
Submitted:
03 May 2023
Posted:
08 May 2023
You are already at the latest version
Abstract
Despite the fact that strong routine separation methodologies can give reliable specificity and validity at usual working pharmaceuticals concentrations, they may fail at very low concentration levels. This poses considerable challenges for researchers inves-tigating product purity and therapeutic drug monitoring. Sensitivity enhancement pro-cedures are thus required to maximize the performance of separation techniques. Large volume injection, solid phase extraction/solid phase enrichment (SPE/SPEn), pre-, post-, and in-column derivatization, as well as the use of sensitive detection devices are the simplest strategies for improving sensitivity of the separation-based analytical techniques. Large volume injection of samples with online SPE/SPEn coupled with separation techniques increased sensitivity and improved detection as well as quantification limits without affecting peak shape and system performance. Although the primary purpose of derivatization is to improve sensitivity and selectivity, greener derivatization is growing in popularity and should be considered in analytical chemistry. In general, two strategies are essential for accomplishing greener derivatization goals. The first is the search for and use of ecologically acceptable derivatizing reagents, solvents, and reaction conditions. The second is miniaturization and automation of analytical methods. This review discusses significant advances in separation-based analytical techniques, specifically enrichment approaches and detector signal improvement for pharmaceutical quantification in various matrices at very low concentration levels. As a result of improved analytical systems setup in drug assays, the possibility of high-throughput analyses was also highlighted.
Keywords:
separation-based analytical techniques
; large volume injection
; solid phase enrichment
; solid phase analytical derivatization
; packed reactor
; trace analysis
1. Introduction
The development of a sufficiently sensitive procedure ensures the technology's viability for its intended purpose. Despite many advances in chromatographic technologies such as liquid chromatography (LC), gas chromatography (GC) and capillary electrophoresis (CE), sensitivity concerns continue to pose significant difficulties for compounds with poor detection limits. Highly sensitive analytical separation-based techniques play important roles in contamination assessments, environmental analysis and therapeutic drug monitoring (TDM) [1,2]. In recent years, there has been a significant increase in the participation of researchers in the sensitivity improvement of quantitative techniques. In this area, a wide range of approaches have been described.
Analysis of pharmaceuticals in bio-fluids possesses numerous analytical challenges. The medications in a complex matrix such as serum or plasma are frequently bound to the protein’s contents or present as free forms in different ratios. Because it can be used to separate and isolate medications and their metabolites from biological fluids, high performance liquid chromatography (HPLC) has emerged as a very helpful technology. Reversed-phase (RP) HPLC is widely used to separate various pharmaceuticals [3,4,5,6,7,8,9,10,11,12]; however, serum and plasma samples cannot be directly injected onto RP columns due to the presence of high-molecular-mass proteins. Bio-molecules such as proteins may have many affinities mechanisms by which they are adsorbed onto surfaces they encounter because of their complex chemistry and amphoteric nature [13]. Each protein molecule generally contains a large number of hydrophobic moieties, as well as multiple positive and negative charges. Hence, there may be a variety of points of interaction that cause protein to adhere to surfaces. The complexity of such macromolecules can lead to non-specific adhesion to surfaces. This can have an adverse effect on the chromatographic analysis in addition to carryover and instrument problems. As proteins can accumulate and plug the columns and instruments, they could make separation by HPLC troublesome. Thus, the LC system will certainly be affected after direct application of protein-rich complex matrices such as serum and plasma. For instance, the porosity of the columns will decrease if a lot of proteinaceous materials are absorbed onto their surfaces, resulting in increasing backpressure. The quick pressure rise at the column's head brought on by protein denaturation and precipitation. The high concentration of organic solvents in the mobile phases used to elute the tested analytes from the RP columns, particularly the ODS columns, causes protein denaturation and precipitation. Moreover, the analyte's distribution between the solid and mobile phases may be affected resulting in alteration of the capacity factor. Thus, sample preparation is an essential component of analytical technique development, regardless of whether the analysis is for pharmaceutical, bio-analytical, environmental, or other applications. The fundamental purpose of sample preparation is to eliminate or reduce the impact of interferences caused by the many matrix components in complicated samples. Moreover, sample preparation can be utilized to simplify other elements of analysis, such as pre-concentration or derivatization, in order to improve sensitivity or selectivity.
In many circumstances, sample preparation consumes the most time and resources throughout the analytical process. Generally, analysts spend almost two-thirds of their times doing the sample preparation steps during the entire procedure [14]. Additionally, the offline sample preparation approach is anticipated to cause more than one-third of the analytical error [15]. Conventional sample work-up processes in bio-analysis frequently entail a variety of pretreatment stages to purify the protein-rich samples before their loaded into the LC, but three of the most frequent are solid phase extraction (SPE) [3,4], liquid-liquid extraction (LLE) [16,17,18], and protein precipitation [19].
Despite their success, classical methods are generally regarded as having limited selectivity and/or poor recovery, as well as being more expensive for target analyte measurement. The equipment costs and development times of such approaches are only justifiable in situations where large sample throughput over an extended period of time is anticipated, despite the fact that using robotics can enable sophisticated sample preparation to be carried out with great precision and minimal labor cost. Thus, there is a huge need for simplified analytical procedures because the sample throughput in bio-analysis, for instance, in the TDM and pharmaceutical industry, has increased significantly. Therefore, developing new sample preparation procedures to accommodate for the various types of samples and conditions encountered during analysis is a research field that requires continuing development.
2. Solid Phase Extraction/Pre-Concentration Strategies for Drug Analysis
Because of the lower concentration levels of pharmaceuticals and the high levels of interferences present in bio-fluid samples, sample clean-up and enrichment processes are critical prior to chromatographic analysis to optimize technique sensitivity, recovery, and accuracy. It is difficult to justify extraction procedures that employ large amounts of hazardous organic solvents in the sample preparation steps in an era when it is recommended to implement green chemistry principles in analytical laboratories. Of the different purification methods, SPE is a quick, low-cost, and extensively applicable technology. Furthermore, it is broadly applicable for enrichment of pharmaceuticals and extraction of biological interferences with high removal effectiveness. SPE is seen as a good substitute for LLE because it moves past many of the shortcomings of the LLE [16,17,18]. Moreover, the entire procedure can be automated. Besides that, SPE does not require phase separation, as LLE does, which eliminates errors related to inaccurately estimated extract volumes, one of the primary reasons of error observed in the analysis of extracts obtained by LLE. Thus, major efforts have been made to design and evaluate innovative formats and efficient sorbent materials in order to improve their selectivity, specificity and sorptive capacity towards target analytes, and enhance physicochemical or mechanical stability, among other SPE-related properties. Solventless sample preparation approaches based on analyte extraction and enrichment by online SPE, using phosphate buffers as washing solvents, have proven to be viable and environmentally friendly alternatives to conventional solvent extraction techniques [20,21]. Fluconazole in serum has been directly quantified by trapping on a pre-column and subsequently separating on an analytical column utilizing regular on through elution mode by on-line SPE and HPLC method (Figure 1) [21]. Because of its simplicity of use, flexibility, rapid extraction time, safety, minimal organic solvent consumption, and high enrichment factor, solid phase microextraction (SPME) is a promising sample pretreatment approach [22]. To boost sensitivity in separation techniques, large volume injection approach and online sample pre-concentration have been widely employed. Highly sensitive and selective HPLC methodologies with less costly fluorescence (FL) detection system are also required. Thus, the derivatization technique is necessary to enable sensitivity enhancement by converting non- or weakly native fluorescent compounds into highly fluorescence derivatives. The combination of pre- or post-column derivatization procedures with the chromatographic systems, as well as FL detection made it possible for determining different medications at low concentration levels. One of the primary goals of the present review is to achieve greener derivatization developments. As stated, automation and miniaturization are two critical factors in developing greener derivatization techniques. When compared to conventional techniques, the amounts of chemicals needed and wastes generated are reduced. On-column/in-capillary [23,24] derivatization with HPLC and CE are two greener derivatization approaches. Because derivatization takes place during the separation process, these methodologies are superior to the most common pre-column and pre-capillary offline modes of derivatization in that the sampleand derivatizing agent consumption is low, and full automation arises without the need for additional equipment.
2.1. Off-Line Solid Phase Extraction/Enrichment
An advanced sorbents technologies reorient SPE materials with various functionalities according to their structures, such as RP, normal-phase (NP), cation exchange (CEx), anion exchange (AEx), and mixed-mode types. Offline SPE in conjunction with RP-HPLC approach for highly sensitive determination of ciprofloxacin, acetaminophen, caffeine, benzophenone, and irgasan in aquatic environment has been reported [25]. The SPE was conducted prior to the analysis using a RP-C18 cartridge to pre-concentrate the tested analytes from the ecological water samples. Dawson et al. reported a simple and reliable assay for nicotine and its main metabolite, cotinine, in plasma [26]. On the silica columns, an extraction/pre-concentration approach compatible with RP-HPLC separation was devised followed by quantification on ODS column using UV detection. Torre et al. simplified SPE process for determining risperidone and 9-hydroxyrisperidone in human plasma using polymeric RP sorbents [27]. SPE-HPLC approach for assessing melamine in liquid milk has been reported to meet the detection demand for melamine contaminated milk [28]. The developed method was validated using LC tandem mass spectrometry (LC-MS/MS) and has been successfully applied for routine melamine measurement in a variety of milk samples. Also, CEx resin column is utilized for separation and pre-concentration purposes in LC-MS to analyze melamine in egg samples [29]. He et al. and Wang et al. described rapid and efficient SPE procedures followed by HPLC-UV methods for the determination of melamine in aqueous and milk formula samples, respectively [30,31].
2.2. Off-Line Solid-Phase Microextraction
The application of the twelve green chemistry principles to laboratory practice surely fueled the search for innovative methodological approaches to guarantee an improvement in results quality while enhancing environmental friendliness. Since the concept of SPE of target analytes was developed, substantial advancements in this technology have been noted, including the original concept's simplification, automation, and miniaturization. The method put forth in 1951 by Braus and colleagues, which was based on the insertion of up to 1.2-1.5 kg of granular activated carbon into an iron cylinder, is fundamentally different from the SPE formats currently employed in laboratory practice [32]. Because of undeniable advances in both the adsorption process and the large-scale production of new classes of materials, practical solutions for achieving high rates of recovery and enrichment while using significantly less sorbents and organic solvents have become possible for trapping various types of analytes. SPME is presently gaining popularity in a variety of fields of investigation, including dietary, biological, and pharmaceutical products [33]. SPME provides several advantages, including ease of use, low cost, compatibility with analytical systems, automation, and solvent less extraction procedure. In recent years, SPME has been employed prior to LC and CE, in addition to its application with GC. Enrichments of pharmaceuticals from various samples with complex matrices make it necessary to produce unique SPME fiber coatings such as metal organic frameworks, covalent organic frameworks, carbon, polymer, ionic liquids, metal/metal oxide nanoparticles and silica [34,35].
The majority of pyrethroid metabolites are extracted from urine in the literatures using SPE or LLE, before being subjected to GC-MS or LC-MS [36,37,38]. As a pre-treatment procedure to meet the demands for rapid and environmentally friendly extraction protocols, SPME is becoming more important. There have been various SPME methods reported in the literature [39,40], but they are not sensitive enough to measure the trace concentrations of pyrethroid metabolites that are important for evaluating environmental exposure. For the analysis of pyrethroid metabolites in urine samples, a green analytical method by a packed sorbent coupled to large volume injection and GC-MS has been developed using off line SPME technique [41]. With the aid of direct MS technology, quantification of cocaine, methamphetamine, 3,4-Methyl-enedioxy-methamphetamine, and lysergic acid diethylamide from oral fluid and urine samples has been conducted using offline SPME [42].Quinine, naproxen, haloperidol, ciprofloxacin, and paclitaxel have all been extracted using multiple SPME [43]. After SPME, desorption was carried out offline, and each drug was then analyzed using HPLC with UV or FL detection. Cantúet al. reported an off-line SPME technique for analyzing anticonvulsants and tricyclic antidepressants in human plasma for TDM purposes by HPLC-UV [44].
3. Column Switching and Large Volume Sample Injection
The offline SPE techniques utilized different types of organic solvents which have negative impacts on the environment are the most time consuming steps of the whole analytical procedures. The scientists then consider how to simplify and accelerate the measurement process during chromatographic analysis while simultaneously minimizing analytical errors generated by the off line sample preparation techniques. Aside from developing novel efficient sample preparation approach, online coupling of SPE with subsequent chromatographic measurement is another strategy for improving analytical sensitivity and accuracy. The online coupling approach of sample preparation and chromatographic separation has the following benefits: (1) enhancing sensitivity by reducing sample loss; (2) reducing sample preparation process time; (3) lowering contamination of samples and possible analyte degradation due to online sample preparation in a closed system; (4) enhancing process reproducibility and further improving analytical precision and accuracy with fewer human errors; and (5) potentially reducing organic solvent and sample consumables. The online coupling of sample preparation methods with LC may be traced back to the early 1980s, when SPE was hyphenated to LC [45]. Since then, online coupling approaches have gained a lot of attention and have been used in a lot of analytical studies. During the past twenty years, there have been numerous published articles on online sample preparation techniques combined with LC.
Column switching serves a variety of purposes in online SPE and SPME strategies, which are widely used in many automated LC methods. Several successful on-line SPE approaches, followed by HPLC analysis with UV and FL detection systems using an isocratic elution mode have been described [46,47]. These systems were designed with second pumps and six-port switching valves for better methods development. Small changes in the flow scheme of the traditional on-through elution SPE modes by reversing the flowing direction of the mobile phase into the SPE columns in the back flush modes, producing on-line SPE systems with dual functions (i.e., trapping the analytes and separating them from interferences proteinaceous materials prior to the analytical columns) that eliminate interferences and achieve more narrow analytes transferring zones. After inserting large volume of filtered plasma or serum samples into the sample loops, bio-fluid samples were conveyed to the pre-columns by washing mobile phase (without mixing with the analytical mobile phase), and the analytes were attached to the front-ends of the pre-columns due to the low elution performance of the solvent-free mobile phase. Levofloxacin in serum was trapped on the pre-column and then separated on an analytical column using this on-line SPE technique and back flushed elution mode [20] (Figure 2). This peak focusing method based on trapping analytes at the head of the columns using eluents with weak solvents can effectively reduce the eluotropic strength of eluent, allowing analyte to be focused on the stationary phase at the column head. In comparison to the traditional off-line SPE approaches, the entire analytical procedures are completed in few minutes and provide the advantages of full extraction automation, lack of operator interferences, and precise processes control.
3.1. Online Solid Phase Extraction and Solid Phase Enrichment
In the monitoring of pharmaceuticals at the trace levels, detection and quantification limits are very important. Selectivity is also very common additional requirement that may necessitate chromatographic separation. Then, the detectable level will also be substantially influenced by chromatographic variables, especially in the sample pre-treatment procedures. The nature of the samples or analytes of interest greatly influences the choice of sorbent for the solid phase and solvents for the mobile phase. The primary polarities and charge properties of the analytes—whether they are polar, nonpolar, or cations or anions—will aid in determining the proper stationary phases. Pairing this with the appropriate solvent hydrophobicity, depending on whether the analyte must pass through or be retained by the column, will result in the most effective set of SPE sorbents.
