
Submitted:
05 May 2023
Posted:
08 May 2023
You are already at the latest version
Abstract
Ribosome assembly is one of the most fundamental processes in gene expression and has served as a playground to investigate the molecular mechanisms of how protein-RNA complexes (RNPs) assemble. The bacterial ribosome is composed of around 50 ribosomal proteins several of which are co-transcriptionally assembled on a ~4,500 nucleotides long pre-rRNA transcript that is further processed and modified during transcription, the entire process taking around 2 minutes in vivo and assisted by dozens of assembly factors. How this complex molecular process works so efficiently to produce an active ribosome has been investigated over decades and has resulted in the development of a plethora of novel approaches that can also be used to study the assembly of other RNPs. Here we review biochemical, structural and biophysical methods that have been developed and integrated to provide a detailed and quantitative understanding of this complex and intricate molecular process of assembly. We also discuss emerging cutting-edge approaches that could be used in the future to study how transcription, rRNA processing, cellular factors and the native cellular environment shape ribosome assembly and RNP assembly at large.
Keywords:
RNP assembly
; ribosome assembly
; protein-RNA interactions
; RNA folding
; assembly intermediates
; in vitro reconstitutions
; mass spectrometry
; single-molecule fluorescence microscopy
; cryo-electron microscopy
; RNA structure probing
1. Introduction
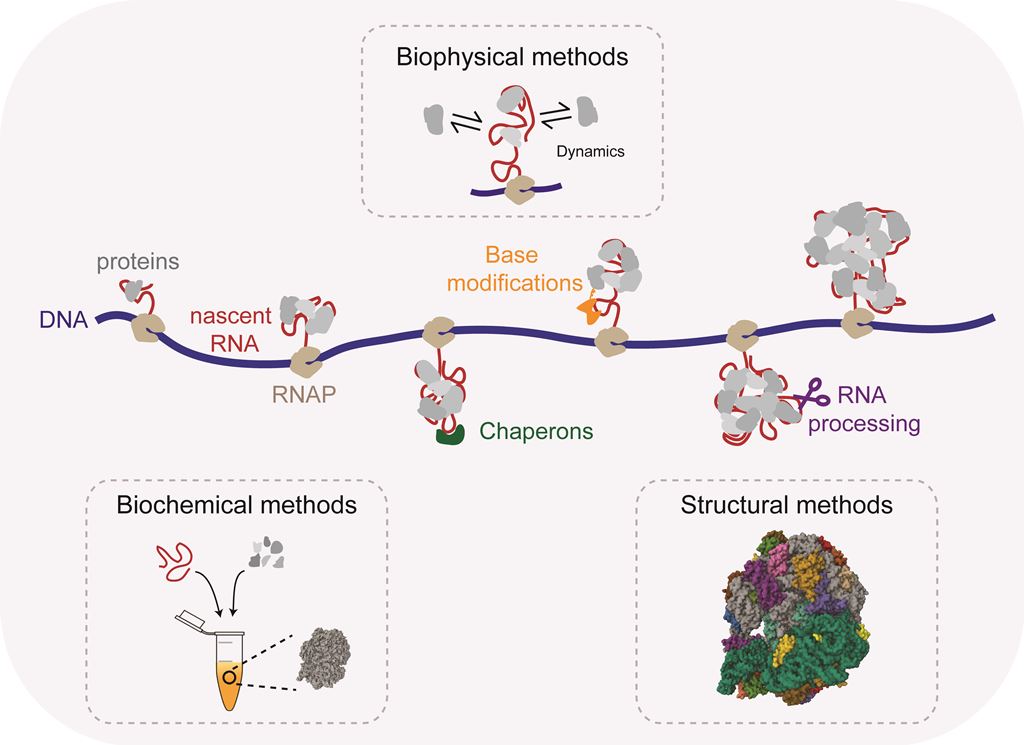
The ribosome is responsible for protein synthesis and is one of the largest and most complex macromolecular machines in the cell. The prokaryotic ribosome is made up of a large subunit (LSU or 50S) and a small subunit (SSU or 30S). The Escherichia coli (E. coli) LSU consists of the 23S and 5S ribosomal RNAs (rRNA) bound by 33 ribosomal proteins (r-proteins) while the SSU consists of 16S rRNA and 21 r-proteins [1]. The assembly of the ribosome is a very complex and multi-step process that consumes about 40 % of the cellular energy [2]. Assembly is initiated by the transcription of a primary rRNA transcript of ~4,500 nucleotides. Transcription is assisted by the rRNA Transcription Antitermination Complex (rrnTAC), which reduces transcription pausing and prevents early termination [3,4,5]. The primary transcript is co-transcriptionally processed by multiple specific RNases to form the three rRNA fragments (16S, 23S and 5S rRNAs) [6,7,8,9] that simultaneously fold into secondary and tertiary RNA structure [10,11,12]. Co-transcriptional rRNA folding follows the vectorial (5' to 3') direction and allows sequential binding of r-proteins [13,14,15,16,17,18]. Co-transcriptional rRNA processing, rRNA folding and r-protein binding is accompanied by the introduction of base modifications such as pseudouridinylations and methylations [19,20]. Furthermore, these processes are assisted by multiple assembly factors such as GTPases, helicases and maturation factors [1,21]. Remarkably, it takes only about 2 minutes for the cell to assemble a functional bacterial ribosome [22]. Consequently, the assembly intermediates of this process are short-lived and contribute to only ~2 % of the total ribosome population [23], making them difficult to study.
Ribosome assembly, and RNP assembly in general, is very difficult to investigate. Apart from the complexity of the process and the low abundance of assembly intermediates, many biomolecular interactions that are forming during assembly are transient and dynamic in nature and therefore difficult to capture biochemically and structurally. Furthermore, the assembly processes are often very heterogeneous and consist of multiple parallel assembly pathways.
Ribosome assembly has been studied over many decades and despite its complexity and technical limitations various aspects of the process are well understood. There are several reviews that provide a detailed overview of various aspects of the assembly process [1,20,21,25,26,27,28,29,30,31]. Here, we aim to provide a methods perspective to study ribosome assembly and the assembly of other RNPs such as the spliceosome, various mRNPs and large non-coding RNPs. We summarize various biochemical, structural and biophysical methods employed over the years to study different facets of the ribosome assembly mechanism, with a focus on bacterial ribosome assembly. The review highlights the exciting parallel between the evolution of our understanding of ribosome assembly and the technological advancements leading to the development of new methods (Figure 1). We start by discussing in vitro reconstitutions that employ a bottom-up approach using minimal components to understand the assembly process in a very controlled manner. Time-resolved mass-spectrometry, RNA structural probing and cryo-electron microscopy have provided information on the kinetics of assembly and have permitted the structural visualization of the assembly process at high resolution. Single-molecule experiments have become instrumental to understand how the different processes are functionally coupled to each other as they allow us to follow complicated multi-step processes in real-time. We conclude our review by discussing approaches that we think will be required in the future to understand how the ribosome and other complex protein-RNA machineries are assembled so fast and efficiently in vivo.
2. Biochemical Reconstitutions
2.1. In Vitro Reconstitutions
In the early days of studying ribosome assembly, it was evident that the ribosome is a very complex machinery composed of multiple r-proteins interacting with rRNA. In order to understand the assembly, the two subunits of the ribosome were studied separately. In vitro reconstitution/omission experiments were performed by mixing purified rRNA with different sets of r-proteins and then purifying the resulting assembly intermediates using ultracentrifugation of sucrose gradients [32]. Initial attempts to reconstitute these subunits indicated that the 30S can be reconstituted in a single step [17], while several heating steps and various Mg2+ concentrations were required to reconstitute the 50S [33,34]. The reconstituted ribosomes were tested for their ability to read polyU templates [35,36], form peptide bonds [37] or to bind tRNA [38] suggesting that these in vitro reconstitutions provide active ribosomes. Reconstitution experiments indicated that the binding of r-proteins occurred in a sequential order and allowed organizing ribosome assembly into assembly maps (Nomura map for 30S and Nierhaus map for 50S) containing the thermodynamic binding dependencies of the various r-proteins [16,17,18]. In vivo experiments using cold sensitive mutant strains and strains lacking r-proteins validated the assembly maps derived from in vitro reconstitution methods [39].
2.2. In Vivo Mimicry
While these reconstitution efforts were successful in describing the in vitro thermodynamic assembly pathway, assembly was much less efficient, required unphysiological heating steps and buffer conditions. Furthermore, the reconstituted ribosomes were not tested for their ability to translate a complete mRNA [40]. Importantly, these experimental conditions did not properly mimic the in vivo situation. Inside cells, the rRNA is efficiently transcribed and co-transcriptionally processed, modified and bound by r-proteins simultaneously [41,42,43,44]. This entire process is assisted by multiple assembly factors. Developments in the field of cell free systems spearheaded by the Jewett lab were used to reconstitute ribosomes with high activity in near native assembly conditions. The Integrative ribosome Synthesis, Assembly and Translation (iSAT) assay combines co-transcriptional ribosome assembly and subsequent translation of mRNA by the assembled ribosome in a single reaction, with GFP as a readout for the successful assembly of an active ribosome (Figure 2A–C) [40]. The iSAT reaction consists of a plasmid containing the entire rRNA operon initiated by the T7 promoter sequence, T7 RNA polymerase, all r-proteins purified from native ribosomes (TP70), a second plasmid coding for the reporter mRNA sequence (GFP) and cell extract (S150) containing all the cellular factors required for ribosome assembly and translation (Figure 2A). The cell extract allows the correct processing [45] and modification [46] of the rRNA. Since all the key components required in ribosome assembly as well as other components such as assembly factors that assist ribosome assembly are present, the assembly of the ribosome is expected to proceed In a native state i.e. the processes of transcription, rRNA processing, r-protein binding and base modifications are expected to occur simultaneously and assisted by assembly factors. While earlier iSAT reactions had translational efficiencies of 20 % when compared to in vivo purified ribosomes [45], the efficiency could be improved to 70 % by addition of crowding and reducing agents to the iSAT reactions [47]. iSAT reactions were further extended to include the synthesis of individual r-proteins [48], yet the assay needs to be further developed to have all r-proteins synthesized in the same reaction. Of note, iSAT reactions work efficiently despite using T7 RNAP instead of the native E. coli RNAP. Better mimicking the in vivo situation, future adaptions of iSAT should include the native E. coli RNAP in order to also properly reproduce the native rRNA transcription speed and pausing behaviour, which is assisted by the rrnTAC.
Inspired from the lab-on-a-chip approach, the one-pot iSAT reaction assay was also performed on a chip to reconstitute 30S subunits in near native conditions (Figure 2D) [49]. Genes encoding for r-proteins and rRNA were immobilized on a chip surface as DNA brushes along with anti-HA antibodies (Figure 2D, right panel). One of the r-proteins at a time was designed as a fusion protein with a HA tag and the rRNA was modified to include a Broccoli aptamer. All genes were transcribed and r-proteins were translated locally at the surface. The resultant increase in fluorescence signal from the broccoli aptamer on the regions of the chip coated with anti-HA antibodies indicated that the rRNA was bound by the HA-tagged r-protein and all upstream binding r-proteins according to the Nomura assembly map (Figure 2D bottom, right panel). Using this approach they could recapitulate the r-protein binding dependencies (Nomura map) and their binding kinetics (Figure 2E). They were also able to monitor late stages of the 30S assembly including the binding of the mature 30S to the 50S.
In summary, biochemical reconstitutions are a powerful tool to investigate intricate details of a specific process using a minimalistic system. Recent ribosome reconstitutions mimicking native conditions have enabled to study mutant ribosomes [50], to incorporate non-canonical amino acids [51] and to investigate the process of evolution in context of ribosome assembly and function [52]. Furthermore, these methods can enable the investigation of the role of various assembly factors in wild type versus mutant ribosomes and to engineer new ribosomes with specific functions.
3. Mass Spectrometry
While the in vitro reconstitution/omission experiments allowed the construction of the ribosome assembly maps that summarize the thermodynamic protein binding dependencies, they do not contain any information on the protein binding kinetics during assembly. By combining quantitative mass spectrometry (qMS) with pulse-chase experiments using stable isotope labelling it became possible to complement the thermodynamic r-protein binding dependencies with r-protein binding rates.
3.1. In Vitro Mass Spectrometry for R-Proteins
Pulse-chase qMS (PC-qMS) allows the tracking of the binding rates of all r-proteins to the rRNA in a single experiment [15]. The rRNA is incubated with heavy isotope labelled r-proteins for a specific amount of time, followed by a chase with an excess of light isotope labelled r-proteins to complete assembly (Figure 3A, left panel). Completely assembled subunits are then isolated on a sucrose gradient and the abundance of heavy to total protein ratio for each protein is determined by mass-spectrometry and plotted as a function of time (Figure 3A, centre panel). The resulting binding curves provide the average binding rates for the individual r-proteins (Figure 3A, centre and right panels). By repeating these experiments at different protein concentrations and temperatures the authors demonstrated that 1) RNA folding and protein binding occurs at similar rates, 2) the rate-limiting steps for the different proteins is similar at low or high temperature and 3) the final steps of 30S synthesis are limited by many different transitions. Similar experiments were performed with a pre-folded 16S rRNA that was pre-bound with a subset of r-proteins [53]. They observed multiphase binding kinetics of r-proteins suggesting further complexity in the assembly pathway. Their observations also indicated the presence of multiple assembly pathways and a delicate interplay between thermodynamic dependency and kinetic cooperativity. PC-qMS was also used to investigate the influence of assembly factors for the assembly of the 30S, for example, showing that RimP allows faster binding of S9 and S19 but prevents the binding of S12 and S13, potentially by blocking their binding sites [54].
3.2. In Vivo Mass Spectrometry for R-Proteins and Assembly Factors
qMS based methods were also applied to recapitulate the assembly pathway in vivo and for the identification of multiple assembly factors [22] (Figure 3B,C). The authors used an in vivo stable isotope pulse labelling approach to characterize the exact r-protein composition of various populations of intermediates (Figure 3B). The cells were grown in heavy isotope media and pulse labelled with light isotope media. Various fractions from the sucrose gradient corresponding to assembly intermediates were digested by trypsin and subjected to qMS. The resultant in vivo data validated the presence of 4 assembly intermediates of 30S particles as observed by Mulder et al. using in vitro reconstitutions. 50S assembly was more continuous in cells and revealed 6 assembly intermediates which indicated a general pathway where the 50S assembly starts opposite to the peptidyl transferase centre and forms intermediates where r-proteins are added globally to the whole structure and ends with the formation of the central protrusion. Likewise, subjecting the fractions of a sucrose gradient to qMS analysis led to the identification of 15 known and 6 unknown assembly factors that were co-occurring with specific assembly intermediates indicating their role in that particular stage of assembly (Figure 3C).
MS was also used to understand the effect of cellular knockouts of assembly factors on the composition of ribosome assembly intermediates. Experiments with strains lacking specific assembly factors showed a slower growth rate and an accumulation of assembly intermediates [54,55,56,57]. Investigation of in vivo assembly intermediates from mutant strains, for example, lacking assembly factors LepA or RsgA showed reduced levels of late binding r-proteins suggesting the role of these assembly factors in late stages of assembly [58].
Apart from quantifying the composition of assembly intermediates, MS can also be used to investigate post-translational modifications of r-proteins during assembly. For example, the Woodson lab used MS to understand the extent of S5 and S18 acetylation during in vivo ribosome assembly and its effect on the formation of specific rRNA contacts [59].
qMS methods were also applied to study eukaryotic ribosome assembly. For example, Sailer et. al, used multiple different affinity tagged assembly factors to pull-down and crosslink different intermediates of pre-60S particles from Saccharomyces cerevisiae [60]. The mass spectrometry analysis of the crosslinked peptides produced a protein-protein interaction map which identified the localization of 22 unmapped assembly factors. The association based on relative abundances of the newly mapped assembly factors with specific intermediates indicated the approximate time at which they act in the assembly pathway.
