
Submitted:
04 May 2023
Posted:
05 May 2023
You are already at the latest version
Abstract
We developed and applied a method for characterizing bacterial promoters genome-wide by in vitro transcription coupled to transcriptome sequencing specific for native 5’-ends of transcripts. This method called ROSE (Run-Off transcription/RNA-SEquencing), only requires chromosomal DNA, ribonucleotides, RNA polymerase (RNAP) core enzyme, and a specific sigma factor, recognizing the corresponding promoters, which have to be analyzed. ROSE was performed on E. coli K-12 MG1655 genomic DNA using E. coli RNAP holoenzyme (including σ70) and yielded 3,226 transcription start sites, 2,167 of which were also identified in in vivo studies, and 598 were new. Many new promoters not yet identified by in vivo experiments might be repressed under the tested conditions. Complementary in vivo experiments with E. coli K-12 strain BW25113 and isogenic transcription factor gene knockout mutants of fis, fur, and hns were used to test this hypothesis. Comparative transcriptome analysis demonstrated that ROSE could identify bona fide promoters that were apparently repressed in vivo. In this sense, ROSE is well-suited as a bottom-up approach for characterizing transcriptional networks in bacteria and ideally complementary to top-down in vivo transcriptome studies.
Keywords:
RNA-Seq
; run-off in vitro transcription
; RNA polymerase
; sigma factor
; TSS
; promoter
1. Introduction
Most bacteria must quickly adapt to changing environmental conditions like temperature, pH, osmolarity, or nutrient availability. Gene expression can be regulated at every step, from transcription to post-translational processing. However, transcription and, more accurately, transcription initiation is the first and most widely regulated step in bacteria. [1] One important mechanism of regulating transcription initiation is either repression or activation by DNA-binding transcription factors (TFs). Another widely found mechanism of transcription regulation is using multiple forms of RNA polymerase consisting of RNA polymerase core enzyme and different sigma factors, each allowing specific recognition of distinct promoter sequences. Bacteria can quickly reprogram their transcriptional landscape Using several sigma factors to cope with extracellular and intracellular stress factors and the changing environment. [2]
In Escherichia coli, seven sigma factors have been described: The primary sigma factor σ70 and six alternative sigma factors (σ54, σ38, σ32, σ28, σ24, and σ19). σ70 is the housekeeping sigma factor responsible for the transcription of most growth-related genes. The consensus sequence of σ70-dependent promoters (5′-TTGACA-17-TATAAT) is well-known and highly conserved among σ70-dependent promoters. However, the -10 region, which occurs about seven base pairs upstream of the transcription start site, shows a higher level of conservation than the -35 region in E. coli [3].
The ROMA method has been described for genome-wide analysis of transcription regulated by sigma factors [4]. Here, purified RNA polymerase holoenzyme is used for in vitro transcription on fragmented genomic DNA. Transcribed mRNA species are then identified by DNA microarray hybridization representing all genes of the respective genome. After the initial ROMA experiments conducted for transcriptional profiling of Bacillus subtilis, the ROMA method was successfully applied to E. coli and was used to disentangle the overlapping σ70 and σ38 regulons [5]. ROMA allows investigating the direct effects of different sigma factors without regulatory proteins.
ROMA also has limitations, including the lack of single-nucleotide resolution and transcriptional read-through at convergently oriented genes, possibly leading to false-positive signals. MacLellan et al. observed extended transcription of up to 10 kb downstream of the active promoters [6]. Although co-transcription of convergent genes regularly occurs in vivo, the frequency is significantly higher in vitro. Therefore, specifically activated promoters identified by ROMA must be confirmed by alternative methods like in vivo reporter fusions or single-promoter in vitro transcription [4].
We developed ROSE (Run-Off transcription/RNA-SEq) to overcome these limitations. ROSE employs genome-wide in vitro transcription with isolated RNA polymerase and genomic DNA. In vitro transcribed RNA analysis includes the preparation of native 5′-end-specific transcript libraries [7] and subsequent transcriptome sequencing. Mapping the sequenced 5′-ends to the genome provides distinct read stacks at the transcription start site of a given mRNA, which enables the detection of promoter sequences with single-nucleotide resolution. The method was initially developed in the frame of a Ph.D. thesis in 2013, by combining the E. coli core RNA polymerase with the ECF sigma factors of Corynebacterium glutamicum [8]. Only recently, a similar technique, RIViT-seq, was described [9]. RIViT-seq was used to identify new target genes of 11 different sigma factors in Streptomyces coelicolor. Since there are a number of technical differences between ROSE and RIViT-seq, especially with the preparation of the primary transcript libraries and the determination of transcription start sites (TSS), we like to keep the name ROSE and refer to the technical differences compared to RIViT-seq and their implications in more detail below. ROSE and RIVit-seq can be regarded as different flavours of a ‘bottom-up’ approach to transcription regulatory networks in bacteria. Therefore, they are ideally complementary to ‘top-down’ transcriptome analysis in vivo, e.g., by comparing wildtype and transcription factor mutant strains.
In this study, we demonstrate the power of ROSE by using the native E. coli RNA polymerase holoenzyme with σ70 to characterize bacterial transcriptional regulatory networks of E. coli.
2. Materials and Methods
2.1. In vitro transcription on genomic DNA fragments
Genomic DNA from E. coli K-12 MG 1655, cultivated overnight on solid lysogeny broth medium [10], was isolated with three different approaches, the Quick-DNA Universal Kit (Zymo Research), the NucleoSpin Microbial DNA Kit (Macherey-Nagel) and a Phenol-Chloroform Isoamyl alcohol DNA extraction. Each isolation method was used twiceto get three biological and two technical replicates for ROSE. For the Phenol-Chloroform extraction, the cells were treated with lysozyme, RNAse H, and proteinase K to open the cells before the phenol extraction of the DNA. All three isolation methods were made with two technical replicates. DNA was fragmented randomly to an average size of 6 kb using gTubes (Covaris). Size distribution of the fragmented template DNA was checked by Agilent Bioanalyzer using the high-sensitivity DNA kit. 1 µg of template DNA was used for a single in vitro run-off transcription reaction. In vitro run-off transcription was performed in E. coli RNA Polymerase Buffer (New England Biolabs). Template DNA and reconstituted RNAP holoenzyme were incubated at 37 °C for 15 minutes, followed by the addition of NTPs to a final concentration of 200 nM each to start the transcription reaction. After 60 minutes, in vitro run-off transcription was terminated by five-minute incubation at 65 °C. Template DNA was digested with DNase I (Roche) immediately after in vitro transcription (30 minutes, 25 °C). In vitro transcribed RNA was purified using Qiagen RNeasy MinElute Kit, including a second DNase digestion, and eluted in nuclease-free water. To obtain sufficient RNA for transcriptome sequencing, at least three independent in vitro transcription reactions were combined. Nucleic acid concentration and purity were determined with an Xpose spectrophotometer. RNA Quality and size distribution were checked on Agilent Bioanalyzer 2100 using the RNA Pico assay. PCR amplification has ruled out residual DNA contamination with specific primers binding to the E. coli genome.
2.2. Cultivation of the E. coli knockout strains
The following transcription factor deletion mutants were derived from Coli Genetics Stock Center (CGSC): #7636 (BW25113), #8758 (Δfur), #10443 (Δfis), #9111 (Δhns). Deletion strains were cultivated in liquid LB broth containing 50 mg/mL kanamycin in shaking flasks (37 °C, 180 rpm). After inoculation to an OD600 of 0.05 from an overnight liquid culture, 2 mL cell suspensions were harvested at an OD600 of 0.8 in the exponential growth phase and immediately frozen in liquid nitrogen and stored at -80 °C. For the wildtype strain, another sample was taken after cells reached the stationary growth phase.
2.3. Construction of primary transcript libraries
A previously established procedure has been used to prepare the mRNA for sequencing and to enrich primary, unprocessed transcripts [7]. Shortly, 100 ng purified in vitro-transcribed RNA was fragmented to an average size of 500 nt. For in vivo-based libraries, 5 µg total RNA was initially subjected to Ribo-Zero treatment (Illumina, San Diego, USA) to deplete rRNA before fragmentation. Next, transcripts harboring a 5′ di- or monophosphate were digested by terminator exonuclease (Epicentre). RNA index adapters were ligated for noise reduction in the sequencing. Furthermore, the indexed transcripts were treated with RNA 5′-polyphosphatase to enable the ligation of specific sequencing adapters. RNA adapters were ligated, and RNA was then reverse-transcribed to cDNA using a sequence-independent loop adapter. With 18 cycles of PCR, cDNA was amplified using barcoded primers to generate a multiplexed cDNA library ready for sequencing with Illumina technology.
2.4. Sequencing and data processing
High throughput single-end sequencing (1x75bp) was conducted on Illumina MiSeq (San Diego, USA). Reads were quality trimmed using Trimmomatic v0.3.5 [11] with the parameters: TRAILING:3, MINLEN:39. Forward reads were mapped to the respective Escherichia coli K-12 MG1655 (U00096.3) genome sequence using bowtie2 v2.3.0 [12] in single-end mode, as the 5′-ends of transcripts were of particular interest for TSS identification.
