A.M.; Garin-Chesa, P.; Finn, R.D.; Graham, M.; Carswell, E.A.; Cohen, A.; Larson, S.M.; Old, L.J. Antibody Targeting in Metastatic Colon Cancer: A Phase I Study of Monoclonal Antibody F19 against a Cell-Surface Protein of Reactive Tumor Stromal Fibroblasts. JCO 1994, 12, 1193–1203. [Google Scholar] [CrossRef]
- Riet, T.; Abken, H. Chimeric Antigen Receptor T Cells: Power Tools to Wipe out Leukemia and Lymphoma. Expert Review of Hematology 2015, 8, 383–385. [Google Scholar] [CrossRef]
- Sheykhhasan, M.; Manoochehri, H.; Dama, P. Use of CAR T-Cell for Acute Lymphoblastic Leukemia (ALL) Treatment: A Review Study. Cancer Gene Ther 2022, 29, 1080–1096. [Google Scholar] [CrossRef]
- Wen, Y.; Wang, C.-T.; Ma, T.-T.; Li, Z.-Y.; Zhou, L.-N.; Mu, B.; Leng, F.; Shi, H.-S.; Li, Y.-O.; Wei, Y.-Q. Immunotherapy Targeting Fibroblast Activation Protein Inhibits Tumor Growth and Increases Survival in a Murine Colon Cancer Model. Cancer Science 2010, 101, 2325–2332. [Google Scholar] [CrossRef]
- Tsai, T.-Y.; Yeh, T.-K.; Chen, X.; Hsu, T.; Jao, Y.-C.; Huang, C.-H.; Song, J.-S.; Huang, Y.-C.; Chien, C.-H.; Chiu, J.-H.; et al. Substituted 4-Carboxymethylpyroglutamic Acid Diamides as Potent and Selective Inhibitors of Fibroblast Activation Protein. J. Med. Chem. 2010, 53, 6572–6583. [Google Scholar] [CrossRef]
- Ryabtsova, O.; Jansen, K.; Van Goethem, S.; Joossens, J.; Cheng, J.D.; Lambeir, A.-M.; De Meester, I.; Augustyns, K.; Van der Veken, P. Acylated Gly-(2-Cyano)Pyrrolidines as Inhibitors of Fibroblast Activation Protein (FAP) and the Issue of FAP/Prolyl Oligopeptidase (PREP)-Selectivity. Bioorganic & Medicinal Chemistry Letters 2012, 22, 3412–3417. [Google Scholar] [CrossRef]
- Jansen, K.; Heirbaut, L.; Verkerk, R.; Cheng, J.D.; Joossens, J.; Cos, P.; Maes, L.; Lambeir, A.-M.; De Meester, I.; Augustyns, K.; et al. Extended Structure–Activity Relationship and Pharmacokinetic Investigation of (4-Quinolinoyl)Glycyl-2-Cyanopyrrolidine Inhibitors of Fibroblast Activation Protein (FAP). J. Med. Chem. 2014, 57, 3053–3074. [Google Scholar] [CrossRef]
- Hu, Y.; Ma, L.; Wu, M.; Wong, M.S.; Li, B.; Corral, S.; Yu, Z.; Nomanbhoy, T.; Alemayehu, S.; Fuller, S.R.; et al. Synthesis and Structure–Activity Relationship of N-Alkyl Gly-Boro-Pro Inhibitors of DPP4, FAP, and DPP7. Bioorganic & Medicinal Chemistry Letters 2005, 15, 4239–4242. [Google Scholar] [CrossRef]
- Rosenblum, J.S.; Kozarich, J.W. Prolyl Peptidases: A Serine Protease Subfamily with High Potential for Drug Discovery. Current Opinion in Chemical Biology 2003, 7, 496–504. [Google Scholar] [CrossRef]
- Edosada, C.Y.; Quan, C.; Wiesmann, C.; Tran, T.; Sutherlin, D.; Reynolds, M.; Elliott, J.M.; Raab, H.; Fairbrother, W.; Wolf, B.B. Selective Inhibition of Fibroblast Activation Protein Protease Based on Dipeptide Substrate Specificity. Journal of Biological Chemistry 2006, 281, 7437–7444. [Google Scholar] [CrossRef]
- Edosada, C.Y.; Quan, C.; Tran, T.; Pham, V.; Wiesmann, C.; Fairbrother, W.; Wolf, B.B. Peptide Substrate Profiling Defines Fibroblast Activation Protein as an Endopeptidase of Strict Gly 2 -Pro 1 -Cleaving Specificity. FEBS Letters 2006, 580, 1581–1586. [Google Scholar] [CrossRef]
- Jansen, K.; Heirbaut, L.; Cheng, J.D.; Joossens, J.; Ryabtsova, O.; Cos, P.; Maes, L.; Lambeir, A.-M.; De Meester, I.; Augustyns, K.; et al. Selective Inhibitors of Fibroblast Activation Protein (FAP) with a (4-Quinolinoyl)-Glycyl-2-Cyanopyrrolidine Scaffold. ACS Med. Chem. Lett. 2013, 4, 491–496. [Google Scholar] [CrossRef]
- Tran, T.; Quan, C.; Edosada, C.Y.; Mayeda, M.; Wiesmann, C.; Sutherlin, D.; Wolf, B.B. Synthesis and Structure–Activity Relationship of N-Acyl-Gly-, N-Acyl-Sar- and N-Blocked-BoroPro Inhibitors of FAP, DPP4, and POP. Bioorganic & Medicinal Chemistry Letters 2007, 17, 1438–1442. [Google Scholar] [CrossRef]