3.3. In Vivo Mass Spectrometry for RNA Modifications
rRNA is modified by methylations as well as pseudouridinylations [19]. These modifications are deposited site-specifically by multiple different modification enzymes during the course of the assembly process. Traditionally modifications are detected using reverse transcriptase primer extension techniques [61], P1 nuclease digestion followed by Thin-Layer Chromatography (TLC) or High Performance Liquid Chromatography (HPLC) [62,63]. Although these are very sensitive methods, they are tedious as they allow the observation of only one modification at a time and are suitable to detect only specific modifications. qMS analysis of RNA enables the detection of multiple site-specific modifications simultaneously. Typically, isotope-based labelling is used to detect the fraction of RNA molecules that is site-specifically methylated. However, accurate quantification of lowly abundant modifications can be challenging. Furthermore, since pseudouridine is a structural isomer of uridine, it cannot be detected. Popova et al. used a metabolic labelling approach to validate methylations and detect pseudouridinylated residues [64]. CD3-methionine (precursor for SAM) leads to a +3 Da mass shift that can be distinctly and confidently annotated. Similarly, 5,6-D-uracil leads to a -1 Da mass shift for a pseudouridinylated residue. Using this approach on assembly intermediates purified from cells, the authors were able to characterize the stages at which each residue is modified during the assembly process. For example, most of the modifications on the 23S rRNA occur early during assembly, as opposed to the 16S where the modifications are incorporated from a 5’ to 3’ direction, in agreement with a co-transcriptional rRNA modification process. Another study used qMS on S-adenosylmethionine (SAM; methyl donor used by methyltransferases) depleted cells to study the importance of RNA modification on ribosome assembly [65].
Overall, mass spectrometry is a highly sensitive and quantitative method to determine binding kinetics of r-proteins to rRNA as well as to study when multiple r-protein or rRNA chemical modifications are introduced during assembly.
4. Electron Microscopy
Electron microscopy (EM) has proven instrumental in providing high-resolution structural information of ribosome assembly intermediates. Both for negative-stain and cryogenic EM, ribosome assembly intermediates either from in vitro reconstitutions or purified from cells are applied to a grid for imaging. Optimally, the individual particles to be imaged are present in multiple different orientations to reconstruct a 3D image [66]. Seminal work by the Williamson lab in 2010 demonstrated the potential of using structural information derived from a heterogenous population of assembly intermediates to understand the mechanisms of ribosome assembly [67]. They performed time-resolved low-resolution negative-stain EM after mixing 16S rRNA with all 30S r-proteins and then freezing at different time-points. They were able to visualize 14 different assembly intermediates, which were classified into 4 major groups (Figure 4A): the population of the first group, representing the smallest assembly intermediate, decreased over time. The second group peaked at several minutes, while the third and fourth groups appeared only at later time points. In combination with PC-qMS, they were able to reconstruct a detailed assembly pathway for the 30S subunit in vitro, demonstrating multiple parallel assembly pathways (Figure 4A).
The resolution revolution in 2013 led to significant improvements in electron detection technology and reconstruction algorithms [70,71]. This enabled its use for investigating more heterogeneous populations of complexes present in the same sample, providing the basis for imaging multiple assembly intermediates that are populating the 50S assembly pathway both in vitro and in vivo.
In vitro reconstitution of the 50S is a 2-step process that leads to activation of the 50S [72]. High-resolution cryoEM of the 2-step reconstitution process displayed 5 main classes (subpopulations) resulting from the step 1 and a mature 50S structure resulting from step 2. The 50S assembly initiates at the core, followed by the L1 protuberance and the central protuberance (CP). Interestingly the main difference between the last class from step 1 and the fully mature 50S is a structural rearrangement of the rRNA that leads to the maturation of the peptidyl transferase center.
In order to perform experiments in native conditions and get a perspective on ribosome assembly in vivo, Davis et. al, used high-resolution cryoEM of assembly intermediates isolated from a bL17 (r-protein of 50S) depleted strain to enrich intermediates [73]. Sub-population averaging revealed that, similar to in vitro experiments, the in vivo 50S assembly consists of a heterogeneous ensemble of intermediates. The different subpopulations that progressively evolved into a more mature complex could be further grouped together. Thus, providing structural evidence of parallel pathways of 50S assembly. Interestingly, reanalysis of this compositionally and conformationally heterogenous data using a neural network based framework called CryoDRGN revealed a previously unreported assembly intermediate [74]. CryoDRGN is a powerful tool that enables automated classification of various states which is typically done using multiple manual and expert-guided rounds of hierarchical 3D classification.
Correlative analysis using qMS and cryoEM data from bL17 depleted cells also indicated that the unidentified densities in subpopulations from one of the assembly pathways is corresponding to assembly factor YjgA [73]. Putative YjgA binding blocked the docking of a helix crucial for inter-subunit bridge formation suggesting that YjgA acts as a late-stage assembly factor for maturation. Recent evidence suggests that the presence of assembly factors in vivo directly affects the order of maturation of specific regions. For example, in contrast to in vitro assembly [72], the core and the central protuberance formation were suggested to be interdependent in vivo [68,75]. Another detailed characterisation of pre-50S assembly intermediates revealed a network of assembly factors such as ObgE, RsfS, YjgA, RldU and YhbY that orchestrate 50S maturation (Figure 4B) [68]. Several other studies have used cryo-EM to determine structures of bacterial ribosome assembly intermediates to understand the function of assembly factors but are not further reviewed here [76,77,78,79,80,81,82,83].
Apart from structurally characterizing later assembly intermediates that are formed once transcription is already completed and the majority of the rRNA is already processed, recent structural work has also provided information on the process of early rRNA transcription by the rRNA Transcription Antitermination Complex and on the mechanism of initial rRNA processing. The rrnTAC is the macromolecular machinery responsible for efficient transcription of rRNA in the cell [3,4,5]. The rrnTAC assembles on the RNA polymerase (RNAP) and reduces NusA-mediated transcriptional pausing, R-loop formation and polymerase backtracking, intrinsic as well as Rho-dependent termination, enables chaperone-mediated rRNA folding and the formation of long-range rRNA-rRNA interactions. The high-resolution cryoEM structures of an in vitro reconstituted rrnTAC-associated transcription complex revealed the presence of NusA, NusB, NusE, NusG, SuhB and S4 (Figure 4C) [69]. Interestingly, in the rrnTAC, NusA is repositioned to prevent pausing caused by hairpin stabilisation as well as intrinsic termination. Similarly, the presence of NusG in the rrnTAC suppresses RNAP backtracking. The interactions of NusA, NusE and SuhB with the C-terminus of NusG prevents it from recruiting Rho. Furthermore, the formation of a ring-like structure made by SuhB and S4 around the E. coli polymerase exit channel prevents Rho from directly interacting with the exit channel, therefore preventing Rho-dependent termination. Finally, the authors demonstrated that the 5’ end of RNA is bound by S4 and the emerging 3’ end of RNA is bound by Nus factors along with SuhB on the ring. This brings the distal regions of RNA close in space to form long-range interactions that are required for creating the substrate for rRNA processing by RNase III [84].
The 4,500 nucleotides long primary rRNA transcript is initially processed by dsRNA-specific RNases to generate the pre-rRNA fragments that further mature into 16S, 23S and 5S [1,8,9]. In B. subtilis, the mature 23S is obtained by Mini-III and the mature 5S by M5 processing [85,86]. Structural characterisation of these RNases with their respective substrates revealed that the Mini-III binds pre-23S ds-rRNA as a dimer where each subunit cleaves one of the strands [87]. In contrast, the N-terminal domain of M5 binds to the 3' strand of the ds-rRNA and cleaves it. This then leads to structural rearrangements enabling the C-terminal domain of M5 to bind and cleave the 5' strand. Mini-III and M5 are assisted by r-proteins such as uL3 and uL18 that bind to the respective substrates and keep them in a conformation that can be recognised by the enzymes.
The above examples highlight the role of cryoEM as a powerful structural method for increasing our understanding about the structural and mechanistic details of ribosome assembly including providing time-resolved information.
5. RNA Structure Probing
Multiple different studies have indicated the role of rRNA secondary and tertiary structure in binding of r-proteins. Simple chemical and enzymatic probing of rRNA structure are powerful methods but have limited time-resolution and throughput [88]. Structure determination of rRNA during assembly is also difficult due to the presence of a heterogeneous set of conformations and the difficulty in resolving flexible regions. Hydroxyl Radical Footprinting (HRF) and DMS/SHAPE probing are two complementary methods, providing information on RNA tertiary and secondary structure, respectively. These methods overcome some of the above-mentioned limitations and have successfully been used to obtain more detailed and better-resolved information on rRNA folding.
5.1. In Vitro RNA Structure Probing
RNA secondary structure information can be obtained using chemical reagents such as DMS and many other probes (e.g. glyoxal, 1m7, 1m6, NMIA, BzCN, NAI) which react specifically with single-stranded RNA but do not chemically modify dsRNA [89]. The introduced adducts result in a stop when read by a reverse transcriptase. The resulting fragments are detected using primer extension. This approach has been used to predict 16S rRNA secondary structure with 97 % accuracy [90]. In order to increase throughput, an alternate strategy, termed SHAPE-MaP, uses manganese ions during the reverse transcription step which causes reverse transcriptase to introduce a mutation into the cDNA rather than stopping at the modified sites [91]. The cDNA is sequenced and the percentage of the underlying mutations is used to generate a reactivity profile to predict the secondary structure (Figure 5A). SHAPE-MaP was used to track rRNA structure during ribosome assembly. For example, SHAPE-MaP based structure probing of the 23S rRNA in presence and absence of r-proteins showed very similar reactivity profiles suggesting that the 23S rRNA assumes its secondary structure even in absence of r-proteins [73].
While most of the chemical reagents introduced above require seconds to several minutes to react with their RNA substrate, and therefore limit the time-resolution, Hydroxyl Radical Footprinting (HRF) provides information at few milliseconds resolution, and therefore allows the study of very early rRNA folding events and formation of protein-RNA interactions [92,93]. In HRF, rRNA is exposed to short pulses of hydroxyl radicals generated by X-rays. These hydroxyl radicals react with the unprotected RNA backbone and thereby cleave the RNA into smaller fragments. Site-specific primer extension is used to amplify these fragments. The probability of cleavage depends on solvent accessibility, and therefore reports on RNA tertiary structure and/or the interaction with proteins. HRF experiments were performed in a time-resolved manner by mixing 16S rRNA with all r-proteins and exposing the reaction to an X-ray pulse at different time-points after mixing, thus providing the first time-point as early as 20 ms after mixing (Figure 5B–D) [92]. Apart from validating the kinetics of early and late r-protein binding as determined using PC-qMS, these experiments showed that 30S assembly nucleates from different points along the rRNA (Figure 5C). Additionally, the authors observed that initial encounter complexes refold during assembly. For example, S7 initially binds in a non-native conformation (protecting only H43 within 20-50 ms) and adapts a native conformation (protecting H29, H37, H41) only much later in assembly (Figure 5D). These experiments demonstrate the potential of HRF to provide information on RNA structural changes at the nucleotide resolution and at the milliseconds to second times-scale.
rRNA folds into a very heterogeneous set of conformations during ribosome assembly [10,11,94,95]. Therefore, RNA structural probing will provide an average of all conformations present in the sample to be probed. While this has not been done yet for studying rRNA folding during ribosome assembly, recent analysis pipelines have shown the potential to dissect RNA heterogeneity by using the property of DMS to achieve multi-hit kinetics and using single-molecule sequencing as a read-out [96,97,98,99,100].
5.2. Co-Transcriptional RNA Structure Probing
RNA probing assays were performed on pre-transcribed rRNA but the rRNA folds co-transcriptionally in vivo and is affected by the speed of transcription [103]. While it has been shown that co-transcriptional rRNA folding is different than folding of a pre-transcribed RNA [10,11,95], structural probing of rRNA has not been done in the context of transcription yet. However, co-transcriptional probing has been employed to study relatively simpler systems that undergo ligand induced conformational changes such as the fluoride riboswitch and the SRP RNA [102]. For this, DMS/SHAPE based structure probing was adapted by designing roadblocks on the 3’-end of the DNA transcription template. The roadblock prevents the polymerase from transcribing further. The reactivity profiles of RNAs transcribed from different lengths of DNA templates (made by placing roadblocks at different positions) allowed the authors to mimic the co-transcriptional RNA folding pathways, however simulating an infinitesimally slow transcription rate (Figure 5E).
5.3. In Vivo RNA Structure Probing
RNA structural probing was also performed in vivo to understand how rRNA folds in the native cellular environment and in the natural context of rRNA transcription. Soper et. al. used HRF to study how assembly factors affect rRNA structure formation during assembly [59]. They compared protection resulting from mutant strains lacking assembly factors such as RbfA and RimM to wildtype strains to determine the putative binding site of these assembly factors. Furthermore, closely analyzing the assembly intermediates from mutant strains helped discover the role of these assembly factors in the assembly pathway. These assembly factors lead to global structural changes at late time points in assembly, binding to the 50S inter-subunit interface and thus acting as a checkpoint for quality control.
In order to obtain information on the early co-transcriptional rRNA folding in vivo, a protocol was developed that uses metabolic labelling of cells to separate nascently transcribing rRNA intermediates from the total pool of rRNA [104]. Transcriptionally inactive cells (in nutrient poor media) are labelled with 4-thiouridine (4sU) right before feeding with rich media. This allows the isolation of nascent transcribed rRNA which can be probed by DMS or HRF. The results of in vivo DMS probing of nascent 16S rRNA recapitulated the general vectorial folding pathway and specifically provided information on the rRNA interactions at 30 seconds resolution. The results from in vivo HRF probing of nascent rRNA is expected to provide exciting insights into rRNA tertiary structure formation at millisecond resolution. Furthermore, the approach can potentially be extended by developing cell-compatible and faster acting probes to get milliseconds to seconds resolution for secondary structure probing [105].
6. Single-Molecule Methods
The so far discussed ensemble biochemical, biophysical and structural methods lead to detailed characterization of the mechanism of ribosome assembly including the order and kinetics of r-protein binding, the dynamics of RNA structure formation and the structural characterization of assembly intermediates formed at various stages during assembly. However, these ensemble methods provide an average over the individual molecules. The need for averaging leads to the following major challenges: 1) the heterogeneity of the ribosome assembly process cannot be sufficiently resolved, i.e. it is not possible to separately monitor the trajectories along the reaction coordinates of the individual assembly pathways, 2) it is not possible to dissect how different molecular processes such as transcription progression, RNA folding and protein binding are functionally coupled to each other and 3) dynamic structural changes may not be resolved.
Single-molecule methods instead allow tracking the activity of individual molecules over long time scales at high temporal resolution, thereby directly following multistep processes in real-time and dissecting the heterogeneity. To observe a single-molecule for minutes to hours, the molecules of interest are immobilized to a chemically functionalized surface of a glass coverslip, typically using a biotin-streptavidin/neutravidin interaction (Figure 6A) [106]. Fluorescently labelling the molecule on the surface or the ligands that can bind to the surface-immobilized molecules allows the monitoring of conformational changes, binding events and enzymatic activities of the molecules in real-time using Total Internal Reflection Fluorescence (TIRF) microscopy to reduce the fluorescence background. Fluorescent Resonance Energy Transfer (FRET) can be used to directly measure distance changes between a donor and one or several acceptor dyes [107] and thereby, for example, inform on conformational changes as they happen in real-time [108].
6.1. Multi-Color Single-Molecule Fluorescence Microscopy
Some of the initial single-molecule experiments investigated the folding of the H20-H21-H22 three-way junction of the 16S rRNA upon S15 binding [109]. The three-way junction was immobilized using one of the helices and the other two helices (H22 and H21) were labelled with a donor and acceptor dye, respectively. In absence of S15, the 3 helices adapt a planar conformation resulting in limited transfer of energy from donor to acceptor (low FRET). However, in presence of S15, the helices form a non-planar tertiary structure that brings the two dyes closer leading to high FRET efficiency. Further, using a fast buffer-exchanging system, the authors titrated the levels of Mg2+ ions to determine that the three-way junction reacts instantaneously to Mg2+ ion levels.