2.5. Sequence analysis
After identification of transcription start site (TSS) positions with ReadXplorer [13], upstream sequences (-1 to -49) were subjected to motif enrichment analysis using either Improbizer [14] (available at: https://users.soe.ucsc.edu/~kent/improbizer/improbizer.html) or MEME v4.1.2 [15]. Finally, sequences were aligned at conserved motif positions, and sequence logos were generated using WebLogo v3.7.0 [16]. For higher accuracy, only TSS positions detected in at least four of the six ROSE approaches were considered for sequence analysis.
3. Results
3.1. Development of ROSE and application to the analysis of σ70-dependent promoters in E. coli
The ROSE method was developed based on ROMA [4], which used DNA microarrays for genome-wide run-off transcription analysis. Accordingly, run-off transcription assays were performed employing the commercially available σ70-saturated form of E. coli RNA polymerase (Eσ70). In contrast to ROMA and to RIViT-seq [9], template DNA has been fragmented to an average size of 6 kb by shearing instead of restriction enzyme treatment to avoid bias by unequal distribution of restriction enzyme recognition sites or by cutting within a promoter. In addition, RNA yield was increased by using commercially available Tris HCl buffer (NEB, Ipswich, USA) instead of the potassium glutamate-based buffer system previously used in ROMA (data not shown). Although ROSE and RIViT-seq are both aiming for a genome wide in vitro-transcriptome, there are distinct technical differences in the two approaches (See Supplemental Table S1 for a complete list). The focus of ROSE is the construction of high-quality primary transcript libraries. Therefore, the digestion of RNA having a 5′ di- or monophosphate is necessary to maximize the purification of unprocessed, primary bacterial transcripts. Moreover, ROSE uses index adapter ligation to reduce the noise in the sequencing, and the TSS were identified in an automated fashion using the software ReadXplorer [13,17].
Before sequencing, in vitro transcribed mRNA was subjected to native 5′-end-specific transcript library preparation [7]. Sequencing on Illumina MiSeq yielded around 2 million reads per library (See Supplemental Table S2), which were quality-filtered and mapped to the respective reference genome (U00096.3). Three different approaches were tested for the isolation of chromosomal DNA of E. coli K-12 MG1655. The different isolation methods did not result in notable differences of the quality or the distribution of reads (See Supplemental Figure S1).
Mapped reads were visualized using the ReadXplorer software [17], and transcription start site (TSS) detection was performed with the same tool and automatic parameter estimation (See Supplemental Table S3). The automatic TSS detection was able to identify 3,226 possible TSS detected in at least four of the six ROSE runs. Depending on their location relative to known genes, the TSS were classified into four categories according to Sharma et al. [18]: primary TSS (44.6%), intragenic TSS (24.4%), antisense TSS (27.1%) and orphan TSS (3.9%). Primary TSS comprises all TSS located in a suitable distance and direction to a protein-coding region or a known transcript. Intragenic TSS are located within a coding sequence. In sense orientation, antisense TSS are situated on the opposite strand of a protein-coding region up to 100 bases upstream or downstream (± 100 nt), and orphan TSS do not meet any of these criteria.
To validate the suitability of the ROSE method for promoter identification, upstream sequences of 50 nt lengths (positions -1 through -49 relative to the TSS) were extracted for further analysis. All 3,226 putative promoter sequences were subjected to motif enrichment analysis using Improbizer [14]. Two distinct motifs corresponding to -10 and -35 regions of σ70-dependent promoters were detected independently (Figure 1). As expected from previous studies, the -10 region shows a considerably higher level of conservation [19]. 3,128 putative promoter sequences contained a region similar to the σ70 -10 consensus.
In contrast, a -35 consensus motif, namely a conserved ttGA about 35 nucleotides upstream of the transcription start site, was derived from 2,922 promoter sequences. A total of 2,838 putative promoter sequences contained both -10 and -35 regions. Only eleven sequences did not resemble either the -10 or the -35 consensus sequence.
It is apparent that Eσ70 recognizes natural σ70-dependent promoters in vitro with high specificity and initiates transcription at well-defined nucleotides. Transcription initiation occurred preferentially at purine bases (A/G), which was observed in 81.0% (50.3% A and 30.7% G) of the detected promoters. Interestingly, the base directly upstream of the TSS at position -1 prefers pyrimidine bases, with 77.3% of the promoters harboring T (41.3%) or C nucleotides (36.0%) at the respective position. Both findings align with in vivo transcriptional profiling studies, reporting similar nucleotide preferences of 78.6% purine bases at +1 and 80.2% pyrimidine bases at -1 [19,20].
3.2. Detailed Promoter Analysis by Comparison to Experimentally Characterized Promoters Listed in RegulonDB
The genome of Escherichia coli K-12 MG1655 contains 4,146 genes organized in 2,376 transcription units. 1,523 transcription units are monocistronic, whereas 853 operons have more than one gene [21]. Thus, at least 2,376 primary TSS are expected to be found, possibly except for TSS of promoters that need to be activated by factors not contained in the in vitro transcription assay. The RegulonDB database [22] includes the most comprehensive information regarding the transcriptional regulation of E. coli, including experimentally determined transcriptional start sites of the strain K-12 MG1655. In a subset of the database, TSS are assigned to the different sigma factors and provided with a level of evidence (Confirmed, Strong, or Weak), depending on the informative value of the method for TSS identification. For the following comparison, only those TSS were considered that belong to the classes “Confirmed” or “Strong”. In addition, to cope with different experimental methods of TSS identification and issues of the template, such as the degree of supercoiling, a deviation of three nucleotides in either upstream or downstream direction has been allowed to compare two TSS positions. The mapped TSS show a clear peak, with 64.7% having zero and 7.0% having one nucleotide deviation in either direction (See Supplemental Figure S2).
In RegulonDB, 881 TSS are classified as σ70-dependent; thereof, 352 (40.0 %) were also identified in the ROSE-Eσ70 experiment. A total of 30 TSS found in our ROSE experiment are assigned to other sigma factors in the database with no affiliation to σ70. 25 of these TSS are classified as σ38- and the other five as σ32-dependent promoters. However, it is known that the consensus sequence of σ38-dependent promoters is similar to the σ70 consensus sequence, and a clear distinction between both promoter sets cannot be made [23]. Therefore, promoter sequences identified by ROSE-Eσ70 but listed as σ38-dependent in RegulonDB were compared to those of σ38-dependent promoters that ROSE did not recognize. Again 50 nt upstream of the TSS have been extracted and analyzed for conserved motifs. Comparing the resulting motifs clearly shows differences, mainly in the -10 regions. The presumed σ38-dependent promoters show conserved bases at all positions from -12 through -7 (TATACT), whereas in the σ38-dependent promoters not detected by ROSE-Eσ70 only the bases at -12, -11, and -7 are conserved (TANNNT). Additionally, there is a C at position -13, upstream of the -10 region, described earlier as a distinct sequence characteristic in σ38-dependent promoters [24]. Another distinguishable feature of the exclusively σ38-dependent promoters is a highly conserved GC at positions -33/-32 (ttGC), occurring in most σ38-dependent promoter sequences, with a higher conservation of the TT at position -35/-34 in the promoters present in ROSE-E Eσ70 (Figure 2).
Following the same reasoning, five predicted false-positive σ32-dependent promoters were compared to 66 σ32-dependent promoters from RegulonDB that ROSE did not detect. Due to the low number of five false-positive promoters, no precise consensus sequence could be identified in the -10-region (data not shown). However, the similarity of the -10-region of these false-positive σ38-dependent promoters to those of σ70-dependent promoters suggests that ROSE-Eσ70 falsely identifies these promoters as σ70 promoters, possibly due to in vivo regulatory mechanisms in the in vitro ROSE-Eσ70 system.
3.3. Comparison of the ROSE Data to Existing Comprehensive Genome-wide in vivo RNA-Seq Data Sets of E. coli K-12 MG1655
To date, genome-wide transcription start site determination is mainly done by analyzing in vivo transcribed mRNA via approaches like dRNA-Seq [18,25]. To assess the sensitivity and selectivity of ROSE, results were compared to a transcriptome study by Thomason et al. [20] and another high-throughput transcription initiation mapping study included in RegulonDB [22]. Both studies were conducted on shaking flask cultivations of Escherichia coli MG1655 in different media. After enriching 5′ triphosphorylated RNA species and high-throughput sequencing, they detected 14,865 TSS and 5,197 TSS, respectively [20,22]. Although both studies relied on transcriptome sequencing for TSS identification, their suitability for validating ROSE is limited because no specific sigma factor-promoter interaction can be examined. However, as we performed ROSE using the primary sigma factor σ70, it was assumed that there was reasonable overlap in detected TSS.
Comparing the three TSS datasets showed that 2,006 (62.2%) of the TSS detected by ROSE were also determined by Thomason et al., while 168 further TSS are confirmed by the study included in RegulonDB. A set of 755 TSS was contained in all three datasets (Figure 3). Again, a deviation of three nucleotides has been allowed to compare two TSS positions. Here, ROSE-Eσ70 and RegulonDB exactly matched in 76.0% (±1 bp: 13.9%) of the overlapping TSS, while ROSE-Eσ70 and Thomason et al. had an exact match at 86.2% (±1 bp: 7.6%) of the TSS (data not shown).