- Poplawski, S.E.; Lai, J.H.; Li, Y.; Jin, Z.; Liu, Y.; Wu, W.; Wu, Y.; Zhou, Y.; Sudmeier, J.L.; Sanford, D.G.; et al. Identification of Selective and Potent Inhibitors of Fibroblast Activation Protein and Prolyl Oligopeptidase. J. Med. Chem. 2013, 56, 3467–3477. [Google Scholar] [CrossRef]
- Meletta, R.; Müller Herde, A.; Chiotellis, A.; Isa, M.; Rancic, Z.; Borel, N.; Ametamey, S.; Krämer, S.; Schibli, R. Evaluation of the Radiolabeled Boronic Acid-Based FAP Inhibitor MIP-1232 for Atherosclerotic Plaque Imaging. Molecules 2015, 20, 2081–2099. [Google Scholar] [CrossRef]
- Trujillo-Benítez, D.; Luna-Gutiérrez, M.; Ferro-Flores, G.; Ocampo-García, B.; Santos-Cuevas, C.; Bravo-Villegas, G.; Morales-Ávila, E.; Cruz-Nova, P.; Díaz-Nieto, L.; García-Quiroz, J.; et al. Design, Synthesis and Preclinical Assessment of 99mTc-IFAP for In Vivo Fibroblast Activation Protein (FAP) Imaging. Molecules 2022, 27, 264. [Google Scholar] [CrossRef]
- Coria-Domínguez, L.; Vallejo-Armenta, P.; Luna-Gutiérrez, M.; Ocampo-García, B.; Gibbens-Bandala, B.; García-Pérez, F.; Ramírez-Nava, G.; Santos-Cuevas, C.; Ferro-Flores, G. [99mTc]Tc-IFAP Radioligand for SPECT/CT Imaging of the Tumor Microenvironment: Kinetics, Radiation Dosimetry, and Imaging in Patients. Pharmaceuticals 2022, 15, 590. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jäger, D.; Mier, W.; Haberkorn, U. Development of Quinoline-Based Theranostic Ligands for the Targeting of Fibroblast Activation Protein. J Nucl Med 2018, 59, 1415–1422. [Google Scholar] [CrossRef]
- Loktev, A.; Lindner, T.; Burger, E.-M.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Marmé, F.; Jäger, D.; Mier, W.; et al. Development of Fibroblast Activation Protein–Targeted Radiotracers with Improved Tumor Retention. J Nucl Med 2019, 60, 1421–1429. [Google Scholar] [CrossRef]
- Loktev, A.; Lindner, T.; Mier, W.; Debus, J.; Altmann, A.; Jäger, D.; Giesel, F.; Kratochwil, C.; Barthe, P.; Roumestand, C.; et al. A Tumor-Imaging Method Targeting Cancer-Associated Fibroblasts. J Nucl Med 2018, 59, 1423–1429. [Google Scholar] [CrossRef]
- Giesel, F.L.; Kratochwil, C.; Lindner, T.; Marschalek, M.M.; Loktev, A.; Lehnert, W.; Debus, J.; Jäger, D.; Flechsig, P.; Altmann, A.; et al. 68 Ga-FAPI PET/CT: Biodistribution and Preliminary Dosimetry Estimate of 2 DOTA-Containing FAP-Targeting Agents in Patients with Various Cancers. J Nucl Med 2019, 60, 386–392. [Google Scholar] [CrossRef]
- Meyer, C.; Dahlbom, M.; Lindner, T.; Vauclin, S.; Mona, C.; Slavik, R.; Czernin, J.; Haberkorn, U.; Calais, J. Radiation Dosimetry and Biodistribution of 68 Ga-FAPI-46 PET Imaging in Cancer Patients. J Nucl Med 2020, 61, 1171–1177. [Google Scholar] [CrossRef]
- Lindner, T.; Altmann, A.; Krämer, S.; Kleist, C.; Loktev, A.; Kratochwil, C.; Giesel, F.; Mier, W.; Marme, F.; Debus, J.; et al. Design and Development of 99m Tc-Labeled FAPI Tracers for SPECT Imaging and 188 Re Therapy. J Nucl Med 2020, 61, 1507–1513. [Google Scholar] [CrossRef]
- Hu, K.; Wang, L.; Wu, H.; Huang, S.; Tian, Y.; Wang, Q.; Xiao, C.; Han, Y.; Tang, G. [18F]FAPI-42 PET Imaging in Cancer Patients: Optimal Acquisition Time, Biodistribution, and Comparison with [68Ga]Ga-FAPI-04. Eur J Nucl Med Mol Imaging 2022, 49, 2833–2843. [Google Scholar] [CrossRef]
- Giesel, F.L.; Adeberg, S.; Syed, M.; Lindner, T.; Jiménez-Franco, L.D.; Mavriopoulou, E.; Staudinger, F.; Tonndorf-Martini, E.; Regnery, S.; Rieken, S.; et al. FAPI-74 PET/CT Using Either 18 F-AlF or Cold-Kit 68 Ga Labeling: Biodistribution, Radiation Dosimetry, and Tumor Delineation in Lung Cancer Patients. J Nucl Med 2021, 62, 201–207. [Google Scholar] [CrossRef]