Pre-concentration of the drugs and cleaning of complicated biological specimens thus become parts of the chromatographic process and are accomplished through valve switching. As a result, preliminary sample preparation can be simplified. In the early 1970s, the only method for determining trace quantities was to introduce large volumes of sample. Karger et al. addressed the effect of sample volume on the resolution and column efficiency [48]. Little and Fallickproposed a technique termed "trace enrichment" in 1975 for concentrating nonpolar organic compounds from polar sources (river water or food matrices) for quantification analysis by RP-C18 column [49]. Schauwecker et al. published a trace enrichment approach utilizing RP sorbent materials [50]. They employed RP-C8 and RP-C18 to enrich ergot alkaloids and peptides like cyclosporine A and oxytosine from urine samples of 2 ml or more. Huber and Becker used adsorbent silica gel such as LiChrosorb Si-60 as well as RP-C18 with hydrophilic and hydrophobic surfaces to transfer the analyte to a smaller volume than the original one, and obtained enrichment factors of the order of ten thousand for both polar and nonpolar compounds [51]. Lankelma and Poppe employed a pre-column of RP-C8 silica for concentration and an ion exchange (IEx) analytical column for separation of methotrexate (MTX) from plasma [52]. The SPE column was placed at the position of the loop in an injection valve, allowing pre-concentration to be controlled by simple solvent switching. The back flushing mode was employed by this system to reduce accumulating contamination on the trapping column.
Most pre-columns have been packed using silica-based bonded phase sorbent materials, which limit the pH of aqueous solvents that can be pumped through them to be less than 8. Amberlite XAD-2 was an excellent adsorbent for on-line trace enrichment of lonazolac from 100 µL sample volume of plasma specimen at higher pH values [53]. A pre-column packed with the polymeric RP sorbent was connected to a NP-LC system in real time for the analysis of chlorophenol [54]. A large volume (450 µl) of chlorophenol-containing aqueous sample was run through the pre-column. The retained tested compound was desorbed by the NPLC mobile phase, transferred to the separation column, and analyzed after a brief period of nitrogen flushing to remove the leftover water from this column. Wang et al. established an on-line SPE-HPLC technique for simultaneous enrichment and measurement of β-sitosterol in five different edible oil samples by using poly(NMA-ST-co-TAIC-co-EDMA) monolith as absorbent [55]. The first step in studying the glycoproteome may entail glycopeptide enrichment to raise the amount of glycosylated peptides (glycopeptides) [56]. Hydrophilic interaction liquid chromatography (HILIC) was frequently utilized for enrichment of intact glycopeptides based on the hydrophilic characteristics of glycopeptideglycans [57]. HILIC has been shown in many investigations to enrich N-linked glycopeptides (N-glycopeptides) [58,59]. However, it was stated that HILIC could not be a viable enrichment approach for O-linked glycopeptides (O-glycopeptides) [60,61]. It was reported that employing columns containing materials for strong AEx chromatography increased the yield and improved identification of N- and O-linked glycopeptides. Isobaric tag labeled glycopeptides could be efficiently enriched by AEx cartridges allowing quantitative measurement of glycoproteomics [62].
3.2. Immobilized Metal Affinity Chromatography
Immobilized metal affinity chromatography (IMAC) was originally designed for protein affinity purification using the interactions of histidine and cysteine residues with the IMAC resin [63,64], but the binding of phosphoproteins and phosphoamino acids to metal ions reported by Andersson and Porath added a new dimension to this approach [65]. The IMAC technique was extended by Neville et al. to the enrichment of phosphopeptides derived from proteolytically degraded proteins [66]. Because many of the proteins targeted for phosphorylation are low in quantity, phosphopeptides enrichment prior to MS analysis is thus essential for their identification. Since then, the IMAC approach has been widely utilized to enrich phosphorylated peptides prior to MS analysis and sequencing [67,68,69,70,71]. Immobilized titanium ion (Ti(IV)) affinity chromatography (Ti(IV)-IMAC) has been designed for enriching phosphopeptides [72]. An IMAC adsorbent has been successfully applied for the analysis of mouse liver phosphoproteome via zirconium (Zr(IV)) chelation to the phosphonate-modified poly(glycidyl methacrylate-co-ethylene dimethacrylate) polymer beads [73]. Monodisperse microsphere-based IMAC resins with flexible linker endings with phosphonate groups that bind either Zr(IV) or Ti(IV) ions have proven to be an efficient method for phosphopeptides enrichment [74]. IMAC method has also been utilized for the comprehensive analysis of salivary phosphoproteome [75]. The necessity for effective sample preparation methodologies that allow for in-depth investigation of protein phosphorylation has resulted in the development of a number of selective affinity techniques that allow for the enrichment of phosphopeptides from highly complicated peptide mixtures. Metal oxide affinity (MOA) materials have grown in popularity, and MOA chromatography (MOAC) is currently one of the most extensively utilized phosphoproteomics techniques [76]. Hybrid materials of IMAC and MOAC were successfully manufactured for the enrichment of phosphopeptides, with the goal of combining their benefits for enriching both mono- and multi-phosphorylated species [77]. The use of MOAC in combination with metal hydroxide (Al(OH)3)has been reported for the selective and effective enrichment of phosphorylated proteins and peptides [78].
3.3. Immobilized Protein Reversed Phase Columns for Solid Phase Extraction/Enrichment
Direct injection of biological fluids including proteins is thought to impair the performance or properties of the analytical column in conventional HPLC. Off-line deproteinization is hence the first conventional step in HPLC analysis. These procedures, nevertheless, can occasionally be cumbersome and aren't always reproducible. It is desirable to develop an HPLC method that can directly analyze biological samples comprising proteins for the sake of simplicity and reproducibility of chromatographic analysis. As mentioned briefly in several research articles [4,5,46,47,79,80,81,82,83,84], RP materials coated with denatured plasma proteins ("protein-coated (PC) columns") exhibited RP properties for hydrophobic small molecules even though they lost their affinity for plasma proteins. Protein immobilization integrates various chromatographic modes in packed porous particles (most commonly, a size exclusion mode caused by small pores as well as nonretentive outer layers with a RP mode of a retentive inner pore surface) to accommodate matrix’s macromolecular components extraction and analytes enrichment prior to HPLC separation methodologies.
Although the magnitude of the direct large-volume injection is required for measuring medications at very low concentration levels, it introduces large amounts of interfering components. As a result, adsorption onto SPE using small PC RP pre-columns (PC-RP-pre-columns) is the technique of choice for concentrating analytes from bio-fluid samples while removing the majority of interfering components. A PC-columns methodology was successfully employed to conduct on-line SPE that, when combined with HPLC could determine various drugs in serum and plasma. The components in the plasma and serum samples must be compatible with the chromatographic phases when designing HPLC systems for direct injection of these matrices. PC-RP-pre-columns are compatible with serum and plasma matrices in the presence of phosphate buffer mobile phases, overcoming the limits of conventional native RP sorbent columns. As a result, the PC-column is an effective SPE tool for pre-concentrating pharmaceutical compounds from large-volume of bio-fluid samples without the need of organic solvents. Protein immobilization on RP silica sorbents as a pre-column considerably protects the analytical column's efficiency for TDM; however, the most straightforward signal-column technique is still helpful in many situations [85]. The signal-column approach has been applied to analyze bupivacaine in human plasma. The use of a single column chromatography has further simplified sample preparation by obviating the necessity for a second pump, pre-column and switching valve.
3.4. Restricted Access Media Solid Phase Extraction/Enrichment
Restricted access materials (RAM) or media are another type of SPE. This type of SPE is composed of porous materials that can limit macromolecule accessibility from the sample to the active sites of the SPE materials. RAMs have beneficial synergistic separation capabilities in that their macromolecular size exclusion features are based on characteristics of the hydrophilic outer surface of the silica resin particles, but its varied retention characteristics for smaller molecules arise from varying modifications on the inner surface of the silica [86,87,88,89,90,91,92]. These separation capabilities can be accomplished through (1) size exclusion, (2) size exclusion combined with hydrophobic interaction, (3) size exclusion combined with hydrophilic interaction, and (4) size exclusion associated with IEx interaction.
On-line SPE approaches with RAM columns have been employed for the extraction and enrichment of small analytes in bio-fluid samples. In this instance, direct high throughput online analysis of biological materials is made possible by RAM combined with an LC system. This can achieve by direct injection of large volume of biological samples while minimizing the process of clean up and extraction [93,94,95,96]. Pharmaceutical, clinical, omics, and toxicological research always encounter the challenge of trace analysis of medications as well as biomarkers in biological matrixes. Given the physicochemical characteristics of the aforementioned analytes, MS coupled with separation techniques has emerged as the most sensitive and selective analytical tool for their measurement in the complex samples. For the extraction, pre-concentration and cleanup of numerous endocrine-disrupting chemicals in honey, a strategy based on the combined use of RAMs and polymeric SPE sorbents was developed. Following online sample preparation, the analytes were separated and analyzed using CE-MS [97].The potential of the RAM column to enrich benazepril hydrochloride in plasma online was investigated using a column switching technique coupled HPLC-UV. This system was made up of a RAM column as an enrichment column and a RP-C18 as an analytical column [98]. Zhang et al. [99] and Wu et al. [100] developed and optimized column-switching HPLC-UV approaches for the quantitative measurement of rifampicin in rat plasma samples utilizing RAM columns. In the single column configuration, the RAM column was connected directly to a detector. Following injection, the analytes were eluted from the column and transferred directly to the detector. When used in conjunction with MS detector, the single column technique has more potential applications because of the high selectivity of this detector compared to UV, or FL detectors. RAM-MS approach, where RAM column directly connected to MS systems, has been developed for the analysis of fluoroquinolones [103].
3.5. Surface Modified Restricted Access Media
RAMs, as previously indicated, have sparked considerable attention in the field of SPE as specific types of materials [104] with internal phases that hinder small molecules and external hydrophilic layers that eliminate proteins. Protein exclusion is frequently achieved through the use of physical barrier layers based on pore size or a chemical diffusion barrier composed of hydrophilic polymer networks. The modification of adsorbent surfaces with hydrophilic polymers for the manufacture of modified RAMs has received more attention [105,106,107,108]. Several kinds of RAMs as SPEs have been designed that utilized different functional groups on the solid particles' outer surface. To obtain distinct matrix exclusion capabilities, bovine serum albumin (BSA) has been employed as a protein functional group bound on the outer surfaces of RAM (RAM-BSA) [109,110,111,112]. RAM-BSA columns exhibit good protein exclusion efficiency and suitable sorption retention for acidic, neutral, and basic chemicals [113]. In terms of removing specific matrix components, the RAM-BSA trap column outperforms other RAM, notably in the analysis of milk-based specimens. Lopes et al. [114] successfully demonstrated the utilization of a RAM-BSA trap column for measuring carbamazepine and its metabolites in human milk. Human milk and other dairy products have special difficulties not encountered in plasma or serum. In addition to protein matrix components, milk, like other food-type matrices, has a high lipid composition. Lipids may gain access to the RAM's inner layers and hinder the effective retention of the tested small-molecule analytes. Surface modified RAM can overcome this problem. To ensure that the human milk protein content was efficiently removed by the RAM-BSA trap column, an in-depth matrix impact evaluation was investigated [114]. Aliquots of 500 µL of milk sample were diluted with water to a final volume of 2 mL and then injected through the trap column at a flow rate of 1.0 mL/min. To quantify protein concentration, Bradford's technique was applied to the collected eluent fractions. It was revealed that protein removal of 92% could be accomplished in 3 minutes. Moreover, protein standards of β-casein and ɑ-lactalbumin, which are the principal protein components of human milk, were introduced into milk samples to evaluate the RAM-BSA column's molecular-mass cutoff. The tested protein standards were eluted from the trap column in 1.5 minutes. RAM-BSA extraction efficiency ranged from 78.2 to 105.3% for β-casein and ɑ-lactalbumin, respectively. Protein coating RAM phases are also made up of a protein-like component, such as acid glycoprotein (AGP), which is covalently attached to the sorbents (RAM-AGP). As a result of this covalent attachment, the protein network on the particles' external hydrophilic surface renders RAM-AGP compatible with proteinaceous samples that cannot fit through the tiny pores. Online LC-MS was developed to determine free non-cholesterol sterols in human serum using RAM-AGP [115]. A multidimensional HPLC method for the simultaneous determination of sulfamethoxazole and trimethoprim in whole eggs samples with UV detection has been described, using a RAM-BSA column in the first dimension and an analytical ODS column in the second dimension [116].
3.6. Restricted Access Media Molecularly Imprinted Polymers
The use of typical molecularly imprinted polymers (MIPs) for bio-fluid sample analysis is a problematic technique because of the presence of high concentration levels of proteins, which can block selective binding sites, reduce adsorption capabilities, and affect analytical validation. Modifications of typical MIPs have been developed to provide them with the capacity to eliminate macromolecules. Surface coatings based on hydrophilic groups and/or proteins have been the key methods in the development of restricted access MIPs (RAMIPs). Thus, RAMIP is another good sort of RAM that combines restricted access with molecular recognition, similar to MIP [117]. The interior part of the porous particles may be hydrophobic while having a hydrophilic surface and a relatively narrow pore size that prevents macromolecular compounds from entering the inner pores. These properties are responsible for the retention and isolation of low molecular weight molecules from complicated samples. Thus, RAMIPs have been successfully employed to selectively extract and enrich small molecular analytes from untreated bio-fluid matrices (e.g., plasma, serum, and milk) without the necessity of pre-deproteinization. An online RAMIP coated with BSA (RAMIP-BSA) connected to an LC-MS system has been developed for the simultaneous measurement of serum bile acids as well as their taurine and glycine conjugates [118]. Restricted RAMIP coupled with MS detector [101,102], where RAMIP column directly connected to the detection system, has been developed for measuring tricyclic antidepressants.
3.7. N-Rich Twin-Column Continuous Chromatography
Isolation of product-related impurities prior to characterization is currently performed utilizing inefficient and time-consuming procedures. The use of linear scale RP or IEx chromatographic technique, either employing analytical LC or preparative systems coupled to fraction collectors, is a standard strategy [119,120]. These techniques rely in the linear range of the adsorption isotherm, achieving great resolution and minimizing components overlapping, and yielding high purity outcomes. This, however, comes at the expense of a limited injection volume per cycle. Thus, very low final target concentrations, and longer processing durations due to the necessity to pool target material from numerous chromatographic runs were stated. Given the limitations of the conventional methods, we might potentially alleviate bottlenecks by using the N-Rich process, a twin-column continuous chromatography technology, to provide larger amounts of high purity product-related impurities in a short operation time [121,122,123]. This approach has already been employed to successfully concentrate antibody isoforms and peptide impurities. Thus, continuous chromatography was becoming more popular among industrial separation professionals as a scalable manufacturing technology capable of minimizing bottlenecks in biopharmaceutical downstream processing applications such as capturing and polishing [124,125,126,127]. N-Rich is relatively promising automated technique that enriches and purifies a desired molecule from a complicated mixture utilizing two columns. Common resin materials can be employed for N-Rich separations such as IEx, hydrophobic interaction (HIC), or RP chromatography (e.g., ODS). N-Rich can be configured to target a single molecule or a region of the chromatogram that contains multiple molecules. The first phase (startup) starts by loading feed material into the first column and eluting it using a linear gradient. During the second phase (enrichment), a section of the chromatogram containing target impurity is moved from the first column and re-adsorbed onto the second column using in-line dilution. Any fraction not intended for recycling, including the primary products as well as additional side-impurities, can be dumped in waste or separately collected for other purposes if worthwhile. Meanwhile, fresh feed is loaded onto the second column alongside the recycled target. This procedure causes the target molecules to be enriched in comparison to the other molecules in the mixture. The process step is repeated cyclically between the two columns, raising the concentration of the target impurities progressively. The third phase (depletion) is a single switch with no fresh feed added. Before the final elution phase, this step depletes non-target molecules while internally recycling the accumulated target impurities. The depletion process significantly increases final quality of impurities that elute with the product peak. Finally, in the phase four (final fractionation), the pre-concentrated target molecule is eluted using a shallow gradient over two columns in series, and then recovered using fine fractionation. The N-Rich technique improves the resolution of enriched compounds and recovers pure target material at higher concentration levels than batch methodology. Monoclonal antibodies are important biopharmaceutical medications with a high commercial value [128]. Other monoclonal antibody heterogeneities, including enzyme catalysis degradation and modification, could, however, be produced throughout the production process [129]. Jing et al. developed an innovative twin-column continuous chromatography method for isolating and enriching monoclonal antibody charge variants [130]. When compared to typical analytical and preparative chromatography techniques, Richard and Thomas found that N-Rich approach performed better for isolating Angiotensin II peptide contaminants [131].
