
Submitted:
05 May 2023
Posted:
06 May 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Macrophage-derived chemokine belongs to the CC subfamily. It is produced by dendritic cells (DCs) and macrophages with or without external stimulation. We have previously shown a statistically significant depletion of MDC/CCL22 concentrations in a number of studies concerning COVID-19. These shifts in concentrations demonstrated stability unrelated to the SARS-CoV-2 genetic variant and remained noticeable even in convalescent patients. In this work, we analyze MDC/CCL22 dynamics in various diseases, including those that manifest with inflammation in lung tissue. In addition, we provide our hypothesis on such a decrease in MDC/CCL22 concentrations in COVID-19. If its secretion by producer cells is unperturbed, then it is possible for viral products to bind to this chemokine and to block its functional activity. There is, however, another possible explanation directly linked to depletion in DC subpopulations and the inhibition of their function. We also discuss MDC/CCL22's role in the immunology of novel coronavirus infection, based on both our own data and other studies.
Keywords:
macrophage-derived chemokine
; MDC/CCL22
; chemokines
; novel coronavirus infection
; COVID-19
; post-COVID
1. Introduction
COVID-19 is an acute infectious disease caused by the RNA-based SARS-CoV-2 virion of the genus Betacoronavirus. The first cases of COVID-19 were registered in Wuhan, China, in December 2019. By March 2020, the World Health Organization (WHO) had officially declared COVID-19 a global pandemic [1,2,3]. As do most viral infections, COVID-19 begins with the virus entering the cell via interactions between viral proteins and complementary receptors on host cellular membranes. SARS-CoV-2 has two ways of entering cells, either through membrane fusion or viral particle uptake. This can activate various innate immune PRRs depending on the mode of entry. For membrane fusion to occur, the cellular receptors ACE2, TMPRSS2, and sometimes NRP1 are necessary. Once fusion happens, the virus's genetic material is released into the host cell's cytoplasm. In COVID-19, angiotensin-converting enzyme 2 (ACE 2) receptor usually acts as the major entry point for the virus. It is widely represented in human cells: pneumocytes, enterocytes, vascular endothelial cells, smooth muscle cells, and even glia [4,5,6].
Most SARS-CoV-2 target cells are located in the upper and lower parts of the respiratory system. In addition to inflammation caused by interactions between viral spike protein (S protein) and the ACE 2 receptor, there is also the subsequent inhibition of this receptor. This leads to the enhanced expressions of genes responsible for angiotensin II secretion. In turn, angiotensin II binds to Toll-like receptors (type II), which activates inflammatory transcription factors (IRF, IkBɑ, NF-kB). Due to the nuances of COVID-19, such conditions can lead to enhanced inflammation which can potentially activate a so-called ‘cytokine storm’.
In addition, there is an activation of innate immunity (i.e., DCs, macrophages, granulocytes) that precedes the inclusion of the main effector lymphocytes of adaptive immunity [7,8]. A simplified representation of immune interactions at the moment of SARS-CoV-2 entry into the cell is presented in Figure 1.
COVID-19 immunity is often followed by the development of cytokine storm, a pathological hyperinflammatory reaction with increased cytokine secretion, directly linked with COVID-19 severity. It is also an important cause of death in COVID-19 patients [9]. Cytokine storms are often mediated by СС-chemokines, e.g., MCP-1/CCL2, MIP-1α/CCL3, MCP-3/CCL7, eotaxin-3/CCL26 [9,10]. Specifically, MDC/CCL22 is involved in immunity against COVID-19.
2.1. Immunology of macrophage derived chemokine
Macrophage derived chemokine belongs to the CC family. Via this classification, it holds the double name MDC/CCL22. MDC/CCL22 is produced by macrophages and dendritic cells, with or without external stimuli. Such stimuli may include bacteria, viruses and parasites and associated substances, such as lipopolysaccharides, peptidoglycans, CpG motifs, flagella and viral nucleic acids [11]. The gene for MDC/CCL22 is located in the long arm of human chromosome 16 in a cluster with chemokines Fractalkine/CX3CL1 and TARC/CCL17. MDC/CCL22 is a potent chemoattractant for CD4 and CD8 T cells, IL-2–activated natural killer cells, as well as for DCs expressing the CCL22 receptor CCR4. It is involved in chronic inflammation mediated by the continuous homing of DCs and lymphocytes [12]. Its function is directly linked to the CCR4 molecule, a receptor widely present on Th2 cells. CCR4 receptors on CD4+ lymphocytes in the bone marrow, like MDC/CCL22, mediate cellular growth and maturation. Activation of cellular migration and Th1/Th2 polarization are coordinated with the help of MDC/CCL22. In vitro, MDC/CCL22 is produced primarily by maturing DC and activated monocytes/macrophages [12]. In vivo, MDC/CCL22 is constitutively produced by epithelial cells of human thymic medulla [13] and interdigitating DC within T-cell zones of mouse lymph nodes [14].
Macrophage-derived chemokine is not homologous to most chemokines. It is most similar to thymus- and activation-regulated chemokine (TARC), with 37% identity [15], which explains its active participation in thymocyte migration.
2.2. Changes in MDC/CCL22 concentrations in COVID-19 in vitro and in vivo
Adequate regulation of the immune response is the key factor in disease severity. As COVID-19 progresses, it is often accompanied by immunity dysregulation, from both cellular and humoral links of the immune responses. Therefore, studying regulatory molecules (cytokines, chemokines) included in this process is vital for understanding COVID-19 immunity.
Within our work, we have studied both cytokines and chemokines in COVID-19, and we also took different approaches to experimental design and data analysis. As the pandemic progressed, newer points of interest emerged: for instance, we investigated the value of cytokine profiling in acute infection and persisting patterns in convalescent patients [16]. We also explored predictors of disease severity among cytokines/chemokines [17], and suggested a model for prognosis of the disease outcome. However, as the virus changed, we have realized that the immunological landscape went through certain changes as well. Therefore, our study group decided to pay closer attention to the role of viral genetics in COVID-19 immunity [18].
Among the biological substances highlighted in our previous studies (CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, CCL7/MCP-3, CCL11/Eotaxin, CCL22/MDC, CXCL1/GROα, CXCL8/IL-8, CXCL9/MIG, CXCL10/IP-10, and CX3CL1/Fractalkine, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17A/CTLA8, IL-18, IL-22, IL-27, IFNα2, IFNγ, TNFα, TNFβ/Lymphotoxin-α (LTA), IL-1ra, IL-10, EGF, FGF-2/FGF-basic, Flt3 Ligand, G-CSF, M-CSF, GM-CSF, PDGF-AA, PDGF-AB/BB, TGF-α, and VEGF-A), the most prominent and unexpected role belongs to one of the CC chemokines, macrophage-derived chemokine (MDC/CCL22). Specifically, significantly lower MDC/CCL22 concentrations were seen in COVID-19 patient plasma, independent of SARS-CoV-2 genetic variant (original Wuhan strain, Alpha, Delta or Omicron variant). This finding was especially interesting in comparison to other chemokines, as their concentrations showed a tendency to rise in the blood plasma of COVID-19 patients in comparison with healthy donors (HD). A shortened version of our original study comparing four genetic variants of SARS-CoV-2 (Wuhan strain, Alpha, Delta, Omicron variants) in terms of MDC/CCL22 concentration is presented in Figure 2 [18].
At early stages of the pandemic, when we studied patterns of cytokine profiling in acute phase patients and convalescents, we discovered a note-worthy tendency concerning MDC/CCL22 levels. Interestingly, convalescents of the original Wuhan viral strain had significantly lower MDC/CCL22 concentrations, not only compared to healthy donors, but also in comparison with those in the acute phase of infection. The results of previous studies on the matter are presented in Figure 3 [16].
As seen, the levels of MDC/CCL22 have been observed to decrease in patients with acute COVID-19, and surprisingly, they remain low in convalescents as well. This notion suggests that the impact of the virus on the immune system may be more profound than previously thought. It is unclear whether this decrease is solely due to the virus's effects on DCs. However, recent studies indicate that MDC/CCL22 may be crucial for regulating immune responses and preventing excessive inflammation by recruiting regulatory T cells. Thus, the deficiency of MDC/CCL22 in COVID-19 patients may contribute to immune dysregulation and severe pulmonary pathology. Further research is necessary to fully comprehend the complex interactions between MDC/CCL22, DCs, platelets, and immune regulation in COVID-19.