- Robin, M. Hallett SEP, Kyle E. Novakowski, Mark H. Dornan, Shin Hye Ahn, Shuang Pan, Wu Wengen, Liu Yuxin, David G., Sanford VSH, Quang-De Nguyen, Anthony P. Belanger, Jack H. Lai, William W. Bachovchin, Joe A. B. McCann. Pre-clinical characterization of the novel Fibroblast Activation Protein (FAP) targeting ligand PNT6555 for the imaging and therapy of cancer. Cancer Res 2022, 82, 1–6403. [Google Scholar]
- Robin Hallett, Sarah Poplawski, Kyle Novakowski, Mark Dornan, Shin Hye Ahn, Shuang Pan, Wengen Wu, Yuxin Liu, David Sanford, Valerie Hergott, Quang-De Nguyen, Anthony Belanger, Jack Lai, William Bachovchin and Joe McCann. J Nucl Med 2022, 63, 4028.
- FAPi Radioligand OpeN-Label, Phase 1 Study to Evaluate Safety, Tolerability and DosImetry of [Lu-177]-PNT6555; A Dose Escalation Study for TReatment of Patients With Select Solid Tumors (FRONTIER) (NCT05432193).
- Toms, J.; Kogler, J.; Maschauer, S.; Daniel, C.; Schmidkonz, C.; Kuwert, T.; Prante, O. Targeting Fibroblast Activation Protein: Radiosynthesis and Preclinical Evaluation of an 18F-Labeled FAP Inhibitor. J Nucl Med 2020, 61, 1806–1813. [Google Scholar] [CrossRef]
- Kelly, J.M.; Jeitner, T.M.; Ponnala, S.; Williams, C.; Nikolopoulou, A.; DiMagno, S.G.; Babich, J.W. A Trifunctional Theranostic Ligand Targeting Fibroblast Activation Protein-α (FAPα). Mol Imaging Biol 2021, 23, 686–696. [Google Scholar] [CrossRef]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.-C.R.; Restifo, N.P.; Rosenberg, S.A. Immune Targeting of Fibroblast Activation Protein Triggers Recognition of Multipotent Bone Marrow Stromal Cells and Cachexia. Journal of Experimental Medicine 2013, 210, 1125–1135. [Google Scholar] [CrossRef]
- Kessler, L.; Ferdinandus, J.; Hirmas, N.; Zarrad, F.; Nader, M.; Kersting, D.; Weber, M.; Kazek, S.; Sraieb, M.; Hamacher, R.; et al. Pitfalls and Common Findings in 68 Ga-FAPI PET: A Pictorial Analysis. J Nucl Med 2022, 63, 890–896. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, X.; Xu, X.; Ding, J.; Liu, T.; Jiang, J.; Li, N.; Zhu, H.; Yang, Z. Dynamic PET/CT Imaging of 68Ga-FAPI-04 in Chinese Subjects. Front. Oncol. 2021, 11, 651005. [Google Scholar] [CrossRef]
- Bendre, S.; Zhang, Z.; Colpo, N.; Zeisler, J.; Wong, A.A.W.L.; Bénard, F.; Lin, K.-S. Synthesis and Evaluation of 68Ga-Labeled (2S,4S)-4-Fluoropyrrolidine-2-Carbonitrile and (4R)-Thiazolidine-4-Carbonitrile Derivatives as Novel Fibroblast Activation Protein-Targeted PET Tracers for Cancer Imaging. Molecules 2023, 28, 3481. [Google Scholar] [CrossRef]
- Coutts, S.J.; Kelly, T.A.; Snow, R.J.; Kennedy, C.A.; Barton, R.W.; Adams, J.; Krolikowski, D.A.; Freeman, D.M.; Campbell, S.J.; Ksiazek, J.F.; et al. Structure−Activity Relationships of Boronic Acid Inhibitors of Dipeptidyl Peptidase IV. 1. Variation of the P 2 Position of X aa -BoroPro Dipeptides. J. Med. Chem. 1996, 39, 2087–2094. [Google Scholar] [CrossRef]
- Lin, K.-S.; Pan, J.; Amouroux, G.; Turashvili, G.; Mesak, F.; Hundal-Jabal, N.; Pourghiasian, M.; Lau, J.; Jenni, S.; Aparicio, S.; et al. In Vivo Radioimaging of Bradykinin Receptor B1, a Widely Overexpressed Molecule in Human Cancer. Cancer Research 2015, 75, 387–393. [Google Scholar] [CrossRef]
- Verena, A.; Zhang, Z.; Kuo, H.-T.; Merkens, H.; Zeisler, J.; Wilson, R.; Bendre, S.; Wong, A.A.W.L.; Bénard, F.; Lin, K.-S. Synthesis and Preclinical Evaluation of Three Novel 68Ga-Labeled Bispecific PSMA/FAP-Targeting Tracers for Prostate Cancer Imaging. Molecules 2023, 28, 1088. [Google Scholar] [CrossRef]
- Bachovchin, W.W.; Lai, H.S. U.S. Patent 2007, PCT/US2006/026258.