4. Enhancement of Sensitivity through Chemical Modification
Chemical derivatization is known to be employed for various groups of analytes to enhance their detectability as well as stability and improve chromatographic selectivity. The derivatization reaction should be selected with the purpose of analysis and the intended method of detection. Derivatizations are commonly used to increase the detectability of analytes by introducing chromophores and fluorophores into weakly detectable compounds. These choices are influenced by different processing variables, including the target compounds' functional group(s), the complexity of the sample matrix, the detection technique, and the quantity of resulting derivatives. Kinetic variables and derivatization reaction mechanisms play important roles on selectivity and can influence statistical values such as precision and accuracy [132]. The frequency of samples is also an important issue for such modification [133]. Another point of view is the experimental framework in which the derivatization is performed. Taking into account that bio-analytical chromatographic-based methods generally have difficult tasks in the isolation and concentration of target analytes from complex matrices. Thus, chemical modification of the target analytes is depending on matrices removal or analytes extraction. Derivatization can take the form of simple reactions between analytes and reagents, in which functional groups are substituted or condensed using various reactants. Derivatization can also be conducted through complex routes involving many functional groups, as a result of many successive reactions involving the elimination of parts of small molecules and the formation of new moieties. By enhancing the method's selectivity, interference from components that chromatographic separation cannot adequately separate can be avoided. Pre-column [134,135], post-column [136], online, and offline settings can all be utilized for chemical modifications. Although pre-column derivatization approach is simple, it can alter the performances of some chromatographic components. Furthermore, pre-column derivatization errors are more likely to occur since the reactions take place in samples matrices. As a result, post-column derivatization is frequently chosen. Post-column derivatization has been carried out online in segmented flow reactors, packed bed reactors, and open tubular reactors [137]. The design and functionality of these kinds of chemical reactors for chromatography have been addressed in a variety of studies. The open tube reactor, which consists of a small tube through which the effluent-reagent mixture passes, is the most popular. The most serious concern when employing any type of post-column reactor is the additional peak broadening that happens within the reactor itself. A well-designed post-column reactor should be implemented in order to decrease the inevitable loss of sharpness due to the peak broadening.
To produce specific reaction products using a flow reactor, several conditions must be fulfilled: (1) the detecting systems shouldn't be hampered by solvents and chemicals; (2) the reaction kinetics must be rather quick to avoid high peak broadening brought on by large capacity reactors; (3) the reaction yield should be high in order to achieve a low detection limit; and (4) the reaction product should be stable over the course of the experiment. Offline post-column chemical reactions are possible by fractionally collecting the column eluates, applying derivatization agents, and quantifying the reaction products.
Although the primary goal of derivatization is to improve sensitivity, selectivity, and detectability, greener derivatizations are receiving significant attention in analytical chemistry [138]. The reason for this concern is that the majority of regularly used derivatizing reagents are corrosive, persistent, and poisonous. Furthermore, typical derivatizations require unusually large amounts of derivatizing reagents and solvents and are time-consuming. When converting analytes into highly detectable forms is becoming a fundamental component of the analytical methodology, it is necessary to focus on the green analytical chemistry (GAC) issue and conduct a series of experiments in accordance with green chemistry and GAC [139], to provide the ideal environmental conditions for analyte modification. However, derivatization is explicitly forbidden by GAC's sixth principle. In general, two strategies are critical for achieving the goals of greener derivatizations: searching for and employing of environmentally friendly derivatizing reagents, solvents, reaction conditions, and energy sources, and miniaturization as well as automation of the analytical technique [140]. Because of the advancement of automated and/or miniaturized technology, concerns about additional procedures and time requirements are not always to worry about [141]. As a result of this strategy, reagent waste is reduced, and so the amount of waste generated is reduced. The use of microextraction methods in association with derivatization effectively fulfills the requirements.Other strategies to conduct derivatization processes in a "green" manner involve the use of environmentally friendly solvents and reagents, the use of eco-friendly variables such as microwaves, UV radiation, and ultrasound. It is obvious that using analytical derivatizations correctly can result in improved sensitivity and selectivity. Certain instrumental setups are another option for greening the derivatization technique. On-column or in-capillary derivatization [142,143,144,145] using LC and CE, respectively, are of utmost significance in such cases. In these circumstances, the derivatization process occurs during the separation step, making them superior to the most common offline derivatization modes because sample and derivatizing agent consumption is low, and full automation occurs without the requirement of additional equipment [146]. Furthermore, in-port derivatization (introduction of sample and derivatization agent in the injection port), which is primarily used in GC, allows for simplified sample preparation. It also reduces solvent consumption, as well as avoids hazardous circumstances and waste generation [147].
4.1. Solid Phase Analytical Derivatization
High sensitivity, high sample frequency, ease of use, and automation are all criteria for modern analytical procedures, in addition to statistical characteristics like accuracy and precision. Although some of these needs are met by instruments, sample preparation prior to instrumental analysis is still a work in progress. Despite the additional reagent stages, analytical derivatization (AD) of the analyte during sample preparation can significantly increase chromatographic selectivity for good separation as well as sensitivity for measurement of analytes at low concentration levels [148]. Even with the most modern detector, MS, AD boosts sensitivity by one to three orders of magnitude [149]. This technique's application is limited since it requires an additional step in sample preparation that might be time consuming. Despite this disadvantage, the benefits of AD prompted efforts to simplify, minimize the solvent usage, and automate the analytical technique [150,151].
SPEs are commonly used sample preparation protocols for determining analytes from bio-matrices using chromatographic techniques; however, they can be used as solid-phase analytical derivatization (SPAD) tools to produce highly detectable species. Extraction and derivatization are combined into a single process by SPAD approach simplifying sample preparation. The solid-phase retains both reagents and derivatives and frequently allows for the simple separation of excess reagent or selective elution of the target products. SPAD is a promising technique for determining a wide range of pharmaceuticals in complex bio-fluid and environmental samples. SPAD has grown in popularity due to its minimal organic solvent usage, simplicity, low price, and high efficiency [152]. As time becomes more crucial in sample preparation procedures, more emphasis on automation can be expected in SPAD method development. More SPAD variations are expected in analytical chemistry literatures as a result of immerging derivatizing reagents. The previous attractive characteristics will ensure SPAD's importance as a technique for preparing samples in both research and industrial settings. SPAD is also classified as a microextraction/derivatization process and could achieve hydrophobicity modification [153]. Analytes are transformed into their derivatization products on the surface of adsorbents or at the liquid-solid interface using the SPAD approach.
Two recognized lung cancer biomarkers, hexanal and heptanal, have been analyzed in human urine samples by HPLC-DAD using a combination of derivatization and bar adsorptive microextraction for sample preparation [154]. 2,4-Dinitrophenylhydrazine (2,4-DNPH) as a derivatization agent was adsorbed on the surface of the bar containing cork powder as a SPAD for the determination of the tested aldehydes. Another approach for determining hexanal and heptanal in human urine by HPLC-DAD utilizing 2,4-DNPH adsorbed onto the surface of magnetite/silica/poly(methacrylic acid-co-ethylene glycol dimethacrylate) has been reported [155]. The structures of the compounds arising from SPAD approach include diverse chromophores, fluorophores, and moieties for facilitating mass spectral identification [156]. The reaction products are removed from the surface thermally or by a solvent after derivatization for column chromatography. The entire technique is typically automated, leading to a high-throughput capacity for bioanalysis. Derivatization of urine samples using 2,4-DNPH impregnated on a LiChrolut EN SPE column yielded highly detected low-molecular-weight aldehydes for analysis by LC-MS/MS [157]. Xia et al. designed a one-step membrane protected micro-SPE and derivatization approach for selectively determining trace aliphatic aldehydes from complex cosmetic and food samples using HPLC and DAD [158].
4.2. Solid-Phase Permethylation
Solid phase premethlation (SPPM) technique has been widely employed for carbohydrate derivatization, particularly with complicated oligosaccharides and glycolipids. SPPM produces extremely permethylation of low amounts of glycans produced from glycoproteins found in biological materials to improve MS analysis at very low concentration levels [159,160,161,162]. This permethylation approach has also been used to characterize sulfated glycan structures [163,164].
Ginseng oligosaccharide extracts have been permethylated using a SPPM technique before being analyzed using LC-MS [165]. The permethylation step was carried out by injecting 400 µL of sample solution into the prepared SPPM column, then washing with 800 µL of dimethyl sulfoxide and collecting the eluent in a centrifuge tube. The reaction was then stopped using a 5% acetic acid solution. Dichloromethane was used four times to extract permethylated oligosaccharides followed by evaporated. For MS measurement, residues were reconstituted in acetonitrile/water (1:1). The potential of miniaturizing SPPM for permethylated N-glycans obtained from model glycoproteins and human blood serum has been investigated with the aim of minimizing sample loss and yielding more sensitive results through MS study. Permethylated N-glycans were purified online utilizing RP-C18 trapping column prior to LC-MS/MS analysis. The feasibility of this arrangement and its ability to be used for glycomics can allow for high-throughput analysis [166].
4.3. Packed Oxidant Reactors
As part of pharmaceutical quality control, determining low drug levels is a crucial concern. The development of flow injection analysis (FIA) methods for such analysis is possible with the proper pre-concentration strategy, and using appropriate detection systems. The primary drawback of all continuous FIA systems [167,168,169,170,171] is their high reagent consumption, which occurs even when there is no sample in the instrument. Figure 3 illustrates three channels (1, 2, and 3) FIA system operating continuously for measuring MTX in dosage forms. As a result, innovative solutions have been developed to reduce reagent consumption and waste production while maintaining analytical performance. Solid-phase reagents (SPRs) can be effectively exploited for chemical derivatization in flow analysis [172]. The strategy relies on the reproducible experimental conditions, which ensure a reproducible analyte interaction with the SPRs. Advantages include a low reagent consumption (only the amount needed for the reaction is consumed), avoidance of dilution effects, and manifold simplification. Increased reaction rate due to SPR excess, the ability to use sparingly soluble reagents, and the existence of additional reactors in the form of solid particles are all advantages.
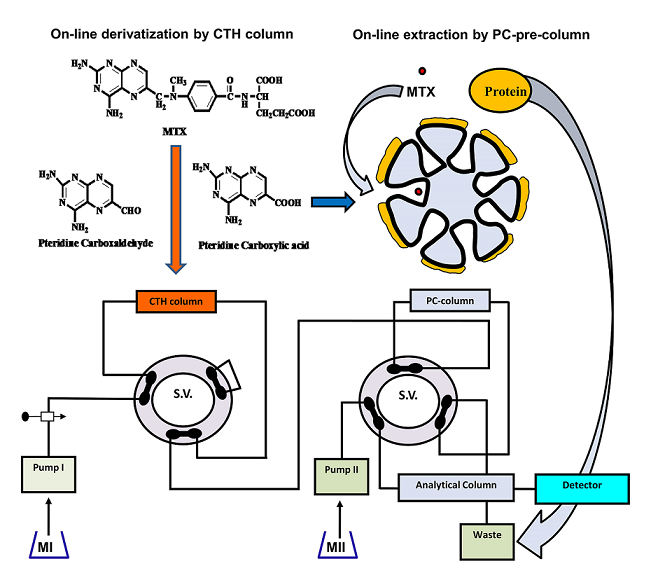
While it is beneficial to react each sample with a fresh amount of reagent, this technique is not always practical if the reagent is only obtainable in solid form. In this situation, the reagent's solid particles should be packed in a small column through which the sample zone passes, transported by a stream of suitable carrier. Particles are employed instead of soluble reagents because they are more convenient and affordable, or because the reagents are not available in soluble forms, such as metallic zinc reductant [173]. For the purpose of determining nitrazepam in pharmaceutical tablets, a simple and fast reverse FIA system has been proposed [174]. This system included a solid-phase reactor comprising PbO2 and spectrophotometric detection. Phloroglucinol reagent was subjected to oxidation using PbO2 immobilized in a polymeric matrix as part of the procedure, which was then paired with reduced nitrazepam in aqueous medium. At 530 nm wavelength, measurement was conducted for the resulting pink product. Green analytical techniques for measuring folic acid (FA) and MTX have been developed and validated using on-line derivatization in conjunction with column switching and a RP-C18 short column for SPEn [175,176]. The procedures employed a packed reactor containing cerium (IV) trihydroxyhydroperoxide (CTH) to oxidatively break down FA and MTX into highly fluorescent 2-amino-4-hydroxypteridine-6-carboxylic acid and 2,4-diaminopterdine-6-carboxylic acid, respectively (Scheme 1 and Scheme 2), then enriching the reaction products on short RP-C18 columns. A 400-μL aliquot of FA and MTX samples was loaded into the injection valve and injected into phosphate buffer. FA and MTX moving zones flowed through the CTH packed reactor at a rate of 0.25 mL/min (pump I). The oxidative cleavage of FA and MTX occurs during the movement of the drugs-containing phosphate buffer through the CTH column. The fluorescent products were pre-concentrated by flowing them from the packed oxidant reactor to the short RP-C18 column head. The valve was switched to position B after 4 minutes (Figure 4). At this position, a green mobile phase with a 5:95 (v/v) ratio of ethanol and phosphate buffer could pass through the short RP-C18 column, where the fluorescent products of FA and MTX were eluted in a back-flush mode to the fluorescence detection system. The flow rate was kept constant at 1 mL/min, and the fluorescence intensity of the eluting compounds was measured at 463 and 367 nm for emission and excitation, respectively. The valve was switched to position A five minutes after injection. This packed reactor approach avoids costly method by allowing the sample zones to easily move into the CTH reactor while the remainder of the system is flooded with phosphate buffer. The CTH packed reactor also offers improved mixing conditions and manifold simplifications. The CTH solid reactor was chosen for the derivatization of FA and MTX in accordance with previous studies of MTX since this reagent demonstrated good reproducibility [177]. Although the conventional FIA technique is promising for measuring FA in pharmaceutical formulations, the implementation of on-line SPEn with FIA-manifold offers enhanced sensitivity.
Emara et al. also used the CTH packed reactor for pre-column oxidation combined by column switching with RP-C18 analytical column for sample enrichment and separation to analyze MTX and FA [178,179]. In these studies, the automation has been expanded by incorporating online oxidation and pre-concentration steps before the HPLC separation of the tested analytes using column switching technique (Figure 5). The separations were conducted at room temperature with an environmentally friendly mobile phase composed of ethanol and phosphate buffer in a 10:90 (v/v) ratio for MTX and a 90:10 (v/v) mixture of phosphate buffer and acetonitrile for FA.