1.5 decades later, more sophisticated multi-color experiments allowed the visualization of multiple processes at the same time, specifically the simultaneous tracking of rRNA folding and r-protein binding. Kim et. al, investigated the binding of S4 (primary binding r-protein) to a 5-way junction (5WJ) in the 5' domain of 16S rRNA (Figure 6A) [94]. They used a similar helix labelling system as described above for H3 and H16 and additionally labelled the r-protein S4 with another acceptor. Using this approach, they showed that S4 initially binds in a low FRET state (non-native conformation) and then later transitions into a high FRET state (native conformation) (Figure 6B,C). Performing similar experiments on the 5WJ indicated that helix H3 initially adopts a flipped conformation that recruits S4. This then enables H3 to dock onto S4 assuming a native conformation, suggesting that S4 guides rRNA folding (Figure 6D).
Interestingly, similar experiments applied to the initial binding of S15 to the central domain H20-H21-H22 junction showed that binding of S15 leads directly into a high FRET state which does not change over time [95]. This suggest that unlike for S4, the S15 binding site immediately folds into its native conformation upon recruitment of the primary binding protein S15.
Further multi-color experiments on the 5’domain system highlighted that the r-proteins can efficiently change the rRNA folding landscape [110]. Monitoring recruitment of S4, S20 and S16 showed that S16 can be stably recruited to a complex consisting of S4 and S20. Stable recruitment of S16 leads to conformational changes that enable H12 to interact with H3 which prevent H3 from flipping out and stabilizing the native conformation. Overall, these experiments showed that r-protein binding changes the energy landscape such that only certain barriers can be crossed and thus, limits the conformational search space.
6.2. Co-Transcriptional Single-Molecule Imaging
Ribosome assembly occurs co-transcriptionally and thus, the processes of rRNA folding and r-protein binding are linked to transcription [41,42,43,44,111]. Duss et. al developed a method to simultaneously monitor the process of transcription elongation and r-protein binding to the nascent rRNA directly emerging from the RNAP [11,95]. To this end, a stalled transcription elongation complex was formed which consists of a DNA template labelled with dyes at the 3’ end, native E. coli RNAP and nascent rRNA (Figure 6E, left panel). This stalled complex was obtained by initiating transcription using only 3 out of the 4 NTPs on a sequence missing the 4th nucleotide. The stalled transcription complex was then immobilized to the imaging surface through the 5’-end of its nascent RNA by using a complementary biotinylated probe. The experiment was initiated by the addition of all 4 NTPs. The progression of transcription brings the fluorescently labelled 3’ end of the DNA template closer to the surface which leads to an exponential increase in signal intensity (Figure 6E, left panel) (as a result of exponentially increasing excitation in the evanescent field generated by total internal reflection when moving closer to the surface). A plateau in fluorescence intensity during transcription termination demonstrated that the RNAP can stall for a few seconds before dissociation from the DNA template which shows as a sudden signal intensity drop to zero [95]. The authors then monitored in real-time transcription elongation of the 16S rRNA H20-H21-H23 three-way junction and simultaneously the binding kinetics of S15 to the nascent RNA (Figure 6E, centre panel). They found that S15 can only bind once the full-length 3-way junction RNA has been transcribed. Detailed characterization of the S15 binding events revealed 3 populations of nascent RNA molecules: 1) natively folded RNA molecules that bound S15 stably immediately upon transcription of the full-length 3-way junction, 2) partially folded RNA molecules that bound S15 transiently and 3) misfolded RNA molecules that did not bind S15 at all. They further showed that pre-transcribed RNA has distinct properties as compared to co-transcriptionally folded RNA [95].
While this study indirectly reported on RNA folding using protein binding kinetics as a read-out, direct information on rRNA folding was missing. In a follow up study, the authors developed an approach which allows simultaneous tracking of 1) transcription elongation, 2) co-transcriptional folding of the nascent RNA and 3) the binding of one or two proteins to the nascent RNA (Figure 6E) [11]. Studying the 3’ domain of 16S rRNA showed that the primary binding r-protein S7 first engages transiently with the nascent RNA before becoming stably incorporated, which happens upon binding of the secondary and tertiary binding proteins. Furthermore, the authors observed that the binding of S7 was more efficient on smaller constructs as opposed to the full-length 3’ domain indicating the higher tendency of longer rRNA to misfold and thereby preventing r-protein binding. Four-color experiments then showed that the binding of S7 directly depends on the formation of a long-range helix (H28), which forms more efficiently if less RNA needs to be transcribed before the 5’-and 3’-halves of this helix can meet to form the long-range helix (Figure 6F,G). This directly demonstrated that the formation of long-range RNA interactions are impeded by the 5’ to 3’ directional process of transcription [112]. Remarkably, the rRNA folding efficiency increased in the presence of 3’ domain binding r-proteins indicating that the r-proteins can chaperone rRNA folding and guide the energy landscape of ribosome assembly.
A similar study on the 5’domain of the 16S rRNA showed that primary binding r-protein S4 binds transiently to the transcribing rRNA whereas S4 could bind stably to pre-transcribed rRNA [10]. This suggested that structures formed early during transcription are not competent to stably recruit S4. They also found that addition of secondary binding r-proteins led to more long-lived S4 binding events. These studies together suggest that r-protein binding based rRNA remodelling is a general mechanism of ribosome assembly.
Other approaches to track co-transcriptional RNA folding have also been developed, but have not been applied yet to study co-transcriptional ribosome assembly. For example, forming an artificial transcription bubble, the authors were able to introduce two different fluorescent labels site-specifically into the nascent RNA [113], to study co-transcriptional folding of the thiamine pyrophosphate (TPP) riboswitch. A FRET signal was used to study different conformational states of the aptamer assumed during transcription, in presence and absence of the TPP ligand. In a similar approach an azido UTP was site-specifically introduced into the RNA and linked to a dye using copper free click chemistry [114]. This approach revealed the inverse relationship between transcription speed and metabolite dependent folding of the TPP riboswitch.
In another elegant study, a superhelicase was used to simulate and study co-transcriptional folding of an RNA ribozyme [115]. First, a fully transcribed RNA, site-specifically labelled with two dyes, was hybridized to a complementary strand of DNA. This RNA/DNA hybrid was then immobilized to the surface for single-molecule imaging in presence of the superhelicase. Transcription was mimicked by addition of ATP which triggered the helicase activity making the RNA single-stranded in the direction from the 5’ to 3’. They were able to investigate the RNA transitioning from a single-stranded state (low FRET) to a secondary folded (intermediate FRET) and a tertiary folded state (high FRET). The helicase activity can potentially be matched to transcription speed, but it still lacks the native transcriptional pausing that can directly influence RNA folding.
6.3. Optical Tweezers
While single-molecule fluorescence microscopy studies are powerful for tracking co-transcriptional RNA folding and the binding of proteins simultaneously at relatively high throughput, they lack the ability of tracking transcription elongation at single nucleotide resolution. Optical tweezers instead can trap biomolecules, for example, transcription complexes between two beads and allow the observation of transcription progression [116] and RNAP pausing at single nucleotide resolution [117]. Optical tweezers have been used to characterize real-time co-transcriptional RNA folding to understand the switching function of the adenine riboswitch and the resultant changes in RNA conformation upon ligand addition [118]. Optical tweezers also provide information on forces exerted by biomolecules. For example, to understand how r-proteins stabilize rRNA structure, they mechanically unfolded and folded an irregular stem in domain II of the 23S rRNA [119] in presence and absence of r-protein L20. They found that L20 made the rRNA more resistant to mechanical unfolding by acting as a clamp around both strands of the rRNA stem.
Overall single-molecule methods are very sensitive and provide direct and quantitative information. They inherently resolve biological heterogeneity and provide high temporal resolution to track small and fast conformational changes of flexible regions that are averaged-out by conventional structural methods. Importantly, they provide information on how several different processes are functionally coupled to each other and how different assembly intermediates are placed along a reaction coordinate.
7. Integrative Methods
Multiple different biochemical, structural and biophysical methods have been employed to study the complex multistep process of ribosome assembly. Yet, none of the methods can independently provide information on the entire process. Here, we highlight a few selected examples that use the power of integrating various methods.
In order to study the assembly mechanism of the bacterial 50S subunit in vivo, Davis et. al, used a depleted bL17 strain to accumulate 50S assembly intermediates [73]. High-resolution cryoEM was used to determine the structures of 13 assembly intermediates. However, missing densities in the structures of these immature particles precludes to obtain information on RNA structure and associated proteins in these presumably dynamic regions. They used SHAPE-MaP based chemical structure probing data to determine that in these assembly intermediates the 23S rRNA had native secondary structure. Interestingly, the sequencing reads also showed that some of the rRNA was not completely processed in the assembly intermediates. This was in agreement with the previous reports that final rRNA maturation occurs very late in assembly [1,120,121]. In order to provide information on the protein composition of the structural blocks that were missing in the cryoEM maps, they performed qMS showing that the majority of the r-proteins are already bound to these dynamic regions and these blocks just need to be docked to the rest of the subunit to become a mature 50S subunit. Finally, one of the major drawbacks of structural methods is their inability to give direct information on function. In this case, to determine if the assembly intermediates are capable of maturing into functional subunits, Davis et. al, pulse-labelled the bL17 depleted cells with heavy labelled media and simultaneously induced the bL17 production. As expected, the peak in the sucrose gradient of the bL17 depleted assembly intermediates disappeared completely and the native 70S peak increased in intensity. This native 70S peak had heavy labelled bL17 incorporated indicating that addition of bL17 can rescue the intermediate and complete the maturation.
In another study, Soper et. al used a combination of hydroxyl radical footprinting and qMS to understand the role of cellular factors in RNA folding and ribosome assembly quality control [59]. Hydroxyl radical footprinting experiments showed how the assembly factor RimM reduces misfolding of the 16S head during transcription in vivo. qMS instead allowed them to confirm that in absence of RimM/RbfA, some tertiary r-proteins were missing in the assembly intermediates. Further, they observed that the acetylation state of S18 directly correlated with the folding of rRNA and the formation of specific RNA-protein contacts during assembly.
A more recent study used native co-transcriptional in vitro reconstitutions in cell extract (iSAT) and characterized the ribosome assembly intermediates using time-resolved cryoEM and qMS to both quantify r-protein composition and the status of rRNA modifications during assembly [46]. The structures derived from the iSAT reaction were highly heterogenous. 13 structures were classified spanning from one of the smallest known assembly intermediates detected to date (made of 600nts and 3 r-proteins) to the latest stages of assembly with the nearly complete 50S subunit. Remarkably, studies doing in vitro reconstitutions from purified components [72,122], co-transcriptional in vitro reconstitutions with cell extract [46] and characterizing intermediates in vivo [73] show similar assembly intermediates providing a general consensus on the mechanism of the 50S assembly.
Overall, integrating multiple methods is very powerful and crucial to mechanistically understand ribosome assembly and the assembly of other RNPs in detail.
8. Future Methods
A combination of different biochemical, biophysical and structural approaches has allowed us to understand in great detail how the very complex process of ribosome assembly works at the molecular level. Moving forward, the major challenges to solve are to 1) understand how different processes in ribosome assembly are functionally coupled to each other and 2) visualizing the structure and dynamics of ribosome assembly in the dense native cellular environment. In the following section we will discuss emerging methods that we think will help to address these challenges.
8.1. Multi-Color and Multiscale Single-Molecule Methods
Single-molecule methods are uniquely suited to understand how different processes are functionally coupled to each other. The above discussed multi-color single-molecule fluorescence microscopy approaches demonstrate the potential to track simultaneously multiple processes, for example, they allowed us to understand how transcription, RNA folding and protein binding are directly interconnected [11]. Moving forward, more complex in vitro reconstitutions including more factors and processes will become accessible. Furthermore, experiments in cell extract containing all the cellular factors will bridge the gap to in vivo experiments.
Apart from developing more complex multi-color single-molecule fluorescence experiments, the future will also include combining single-molecule experiments with force experiments such as optical tweezers. For example, combining the two single-molecule modalities may allow tracking transcription elongation and RNA folding at single nucleotide resolution and in addition correlate the binding of one or two proteins to the co-transcriptionally folding rRNA. In a recent study, the authors used force changes as readout to monitor individual codon translocation of ribosomes on mRNA or the unwinding of mRNA secondary structure by ribosomes and simultaneously monitored the binding of fluorescently labelled elongation factor EF-G (Figure 7A) [123]. As a further extension of this technology LUMICKS has extended the imaging part from single-color to multi-color fluorescent microscopy [124]. However, despite its power to study multiple processes simultaneously, the method lacks throughput. The optical tweezer technology can only study one complex at a time. To study very complex and heterogenous systems like ribosome assembly, efforts will be required to increase the throughput and automation such as microfluids as commercially introduced by LUMICKS.
Mass photometry imaging is another single-molecule method that uses interferometric scattering to determine the mass of individual molecules [125]. This in combination with other methods could be useful to study the size distribution of assembly intermediates during different stages of assembly.
Recent advancements in the field of single-molecule nanopore direct RNA sequencing may provide new opportunities to understand how and when RNA modifications are introduced during ribosome assembly. In this technique, voltage is applied to a pore located in a membrane and the resulting ionic current can be detected [126]. When the RNA passes through the pore, the detected current changes depending on which nucleotide is passing through the pore. Similarly, modified nucleotides also lead to a change in current, specific for each RNA modification. In principle, this allows the direct detection of all the modifications present on a single-molecule of the RNA. Direct sequencing of 16S rRNA successfully detected the presence of m7G and pseudouridine at the population level [127]. Current advances in data analysis methods have allowed the study of multiple other modifications such as and not limited to m6A, m5C, m1G, m62A, I, Nm and 2’-OMe [128,129,130]. Recent developments highlight the potential of nanopore sequencing for detecting multiple RNA modifications on the same molecule at single transcript resolution [128,131,132]. This opens up the avenue to investigate if there is a specific order in which RNA modifications are introduced. Chemical probing of RNA followed by direct RNA nanopore sequencing was used to predict RNA secondary structure [133]. Combining base modification detection with chemical probing based RNA structure determination could allow investigating how RNA modification and RNA structure formation are functionally coupled in RNP assembly [134].
8.2. In Vivo Single-Molecule Tracking
For in vivo single-molecule tracking, individual molecules are not tethered to the coverslip but the molecules of interest are endogenously tagged with a fluorescent reporter and tracked in-real time while they are moving within a cell [137]. The majority of the molecules are much too abundant in the cell to be tracked all at once due to the diffraction limit of light. Therefore, a small subset of the molecules can be photoactivated first and then excited with a different wavelength for tracking. One common endogenous tag, which can be linked to the protein of interest is mMaple3 [138]. This photoconvertible protein is activated by illuminating at 405 nm and can then be imaged by exciting the protein at 561 nm. Some initial studies looked at clustering of RNA polymerase (RNAP) using in vivo single-molecule localization to characterize the RNAP organization inside cells. Interestingly RNAP localization experiments showed that the spatial clustering of RNAP is independent to rRNA transcription activity as opposed to what was suggested earlier but rather dependent on the underlying nucleoid structure [139]. Pushing this further, the transcription factor NusA, which is part of the rrnTAC involved in early ribosome assembly, was tracked in vivo [136] (Figure 7B, top panel). By evaluating the different single-molecule tracks and converting them to apparent diffusion coefficients, the authors found that NusA diffuses in three states: slow-moving molecules were assigned to NusA molecules associated with the transcription complex, fast-moving molecules as freely diffusing and a third class with intermediate mobility was assigned to NusA molecules present in a transcription condensate, which likely forms by liquid-liquid phase separation. The individual components can freely diffuse in and out of these clusters indicating that the droplets are dynamic (Figure 7B, bottom panel). These studies provided evidence that not only eukaryotic ribosome assembly occurs in a biomolecule condensate (nucleolus) but that a similar condensed state may also organize bacterial ribosome assembly. Such a mechanism could explain the much higher ribosome assembly efficiency in vivo compared to in vitro reconstitutions.
Similar experiments were also applied to study eukaryotic ribosome assembly, for example, to track the export of pre-60S particles to the cytoplasm through the nuclear pore complex [140]. The authors observed that transport is a single rate limiting step and takes about 24 ms in average. Furthermore, quantification of export from single pores revealed that only a one third of the export attempts are successful and overall mass flux can be as high as 125 MDa per second.
