
Submitted:
05 May 2023
Posted:
08 May 2023
You are already at the latest version
Abstract
Repeated exposure to environmental ozone causes a chronic state of oxidative stress. This state is present in chronic degenerative diseases and induces a loss of control of the inflammatory response. The dysfunction of redox system and failures in control the inflammatory responses are involved in a vicious circle that maintains and increases the degenerative process. Intestine also responds to secondary reactive species formed by exposure to ozone doses, generating noxious stimuli that increase degenerative damage. This review aims to elucidate how environmental pollution, mainly by ozone, induces a state of chronic oxidative stress with the loss of regulation of the inflammatory response, both in the intestine and in the brain, where the functionality of both structures is altered and plays a determining role in some neurodegenerative and chronic degenerative diseases. For this purpose, we searched for information on sites like the Cochrane Library Database, PubMed, Scopus, and Medscape. Reviewing the data published, we can conclude that environmental pollutants are a severe health problem. Ozone pollution has different pathways of action, both molecular and systemic, and participates in neurodegenerative diseases such as Parkinson's and Alzheimer's disease as well in bowel diseases as Inflammatory Bowel Disease, Crohn's Disease and Irritable Bowel Syndrome.
Keywords:
Ozone
; Oxidative stress
; Bowel
; Brain
; Degenerative disease
1. Environmental pollution and disease
Environmental pollution is one of the most urgent public health problems to solve. The World Health Organization (WHO) estimates that 4.2 million deaths per year can be attributed to air pollution, of which 237,000 deaths would be among children under six [1]. The deterioration of the environment due to the presence of materials and compounds such as carbon dioxide (CO2), carbon monoxide (CO), nitrogen oxides (NOx), sulfur dioxide (SO2), volatile organic compounds (VOCs), nitrogen dioxide (NO2), ozone (O3), in amounts more significant than natural ones are an anthropogenic phenomenon [2]. The increase in these compounds is correlated with the rise in chronic degenerative diseases. Of the wide variety of pollutants that affect the quality of life of organisms, particulate matter (PM) stands out, specific microparticles with an aerodynamic diameter of less than 2.5µm (PM2.5) and particles equal or less than 10µm (PM10). Epidemiological studies have shown that the fraction of fine particles of PM2.5 has a more significant impact on health than PM10 [3]. PM2.5 is generated mainly in combustion processes, which produce carbon particles that, within their structure, may contain nitrates, sulfates, polycyclic aromatic hydrocarbons, and reactive metals such as iron, copper, nickel, zinc, and vanadium [4]. The physicochemical properties of the particles, such as size, structure, chemical composition, reactivity and solubility, as well as the ability to enter the cell, determine their impact on health and the mechanisms by which PM induces this variety of effects [5]. It is estimated that O3 could be responsible for almost 17,000 premature deaths in Europe each year [6]. Also tropospheric ozone, as well to affecting health, has adverse effects on ecosystems, particularly forests and crops.
The O3 concentrations are significant because its molecular conformation gives it highly reactive and oxidizing properties, thus producing an oxidizing environment, which when inhaled, alters the balance of the redox balance in the body. Biological systems must maintain the oxidation-reduction balance since a large part of cell signaling depends on redox signals. The loss of oxidation-reduction balance in the organism, caused by excess oxidants, produces a state of oxidative stress. When chronic, this state plays an essential role in developing degenerative diseases, such as autoimmune diseases, cancer, heart disease and diabetes. Still, it also plays a crucial role in developing neurodegenerative immune disorders such as Alzheimer's disease (AD) [5], Parkinson's disease (PD) [7], Huntington's disease [8], amyotrophic lateral sclerosis [9] and other processes related to pathological aging [10] (Figure 1).
2. Environmental pollution and intestine
Mucous membranes are body barriers that are in contact with the environment and constitute different habitats where microbes interact with the host. The digestive tract, respiratory tract, genitourinary tract, and skin are sites where the association with these microorganisms is essential [11]. The largest population of these microbes is found in the gastrointestinal tract, where one of their functions is to make food and water digested and absorbed into the body [12,13]. Moreover, the gut houses most of the mucosa-associated lymphoid tissue in the human body. The intestinal mucosa contains more lymphocytes than any other lymphatic organ [14]. There is a mutualistic relationship between the gut microbiota and the human host; the intestine provides a niche for microbes to inhabit, while the host benefits from an increase in digestive capacity favored by the microbiota and the generation of metabolites that stimulate the immune system, they also occupy a space within the intestine that prevents accumulation of potentially pathogenic organisms [15]. Variations in the microbial species and densities of the gut microbiota have been seen to modulate the immune response and susceptibility to different diseases [16]. In humans, environmental factors such as diet, pollutants, antibiotic treatments, and stress can change the composition of the gut microbiota towards a decrease in the abundance of beneficial bacterial species accompanied by the growth of pathogenic species [16]. The disturbance in microbial structure and function is known as dysbiosis, which disrupts immune and tissue homeostasis and is associated with various inflammatory diseases inside and outside the gastrointestinal tract [17]. There are bacterial species of the microbiota in the intestine that can counteract systemic inflammation. The gut microbiota is essential for host digestion and nutrition, as it can generate nutrients from substrates that the host could not otherwise digest. Intestinal microorganisms release short-chain fatty acids (SCFAs) from indigestible dietary fibers, an important energy source for intestinal mucosal cells and essential for modulating immune responses emanating from the intestine [17]. Therefore, gut bacteria help protect against pathogenic infections through competition, secretion of antimicrobial peptides, stimulation of innate immune cells, development of lymphoid tissue, antibody production and T-cell differentiation [18]. The gut microbiome has been shown to play a critical role in mediating immune and anti-inflammatory responses at distant sites by releasing and moving microbial metabolites such as SCFAs, cytokines, and hormones into the bloodstream [16]. The gut mucosa and associated immune system are primed to interact with food, microbiota, potentially harmful antigens, and inflammatory signals from other organs to maintain a fine homeostatic balance and promote an appropriate response, as well to ensure the presence of harmless microbes [14,19].
The intestine is exposed to air pollutants through direct or indirect routes. In gas exchange in the lungs, during exhalation, the lung mucus layer can reach the oropharynx and be swallowed and enter the tract. Another mechanism by which contaminants have access to the gastrointestinal tract is through direct ingestion of food and water that contain air pollutants [20]. Blood flow is important for distributing reactive oxygen species generated in the lungs, secondarily after ozone exposure. Mainly, the gastrointestinal tract is very susceptible to tobacco smoke and exposure to air pollution. These contaminants exacerbate systemic inflammation and cause oxidative damage to gut mucosa and microbiota [21]. Alterations in intestinal bacterial species, populations and microbial metabolites have been linked to changes in immune responses and inflammation in different organs [22]. Therefore, the evidence obtained in recent decades strongly suggests that air pollutants, ozone included, generate adverse effects on the digestive tract and that they are participating in the development of Inflammatory Bowel Disease (IBD), Crohn's Disease (CD), appendicitis and Irritable Bowel Syndrome (IBS) [23,24], immune diseases, chronic degenerative and neurodegenerative diseases such as Parkinson's and Alzheimer's disease, in which, during their pathophysiology, an environment is generated proinflammatory that affects the rest of the organism (Figure 2).
3. Intestinal alterations and neurodegeneration
The architecture of the intestinal epithelium is very complex; it is made up of a monolayer of cells connected by different cell junctions, such as tight junctions, gap junctions, and anchor junctions.
The surface of these cells is covered by mucosa, which separates the internal environment from the external one, blocking the passage of substances, which makes it very selective. One of the main functions of these cells is the absorption of nutrients and some electrolytes, which is why it is considered a highly communicated and coordinated semi-permeable barrier to prevent the free passage of certain toxins, antigens, and microbial products. In this way, a balance is generated between these transport mechanisms, the immune system, the metabolites products of the diet, and the intestinal microbiota. An alteration in this dynamic communication would cause a loss in this intestinal barrier, which would generate an imbalance and, therefore, the free passage of some substances and the development of various immune responses [25]. Some diseases, such as IBD, UC, and CD, whose main characteristic is the presence of a chronic inflammatory process, in which the immune response plays a fundamental role as part of the etiopathogenesis of these diseases, especially the response of T lymphocytes helper 17 (Th17) [26,27] and autophagy processes. It is known that patients with IBD have altered levels of cytokines, so they have focused on finding an association between polymorphisms of these cytokines and the relationship between promoters such as tumor necrosis factor-alpha (TNFα) and interleukin 10 (IL-10), as well as the relationships found between IL-1 and its receptor or between the IL-6 or IL-23 receptor [28]. It has been described that the function of Th17 is to protect the intestinal mucosa and maintain the immune microenvironment, in addition to increasing the inflammatory response through proinflammatory cytokines [29]; this function is maintained by IL-23 since it acts on the innate response and facilitates the maintenance of Th17 cells [30]. The cellular organization of the intestine that allows permeability maintenance can be compromised and facilitate the development of certain diseases. Proteins associated with granule exocytosis pathways in Paneth cells [31] and by transcription factors, such as XBP1, are known to respond to endoplasmic reticulum stress and are required for maintenance of the secretory cells [32], both mechanisms generate more susceptibility to intestinal inflammation, generating the loss of intestinal permeability (Figure 3).