- Bu, W.; Akama, T.; Chanda, S.; Sullivan, D.; Ciaravino, V.; Jarnagin, K.; Freund, Y.; Sanders, V.; Chen, C.-W.; Fan, X.; et al. Early Rapid Identification of in Vivo Rat Metabolites of AN6414, a Novel Boron-Containing PDE4 Inhibitor by QTRAP LC/MS/MS to Support Drug Discovery. Journal of Pharmaceutical and Biomedical Analysis 2012, 70, 344–353. [Google Scholar] [CrossRef]
- Graham, B.J.; Windsor, I.W.; Gold, B.; Raines, R.T. Boronic Acid with High Oxidative Stability and Utility in Biological Contexts. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2013691118. [Google Scholar] [CrossRef]
- Marco-Dufort, B.; Tibbitt, M.W. Design of Moldable Hydrogels for Biomedical Applications Using Dynamic Covalent Boronic Esters. Materials Today Chemistry 2019, 12, 16–33. [Google Scholar] [CrossRef]
- Lau, J.; Rousseau, E.; Zhang, Z.; Uribe, C.F.; Kuo, H.-T.; Zeisler, J.; Zhang, C.; Kwon, D.; Lin, K.-S.; Bénard, F. Positron Emission Tomography Imaging of the Gastrin-Releasing Peptide Receptor with a Novel Bombesin Analogue. ACS Omega 2019, 4, 1470–1478. [Google Scholar] [CrossRef]
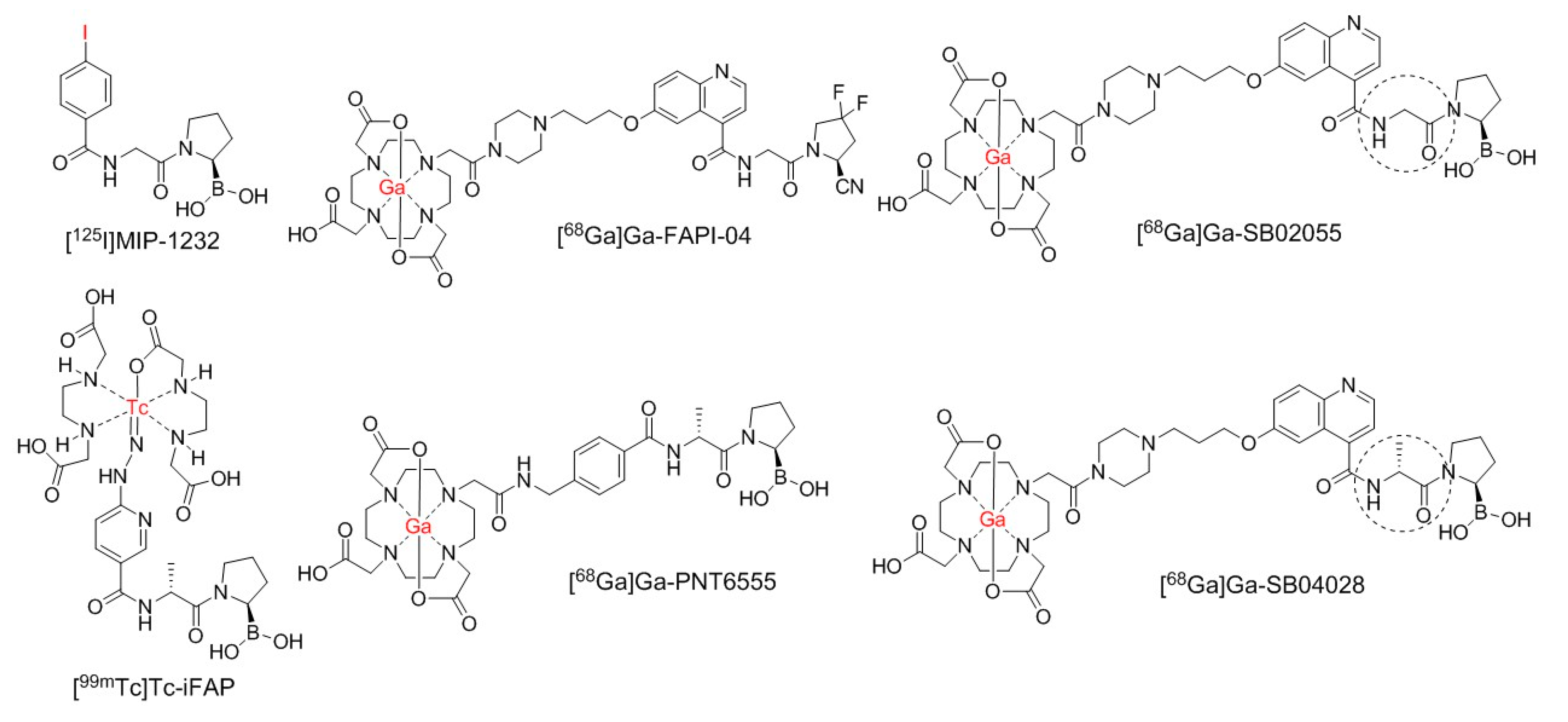
Figure 1.