Similar experiments could in future allow us to track the dynamics of individual r-proteins or assembly factors to gain a better understanding of ribosome assembly in vivo. While single-molecule tracking can be extended to more than one color and recent break-throughs with the MINFLUX technology are maximizing spatiotemporal resolution to nanometer spatial and submillisecond temporal resolution [141,142,143], the requirement for stochastic activation of single fluorophores in an ocean of otherwise unlabelled molecules makes it very unlikely that two differently labelled molecules would interact with each other. Therefore, directly tracking individual protein-RNA interactions or macromolecular conformational changes in vivo will require new technologies to be developed.
8.3. Cryo-Electron Tomography
Cryo-Electron Tomography (cryoET) is an emerging method to gain structural understanding directly in native cellular context. CryoET uses the same basic idea as single-particle cryoEM to reconstruct 3D images. The main difference is that in tomography an image is acquired by tilting the sample at multiple different angles [144]. This provides images of the sample at multiple different orientations which can be used to reconstruct a 3D image for each individual particle. This is in contrast to single-particle cryoEM that typically uses averaged information from hundreds of thousands of particles present in different orientations [66]. Thus, cryoET can be used to look at individual complexes inside whole cells or sections of cells, thereby preserving the native structure.
For example, Xue et al. were able to identify Mycoplasma pneumoniae ribosomes during various stages of translation and to provide a detailed map of the translation elongation cycle within a single cell [24]. Importantly, they were able to assign for each ribosome in the cell the specific translation state, providing spatial functional information on the translation status. They were able to quantitatively show that 26 % of all ribosomes were polysomes and determine the orientation of each ribosome in the polysome with respect to each other and their overall packing density. Comparing the individual ribosomes within a polysome, they could identify that r-protein L9 of the leading ribosome adopts an extended conformation protruding into the binding site of the translation elongation factors of the trailing ribosome thereby, providing a mechanism to prevent ribosome collisions. Applying similar approaches to study bacterial ribosome assembly in cellular context will be challenging due to the low abundance of ribosome assembly intermediates as compared to fully assembled ribosomes. Imaging cells treated with antibiotics to accumulate ribosome assembly intermediates could be the first step to tackle this challenging problem.
In another study from the Mahamid lab, structures of the RNAP-ribosome supercomplex, termed expressome, were visualized in situ by combining cryoET with cross-linking mass spectrometry (Figure 7C, left panel) [135]. The structures showed for the first time how transcription-translation coupling is structurally organized in vivo (Figure 7C, right panel). They could show that the transcription factor NusA mediates coupling by physically linking the RNAP with the ribosome in M. pneumoniae. Furthermore, they visualized at high-resolution a state in which the ribosome is collided with the RNAP in presence of an antibiotic stalling the RNAP. Similar approaches could be used to visualize how bacterial ribosome assembly is coupled to transcription.
Eukaryotic ribosome assembly is separated from translation and takes place inside the nucleolus, which is a multi-phasic biomolecular condensate and spatially organizes maturing ribosome assembly intermediates [145]. The Baumeister lab used cryoET on native nucleoli of Chlamydomonas reinhardtii to show that the pre-60S (LSU precursor) and SSU processome (SSU precursor) have different spatial localization patterns. Furthermore, they could classify three low-resolution structural assembly intermediates of each pre-60S and SSU processome. The maturation of these intermediates followed a gradient from the inside to the outside of the granular component [146].
Overall, these pioneering studies provide a starting point and demonstrate the potential to study the complex process of ribosome assembly at high-resolution in the native cellular context. Studying in vivo ribosome assembly could potentially answer questions such as the number of alternate pathways present in the assembly process and quantify the percentage flux in each of these pathways.
9. Conclusions
The assembly of the ribosome is a very complicated process involving transcription, folding, modification and processing of the rRNA and the binding of dozens of r-proteins to the nascent rRNA, assisted by dozens of assembly factors. Remarkably, the entire assembly process is completed within 2 minutes in the dense cellular environment. A plethora of biochemical, biophysical and structural methods have helped to understand this process in a quantitative manner: sophisticated in vitro reconstitution systems in cell extract that closely mimic the native process have been developed to bridge the gap between in vitro reconstitution from purified components and the assembly in vivo. Pulse-chase quantitative mass spectrometry, time-resolved cryo-electron microscopy and time-resolved RNA structure probing approaches have provided compositional and high-resolution structural data to understand the kinetics of ribosome assembly and were instrumental to characterize multiple assembly intermediates along the parallel assembly pathways. Recent multi-color single-molecule fluorescence experiments have shown the potential to follow in real-time how individual RNAs transcribe, simultaneously fold and start to assemble into protein-RNA complexes, providing information on how multiple different processes are functionally coupled to each other. Moving forward, in vivo single-molecule tracking as well as cryo-electron tomography will provide us the much-needed understanding of how the ribosome assembles in the dense native cellular environment. Combining our efforts of developing bottom-up reconstitutions of active systems with ever increasing complexity with biophysical and structural approaches to visualize systems in vivo will bring us closer to understand and importantly, generate predictive models of how complex cellular processes work in a living cell [147].
Author Contributions
Conceptualization, O.D. and K.G.; writing—original draft preparation, review and editing, K.G. and O.D.; visualization, K.G.; supervision, O.D.; funding acquisition, O.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the European Molecular Biology Laboratory.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the entire Duss lab for helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shajani, Z.; Sykes, M.T.; Williamson, J.R. Assembly of bacterial ribosomes. Annu Rev Biochem 2011, 80, 501–526. [Google Scholar] [CrossRef]
- Nierhaus, K.H. The assembly of prokaryotic ribosomes. Biochimie 1991, 73, 739–755. [Google Scholar] [CrossRef]
- Huang, Y.H.; Said, N.; Loll, B.; Wahl, M.C. Structural basis for the function of SuhB as a transcription factor in ribosomal RNA synthesis. Nucleic Acids Res 2019, 47, 6488–6503. [Google Scholar] [CrossRef]
- Singh, N.; Bubunenko, M.; Smith, C.; Abbott, D.M.; Stringer, A.M.; Shi, R.; Court, D.L.; Wade, J.T. SuhB Associates with Nus Factors To Facilitate 30S Ribosome Biogenesis in Escherichia coli. MBio 2016, 7, e00114. [Google Scholar] [CrossRef]
- Torres, M.; Condon, C.; Balada, J.M.; Squires, C.; Squires, C.L. Ribosomal protein S4 is a transcription factor with properties remarkably similar to NusA, a protein involved in both non-ribosomal and ribosomal RNA antitermination. EMBO J 2001, 20, 3811–3820. [Google Scholar] [CrossRef]
- Li, Z.; Pandit, S.; Deutscher, M.P. Maturation of 23S ribosomal RNA requires the exoribonuclease RNase T. RNA 1999, 5, 139–146. [Google Scholar] [CrossRef]
- Li, Z.; Pandit, S.; Deutscher, M.P. RNase G (CafA protein) and RNase E are both required for the 5' maturation of 16S ribosomal RNA. EMBO J 1999, 18, 2878–2885. [Google Scholar] [CrossRef]
- Ginsburg, D.; Steitz, J.A. The 30 S ribosomal precursor RNA from Escherichia coli. A primary transcript containing 23 S, 16 S, and 5 S sequences. J Biol Chem 1975, 250, 5647–5654. [Google Scholar] [CrossRef]
- King, T.C.; Schlessinger, D. S1 nuclease mapping analysis of ribosomal RNA processing in wild type and processing deficient Escherichia coli. J Biol Chem 1983, 258, 12034–12042. [Google Scholar] [CrossRef]
- Rodgers, M.L.; Woodson, S.A. Transcription Increases the Cooperativity of Ribonucleoprotein Assembly. Cell 2019, 179, 1370–1381. [Google Scholar] [CrossRef]
- Duss, O.; Stepanyuk, G.A.; Puglisi, J.D.; Williamson, J.R. Transient Protein-RNA Interactions Guide Nascent Ribosomal RNA Folding. Cell 2019, 179, 1357–1369. [Google Scholar] [CrossRef]
- Herschlag, D. RNA chaperones and the RNA folding problem. J Biol Chem 1995, 270, 20871–20874. [Google Scholar] [CrossRef]
- Adilakshmi, T.; Ramaswamy, P.; Woodson, S.A. Protein-independent folding pathway of the 16S rRNA 5' domain. J Mol Biol 2005, 351, 508–519. [Google Scholar] [CrossRef]
- Powers, T.; Daubresse, G.; Noller, H.F. Dynamics of in vitro assembly of 16 S rRNA into 30 S ribosomal subunits. J Mol Biol 1993, 232, 362–374. [Google Scholar] [CrossRef]
- Talkington, M.W.; Siuzdak, G.; Williamson, J.R. An assembly landscape for the 30S ribosomal subunit. Nature 2005, 438, 628–632. [Google Scholar] [CrossRef]
- Held, W.A.; Ballou, B.; Mizushima, S.; Nomura, M. Assembly mapping of 30 S ribosomal proteins from Escherichia coli. Further studies. J Biol Chem 1974, 249, 3103–3111. [Google Scholar] [CrossRef]
- Mizushima, S.; Nomura, M. Assembly mapping of 30S ribosomal proteins from E. coli. Nature 1970, 226, 1214. [Google Scholar] [CrossRef]
- Rohl, R.; Nierhaus, K.H. Assembly map of the large subunit (50S) of Escherichia coli ribosomes. Proc Natl Acad Sci U S A 1982, 79, 729–733. [Google Scholar] [CrossRef]
- Decatur, W.A.; Fournier, M.J. rRNA modifications and ribosome function. Trends Biochem Sci 2002, 27, 344–351. [Google Scholar] [CrossRef]
- Rodgers, M.L.; Woodson, S.A. A roadmap for rRNA folding and assembly during transcription. Trends Biochem Sci 2021, 46, 889–901. [Google Scholar] [CrossRef]
- Britton, R.A. Role of GTPases in bacterial ribosome assembly. Annu Rev Microbiol 2009, 63, 155–176. [Google Scholar] [CrossRef]
- Chen, S.S.; Williamson, J.R. Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J Mol Biol 2013, 425, 767–779. [Google Scholar] [CrossRef]
- Lindahl, L. Intermediates and time kinetics of the in vivo assembly of Escherichia coli ribosomes. J Mol Biol 1975, 92, 15–37. [Google Scholar] [CrossRef]
- Xue, L.; Lenz, S.; Zimmermann-Kogadeeva, M.; Tegunov, D.; Cramer, P.; Bork, P.; Rappsilber, J.; Mahamid, J. Visualizing translation dynamics at atomic detail inside a bacterial cell. Nature 2022, 610, 205–211. [Google Scholar] [CrossRef]
- Davis, J.H.; Williamson, J.R. Structure and dynamics of bacterial ribosome biogenesis. Philos Trans R Soc Lond B Biol Sci 2017, 372. [Google Scholar] [CrossRef]
- Naganathan, A.; Culver, G.M. Interdependency and Redundancy Add Complexity and Resilience to Biogenesis of Bacterial Ribosomes. Annu Rev Microbiol 2022, 76, 193–210. [Google Scholar] [CrossRef]
- Oborska-Oplova, M.; Gerhardy, S.; Panse, V.G. Orchestrating ribosomal RNA folding during ribosome assembly. Bioessays 2022, 44, e2200066. [Google Scholar] [CrossRef]
- Sykes, M.T.; Williamson, J.R. A complex assembly landscape for the 30S ribosomal subunit. Annu Rev Biophys 2009, 38, 197–215. [Google Scholar] [CrossRef]
- Williamson, J.R. Biophysical studies of bacterial ribosome assembly. Curr Opin Struct Biol 2008, 18, 299–304. [Google Scholar] [CrossRef]
- Woodson, S.A. RNA folding and ribosome assembly. Curr Opin Chem Biol 2008, 12, 667–673. [Google Scholar] [CrossRef]
- Woodson, S.A. RNA folding pathways and the self-assembly of ribosomes. Acc Chem Res 2011, 44, 1312–1319. [Google Scholar] [CrossRef]
- Culver, G.M.; Noller, H.F. Efficient reconstitution of functional Escherichia coli 30S ribosomal subunits from a complete set of recombinant small subunit ribosomal proteins. RNA 1999, 5, 832–843. [Google Scholar] [CrossRef]
- Aoyama, R.; Masuda, K.; Shimojo, M.; Kanamori, T.; Ueda, T.; Shimizu, Y. In vitro reconstitution of the Escherichia coli 70S ribosome with a full set of recombinant ribosomal proteins. J Biochem 2022, 171, 227–237. [Google Scholar] [CrossRef]
- Nierhaus, K.H.; Dohme, F. Total reconstitution of functionally active 50S ribosomal subunits from Escherichia coli. Proc Natl Acad Sci U S A 1974, 71, 4713–4717. [Google Scholar] [CrossRef]
- Rheinberger, H.J.; Nierhaus, K.H. Partial release of AcPhe-Phe-tRNA from ribosomes during poly(U)-dependent poly(Phe) synthesis and the effects of chloramphenicol. Eur J Biochem 1990, 193, 643–650. [Google Scholar] [CrossRef]
- Tamaru, D.; Amikura, K.; Shimizu, Y.; Nierhaus, K.H.; Ueda, T. Reconstitution of 30S ribosomal subunits in vitro using ribosome biogenesis factors. RNA 2018, 24, 1512–1519. [Google Scholar] [CrossRef]
- Semrad, K.; Green, R. Osmolytes stimulate the reconstitution of functional 50S ribosomes from in vitro transcripts of Escherichia coli 23S rRNA. RNA 2002, 8, 401–411. [Google Scholar] [CrossRef]
- Maki, J.A.; Culver, G.M. Recent developments in factor-facilitated ribosome assembly. Methods 2005, 36, 313–320. [Google Scholar] [CrossRef]
- Nomura, M.; Traub, P.; Guthrie, C.; Nashimoto, H. The assembly of ribosomes. J Cell Physiol 1969, 74, Suppl–1:241. [Google Scholar] [CrossRef]
- Jewett, M.C.; Fritz, B.R.; Timmerman, L.E.; Church, G.M. In vitro integration of ribosomal RNA synthesis, ribosome assembly, and translation. Mol Syst Biol 2013, 9, 678. [Google Scholar] [CrossRef]
- Miller, O.L., Jr.; Hamkalo, B.A.; Thomas, C.A., Jr. Visualization of bacterial genes in action. Science 1970, 169, 392–395. [Google Scholar] [CrossRef]
- Miller, O.L., Jr.; Beatty, B.R. Visualization of nucleolar genes. Science 1969, 164, 955–957. [Google Scholar] [CrossRef]
- Hofmann, S.; Miller, O.L., Jr. Visualization of ribosomal ribonucleic acid synthesis in a ribonuclease III-Deficient strain of Escherichia coli. J Bacteriol 1977, 132, 718–722. [Google Scholar] [CrossRef]
- Gotta, S.L.; Miller, O.L., Jr.; French, S.L. rRNA transcription rate in Escherichia coli. J Bacteriol 1991, 173, 6647–6649. [Google Scholar] [CrossRef]
- Fritz, B.R.; Jewett, M.C. The impact of transcriptional tuning on in vitro integrated rRNA transcription and ribosome construction. Nucleic Acids Res 2014, 42, 6774–6785. [Google Scholar] [CrossRef]
- Dong, X.; Doerfel, L.K.; Sheng, K.; Rabuck-Gibbons, J.N.; Popova, A.M.; Lyumkis, D.; Williamson, J.R. Near-physiological in vitro assembly of 50S ribosomes involves parallel pathways. Nucleic Acids Res 2023. [Google Scholar] [CrossRef]
- Fritz, B.R.; Jamil, O.K.; Jewett, M.C. Implications of macromolecular crowding and reducing conditions for in vitro ribosome construction. Nucleic Acids Res 2015, 43, 4774–4784. [Google Scholar] [CrossRef]
- Shimojo, M.; Amikura, K.; Masuda, K.; Kanamori, T.; Ueda, T.; Shimizu, Y. In vitro reconstitution of functional small ribosomal subunit assembly for comprehensive analysis of ribosomal elements in E. coli. Commun Biol 2020, 3, 142. [Google Scholar] [CrossRef]
- Levy, M.; Falkovich, R.; Daube, S.S.; Bar-Ziv, R.H. Autonomous synthesis and assembly of a ribosomal subunit on a chip. Sci Adv 2020, 6, eaaz6020. [Google Scholar] [CrossRef]
- d'Aquino, A.E.; Azim, T.; Aleksashin, N.A.; Hockenberry, A.J.; Kruger, A.; Jewett, M.C. Mutational characterization and mapping of the 70S ribosome active site. Nucleic Acids Res 2020, 48, 2777–2789. [Google Scholar] [CrossRef]
- Liu, Y.; Davis, R.G.; Thomas, P.M.; Kelleher, N.L.; Jewett, M.C. In vitro-Constructed Ribosomes Enable Multi-site Incorporation of Noncanonical Amino Acids into Proteins. Biochemistry 2021, 60, 161–169. [Google Scholar] [CrossRef]