The observed concentration dynamics can be explained by a possible ‘depletion’ of MDC/CCL22 production, even in the next few months after recovery. It is worth mentioning that, in other studies concerning other inflammation prone illnesses, MDC/CCL22 rarely shows such a decrease, even in diseases affecting the respiratory tract. Therefore, such consistently lower concentrations may be specific to COVID-19 and require thorough investigation.
Our previous studies showed that COVID-19 recovery is often associated with decreases in several cytokines, MDC/CCL22 being only one of them [16,17,18]. Such a depletion and dysregulation in the cytokine branch has been described by other researchers in works concerning the first 8 months since the recovery [19,20]. At the same time, we noted changes affecting cellular immunity [21] even more than 6 months after recovery: for instance, decrease in CD8+ effector subsets and higher frequencies of CD8+ T cell subsets expressing lung tissue and mucosal tissue homing molecules (Tc2, Tc17, and Tc17.1). This finding was confirmed by other researchers [22,23]. It therefore proves that COVID-19 has a certain effect on immune cells and their mediators even after recovery.
In the study by Ling et al., a statistically significant decrease in MDC/CCL22 was noted not only during acute COVID-19, but also a year after the infection [24]. This data overlaps with our own findings: in convalescent patients we noted a statistically significant decrease of MDC/CCL22 between 30 and 100 days since the onset of the disease. Tufa et al. confirmed our finding concerning lower levels of MDC/CCL22 in patients with COVID-19, as their study highlighted the same tendency [23]. Noteworthy is the fact that they reported negative correlation between MDC/CCL22 and the severity of COVID-19.
Recent research has suggested that the deficiency of MDC/CCL22, a protein that plays a crucial role in regulating inflammation, could potentially contribute to the persistent lymphopenia observed in COVID-19 patients. This phenomenon has been reported to persist even after patients have recovered from the virus [24,25]. Several in vitro studies have shown the importance of MDC/CCL22 in regulation of inflammation [9,26,27]. Its presence complemented regulatory T cell activation and restricted enhanced inflammation with type I helper T cells. Therefore, MDC/CCL22 plays the role of ambivalent inflammation regulator, from both activating and inhibiting standpoints.
Such findings provide grounds for discussing changes in immunity both in acute phase infection and in COVID-19 convalescents. In 2022, the International Statistical Classification of Diseases and Related Health Problems (ICD) registered a new diagnosis of 'post-COVID syndrome', which included a wide spectrum of symptoms noticeable long after COVID-19 recovery [27]. These symptoms include fatigue, shortness of breath and chronic cough, muscle and joint pains, heart palpitations, persistent vascular changes, i.e. recurrent thrombosis, and mental health issues (anxiety, depression, memory and concentration loss). The reasons behind these symptoms are yet to be investigated.
Studying post-COVID is a new challenge brought by the pandemic, and this challenge has yet to be overcome by global scientific efforts. As the consequences and complications of this novel phenomenon are being discovered, the mechanisms behind post-COVID are still unknown. Even now, however, it is clear that cytokines (and chemokines) play a pivotal role in the immune responses behind this process. Keeping in mind the knowledge already collected and described in the past, we may presume that MDC/CCL22 plays a bigger role in post-COVID than currently understood.
2.3. Macrophage-derived chemokine MDC/CCL22 in various pathologies
2.3.1. MDC/CCL22 in oncology of non-respiratory organs
Novel areas of research in oncology provide the most thorough insight into the role of MDC/CCL22 in the disease development. The chemokine MDC/CCL22 may be a biomarker of macrophage/DC compartment of immune suppression and provide relevant information about anti-tumor responses elicited by the host that may be negatively impacted by tumor cells or immunosuppressive treatments.
MDC/CCL22 accumulation in solid tumors has been shown to lead to T regulatory cells infiltration in melanoma, and ovarian, prostate or breast carcinomas [28,29,30]. Circulating MDC/CCL22 levels are related to both glioma risk and survival duration independent of age, histology, grade and IDH mutation status. CCL22 should be considered a marker of immune status with potential prognostic value [31]. In contrast, it was shown that tumors that lacked MDC/CCL22 expression were not infiltrated by Tregs, regardless of whether they produced other CCR4-binding chemokines, such as CCL17 [32].
MDC/CCL22 is a chemokine that has been extensively studied in the field of oncology as a potential target for monoclonal antibody therapy. This protein is known to play a critical role in the recruitment of immune cells to the site of tumors, and blocking its activity may represent a promising strategy for treating cancer. However, despite its importance in cancer biology, the precise function of MDC/CCL22 in the immune system remains poorly understood. Researchers are actively working to unravel the complex signaling pathways and interactions that underlie this protein's activity, with the goal of developing new therapeutic approaches to a range of diseases.
2.3.2. Macrophage-derived chemokine MDC/CCL22 in autoimmunity and atopic diseases
Like many other respiratory infections, COVID-19 is followed by extensive inflammatory reactions. These inflammatory reactions, however, can ignite not only in the lung tissue, but anywhere in the body, as long as SARS-CoV-2 can find a potential target for binding.
Therefore, it is important to study how MDC/CCL22 acts in other pathologies followed by inflammation. For instance, autoimmune and atopic diseases are characterized by inadequate recruitment of immune cells to target tissues as well as intense expression of pro-inflammatory markers. In the studies concerning MDC/CCL22 in autoimmune diseases and atopic dermatitis (AD) it shows prominent increase. For instance, MDC/CCL22 is usually overexpressed in the lesional skin of atopic dermatitis (AD). Th2 cells in lesions of AD express more IL-4 and IL-13 than unaffected Th2 cells. Dendritic cells respond to IL-4 and IL-13 by secreting MDC/CCL22 (as well as TARC/CCL17 and MIP-4/CCL18) [33]. Serum levels of MDC/CCL22 and TARC/CCL17 are significantly augmented in patients with AD compared to those in healthy controls and are associated with disease severity in AD.
Interestingly, MDC/CCL22 has also been found to play a role in other inflammatory conditions. For example, MDC/CCL22-producing resident macrophages have been associated with the inflammatory profile of patients with autoimmune Sjögren's Syndrome [34], an autoimmune disease, affecting secretory glands in the body.
In vitro models of autoimmune encephalitis show increased levels of MDC/CCL22, but in this context, MDC/CCL22 mediates chronic inflammatory processes by recruiting T regulatory cells to glia [35]. This suggests that MDC/CCL22 may have a broader role in modulating immune responses than simply intensifying inflammation.
2.3.3. Macrophage-derived chemokine MDC/CCL22 in respiratory disease
Since coronavirus infection is primarily accompanied by damage to the upper and lower respiratory tract, the role of MDC/CCL22 in the development of pulmonary pathology is of interest. In tuberculosis, MDC/CCL22 shows lower concentrations in vitro in cells and autologous platelets taken from patients infected with Mycobacterium tuberculosis. It is known, however, that COVID-19 is often accompanied by thrombosis and elevation of platelet-derived factors [36].
Nakanishi et al. showed that in patients with lung cancer (adenocarcinoma, squamous cell carcinoma), high levels of MDC/CCL22 are associated with positive post-surgical outcomes. Moreover, MDC/CCL22 levels reflected a negative correlation with the risk of recurrent malignancy [37]. In the post-surgery period, patients with malignant tumors in the lung show higher survival rates when the immune cells infiltrate the lung tissue. In such cases, MDC/CCL22 acts as a mediator and a regulator for recruitment of the cells to the lung tissue.
In murine models, it has been shown that i.v. introduction of recombinant MDC can enhance inflammation in lung tissue [38]. One of the possible explanations is an intensive mobilization of certain immune cells: macrophages and type II helper T cells. Additional study performed by the same group demonstrated that blockage of NFκB signaling leads to decrease in MDC/CCL22 gene expression in response to proinflammatory stimuli [39]. In the post-surgical period, patients with malignant lung tumors show higher survival rates when immune cells infiltrate lung tissue [40]. In such cases, MDC/CCL22 acts as a mediator and a regulator for recruitment of cells to the lung tissue.
In most cases, when MDC/CCL22 levels are compared between healthy individuals and those with other illnesses such as cancer or autoimmune diseases, a significant increase is typically observed. However, in the case of COVID-19, the opposite trend is witnessed as MDC/CCL22 levels decrease. In the following sections, we will explore potential reasons for this phenomenon.
2.6. Possible mechanisms behind the decrease in MDC/CCL22 concentrations in COVID-19
We believe that the drop in MDC/CCL22 concentration in COVID-19 patients is worthy of attention. Since the reasons behind this occurence are yet unclear, we present hypothetical explanations for the phenomenon in question in Figure 4.
Our hypothesis implies two possible mechanisms for lower MDC/CCL22 concentrations in COVID-19.