3.4. Transcription Start Sites of Promoters That are Repressed Under Standard in vivo Assay Conditions are Comprehensively Identified in ROSE Experiments
By design, ROSE should be able to identify two classes of promoters not represented in RegulonDB. The first class comprises those present in the E. coli genome but not described in existing TSS mapping studies. The second class includes repressed or not activated under standard in vivo testing conditions. In total, 2,303 transcriptional start sites detected by ROSE-Eσ70 are yet undescribed, according to RegulonDB. Thomason et al. identified 1,254 of those TSS in vivo. The remaining 1,049 upstream regions were subjected to motif enrichment analysis using Improbizer [14]. To remove possible background signals, the sequences have been sorted by the -10-region score given by Improbizer, which corresponds to the similarity of a given sequence to the detected consensus motif. A randomized control run yielded a 95% confidence score of 6.20 for a given sequence. After filtering with this value as a cut-off, 598 sequences remained, containing a precise σ70 consensus sequence. Due to this, it can be speculated that these promoters were repressed under the conditions tested in the in vivo studies. Manual inspection showed regulator binding sites around many of these promoters, suggesting that transcription from those promoters is prevented in vivo by known transcriptional regulators such as H-NS, Fur, or Fis. In the following, we describe two exemplary promoter regions for each transcriptional regulator, H-NS, Fur, or Fis, in more detail. We performed in vivo experiments for each regulator with defined transcription factor knockout mutants from the KEIO collection [26] to validate the results observed with ROSE. The knockout mutants were JW1225-2 for Δhns, JW0669 for Δfur, and JW3229-1 for Δfis. Sequencing on Illumina MiSeq yielded, on average, 0.91 million reads per library (See Supplemental Table S4). The mapped reads were visualized using the ReadXplorer software [17], and transcription start site (TSS) detection was performed with the same tool and automatic parameter estimation (See Supplemental Table S5).
The genes stpA (b2669) and ftnA (b1905) are both negatively regulated by H-NS, a global transcriptional silencer [28], which is involved in the regulation of 5% of all E. coli genes [29]. In both cases, H-NS binds upstream of the TSS and leads to a repression of transcription [30,31] (Figure 4A). The gene stpA has a TSS at position 2,798,556 and a perfect σ70-like -10 region (TATAAT). The gene ftnA with a TSS at position 1,988,682 has a complete σ70-like promoter (TTGCAA-16-TATAGT). Both genes showed no transcription in the wildtype strain, but transcriptional activity was measured in the Δhns knockout strain and in the ROSE approach (See Supplemental Figures S3 and S4). Moreover, both genes were also described by Thomason et al. and RegulonDB.
The TSS of yjjZ (b4567) has already been described for genomic position 4,605,777 in the E. coli MG1655 genome. According to EcoCyc, there are two ferric uptake regulator (Fur) binding sites in the vicinity of the transcription start site of yjjZ [27,32] (Figure 4B). Although the respective promoter harbors a σ70-like consensus sequence (TTGCAA-18-TATGAT), Thomason et al. did not detect a transcription start site for yjjZ, suggesting efficient transcriptional repression in vivo. This has been validated in our in vivo experiment, where the wildtype strain has shown no activity of the yjjZ gene. However, in the Δfur knockout strain, transcription from the σ70 promoter is measurable. Moreover, the TSS has been identified clearly in vitro (1,494 read starts) with the ROSE method. (See Supplemental Figure S5). Another example of a gene activated by the regulator Fur is the gene fepA (b0584) [33,34](Figure 4B). The fepA promoter has a σ70-like consensus sequence (TTGCAG-14-TATTAT) and was not detectable in vivo in the wildtype strain. However, both the Δfur knockout strain (508 read starts, in vivo) and ROSE (348 read starts, in vitro) show transcriptional activity for the gene fepA (See Supplemental Figure S6).
The gene for the DNA-binding transcriptional dual regulator GlcC has a TSS at position 3,128,206. It has an unusual -10 region (CATAAT) and a -35 sequence (TTAACT). As stated in EcoCyc, the gene’s promoter region has four binding sites for the global regulator Fis [35] (Figure 4C), which causes gene repression. This repression has been validated in the in vivo experiment with the wildtype strain and the Δfis knockout mutant. The wildtype strain showed minimal read starts (7 read starts) for the gene in vivo. In the knockout strain, the amount of read starts was five times higher than in the wildtype strain, demonstrating the higher transcription of glcC in the absence of the Fis regulator. However, the most read starts and the strongest transcription of the gene were identified by the in vitro approach ROSE (493 read starts) (See Supplemental Figure S7). Another exciting gene is aer (b3072), which shows a clear transcription start site in ROSE and the Δfis knockout strain at position 3,219,346 and harboring a σ70-like consensus sequence (TTGTGC-19-TAACAT). This transcription start site is also described in the publication of Thomason et al. but is not defined in RegulonDB. Nevertheless, RegulonDB contains a Fis binding site with an unknown function upstream of the aer gene (Figure 4C). The fact that ROSE and the Δfis knockout strain showed transcriptional activity, but there was no transcription in the wildtype strain suggests that Fis is a transcriptional repressor of aer (See Supplemental Figure S8).
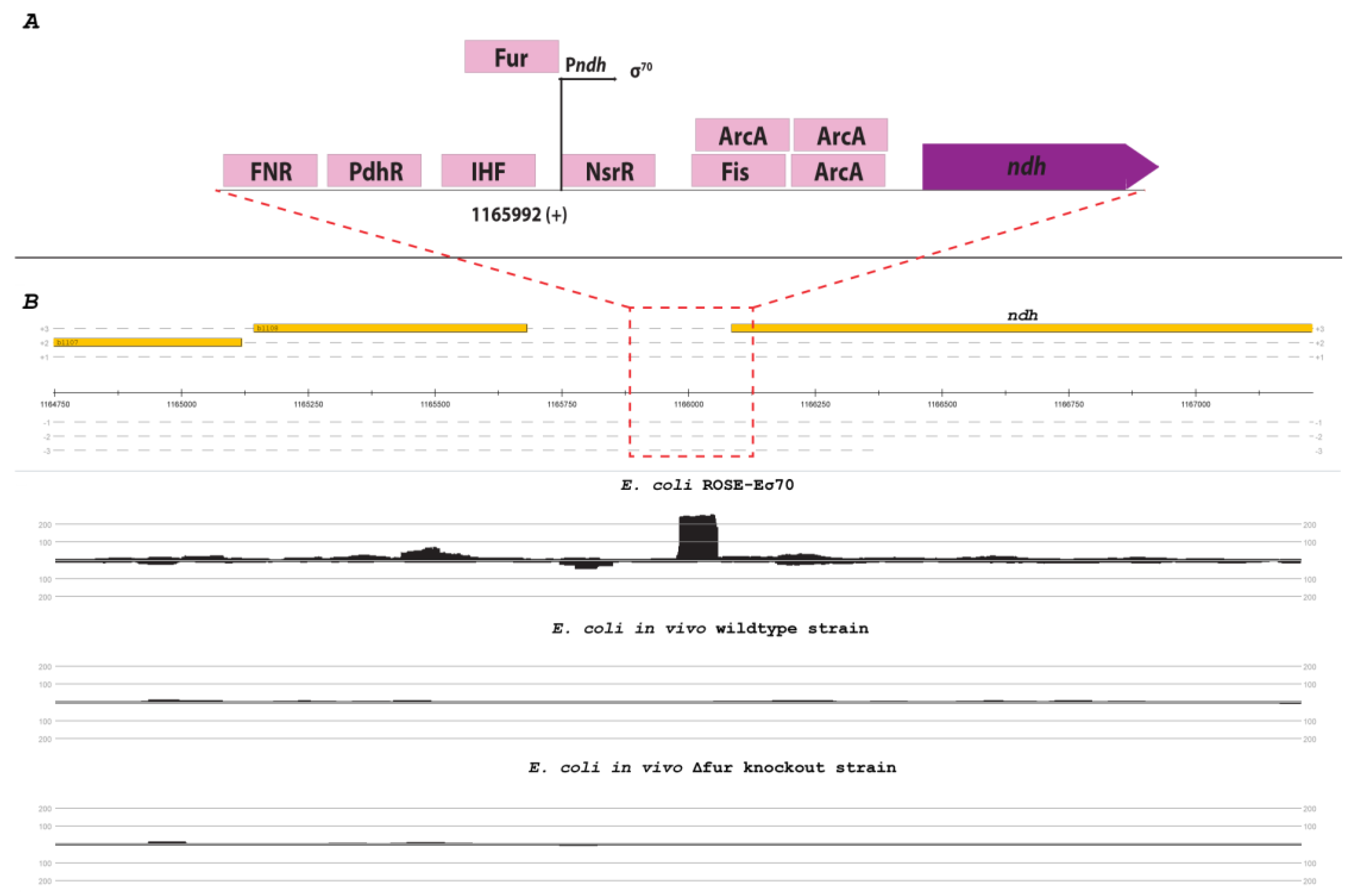
The gene ndh of E. coli expresses the NADH dehydrogenase II. The corresponding promoter Pndh is located at position 1,165,992 of the genome and is harboring a standard σ70-like consensus sequence (TTGGTA-21-TATTCT). This gene is negatively regulated by multiple transcription factors like FNR [36], Fur-Fe2+ [37], and NsrR [38]. Due to the high number of different repressors of ndh, no transcription was detectable in the E. coli wildtype strain or the single knockout strains in vivo. However, the ROSE method showed a distinct TSS at the known position of Pndh with over 200 read starts (Figure 5).