Other study has been attempted for the quantitative analysis of MTX in bio-fluid sample using on-line pre-column derivatization coupled with LC technique [180]. CTH was used as a packed oxidant in this study. To achieve minimal background fluorescence signals, as many endogenous plasma constituents as possible must be removed. As a result, employing a precipitating agent like acetone to separate proteins from plasma samples was essential. This approach allowed for the HPLC measurement of plasma MTX in 14 minutes. According to the obtained results, on-line oxidative cleavage with florescence detection was six to eight times more sensitive than UV detection.MTX in human plasma was also measured by HPLC with the integration of on-line pre-column derivatization and automated SPE utilizing packed oxidant and PC-RP-pre-columns, respectively (Figure 6) [181]. The goal of this work was to reduce analytical errors by developing a direct injection chromatographic technique for derivatization, extraction, separation, and quantification of the tested analyte in plasma. Two solvent-delivery pumps comprised the system manifold. A manual injector was used, as well as two switching valves. The time programmes for flow direction switching and isocratic elution were controlled by a sequence programmer. This system was equipped with a CTH column for oxidative-cleavage of MTX into highly fluorescent 2,4-diaminopteridine-6-carboxaldehyde and the corresponding carboxylic acid derivatives, as well as a short PC-RP-C18 pre-column for deproteinization and trapping of the fluorescence derivatives. The trapped compounds were desorbed from the pre-column and isocratic eluted onto the end capped RP-C18 analytical column for further separation and quantification.
4. Conclusions
In this review, we have summarized the development of sample enrichment techniques prior to separation-based analytical methods, and attempted to provide a comprehensive overview of two strategies, large volume injection and analytederivatization. Online SPE/SPEn approaches are gaining huge interest because they integrate several sample preparation steps into a single step, demonstrating rapid, simple, solvent-saving and sensitive separation techniques. Additionally, the use of PC-RP-pre-columns, especially in the online SPE technology, can allow rapid investigation of optimal extraction conditions in a small number of analyses, resulting in quick, solvent-saving, and hence environmentally friendly quantification procedures. The versatility and capability of the online separation-based analytical techniques using powerful detection systems like FL and MS are also key benefits.
Because of the benefits it gives for improving sensitivity and selectivity, derivatization is still frequently utilized in bio-chromatographic analysis. Derivatization is not always desired; however, it is usually required to overcome the problem for measuring target analytes at low concentration levels from bio-matrices. Although derivatization complicates the analytical technique by adding more steps to the sample preparation process,it is obvious that employing analytical derivatization correctly results in improved sensitivity and selectivity. As GAC has become a popular concept nowadays, it requires analytical chemists to develop techniques and tools, particularly those with a high degree of automation, that take advantage of increased sensitivity and specificity while reducing or eliminating the frequent downsides of derivatization. Due to advances in automated and/or miniaturized of the packed reagent technology, there is no longer need to worry about additional procedures and time requirements. Thus, miniaturization and automation are two important factors to consider while designing a "green" procedure that employs packed reactor derivatization for analysis. As a result of this approach, reagent waste is reduced, lowering the amount of waste generated. Many applications focusing on combined techniques of online derivatization and online enrichment have also emerged to significantly improve sensitivity in HPLC.
5. Challenges and Perspectives
The primary goal and challenge of the separation-based analytical techniques is to maximize sensitivity while minimizing analysis times. When analyzing large numbers of samples or when sample measurement demands the employment of multiple techniques and procedures, boosting analysis speed is also crucial. The second is the cleanliness after preparing the complex sample matrices. For SPE of matrix’s interferences and SPEn of analytes as well as measurement, highly selective extraction supplies and procedures are thus required. The third consideration is the importance of environmentally friendly strategies, which necessitate reducing or eliminating the usage of organic solvents. More SPE tools have to be designed to meet the requirement of environmental friendliness, and solvent-free SPE/SPEn techniques. Hyphenation technology, such as HPLC-FIA, represents a promising future route towards integrated and robust approaches for quick analysis. Deep advancements in such hyphenation techniques are undoubtedly required to boost sensitivity and selectivity in many different areas of pharmaceutical analyses.
Author Contributions
H.F.: conceptualization and supervision; H.E., H.S. and M.M.: writing—original draft and investigation; M.K.: N.I. A.H. and W.Z.: writing—original draft and English editing; A.E. and S.E.: conceptualization and supervision. All authors have read and agreed to the published version of the manuscript
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the manuscript.
Conflicts of Interest
The authors declare no conflict of interest
References
- Aydin, D.C.; Pineres, J.Z.; Al-Manji, F.; Rijnaarts, H.; Grotenhuis, T. Direct Analysis ofAromatic Pollutants Using anHPLC-FLD/DAD Method for Monitoring Biodegradation Processes. Anal. Methods 2021, 13, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Chiriac, U.; Rau, H.; Frey, O.R.; Röhr, A.C.; Klein, S.; Meyer, A. L.; Morath, B. Validation and Application ofan HPLC-UV Method for Routine Therapeutic Drug Monitoring of Dalbavancin. Antibiotics 2022, 11, 541. [Google Scholar] [CrossRef] [PubMed]
- Al-Sanea, M.M.; Gamal, M. Critical Analytical Review: Rare and Recent Applications ofRefractive Index Detector in HPLC Chromatographic Drug Analysis. Microchem. J. 2022, 178, 107339. [Google Scholar] [CrossRef]
- Marzouk, H.M.; Rezk, M.R.; Gouda, A.S.; Abdel-Megied, A.M. A Novel Stability-Indicating HPLC-DAD Method for Determination of Favipiravir, a Potential Antiviral Drug for COVID-19 Treatment; Application to Degradation Kinetic Studies and In-Vitro Dissolution Profiling. Microchem. J. 2022, 172, 106917. [Google Scholar] [CrossRef] [PubMed]
- El-Gindy, A.; Emara, S.; Mesbah, M.K.; Hadad, G.M. Liquid Chromatography Determination ofCitalopram Enantiomers Using beta-Cyclodextrin as a Chiral Mobile Phase Additive. J. AOAC Int. 2006, 89, 65–70. [Google Scholar]
- El-Gindy, A.; Emara, S.; Mesbah, M.K.; Hadad, G.M. Liquid Chromatography and Chemometric-Assisted Spectrophotometric Methods for the Analysis of Two Multicomponent Mixtures Containing Cough Suppressant Drugs. J. AOAC Int. 2005, 88, 1069–1080. [Google Scholar] [CrossRef]
- Hadad, G.M.; Emara, S.; Mahmoud, W.M.M. Optimization and Validation of an LC Method for the Determination of Cefdinir in Dosage form and Human Urine. Chromatographia. 2009, 70, 1593–1598. [Google Scholar] [CrossRef]
- EL-Gindy, A.; Emara, S.; Shaaban, H. Stability-Indicating Method for Determination of Oxyphenonium Bromide and its Degradation Product by High-Performance Liquid Chromatography. J. AOAC Int. 2007, 90, 1250–1257. [Google Scholar] [CrossRef]
- Mostafa, A.; El-Gindy, A.; Emara, S. Development, Application and Validation of RP-HPLC Method for the Simultaneous Determination of Butamirate Citrate and its Main Degradation Product in Pharmaceutical Dosage forms. Anal. Methods. 2011, 3, 1643–1651. [Google Scholar] [CrossRef]
- El-Gindy, A.; Emara, S.; Mostafa, A. HPLC and Chemometric-Assisted Spectrophotometric Methods for Simultaneous Determination of Atenolol, Amiloride Hydrochloride and Chlorthalidone. Il Farmaco 2005, 60, 269–278. [Google Scholar] [CrossRef]
- Hadad, G.M.; Salam, R.A.A.; Emara, S. Validated Stability—Indicating HPTLC and HPLC Methods for Determination of Pipazethate and its Degradant. J. Liq. Chromatogr. Relat. Technol. 2011, 34, 1850–1869. [Google Scholar] [CrossRef]
- Hadad, G.M.; Emara, S.; Mahmoud, W.M.M. Development and Validation ofa Stability-Indicating RP-HPLC Method for the Determination ofParacetamol with Dantrolene or/and Cetirizine and Pseudoephedrine in Two Pharmaceutical Dosage forms. Talanta 2009, 79, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Rabe, M.; Verdes, D.; Seeger, S. Understanding Protein Adsorption Phenomena at Solid Surfaces. Adv. Colloid Interface Sci. 2011, 162, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.W.; Subrahmanyam, S.; Piletsky, S.A. Analytical Methods for Determination of Mycotoxins: A Review. Anal. Chim. Acta 2009, 632, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, E.d. Sample Preparation for Atomic Spectroscopy: Evolution and Future Trends. J. Braz. Chem. Soc. 2003, 14, 174–182. [Google Scholar] [CrossRef]
- Urge, A.Y.; Pampanin, D.M.; Martino, M.E.; Knudsen, D.L.; Brede, C. Salting-Out Assisted Liquid-Liquid Extraction for UPLC-MS/MS Determination of Thyroxine and Steroid Hormones in Human Serum and Fish Plasma. Separations 2023, 10, 240. [Google Scholar] [CrossRef]
- Zygler, A.; Wasik, A.; Namieśnik, J. Retention Behaviour of Some High-Intensity Sweeteners on Different SPE Sorbents. Talanta 2010, 82, 1742–1748. [Google Scholar] [CrossRef]
- Badawy, M.E.I.; El-Nouby, M.A.M.; Kimani, P. K.; Lim, L.W.; Rabea, E.I. Review of the Modern Principles and Applications of Solid-Phase Extraction Techniques in Chromatographic Analysis. Anal. Sci. 2022, 38, 1457–1487. [Google Scholar] [CrossRef]
- Hadad, G.M.; Abdel Salam, R.A.; Emara, S. Validated and Optimized High-Performance Liquid Chromatographic Determination of Tizoxanide, the Main Active Metabolite of Nitazoxanide in Human Urine, Plasma and Breast Milk. J. Chromatogr.Sci. 2012, 50, 509–515. [Google Scholar] [CrossRef]
- Ebrahim, H.; Sonbol, H.; Malak, M.; Ali, A.; Aboulella, Y.; Hadad, G.; Zarad, W.; Emara, S.; Bazan, L. Green Automated Solid Phase Extraction to Measure Levofloxacin in Human Serum Via Liquid Chromatography with Fluorescence Detection for Pharmacokinetic Study. Separations 2023, 10, 136. [Google Scholar] [CrossRef]
- Sonbol, H.; Ebrahim, H.; Malak, M.; Ali, A.; Aboulella, Y.; Hadad, G.; Emara, S.; Shawky, A. Application of a Small Protein-Coated Column to Trap, Extract and Enrich Carbamazepine Directly from Human Serum for Direct Chromatographic Analysis. Separations 2023, 10, 71. [Google Scholar] [CrossRef]
- Abughrin, S. E.; Alshana, U.; Bakirdere, S. Magnetic Nanoparticle-Based Dispersive Solid-Phase Microextraction of Three UV Blockers Prior to their Determination by HPLC-DAD. Int. J. Environ. Res. Public Health. 2022, 19, 6037. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Namera, A.; Yashiki, M.; Kojima, T. On-Column Derivatization for Determination of Amphetamine and Methamphetamine in Human Blood by Gas Chromatography–Mass Spectrometry. Forensic Sci. Int. 2002, 125, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Malak, M.; Ebrahim, H.; Sonbol, H.; Ali, A.; Aboulella, Y.; Hadad, G.; Emara, S. Highly Sensitive In-Capillary Derivatization and Field Amplified Sample Stacking to Analyze Narcotic Drugs in Human Serum by Capillary Zone Electrophoresis. Separations 2023, 10, 58. [Google Scholar] [CrossRef]
- Archana, G.; Dhodapkar, R.; Kumar, A. Offline Solid-Phase Extraction for Preconcentration of Pharmaceuticals and Personal Care Products in Environmental Water and their Simultaneous Determination Using the Reversed Phase High-Performance Liquid Chromatography Method. Environ. Monit. Assess. 2016, 188, 512. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.; Messina, S.M.; Stokes, C.; Salyani, S.; Alcalay, N.; De Fiebre, N.C.; De Fiebre, C.M. Solid-Phase Extraction and HPLC Assay ofNicotine and Cotinine in Plasma and Brain. Toxicol. Mech. Methods 2002, 12, 45–58. [Google Scholar] [PubMed]
- Torres, V.P.; Sepúlveda, C.M. J.; Von Plessing, R.C. Pharmacokinetic Study of Risperidone: Application of an HPLC Method with Solid Phase Extraction. j. Chil. Chem. Soc. 2011, 56, 606–609. [Google Scholar] [CrossRef]
- Sun, H.; Wang, L.; Ai, L.; Liang, S.; Wu, H. A Sensitive and Validated Method for Determination of Melamine Residue in Liquid Milk by Reversed Phase High-Performance Liquid Chromatography with Solid-Phase Extraction. Food Control. 2010, 21, 686–691. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, L.; Wang, H.; Zhang, X.; Zeng, Q.; Xu, H.; Sun, L.; Zhao, Q.; Ding, L. Preparation of Magnetic Strong Cation Exchange Resin for the Extraction of Melamine from Egg Samples Followed by Liquid Chromatography–Tandem Mass Spectrometry. Anal. Chim. Acta. 2010, 661, 35–41. [Google Scholar] [CrossRef]