The first mechanism implies possible binding of SARS-CoV-2 viral proteins with MDC/CCL22 due to potential affinity with, or mimicry of, MDC/CCL22's main ligands. In such cases, MDC/CCL22 production by producer cells (i.e., DCs and macrophages) is unperturbed. Yet, the selective binding of this chemokine makes it undetectable for commercial kits as it changes its antigenic structure. Moreover, it is possible that its functional activity reduces due to this process. This hypothesis is supported by the fact that other cytokines and chemokines, produced by DCs and macrophages, show enhanced expression when compared to healthy donors [9,10,11,12,19,40]. The infectious agent requires mechanisms of evading the immune responses, and one of the potential ways is to block chemoattraction by inactivating chemokines [41]. Although the concept of chemokine binding proteins is not entirely new, the nature of these peptides or proteins and their specific properties are yet to be discovered, and the whole concept of protein-based binding in COVID-19 requires in vitro experiments. Proteomics studies concerning cytokine dysregulation in presence of SARS-CoV-2 proteins have been previously performed [42], yet there is no information covering potential interactions between MDC/CCL2 and viral proteins. Among known proteins, ORF (open reading frame) 8 is known to bind with the dendritic cells and alternate cytokine expression [43]. At the same time, S protein of SARS-CoV-2 was shown to play a part in activation of several proinflammatory cytokines [44]. Within the same study, no such inflammatory response was observed in response to membrane (M), envelope (E), and nucleocapsid (N) proteins. However, S protein is known to be less conservative [45]: it undergoes changes in its structure with each genetic change in the virion proteins’ structure. N-protein structure is believed to be more stable, and as MDC/CCL22 shows stable concentrations independent of the genetic variant, it is more likely to be a target for MDC/CCL22 binding. In the study by López-Muñoz it is shown that N-protein binds chemokines through its GAG-binding domain and inhibits in vitro chemokine-mediated leukocyte migration [46]. In other viral infections such binding of chemokines to viral proteins is well-known and previously described [47].
There is, however, an opposite hypothesis, implying that COVID-19 can actually affect functional activity of macrophage-derived chemokine producer cells. Specifically, researchers highlighted a significant shortage of DCs in COVID-19 patients, both in acute and post-recovery periods [49,50,51]. Such shortage is seen not only in quantitative, but also in qualitative way: specifically, in vitro studies showed depletion of functional activity in dendritic cells [52]. Moreover, other studies have highlighted the relationship between disease severity and dendritic cell properties, both quantitative and qualitative. This, however, for some reason does not affect other cytokines and chemokines, produced by DCs (IL-1α, IL-1β, IL-6, IL-7, IL-12 (p35 and p40), IL-15, IL-18, TNF-α, TGF-β, macrophage CSF, and granulocyte-macrophage CSF, but not IL-2, IL-3, IL-4, IL-5, IL-9, and IFN-γ transcripts) [53]. In our studies, some of these cytokines showed a typical pro-inflammatory profile and an actual increase in concentrations [10,11,12]. Other studies exploring the role of DCs in coronavirus infections highlight their participation in viral dissemination [54]. Notably, not only in SARS-CoV-2 associated diseases, but also in SARS and MERS [55], DCs are named ‘the missing link’ between antiviral innate and adaptive responses. As SARS-CoV-2 induces activation of specific plasmacytoid IFN-producing cells (pDCs) [56], it may be possible that this subpopulation of DCs displaces conventional DCs (cDCs) and monocyte-derived DCs (MoDCs) [57,58]. Plasmacytoid dendritic cells are characterized by CD123+CD303+ phenotype, their main role is enabling type I IFN expression. Moreover, the severity of the disease correlates with the responsiveness of pDCs [59]. Although there is little information on pDCs ability to produce MDC/CCL22, there is a possibility that it is not as prominent as in other types of dendritic cells (e.g., MoDCs). If this assumption is correct and pDCs production of MDC/CCL22 is in any way impaired, this may explain differences in chemokine production associated with the coronavirus infection.
Theoretically, this phenomenon may explain a defect in MDC/CCL22 production and its deficiency in the blood plasma of COVID-19 patients in comparison with healthy donors. While the precise mechanisms by which SARS-CoV-2 suppresses DC function and MDC/CCL22 production are not yet fully understood, the deficiency of MDC/CCL22 in the blood plasma of COVID-19 patients compared to healthy donors suggests that this chemokine may play a critical role in the pathogenesis of the disease. Further research is needed to fully elucidate the complex interactions between SARS-CoV-2, DCs, and MDC/CCL22, with the ultimate goal of developing new therapeutic strategies to combat COVID-19 and other infectious diseases.
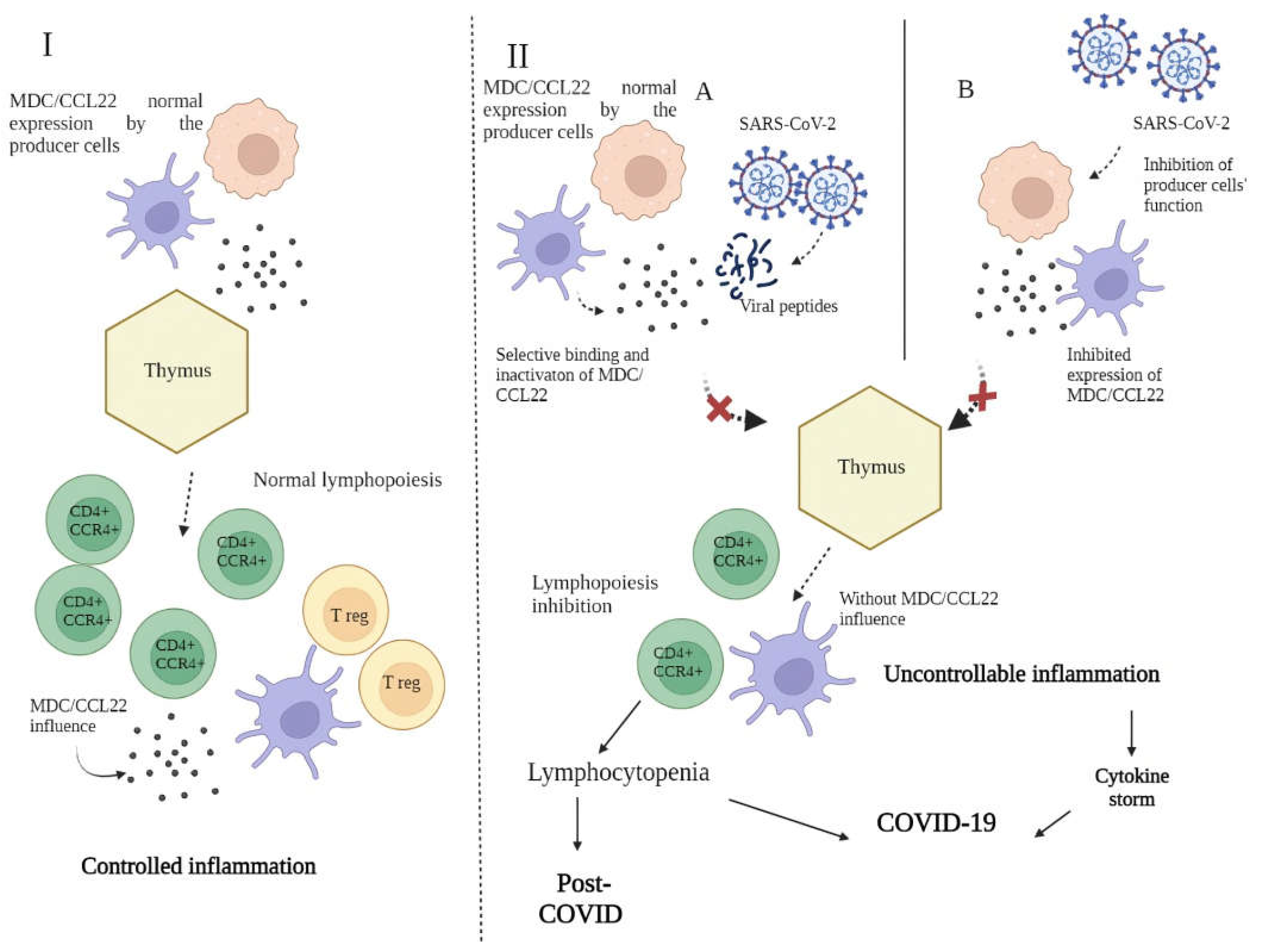
In any case, both hypotheses prove the role of the SARS-CoV-2 infectious process in the suppression of DCs. Potential mechanisms for inflammatory dysfunction in patients with lower MDC/CCL22 are presented in Figure 5.