These findings underline that the bottom-up approach employed within ROSE aids the identification of previously undetected TSS, especially those that are repressed or not activated under a given in vivo testing condition.
3.5. Promoters activated by transcriptional regulators in vivo are not identified in vitro
A different type of σ70-dependent promoters comprises those specifically activated by transcriptional regulators in vivo, possibly allowing for lesser conservation of promoter motifs. For example, the well-known promoter of the araBAD operon (CTGACG-18-TACTGT) of E. coli can be activated and repressed by the transcriptional regulator AraC in vivo, depending on the availability of arabinose [39,40]. It is furthermore activated by the cAMP receptor protein (CRP) in vivo [41,42,43]. Since none of these regulators are included in the ROSE in vitro transcription assay, neither activation nor repression of pBAD should occur. Interestingly, no TSS has been identified upstream of the araBAD operon by ROSE-Eσ70, suggesting that activation by CRP and/or AraC is indeed critical for transcription initiation at pBAD. Another instance is the σ70-dependent promoter of csiE (b2535), known to be activated by both CRP and H-NS in vivo [44,45]. Dual activation allows for relatively weak -10 and -35 hexamers (TTCCCT-18-AACTTT). Consequently, the respective TSS at position 2,665,401 is included in both in vivo-based studies but was not detected by ROSE-Eσ70. The σ70-dependent promoter of alkA is activated upon binding Ada, a DNA repair protein, which is a critical component of the adaptive response [46,47]. The promoter of its TSS at position 2,147,559 harbors a well-conserved -10 region (TATGCT) but has no -35 region. In contrast to both in vivo studies, it is not detected by ROSE-Eσ70, obviously requiring activation by Ada. In conclusion, ROSE robustly and comprehensively identifies bona fide promoters and those potentially repressed under in vivo conditions. It also allows drawing conclusions from negative results, predicting efficient activation in vivo.
4. Discussion
In this study, we developed the ROSE method for genome-wide in vitro transcriptional profiling and validated it by exploring the σ70 regulon of E. coli K-12 MG1655.
Like ROMA [4], ROSE is a bottom-up approach aiming to assemble the transcriptional machinery from a few simple parts. It perfectly complements in vivo transcriptome profiling, which can be regarded as a top-down approach. The latter represents the much more complex situation that includes indirect interactions, making such data harder to interpret.
In vitro transcription analyzed by genome-wide methods, as in ROMA [4], RIViT-seq [9] or ROSE, provides several benefits compared to traditional single gene-oriented approaches. The in vitro methods are free from transcriptional repression, allowing for the detection of promoters negatively regulated at standard cultivation conditions. These simple bottom-up approaches enable the precise dissection of overlapping sigma factor networks by employing single sigma factor proteins in the assay, thereby focusing the observation on the direct effects of the respective regulators. It has furthermore been shown by the pioneering work of MacLellan et al. [4] that linear DNA conformation and relatively low complexity of in vitro systems maintain the specificity of transcription initiation. In contrast to ROMA, the particular RNA-Seq protocol used here provides clear evidence that even the transcriptional start nucleotide is the same as in vivo. Single-nucleotide resolution furthermore allows direct TSS identification and, consequently, the derivation of promoter sequences and their consensus motifs. Technically, ROSE has some additional features to the RIViT-seq technique. First is the shearing of the DNA, avoiding bias by restriction enzyme digestion. Second is the focus on establishing an enriched unprocessed primary transcript library by removal of transcripts having 5′ di- and monophosphate ends. With the usage of the index adapter before the Illumina adapter ligation, some noise in the sequencing is reduced, leading to a higher quality of the sequenced library. Whereas ROSE is optimized for high accuracy TSS detection, RIViT-seq has an advantage in differential expression analysis by using whole transcriptomics as an additional data set to its primary transcript libraries. As such, it might also detect 5′-ends of transcripts that are prone to very fast 5′-end decay. Moreover, ROSE and RIViT-seq have their individual approaches to identify transcription start sites. Therefore, a combination of both techniques could result in a more comprehensive determination of novel TSS and in a more complete identification of target genes of interesting transcription factors.
The ROSE analysis of the E. coli σ70 regulatory network proved consistent with in vivo-based transcriptome studies. Accordingly, 2,174 of 3,226 TSS (67.4%) identified by ROSE were also described earlier in comprehensive reference studies [20,22], while ROSE-Eσ70 additionally identified 598 promoters with conserved σ70 motifs. One major cause for differences likely is the simple composition of the ROSE bottom-up in vitro transcription assay, which does not resemble the complex in vivo situation by design. Nonetheless, genome-wide in vitro transcription using homologous E. coli RNAP showed high specificity with only eleven detected TSS lacking a typical σ70 promoter motif. Interestingly, ROSE-Eσ70 data also contained TSS earlier assigned to other sigma factors (σ38, σ32). Apart from possible dual recognition in vivo, linear template DNA conformation could have facilitated this issue, as the σ70-containing holoenzyme is known to preferentially initiate transcription on more highly supercoiled DNA [48,49]. However, as proposed earlier and confirmed by recent studies, actively transcribing RNA polymerase produces a (+) supercoiling domain ahead and a (-) supercoiling domain behind it, even on linear template DNA [50,51,52]. This activates supercoiling-dependent promoters like the leu-500 promoter from E. coli [52] and suggests that the linear template within the ROSE assay exhibits a certain degree of supercoiling and, therefore, supercoiling-dependent promoters should be, in principle, identified in ROSE experiments. Furthermore, complementary in vivo experiments were used to demonstrate the identification of promoters, which are repressed under standard testing conditions in vivo with ROSE. In vivo knockout strains showed no expression of the respective knockout genes, indicating the knockout’s functionality. The in vivo experiments demonstrated that the three tested transcription factors, Fur, Fis, and H-NS, lead to a repression of specific genes, which could only be identified with the transcription factor knockout strain in vivo. Nevertheless, we demonstrated that ROSE enables the identification of TSS that are detectable in vivo only in specific knockout strains. Furthermore, we found many genes like yjjZ, described as regulated by one of the three tested transcription factors, which do not show any read starts in the in vivo experiments but have noticeable TSS in ROSE. These are more complex promoters with multiple repressor binding sites, and numerous knockouts would be needed to identify this TSS in vivo. For example, the promoter Pndh of the gene ndh is repressed by different regulators like FNR [36], Fur-Fe2+ [37], and NsrR [38] and showed no activity in all strains in vivo. However, ROSE identified a TSS for the ndh gene and demonstrated the method’s power due to its minimalistic construction (Figure 5). But the minimalistic structure of the system leads to limitations regarding more complex regulatory systems. This results in a lack of identification of promoters that need activators, like the promoter of the araBAD operon or the promoter of the gene csiE.
Therefore, expanding ROSE appears possible by adding regulators, such as transcription factors, or metabolite effectors to directly investigate their influence on transcriptional regulation. This has, for instance, been demonstrated for the regulator protein DksA [53] and the small alarmone ppGpp [54,55] in single-promoter in vitro transcription assays. Moreover, it became obvious that genome-wide in vitro transcription studies are not limited to E. coli genomes. For example, the E. coli RNAP holoenzyme has also been used successfully for in vitro transcription of promoters from other bacteria [56,57]. Furthermore, RIViT-seq [9] demonstrated that a reconstitution of the E. coli RNAP core enzyme with sigma factors of Streptomyces coelicolor is possible for genome-wide in vitro transcription studies. However, problems might arise if the interaction of the organism-specific RNAP core enzyme with distinct promoter motifs or sigma factors is crucial for the transcription. Therefore, homologous RNAP complexes have been isolated and functionally tested for a broad spectrum of bacteria like Bacillus subtilis [58], Pseudomonas aeruginosa [59], Mycobacterium tuberculosis [60] or Corynebacterium glutamicum [61] and can be used in in vitro transcription systems. Therefore, ROSE and RIViT-seq could be applied to almost any other bacteria, including those with highly complex sigma factor networks, bacteria without developed genetic engineering technologies, or highly pathogenic ones.