4. Intestinal permeability and neurodegeneration
Paneth cells, participating in granule exocytosis pathways, have the function of producing antimicrobial peptides that control populations of bacteria harmful to the intestinal microbiota [33]. There is evidence that endotoxins, which are lipopolysaccharides (LPS) components of the cell wall of Gram-negative bacteria, generate a proinflammatory response mediated by TNFα, IL-1β or nitric oxide (NO) (Figure 3) [34]. In a mouse model, inhalation of low levels of endotoxin after exposure to ozone promoted an increase in various factors such as IL-1β, IL-1α, and IL-6, potentiating lung lesions in mice [35]. In addition, in clinical studies, Angelico Mendy et al. 2019 concluded a synergistic association between co-exposure to endotoxin and environmental contamination, increasing the recurrence of hospitalization due to asthma attacks in children [36].
Recent studies point to the relationship between intestinal microbiota and some neurodegenerative diseases. The brain-gut-microbiota axis is a bidirectional system that allows metabolic, immune, endocrine, and neuronal signaling [37] in both directions. Some evidence links endotoxins directly to neurodegeneration through systemic inflammation and microglia activation. Systemic LPS promotes IL-1β activation of microglia, chemokines, IL-6, and acute cognitive loss in mice [38]. LPS injected intraperitoneally promotes the accumulation of Aβ 1-42 in the hippocampus and cerebral cortex in mice, generating neuronal pyroptosis [39], hyperphosphorylation of the tau protein [40] and an increase in α-synuclein [41]. On the other hand, Alzheimer's disease has been characterized mainly as a disease in which extracellular deposits of the β-amyloid (Aβ) protein form neuritic plaques, which eventually lead to the intracellular accumulation of abnormal tau proteins and the subsequent formation of neurofibrillary tangles [42,43]. Molecular mimicry is a concept that allows us to explain how certain similarities occur in the tertiary structure of an exogenous and pathogenic protein and some human proteins [44]. This hypothesis reminds us of the formation of prions in the central nervous system and the generation of certain diseases. Particularly in Parkinson's disease, patients have a recurrent history of gastrointestinal dysfunctions before diagnosis [45]. Various studies have generated the "seed" of α-synuclein protein that recruits other endogenous α-synuclein proteins and subsequently becomes insoluble, hyperphosphorylated, and ubiquitinated pathological proteins [46]. Different models of α-synuclein "seeding" demonstrated the induction of pathological α-synuclein from the peripheral nervous system to the central nervous system following the injection of intramuscular α-synuclein in transgenic mice [47].
Some studies show how some species of bacteria produce extracellular amyloid protein fibers, which are helpful for environmental protection and surface adherence [48]. This protein, formed from the CsgG protein, allows a flow of macromolecules since it forms a functional nanopore [49]. Through a complex transport system, this system creates a "curli" fiber within the system [50]. Cellular pathways activated by bacterial amyloid in neurodegeneration processes include the TLR 2/1, CD14, and NFκB system [51].
Therefore, the structural complexity of the intestinal barrier is compromised when bowel permeability fails. The mechanisms underlying this mechanism range from a loss in the transport of granules, a decrease in the mucus layer, changes in the intestinal microbiota, and an alteration in the production of specific metabolites, such as LPS and proteins, such as α-synuclein or amyloid protein. All these changes activate signaling pathways that increase free radicals, as well as the activation of antioxidant and immune response systems, which, when they lose regulation, establishes a chronic state of oxidative stress and loss of regulation of the inflammatory response, establishing a vicious circle, which is present in chronic-degenerative diseases.
5. Parkinson's disease and intestine
Parkinson's disease (PD) is a neurodegenerative disease with a chronic state of oxidative stress and loss of regulation of the inflammatory response. These alterations give rise to oxidation of dopamine and formation of metabolites of this neurotransmitter, such as dopamine quinones [52]. Neuromelanin is found in catecholaminergic neurons of the substantia nigra pars compacta and locus coeruleus and protects neurons from oxidation processes. Loss of neuromelanin and subsequent depigmentation of these brain regions is a hallmark of PD. There is evidence to suggest that neuromelanin-abundant substantia nigra pars compacta dopaminergic neurons are more sensitive to cell death [53,54]. It has been proposed that there is a bidirectional communication of the brain-gut axis since there is evidence of accumulation of α-synuclein outside the brain, the abnormal accumulation of α-synuclein in the esophagus and the submucosal in the myenteric plexus and nerve fibers in the enteric nervous system has also been demonstrated [55].
It has been described alterations in gastrointestinal tract may precede changes in the brain; among these are dysphagia, increased salivation, delayed gastric emptying, and constipation [56,57], also alterations in the microbiota and abnormal accumulation of α-synuclein in the gastrointestinal system. The intestine has an essential role as a barrier [58] since it prevents the release of food antigens and inflammatory bacteria, preventing them from entering the circulation; however, when the barrier is altered, it becomes very permeable, releasing it into the torrent blood proinflammatory molecules [59] which causes the lymphoid follicles of the mucosa to become activated; T and B lymphocytes found in Peyer's patches adjacent to M cells proliferate as a process of stimulation by a specific antigen, releasing into the bloodstream, from there, they migrate to the lamina propria, and this is where the lymphocytes B are transformed into plasma cells, which produce specific secretory IgA against different antigens. IgA produced in the mucosa does not activate the complement pathway and does not cause inflammation [60]. Also, an inflammatory process is induced when foods rich in antigens from pathogenic organisms such as bacteria are ingested. Matthew Stephens and Pierre-Yves von der Weid (2020) demonstrated that LPS from specific species of the bacteria S. macescens, P. aeruginosa, K. pnemoniae, and S. enterica could differentially modulate TNFα through factor activation—core kB (NFkB) [61]. The intestine can distinguish between its own microbiota and pathogenic organisms. It is known that alterations between resident and pathogenic bacteria lead to an immune imbalance producing inflammation. It is accompanied by a loss of control of intestinal permeability that allows the release of bacterial LPS that reach the enteric nervous system, maintaining an inflammatory state as occurs in CD and PD. Otherwise, it has been observed that patients with Parkinson's disease have increased permeability in the colon (Figure 4), contributing to the systemic inflammatory process in patients with said disease.
6. Alzheimer's disease and intestine
One of the characteristics of Alzheimer's disease is the extracellular accumulation of β-amyloid peptide (βA), mainly the βA1-42 isoform, which forms plaques in various regions of the cerebral cortex and intracellular neurofibrillary tangles of hyperphosphorylated tau protein in the hippocampus, medial temporal lobe, isocortical temporal, parietal areas, and frontal lobes, producing loss of regulation of neuronal metabolism such as glutamatergic, cholinergic, and GABAergic neurotransmission [62]. Oxidative stress causes a change in the conformation of this peptide, making it more insoluble and impossible to eliminate [63]. The decrease in its clearance of βA leads to the loss of neuronal homeostasis [64]. However, one of the roles that βA plays in "normal" functioning in the brain is associated with glucose metabolism, specifically in the internalization of glucose [65]. Glucose uptake in neurons is mainly carried out by the glucose transporter 3 (GLUT3) [66]. In regions such as the hippocampus, one of the main areas compromised in AD, transport is carried out through the expression of GLUT4 in neurons by binding insulin or βA1-40 to the insulin receptor. Which activates the PI3K/Akt pathway, whose anti-apoptotic processes promote neuronal survival, as well as receptor translocation [65]. The pathogenesis associated with AD involves decreased hippocampal volume, cerebral hypoxia, neuroinflammation and neuronal ischemic events, which is associated with the gradual loss of episodic memory in patients [64]. It has been observed that the oxidative damage generated by the increase in reactive oxygen species (ROS) due to exposure to O3 influences the density and functionality of neurons and glia in the hippocampus, reflecting the loss of synapses and with it a deficit in the functionality of the hippocampus, which could be reflected in the decrease in attention or short-term memory [67,68,69]. Studies in rats exposed to O3 found that the increase in ROS is directly related to the rise in the levels of peroxidized lipids and proteins, which promote apoptosis in the hippocampus [70]; specifically in the CA1, CA3, and dentate gyrus regions; increasing the production and accumulation of the βA1-42 peptide in cells in the dentate gyrus [71].