- He, L.; Su, Y.; Shen, X.; Zheng, Y.; Guo, H.; Zeng, Z. Solid-Phase Extraction of Melamine from Aqueous Samples Using Water-Compatible Molecularly Imprinted Polymers. J. Sep. Sci. 2009, 32, 3310–3318. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, Y.; Ma, J.; Xuan, R.; Gao, H.; Liang, Z.; Zhang, L.; Zhang, Y. Hydrophilic Solid-Phase Extraction of Melamine with Ampholine-Modified Hybrid Organic–Inorganic Silica Material. J. Sep. Sci. 2015, 38, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Braus, H.; Middleton, F.; Walton, G. Organic Chemical Compounds in Raw and Filtered Surface Waters. Anal. Chem. 1951, 23, 1160–1164. [Google Scholar] [CrossRef]
- Theodoridis, G.; Koster, E. H.M.; de Jong, G.J. Solid-Phase Microextraction for the Analysis of Biological Samples. J. Chromatogr. B. 2000, 745, 49–82. [Google Scholar] [CrossRef]
- Zheng, J.; Huang, J.; Yang, Q.; Ni, C.; Xie, X.; Shi, Y.; Sun, J.; Zhu, F.; Ouyang, G. Fabrications of Novel Solid Phase Microextraction Fiber Coatings Based on New Materials for High Enrichment Capability. Trends Anal. Chem. 2018, 108, 135–153. [Google Scholar] [CrossRef]
- Gao, Y.; Sheng, K.; Bao, T.; Wang, S. Recent Applications of Organic Molecule-Based Framework Porous Materials in Solid-Phase Microextraction for Pharmaceutical Analysis. J. Pharm. Biomed. Anal. 2022, 221, 115040. [Google Scholar] [CrossRef] [PubMed]
- Arrebola, F.J.; Martı́nez-Vidal, J.L.; Fernández-Gutiérrez, A.; Akhtar, M.H. Monitoring of Pyrethroid Metabolites in Human Urine Using Solid-Phase Extraction Followed by Gas Chromatography-Tandem Mass Spectrometry. Anal. Chim. Acta 1999, 401, 45–54. [Google Scholar] [CrossRef]
- Le Grand, R.; Dulaurent, S.; Gaulier, J. M.; Saint-Marcoux, F.; Moesch, C.; Lachâtre, G. Simultaneous Determination of Five Synthetic Pyrethroid Metabolites in Urine by Liquid Chromatography–Tandem Mass Spectrometry: Application to 39 Persons without Known Exposure to Pyrethroids. Toxicol. Lett. 2012, 210, 248–253. [Google Scholar] [CrossRef]
- Wielgomas, B.; Nahorski, W.; Czarnowski, W. Urinary Concentrations of Pyrethroid Metabolites in the Convenience Sample ofan Urban Population of Northern Poland. Int. J. Hyg. Environ. Health 2013, 216, 295–300. [Google Scholar] [CrossRef]
- Lin, C.-H.; Yan, C.-T.; Kumar, P.V.; Li, H.-P.; Jen, J.-F. Determination of Pyrethroid Metabolites in Human Urine Using Liquid Phase Microextraction Coupled In-Syringe Derivatization Followed by Gas Chromatography/Electron Capture Detection. Anal. Bioanal. Chem. 2011, 401, 927–937. [Google Scholar] [CrossRef]
- Bartosz, W.; Marcin, W.; Wojciech, C. Development of Hollow Fiber-Supported Liquid-Phase Microextraction and HPLC-DAD Method for the Determination of Pyrethroid Metabolites in Human and Rat Urine. Biomed. Chromatogr. 2014, 28, 708–716. [Google Scholar] [CrossRef]
- Klimowska, A.; Wielgomas, B. Off-Line Microextraction by Packed Sorbent Combined withon Solid Support Derivatization and GC-MS: Application for the Analysis of Five Pyrethroid Metabolites in Urine Samples. Talanta 2018, 176, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R.A.; da Silva, L.C.; Queiroz, M.E.C.; Vaz, B.G.; Chaves, A.R. Lab-Made Solid Phase Microextraction Phases for off Line Extraction and Direct Mass Spectrometry Analysis: Evaluating the Extraction Parameters. J. Chromatogr. A. 2019, 1603, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Theodoridis, G.; Aikaterini Lontou, M.; Michopoulos, F.; Sucha, M.; Gondova, T. Study of Multiple Solid-Phase Microextraction Combined off-Line with High Performance Liquid Chromatography: Application in the Analysis of Pharmaceuticals in Urine. Anal. Chim. Acta. 2004, 516, 197–204. [Google Scholar] [CrossRef]
- Cantú, M. D.; Toso, D. R.; Lacerda, C. A.; Lanças, F. M.; Carrilho, E.; Queiroz, M. E. C. ; Optimization of Solid-Phase Microextraction Procedures for the Determination of Tricyclic Antidepressants and Anticonvulsants in Plasma Samples by Liquid Chromatography. Anal. Bioanal. Chem. 2006, 386, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Majors, R. E. Multidimensional High Performance Liquid Chromatography. J.Chromatogr. Sci. 1980, 18, 571–579. [Google Scholar] [CrossRef]
- El-Gendy, H.; Zarad, W.; Bazan, L.; Ali, A.; Aboulella, Y.; Kamal, M.; Emara, S.; Shawky, A. ; Rapid Back Flushed Direct Sample Injection Bio-Analytical HPLC-UV Method for therapeutic Drug Monitoring of Terbinafine. Anal. Biochem. 2022, 659, 114951. [Google Scholar] [CrossRef]
- Emara, S.; Kamal, M.; Hadad, G.; ZaaZaa, H.; Kawi, M.A. Back-Flush Column-Switching Technique for On-Line Sample Cleanup and Enrichment to Determine Guaiphenesin in Human Serum. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 15–27. [Google Scholar] [CrossRef]
- Karger, B. L.; Martin, M.; Guiochon, G. Role of Column Parameters and Injection Volume on Detection Limits in Liquid Chromatography. Anal. Chem. 1974, 46, 1640–1647. [Google Scholar] [CrossRef]
- Little, J.N.; Fallick, G.J. New Considerations in Detector-Application Relationships. J. Chromatogr. A. 1975, 112, 389–397. [Google Scholar] [CrossRef]
- Poole, C.F.; Evans, N.J.; Wibberley, D.G. Determination of Selenium in Biological Samples by Gas—Liquid Chromatography with Electron-Capture Detection. J. Chromatogr. A. 1977, 136, 63–72. [Google Scholar] [CrossRef]
- Huber, J.F.K.; Becker, R.R. Enrichment of Trace Components from Liquids by Displacement Column Liquid Chromatography. J. Chromatogr. A. 1977, 142, 765–776. [Google Scholar] [CrossRef]
- Lankelma, J.; Poppe, H. Determination of Methotrexate in Plasma by on-Column Concentration and Ion-Exchange Chromatography. J. Chromatogr. A. 1978, 149, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Kim, E.J.; Zee, O. P.; Lee, Y.J. On-Line Trace Enrichment in High Performance Liquid Chromatography Using XAD-2 Precolumn for the Determination of Lonazolac in Human Plasma. Arch. Pharm. Res. 1989, 12, 108–113. [Google Scholar] [CrossRef]
- Maris, F.A.; Stäb, J.A.; de Jong, G.J.; Brinkman, U.A.T. On-Line Trace Enrichment on a Reversed-Phase Pre-Column for Normal-Phase Liquid Chromatography with Electron-Capture Detection. J. Chromatogr. A. 1988, 445, 129–138. [Google Scholar] [CrossRef]
- Wang, H.; Guo, B.; Pang, X.; Yu, H.; Liu, H.; Yan, H.; Han, D.; Guo, H.; Bai, L. Simultaneous Determination and Enrichment of β-Sitosterol From Edible Oil Samples Using Poly(NMA-ST-Co-TAIC-Co-EDMA) Monolith as Sorbent with On-Line SPE-HPLC. Chromatographia. 2019, 82, 1285–1293. [Google Scholar] [CrossRef]
- Woo, C.M.; Iavarone, A.T.; Spiciarich, D.R.; Palaniappan, K.K.; Bertozzi, C.R. Isotope-Targeted Glycoproteomics (Isotag): A Mass-Independent Platform for Intact N- and O-Glycopeptide Discovery and Analysis. Nat. Methods 2015, 12, 561–567. [Google Scholar] [CrossRef]
- Hägglund, P.; Bunkenborg, J.; Elortza, F.; Jensen, O.N.; Roepstorff, P. A New Strategy for Identification of N-Glycosylated Proteins and Unambiguous Assignment of their Glycosylation Sites Using HILIC Enrichment and Partial Deglycosylation. J. Proteome Res. 2004, 3, 556–566. [Google Scholar] [CrossRef]
- Zauner, G.; Deelder, A.M.; Wuhrer, M. Recent Advances in Hydrophilic Interaction Liquid Chromatography (HILIC) for Structural Glycomics. Electrophoresis. 2011, 32, 3456–3466. [Google Scholar] [CrossRef]
- Shah, P.; Wang, X.; Yang, W.; Eshghi, S.T.; Sun, S.; Hoti, N.; Chen, L.; Yang, S.; Pasay, J.; Rubin, A. Integrated Proteomic and Glycoproteomic Analyses of Prostate Cancer Cells Reveal Glycoprotein Alteration in Protein Abundance and Glycosylation. Mol. Cell. Proteomics 2015, 14, 2753–2763. [Google Scholar] [CrossRef]
- Darula, Z.; Sarnyai, F.; Medzihradszky, K.F. O-Glycosylation Sites Identified from Mucin Core-1 Type Glycopeptides from Human Serum. Glycoconjugate J. 2016, 33, 435–445. [Google Scholar] [CrossRef]
- Qin, H.; Cheng, K.; Zhu, J.; Mao, J.; Wang, F.; Dong, M.; Chen, R.; Guo, Z.; Liang, X.; Ye, M.; Zou, H. Proteomics Analysis of O-Galnac Glycosylation in Human Serum by an Integrated Strategy. Anal.Chem. 2017, 89, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Shah, P.; Hu, Y.; Eshghi, S.T.; Sun, S.; Liu, Y.; Zhang, H. Comparison of Enrichment Methods for Intact N- and O-Linked Glycopeptides Using Strong Anion Exchange and Hydrophilic Interaction Liquid Chromatography. Anal. Chem. 2017, 89, 11193–11197. [Google Scholar] [CrossRef]
- Chaga, G.; Hopp, J.; Nelson, P. Immobilized Metal Ion Affinity Chromatography on Co2+-Carboxymethylaspartate–Agarose Superflow, as Demonstrated by One-Step Purification of Lactate Dehydrogenase from Chicken Breast Muscle. Biotechnol. Appl. Biochem. 1999, 29, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Hochuli, E.; Döbeli, H.; Schacher, A. New Metal Chelate Adsorbent Selective for Proteins and Peptides Containing Neighbouring Histidine Residues. J. Chromatogr. A. 1987, 411, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Porath, J. Isolation of Phosphoproteins by Immobilized Metal (Fe3+) Affinity Chromatography. Anal. Biochem. 1986, 154, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Neville, D.C.; Townsend, R.R.; Rozanas, C.R.; Verkman, A.S.; Price, E.M.; Gruis, D.B. Evidence for Phosphorylation of Serine 753 in CFTR Using a Novel Metal-Ion Affinity Resin and Matrix-Assisted Laser Desorption Mass Spectrometry. Protein Sci. 1997, 6, 2436–2445. [Google Scholar] [CrossRef]
- Figeys, D.; Gygi, S.P.; McKinnon, G.; Aebersold, R. An Integrated Microfluidics-Tandem Mass Spectrometry System for Automated Protein Analysis. Anal. Chem. 1998, 70, 3728–3734. [Google Scholar] [CrossRef]
- Li, S.; Dass, C. Iron (III)-Immobilized Metal Ion Affinity Chromatography and Mass Spectrometry forthe Purification and Characterization of Synthetic Phosphopeptides. Anal. Biochem. 1999, 270, 9–14. [Google Scholar] [CrossRef]
- Nuhse, T.S.; Stensballe, A.; Jensen, O.N.; Peck, S.C. Large-Scale Analysis ofin Vivo Phosphorylated Membrane Proteins by Immobilized Metal Ion Affinity Chromatography and Mass Spectrometry. Mol. Cell. Proteomics 2003, 2, 1234–1243. [Google Scholar] [CrossRef]
- Posewitz, M.C.; Tempst, P. Immobilized Gallium (III) Affinity Chromatography of Phosphopeptides. Anal. Chem. 1999, 71, 2883–2892. [Google Scholar] [CrossRef]
- Stensballe, A.; andersen, S.; Jensen, O.N. Characterization ofPhosphoproteins from Electrophoretic Gels by Nanoscale Fe (III) Affinity Chromatography with off-Line Mass Spectrometry Analysis. Proteomics 2001, 1, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ye, M.; Dong, J.; Han, G.; Jiang, X.; Wu, R.; Zou, H. Specific Phosphopeptide Enrichment with Immobilized Titanium Ion Affinity Chromatography Adsorbent for Phosphoproteome Analysis. J. Proteome Res. 2008, 7, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Ye, M.; Zhou, H.; Jiang, X.; Jiang, X.; Zou, H.; Gong, B. Immobilized Zirconium Ion Affinity Chromatography for Specific Enrichment of Phosphopeptides in Phosphoproteome Analysis. Mol. Cell. Proteomics. 2007, 6, 1656–1665. [Google Scholar] [CrossRef]
- Zhou, H.; Ye, M.; Dong, J.; Corradini, E.; Cristobal, A.; Heck, A.J.; Zou, H.; Mohammed, S. Robust Phosphoproteome Enrichment Using Monodisperse Microsphere–Based Immobilized Titanium (Iv) Ion Affinity Chromatography. Nat. Protoc. 2013, 8, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Qian, L.; Ji, L.; Liu, S.; Wahid, A.; Jiang, X.; Sohail, A.; Ji, Y.; Zhang, Y.; Wang, P. Affinity Chromatography Assisted Comprehensive Phosphoproteomics Analysis of Human Saliva forLung Cancer. Anal. Chim. Acta. 2020, 1111, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tang, N.; Brock, J.W.; Mottaz, H.M.; Ames, J.M.; Baynes, J.W.; Smith, R.D.; Metz, T.O. Enrichment and Analysis of Nonenzymatically Glycated Peptides: Boronate Affinity Chromatography Coupled with Electron-Transfer Dissociation Mass Spectrometry. J. Proteome Res. 2007, 6, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-S.; Ding, X.-Y.; Min, H.-P.; Li, B.; Su, M.-X.; Niu, M.-M.; Di, B.; Yan, F. Design andSynthesis ofan Immobilized Metal Affinity Chromatography and Metal Oxide Affinity Chromatography Hybrid Material for Improved Phosphopeptide Enrichment. J. Chromatogr. A. 2017, 1505, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Wolschin, F.; Wienkoop, S.; Weckwerth, W. Enrichment of Phosphorylated Proteins andPeptides from Complex Mixtures Using Metal Oxide/Hydroxide Affinity Chromatography (MOAC). Proteomics 2005, 5, 4389–4397. [Google Scholar] [CrossRef]
- Zarad, W.; El-Gendy, H.; Bazan, L.; Ali, A.; Aboulella, Y.; Kamal, M.; Emara, S.; Shawky, A. Bio-Analytical Liquid Chromatographic-Based Method with a Mixed Mode Online Solid Phase Extraction for Drug Monitoring of Fluconazole in Human Serum. J. Chromatogr. B. 2021, 1187, 123045. [Google Scholar] [CrossRef]
- Emara, S.; Kamal, M.; Abdel Kawi, M. On-Line Sample Cleanup andEnrichment Chromatographic Technique for the Determination of Ambroxol in Human Serum. J. Chromatogr. Sci. 2012, 50, 91–96. [Google Scholar] [CrossRef]