5. Conclusions
The global in vitro transcription method ROSE presented in this study is the perfect addition to classical global in vivo and local in vitro transcription assays due to its simplicity and wide range of possible applications. It can be used to identify the primary effects of different sigma factors and their binding motifs with single-nucleotide resolution. We are expanding the technology by transferring it to other bacteria and by adding regulatory proteins and small molecules.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1. Technical comparison between the two genome-wide in vitro transcription techniques ROSE and RIViT-seq. Table S2: Mapping statistics for all six 5′-end specific ROSE- Eσ70 libraries, Table S3: Mapping statistics for four 5′-end specific in vivo libraries, Table S4: Transcription start site detection parameters for ROSE-Eσ70 libraries, Table S5: Transcription start site detection parameters for in vivo libraries, Figure S1: Mapped reads of the 5′-end-specific transcript library, Figure S2: Distribution of the identified TSS by ROSE and by Thomason et al. in relation to their distance to the published TSS in RegulonDB, Figure S3: Coverage of mapped reads on the reference genome with focus on gene stpA, Figure S4: Coverage of mapped reads on the reference genome with an emphasis on gene ftnA, Figure S5: Coverage of mapped reads on the reference genome with an emphasis on gene yjjZ, Figure S6: Coverage of mapped reads on the reference genome with focus on gene fepA, Figure S7: Coverage of mapped reads on the reference genome with an emphasis on gene glcC, Figure S8: Coverage of mapped reads on the reference genome with focus on gene aer.
Author Contributions
Conceptualization, P.S. and D.B.; methodology, P.S and D.B..; software, P.S and D.B..; validation, P.S. and D.B.; formal analysis, P.S. and D.B.; investigation, P.S.; resources, J.K.; data curation, P.S. and D.B.; writing—original draft preparation, P.S. and D.B.; writing—review and editing, P.S., D.B., T.B., and J.K.; visualization, P.S.; supervision, T.B. and J.K.; project administration, P.S., D.B., T.B., and J.K..; funding acquisition, J.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported initially by grant Ka1722/1-1 from the Deutsche Forschungsgemeinschaft (DFG). In addition, TB, DB, and JK gratefully acknowledge support from the “European Regional Development Fund (EFRE)” through the project “Cluster Industrial Biotechnology (CLIB) Kompetenzzentrum Biotechnologie (CKB)” (34.EFRE-0300095/1703FI04). We further acknowledge support for the Article Processing Charge by the DFG and the Open Access Publication Fund of Bielefeld University.
Data Availability Statement
Coverage tracks imported into the UCSC genome browser session (only for access during reviewing period): https://genome.ucsc.edu/s/dbrandt/schmidt_brandt_K-12_MG1655The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus [62] and are accessible through GEO Series accession number GSE159312 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE159312). To review GEO accession GSE159312: Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE159312 Enter token mtgpyqmullwlrcv into the box.
Acknowledgments
We acknowledge Anika Winkler and Katharina Hanuschka (Center for Biotechnology, Bielefeld University) for assistance during next-generation Illumina sequencing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Browning, D.F.; Busby, S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004, 2, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Browning, D.F.; Busby, S.J.W. Local and global regulation of transcription initiation in bacteria. Nat. Rev. Microbiol. 2016, 14, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Shultzaberger, R.K.; Chen, Z.; Lewis, K.A.; Schneider, T.D. Anatomy of Escherichia coli sigma70 promoters. Nucleic Acids Res. 2007, 35, 771–788. [Google Scholar] [CrossRef] [PubMed]
- Maclellan, S.R.; Eiamphungporn, W.; Helmann, J.D. ROMA: an in vitro approach to defining target genes for transcription regulators. Methods 2009, 47, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Maciag, A.; Peano, C.; Pietrelli, A.; Egli, T.; Bellis, G. de; Landini, P. In vitro transcription profiling of the σS subunit of bacterial RNA polymerase: re-definition of the σS regulon and identification of σS-specific promoter sequence elements. Nucleic Acids Res. 2011, 39, 5338–5355. [Google Scholar] [CrossRef] [PubMed]
- Maclellan, S.R.; Wecke, T.; Helmann, J.D. A previously unidentified sigma factor and two accessory proteins regulate oxalate decarboxylase expression in Bacillus subtilis. Mol. Microbiol. 2008, 69, 954–967. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer-Sancar, K.; Mentz, A.; Rückert, C.; Kalinowski, J. Comprehensive analysis of the Corynebacterium glutamicum transcriptome using an improved RNAseq technique. BMC Genomics 2013, 14, 888. [Google Scholar] [CrossRef] [PubMed]
- Busche, T. Analyse von Regulationsnetzwerken der Extracytoplasmic Function (ECF)-Sigmafaktoren in Corynebacterium glutamicum. Universität Bielefeld 2013. [Google Scholar]
- Otani, H.; Mouncey, N.J. RIViT-seq enables systematic identification of regulons of transcriptional machineries. Nat. Commun. 2022, 13, 3502. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Molecular cloning : a laboratory manual., 4th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, N.Y, 2012. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Hilker, R.; Stadermann, K.B.; Doppmeier, D.; Kalinowski, J.; Stoye, J.; Straube, J.; Winnebald, J.; Goesmann, A. ReadXplorer--visualization and analysis of mapped sequences. Bioinformatics 2014, 30, 2247–2254. [Google Scholar] [CrossRef]
- Ao, W.; Gaudet, J.; Kent, W.J.; Muttumu, S.; Mango, S.E. Environmentally induced foregut remodeling by PHA-4/FoxA and DAF-12/NHR. Science 2004, 305, 1743–1746. [Google Scholar] [CrossRef]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–73. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: a sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Hilker, R.; Stadermann, K.B.; Schwengers, O.; Anisiforov, E.; Jaenicke, S.; Weisshaar, B.; Zimmermann, T.; Goesmann, A. ReadXplorer 2-detailed read mapping analysis and visualization from one single source. Bioinformatics 2016, 32, 3702–3708. [Google Scholar] [CrossRef]
- Sharma, C.M.; Hoffmann, S.; Darfeuille, F.; Reignier, J.; Findeiss, S.; Sittka, A.; Chabas, S.; Reiche, K.; Hackermüller, J.; Reinhardt, R.; et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010, 464, 250–255. [Google Scholar] [CrossRef]
- Kim, D.; Hong, J.S.-J.; Qiu, Y.; Nagarajan, H.; Seo, J.-H.; Cho, B.-K.; Tsai, S.-F.; Palsson, B.Ø. Comparative analysis of regulatory elements between Escherichia coli and Klebsiella pneumoniae by genome-wide transcription start site profiling. PLoS Genet. 2012, 8, e1002867. [Google Scholar] [CrossRef]
- Thomason, M.K.; Bischler, T.; Eisenbart, S.K.; Förstner, K.U.; Zhang, A.; Herbig, A.; Nieselt, K.; Sharma, C.M.; Storz, G. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J. Bacteriol. 2015, 197, 18–28. [Google Scholar] [CrossRef]
- Mao, F.; Dam, P.; Chou, J.; Olman, V.; Xu, Y. DOOR: a database for prokaryotic operons. Nucleic Acids Res. 2009, 37, D459–63. [Google Scholar] [CrossRef]
- Santos-Zavaleta, A.; Salgado, H.; Gama-Castro, S.; Sánchez-Pérez, M.; Gómez-Romero, L.; Ledezma-Tejeida, D.; García-Sotelo, J.S.; Alquicira-Hernández, K.; Muñiz-Rascado, L.J.; Peña-Loredo, P.; et al. RegulonDB v 10.5: tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12. Nucleic Acids Res. 2019, 47, D212–D220. [Google Scholar] [CrossRef]
- Huerta, A.M.; Collado-Vides, J. Sigma70 promoters in Escherichia coli: specific transcription in dense regions of overlapping promoter-like signals. J. Mol. Biol. 2003, 333, 261–278. [Google Scholar] [CrossRef]
- Typas, A.; Becker, G.; Hengge, R. The molecular basis of selective promoter activation by the sigmaS subunit of RNA polymerase. Mol. Microbiol. 2007, 63, 1296–1306. [Google Scholar] [CrossRef]
- Sharma, C.M.; Vogel, J. Differential RNA-seq: the approach behind and the biological insight gained. Curr. Opin. Microbiol. 2014, 19, 97–105. [Google Scholar] [CrossRef]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006, 2, 2006–0008. [Google Scholar] [CrossRef]
- Keseler, I.M.; Mackie, A.; Peralta-Gil, M.; Santos-Zavaleta, A.; Gama-Castro, S.; Bonavides-Martínez, C.; Fulcher, C.; Huerta, A.M.; Kothari, A.; Krummenacker, M.; et al. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2013, 41, D605–12. [Google Scholar] [CrossRef]