2.7. Conclusions
As explained earlier, MDC/CCL22 is a mediator for both Th2 immune responses and for regulatory T cells. Presumably, in the absence of this chemokine, there would be a shift in the inflammatory response towards hyperactivation. Noticeable depletion of MDC/CCL22 concentrations in blood plasma of acute COVID-19 patients may explain the relatively greater severity of COVID-19 in comparison with other respiratory viral infections.
Keeping in mind preexisting data on several related topics (macrophage-derived chemokine, COVID-19 and concomitant hyperinflammatory profiles), we have grounds to presume that MDC/CCL22 may be a missing link in the chain of yet unexplained processes in COVID-19. It is important to keep in mind its role in the formation of vaccine-associated immunity, especially in individuals who have survived severe COVID-19. As its concentrations tend to decrease in association with COVID-19, hyperinflammatory profile and inadequate reaction to external stimuli may present a potential problem in immunity formation.
Due to the aforementioned features of coronavirus infection, the role of MDC/CCL22 still requires further research.
Acknowledgements
Special thanks go to Edward S. Ramsay for his useful suggestions and help with editing and language. We thank the staff of Core facility centre ‘Cytometry and biomarkers’ in Pasteur Institute, Russia, for helping with experimental parts of our work.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Liu YC, Kuo RL, Shih SR. COVID-19: The first documented coronavirus pandemic in history. Biomed J. 2020 Aug;43(4):328-333. https://doi.org/10.1016/j.bj.2020.04.007. Epub 2020 May 5. PMID: 32387617; PMCID: PMC7199674. [CrossRef]
- Borczuk, A.C., Yantiss, R.K. The pathogenesis of coronavirus-19 disease. J Biomed Sci 29, 87 (2022). https://doi.org/10.1186/s12929-022-00872-53. [CrossRef]
- Boechat JL, Chora I, Morais A, Delgado L. The immune response to SARS-CoV-2 and COVID-19 immunopathology - Current perspectives. Pulmonology. 2021 Sep-Oct;27(5):423-437. https://doi.org/10.1016/j.pulmoe.2021.03.008. Epub 2021 Apr 9. PMID: 33867315; PMCID: PMC8040543. [CrossRef]
- Merad M, Blish CA, Sallusto F, Iwasaki A. The immunology and immunopathology of COVID-19. Science. 2022 Mar 11;375(6585):1122-1127. https://doi.org/10.1126/science.abm8108. Epub 2022 Mar 10. PMID: 35271343. [CrossRef]
- Salamanna F, Maglio M, Landini MP, Fini M. Body Localization of ACE-2: On the Trail of the Keyhole of SARS-CoV-2. Front Med (Lausanne). 2020 Dec 3;7:594495. https://doi.org/10.3389/fmed.2020.594495. PMID: 33344479; PMCID: PMC7744810. [CrossRef]
- Shirbhate E, Pandey J, Patel VK, Kamal M, Jawaid T, Gorain B, Kesharwani P, Rajak H. Understanding the role of ACE-2 receptor in pathogenesis of COVID-19 disease: a potential approach for therapeutic intervention. Pharmacol Rep. 2021 Dec;73(6):1539-1550. https://doi.org/10.1007/s43440-021-00303-6. Epub 2021 Jun 27. PMID: 34176080; PMCID: PMC8236094. [CrossRef]
- Xu J, Lazartigues E. Expression of ACE2 in Human Neurons Supports the Neuro-Invasive Potential of COVID-19 Virus. Cell Mol Neurobiol. 2022 Jan;42(1):305-309. https://doi.org/10.1007/s10571-020-00915-1. Epub 2020 Jul 4. PMID: 32623546; PMCID: PMC7334623. [CrossRef]
- Boechat JL, Chora I, Morais A, Delgado L. The immune response to SARS-CoV-2 and COVID-19 immunopathology - Current perspectives. Pulmonology. 2021 Sep-Oct;27(5):423-437. https://doi.org/10.1016/j.pulmoe.2021.03.008. Epub 2021 Apr 9. PMID: 33867315; PMCID: PMC8040543. [CrossRef]
- Rodriguez L., Brodin P. Unraveling the immune response in severe COVID-19. J. Clin. Immunol., 2020, Vol. 40, no. 7, pp. 958-959. [CrossRef]
- Wiech M, Chroscicki P, Swatler J, Stepnik D, De Biasi S, Hampel M, Brewinska-Olchowik M, Maliszewska A, Sklinda K, Durlik M, Wierzba W, Cossarizza A, Piwocka K. Remodeling of T Cell Dynamics During Long COVID Is Dependent on Severity of SARS-CoV-2 Infection. Front Immunol. 2022 Jun 10;13:886431. https://doi.org/10.3389/fimmu.2022.886431. PMID: 35757700; PMCID: PMC9226563. [CrossRef]
- Wallet MA, Sen P, Tisch R. Immunoregulation of dendritic cells. Clin Med Res. 2005 Aug;3(3):166-75. https://doi.org/10.3121/cmr.3.3.166. PMID: 16160071; PMCID: PMC1237158. [CrossRef]
- Godiska R, Chantry D, Raport CJ, Sozzani S, Allavena P, Leviten D, Mantovani A, Gray PW. Human macrophage-derived chemokine (MDC), a novel chemoattractant for monocytes, monocyte-derived dendritic cells, and natural killer cells. J Exp Med. 1997; 185:1595–1604. [PubMed: 9151897]. [CrossRef]
- D. Chantry, P. Romagnani, C.J. Raport, C.L. Wood, A. Epp, P.W. Gray Macrophage-derived chemokine is localized to thymic medullary epithelial cells and is a chemoattractant for CD3(+), CD4(+), CD8(low) thymocytes Blood, 94 (1999), pp. 1890-1898.
- Andrew DP, Chang MS, McNinch J, Wathen ST, Rihanek M, Tseng J, Spellberg JP, Elias CG 3rd. STCP-1 (MDC) CC chemokine acts specifically on chronically activated Th2 lymphocytes and is produced by monocytes on stimulation with Th2 cytokines IL-4 and IL-13. J Immunol. 1998 Nov 1;161(9):5027-38. PMID: 9794440.