The increase in lipids and proteins oxidized by ROS in these hippocampal regions promotes the expression of βA1-42, which results from the oxidation of the sulfhydryl radical of βA1-40, specifically methionine at position 35, promoting a more significant interaction between βA and the unsaturation of the lipid chains of the membrane of neurons, initiating lipid peroxidation, generating highly reactive products such as 4-hydroxy-2-nonenal (4HNE) and acrolein, which amplifies the oxidative effect of βA1 -42 [72], this causes accumulation of βA1-42, generating oligomers, which, when bound to IR, prevent its phosphorylation, decreasing glucose metabolism and inactivating the PI3K/Akt pathway, compromising survival neuron of the hippocampus [65].
Besides this, to try to eliminate these βA adducts, astrocytes and microglia, by binding to βA activate the MyD88 pathway through their toll-like 4 receptors (TLR4), promoting the expression of the receptor for tumor necrosis factor associated with factor 6 (TRAF6) and the CD33 receptor in the hippocampus and the cerebral cortex, causing neuroinflammation due to overexpression of glial fibrillary acidic protein (GFAP), generating astrogliosis [73,74], as well as the increase in NFkB which promotes the expression of proinflammatory cytokines such as IL-1β, IL-6, and TNFα, atrophying hippocampal communication between neurons and the glia [70].
In murine models, it has been shown that exposure to O3 promotes the production of NO, and this, in the presence of superoxide ion, generates the peroxynitrite ion (ONOO-). These highly oxidizing species together induce neurotoxicity in the hippocampus and cortical areas. The generation of NO in microglia causes high oxidation of lipids, proteins, and genetic material that favors the activation of different signaling pathways such as the Janus kinase complex (JAK's), protein tyrosine kinase (TK's), protein kinase C (PKC), mitogen-activated protein kinases (MAPK's), as well as NFκB and activator protein 1 (AP-1), prooxidant enzymes such as inducible nitric oxide synthase (iNOS), cyclooxygenase 2 (COX-2) and lipoxygenase 5 (LPO-5) [75]. In addition to the activation of these pathways, there is also an expression of proinflammatory cytokines such as IL-6, IL-1α, IL1β, TNFα factor and granulocyte-macrophage colony-stimulating factor that stimulate a state of neuronal stress. This state is mediated by high levels of ROS and inflammatory mediators, resulting in neuronal death and expression of AD pathogenesis [64,75]. RAMAN spectrometry studies show that repeated exposure to O3 causes conformational changes in the secondary amino acids of the βA1-40 peptide, taking it from an alpha helix conformation to a beta-sheet structure, which is insoluble and impossible to eliminate [63]. Therefore, in urbanized cities such as ours, exposure to O3 from our childhood can affect our learning and even be a promoter of the oxidation of proteins, lipids, and genetic material that in our adult life are related to the overexpression of the βA1-42 peptide and neurofibrillary tangles that could lead to the development of Alzheimer's disease.
Additionally, an increase in cholesterol ester hydroperoxides (CE-OOH) has been observed in patients with AD. These hydroperoxides are products of lipoprotein oxidation, such as chylomicrons, high-density lipoproteins (HDL), and intermediate-density lipoproteins (IDL), low-density lipoproteins (LDL) and very-low-density lipoproteins (VLDL), specifically those whose structure includes apolipoproteins AI, B48/B100, D, E, and J/clusterin, which participate in lipoprotein binding to the anchoring sites associated with ATP, in lipid transport, in cholesterol metabolism, as well as defense against oxidative damage, increasing their levels in plasma, promoting a state of oxidized hypercholesterolemia [76], which increases the expression of proinflammatory cytokines as well as inducing cell damage and due to this increase in peroxidized lipids the RedOx balance is compromised. Therefore, antioxidant proteins such as paraoxonases (PON) protect lipoproteins from oxidative stress, reducing the damage caused by ROS [77]. However, it has been observed that in patients and mouse models of AD, PON1, and PON3, located in the systemic circulation, and PON2, located throughout the intestinal tract from the duodenum to the distal colon, in different subcellular compartments such as mitochondria, lysosomes, and microsomes decrease their activity, promoting intestinal inflammation [78]. Therefore, in urbanized cities like ours, exposure to O3 from our childhood can have repercussions on our learning and even be a promoter of the oxidation of genetic material, proteins, and lipids, as well as the decrease in our antioxidant proteins that protect us from damage associated with oxidation; which in our adult life can be related to intestinal problems associated with an increase in plasma cholesterol and the overexpression of the βA1-42 peptide and neurofibrillary tangles that could lead to the development of Alzheimer's disease.
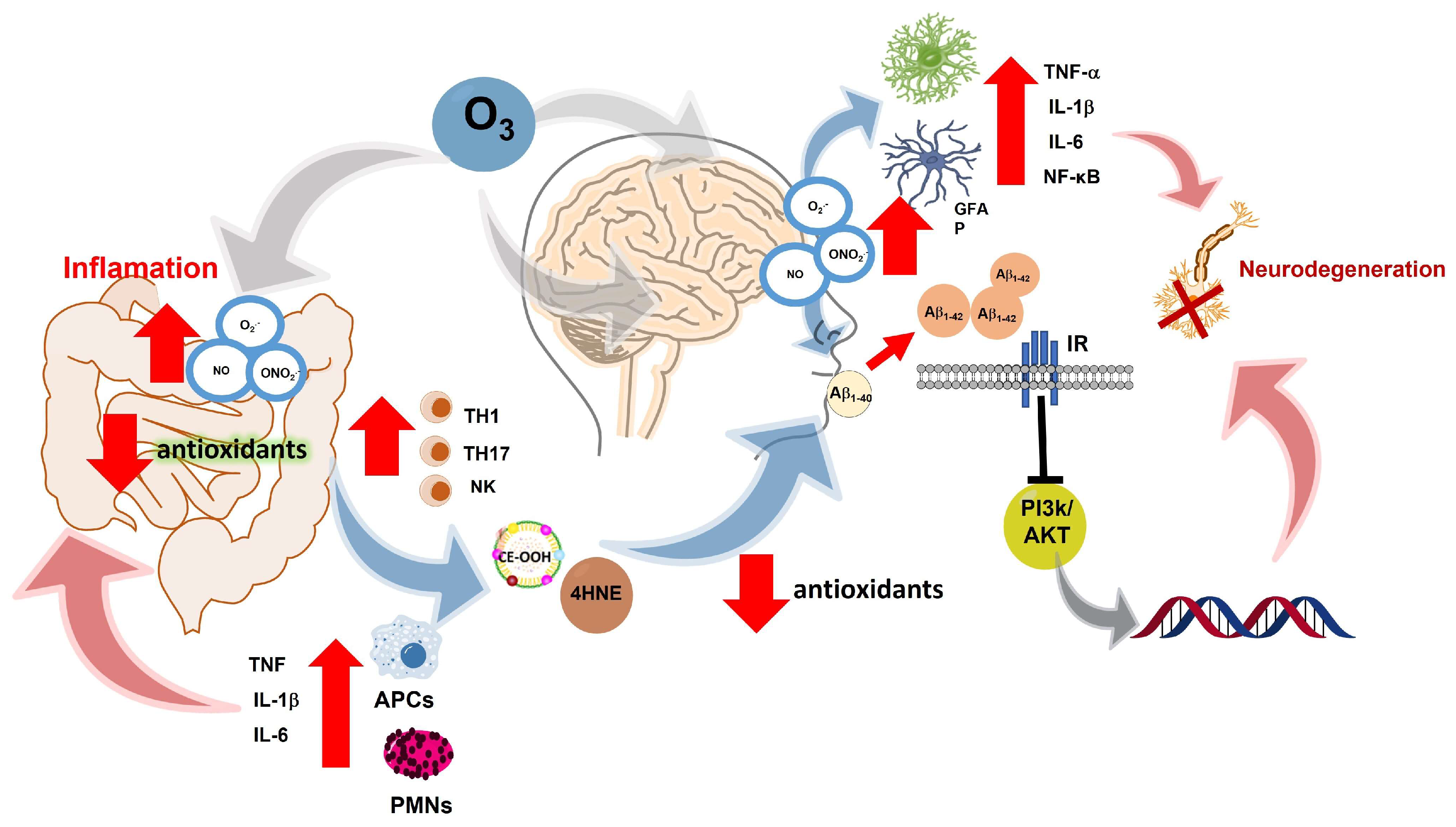
Figure 5.