- Dorman, C.J. H-NS: a universal regulator for a dynamic genome. Nat. Rev. Microbiol. 2004, 2, 391–400. [Google Scholar] [CrossRef]
- Hommais, F.; Krin, E.; Laurent-Winter, C.; Soutourina, O.; Malpertuy, A.; Le Caer, J.P.; Danchin, A.; Bertin, P. Large-scale monitoring of pleiotropic regulation of gene expression by the prokaryotic nucleoid-associated protein, H-NS. Mol. Microbiol. 2001, 40, 20–36. [Google Scholar] [CrossRef]
- Free, A.; Dorman, C.J. The Escherichia coli stpA gene is transiently expressed during growth in rich medium and is induced in minimal medium and by stress conditions. J. Bacteriol. 1997, 179, 909–918. [Google Scholar] [CrossRef]
- Nandal, A.; Huggins, C.C.O.; Woodhall, M.R.; McHugh, J.; Rodríguez-Quiñones, F.; Quail, M.A.; Guest, J.R.; Andrews, S.C. Induction of the ferritin gene (ftnA) of Escherichia coli by Fe(2+)-Fur is mediated by reversal of H-NS silencing and is RyhB independent. Mol. Microbiol. 2010, 75, 637–657. [Google Scholar] [CrossRef]
- Bagg, A.; Neilands, J.B. Ferric uptake regulation protein acts as a repressor, employing iron (II) as a cofactor to bind the operator of an iron transport operon in Escherichia coli. Biochemistry 1987, 26, 5471–5477. [Google Scholar] [CrossRef]
- Hunt, M.D.; Pettis, G.S.; McIntosh, M.A. Promoter and operator determinants for fur-mediated iron regulation in the bidirectional fepA-fes control region of the Escherichia coli enterobactin gene system. J. Bacteriol. 1994, 176, 3944–3955. [Google Scholar] [CrossRef]
- Zhang, Z.; Gosset, G.; Barabote, R.; Gonzalez, C.S.; Cuevas, W.A.; Saier, M.H. Functional interactions between the carbon and iron utilization regulators, Crp and Fur, in Escherichia coli. J. Bacteriol. 2005, 187, 980–990. [Google Scholar] [CrossRef]
- Bradley, M.D.; Beach, M.B.; Koning, A.P.J. de; Pratt, T.S.; Osuna, R. Effects of Fis on Escherichia coli gene expression during different growth stages. Microbiology (Reading) 2007, 153, 2922–2940. [Google Scholar] [CrossRef]
- Green, J.; Guest, J.R. Regulation of transcription at the ndh promoter of Escherichia coli by FNR and novel factors. Mol. Microbiol. 1994, 12, 433–444. [Google Scholar] [CrossRef]
- Kumar, R.; Shimizu, K. Transcriptional regulation of main metabolic pathways of cyoA, cydB, fnr, and fur gene knockout Escherichia coli in C-limited and N-limited aerobic continuous cultures. Microb. Cell Fact. 2011, 10, 3. [Google Scholar] [CrossRef]
- Partridge, J.D.; Bodenmiller, D.M.; Humphrys, M.S.; Spiro, S. NsrR targets in the Escherichia coli genome: new insights into DNA sequence requirements for binding and a role for NsrR in the regulation of motility. Mol. Microbiol. 2009, 73, 680–694. [Google Scholar] [CrossRef]
- Niland, P.; Hühne, R.; Müller-Hill, B. How AraC interacts specifically with its target DNAs. J. Mol. Biol. 1996, 264, 667–674. [Google Scholar] [CrossRef]
- Schleif, R. Regulation of the L-arabinose operon of Escherichia coli. Trends Genet. 2000, 16, 559–565. [Google Scholar] [CrossRef]
- Lobell, R.B.; Schleif, R.F. AraC-DNA looping: orientation and distance-dependent loop breaking by the cyclic AMP receptor protein. J. Mol. Biol. 1991, 218, 45–54. [Google Scholar] [CrossRef]
- Stoltzfus, L.; Wilcox, G. Effect of mutations in the cyclic AMP receptor protein-binding site on araBAD and araC expression. J. Bacteriol. 1989, 171, 1178–1184. [Google Scholar] [CrossRef]
- Zhang, X.; Schleif, R. Catabolite gene activator protein mutations affecting activity of the araBAD promoter. J. Bacteriol. 1998, 180, 195–200. [Google Scholar] [CrossRef]
- Barth, M.; Marschall, C.; Muffler, A.; Fischer, D.; Hengge-Aronis, R. Role for the histone-like protein H-NS in growth phase-dependent and osmotic regulation of sigma S and many sigma S-dependent genes in Escherichia coli. J. Bacteriol. 1995, 177, 3455–3464. [Google Scholar] [CrossRef]
- Marschall, C.; Hengge-Aronis, R. Regulatory characteristics and promoter analysis of csiE, a stationary phase-inducible gene under the control of sigma S and the cAMP-CRP complex in Escherichia coli. Mol. Microbiol. 1995, 18, 175–184. [Google Scholar] [CrossRef]
- Wyatt, M.D.; Pittman, D.L. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006, 19, 1580–1594. [Google Scholar] [CrossRef]
- Landini, P.; Busby, S.J. Expression of the Escherichia coli ada regulon in stationary phase: evidence for rpoS-dependent negative regulation of alkA transcription. J. Bacteriol. 1999, 181, 6836–6839. [Google Scholar] [CrossRef]
- Bordes, P.; Conter, A.; Morales, V.; Bouvier, J.; Kolb, A.; Gutierrez, C. DNA supercoiling contributes to disconnect sigmaS accumulation from sigmaS-dependent transcription in Escherichia coli. Mol. Microbiol. 2003, 48, 561–571. [Google Scholar] [CrossRef]
- Kusano, S.; Ding, Q.; Fujita, N.; Ishihama, A. Promoter selectivity of Escherichia coli RNA polymerase E sigma 70 and E sigma 38 holoenzymes. Effect of DNA supercoiling. J. Biol. Chem. 1996, 271, 1998–2004. [Google Scholar] [CrossRef]
- Nelson, P. Transport of torsional stress in DNA. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 14342–14347. [Google Scholar] [CrossRef]
- Kouzine, F.; Liu, J.; Sanford, S.; Chung, H.-J.; Levens, D. The dynamic response of upstream DNA to transcription-generated torsional stress. Nat. Struct. Mol. Biol. 2004, 11, 1092–1100. [Google Scholar] [CrossRef]
- Zhi, X.; Dages, S.; Dages, K.; Liu, Y.; Hua, Z.-C.; Makemson, J.; Leng, F. Transient and dynamic DNA supercoiling potently stimulates the leu-500 promoter in Escherichia coli. J. Biol. Chem. 2017, 292, 14566–14575. [Google Scholar] [CrossRef]
- Mechold, U.; Potrykus, K.; Murphy, H.; Murakami, K.S.; Cashel, M. Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res. 2013, 41, 6175–6189. [Google Scholar] [CrossRef]
- Barker, M.M.; Gaal, T.; Josaitis, C.A.; Gourse, R.L. Mechanism of regulation of transcription initiation by ppGpp. I. Effects of ppGpp on transcription initiation in vivo and in vitro. J. Mol. Biol. 2001, 305, 673–688. [Google Scholar] [CrossRef]
- Jishage, M.; Kvint, K.; Shingler, V.; Nyström, T. Regulation of sigma factor competition by the alarmone ppGpp. Genes Dev. 2002, 16, 1260–1270. [Google Scholar] [CrossRef]
- Walker, S.L.; Hiremath, L.S.; Galloway, D.R. ToxR (RegA) activates Escherichia coli RNA polymerase to initiate transcription of Pseudomonas aeruginosa toxA. Gene 1995, 154, 15–21. [Google Scholar] [CrossRef]
- Pátek, M.; Muth, G.; Wohlleben, W. Function of Corynebacterium glutamicum promoters in Escherichia coli, Streptomyces lividans, and Bacillus subtilis. J. Biotechnol. 2003, 104, 325–334. [Google Scholar] [CrossRef]
- Fujita, M. Identification of new sigma K-dependent promoters using an in vitro transcription system derived from Bacillus subtilis. Gene 1999, 237, 45–52. [Google Scholar] [CrossRef]
- Fujita, M.; Sagara, Y.; Aramaki, H. In vitro transcription system using reconstituted RNA polymerase (Esigma(70), Esigma(H), Esigma(E) and Esigma(S)) of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2000, 183, 253–257. [Google Scholar] [CrossRef]
- Jacques, J.-F.; Rodrigue, S.; Brzezinski, R.; Gaudreau, L. A recombinant Mycobacterium tuberculosis in vitro transcription system. FEMS Microbiol. Lett. 2006, 255, 140–147. [Google Scholar] [CrossRef]
- Holátko, J.; Silar, R.; Rabatinová, A.; Sanderová, H.; Halada, P.; Nešvera, J.; Krásný, L.; Pátek, M. Construction of in vitro transcription system for Corynebacterium glutamicum and its use in the recognition of promoters of different classes. Appl. Microbiol. Biotechnol. 2012, 96, 521–529. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–5. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Distribution of nucleotides within the −35 and −10 core regions of Escherichia coli σ70-dependent promoters detected via ROSE-Eσ70. Upstream sequences of 3,226 TSS (-1 to -49 nt) have been analyzed for enriched sequence motifs using Improbizer [14]. Sequence logos were derived using WebLogo v3.7.4 [16]. (A) 3,128 putative promoter sequences were aligned at conserved -10 regions. (B) 2,922 putative promoter sequences were aligned at conserved -35 regions.
Figure 1.
Distribution of nucleotides within the −35 and −10 core regions of Escherichia coli σ70-dependent promoters detected via ROSE-Eσ70. Upstream sequences of 3,226 TSS (-1 to -49 nt) have been analyzed for enriched sequence motifs using Improbizer [14]. Sequence logos were derived using WebLogo v3.7.4 [16]. (A) 3,128 putative promoter sequences were aligned at conserved -10 regions. (B) 2,922 putative promoter sequences were aligned at conserved -35 regions.