- Imai T, Chantry D, Raport CJ, Wood CL, Nishimura M, Godiska R, Yoshie O, Gray PW. Macrophage-derived chemokine is a functional ligand for the CC chemokine receptor 4. J Biol Chem. 1998 Jan 16;273(3):1764-8. https://doi.org/10.1074/jbc.273.3.1764. PMID: 9430724. [CrossRef]
- Arsentieva N.A., Liubimova N.E., Batsunov O.K., Korobova Z.R., Stanevich O.V., Lebedeva A.A., Vorobyov E.A., Vorobyova S.V., Kulikov A.N., Lioznov D.A., Sharapova M.A., Pevtcov D.E., Totolian A.A. Plasma cytokines in patients with COVID-19 during acute phase of the disease and following complete recovery. Medical Immunology (Russia). 2021;23(2):311-326. (In Russ.) https://doi.org/10.15789/1563-0625-PCI-2312. [CrossRef]
- Arsentieva N.A., Liubimova N.E., Batsunov O.K., et al. Predictive value of specific cytokines for lethal COVID-19 outcome // Russian Journal of Infection and Immunity. - 2022. - Vol. 12. - N. 5. - P. 859-868. https://doi.org/10.15789/2220-7619-PVO-2043. [CrossRef]
- Korobova ZR, Arsentieva NA, Liubimova NE, Batsunov OK, Dedkov VG, Gladkikh AS, Sharova AA, Adish Z, Chernykh EI, Kaschenko VA, Ratnikov VA, Gorelov VP, Stanevich OV, Kulikov AN, Pevtsov DE, Totolian AA. Cytokine Profiling in Different SARS-CoV-2 Genetic Variants. Int J Mol Sci. 2022 Nov 16;23(22):14146. https://doi.org/10.3390/ijms232214146. PMID: 36430621; PMCID: PMC9692520. [CrossRef]
- Fergie J, Srivastava A. Immunity to SARS-CoV-2: Lessons Learned. Front Immunol. 2021 Mar 19;12:654165. https://doi.org/10.3389/fimmu.2021.654165. PMID: 33815415; PMCID: PMC8018176. [CrossRef]
- Yang B, Yang J, Zhou L, Xue C, Li H, Hu W, Liu N. Inflammatory cytokine depletion in severe coronavirus disease 2019 infectious pneumonia: A case report. Medicine (Baltimore). 2020 Dec 4;99(49):e23449. https://doi.org/10.1097/MD.0000000000023449. PMID: 33285740; PMCID: PMC7717847. [CrossRef]
- Kudryavtsev IV, Arsentieva NA, Korobova ZR, Isakov DV, Rubinstein AA, Batsunov OK, Khamitova IV, Kuznetsova RN, Savin TV, Akisheva TV, Stanevich OV, Lebedeva AA, Vorobyov EA, Vorobyova SV, Kulikov AN, Sharapova MA, Pevtsov DE, Totolian AA. Heterogenous CD8+ T Cell Maturation and 'Polarization' in Acute and Convalescent COVID-19 Patients. Viruses. 2022 Aug 28;14(9):1906. https://doi.org/10.3390/v14091906. PMID: 36146713; PMCID: PMC9504186. [CrossRef]
- Wiech M, Chroscicki P, Swatler J, Stepnik D, De Biasi S, Hampel M, Brewinska-Olchowik M, Maliszewska A, Sklinda K, Durlik M, Wierzba W, Cossarizza A, Piwocka K. Remodeling of T Cell Dynamics During Long COVID Is Dependent on Severity of SARS-CoV-2 Infection. Front Immunol. 2022 Jun 10;13:886431. https://doi.org/10.3389/fimmu.2022.886431. PMID: 35757700; PMCID: PMC9226563. [CrossRef]
- Tufa A, Gebremariam TH, Manyazewal T, Getinet T, Webb DL, Hellström PM, Genet S. Inflammatory mediators profile in patients hospitalized with COVID-19: A comparative study. Front Immunol. 2022 Jul 25;13:964179. https://doi.org/10.3389/fimmu.2022.964179. PMID: 35958594; PMCID: PMC9359079. [CrossRef]
- Ling L, Chen Z, Lui G, Wong CK, Wong WT, Ng RWY, Tso EYK, Fung KSC, Chan V, Yeung ACM, Hui DSC, Chan PKS. Longitudinal Cytokine Profile in Patients With Mild to Critical COVID-19. Front Immunol. 2021 Dec 6;12:763292. https://doi.org/10.3389/fimmu.2021.763292. PMID: 34938289; PMCID: PMC8685399. [CrossRef]
- Andrew DP, Chang MS, McNinch J, Wathen ST, Rihanek M, Tseng J, Spellberg JP, Elias CG 3rd. STCP-1 (MDC) CC chemokine acts specifically on chronically activated Th2 lymphocytes and is produced by monocytes on stimulation with Th2 cytokines IL-4 and IL-13. J Immunol. 1998 Nov 1;161(9):5027-38. PMID: 9794440.
- Vulcano M, Albanesi C, Stoppacciaro A, Bagnati R, D'Amico G, Struyf S, Transidico P, Bonecchi R, Del Prete A, Allavena P, Ruco LP, Chiabrando C, Girolomoni G, Mantovani A, Sozzani S. Dendritic cells as a major source of macrophage-derived chemokine/CCL22 in vitro and in vivo. Eur J Immunol. 2001 Mar;31(3):812-2.
- Oronsky B, Larson C, Hammond TC, Oronsky A, Kesari S, Lybeck M, Reid TR. A Review of Persistent Post-COVID Syndrome (PPCS). Clin Rev Allergy Immunol. 2021 Feb 20:1–9. https://doi.org/10.1007/s12016-021-08848-3. Epub ahead of print. PMID: 33609255; PMCID: PMC7896544. [CrossRef]
- H.L. Tang, J.G. Cyster Chemokine up-regulation and activated T cell attraction by maturing dendritic cells Science, 284 (1999), pp. 819-822.
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. https://doi.org/10.1038/nm1093. [CrossRef]
- Klarquist J, Tobin K, Farhangi Oskuei P, Henning SW, Fernandez MF, Dellacecca ER, Navarro FC, Eby JM, Chatterjee S, Mehrotra S, et al. Ccl22 diverts T regulatory cells and controls the growth of melanoma. Cancer Res. 2016;76(21):6230–6240. https://doi.org/10.1158/0008-5472.CAN-16-0618. [CrossRef]
- Zhou M, Bracci PM, McCoy LS, Hsuang G, Wiemels JL, Rice T, Zheng S, Kelsey KT, Wrensch MR, Wiencke JK. Serum macrophage-derived chemokine/CCL22 levels are associated with glioma risk, CD4 T cell lymphopenia and survival time. Int J Cancer. 2015 Aug 15;137(4):826-36. https://doi.org/10.1002/ijc.29441. Epub 2015 Feb 2. PMID: 25604093; PMCID: PMC4478165. [CrossRef]
- Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, Biota C, Doffin AC, Durand I, Olive D, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009;69(5):2000–2009. https://doi.org/10.1158/0008-5472.CAN-08-2360. [CrossRef]
- Furue M, Ulzii D, Vu YH, Tsuji G, Kido-Nakahara M, Nakahara T. Pathogenesis of Atopic Dermatitis: Current Paradigm. Iran J Immunol. 2019 Jun;16(2):97-107. https://doi.org/10.22034/IJI.2019.80253. PMID: 31182684. [CrossRef]
- Ushio A, Arakaki R, Otsuka K, Yamada A, Tsunematsu T, Kudo Y, Aota K, Azuma M, Ishimaru N. CCL22-Producing Resident Macrophages Enhance T Cell Response in Sjögren's Syndrome. Front Immunol. 2018 Nov 8;9:2594. https://doi.org/10.3389/fimmu.2018.02594. PMID: 30467506; PMCID: PMC6236111. [CrossRef]
- Columba-Cabezas S, Serafini B, Ambrosini E, Sanchez M, Penna G, Adorini L, Aloisi F. Induction of macrophage-derived chemokine/CCL22 expression in experimental autoimmune encephalomyelitis and cultured microglia: implications for disease regulation. J Neuroimmunol. 2002 Sep;130(1-2):10-21. https://doi.org/10.1016/s0165-5728(02)00170-4. PMID: 12225884. [CrossRef]
- Fox KA, Kirwan DE, Whittington AM, Krishnan N, Robertson BD, Gilman RH, López JW, Singh S, Porter JC, Friedland JS. Platelets Regulate Pulmonary Inflammation and Tissue Destruction in Tuberculosis. Am J Respir Crit Care Med. 2018 Jul 15;198(2):245-255. https://doi.org/10.1164/rccm.201710-2102OC. PMID: 29420060; PMCID: PMC6058979. [CrossRef]
- Nakanishi, T., Imaizumi, K., Hasegawa, Y. et al. Expression of macrophage-derived chemokine (MDC)/CCL22 in human lung cancer. Cancer Immunol Immunother 55, 1320–1329 (2006). https://doi.org/10.1007/s00262-006-0133-y. [CrossRef]
- Richter JR, Sutton JM, Belizaire RM, Friend LA, Schuster RM, Johannigman TA, Miller SG, Lentsch AB, Pritts TA. Macrophage-derived chemokine (CCL22) is a novel mediator of lung inflammation following hemorrhage and resuscitation. Shock. 2014 Dec;42(6):525-31. https://doi.org/10.1097/SHK.0000000000000253. PMID: 25136780; PMCID: PMC4236272. [CrossRef]
- Berin MC, Dwinell MB, Eckmann L, Kagnoff MF. Production of MDC/CCL22 by human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2001 Jun;280(6):G1217-26. https://doi.org/10.1152/ajpgi.2001.280.6.G1217. PMID: 11352815. [CrossRef]
- Mantovani A, Gray PA, Van Damme J, Sozzani S. Macrophage-derived chemokine (MDC). J Leukoc Biol. 2000 Sep;68(3):400-4. PMID: 10985257.
- Alcami A, Saraiva M. Chemokine Binding Proteins Encoded by Pathogens. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013. Available from: https://www.ncbi.nlm.nih.gov/books/NBK5966/.