Chemical Structures of FAP-targeted ligands [68Ga]Ga-SB02055 and [68Ga]Ga-SB04028, and previously reported [125I]MIP-1232, [99mTc]Tc-iFAP, [68Ga]Ga-FAPI-04 and [68Ga]Ga-PNT6555. The difference between [68Ga]Ga-SB02055 and [68Ga]Ga-SB04028 is indicated by dotted circles. The radioisotopes are in red.
Figure 1.
Chemical Structures of FAP-targeted ligands [68Ga]Ga-SB02055 and [68Ga]Ga-SB04028, and previously reported [125I]MIP-1232, [99mTc]Tc-iFAP, [68Ga]Ga-FAPI-04 and [68Ga]Ga-PNT6555. The difference between [68Ga]Ga-SB02055 and [68Ga]Ga-SB04028 is indicated by dotted circles. The radioisotopes are in red.
Scheme 1.
Synthesis of DOTA-conjugated precursor SB02055.
Scheme 2.
Synthesis of DOTA-conjugated precursor SB04028.
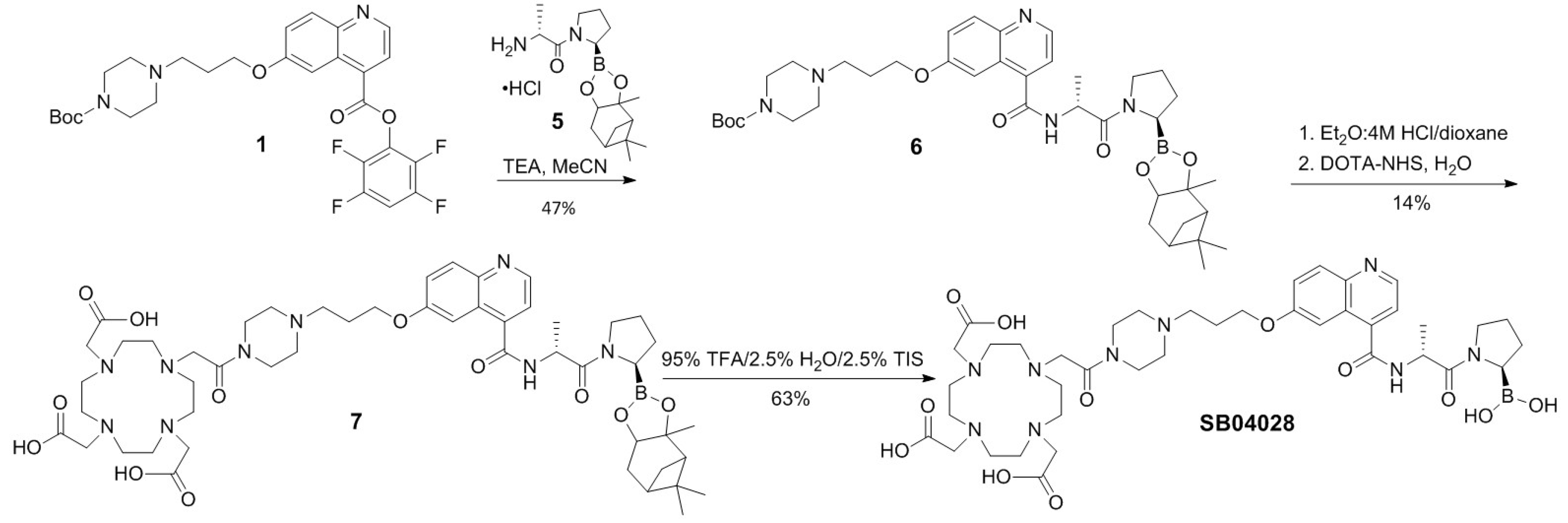
Figure 2.