- Hammerling, M.J.; Fritz, B.R.; Yoesep, D.J.; Kim, D.S.; Carlson, E.D.; Jewett, M.C. In vitro ribosome synthesis and evolution through ribosome display. Nat Commun 2020, 11, 1108. [Google Scholar] [CrossRef]
- Bunner, A.E.; Beck, A.H.; Williamson, J.R. Kinetic cooperativity in Escherichia coli 30S ribosomal subunit reconstitution reveals additional complexity in the assembly landscape. Proc Natl Acad Sci U S A 2010, 107, 5417–5422. [Google Scholar] [CrossRef]
- Bunner, A.E.; Nord, S.; Wikstrom, P.M.; Williamson, J.R. The effect of ribosome assembly cofactors on in vitro 30S subunit reconstitution. J Mol Biol 2010, 398, 1–7. [Google Scholar] [CrossRef]
- Nord, S.; Bylund, G.O.; Lovgren, J.M.; Wikstrom, P.M. The RimP protein is important for maturation of the 30S ribosomal subunit. J Mol Biol 2009, 386, 742–753. [Google Scholar] [CrossRef]
- Bylund, G.O.; Persson, B.C.; Lundberg, L.A.; Wikstrom, P.M. A novel ribosome-associated protein is important for efficient translation in Escherichia coli. J Bacteriol 1997, 179, 4567–4574. [Google Scholar] [CrossRef]
- Inoue, K.; Alsina, J.; Chen, J.; Inouye, M. Suppression of defective ribosome assembly in a rbfA deletion mutant by overexpression of Era, an essential GTPase in Escherichia coli. Mol Microbiol 2003, 48, 1005–1016. [Google Scholar] [CrossRef]
- Gibbs, M.R.; Moon, K.M.; Chen, M.; Balakrishnan, R.; Foster, L.J.; Fredrick, K. Conserved GTPase LepA (Elongation Factor 4) functions in biogenesis of the 30S subunit of the 70S ribosome. Proc Natl Acad Sci U S A 2017, 114, 980–985. [Google Scholar] [CrossRef]
- Clatterbuck Soper, S.F.; Dator, R.P.; Limbach, P.A.; Woodson, S.A. In vivo X-ray footprinting of pre-30S ribosomes reveals chaperone-dependent remodeling of late assembly intermediates. Mol Cell 2013, 52, 506–516. [Google Scholar] [CrossRef]
- Sailer, C.; Jansen, J.; Sekulski, K.; Cruz, V.E.; Erzberger, J.P.; Stengel, F. A comprehensive landscape of 60S ribosome biogenesis factors. Cell Rep 2022, 38, 110353. [Google Scholar] [CrossRef]
- Motorin, Y.; Muller, S.; Behm-Ansmant, I.; Branlant, C. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol 2007, 425, 21–53. [Google Scholar] [CrossRef]
- Siibak, T.; Remme, J. Subribosomal particle analysis reveals the stages of bacterial ribosome assembly at which rRNA nucleotides are modified. RNA 2010, 16, 2023–2032. [Google Scholar] [CrossRef]
- Kellner, S.; Ochel, A.; Thuring, K.; Spenkuch, F.; Neumann, J.; Sharma, S.; Entian, K.D.; Schneider, D.; Helm, M. Absolute and relative quantification of RNA modifications via biosynthetic isotopomers. Nucleic Acids Res 2014, 42, e142. [Google Scholar] [CrossRef]
- Popova, A.M.; Williamson, J.R. Quantitative analysis of rRNA modifications using stable isotope labeling and mass spectrometry. J Am Chem Soc 2014, 136, 2058–2069. [Google Scholar] [CrossRef]
- Ishiguro, K.; Arai, T.; Suzuki, T. Depletion of S-adenosylmethionine impacts on ribosome biogenesis through hypomodification of a single rRNA methylation. Nucleic Acids Res 2019, 47, 4226–4239. [Google Scholar] [CrossRef]
- Murata, K.; Wolf, M. Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim Biophys Acta Gen Subj 2018, 1862, 324–334. [Google Scholar] [CrossRef]
- Mulder, A.M.; Yoshioka, C.; Beck, A.H.; Bunner, A.E.; Milligan, R.A.; Potter, C.S.; Carragher, B.; Williamson, J.R. Visualizing ribosome biogenesis: parallel assembly pathways for the 30S subunit. Science 2010, 330, 673–677. [Google Scholar] [CrossRef]
- Nikolay, R.; Hilal, T.; Schmidt, S.; Qin, B.; Schwefel, D.; Vieira-Vieira, C.H.; Mielke, T.; Burger, J.; Loerke, J.; Amikura, K.; et al. Snapshots of native pre-50S ribosomes reveal a biogenesis factor network and evolutionary specialization. Mol Cell 2021, 81, 1200–1215. [Google Scholar] [CrossRef]
- Huang, Y.H.; Hilal, T.; Loll, B.; Burger, J.; Mielke, T.; Bottcher, C.; Said, N.; Wahl, M.C. Structure-Based Mechanisms of a Molecular RNA Polymerase/Chaperone Machine Required for Ribosome Biosynthesis. Mol Cell 2020, 79, 1024–1036. [Google Scholar] [CrossRef]
- Kuhlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef]
- Callaway, E. 'It opens up a whole new universe': Revolutionary microscopy technique sees individual atoms for first time. Nature 2020, 582, 156–157. [Google Scholar] [CrossRef]
- Nikolay, R.; Hilal, T.; Qin, B.; Mielke, T.; Burger, J.; Loerke, J.; Textoris-Taube, K.; Nierhaus, K.H.; Spahn, C.M.T. Structural Visualization of the Formation and Activation of the 50S Ribosomal Subunit during In Vitro Reconstitution. Mol Cell 2018, 70, 881–893. [Google Scholar] [CrossRef]
- Davis, J.H.; Tan, Y.Z.; Carragher, B.; Potter, C.S.; Lyumkis, D.; Williamson, J.R. Modular Assembly of the Bacterial Large Ribosomal Subunit. Cell 2016, 167, 1610–1622. [Google Scholar] [CrossRef]
- Zhong, E.D.; Bepler, T.; Berger, B.; Davis, J.H. CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat Methods 2021, 18, 176–185. [Google Scholar] [CrossRef]
- Wang, W.; Li, W.; Ge, X.; Yan, K.; Mandava, C.S.; Sanyal, S.; Gao, N. Loss of a single methylation in 23S rRNA delays 50S assembly at multiple late stages and impairs translation initiation and elongation. Proc Natl Acad Sci U S A 2020, 117, 15609–15619. [Google Scholar] [CrossRef]
- Rabuck-Gibbons, J.N.; Popova, A.M.; Greene, E.M.; Cervantes, C.F.; Lyumkis, D.; Williamson, J.R. SrmB Rescues Trapped Ribosome Assembly Intermediates. J Mol Biol 2020, 432, 978–990. [Google Scholar] [CrossRef]
- Razi, A.; Davis, J.H.; Hao, Y.; Jahagirdar, D.; Thurlow, B.; Basu, K.; Jain, N.; Gomez-Blanco, J.; Britton, R.A.; Vargas, J.; et al. Role of Era in assembly and homeostasis of the ribosomal small subunit. Nucleic Acids Res 2019, 47, 8301–8317. [Google Scholar] [CrossRef]
- Thurlow, B.; Davis, J.H.; Leong, V.; Moraes, T.F.; Williamson, J.R.; Ortega, J. Binding properties of YjeQ (RsgA), RbfA, RimM and Era to assembly intermediates of the 30S subunit. Nucleic Acids Res 2016, 44, 9918–9932. [Google Scholar] [CrossRef]
- Guo, Q.; Goto, S.; Chen, Y.; Feng, B.; Xu, Y.; Muto, A.; Himeno, H.; Deng, H.; Lei, J.; Gao, N. Dissecting the in vivo assembly of the 30S ribosomal subunit reveals the role of RimM and general features of the assembly process. Nucleic Acids Res 2013, 41, 2609–2620. [Google Scholar] [CrossRef]
- Jomaa, A.; Stewart, G.; Martin-Benito, J.; Zielke, R.; Campbell, T.L.; Maddock, J.R.; Brown, E.D.; Ortega, J. Understanding ribosome assembly: the structure of in vivo assembled immature 30S subunits revealed by cryo-electron microscopy. RNA 2011, 17, 697–709. [Google Scholar] [CrossRef]
- Leong, V.; Kent, M.; Jomaa, A.; Ortega, J. Escherichia coli rimM and yjeQ null strains accumulate immature 30S subunits of similar structure and protein complement. RNA 2013, 19, 789–802. [Google Scholar] [CrossRef]
- Boehringer, D.; O'Farrell, H.C.; Rife, J.P.; Ban, N. Structural insights into methyltransferase KsgA function in 30S ribosomal subunit biogenesis. J Biol Chem 2012, 287, 10453–10459. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, K.; Zhang, Y.; Li, N.; Ma, C.; Li, Z.; Zhang, Y.; Feng, B.; Liu, J.; Sun, Y.; et al. Structural insights into the function of a unique tandem GTPase EngA in bacterial ribosome assembly. Nucleic Acids Res 2014, 42, 13430–13439. [Google Scholar] [CrossRef]
- Bubunenko, M.; Court, D.L.; Al Refaii, A.; Saxena, S.; Korepanov, A.; Friedman, D.I.; Gottesman, M.E.; Alix, J.H. Nus transcription elongation factors and RNase III modulate small ribosome subunit biogenesis in Escherichia coli. Mol Microbiol 2013, 87, 382–393. [Google Scholar] [CrossRef]
- Redko, Y.; Condon, C. Ribosomal protein L3 bound to 23S precursor rRNA stimulates its maturation by Mini-III ribonuclease. Mol Microbiol 2009, 71, 1145–1154. [Google Scholar] [CrossRef]
- Stahl, D.A.; Pace, B.; Marsh, T.; Pace, N.R. The ribonucleoprotein substrate for a ribosomal RNA-processing nuclease. J Biol Chem 1984, 259, 11448–11453. [Google Scholar] [CrossRef]
- Oerum, S.; Dendooven, T.; Catala, M.; Gilet, L.; Degut, C.; Trinquier, A.; Bourguet, M.; Barraud, P.; Cianferani, S.; Luisi, B.F.; et al. Structures of B. subtilis Maturation RNases Captured on 50S Ribosome with Pre-rRNAs. Mol Cell 2020, 80, 227–236. [Google Scholar] [CrossRef]
- Stiegler, P.; Carbon, P.; Zuker, M.; Ebel, J.P.; Ehresmann, C. Structural organization of the 16S ribosomal RNA from E. coli. Topography and secondary structure. Nucleic Acids Res 1981, 9, 2153–2172. [Google Scholar] [CrossRef]
- Spitale, R.C.; Incarnato, D. Probing the dynamic RNA structurome and its functions. Nat Rev Genet 2023, 24, 178–196. [Google Scholar] [CrossRef]
- Deigan, K.E.; Li, T.W.; Mathews, D.H.; Weeks, K.M. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci U S A 2009, 106, 97–102. [Google Scholar] [CrossRef]
- Siegfried, N.A.; Busan, S.; Rice, G.M.; Nelson, J.A.; Weeks, K.M. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat Methods 2014, 11, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Adilakshmi, T.; Bellur, D.L.; Woodson, S.A. Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature 2008, 455, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Stern, S.; Powers, T.; Changchien, L.M.; Noller, H.F. RNA-protein interactions in 30S ribosomal subunits: folding and function of 16S rRNA. Science 1989, 244, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Abeysirigunawarden, S.C.; Chen, K.; Mayerle, M.; Ragunathan, K.; Luthey-Schulten, Z.; Ha, T.; Woodson, S.A. Protein-guided RNA dynamics during early ribosome assembly. Nature 2014, 506, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Duss, O.; Stepanyuk, G.A.; Grot, A.; O'Leary, S.E.; Puglisi, J.D.; Williamson, J.R. Real-time assembly of ribonucleoprotein complexes on nascent RNA transcripts. Nature communications 2018, 9, 5087. [Google Scholar] [CrossRef]
- Olson, S.W.; Turner, A.W.; Arney, J.W.; Saleem, I.; Weidmann, C.A.; Margolis, D.M.; Weeks, K.M.; Mustoe, A.M. Discovery of a large-scale, cell-state-responsive allosteric switch in the 7SK RNA using DANCE-MaP. Mol Cell 2022, 82, 1708–1723. [Google Scholar] [CrossRef]
- Morandi, E.; Manfredonia, I.; Simon, L.M.; Anselmi, F.; van Hemert, M.J.; Oliviero, S.; Incarnato, D. Genome-scale deconvolution of RNA structure ensembles. Nat Methods 2021, 18, 249–252. [Google Scholar] [CrossRef]
- Tomezsko, P.J.; Corbin, V.D.A.; Gupta, P.; Swaminathan, H.; Glasgow, M.; Persad, S.; Edwards, M.D.; McIntosh, L.; Papenfuss, A.T.; Emery, A.; et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 2020, 582, 438–442. [Google Scholar] [CrossRef]
- Homan, P.J.; Favorov, O.V.; Lavender, C.A.; Kursun, O.; Ge, X.; Busan, S.; Dokholyan, N.V.; Weeks, K.M. Single-molecule correlated chemical probing of RNA. Proc Natl Acad Sci U S A 2014, 111, 13858–13863. [Google Scholar] [CrossRef]
- Khoroshkin, M.; Asarnow, D.; Navickas, A.; Winters, A.; Yu, J.; Zhou, S.K.; Zhou, S.; Palka, C.; Fish, L.; Ansel, K.M.; et al. A systematic search for RNA structural switches across the human transcriptome. bioRxiv 2023, 2023.2003.2011.532161. [CrossRef]
- Mitchell, D., 3rd; Assmann, S.M.; Bevilacqua, P.C. Probing RNA structure in vivo. Curr Opin Struct Biol 2019, 59, 151–158. [Google Scholar] [CrossRef]
- Watters, K.E.; Strobel, E.J.; Yu, A.M.; Lis, J.T.; Lucks, J.B. Cotranscriptional folding of a riboswitch at nucleotide resolution. Nat Struct Mol Biol 2016, 23, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Lewicki, B.T.; Margus, T.; Remme, J.; Nierhaus, K.H. Coupling of rRNA transcription and ribosomal assembly in vivo. Formation of active ribosomal subunits in Escherichia coli requires transcription of rRNA genes by host RNA polymerase which cannot be replaced by bacteriophage T7 RNA polymerase. J Mol Biol 1993, 231, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Hulscher, R.M.; Bohon, J.; Rappe, M.C.; Gupta, S.; D'Mello, R.; Sullivan, M.; Ralston, C.Y.; Chance, M.R.; Woodson, S.A. Probing the structure of ribosome assembly intermediates in vivo using DMS and hydroxyl radical footprinting. Methods 2016, 103, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, S.A.; Weeks, K.M. Time-resolved RNA SHAPE chemistry. J Am Chem Soc 2008, 130, 16178–16180. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat Methods 2008, 5, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.A.; Poyton, M.F.; Ha, T. Multicolor single-molecule FRET for DNA and RNA processes. Curr Opin Struct Biol 2021, 70, 26–33. [Google Scholar] [CrossRef]
- Lerner, E.; Cordes, T.; Ingargiola, A.; Alhadid, Y.; Chung, S.; Michalet, X.; Weiss, S. Toward dynamic structural biology: Two decades of single-molecule Forster resonance energy transfer. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.; Zhuang, X.; Kim, H.D.; Orr, J.W.; Williamson, J.R.; Chu, S. Ligand-induced conformational changes observed in single RNA molecules. Proc Natl Acad Sci U S A 1999, 96, 9077–9082. [Google Scholar] [CrossRef]
- Abeysirigunawardena, S.C.; Kim, H.; Lai, J.; Ragunathan, K.; Rappe, M.C.; Luthey-Schulten, Z.; Ha, T.; Woodson, S.A. Evolution of protein-coupled RNA dynamics during hierarchical assembly of ribosomal complexes. Nature communications 2017, 8, 492. [Google Scholar] [CrossRef]
- Lai, D.; Proctor, J.R.; Meyer, I.M. On the importance of cotranscriptional RNA structure formation. RNA 2013, 19, 1461–1473. [Google Scholar] [CrossRef]
- Zhang, J.; Landick, R. A Two-Way Street: Regulatory Interplay between RNA Polymerase and Nascent RNA Structure. Trends Biochem Sci 2016, 41, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Uhm, H.; Kang, W.; Ha, K.S.; Kang, C.; Hohng, S. Single-molecule FRET studies on the cotranscriptional folding of a thiamine pyrophosphate riboswitch. Proc Natl Acad Sci U S A 2018, 115, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Chauvier, A.; St-Pierre, P.; Nadon, J.F.; Hien, E.D.M.; Perez-Gonzalez, C.; Eschbach, S.H.; Lamontagne, A.M.; Penedo, J.C.; Lafontaine, D.A. Monitoring RNA dynamics in native transcriptional complexes. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Hua, B.; Panja, S.; Wang, Y.; Woodson, S.A.; Ha, T. Mimicking Co-Transcriptional RNA Folding Using a Superhelicase. J Am Chem Soc 2018, 140, 10067–10070. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.D.; Schnitzer, M.J.; Yin, H.; Landick, R.; Gelles, J.; Block, S.M. Force and velocity measured for single molecules of RNA polymerase. Science 1998, 282, 902–907. [Google Scholar] [CrossRef]
- Abbondanzieri, E.A.; Greenleaf, W.J.; Shaevitz, J.W.; Landick, R.; Block, S.M. Direct observation of base-pair stepping by RNA polymerase. Nature 2005, 438, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Frieda, K.L.; Block, S.M. Direct observation of cotranscriptional folding in an adenine riboswitch. Science 2012, 338, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Mangeol, P.; Bizebard, T.; Chiaruttini, C.; Dreyfus, M.; Springer, M.; Bockelmann, U. Probing ribosomal protein-RNA interactions with an external force. Proc Natl Acad Sci U S A 2011, 108, 18272–18276. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Schlessinger, D. Coregulation of processing and translation: mature 5' termini of Escherichia coli 23S ribosomal RNA form in polysomes. Proc Natl Acad Sci U S A 1988, 85, 7144–7148. [Google Scholar] [CrossRef]
- Mangiarotti, G.; Turco, E.; Ponzetto, A.; Altruda, F. Precursor 16S RNA in active 30S ribosomes. Nature 1974, 247, 147–148. [Google Scholar] [CrossRef]
- Qin, B.; Lauer, S.M.; Balke, A.; Vieira-Vieira, C.H.; Burger, J.; Mielke, T.; Selbach, M.; Scheerer, P.; Spahn, C.M.T.; Nikolay, R. Cryo-EM captures early ribosome assembly in action. Nat Commun 2023, 14, 898. [Google Scholar] [CrossRef] [PubMed]
- Desai, V.P.; Frank, F.; Lee, A.; Righini, M.; Lancaster, L.; Noller, H.F.; Tinoco, I., Jr.; Bustamante, C. Co-temporal Force and Fluorescence Measurements Reveal a Ribosomal Gear Shift Mechanism of Translation Regulation by Structured mRNAs. Mol Cell 2019, 75, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- C-TRAP® optical tweezers fluorescence & label-free microscopy. Available online: https://lumicks.com/products/c-trap-optical-tweezers-fluorescence-label-free-microscopy/ (accessed on 6 April 2023).