Exposure to O3 increases the production of ROS, which leads to the development of inflammatory bowel diseases associated with the increase of cytokines IL-6, IL-1β and TNFα, as well as the activation of TH1, TH17 and NK cells responsible for cell maintenance; however, the increase in ROS, as well as the decrease in antioxidant defenses, limits the function of lymphocyte cells, generating peroxidized lipid adducts such as CE-OOH and 4HNE at the systemic level, decreasing antioxidant defenses, causing polypeptides such as βA to oxidize and generating oligomers of βA1-42, which interrupt the internalization of glucose, causing survival pathways such as PI3K/AKT to be inhibited, causing neuronal cells to atrophy, which causes astrocytes and microglia to be activated, releasing proinflammatory cytokines causing neuroinflammation, which chronically leads to neurodegeneration of neurons in the hippocampus and cortical areas that is pathologically associated with gradual loss of movement and memory, finally It is currently believed that discrete degenerative processes at the beginning of AD such as the deposition of highly insoluble βA and the accumulation of tau proteins-damage neurons and provide clear inflammatory stimuli to local microglia, and the hippocampus.
Figure 5.
Exposure to O3 increases the production of ROS, which leads to the development of inflammatory bowel diseases associated with the increase of cytokines IL-6, IL-1β and TNFα, as well as the activation of TH1, TH17 and NK cells responsible for cell maintenance; however, the increase in ROS, as well as the decrease in antioxidant defenses, limits the function of lymphocyte cells, generating peroxidized lipid adducts such as CE-OOH and 4HNE at the systemic level, decreasing antioxidant defenses, causing polypeptides such as βA to oxidize and generating oligomers of βA1-42, which interrupt the internalization of glucose, causing survival pathways such as PI3K/AKT to be inhibited, causing neuronal cells to atrophy, which causes astrocytes and microglia to be activated, releasing proinflammatory cytokines causing neuroinflammation, which chronically leads to neurodegeneration of neurons in the hippocampus and cortical areas that is pathologically associated with gradual loss of movement and memory, finally It is currently believed that discrete degenerative processes at the beginning of AD such as the deposition of highly insoluble βA and the accumulation of tau proteins-damage neurons and provide clear inflammatory stimuli to local microglia, and the hippocampus.
7. Discussion
It has been widely demonstrated that environmental pollution by ozone produces a chronic state of oxidative stress. Increase in ROS secondary to ozone inhalation leads to the loss of regulation of cell functions. The oxidation-reduction processes that regulate cell activity are one of the first signals to appear during evolution. Therefore, they are central to the physiological organization that allows proper cell function. ROS regulate many cellular signals (metabolic, endocrine, reproductive, etc.) and particularly of the immune system. The inflammatory response is key in cell defense and repair, it is a self-limited response and modulated by redox signaling. Hence, the redox balance is necessary for the immune response to maintain its function. However, repeated exposure to environmental pollution by ozone (at low doses) causes a slight increase in ROS, and the antioxidant defenses do not respond adequately. The gradual increase in ROS and the inability of the antioxidant systems induce a chronic state of oxidative stress that leads to a loss of regulation of the inflammatory response, as occurs in non-infectious chronic-degenerative diseases. The chronic state of oxidative stress is an epiphenomenon that affects the entire organism. However, depending on the antioxidant capacity and other factors, whether the genetic or previous presence of disease, the target organ will be more compromised than the others [79,80]. There is a clear association between the gastrointestinal system and neurodegenerative diseases such as Parkinson's disease and Alzheimer's disease. Also, there are intrinsic alterations of the intestine in the face of oxidative stress caused by environmental pollution by ozone. The response of the duodenum and colon seems to be important for gastrointestinal pathologies. Which can be explained as follows:
1) Oxidative stress causes alterations in the intestinal wall, altering permeability and allowing the passage of high molecular weight molecules that will ultimately stimulate the immune system, causing inflammation and disease.
2) Oxidative stress acts on the intestinal microbiome, changing its balance and favoring microorganisms whose metabolism causes toxic molecules that contribute to further increased inflammation and forming a vicious circle between loss of regulation of the inflammatory response, increase intestinal permeability, harmful change of intestinal microorganisms and alteration of the immune response.
Association between gastrointestinal alterations and non-communicable chronic degenerative diseases share a series of characteristics such as the chronic state of oxidative stress, loss of regulation of the immune response, alteration in permeability of both the blood-brain and intestinal barriers allowing passage of high molecular weight substances, formation of toxic metabolites both by the intestinal bacteria as well ROS and accumulation of insoluble proteins such as α-synuclein, amyloid peptides, etc.
8. Conclusions
Environmental pollution by ozone generates a chronic state of oxidative stress that alters the permeability of the blood-brain barrier. The chronic state of oxidative stress is related to the loss of regulation of the inflammatory response; these two factors form a vicious circle that maintains and increases the deterioration of the degenerative disease over time.
Environmental pollution by ozone causes alterations in the intestine, inducing an inflammatory response in the intestinal wall that alters its permeability and the bacterial populations. The passage of macromolecules into the blood stimulates the immune system. The pathological change of the intestinal microbiome contributes to the production of toxic metabolites that increase the degenerative response. Loss of the permeability of the blood-brain barrier increases inflammation and, consequently oxidative stress in the brain. Communication through neurotransmitters, hormones, and proinflammatory metabolites in the blood leads to a close interrelationship between these organs, promoting chronic-degenerative diseases.
Author Contributions
Authors contributed equally to this work.
Funding
This work was funded by grant PAPIIT-DGAPA IN221521 for Selva Rivas-Arancibia. Alfredo Miranda-Martínez receives a CONACyT postdoctoral fellowship, CVU 385286.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Regional Office for, E. WHO guidelines for indoor air quality: selected pollutants. Copenhagen: World Health Organization. Regional Office for Europe; 2010 2010.
- Climate change decreases the quality of the air we breathe.
- Orellano, P.; Reynoso, J.; Quaranta, N.; Bardach, A. , Ciapponi, A. Short-term exposure to particulate matter (PM(10) and PM(2.5)), nitrogen dioxide (NO(2)), and ozone (O(3)) and all-cause and cause-specific mortality: Systematic review and meta-analysis. Environ Int. 2020,142:105876. [CrossRef]
- Harishkumar, K.; Yogesh, K. , Gad, I. Forecasting air pollution particulate matter (PM2. 5) using machine learning regression models. Procedia Computer Science. 2020,171:2057-66. [CrossRef]
- Wang, G.; Pan, X.L.; Cui, P.J.; Wang, Y.; Ma, J.F.; Ren, R.J.; Deng, Y.L.; Xu, W.; Tang, H.D. , Chen, S.D. Association study of the GAB2 gene with the risk of Alzheimer disease in the chinese population. Alzheimer Dis Assoc Disord. 2011,25(3):283-5. [CrossRef]
- de la Salud, O.M. OMS. Convenio Marco de la OMS para el Control del Tabaco Ginebra. 2002, 21. [Google Scholar]
- Sevcsik, E.; Trexler, A.J.; Dunn, J.M. , Rhoades, E. Allostery in a disordered protein: oxidative modifications to α-synuclein act distally to regulate membrane binding. J Am Chem Soc. 2011,133(18):7152-8. [CrossRef]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W. , Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington's disease mice. Acta Neuropathol. 2011,121(4):487-98. [CrossRef]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B. , Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 2011,134(Pt 5):1293-314. [CrossRef]
- Floyd, R.A.; Towner, R.A.; He, T.; Hensley, K. , Maples, K.R. Translational research involving oxidative stress and diseases of aging. Free Radic Biol Med. 2011,51(5):931-41. [CrossRef]
- Fitzgibbon, G. , Mills, K.H.G. The microbiota and immune-mediated diseases: Opportunities for therapeutic intervention. Eur J Immunol. 2020,50(3):326-37. [CrossRef]
- Barrett, K.E. Ganong fisiología médica (24a: McGraw Hill Mexico; 2013.
- Cheng, L.K.; O'Grady, G.; Du, P.; Egbuji, J.U.; Windsor, J.A. , Pullan, A.J. Gastrointestinal system. Wiley Interdiscip Rev Syst Biol Med. 2010,2(1):65-79.
- Baumgart, D.C. , Dignass, A.U. Intestinal barrier function. Curr Opin Clin Nutr Metab Care. 2002,5(6):685-94.