Inhibition of rhFAP-mediated cleavage of a fluorogenic substrate (Suc-Gly-Pro-AMC) by varying concentrations of natGa-SB02055, natGa-SB04028 and natGa-PNT6555.
Figure 2.
Inhibition of rhFAP-mediated cleavage of a fluorogenic substrate (Suc-Gly-Pro-AMC) by varying concentrations of natGa-SB02055, natGa-SB04028 and natGa-PNT6555.
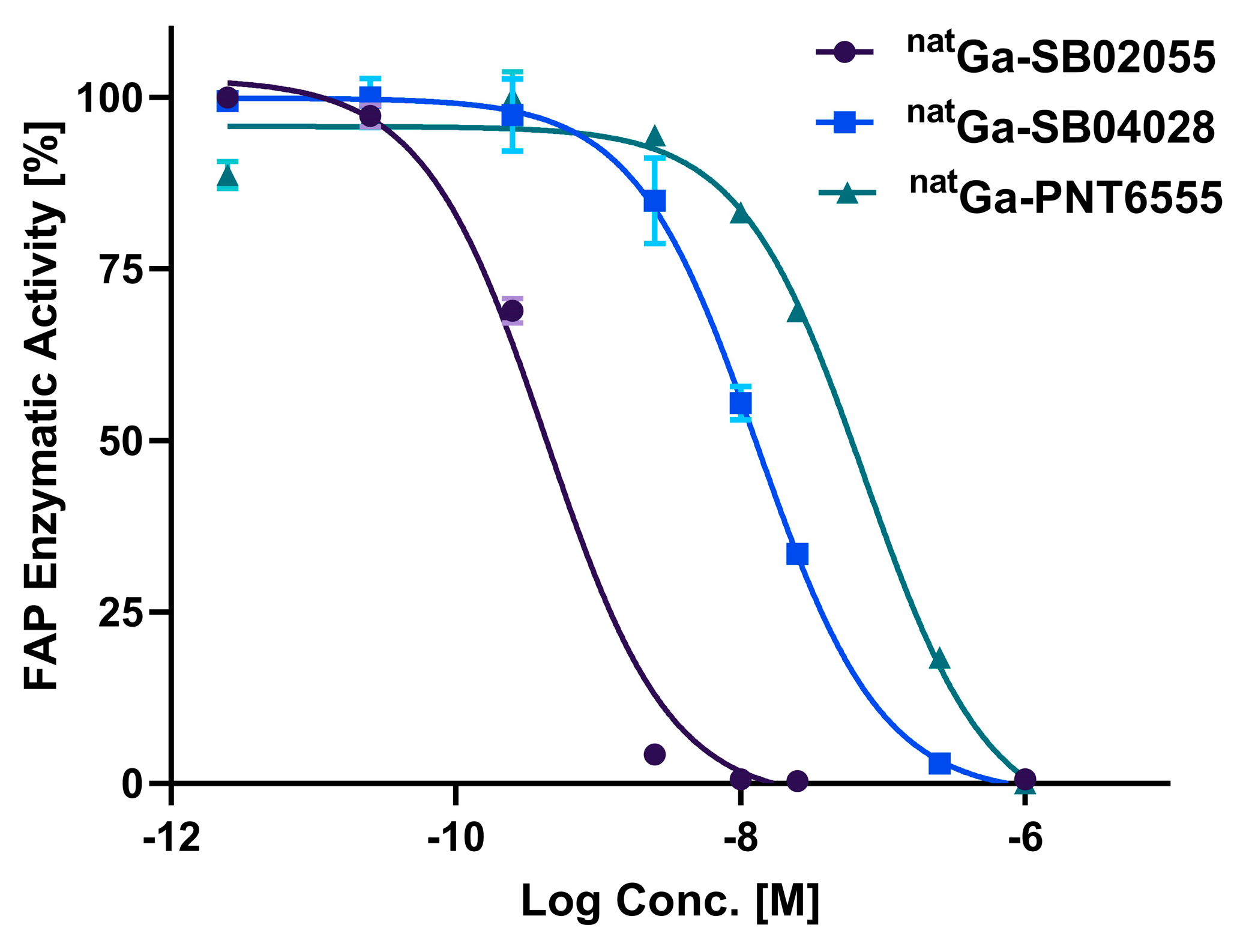
Figure 3.
Representative maximum intensity projection PET/CT images of [68Ga]Ga-SB02055 (unblocked), [68Ga]Ga-SB04028 (unblocked and blocked) and [68Ga]Ga-PNT6555 (unblocked) at 1 h pi in HEK293T:hFAP tumor-bearing male NRG mice. The mice in the blocked group were co-injected with FAPI-04 (0.5 mg/mouse). t: tumor; b: urinary bladder.