- Young, G.; Hundt, N.; Cole, D.; Fineberg, A.; Andrecka, J.; Tyler, A.; Olerinyova, A.; Ansari, A.; Marklund, E.G.; Collier, M.P.; et al. Quantitative mass imaging of single biological macromolecules. Science 2018, 360, 423–427. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Jain, M.; Mulroney, L.; Garalde, D.R.; Akeson, M. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS One 2019, 14, e0216709. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat Commun 2021, 12, 7198. [Google Scholar] [CrossRef]
- Begik, O.; Mattick, J.S.; Novoa, E.M. Exploring the epitranscriptome by native RNA sequencing. RNA 2022, 28, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Begik, O.; Lucas, M.C.; Pryszcz, L.P.; Ramirez, J.M.; Medina, R.; Milenkovic, I.; Cruciani, S.; Liu, H.; Vieira, H.G.S.; Sas-Chen, A.; et al. Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing. Nat Biotechnol 2021, 39, 1278–1291. [Google Scholar] [CrossRef]
- Hendra, C.; Pratanwanich, P.N.; Wan, Y.K.; Goh, W.S.S.; Thiery, A.; Goke, J. Detection of m6A from direct RNA sequencing using a multiple instance learning framework. Nat Methods 2022, 19, 1590–1598. [Google Scholar] [CrossRef]
- Mateos, P.A.; Sethi, A.J.; Ravindran, A.; Guarnacci, M.; Srivastava, A.; Xu, J.; Woodward, K.; Yuen, Z.W.S.; Mahmud, S.; Kanchi, M.; et al. Simultaneous identification of m6A and m5C reveals coordinated RNA modification at single-molecule resolution. bioRxiv, 2003; 2022.2003.2014.484124. [Google Scholar] [CrossRef]
- Aw, J.G.A.; Lim, S.W.; Wang, J.X.; Lambert, F.R.P.; Tan, W.T.; Shen, Y.; Zhang, Y.; Kaewsapsak, P.; Li, C.; Ng, S.B.; et al. Determination of isoform-specific RNA structure with nanopore long reads. Nat Biotechnol 2021, 39, 336–346. [Google Scholar] [CrossRef]
- Wrzesinski, J.; Bakin, A.; Nurse, K.; Lane, B.G.; Ofengand, J. Purification, cloning, and properties of the 16S RNA pseudouridine 516 synthase from Escherichia coli. Biochemistry 1995, 34, 8904–8913. [Google Scholar] [CrossRef] [PubMed]
- O'Reilly, F.J.; Xue, L.; Graziadei, A.; Sinn, L.; Lenz, S.; Tegunov, D.; Blotz, C.; Singh, N.; Hagen, W.J.H.; Cramer, P.; et al. In-cell architecture of an actively transcribing-translating expressome. Science 2020, 369, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Ladouceur, A.M.; Parmar, B.S.; Biedzinski, S.; Wall, J.; Tope, S.G.; Cohn, D.; Kim, A.; Soubry, N.; Reyes-Lamothe, R.; Weber, S.C. Clusters of bacterial RNA polymerase are biomolecular condensates that assemble through liquid-liquid phase separation. Proc Natl Acad Sci U S A 2020, 117, 18540–18549. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Gyparaki, M.T.; Beliu, G.; Schueder, F.; Griffie, J.; Manley, S.; Jungmann, R.; Sauer, M.; Lakadamyali, M.; Zimmer, C. Single-molecule localization microscopy. Nat Rev Methods Primers 2021, 1. [Google Scholar] [CrossRef]
- Wang, S.; Moffitt, J.R.; Dempsey, G.T.; Xie, X.S.; Zhuang, X. Characterization and development of photoactivatable fluorescent proteins for single-molecule-based superresolution imaging. Proc Natl Acad Sci U S A 2014, 111, 8452–8457. [Google Scholar] [CrossRef]
- Weng, X.; Bohrer, C.H.; Bettridge, K.; Lagda, A.C.; Cagliero, C.; Jin, D.J.; Xiao, J. Spatial organization of RNA polymerase and its relationship with transcription in Escherichia coli. Proc Natl Acad Sci U S A 2019, 116, 20115–20123. [Google Scholar] [CrossRef]
- Ruland, J.A.; Kruger, A.M.; Dorner, K.; Bhatia, R.; Wirths, S.; Poetes, D.; Kutay, U.; Siebrasse, J.P.; Kubitscheck, U. Nuclear export of the pre-60S ribosomal subunit through single nuclear pores observed in real time. Nat Commun 2021, 12, 6211. [Google Scholar] [CrossRef]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynna, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef]
- Deguchi, T.; Iwanski, M.K.; Schentarra, E.M.; Heidebrecht, C.; Schmidt, L.; Heck, J.; Weihs, T.; Schnorrenberg, S.; Hoess, P.; Liu, S.; et al. Direct observation of motor protein stepping in living cells using MINFLUX. Science 2023, 379, 1010–1015. [Google Scholar] [CrossRef]
- Wolff, J.O.; Scheiderer, L.; Engelhardt, T.; Engelhardt, J.; Matthias, J.; Hell, S.W. MINFLUX dissects the unimpeded walking of kinesin-1. Science 2023, 379, 1004–1010. [Google Scholar] [CrossRef]
- Turk, M.; Baumeister, W. The promise and the challenges of cryo-electron tomography. FEBS Lett 2020, 594, 3243–3261. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, D.L.J. Birth of Nucleolar Compartments: Phase Separation-Driven Ribosomal RNA Sorting and Processing. Mol Cell 2019, 76, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, P.S.; Hou, Z.; Klumpe, S.; Khavnekar, S.; Beck, F.; Wilfling, F.; Plitzko, J.M.; Baumeister, W. In situ cryo-electron tomography reveals gradient organization of ribosome biogenesis in intact nucleoli. Nat Commun 2021, 12, 5364. [Google Scholar] [CrossRef] [PubMed]
- Earnest, T.M.; Lai, J.; Chen, K.; Hallock, M.J.; Williamson, J.R.; Luthey-Schulten, Z. Toward a Whole-Cell Model of Ribosome Biogenesis: Kinetic Modeling of SSU Assembly. Biophys J 2015, 109, 1117–1135. [Google Scholar] [CrossRef]
Figure 1.
Overview of biochemical, structural and biophysical methods to study ribosome and RNP assembly. Adapted and reproduced from [24], 70S (PDB: 4V6G) and 50S intermediate (PDB: 7BL5).
Figure 1.
Overview of biochemical, structural and biophysical methods to study ribosome and RNP assembly. Adapted and reproduced from [24], 70S (PDB: 4V6G) and 50S intermediate (PDB: 7BL5).
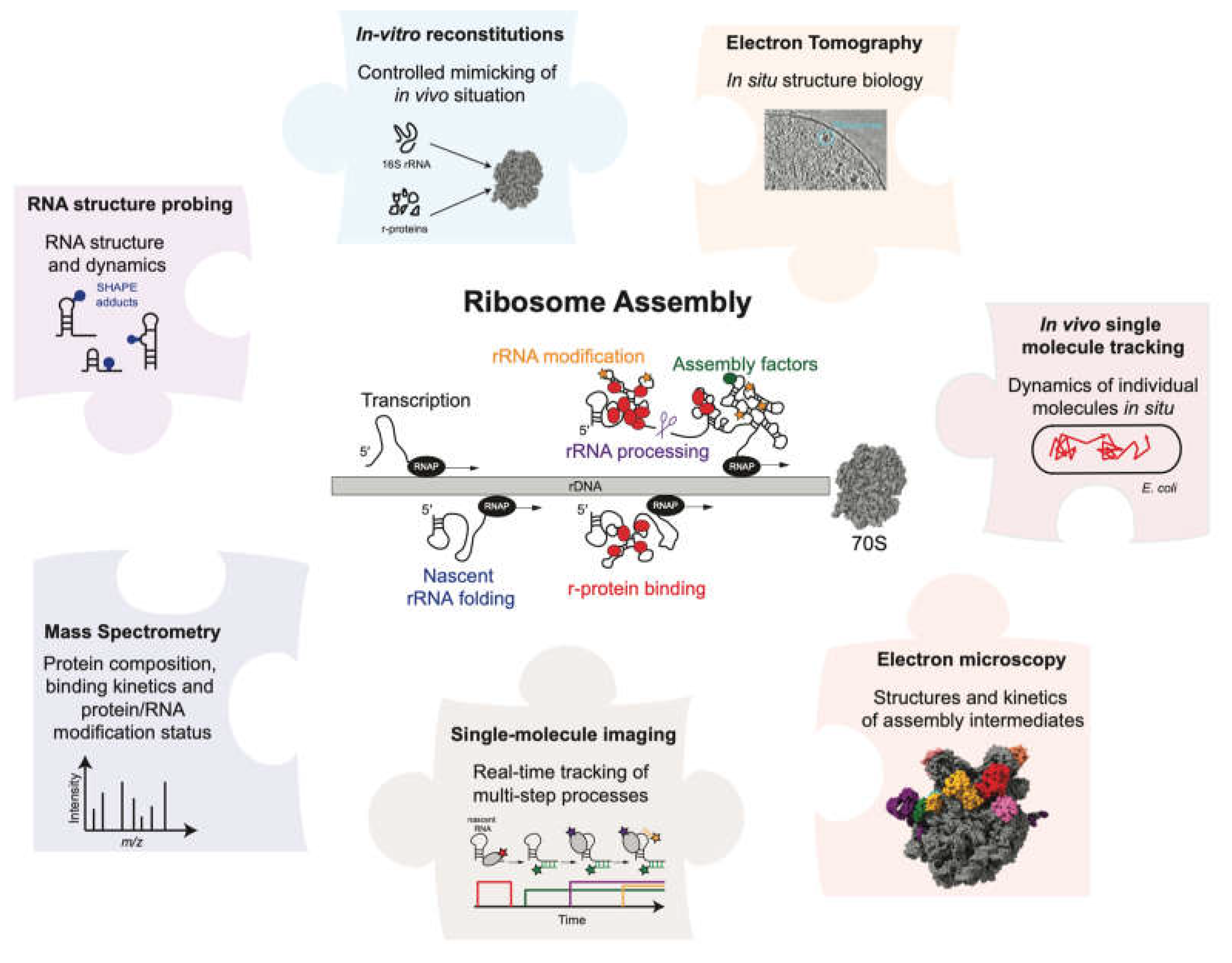
Figure 2.
A-C) Integrated ribosome Synthesis, Assembly and Translation (iSAT): A) Schematic of the one-pot iSAT reaction for synthesis and assembly of ribosomes and translation of a reporter protein; B) Real-time monitoring of fluorescent intensity as a reporter for translation activity of ribosomes produced in iSAT reaction; C) ribosome profiling of the iSAT reaction. D,E) Ribosome assembly on a chip: D) Schematic of chip surface and the distribution of DNA brushes (centre), zoomed-in schematic of one DNA cluster (top), distribution of DNA brushes (right) encoding for rRNA (black) and r-proteins-HA (green), assembly factors (grey) and other r-proteins color-coded as in the Nomura map (bottom right) and schematic of ribosome assembly on chip (bottom centre); E) Time traces of primary, secondary and tertiary r-proteins binding to rRNA during assembly on a chip (top: left to right) and normalized maximum signal from primary (green), secondary (yellow) and tertiary (grey) r-protein-HA (bottom: left to right). A is reproduced with permission from [40]. B,C are reproduced with minor adaptations with permission from [47]. D,E are reproduced from [49].
Figure 2.