- Thursby, E. , Juge, N. Introduction to the human gut microbiota. Biochem J. 2017,474(11):1823-36. [CrossRef]
- Zhang, D.; Li, S.; Wang, N.; Tan, H.Y.; Zhang, Z. , Feng, Y. The Cross-Talk Between Gut Microbiota and Lungs in Common Lung Diseases. Front Microbiol. 2020,11:301. [CrossRef]
- Shreiner, A.B.; Kao, J.Y. , Young, V.B. The gut microbiome in health and in disease. Curr Opin Gastroenterol. 2015,31(1):69-75. [CrossRef]
- McAleer, J.P. , Kolls, J.K. Contributions of the intestinal microbiome in lung immunity. Eur J Immunol. 2018,48(1):39-49. [CrossRef]
- Raftery, A.L.; Tsantikos, E.; Harris, N.L. , Hibbs, M.L. Links Between Inflammatory Bowel Disease and Chronic Obstructive Pulmonary Disease. Front Immunol. 2020,11:2144. [CrossRef]
- Feng, J.; Cavallero, S.; Hsiai, T. , Li, R. Impact of air pollution on intestinal redox lipidome and microbiome. Free Radic Biol Med. 2020,151:99-110. [CrossRef]
- Sobczak, M.; Fabisiak, A.; Murawska, N.; Wesołowska, E.; Wierzbicka, P.; Wlazłowski, M.; Wójcikowska, M.; Zatorski, H.; Zwolińska, M. , Fichna, J. Current overview of extrinsic and intrinsic factors in etiology and progression of inflammatory bowel diseases. Pharmacol Rep. 2014,66(5):766-75. [CrossRef]
- Pascual, I.P.; Martínez, A.R. , de la Fuente Moral, S. Interacciones entre microbiota y huésped. Medicine-Programa de Formación Médica Continuada Acreditado. 2022,13(49):2843-52.
- Marynowski, M.; Likońska, A.; Zatorski, H. , Fichna, J. Role of environmental pollution in irritable bowel syndrome. World J Gastroenterol. 2015,21(40):11371-8. [CrossRef]
- Vignal, C.; Guilloteau, E.; Gower-Rousseau, C. , Body-Malapel, M. Review article: Epidemiological and animal evidence for the role of air pollution in intestinal diseases. Sci Total Environ. 2021,757:143718. [CrossRef]
- Salvo Romero, E.; Alonso Cotoner, C.; Pardo Camacho, C.; Casado Bedmar, M. , Vicario, M. The intestinal barrier function and its involvement in digestive disease. Rev Esp Enferm Dig. 2015,107(11):686-96. [CrossRef]
- Ueno, A.; Jijon, H.; Chan, R.; Ford, K.; Hirota, C.; Kaplan, G.G.; Beck, P.L.; Iacucci, M.; Fort Gasia, M.; Barkema, H.W. , et al. Increased prevalence of circulating novel IL-17 secreting Foxp3 expressing CD4+ T cells and defective suppressive function of circulating Foxp3+ regulatory cells support plasticity between Th17 and regulatory T cells in inflammatory bowel disease patients. Inflamm Bowel Dis. 2013,19(12):2522-34. [CrossRef]
- Cho, J.; Kim, S.; Yang, D.H.; Lee, J.; Park, K.W.; Go, J.; Hyun, C.L.; Jee, Y. , Kang, K.S. Mucosal Immunity Related to FOXP3(+) Regulatory T Cells, Th17 Cells and Cytokines in Pediatric Inflammatory Bowel Disease. J Korean Med Sci. 2018,33(52):e336. [CrossRef]
- Ye, M.; Joosse, M.E.; Liu, L.; Sun, Y.; Dong, Y.; Cai, C.; Song, Z.; Zhang, J.; Brant, S.R.; Lazarev, M. , et al. Deletion of IL-6 Exacerbates Colitis and Induces Systemic Inflammation in IL-10-Deficient Mice. J Crohns Colitis. 2020,14(6):831-40. [CrossRef]
- Yan, J.B.; Luo, M.M.; Chen, Z.Y. , He, B.H. The Function and Role of the Th17/Treg Cell Balance in Inflammatory Bowel Disease. J Immunol Res. 2020,2020:8813558.
- Cătană, C.S.; Berindan Neagoe, I.; Cozma, V.; Magdaş, C.; Tăbăran, F. , Dumitraşcu, D.L. Contribution of the IL-17/IL-23 axis to the pathogenesis of inflammatory bowel disease. World J Gastroenterol. 2015,21(19):5823-30.
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S. , et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008,456(7219):259-63. [CrossRef]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H. , et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008,134(5):743-56. [CrossRef]
- Gubatan, J.; Holman, D.R.; Puntasecca, C.J.; Polevoi, D.; Rubin, S.J. , Rogalla, S. Antimicrobial peptides and the gut microbiome in inflammatory bowel disease. World J Gastroenterol. 2021,27(43):7402-22. [CrossRef]
- Zheng, W.; Zheng, X.; Liu, S.; Ouyang, H.; Levitt, R.C.; Candiotti, K.A. , Hao, S. TNFα and IL-1β are mediated by both TLR4 and Nod1 pathways in the cultured HAPI cells stimulated by LPS. Biochem Biophys Res Commun. 2012,420(4):762-7. [CrossRef]
- Johnston, C.J.; Oberdörster, G.; Gelein, R. , Finkelstein, J.N. Endotoxin potentiates ozone-induced pulmonary chemokine and inflammatory responses. Exp Lung Res. 2002,28(6):419-33. [CrossRef]
- Mendy, A.; Wilkerson, J.; Salo, P.M.; Weir, C.H.; Feinstein, L.; Zeldin, D.C. , Thorne, P.S. Synergistic Association of House Endotoxin Exposure and Ambient Air Pollution with Asthma Outcomes. Am J Respir Crit Care Med. 2019,200(6):712-20. [CrossRef]
- Zhu, X.; Li, B.; Lou, P.; Dai, T.; Chen, Y.; Zhuge, A.; Yuan, Y. , Li, L. The Relationship Between the Gut Microbiome and Neurodegenerative Diseases. Neurosci Bull. 2021,37(10):1510-22. [CrossRef]
- Lopez-Rodriguez, A.B.; Hennessy, E.; Murray, C.L.; Nazmi, A.; Delaney, H.J.; Healy, D.; Fagan, S.G.; Rooney, M.; Stewart, E.; Lewis, A. , et al. Acute systemic inflammation exacerbates neuroinflammation in Alzheimer's disease: IL-1β drives amplified responses in primed astrocytes and neuronal network dysfunction. Alzheimers Dement. 2021,17(10):1735-55.
- Han, C.; Yang, Y.; Guan, Q.; Zhang, X.; Shen, H.; Sheng, Y.; Wang, J.; Zhou, X.; Li, W.; Guo, L. , et al. New mechanism of nerve injury in Alzheimer's disease: β-amyloid-induced neuronal pyroptosis. J Cell Mol Med. 2020,24(14):8078-90.
- Gardner, L.E.; White, J.D.; Eimerbrink, M.J.; Boehm, G.W. , Chumley, M.J. Imatinib methanesulfonate reduces hyperphosphorylation of tau following repeated peripheral exposure to lipopolysaccharide. Neuroscience. 2016,331:72-7. [CrossRef]
- Kim, C.; Lv, G.; Lee, J.S.; Jung, B.C.; Masuda-Suzukake, M.; Hong, C.S.; Valera, E.; Lee, H.J.; Paik, S.R.; Hasegawa, M. , et al. Exposure to bacterial endotoxin generates a distinct strain of α-synuclein fibril. Sci Rep. 2016,6:30891. [CrossRef]
- Hardy, J. , Selkoe, D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002,297(5580):353-6.
- Xie, J.; Van Hoecke, L. , Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer's Disease Pathology. Front Immunol. 2021,12:796867. [CrossRef]
- Segal, Y. , Shoenfeld, Y. Vaccine-induced autoimmunity: the role of molecular mimicry and immune crossreaction. Cell Mol Immunol. 2018,15(6):586-94. [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R. , Jenner, P. Non-motor features of Parkinson disease. Nat Rev Neurosci. 2017,18(7):435-50.
- Luk, K.C.; Song, C.; O'Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q. , Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009,106(47):20051-6. [CrossRef]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; Lee, S.; Regenhardt, R.W.; McGarvey, N.H.; Ayers, J.I.; Notterpek, L.; Borchelt, D.R. , et al. Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci U S A. 2014,111(29):10732-7. [CrossRef]
- Garcia, M.; Lipke, P. , Klotz, S. Pathogenic microbial amyloids: Their function and the host response. OA Microbiol. 2013,1(1).
- Taylor, J.D.; Zhou, Y.; Salgado, P.S.; Patwardhan, A.; McGuffie, M.; Pape, T.; Grabe, G.; Ashman, E.; Constable, S.C.; Simpson, P.J. , et al. Atomic resolution insights into curli fiber biogenesis. Structure. 2011,19(9):1307-16. [CrossRef]
- Taylor, J.D. , Matthews, S.J. New insight into the molecular control of bacterial functional amyloids. Front Cell Infect Microbiol. 2015,5:33.