Figure 3.
Representative maximum intensity projection PET/CT images of [68Ga]Ga-SB02055 (unblocked), [68Ga]Ga-SB04028 (unblocked and blocked) and [68Ga]Ga-PNT6555 (unblocked) at 1 h pi in HEK293T:hFAP tumor-bearing male NRG mice. The mice in the blocked group were co-injected with FAPI-04 (0.5 mg/mouse). t: tumor; b: urinary bladder.
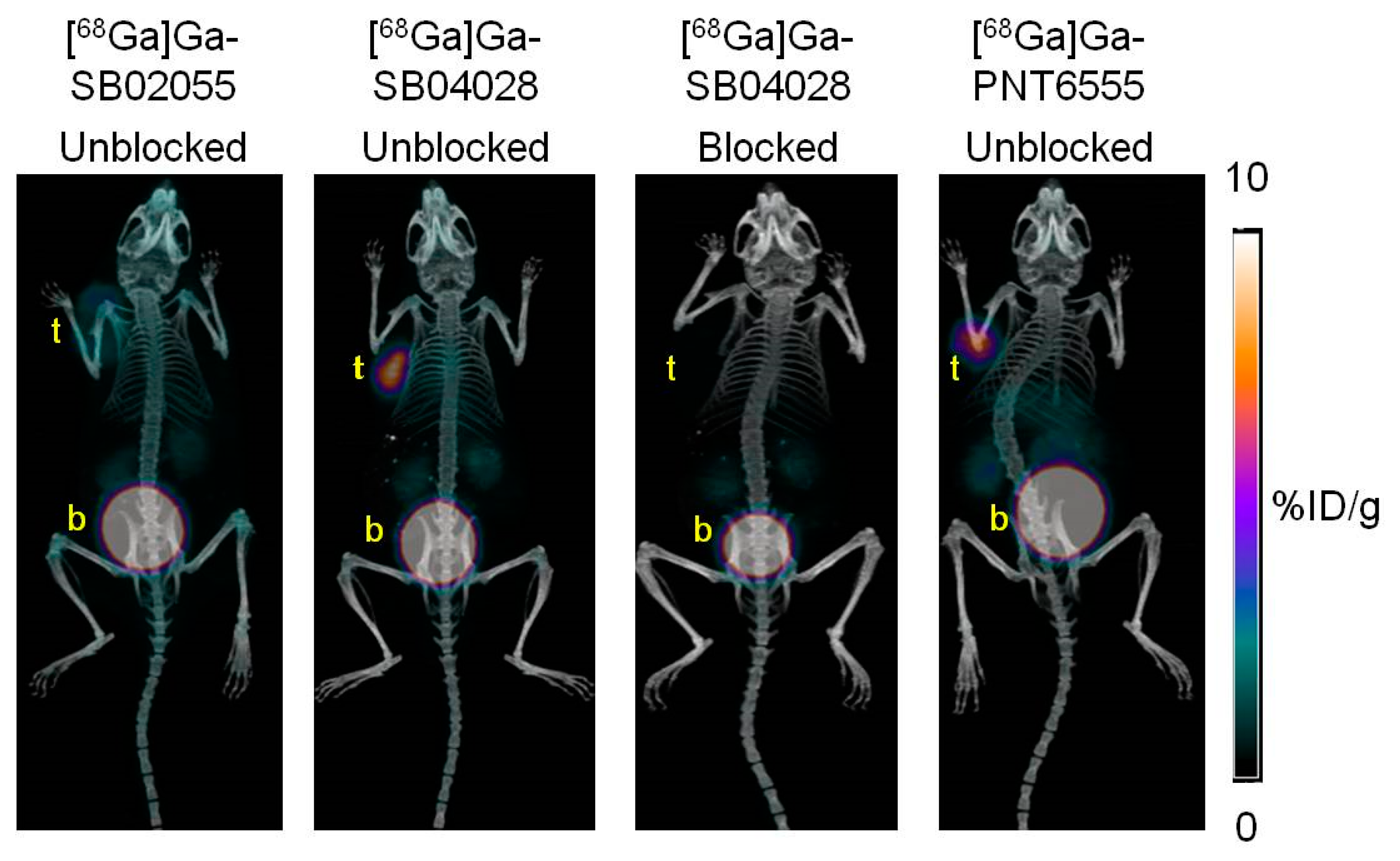
Figure 4.
Ex-vivo biodistribution data of [68Ga]Ga-SB02055, [68Ga]Ga-SB04028 and [68Ga]Ga-PNT6555 in HEK293T:hFAP tumor-bearing male NRG mice at 1 h pi (n = 4). The mice in the blocked group were co-injected with FAPI-04 (0.5 mg/mouse). * p < 0.05; ** p < 0.01, *** p < 0.001.
Figure 4.