A-C) Integrated ribosome Synthesis, Assembly and Translation (iSAT): A) Schematic of the one-pot iSAT reaction for synthesis and assembly of ribosomes and translation of a reporter protein; B) Real-time monitoring of fluorescent intensity as a reporter for translation activity of ribosomes produced in iSAT reaction; C) ribosome profiling of the iSAT reaction. D,E) Ribosome assembly on a chip: D) Schematic of chip surface and the distribution of DNA brushes (centre), zoomed-in schematic of one DNA cluster (top), distribution of DNA brushes (right) encoding for rRNA (black) and r-proteins-HA (green), assembly factors (grey) and other r-proteins color-coded as in the Nomura map (bottom right) and schematic of ribosome assembly on chip (bottom centre); E) Time traces of primary, secondary and tertiary r-proteins binding to rRNA during assembly on a chip (top: left to right) and normalized maximum signal from primary (green), secondary (yellow) and tertiary (grey) r-protein-HA (bottom: left to right). A is reproduced with permission from [40]. B,C are reproduced with minor adaptations with permission from [47]. D,E are reproduced from [49].
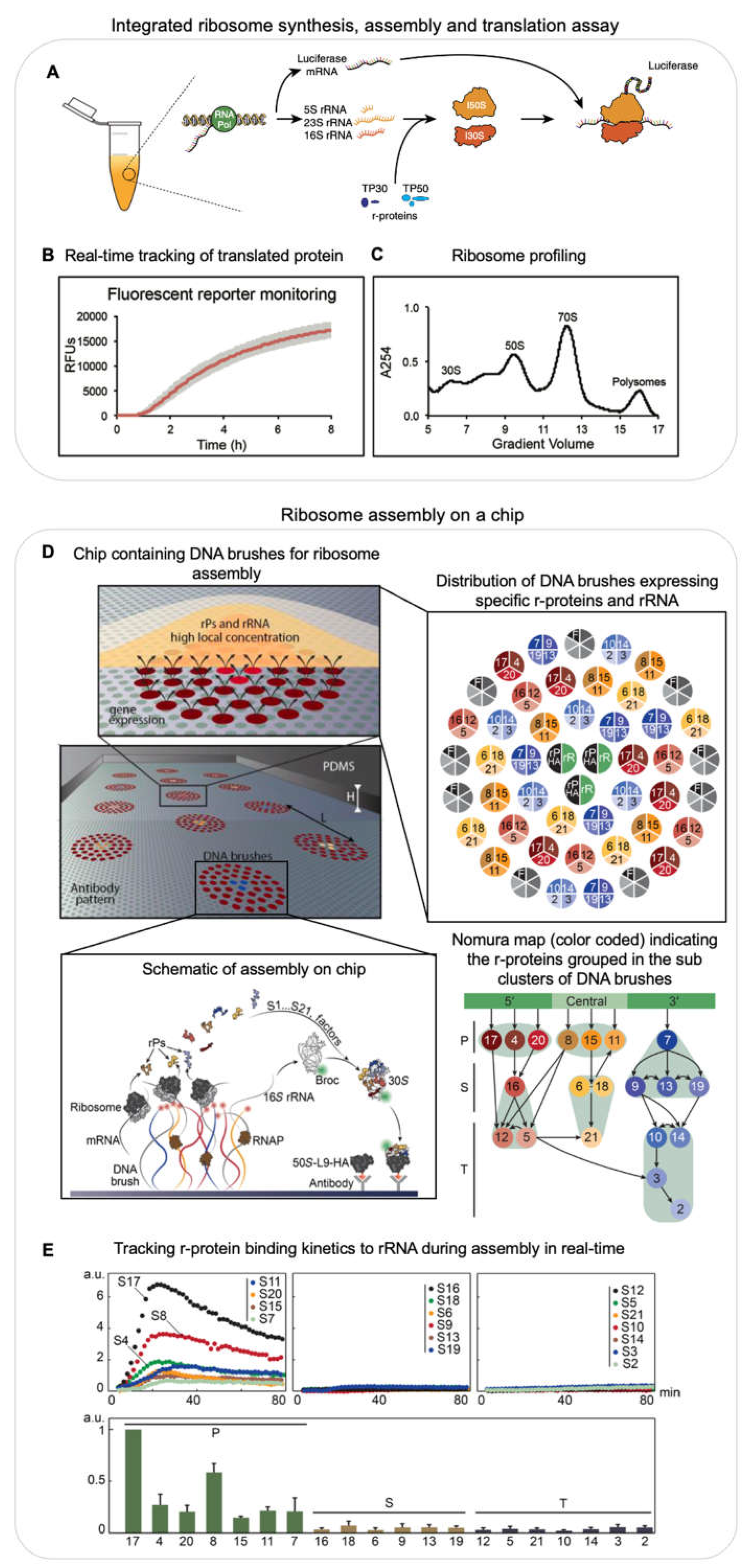
Figure 3.
A) In vitro pulse chase qMS to determine protein binding kinetics: Schematic of pulse-chase qMS workflow (left), r-protein binding curves to 16S rRNA (centre), Nomura assembly map colored according to binding rates derived from pulse-chase qMS (right). B,C) In vivo pulse labelling to determine protein binding kinetics and discovery of new assembly factors bound to the ribosome assembly intermediates: B) Experimental workflow of in vivo pulse labelling and corresponding quantification by MS. C) qMS based identification and discovery of assembly factors and their potential role in assembly of specific subunits (right). A) is reproduced with permission from [15] and B) is adapted and reproduced and C) is reproduced with permission from [22].
Figure 3.
A) In vitro pulse chase qMS to determine protein binding kinetics: Schematic of pulse-chase qMS workflow (left), r-protein binding curves to 16S rRNA (centre), Nomura assembly map colored according to binding rates derived from pulse-chase qMS (right). B,C) In vivo pulse labelling to determine protein binding kinetics and discovery of new assembly factors bound to the ribosome assembly intermediates: B) Experimental workflow of in vivo pulse labelling and corresponding quantification by MS. C) qMS based identification and discovery of assembly factors and their potential role in assembly of specific subunits (right). A) is reproduced with permission from [15] and B) is adapted and reproduced and C) is reproduced with permission from [22].
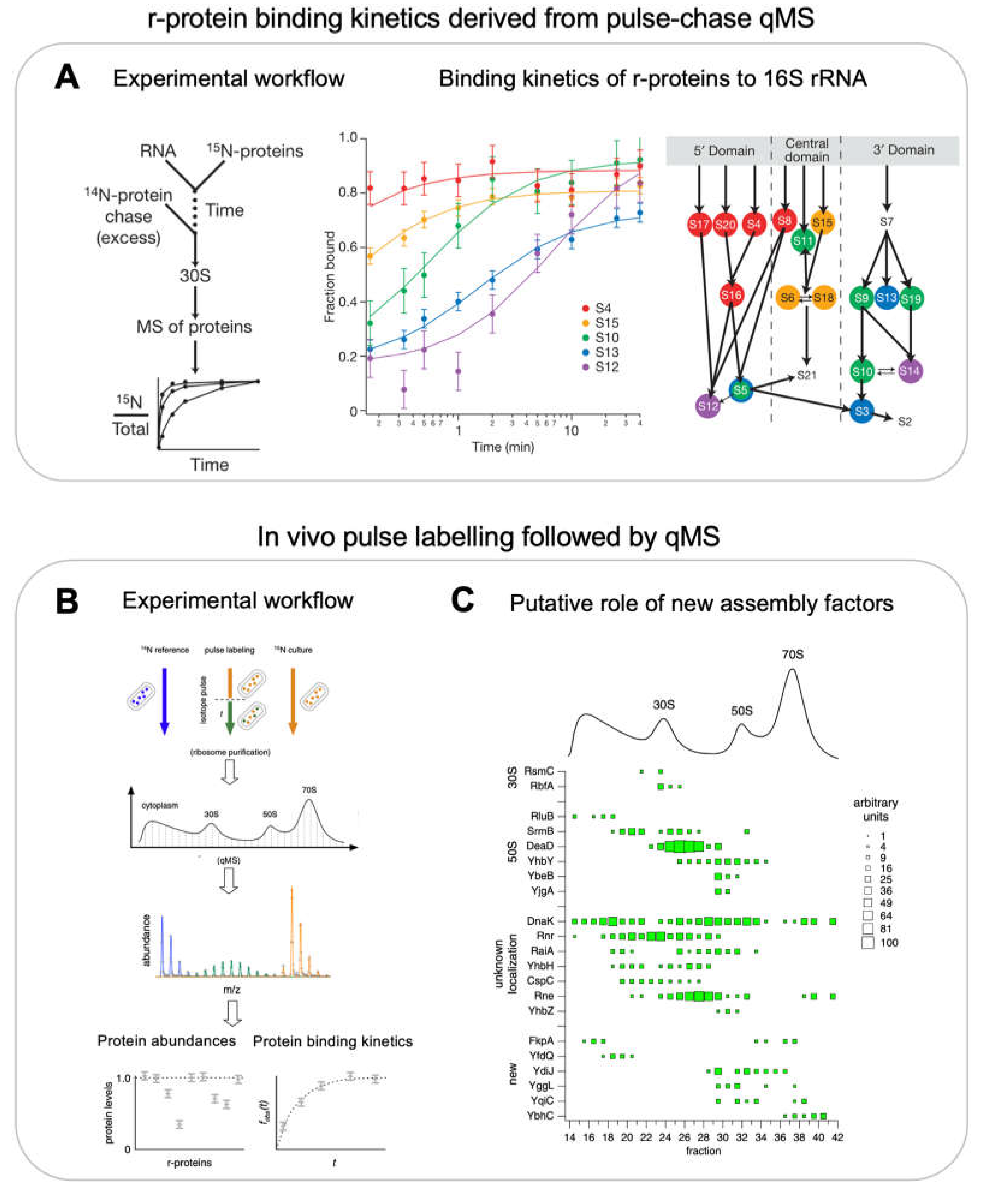
Figure 4.
Electron microscopy: A) Assembly intermediates populating parallel 30S assembly pathways visualized by negative-stain EM. B) Model of how late stage 50S maturation is guided by assembly factors (top), model can be constructed by several high-resolution cryoEM structures of pre-50S bound by assembly factors YjgA, RluD, RsfS and ObgE. C) High-resolution cryoEM structures of the complete rRNA transcription antitermination complex associated transcription machinery responsible for efficient transcription of rRNA. Reproduced with permission: A) from [67], B) from [68] and C) from [69].
Figure 4.
Electron microscopy: A) Assembly intermediates populating parallel 30S assembly pathways visualized by negative-stain EM. B) Model of how late stage 50S maturation is guided by assembly factors (top), model can be constructed by several high-resolution cryoEM structures of pre-50S bound by assembly factors YjgA, RluD, RsfS and ObgE. C) High-resolution cryoEM structures of the complete rRNA transcription antitermination complex associated transcription machinery responsible for efficient transcription of rRNA. Reproduced with permission: A) from [67], B) from [68] and C) from [69].
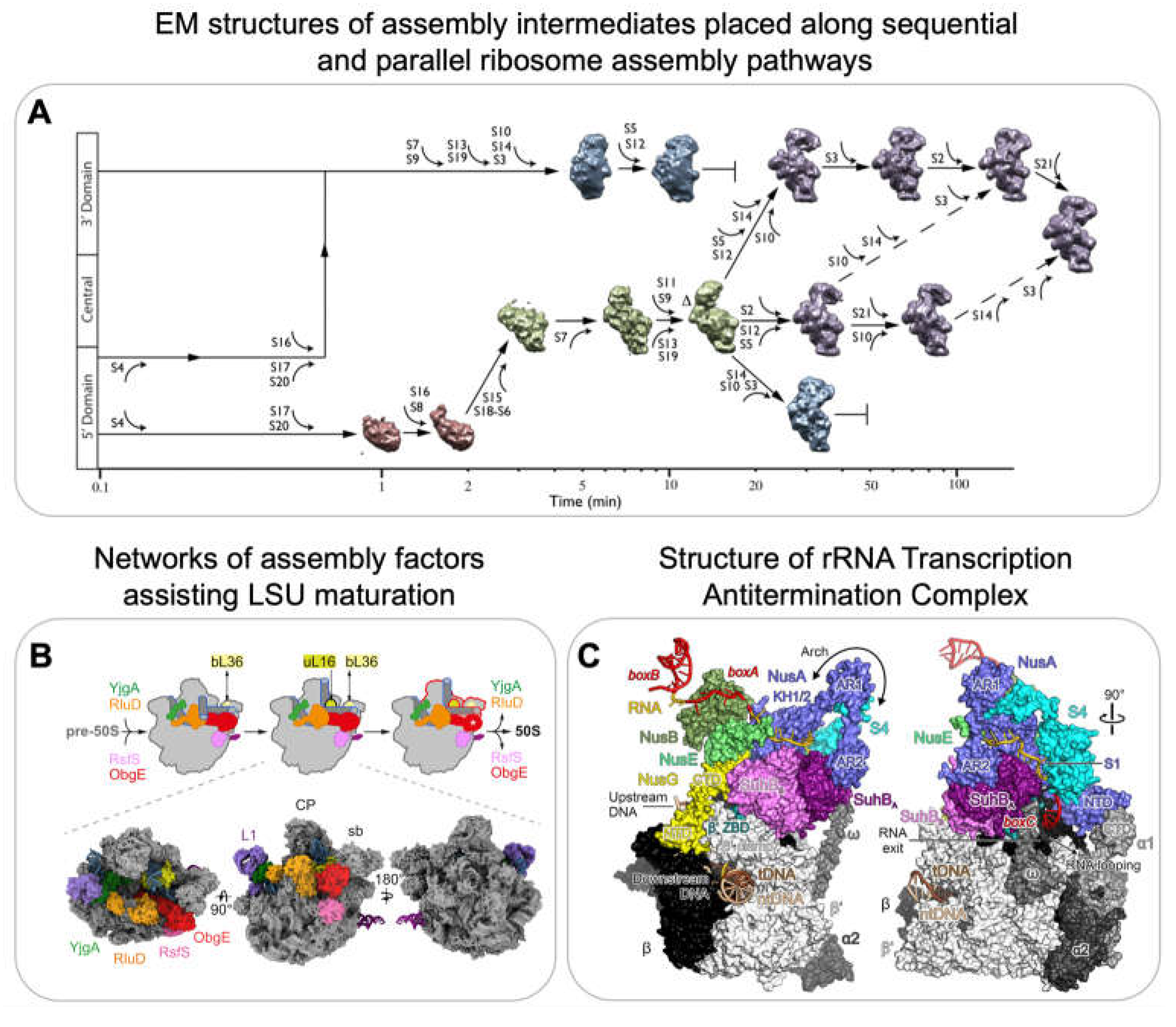
Figure 5.
RNA secondary structure determination using chemical probing and high-throughput sequencing: A) General workflow of RNA secondary structure probing. B-D) Tertiary structure and protein-RNA interaction determination using hydroxyl radical footprinting (HRF): B) Experimental setup of in vitro time-resolved HRF. C) Protection rates of individual residues of the 16S rRNA representing formation of RNA-RNA tertiary contacts as well as RNA-protein contacts. D) Kinetics of rRNA backbone protection as a result of S7 binding represented on secondary (top left) and 3D structure (top right) and normalised fitted curves indicating the protection of residues (y-axis) as a function of time (x-axis) (bottom). Color codes for Figure 5D are same as indicated in Figure 5C. Co-transcriptional RNA folding intermediates: E) Model for co-transcriptional folding of the SRP RNA as determined by co-transcriptional SHAPE-seq. A) is reproduced and adapted with permission from [101]. Reproduced with permission: C),D) from [92], and E) from [102].
Figure 5.
RNA secondary structure determination using chemical probing and high-throughput sequencing: A) General workflow of RNA secondary structure probing. B-D) Tertiary structure and protein-RNA interaction determination using hydroxyl radical footprinting (HRF): B) Experimental setup of in vitro time-resolved HRF. C) Protection rates of individual residues of the 16S rRNA representing formation of RNA-RNA tertiary contacts as well as RNA-protein contacts. D) Kinetics of rRNA backbone protection as a result of S7 binding represented on secondary (top left) and 3D structure (top right) and normalised fitted curves indicating the protection of residues (y-axis) as a function of time (x-axis) (bottom). Color codes for Figure 5D are same as indicated in Figure 5C. Co-transcriptional RNA folding intermediates: E) Model for co-transcriptional folding of the SRP RNA as determined by co-transcriptional SHAPE-seq. A) is reproduced and adapted with permission from [101]. Reproduced with permission: C),D) from [92], and E) from [102].