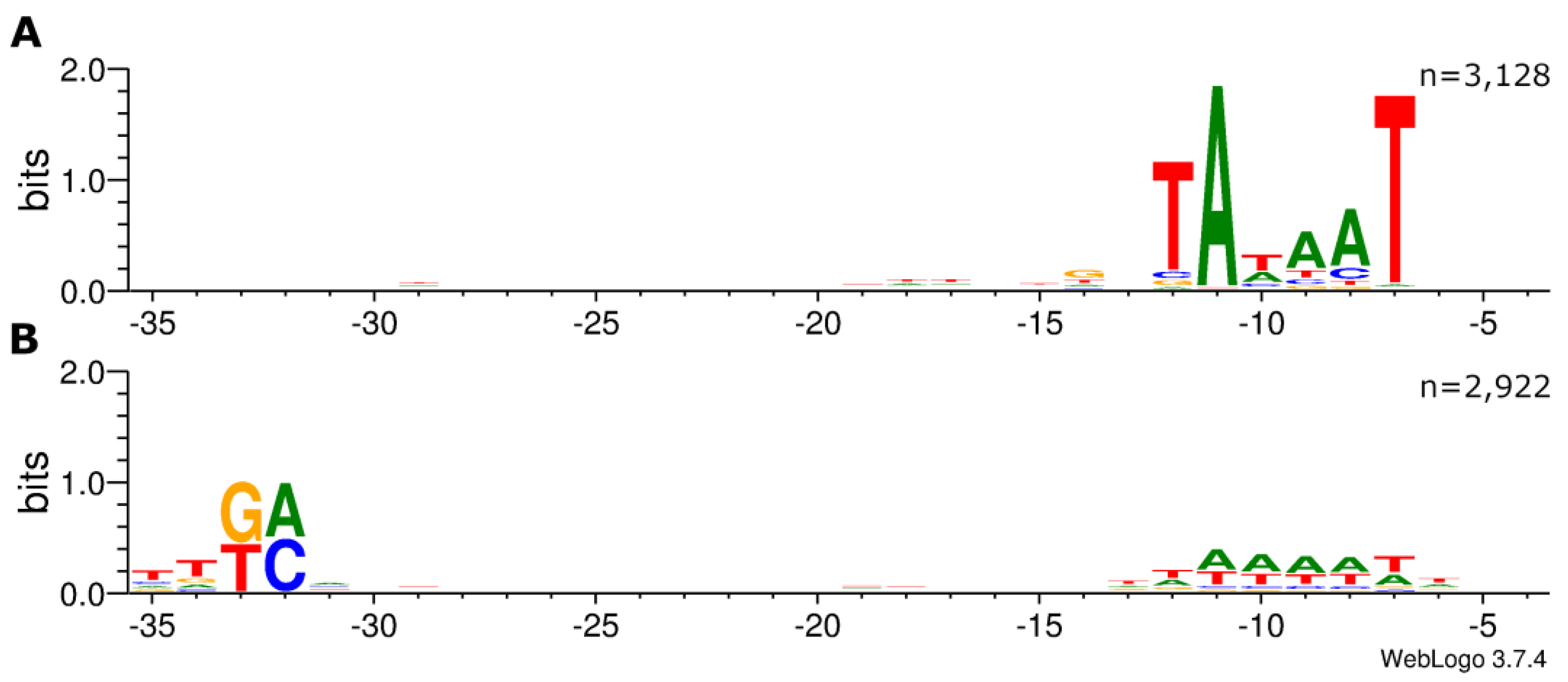
Figure 2.
Distribution of nucleotides within putative σ38-dependent Escherichia coli promoters detected via ROSE-Eσ70. Putative σ38-dependent promoters have been extracted from RegulonDB [22], and promoter motifs upstream of 106 TSS absent (A) and 25 TSS present (B) in the ROSE-Eσ70 dataset were visualized using WebLogo v3.7.4 [16].
Figure 2.
Distribution of nucleotides within putative σ38-dependent Escherichia coli promoters detected via ROSE-Eσ70. Putative σ38-dependent promoters have been extracted from RegulonDB [22], and promoter motifs upstream of 106 TSS absent (A) and 25 TSS present (B) in the ROSE-Eσ70 dataset were visualized using WebLogo v3.7.4 [16].
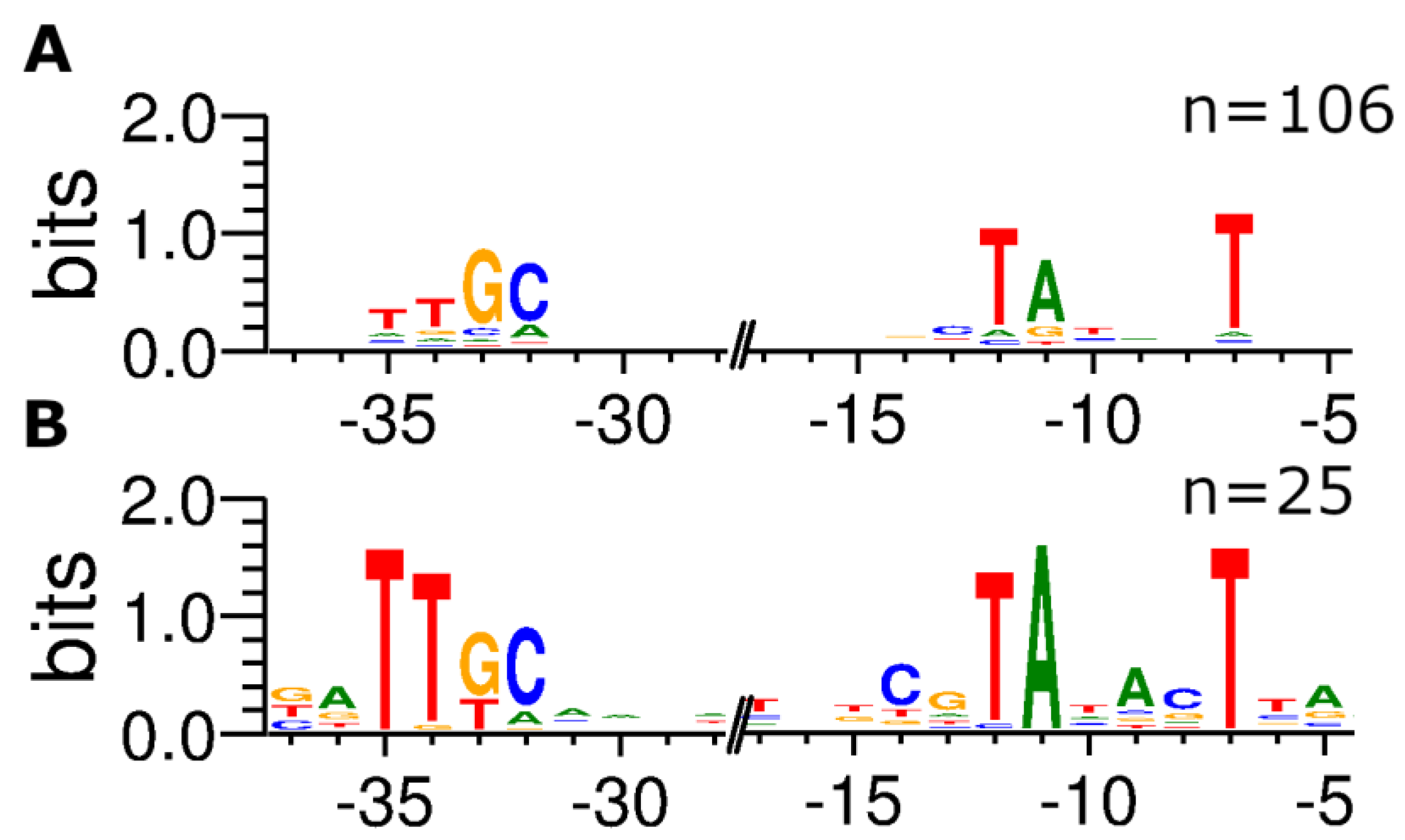
Figure 3.
Comparison of transcription start sites (TSS). TSS positions in the E. coli K-12 MG1655 genome from Thomason et al. [20], RegulonDB [22], and ROSE-Eσ70 (this study) have been compared. A difference of three nucleotides in either direction has been allowed. .