- Emara, S. Simultaneous Determination of Caffeine, theophylline andtheobromine in Human Plasma by On-Line Solid-Phase Extraction Coupled to Reversed-Phase Chromatography. Biomed. Chromatogr. 2004, 18, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Emara, S.; El-Gindy, A.; Mesbah, M.K.; Hadad, G.M. Direct Injection Liquid Chromatographic Technique for Simultaneous Determination of Two Antihistaminic Drugs and their Main Metabolites in Serum. J. AOAC Int. 2007, 90, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Emara, S.; Khedr, A.; Askal, H. Rapid and Specific Precolumn Extraction High-Performance Liquid Chromatographic Assay for Bupivacaine in Human Serum. Biomed. Chromatogr. 1996, 10, 131–134. [Google Scholar] [CrossRef]
- Emara, S.; Hussien, S.A.; Mohamed, F.A. Determination of Cholesterol inEgg Yolk by High Performance Liquid Chromatography Using anAutomated Precolumn-Switching Procedure. J. Liq. Chromatogr. Relat. Technol. 1999, 22, 1225–1246. [Google Scholar] [CrossRef]
- Zarad, W.; El-Gendy, H.; Ali, A.; Aboulella, Y.; Emara, S. Integration ofSolid-Phase Extraction andReversed-Phase Chromatography in Single Protein-Coated Columns for Direct Injection of Bupivacaine in Human Serum. J. Cchromatogr. Sci. 2020, 58, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Desilets, C.P.; Rounds, M.A.; Regnier, F.E. Semipermeable-Surface Reversed-Phase Media forHigh-Performance Liquid Chromatography. J. Chromatogr. A. 1991, 544, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, T.C.; Koeplinger, K.A. Determination of Warfarin-Human Serum Albumin Protein Binding Parameters by an Improved Hummel-Dreyer High-Performance Liquid Chromatographic Method Using Internal Surface Reversed-Phase Columns. Anal. Chem. 1990, 62, 2114–2122. [Google Scholar] [CrossRef]
- Haginaka, J.; Yasuda, N.; Wakai, J.; Matsunaga, H.; Yasuda, H.; Kimura, Y. Internal-Surface Reversed-Phase Silica Support for Direct-Injection Determination of Drugs in Biological Fluids byLiquid Chromatography. Anal. Chem. 1989, 61, 2445–2448. [Google Scholar] [CrossRef]
- Haginaka, J.; Wakai, J.; Yasuda, H. Synthesis of Mixed-Functional-Phase Silica Supports forLiquid Chromatography and their Applications to Assays of Drugs in Serum. J. Chromatogr. A. 1990, 535, 163–172. [Google Scholar] [CrossRef]
- Kimata, K.; Tsuboi, R.; Hosoya, K.; Tanaka, N.; Araki, T. Method for the Preparation of Internal-Surface Reversed-Phase Packing Materials Starting from Alkylsilylated Silica Gels. J. Chromatogr. A 1990, 515, 73–84. [Google Scholar] [CrossRef]
- Vielhauer, S.; Rudolphi, A.; Boos, K.S.; Seidel, D. Evaluation and routine application of the novel restricted-access precolumn packing material Alkyl-Diol Silica: Coupled-Column High-Performance Liquid Chromatographic Analysis of the Photoreactive Drug 8-Methoxypsoralen in Plasma. J. Chromatogr. B 1995, 666, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Račaityt, K.; Lutz, E.S.M.; Unger, K.K.; Lubda, D.; Boos, K.S. Analysis of Neuropeptide Y and its Metabolites byHigh-Performance Liquid Chromatography–Electrospray Ionization Mass Spectrometry andIntegrated Sample Clean-Up with a Novel Restricted-Access Sulphonic Acid Cation Exchanger. J. Chromatogr. A. 2000, 890, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Boos, K.-S.; Rudolphi, A. The Use of Restricted-Access Media in HPLC, Part I―Classification and Review. LC GC 1997, 15, 602–611. [Google Scholar]
- Boos, K.-S.; Grimm, C.-H. High-Performance Liquid Chromatography Integrated Solid-Phase Extraction inBioanalysis Using Restricted Access Precolumn Packings. Trends Anal. Chem. 1999, 18, 175–180. [Google Scholar] [CrossRef]
- Souverain, S.; Rudaz, S.; Veuthey, J.-L. Restricted Access Materials and Large Particle Supports for On-Line Sample Preparation: An Attractive Approach for Biological Fluids Analysis. J. Chromatogr. B 2004, 801, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Cassiano, N.M.; Lima, Vv.; Oliveira, R.V.; De Pietro, A.C.; Cass, Q.B. Development of Restricted-Access Media Supports andtheir Application to the Direct Analysis ofBiological Fluid Samples Via High-Performance Liquid Chromatography. Anal. Bioanal. Chem. 2006, 384, 1462–1469. [Google Scholar] [CrossRef]
- Rodríguez-Gonzalo, E.; Domínguez-Álvarez, J.; García-Gómez, D.; García-Jiménez, M.-G.; Carabias-Martínez, R. Determination ofEndocrine Disruptors in Honey by CZE-MS Using Restricted Access Materials for Matrix Cleanup. Electrophoresis 2010, 31, 2279–2288. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, R.; Xie, H.; Yin, Q.; Li, X.; Jia, Z.; Wu, X.; Zhang, J.; Li, W. Online Enrichment Ability of Restricted-Access Column Coupled with High Performance Liquid Chromatography by Column Switching Technique for Benazepril Hydrochloride. Se Pu 2013, 31, 451–455. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, R.; Xie, H.; Jia, Z.; Li, W.; Zhang, J.; Wang, Y. Determination of Rifampicin in Rat Plasma by Modified Large-Volume Direct Injection RAM-HPLC and its Application to a Pharmcokinetic Study. Biomed. Chromatogr. 2015, 29, 475–480. [Google Scholar] [CrossRef]
- Wu, X.; Wang, R.; Xie, H.; Wang, J.; Yang, P.; Jia, Z.; Zhang, Q.; Wang, X. Preparation and Application of Internal Surface Reversed-Phase Restricted-Access Material. Se Pu 2012, 30, 810–815. [Google Scholar] [CrossRef]
- Santos, M.G.; Tavares, I.M.C.; Barbosa, A.F.; Bettini, J.; Figueiredo, E.C. Analysis of Tricyclic Antidepressants in Human Plasma Using Online-Restricted Access Molecularly Imprinted Solid Phase Extraction Followed by Direct Mass Spectrometry Identification/Quantification. Talanta 2017, 163, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Papp, R.; Mullett, W.M.; Kwong, E.A. . Method for the Direct Analysis of Drug Compounds inPlasma Using a Single Restricted Access Material (RAM) Column. J. Pharm. Biomed. Anal. 2004, 36, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Denadai, M.; Cass, Q.B. Simultaneous Determination of Fluoroquinolones in Environmental Water by Liquid Chromatography–Tandem Mass Spectrometry withDirect Injection: A Green Approach. J. Chromatogr. A. 2015, 1418, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Fan, H.; Classon, R.J.; Schug, K.A. Restricted Access Media as a Streamlined Approach Toward On-Line Sample Preparation: Recent Advancements andApplications. J. Sep. Sci. 2013, 36, 2922–2938. [Google Scholar] [CrossRef] [PubMed]
- Wa, C.; Mallik, R.; Hage, D.S. Development of Immunoaffinity Restricted Access Media for Rapid Extractions of Low-Mass Analytes. Anal. Chem. 2008, 80, 8751–8762. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yamamoto, E.; Takakuwa, S.; Kato, T.; Asakawa, N. Weak Cation-Exchange Restricted-Access Material for On-Line Purification of Basic Drugs in Plasma. J. Chromatogr. A. 2008, 1190, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, F.; Ciogli, A.; D’Acquarica, I.; Misiti, D.; Badaloni, E.; Giorgi, F.; Vigevani, A. Synthesis and Characterization of Novel Internal Surface Reversed-Phase Silica Supports for High-Performance Liquid Chromatography. J. Chromatogr. A. 2007, 1176, 79–88. [Google Scholar] [CrossRef]
- González-Ortega, O.; Porath, J.; Guzmán, R. Adsorption ofPeptides andSmall Proteins withControl Access Polymer Permeation to Affinity Binding Sites. Part I: Polymer Permeation-Immobilized Metal Ion Affinity Chromatography Separation Adsorbents with Polyethylene Glycol andImmobilized Metal Ions. J. Chromatogr. A. 2012, 1227, 115–125. [Google Scholar] [CrossRef]
- Pereira, A.V.; Cass, Q.B. High-Performance Liquid Chromatography Method for the Simultaneous Determination of Sulfamethoxazole and Trimethoprim in Bovine Milk Using An On-Line Clean-Up Column. J. Chromatogr. B. 2005, 826, 139–146. [Google Scholar] [CrossRef]
- Barreiro, J.C.; Vanzolini, K.L.; Madureira, T.V.; Tiritan, M.E.; Cass, Q.B. A Column-Switching Method for Quantification of the Enantiomers of Omeprazole in Native Matrices of Waste and Estuarine Water Samples. Talanta 2010, 82, 384–391. [Google Scholar] [CrossRef]
- Cass, Q.B.; Lima, V.V.; Oliveira, R.V.; Cassiano, N.M.; Degani, A.L.G.; Pedrazzoli Jr, J. Enantiomeric Determination of the Plasma Levels of Omeprazole by Direct Plasma Injection Using High-Performance Liquid Chromatography with a chiral–Chiral Column-Switching. J. Chromatogr. B. 2003, 798, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Cass, Q.B.; Gomes, R.F.; Calafatti, S.A.; Pedrazolli Jr, J. Determination ofAmoxycillin in Human Plasma by Direct Injection andCoupled-Column High-Performance Liquid Chromatography. J. Chromatogr. A. 2003, 987, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, V.F.; Leite, C.P.; Pianetti, G.A.; Fernandes, C. Rapid and Direct Analysis of Statins in Human Plasma by Column-Switching Liquid Chromatography with Restricted-Access Material. J. Chromatogr. B. 2014, 947, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Lopes, B.R.; Barreiro, J.C.; Baraldi, P.T.; Cass, Q.B. Quantification of Carbamazepine and its Active Metabolite by Direct Injection of Human Milk Serum Using Liquid Chromatography Tandem Ion Trap Mass Spectrometry. J. Chromatogr. B. 2012, 889, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Mendiara, I.; Bentayeb, K.; Nerín, C.; Domeño, C. Online Solid-Phase Extraction–Liquid Chromatography–Mass Spectrometry to Determine Free Sterols in Human Serum. Talanta 2015, 132, 690–697. [Google Scholar] [CrossRef] [PubMed]
- De Paula, F.; De Pietro, A.C.; Cass, Q.B. Simultaneous Quantification of Sulfamethoxazole and Trimethoprim in Whole Egg Samples by Column-Switching High-Performance Liquid Chromatography Using Restricted Access Media Column for On-Line Sample Clean-Up. J. Chromatogr. A 2008, 1189, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Rosa, M.A.; Mendes, T.; Figueiredo, E.C. Restricted Access Molecularly Imprinted Polymers. Methods Mol. Biol. 2021, 2359, 53–70. [Google Scholar] [PubMed]
- Silveira, A.T; Barbosa, A.M.C.; Faria, H.D.; Marciano, L.P.A.; Eduardo Costa Figueiredo, E.C.; Martins, I. Online Restricted Access Molecularly Imprinted Solid-Phase Extraction Coupled with Liquid Chromatography-Mass Spectrometry for the Selective Determination of Serum Bile Acids. Analyst 2022, 147, 2779–2792. [Google Scholar] [CrossRef]
- Bigelow, E.; Song, Y.; Chen, J.; Holstein, M.; Huang, Y.; Duhamel, L.; Stone, K.; Furman, R.; Li, Z.J.; Ghose, S. Using Continuous Chromatography Methodology to Achieve High-Productivity and High-Purity Enrichment of Charge Variants for Analytical Characterization. J. Chromatogr. A 2021, 1643, 462008. [Google Scholar] [CrossRef]
- Weldon, R.; Lill, J.; Olbrich, M.; Schmidt, P.; Müller-Späth, T. Purification of A Galnac-Cluster-Conjugated Oligonucleotide by Reversed-Phase Twin-Column Continuous Chromatography. J. Chromatogr. A 2022, 1663, 462734. [Google Scholar] [CrossRef]
- De Luca, C.; Felletti, S.; Lievore, G.; Buratti, A.; Vogg, S.; Morbidelli, M.; Cavazzini, A.; Catani, M.; Macis, M.; Ricci, A. From Batch to Continuous Chromatographic Purification of a therapeutic Peptide Through Multicolumn Countercurrent Solvent Gradient Purification. J. Chromatogr. A 2020, 1625, 461304. [Google Scholar] [CrossRef] [PubMed]
- Aumann, L.; Morbidelli, M. A Continuous Multicolumn Countercurrent Solvent Gradient Purification (Mcsgp) Process. Biotechnol. Bioeng. 2007, 98, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Steinebach, F.; Ulmer, N.; Decker, L.; Aumann, L.; Morbidelli, M. Experimental Design of a Twin-Column Countercurrent Gradient Purification Process. J. Chromatogr. A 2017, 1492, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Botti, C.; Angelo, J.; Xu, X.; Ghose, S.; Li, Z.J.; Morbidelli, M.; Sponchioni, M. Experimental Design of the Multicolumn Countercurrent Solvent Gradient Purification (MCSGP) Unit for the Separation of Pegylated Proteins. Ind. Eng. Chem. Res. 2021, 60, 10764–10776. [Google Scholar] [CrossRef]
- De Luca, C.; Felletti, S.; Lievore, G.; Chenet, T.; Morbidelli, M.; Sponchioni, M.; Cavazzini, A.; Catani, M. Modern Trends in Downstream Processing of Biotherapeutics Through Continuous Chromatography: the Potential of Multicolumn Countercurrent Solvent Gradient Purification. Trends Analyt. Chem. 2020, 132, 116051. [Google Scholar] [CrossRef] [PubMed]
- Zydney, A.L. Continuous Downstream Processing forHigh Value Biological Products: A Review. Biotechnol. Bioeng. 2016, 113, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Challener, C.A. Making the Move to Continuous Chromatography. BioPharm. Int. 2018, 31, 14–18. [Google Scholar]
- Peuker, T.; Bogner, A. Equipment Design and Facility Layout for Flexible Biomanufacturing Processes. Eng. Life Sci. 2011, 11, 443–451. [Google Scholar] [CrossRef]
- Goyon, A.; Dai, L.; Chen, T.; Wei, B.; Yang, F.; andersen, N.; Kopf, R.; Leiss, M.; Mølhøj, M.; Guillarme, D. From Proof of Concept to the Routine Use of An Automated and Robust Multi-Dimensional Liquid Chromatography Mass Spectrometry Workflow Applied for the Charge Variant Characterization of Therapeutic Antibodies. J. Chromatogr. A 2020, 1615, 460740. [Google Scholar] [CrossRef]
- Jing, S.-Y.; Shi, C.; Leong, H.Y.; Yuan, J.-J.; Gao, D.; Wang, H.-B.; Yao, S.-J.; Lin, D.-Q. A Novel Twin-Column Continuous Chromatography Approach for Separation and Enrichment of Monoclonal Antibody Charge Variants. Eng. Life Sci. 2021, 21, 382–391. [Google Scholar] [CrossRef]