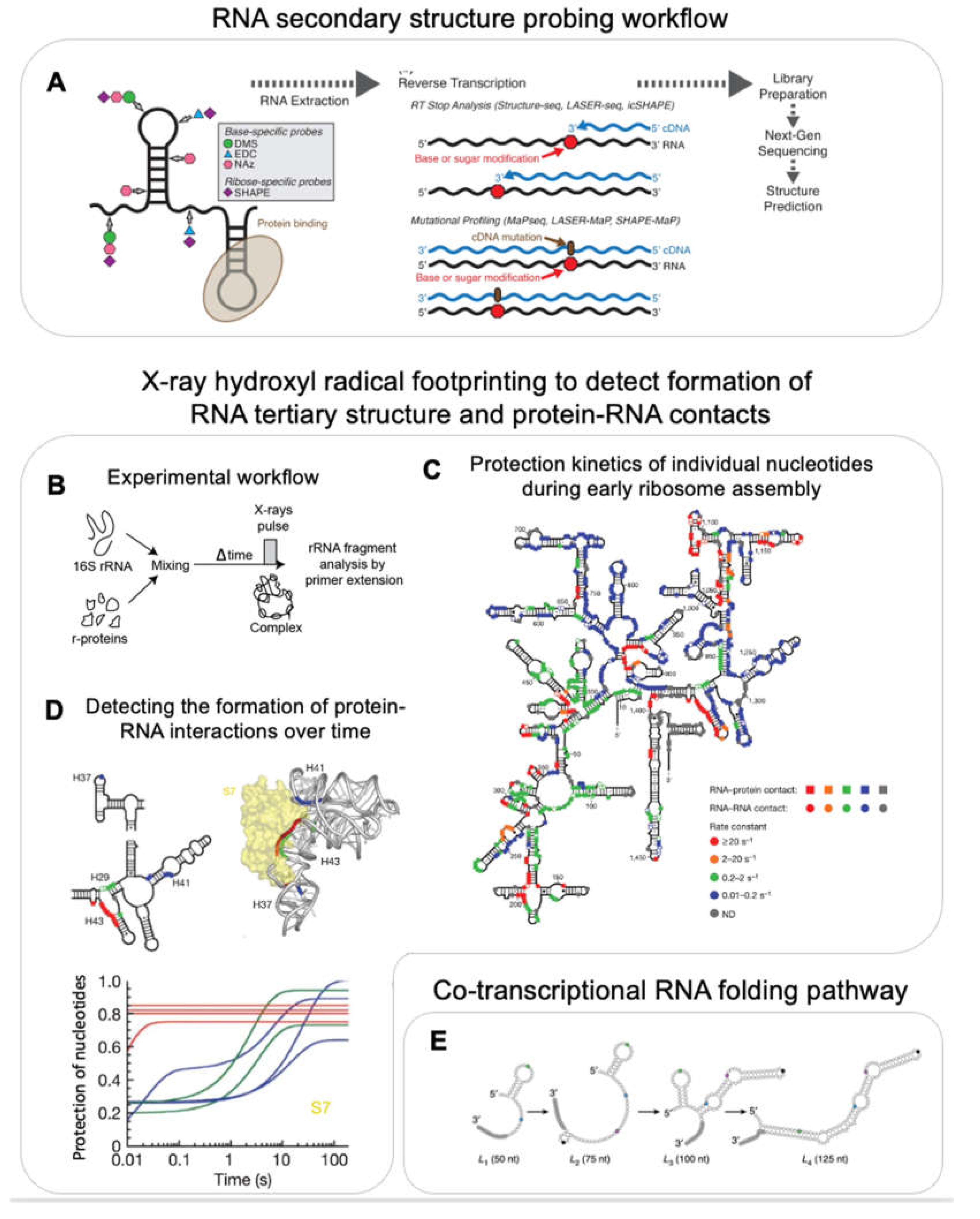
Figure 6.
A-D) Single molecule fluorescence microscopy experiments to track changes in protein-RNA interactions in real-time: A) Experimental setup of typical single-molecule experiments: schematic shows specific binding of S4 to the 5’ domain of 16S rRNA using single-molecule FRET. S4 was labelled with a donor dye (Cy3) and the immobilized RNA by an acceptor dye (Cy5). B) Single-molecule trace of S4 binding to the 5’ domain of the 16S rRNA leading to anti-correlated changes in the Cy3 and Cy5 channels over time. C) Ensemble FRET efficiency plot highlighting a non-native intermediate state of S4 binding (orange box). D) Proposed model of rRNA rearrangements upon S4 binding (bottom panel). E-G) Real-time tracking of multiple processes occurring during co-transcriptional ribosome assembly: E) Experimental setups for simultaneously detecting transcription progression (left), specific protein binding kinetics (centre) and RNA conformational changes (right). F) Multi-color single-molecule trace showing real-time transcription progression, long-range rRNA helix-28 (H28) formation and transient binding of r-protein S7. G) Quantification of single-molecule data of experiments shown in E)/F) under different conditions: the plots are showing the efficiency of H28 formation (top) and the efficiency of S7 binding to the subset of molecules that have H28 formed (bottom). A-D are reproduced with permission from [94] and E-G are reproduced and F is adapted with permission from [11].
Figure 6.
A-D) Single molecule fluorescence microscopy experiments to track changes in protein-RNA interactions in real-time: A) Experimental setup of typical single-molecule experiments: schematic shows specific binding of S4 to the 5’ domain of 16S rRNA using single-molecule FRET. S4 was labelled with a donor dye (Cy3) and the immobilized RNA by an acceptor dye (Cy5). B) Single-molecule trace of S4 binding to the 5’ domain of the 16S rRNA leading to anti-correlated changes in the Cy3 and Cy5 channels over time. C) Ensemble FRET efficiency plot highlighting a non-native intermediate state of S4 binding (orange box). D) Proposed model of rRNA rearrangements upon S4 binding (bottom panel). E-G) Real-time tracking of multiple processes occurring during co-transcriptional ribosome assembly: E) Experimental setups for simultaneously detecting transcription progression (left), specific protein binding kinetics (centre) and RNA conformational changes (right). F) Multi-color single-molecule trace showing real-time transcription progression, long-range rRNA helix-28 (H28) formation and transient binding of r-protein S7. G) Quantification of single-molecule data of experiments shown in E)/F) under different conditions: the plots are showing the efficiency of H28 formation (top) and the efficiency of S7 binding to the subset of molecules that have H28 formed (bottom). A-D are reproduced with permission from [94] and E-G are reproduced and F is adapted with permission from [11].
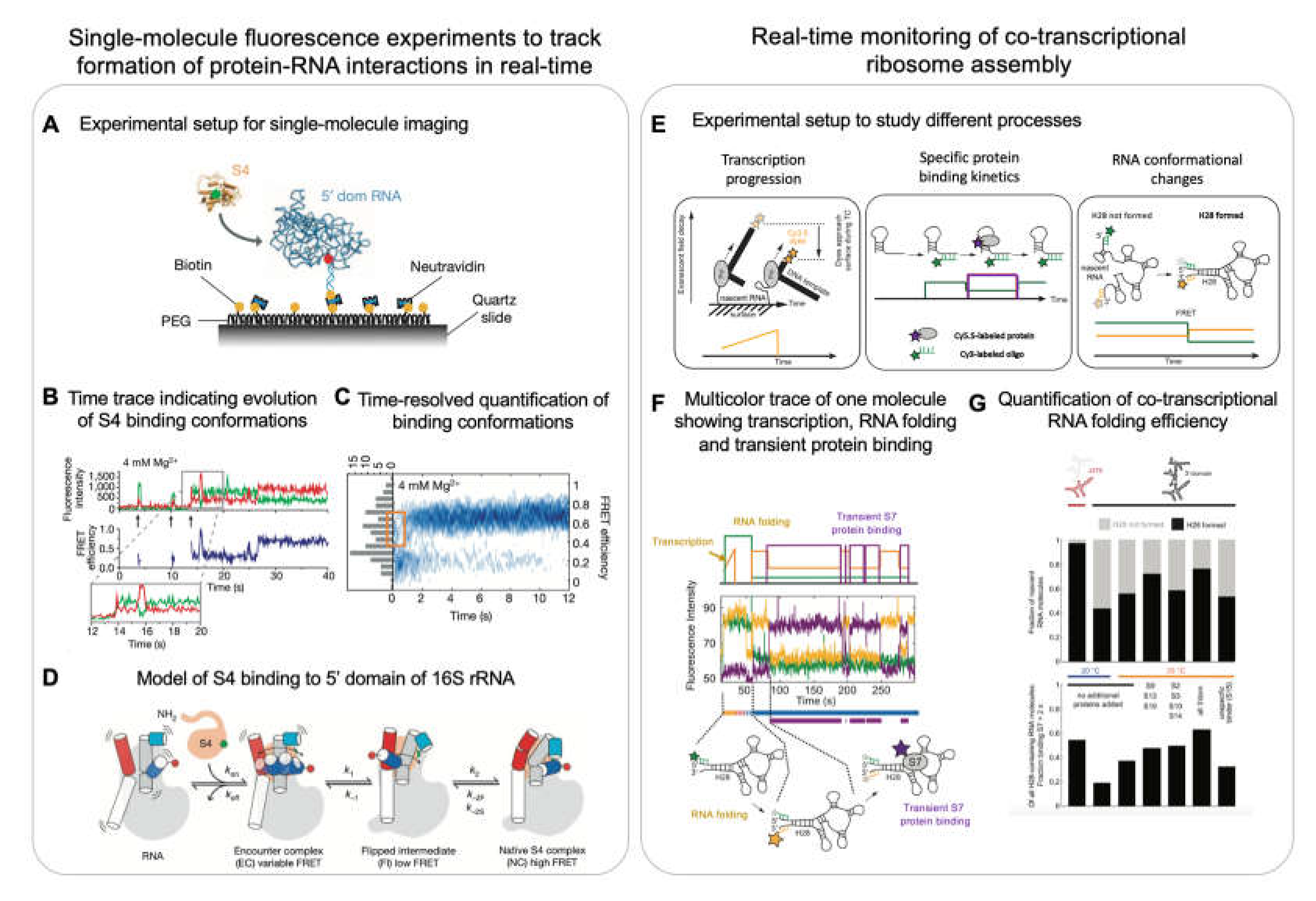
Figure 7.
Multiscale single-molecule methods to study RNP dynamics at nucleotide resolution: A) Experimental setup of optical tweezers combined with fluorescence microscopy to study mRNA unwinding during translation (top); time traces indicating change in distance upon one codon translation (centre), and changes in fluorescence intensity upon elongation factor binding (bottom). In vivo single-molecule tracking to study spatial localization and dynamics: B) Experimental setup of in vivo single-molecule tracking experiment (top); quantification of tracking data by plotting the distribution of the apparent diffusion coefficients indicating dynamic movements of transcription factor NusA within and outside the presumable transcription condensates. In situ structural biology: C) Representative tomographic slice of a M. pneumoniae cell and quantitative classification of ribosome subtomograms (left); resultant structures of 70S (top right) and RNAP-ribosome supercomplex (bottom right). Adapted and reproduced with permission: A) from [123] and C) from [135]. B) is adapted and reproduced from [136].
Figure 7.
Multiscale single-molecule methods to study RNP dynamics at nucleotide resolution: A) Experimental setup of optical tweezers combined with fluorescence microscopy to study mRNA unwinding during translation (top); time traces indicating change in distance upon one codon translation (centre), and changes in fluorescence intensity upon elongation factor binding (bottom). In vivo single-molecule tracking to study spatial localization and dynamics: B) Experimental setup of in vivo single-molecule tracking experiment (top); quantification of tracking data by plotting the distribution of the apparent diffusion coefficients indicating dynamic movements of transcription factor NusA within and outside the presumable transcription condensates. In situ structural biology: C) Representative tomographic slice of a M. pneumoniae cell and quantitative classification of ribosome subtomograms (left); resultant structures of 70S (top right) and RNAP-ribosome supercomplex (bottom right). Adapted and reproduced with permission: A) from [123] and C) from [135]. B) is adapted and reproduced from [136].
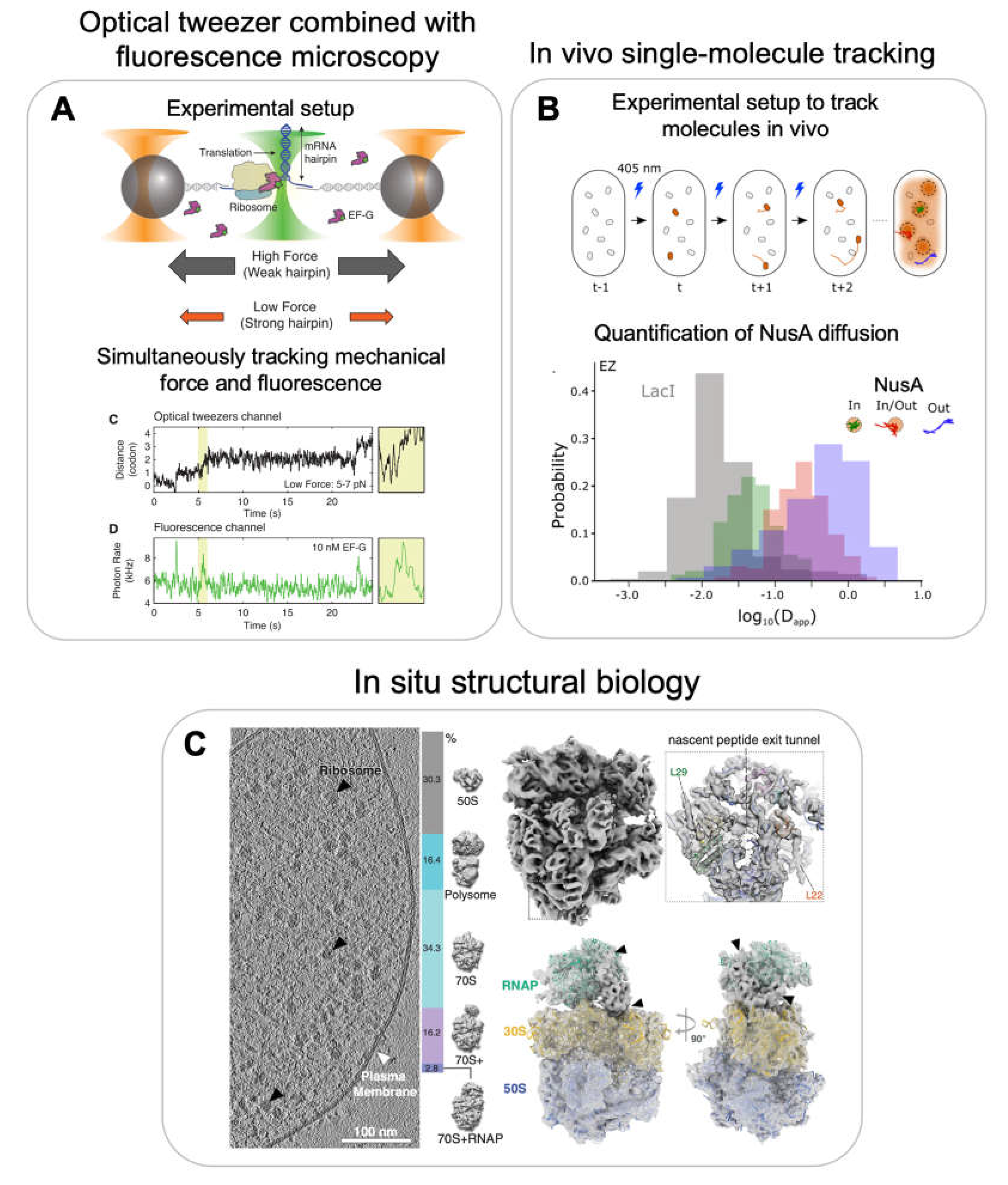
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