- Miraglia, F. , Colla, E. Microbiome, Parkinson's Disease and Molecular Mimicry. Cells. 2019,8(3). [CrossRef]
- Santiago-López, D.; Bautista-Martínez, J.A.; Reyes-Hernandez, C.I.; Aguilar-Martínez, M. , Rivas-Arancibia, S. Oxidative stress, progressive damage in the substantia nigra and plasma dopamine oxidation, in rats chronically exposed to ozone. Toxicol Lett. 2010,197(3):193-200. [CrossRef]
- Zecca, L.; Zucca, F.A.; Wilms, H. , Sulzer, D. Neuromelanin of the substantia nigra: a neuronal black hole with protective and toxic characteristics. Trends Neurosci. 2003,26(11):578-80. [CrossRef]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L. , Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson's disease. Prog Neurobiol. 2017,155:96-119.
- Braak, H.; de Vos, R.A.; Bohl, J. , Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci Lett. 2006,396(1):67-72.
- Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson's disease. Lancet Neurol. 2003,2(2):107-16.
- Fasano, A.; Aquino, C.C.; Krauss, J.K.; Honey, C.R. , Bloem, B.R. Axial disability and deep brain stimulation in patients with Parkinson disease. Nat Rev Neurol. 2015,11(2):98-110. [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009,9(11):799-809. [CrossRef]
- van, I.S.C.D. , Derkinderen, P. The Intestinal Barrier in Parkinson's Disease: Current State of Knowledge. J Parkinsons Dis. 2019,9(s2):S323-s9.
- Saito, H.; Kanamori, Y.; Takemori, T.; Nariuchi, H.; Kubota, E.; Takahashi-Iwanaga, H.; Iwanaga, T. , Ishikawa, H. Generation of intestinal T cells from progenitors residing in gut cryptopatches. Science. 1998,280(5361):275-8. [CrossRef]
- Stephens, M. , von der Weid, P.Y. Lipopolysaccharides modulate intestinal epithelial permeability and inflammation in a species-specific manner. Gut Microbes. 2020,11(3):421-32.
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A. , Jones, D.T. Alzheimer disease. Nat Rev Dis Primers. 2021,7(1):33.
- Rivas-Arancibia, S.; Guevara-Guzmán, R.; López-Vidal, Y.; Rodríguez-Martínez, E.; Zanardo-Gomes, M.; Angoa-Pérez, M. , Raisman-Vozari, R. Oxidative stress caused by ozone exposure induces loss of brain repair in the hippocampus of adult rats. Toxicol Sci. 2010,113(1):187-97.
- Bandyopadhyay, S. Role of Neuron and Glia in Alzheimer's Disease and Associated Vascular Dysfunction. Front Aging Neurosci. 2021,13:653334. [CrossRef]
- Molina-Fernández, R.; Picón-Pagès, P.; Barranco-Almohalla, A.; Crepin, G.; Herrera-Fernández, V.; García-Elías, A.; Fanlo-Ucar, H.; Fernàndez-Busquets, X.; García-Ojalvo, J.; Oliva, B. , et al. Differential regulation of insulin signalling by monomeric and oligomeric amyloid beta-peptide. Brain Commun. 2022,4(5):fcac243. [CrossRef]
- Leino, R.L.; Gerhart, D.Z.; van Bueren, A.M.; McCall, A.L. , Drewes, L.R. Ultrastructural localization of GLUT 1 and GLUT 3 glucose transporters in rat brain. J Neurosci Res. 1997,49(5):617-26.
- Croze, M.L. , Zimmer, L. Ozone Atmospheric Pollution and Alzheimer's Disease: From Epidemiological Facts to Molecular Mechanisms. J Alzheimers Dis. 2018,62(2):503-22.
- Corradi, M.; Alinovi, R.; Goldoni, M.; Vettori, M.; Folesani, G.; Mozzoni, P.; Cavazzini, S.; Bergamaschi, E.; Rossi, L. , Mutti, A. Biomarkers of oxidative stress after controlled human exposure to ozone. Toxicol Lett. 2002,134(1-3):219-25. [CrossRef]
- Rivas-Arancibia, S.; Vazquez-Sandoval, R.; Gonzalez-Kladiano, D.; Schneider-Rivas, S. , Lechuga-Guerrero, A. Effects of ozone exposure in rats on memory and levels of brain and pulmonary superoxide dismutase. Environ Res. 1998,76(1):33-9. [CrossRef]
- Nery-Flores, S.D.; Ramírez-Herrera, M.A.; Mendoza-Magaña, M.L.; Romero-Prado, M.M.J.; Ramírez-Vázquez, J.J.; Bañuelos-Pineda, J.; Espinoza-Gutiérrez, H.A.; Ramírez-Mendoza, A.A. , Tostado, M.C. Dietary Curcumin Prevented Astrocytosis, Microgliosis, and Apoptosis Caused by Acute and Chronic Exposure to Ozone. Molecules. 2019,24(15). [CrossRef]
- Hernández-Zimbrón, L.F. , Rivas-Arancibia, S. Oxidative stress caused by ozone exposure induces β-amyloid 1-42 overproduction and mitochondrial accumulation by activating the amyloidogenic pathway. Neuroscience. 2015,304:340-8. [CrossRef]
- Pocernich, C.B.; Lange, M.L.; Sultana, R. , Butterfield, D.A. Nutritional approaches to modulate oxidative stress in Alzheimer's disease. Curr Alzheimer Res. 2011,8(5):452-69.
- He, Y.; Ruganzu, J.B.; Jin, H.; Peng, X.; Ji, S.; Ma, Y.; Zheng, L. , Yang, W. LRP1 knockdown aggravates Aβ(1-42)-stimulated microglial and astrocytic neuroinflammatory responses by modulating TLR4/NF-κB/MAPKs signaling pathways. Exp Cell Res. 2020,394(2):112166.
- Kamphuis, W.; Middeldorp, J.; Kooijman, L.; Sluijs, J.A.; Kooi, E.J.; Moeton, M.; Freriks, M.; Mizee, M.R. , Hol, E.M. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer's disease. Neurobiol Aging. 2014,35(3):492-510.
- Jung, K.K.; Lee, H.S.; Cho, J.Y.; Shin, W.C.; Rhee, M.H.; Kim, T.G.; Kang, J.H.; Kim, S.H.; Hong, S. , Kang, S.Y. Inhibitory effect of curcumin on nitric oxide production from lipopolysaccharide-activated primary microglia. Life Sci. 2006,79(21):2022-31. [CrossRef]
- Reichert, C.O.; Levy, D. , Bydlowski, S.P. Paraoxonase Role in Human Neurodegenerative Diseases. Antioxidants (Basel). 2020,10(1). [CrossRef]
- Ferretti, G.; Bacchetti, T.; Moroni, C.; Savino, S.; Liuzzi, A.; Balzola, F. , Bicchiega, V. Paraoxonase activity in high-density lipoproteins: a comparison between healthy and obese females. J Clin Endocrinol Metab. 2005,90(3):1728-33. [CrossRef]
- Levy, E.; Trudel, K.; Bendayan, M.; Seidman, E.; Delvin, E.; Elchebly, M.; Lavoie, J.C.; Precourt, L.P.; Amre, D. , Sinnett, D. Biological role, protein expression, subcellular localization, and oxidative stress response of paraoxonase 2 in the intestine of humans and rats. Am J Physiol Gastrointest Liver Physiol. 2007,293(6):G1252-61. [CrossRef]
- Morris, G.; Gevezova, M.; Sarafian, V. , Maes, M. Redox regulation of the immune response. Cell Mol Immunol. 2022,19(10):1079-101. [CrossRef]
- Remigante, A. , Morabito, R. Cellular and Molecular Mechanisms in Oxidative Stress-Related Diseases. Int J Mol Sci. 2022,23(14).
Figure 1.
(A) Ozone plays a protective role in stratosphere since it prevents the excessive passage of ultraviolet radiation to the earth. (B) Ozone acts as an atmospheric pollutant in troposphere. Note the different functions of ozone when it is formed. Yellow arrows indicate ozone production from nitrogen species and solar radiation. Created in BioRender.com.
Figure 1.
(A) Ozone plays a protective role in stratosphere since it prevents the excessive passage of ultraviolet radiation to the earth. (B) Ozone acts as an atmospheric pollutant in troposphere. Note the different functions of ozone when it is formed. Yellow arrows indicate ozone production from nitrogen species and solar radiation. Created in BioRender.com.