- Weldon, R.; Müller-Späth, T. Enrichment and Purification of Peptide Impurities Using Twin-Column Continuous Chromatography. J. Chromatogr. A 2022, 1667, 462894. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.-L.; Liu, P.; Wang, Q.-W.; Cai, W.-J.; Yuan, B.-F.; Feng, Y.-Q. Derivatization for Liquid Chromatography–Mass Spectrometry. Trends Anal. Chem. 2014, 59, 121–132. [Google Scholar] [CrossRef]
- Medvedovici, A.; Bacalum, E.; David, V. Sample Preparation for Large-Scale Bioanalytical Studies Based on Liquid Chromatographic Techniques. Biomed. Chromatogr. 2018, 32, e4137. [Google Scholar] [CrossRef] [PubMed]
- Hadad, G.M.; Abdel-Salam, R.A.; Emara, S. Determination of Glucosamine and Carisoprodol in Pharmaceutical formulations by LC with Pre-Column Derivatization and UV Detection. J. Chromatogr. Sci. 2012, 50, 307–315. [Google Scholar] [CrossRef]
- Tang, T.; Qian, K.; Shi, T.; Wang, F.; Li, J.; Cao, Y.; Hu, Q. Monitoring the Contents of Biogenic Amines in Sufu by HPLC with SPE and Pre-Column Derivatization. Food Control 2011, 22, 1203–1208. [Google Scholar] [CrossRef]
- Emara, S. Development of Highly Sensitive and Specific HPLC Assay for Plasma Morphine Using Direct Injection Technique and Post-Column Derivatization. Biomed. Chromatogr. 1998, 12, 15–20. [Google Scholar] [CrossRef]
- Deelder, R.S.; Kroll, M.G.F.; Beeren, A.J.; Van Den Berg, J.H.M. Post-Column Reactor Systems in Liquid Chromatography. J. Chromatogr. A 1978, 149, 669–682. [Google Scholar] [CrossRef]
- Shin, K-O. ; Park, K.A Newly Developed HPLC-UV/Vis Method Using Chemical Derivatization with 2-Naphthalenethiol for Quantitation of Sulforaphane in Rat Plasma. Molecules 2021, 26, 5473. [Google Scholar] [CrossRef]
- Galuszka, A.; Migaszewski, Z.; Namieśnik, J. The 12 Principles of Green Analytical Chemistry and the Significance Mnemonic of Green Analytical Practices. Trends Anal. Chem. 2013, 50, 78–84. [Google Scholar] [CrossRef]
- Poole, C.F. Microreactions in Separation Science: Reagents and Techniques. J. Chromatogr. A. 2013, 1296, 1–1. [Google Scholar] [CrossRef]
- Keith, L.H.; Gron, L.U.; Young, J.L. Green Analytical Methodologies. Chem. Rev. 2007, 107, 2695–2708. [Google Scholar] [CrossRef] [PubMed]
- Razee, S.; Tamura, A.; Emara, S.; Masujima, T. Application of Capillary Electrophoresis for the Determination of Dissolved Gases in Water Samples: I. Determination of Dissolved Oxygen. Anal. Chim. Acta. 1997, 356, 1–6. [Google Scholar] [CrossRef]
- Zarad, W.; Shawky, A.; Ali, A.; Aboulella, Y.; Kamal, M.; Masujima, T.; Emara, S.; El-Gendy, H. Field Amplified Sample Stacking and In-Capillary Derivatization for forensic Analysis of Morphine and Morphine-6-Glucuronide in Human Urine by Capillary Electrophoresis. Talanta Open. 2021, 3, 100041. [Google Scholar] [CrossRef]
- Emara, S.; Zarad, W.; Kamal, M.; Ali, A.; Aboulella, Y. Sensitivity Enhancement for Direct Injection Capillary Electrophoresis to Determine Morphine in Human Serum Via In-Capillary Derivatization. J. Chromatogr. Sci. 2018, 57, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Emara, S.; Masujima, T.; Zarad, W.; Mohamed, K.; Kamal, M.; Fouad, M.; EL-Bagary, R. Field-Amplified Sample Stacking β-Cyclodextrin Modified Capillary Electrophoresis for Quantitative Determination of Diastereomeric Saponins. J. Chromatogr. Sci. 2013, 52, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Lavilla, I.; Romero, V.; Costas, I.; Bendicho, C. Greener Derivatization in Analytical Chemistry. Trends Anal. Chem. 2014, 61, 1–10. [Google Scholar] [CrossRef]
- Marsol-Vall, A.; Balcells, M.; Eras, J.; Canela-Garayoa, R. Injection-Port Derivatization Coupled to GC–MS/MS for the Analysis of Glycosylated and non-Glycosylated Polyphenols in Fruit Samples. Food Chem. 2016, 204, 210–217. [Google Scholar] [CrossRef]
- Smith, R. M. Before the Injection—Modern Methods of Sample Preparation for Separation Techniques. J.Chromatogr. A 2003, 1000, 3–27. [Google Scholar] [CrossRef]
- Pawliszyn, J.; Lord, H. L. Handbook of Sample Preparation; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 225–245. [Google Scholar]
- Rosenfeld, J. M. Solid-Phase Analytical Derivatization: Enhancement of Sensitivity and Selectivity of Analysis. J. Chromatogr. A. 1999, 843, 19–27. [Google Scholar] [CrossRef]
- Rosenfeld, J. M. Derivatization in the Current Practice of Analytical Chemistry. Trends Anal. Chem. 2003, 22, 785–798. [Google Scholar] [CrossRef]
- Atapattu, S. N.; Rosenfeld, J. M. Solid Phase Analytical Derivatization as a Sample Preparation Method. J. Chromatogr. A. 2013, 1296, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Atapattu, S. N.; Rosenfeld, J. M. Micro Scale Analytical Derivatizations On Solid Phase. Trends Anal. Chem. 2019, 113, 351–356. [Google Scholar] [CrossRef]
- Oenning, A. L.; Morés, L.; Dias, A. N.; Carasek, E. A New Configuration for Bar Adsorptive Microextraction (Baμe) for the Quantification of Biomarkers (Hexanal and Heptanal) in Human Urine by HPLC Providing an Alternative for Early Lung Cancer Diagnosis. Anal. Chim. Acta. 2017, 965, 54–62. [Google Scholar] [CrossRef]
- Liu, J.-F.; Yuan, B.-F.; Feng, Y.-Q. Determination of Hexanal and Heptanal in Human Urine Using Magnetic Solid Phase Extraction Coupled with In-Situ Derivatization by High Performance Liquid Chromatography. Talanta. 2015, 136, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, L. Chemical Derivatization in LC-MS-Based Metabolomics Study. Trends Anal. Chem. 2020, 131, 115988. [Google Scholar] [CrossRef]
- Baños, C. E.; Silva, M. Liquid Chromatography–Tandem Mass Spectrometry forthe Determination of Low-Molecular Mass Aldehydes in Human Urine. J. Chromatogr. B. 2010, 878, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Du, Y.; Xiao, X.; Li, G. One-Step Membrane Protected Micro-Solid-Phase Extraction and Derivatization Coupling to High-Performance Liquid Chromatography for Selective Determination of Aliphatic Aldehydes in Cosmetics and Food. Talanta 2019, 202, 580–590. [Google Scholar] [CrossRef]
- Kang, P.; Mechref, Y.; Kyselova, Z.; Goetz, J. A.; Novotny, M. V. Comparative Glycomic Mapping Through Quantitative Permethylation and Stable-Isotope Labeling. Anal. Chem. 2007, 79, 6064–6073. [Google Scholar] [CrossRef]
- Kang, P.; Mechref, Y.; Novotny, M. V. High-Throughput Solid-Phase Permethylation of Glycans Prior to Mass Spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 721–734. [Google Scholar] [CrossRef]
- Kang, P.; Mechref, Y.; Klouckova, I.; Novotny, M. V. Solid-Phase Permethylation of Glycans for Mass Spectrometric Analysis. Rapid Commun. Mass Spectrom. 2005, 19, 3421–3428. [Google Scholar] [CrossRef]
- Mechref, Y.; Kang, P.; Novotny, M.V. Solid-Phase Permethylation for Glycomic Analysis. Methods Mol. Biol. 2009, 534, 53–64. [Google Scholar] [PubMed]
- Lei, M.; Mechref, Y.; Novotny, M. V. Structural Analysis of Sulfated Glycans by Sequential Double-Permethylation Using Methyl Iodide and Deuteromethyl Iodide. J. Am. Soc. Mass Spectrom. 2009, 20, 1660–1671. [Google Scholar] [CrossRef]
- Lei, M.; Novotny, M. V.; Mechref, Y. Sequential Enrichment of Sulfated Glycans by Strong Anion-Exchange Chromatography Prior to Mass Spectrometric Measurements. J. Am. Soc. Mass Spectrom. 2010, 21, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ma, L.; Guo, Y.; Liu, W.; Wang, Y.; Liu, S. Analysis of Oligosaccharides From Panax Ginseng by Using Solid-Phase Permethylation Method Combined with Ultra-High-Performance Liquid Chromatography-Q-Orbitrap/Mass Spectrometry. J Ginseng Res. 2020, 44, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Desantos-Garcia, J.L.; Khalil, S.I.; Hussein, A.; Hu, Y.; Mechref, Y. Enhanced Sensitivity of LC-MS Analysis of Permethylated N-Glycans Through Online Purification. Electrophoresis 2011, 32, 3516–3525. [Google Scholar] [CrossRef] [PubMed]
- Emara, S. Determination of Methotrexate in Pharmaceutical formulations by Flow Injection Analysis Exploiting the Reaction with Potassium Permanganate. Il Farmaco 2004, 59, 827–833. [Google Scholar] [CrossRef]
- Wang, L.H.; Hung, H.C. Simultaneous Determination of Water-Soluble Vitamins in Human Urine by Fluorescence in a Flow-Injection Analysis. J. Liq. Chromatogr. Relat. Technol. 2006, 29, 329–338. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, P.; Liang, X.; Hua, D.; Ma, T.; Pei, G. A New Potassium Tetrabromoaurate (III)–Luminol Chemiluminescence System for the Determination of Folic Acid in Milk Powder. J. Food Sci. 2012, 77, C102–C106. [Google Scholar] [CrossRef]
- Song, Z.; Wang, L. Chemiluminescence Inhibition Assay for Folic Acid Using Flow Injection Analysis. Phytochem. Anal. 2003, 14, 216–220. [Google Scholar] [CrossRef]
- Hadad, G.; Abdel-Salam, R.; Emara, S. Optimized and Validated Flow-Injection Spectrophotometric Analysis of Topiramate, Piracetam and Levetiracetam in Pharmaceutical formulations. Acta Pharmaceutica. 2011, 61, 377–389. [Google Scholar] [CrossRef]
- Emara, S.; El-Gindy, A.; El-Shorbagi, A.-N.; Hadad, G. Utility of Copper(II) Oxide as a Packed Reactor in Flow Injection Assembly for Rapid Analysis of Some Angiotensin Converting Enzyme Inhibitors. Anal.Chim. Acta 2003, 489, 115–123. [Google Scholar] [CrossRef]
- Erlandsson, M.; Hällbrink, M. Metallic Zinc Reduction of Disulfide Bonds Between Cysteine Residues in Peptides and Proteins. Int. J. Pept. Res. Ther. 2005, 11, 261. [Google Scholar] [CrossRef]
- Jamal, M.; Hadi, H. Determination of Nitrazepamin Pharmaceutical Preparation Using Solid-Phase Reactor–Reverse Flow Injection Analysis. Asian J. Pharm. Clin. Res. 2018, 11, 487–492. [Google Scholar] [CrossRef]
- Emara, S.; Masujima, T.; Zarad, W.; Kamal, M.; El-Bagary, R. On-Line Solid-Phase Enrichment Coupled to Packed Reactor Flow Injection Analysis in A Green Analytical Procedure to Determine Low Levels of Folic Acid Using Fluorescence Detection. Chem. Cent. J. 2012, 6, 155. [Google Scholar] [CrossRef] [PubMed]
- Emara, S.; Zarad, W.; Kamal, M.; EL-Bagary, R. Coupling of On-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate. J. Anal. Methods Chem. 2012, 2, 194–202. [Google Scholar] [CrossRef]
- Emara, S.; Razee, S.; El-Shorbagi, A.-N.; Masujima, T. Flow Injection Method forthe Determination of Methotrexate with A Column-Packed Oxidizing Agent. Analyst 1996, 121, 183–188. [Google Scholar] [CrossRef]
- Emara, S.; Masujima, T.; Zarad, W.; Kamal, M.; El-Bagary, R. On-Line Coupling of Derivatization with Pre-Concentration to Determine Trace Levels of Methotrexate. J. Pharm. Anal. 2013, 3, 28–35. [Google Scholar] [CrossRef]
- Emara, S.; Masujima, T.; Zarad, W.; Kamal, M.; EI-Bagary, R. Online Pre-Column Derivatization with Chromatographic Separation to Determine Folic Acid. J. Chromatogr. Sci. 2012, 51, 544–551. [Google Scholar] [CrossRef]
- Emara, S.; Razee, S.; Khedr, A.; Masujima, T. On-Line Precolumn Derivatization for HPLC Determination of Methotrexate Using a Column Packed Oxidant. Biomed.Chromatogr. 1997, 11, 42–46. [Google Scholar] [CrossRef]
- Emara, S.; Askal, H.; Masujima, T. Rapid Determination of Methotrexate in Plasma by High-Performance Liquid Chromatography with On-Line Solid-Phase Extraction and Automated Precolumn Derivatization. Biomed. Chromatogr. 1998, 12, 338–342. [Google Scholar] [CrossRef]
Figure 1.
Manifold of on through elution HPLC methodology for measuring fluconazole directly from serum samples: Position (A) depicts the system set up for loading, washing, trapping, and pre-concentration of the analyte: Position (B) shows the quantification step, ready for separation and measurement: HPLC circulation (separation and quantification) is segregated from the extraction position: A 6-port high pressure switching valve (S.V.) connects the PC-column to an analytical column.
Figure 1.
Manifold of on through elution HPLC methodology for measuring fluconazole directly from serum samples: Position (A) depicts the system set up for loading, washing, trapping, and pre-concentration of the analyte: Position (B) shows the quantification step, ready for separation and measurement: HPLC circulation (separation and quantification) is segregated from the extraction position: A 6-port high pressure switching valve (S.V.) connects the PC-column to an analytical column.
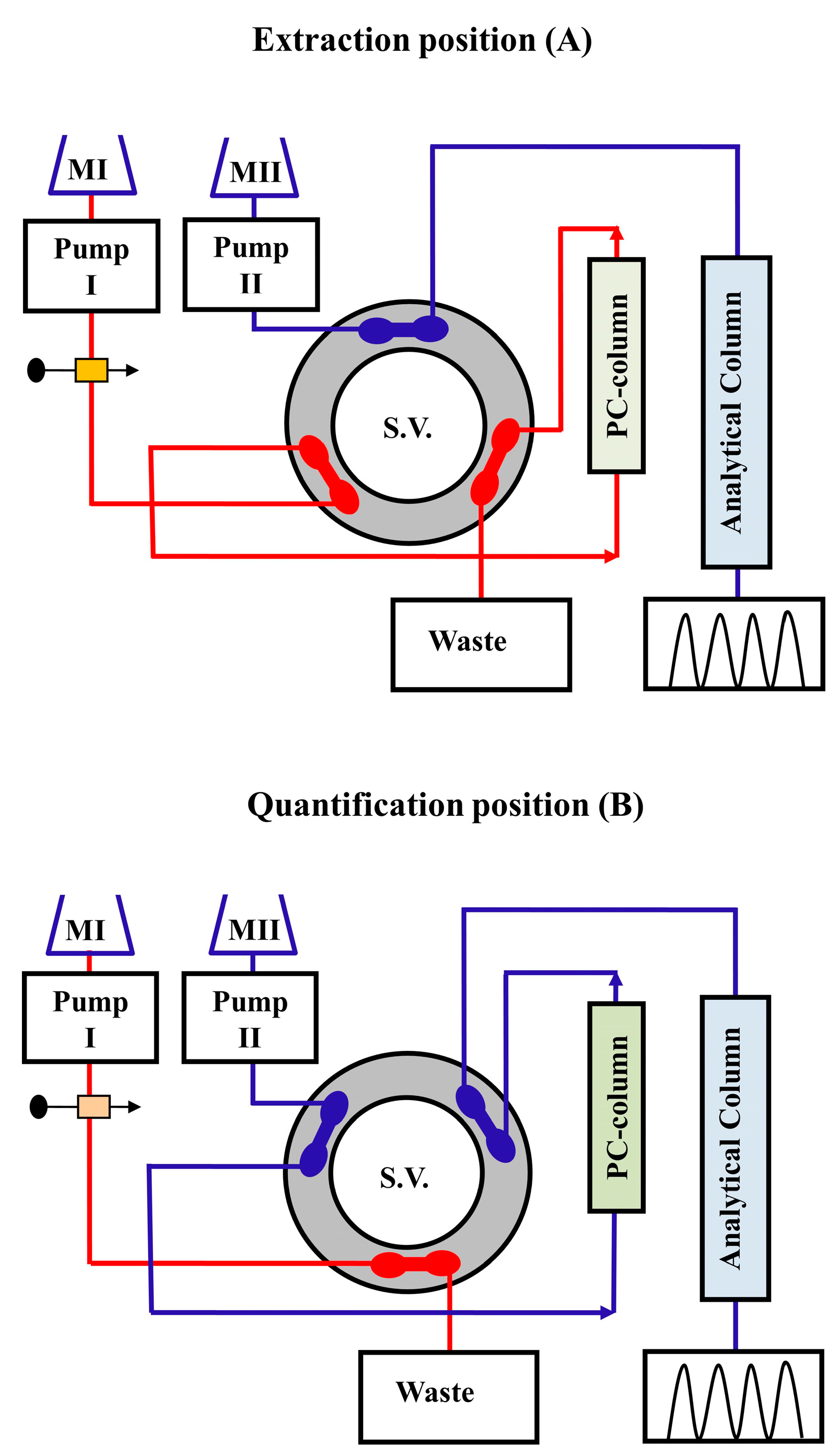
Figure 2.
Manifold of back flushed HPLC methodology for measuring levofloxacin directly from serum samples: Position (A) depicts the system manifold for extraction, capturing as well pre-concentration of the analyte: Position (B) shows the measurement step, ready for separation and quantification: Separation and quantification are segregated from the extraction position: A 6-port high pressure switching valve (S.V.) connects the PC-column to an analytical column.
Figure 2.
Manifold of back flushed HPLC methodology for measuring levofloxacin directly from serum samples: Position (A) depicts the system manifold for extraction, capturing as well pre-concentration of the analyte: Position (B) shows the measurement step, ready for separation and quantification: Separation and quantification are segregated from the extraction position: A 6-port high pressure switching valve (S.V.) connects the PC-column to an analytical column.