Ex-vivo biodistribution data of [68Ga]Ga-SB02055, [68Ga]Ga-SB04028 and [68Ga]Ga-PNT6555 in HEK293T:hFAP tumor-bearing male NRG mice at 1 h pi (n = 4). The mice in the blocked group were co-injected with FAPI-04 (0.5 mg/mouse). * p < 0.05; ** p < 0.01, *** p < 0.001.
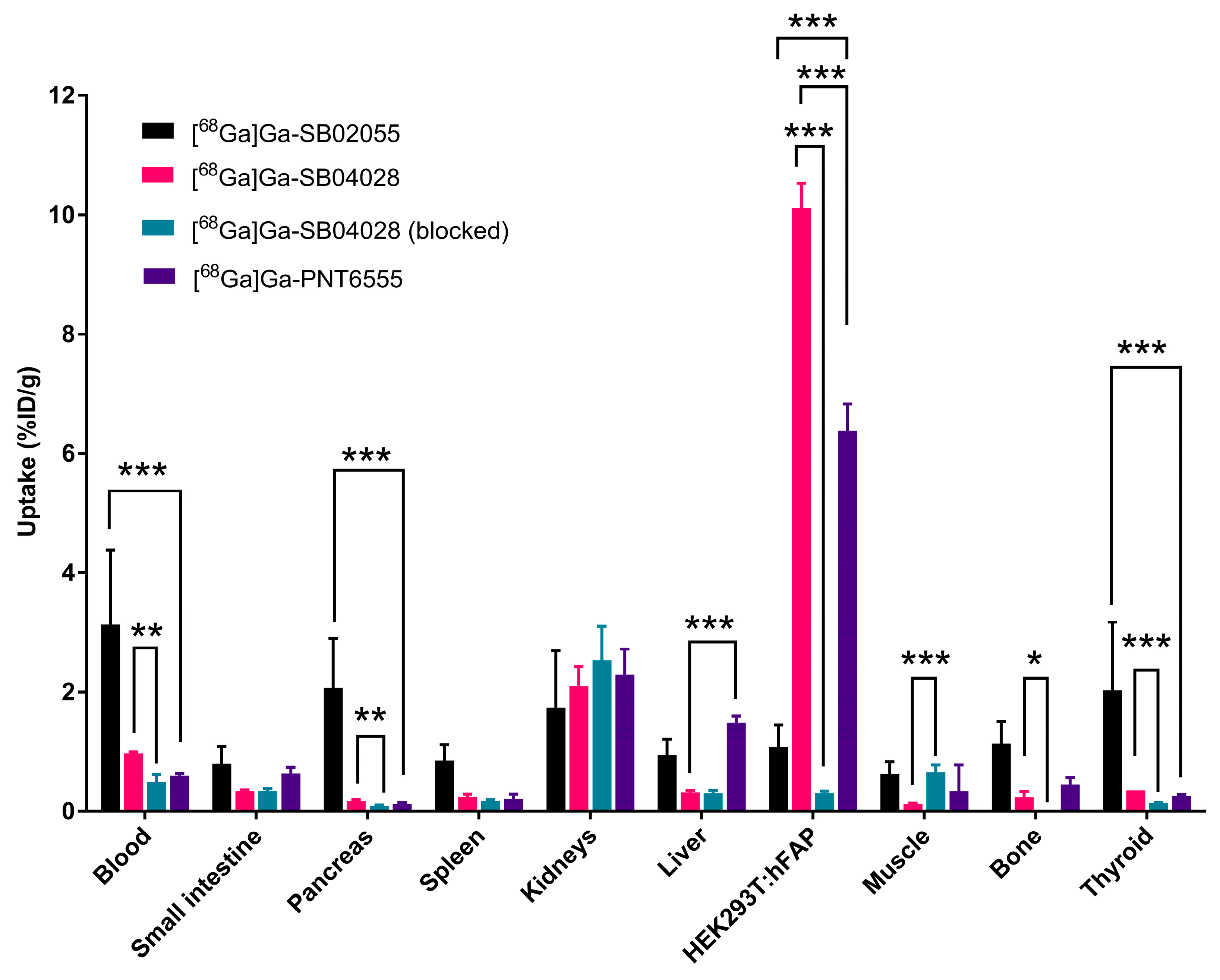
Figure 5.
Tumor/organ uptake ratios of [68Ga]Ga-SB02055, [68Ga]Ga-SB04028 and [68Ga]Ga-PNT6555 in HEK293T:hFAP tumor-bearing male NRG mice at 1 h pi (n = 4). The mice in the blocked group were co-injected with FAPI-04 (0.5 mg/mouse). *** p < 0.001.
Figure 5.
Tumor/organ uptake ratios of [68Ga]Ga-SB02055, [68Ga]Ga-SB04028 and [68Ga]Ga-PNT6555 in HEK293T:hFAP tumor-bearing male NRG mice at 1 h pi (n = 4). The mice in the blocked group were co-injected with FAPI-04 (0.5 mg/mouse). *** p < 0.001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