- Banu S, Nagaraj R, Idris MM. A proteomic perspective and involvement of cytokines in SARS-CoV-2 infection. PLoS One. 2023 Jan 6;18(1):e0279998. https://doi.org/10.1371/journal.pone.0279998. PMID: 36608055; PMCID: PMC9821788. [CrossRef]
- Hamdorf, M., Imhof, T., Bailey-Elkin, B., Betz, J., Theobald, S. J., Simonis, A., Cristanziano, V. D., Gieselmann, L., Dewald, F., Lehmann, C., Augustin, M., Klein, F., Alcazar, M. A., Rongisch, R., Fabri, M., Rybniker, J., Goebel, H., Stetefeld, J., Brachvogel, B., … Bock, F. (2022). The unique ORF8 protein from SARS-COV-2 binds to human dendritic cells and induces a hyper-inflammatory cytokine storm (Preprint). https://doi.org/10.1101/2022.06.06.494969. [CrossRef]
- Shahanshah Khan, Mahnoush S Shafiei, Christopher Longoria, John W Schoggins, Rashmin C Savani, Hasan Zaki (2021) SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway eLife 10:e68563 https://doi.org/10.7554/eLife.68563. [CrossRef]
- Wu, W., Cheng, Y., Zhou, H. et al. The SARS-CoV-2 nucleocapsid protein: its role in the viral life cycle, structure and functions, and use as a potential target in the development of vaccines and diagnostics. Virol J 20, 6 (2023). https://doi.org/10.1186/s12985-023-01968-6. [CrossRef]
- López-Muñoz AD, Kosik I, Holly J, Yewdell JW. Cell surface SARS-CoV-2 nucleocapsid protein modulates innate and adaptive immunity. Sci Adv. 2022 Aug 5;8(31):eabp9770. https://doi.org/10.1126/sciadv.abp9770. Epub 2022 Aug 3. PMID: 35921414; PMCID: PMC9348789. [CrossRef]
- González-Motos V, Kropp KA, Viejo-Borbolla A. Chemokine binding proteins: An immunomodulatory strategy going viral. Cytokine Growth Factor Rev. 2016 Aug;30:71-80. https://doi.org/10.1016/j.cytogfr.2016.02.007. Epub 2016 Mar 4. PMID: 26987612. [CrossRef]
- Tavakolpour S, Rakhshandehroo T, Wei EX, Rashidian M. Lymphopenia during the COVID-19 infection: What it shows and what can be learned. Immunol Lett. 2020 Sep;225:31-32. https://doi.org/10.1016/j.imlet.2020.06.013. Epub 2020 Jun 20. PMID: 32569607; PMCID: PMC7305732. [CrossRef]
- Ho CY, Wong CK, Li EK, Tam LS, Lam CW. Suppressive effect of combination treatment of leflunomide and methotrexate on chemokine expression in patients with rheumatoid arthritis. Clin Exp Immunol. 2003 Jul;133(1):132-8. https://doi.org/10.1046/j.1365-2249.2003.02192.x. PMID: 12823287; PMCID: PMC1808740. [CrossRef]
- Pérez-Gómez, A., Vitallé, J., Gasca-Capote, C. et al. Dendritic cell deficiencies persist seven months after SARS-CoV-2 infection. Cell Mol Immunol 18, 2128–2139 (2021). https://doi.org/10.1038/s41423-021-00728-2. [CrossRef]
- Winheim E, Rinke L, Lutz K, Reischer A, Leutbecher A, Wolfram L, Rausch L, Kranich J, Wratil PR, Huber JE, Baumjohann D, Rothenfusser S, Schubert B, Hilgendorff A, Hellmuth JC, Scherer C, Muenchhoff M, von Bergwelt-Baildon M, Stark K, Straub T, Brocker T, Keppler OT, Subklewe M, Krug AB. Impaired function and delayed regeneration of dendritic cells in COVID-19. PLoS Pathog. 2021 Oct 6;17(10):e1009742. https://doi.org/10.1371/journal.ppat.1009742. PMID: 34614036; PMCID: PMC8523079. [CrossRef]
- Chang T, Yang J, Deng H, Chen D, Yang X, Tang ZH. Depletion and Dysfunction of Dendritic Cells: Understanding SARS-CoV-2 Infection. Front Immunol. 2022 Feb 21;13:843342. https://doi.org/10.3389/fimmu.2022.843342. PMID: 35265087; PMCID: PMC8898834. [CrossRef]
- Montazersaheb, S., Hosseiniyan Khatibi, S.M., Hejazi, M.S. et al. COVID-19 infection: an overview on cytokine storm and related interventions. Virol J 19, 92 (2022). https://doi.org/10.1186/s12985-022-01814-1. [CrossRef]
- Alamri, A.; Fisk, D.; Upreti, D.; Kung, S.K.P. A Missing Link: Engagements of Dendritic Cells in the Pathogenesis of SARS-CoV-2 Infections. Int. J. Mol. Sci. 2021, 22, 1118. https://doi.org/10.3390/ijms22031118. [CrossRef]
- Chu H, Zhou J, Wong BH, Li C, Cheng ZS, Lin X, Poon VK, Sun T, Lau CC, Chan JF, To KK, Chan KH, Lu L, Zheng BJ, Yuen KY. Productive replication of Middle East respiratory syndrome coronavirus in monocyte-derived dendritic cells modulates innate immune response. Virology. 2014 Apr;454-455:197-205. https://doi.org/10.1016/j.virol.2014.02.018. Epub 2014 Mar 7. PMID: 24725946; PMCID: PMC7111975. [CrossRef]
- Onodi F, Bonnet-Madin L, Meertens L, Karpf L, Poirot J, Zhang SY, Picard C, Puel A, Jouanguy E, Zhang Q, Le Goff J, Molina JM, Delaugerre C, Casanova JL, Amara A, Soumelis V. SARS-CoV-2 induces human plasmacytoid pre-dendritic cell diversification via UNC93B and IRAK4. bioRxiv [Preprint]. 2021 Jan 8:2020.07.10.197343. https://doi.org/10.1101/2020.07.10.197343. Update in: J Exp Med. 2021 Apr 5;218(4): PMID: 33442685; PMCID: PMC7805442. [CrossRef]
- Cai G, Du M, Bossé Y, Albrecht H, Qin F, Luo X, Androulakis XM, Cheng C, Nagarkatti M, Nagarkatti P, Christiani DC, Whitfield ML, Amos CI, Xiao F. SARS-CoV-2 Impairs Dendritic Cells and Regulates DC-SIGN Gene Expression in Tissues. Int J Mol Sci. 2021 Aug 26;22(17):9228. https://doi.org/10.3390/ijms22179228. PMID: 34502134; PMCID: PMC8431536. [CrossRef]
- Galati D, Zanotta S, Capitelli L, Bocchino M. A bird's eye view on the role of dendritic cells in SARS-CoV-2 infection: Perspectives for immune-based vaccines. Allergy. 2022 Jan;77(1):100-110. https://doi.org/10.1111/all.15004. Epub 2021 Jul 24. PMID: 34245591; PMCID: PMC8441836. [CrossRef]
- Venet, M., Ribeiro, M.S., Décembre, E. et al. Severe COVID-19 patients have impaired plasmacytoid dendritic cell-mediated control of SARS-CoV-2. Nat Commun 14, 694 (2023). https://doi.org/10.1038/s41467-023-36140-9. [CrossRef]
Figure 1.
Simplified representation of viral binding and the immune response. Key: A - viral entry into cells via interaction with the ACE 2 receptor; B - enhanced expression of the angiotensin II gene, mediating extra stimulation of transcription factors; C - transcription factor activation (IRF, IkBɑ, NFkB); D - proinflammatory gene activation; E - cytokine/chemokine release and chemoattraction of immune cells; F - antigen-presenting cell (APC) arrival and additional release of cytokines with the subsequent activation of adaptive immunity and T-cell cytotoxicity.
Figure 1.
Simplified representation of viral binding and the immune response. Key: A - viral entry into cells via interaction with the ACE 2 receptor; B - enhanced expression of the angiotensin II gene, mediating extra stimulation of transcription factors; C - transcription factor activation (IRF, IkBɑ, NFkB); D - proinflammatory gene activation; E - cytokine/chemokine release and chemoattraction of immune cells; F - antigen-presenting cell (APC) arrival and additional release of cytokines with the subsequent activation of adaptive immunity and T-cell cytotoxicity.