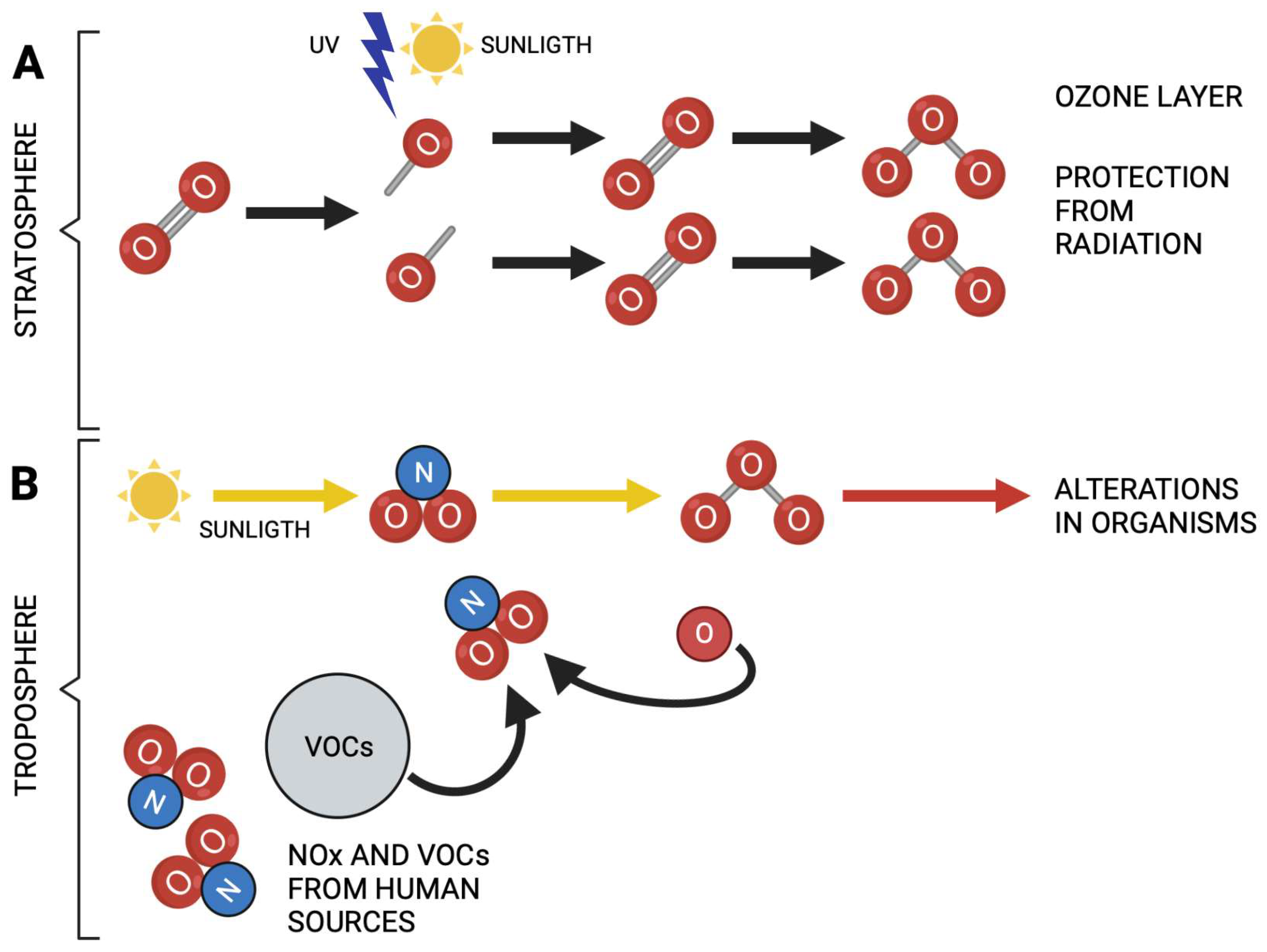
Figure 2.
Environmental pollution is associated with developing various pathologies such as diabetes, obesity, neurodegenerative diseases (Parkinson's, Alzheimer's), cerebral vascular events (CVE), some types of cancer and intestinal alterations as dysbiosis and intestinal diseases. Created in BioRender.com.
Figure 2.
Environmental pollution is associated with developing various pathologies such as diabetes, obesity, neurodegenerative diseases (Parkinson's, Alzheimer's), cerebral vascular events (CVE), some types of cancer and intestinal alterations as dysbiosis and intestinal diseases. Created in BioRender.com.
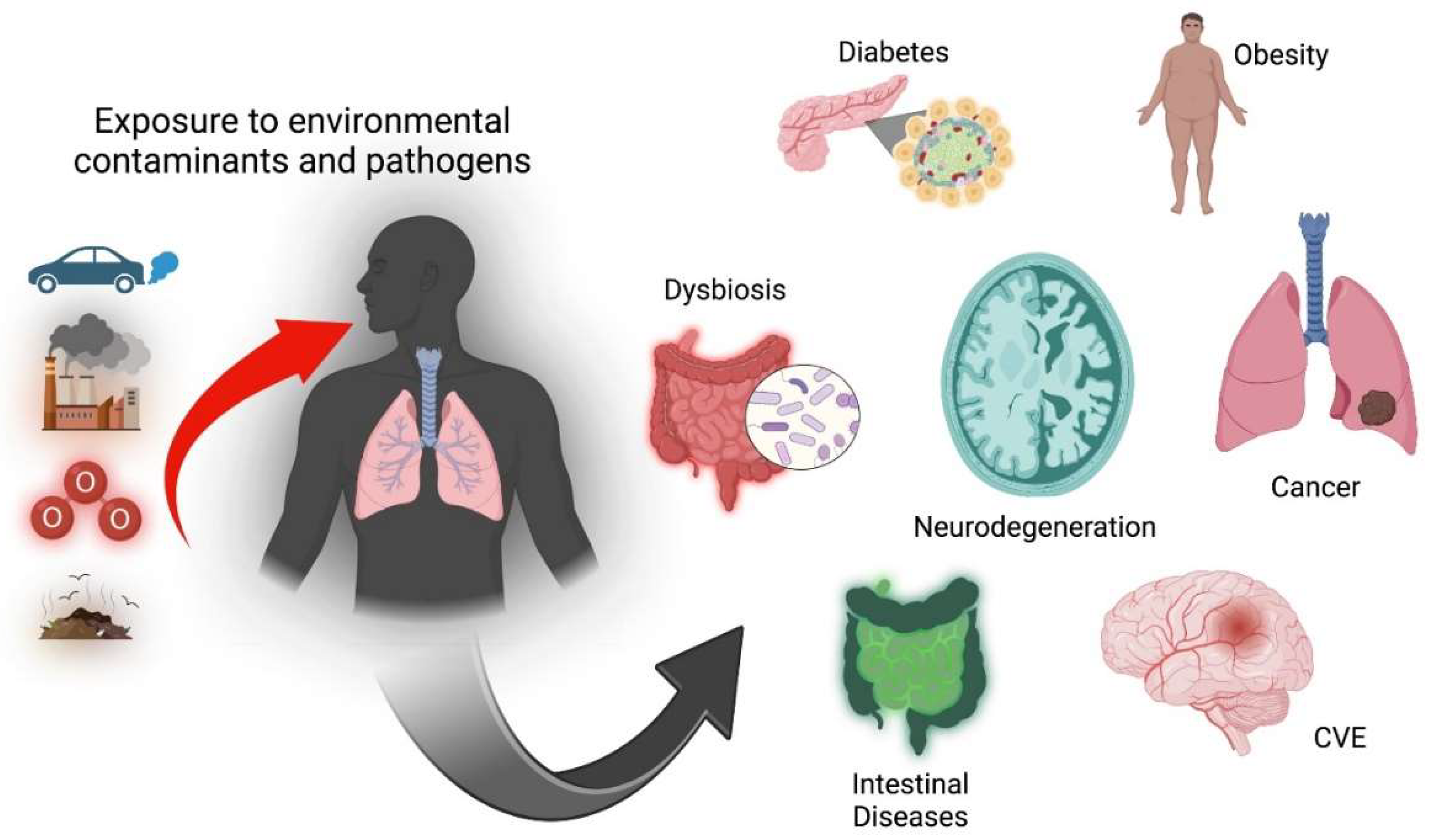
Figure 3.
Epithelial architecture. Maintaining the intestinal barrier maintains the mucosa and the microenvironment of the microbiota. Paneth cells synthesize antimicrobial molecules that control the growth of pathogenic microorganisms. When the functioning of these cells, vesicles they produce and other factors are altered, intestinal permeability is lost, increasing the response of Th17 cells, generating an increase in IL-1β, IL-6, and LPS, among others and establishing an inflammatory process. Created in BioRender.com.
Figure 3.