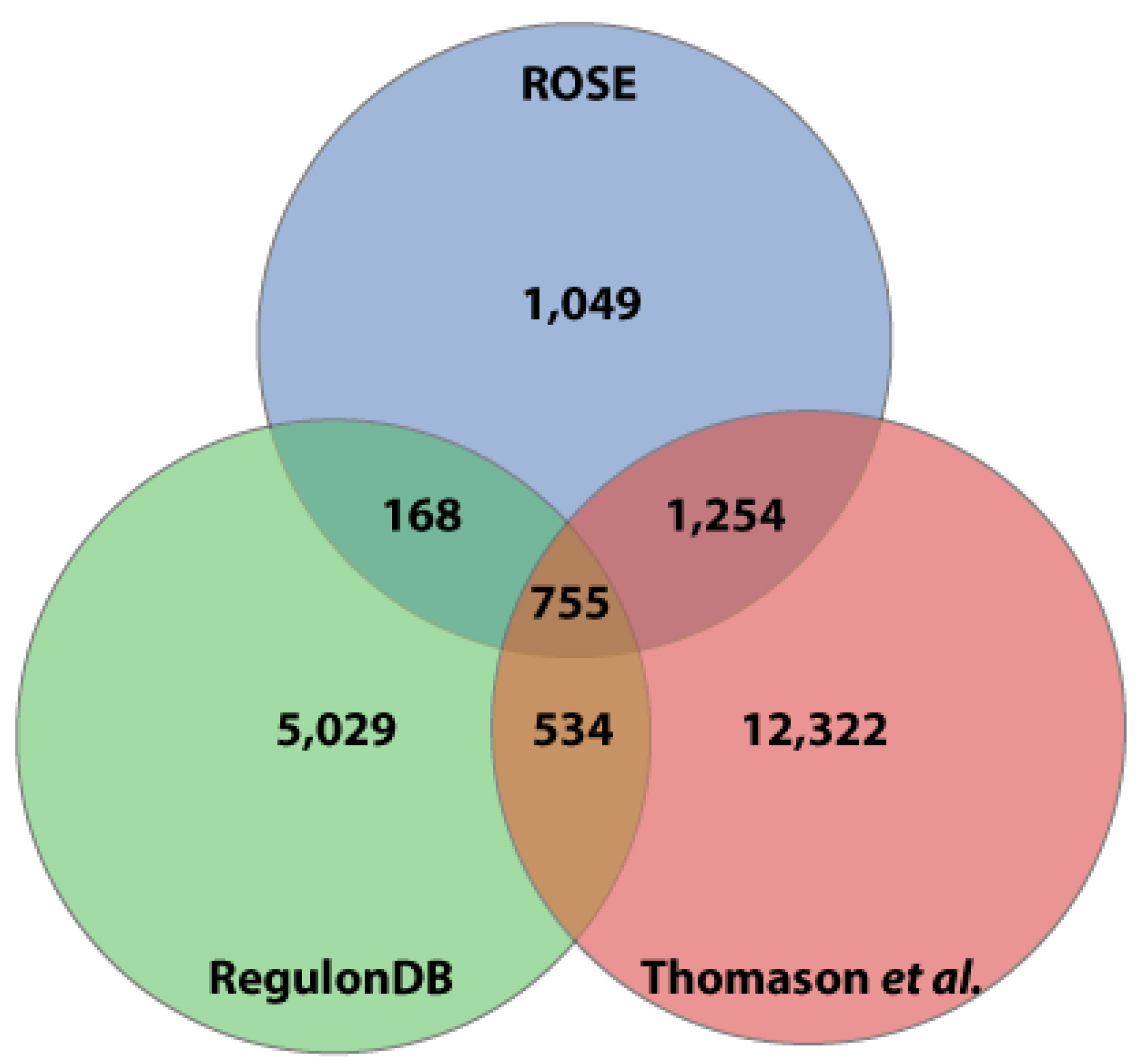
Figure 4.
Promoters from E. coli that were repressed under in vivo conditions and detected by ROSE-Eσ70. The genomic organization of the transcription units is depicted according to the EcoCyc database [27] (not to scale). (A) Promoter region of ftnA and stpA repressed by H-NS. The exact location of the binding site of H-NS in the PstpA2 is unknown. (B) The promoter region of yjjZ and fepA repressed by Fur. (C) The promoter region of glcC repressed by Fis. The gene aer is illustrated with the known σ28-promoter Paer and the newly found TSS (dashed arrow) described by Thomason et al. [20] and identified by ROSE-Eσ70 and the Δfis knockout strain.
Figure 4.
Promoters from E. coli that were repressed under in vivo conditions and detected by ROSE-Eσ70. The genomic organization of the transcription units is depicted according to the EcoCyc database [27] (not to scale). (A) Promoter region of ftnA and stpA repressed by H-NS. The exact location of the binding site of H-NS in the PstpA2 is unknown. (B) The promoter region of yjjZ and fepA repressed by Fur. (C) The promoter region of glcC repressed by Fis. The gene aer is illustrated with the known σ28-promoter Paer and the newly found TSS (dashed arrow) described by Thomason et al. [20] and identified by ROSE-Eσ70 and the Δfis knockout strain.
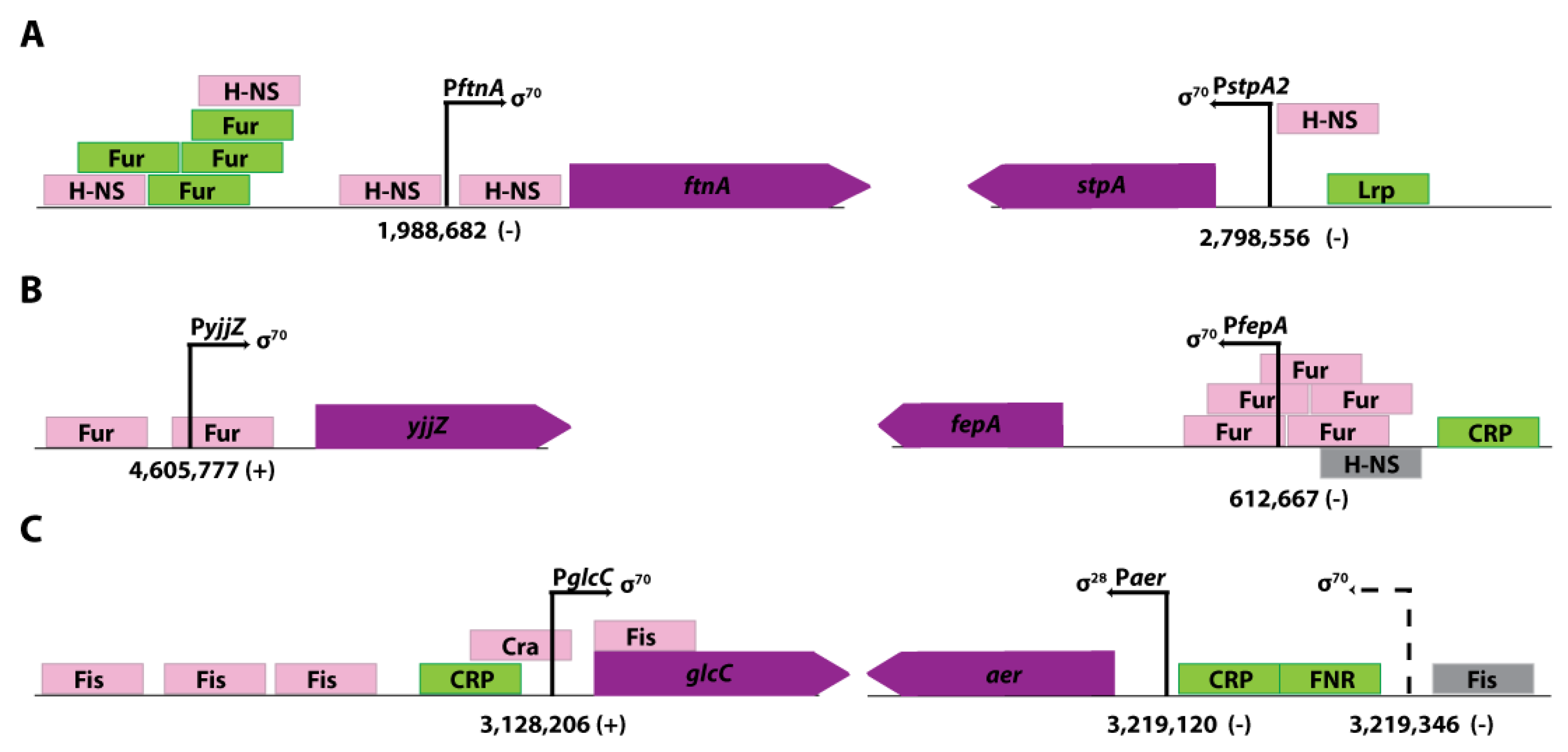
Figure 5.
Promoter region and mapping results of the ndh gene. (A) Genomic organization of the transcription unit of ndh according to RegulonDB database [22] (not to scale). (B) Readcount in the promoter region of ndh from the E. coli ROSE-Eσ70 (top), E. coli in vivo Wildtype strain (middle), and E. coli in vivo Δfur knockout strain. The mapping occurred on the respective reference genome (U00096.3) and is visualized with ReadXplorer [13].
Figure 5.
Promoter region and mapping results of the ndh gene. (A) Genomic organization of the transcription unit of ndh according to RegulonDB database [22] (not to scale). (B) Readcount in the promoter region of ndh from the E. coli ROSE-Eσ70 (top), E. coli in vivo Wildtype strain (middle), and E. coli in vivo Δfur knockout strain. The mapping occurred on the respective reference genome (U00096.3) and is visualized with ReadXplorer [13].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