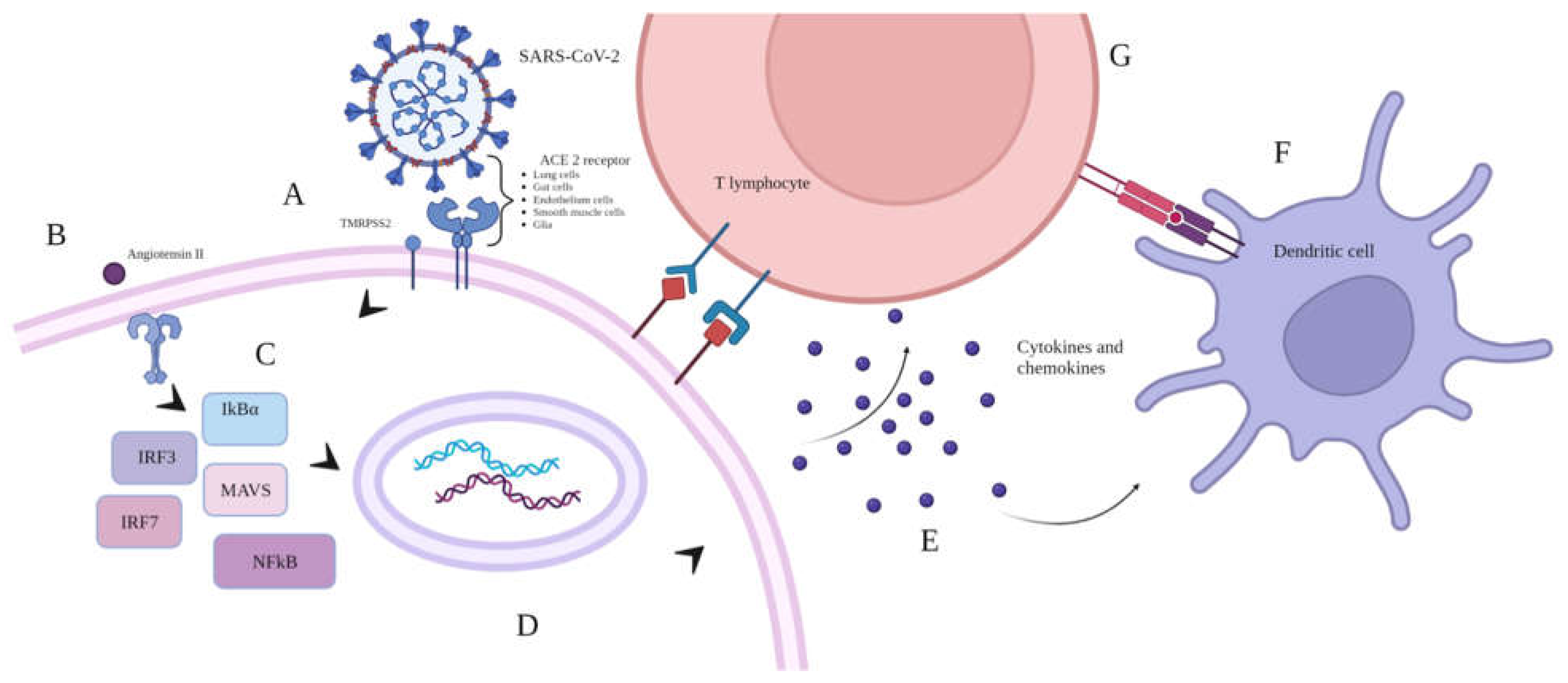
Figure 2.
MDC/CCL22 concentrations in blood plasma of COVID-19 patients infected with the Wuhan strain (n=51), Alpha (n=95), Delta (n=98), or Omicron (n=57) variants in comparison to healthy donors (n=56). Bars represent median concentrations (pg/ml). Whiskers represent the 75th quartile.
Figure 2.
MDC/CCL22 concentrations in blood plasma of COVID-19 patients infected with the Wuhan strain (n=51), Alpha (n=95), Delta (n=98), or Omicron (n=57) variants in comparison to healthy donors (n=56). Bars represent median concentrations (pg/ml). Whiskers represent the 75th quartile.
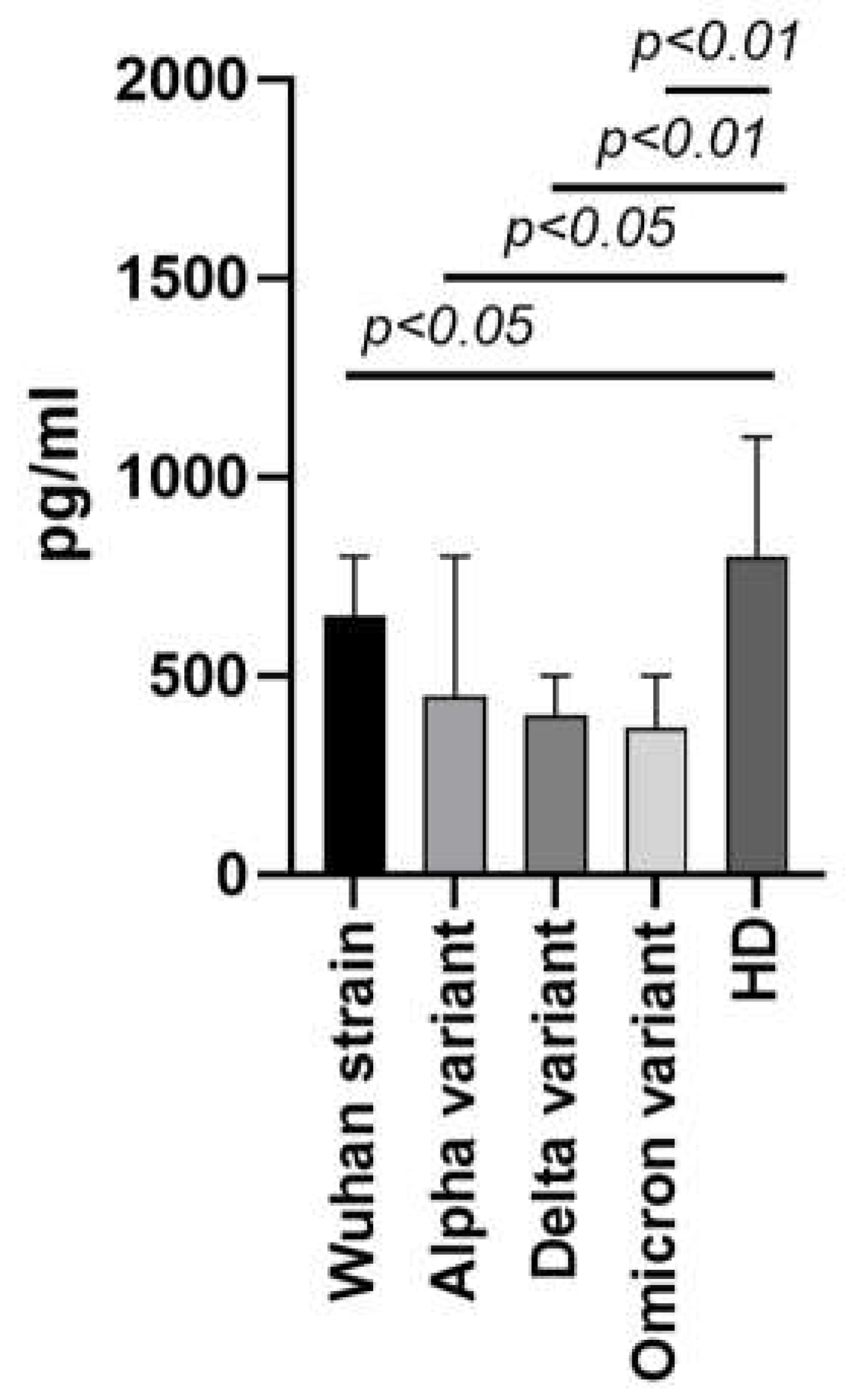
Figure 3.
MDC/CCL22 concentrations in the blood plasma of the first wave COVID-19 convalescents (n=69) in comparison to infected patients in the acute phase (n=51) and healthy donors (n=56). Bars represent median concentrations (pg/ml). Whiskers represent the 75th quartile.
Figure 3.
MDC/CCL22 concentrations in the blood plasma of the first wave COVID-19 convalescents (n=69) in comparison to infected patients in the acute phase (n=51) and healthy donors (n=56). Bars represent median concentrations (pg/ml). Whiskers represent the 75th quartile.