Epithelial architecture. Maintaining the intestinal barrier maintains the mucosa and the microenvironment of the microbiota. Paneth cells synthesize antimicrobial molecules that control the growth of pathogenic microorganisms. When the functioning of these cells, vesicles they produce and other factors are altered, intestinal permeability is lost, increasing the response of Th17 cells, generating an increase in IL-1β, IL-6, and LPS, among others and establishing an inflammatory process. Created in BioRender.com.
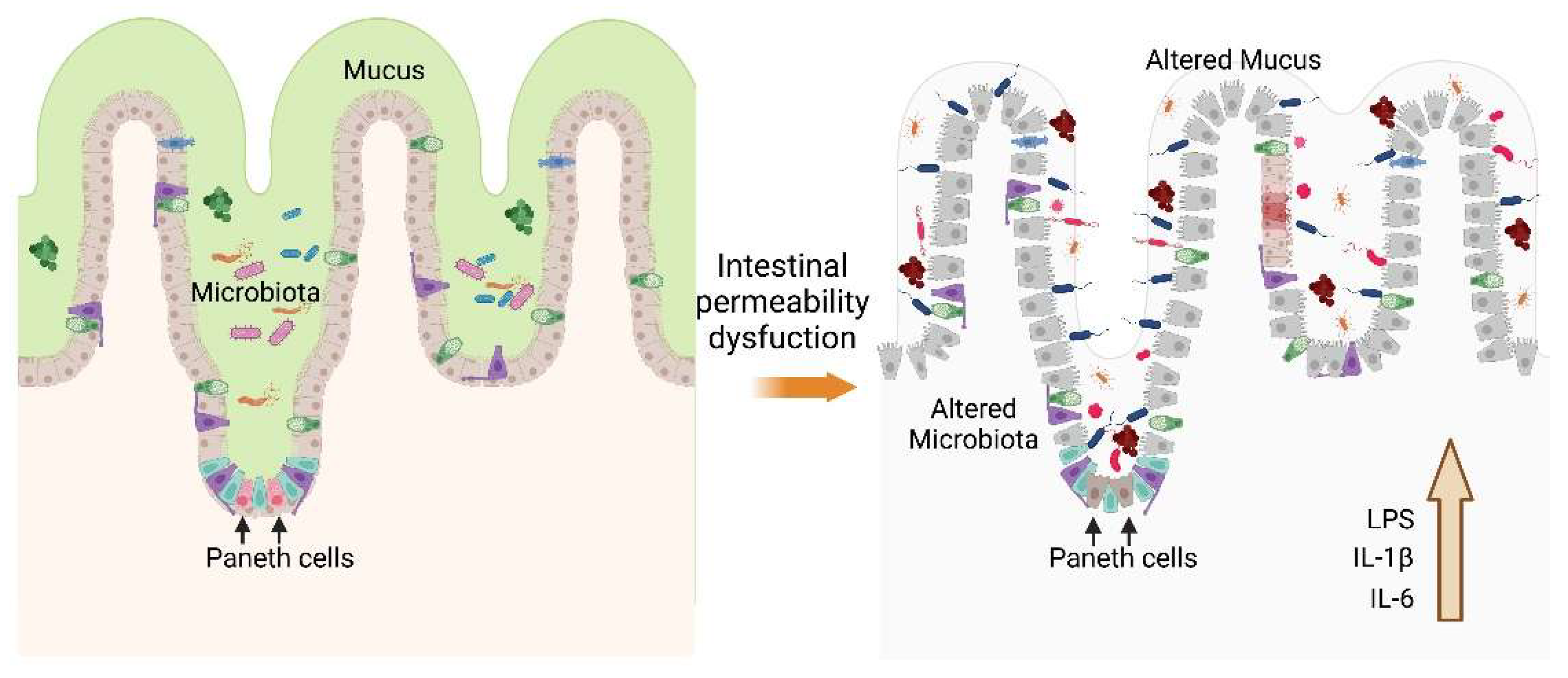
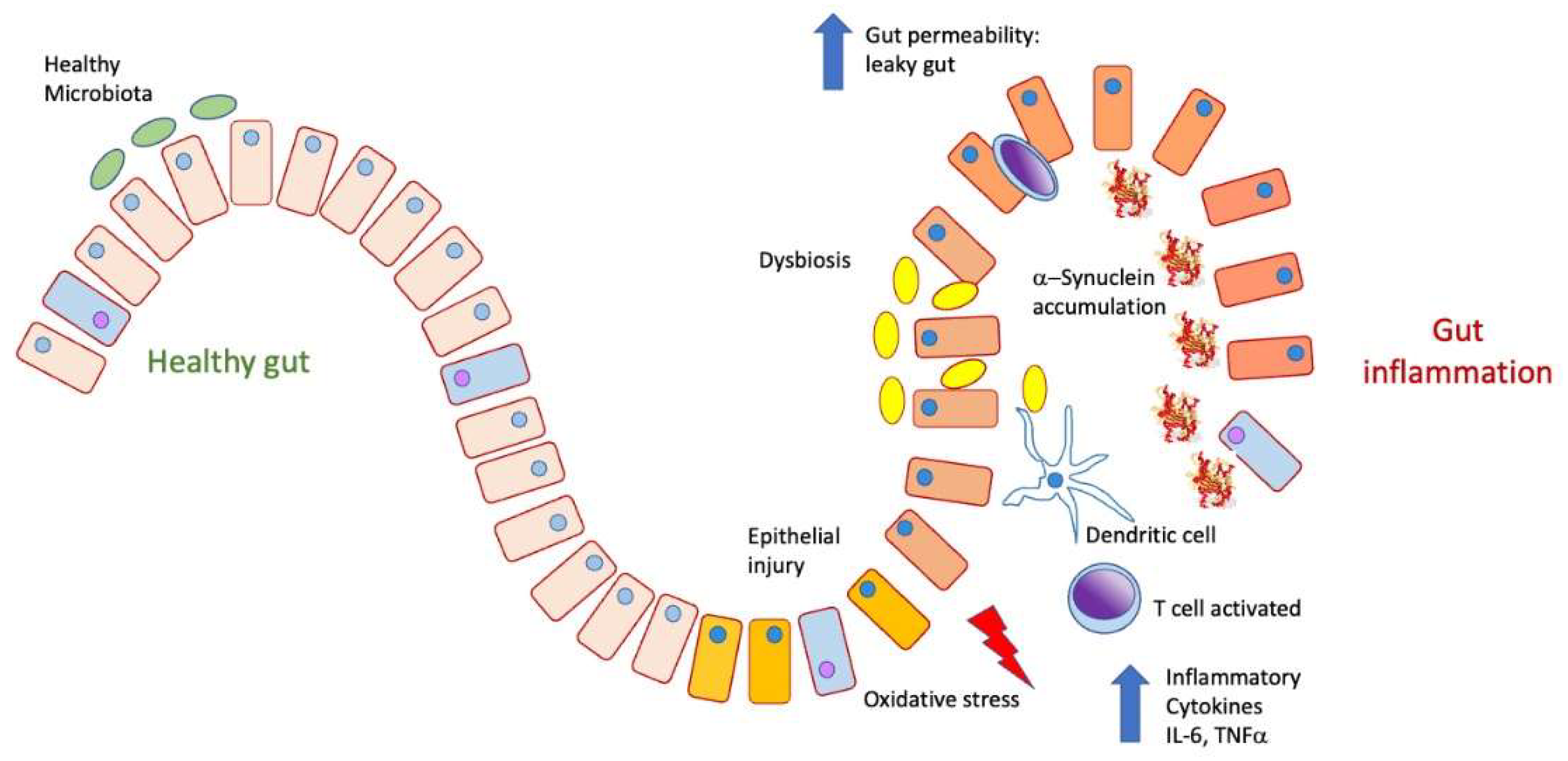
Figure 4.
In a physiological state, the intestine presents a healthy microbiota. It can prevent food antigens and inflammatory cytokines such as TNFα, IL-6, and IL-17 from being released into the bloodstream. Under conditions of inflammation and dysbiosis due to the proliferation of pathological microbiota, the epithelial barrier is lost, increasing permeability to both pathogens and antigens, which produces inflammatory activation, the release of cytokines, and a state of oxidative stress, as well as the accumulation.
Figure 4.
In a physiological state, the intestine presents a healthy microbiota. It can prevent food antigens and inflammatory cytokines such as TNFα, IL-6, and IL-17 from being released into the bloodstream. Under conditions of inflammation and dysbiosis due to the proliferation of pathological microbiota, the epithelial barrier is lost, increasing permeability to both pathogens and antigens, which produces inflammatory activation, the release of cytokines, and a state of oxidative stress, as well as the accumulation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