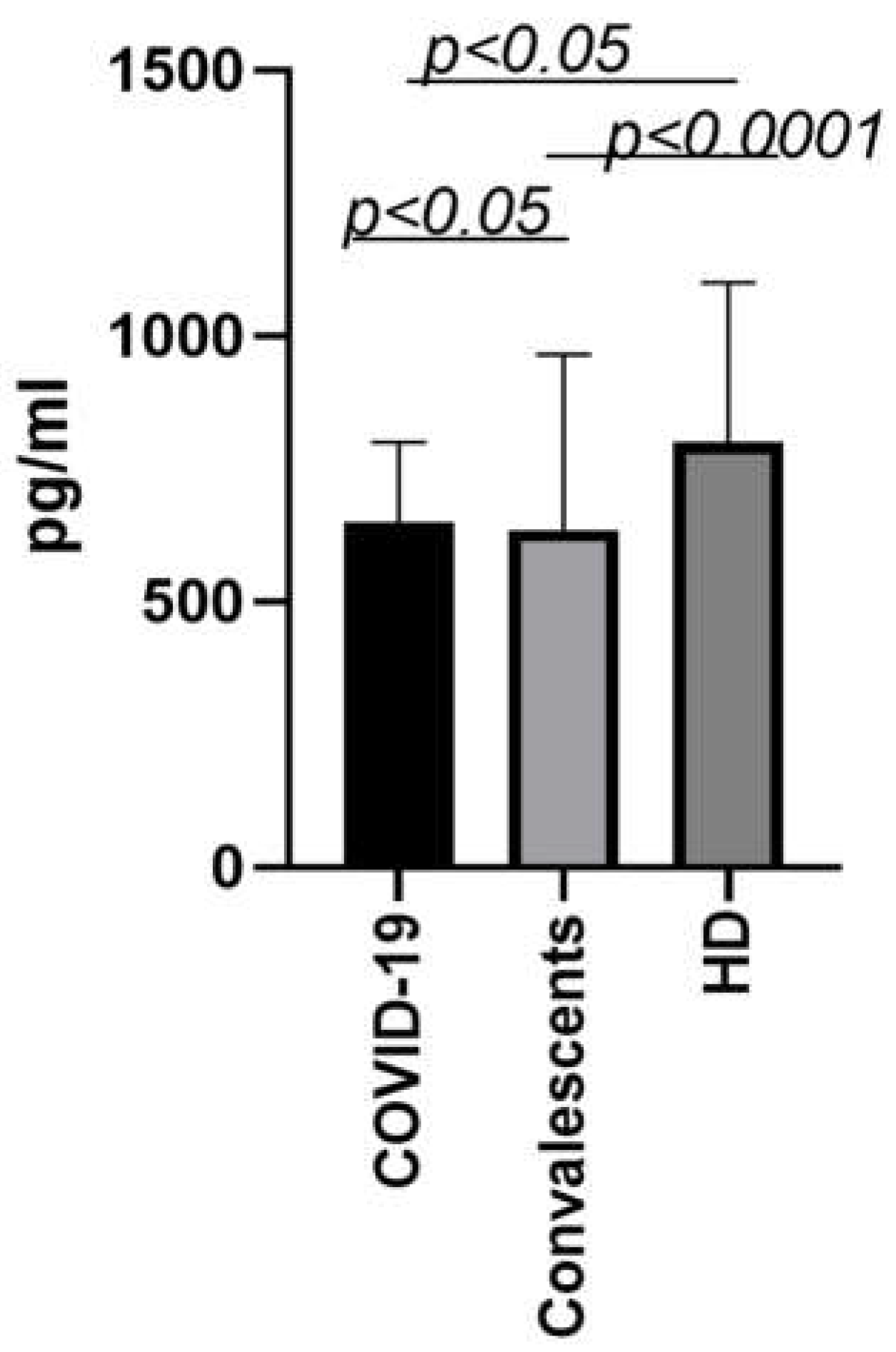
Figure 4.
Possible mechanisms for lower MDC/CCL22 concentrations in COVID-19 patient plasma. I - decrease in MDC/CCL22 concentration associated with selective binding to SARS-CoV-2 viral peptides. II - restriction of MDC/CCL22 secretion by producer cells due to their functional failure.
Figure 4.
Possible mechanisms for lower MDC/CCL22 concentrations in COVID-19 patient plasma. I - decrease in MDC/CCL22 concentration associated with selective binding to SARS-CoV-2 viral peptides. II - restriction of MDC/CCL22 secretion by producer cells due to their functional failure.
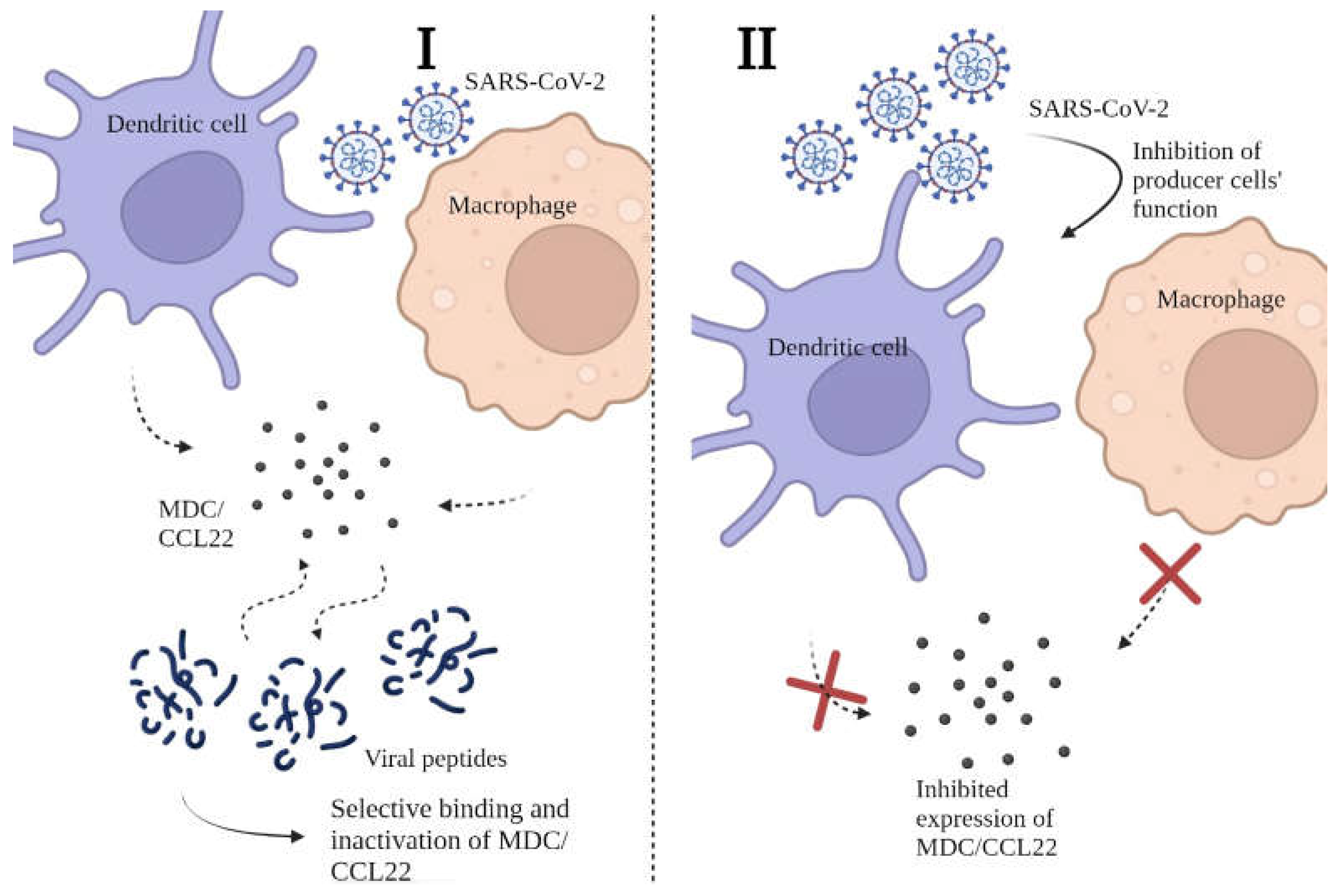
Figure 5.
Role of MDC/CCL22 in immunity and the SARS-CoV-2 infectious process. I - MDC/CCL22 influence on T lymphocyte maturation in thymus via the CCR4 receptor. The presence of this chemokine also mediates an adequate balance between regulatory T cells and helper T cells, thus creating restrictions on inflammatory reactions. II - SARS-CoV-2 influence on T cell maturation in thymus via depletion of MDC/CCL22: A - decrease in MDC/CCL22 concentrations associated with selective binding to SARS-CoV-2 viral peptides; B - restriction of MDC/CCL22 secretion by producer cells due to their functional failure.
Figure 5.
Role of MDC/CCL22 in immunity and the SARS-CoV-2 infectious process. I - MDC/CCL22 influence on T lymphocyte maturation in thymus via the CCR4 receptor. The presence of this chemokine also mediates an adequate balance between regulatory T cells and helper T cells, thus creating restrictions on inflammatory reactions. II - SARS-CoV-2 influence on T cell maturation in thymus via depletion of MDC/CCL22: A - decrease in MDC/CCL22 concentrations associated with selective binding to SARS-CoV-2 viral peptides; B - restriction of MDC/CCL22 secretion by producer cells due to their functional failure.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