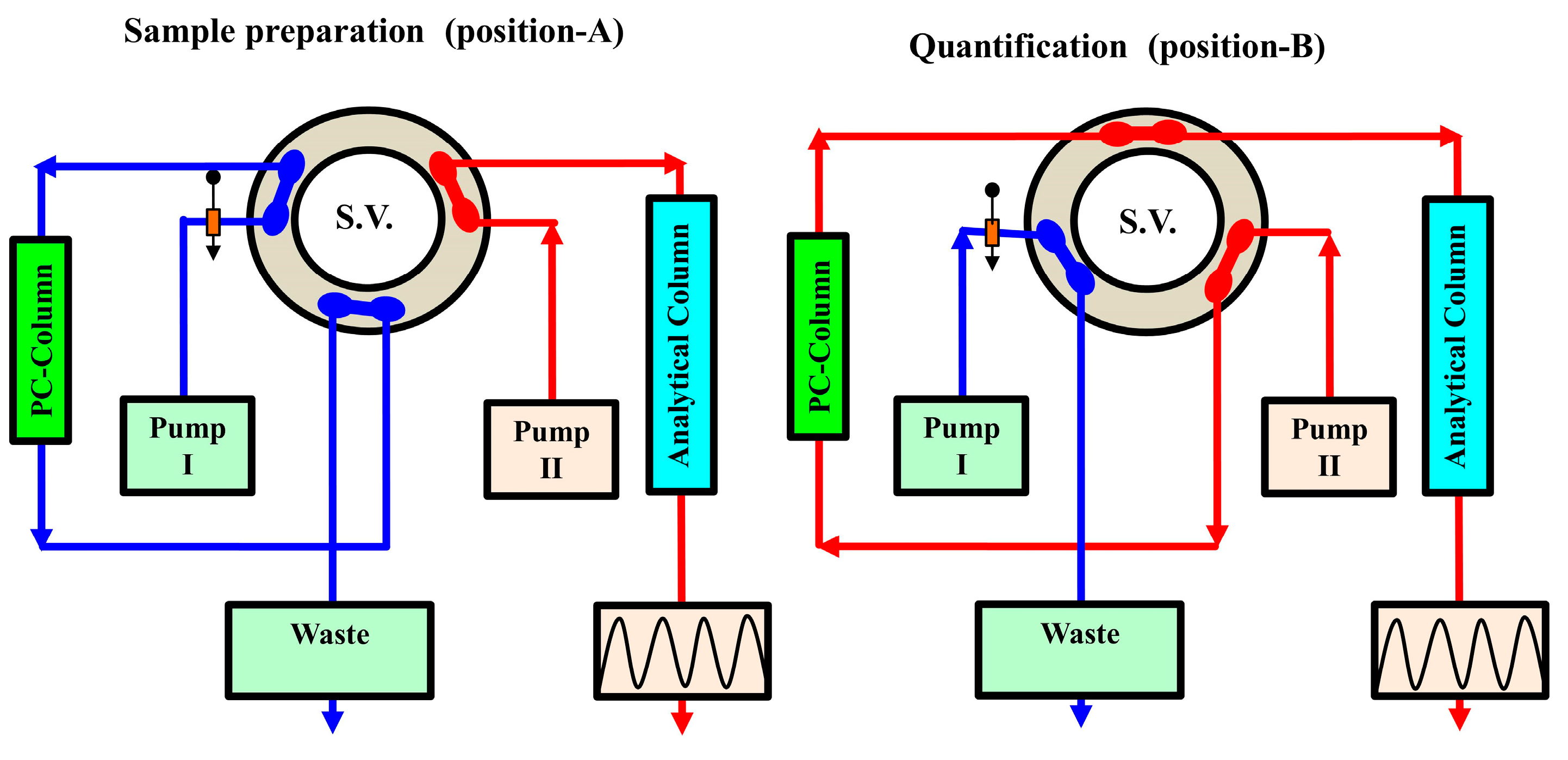
Figure 3.
FIA manifold used to analyze MTX. Channel 1 (1 mL/min), sulfuric acid carrier stream; channel 2 (1 mL/min), potassium permanganate oxidant stream; channel 3 (2 mL/min), sodium sulfite decolorizing stream; R, reaction coil; T, water bath at 65 °C..
Figure 3.
FIA manifold used to analyze MTX. Channel 1 (1 mL/min), sulfuric acid carrier stream; channel 2 (1 mL/min), potassium permanganate oxidant stream; channel 3 (2 mL/min), sodium sulfite decolorizing stream; R, reaction coil; T, water bath at 65 °C..
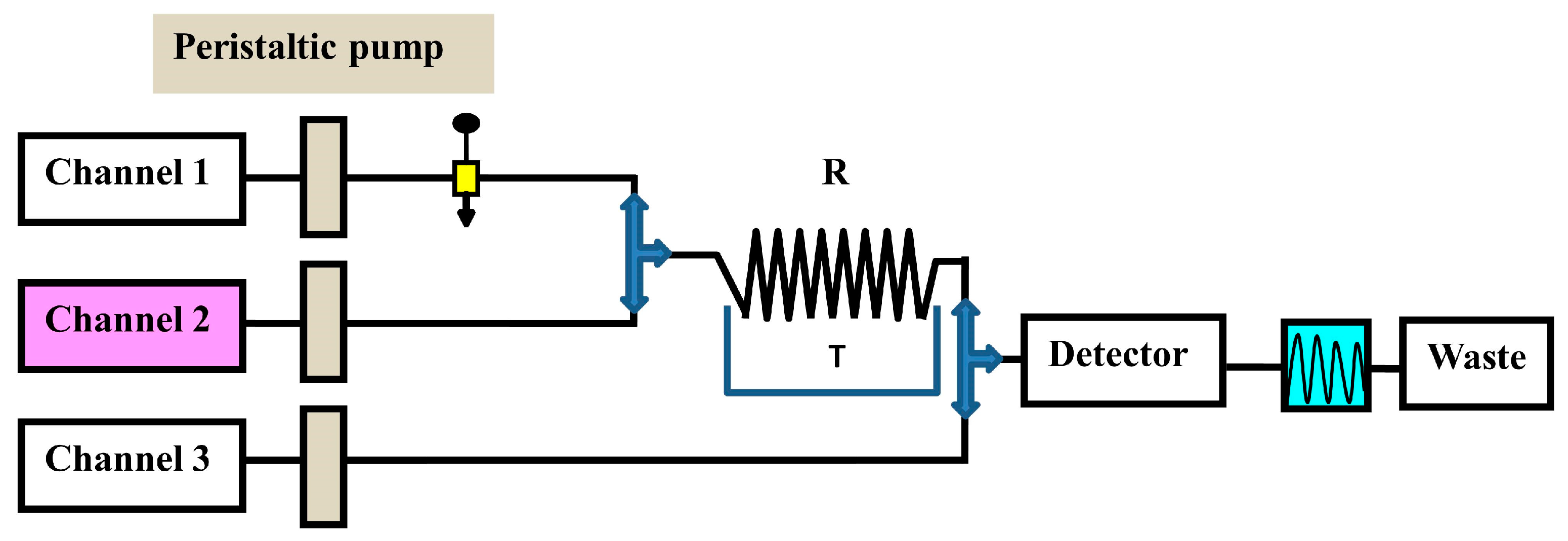
Scheme 1.
Oxidation of FA into highly fluorescent 2-amino-4-hydroxypteridine-6-carboxaldehyde and the corresponding carboxylic acid derivative.
Scheme 1.
Oxidation of FA into highly fluorescent 2-amino-4-hydroxypteridine-6-carboxaldehyde and the corresponding carboxylic acid derivative.
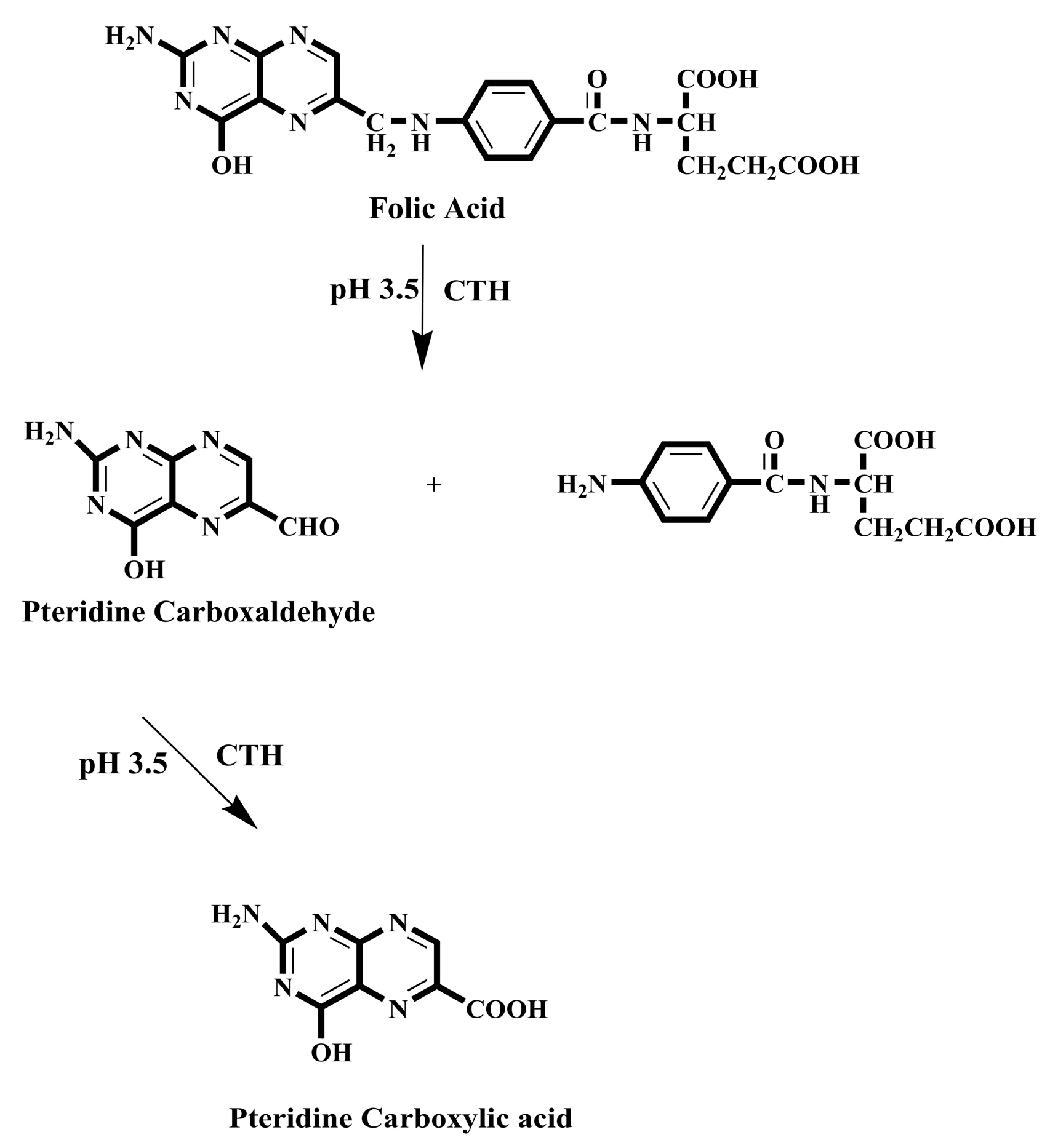
Scheme 1.
Oxidation of MTX into highly fluorescent 2,4-diaminopteridine-6-carboxaldehyde and the corresponding carboxylic acid derivative.
Scheme 1.
Oxidation of MTX into highly fluorescent 2,4-diaminopteridine-6-carboxaldehyde and the corresponding carboxylic acid derivative.
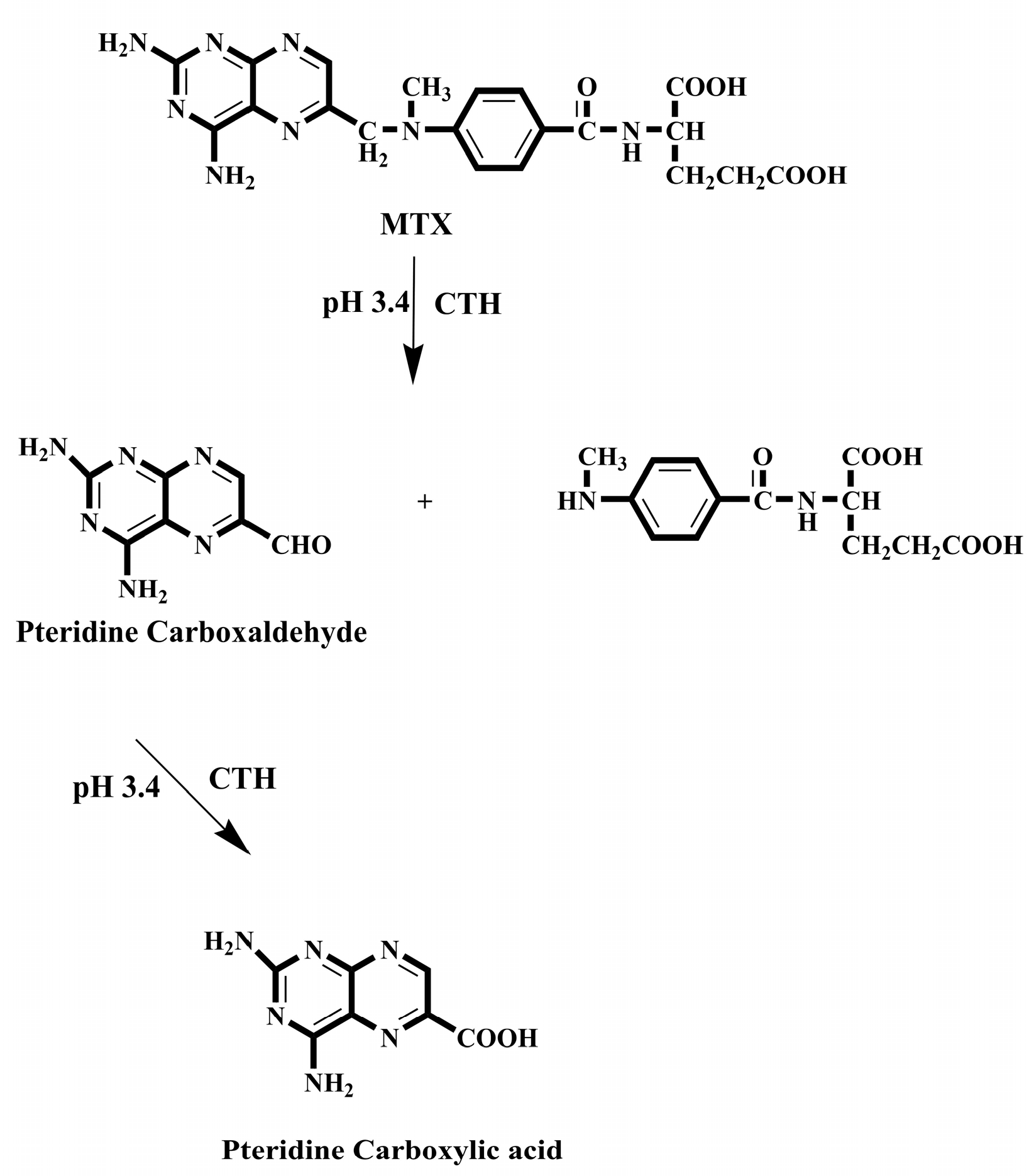
Figure 4.
Schematic diagram of an on-line-SPEn connected to a packed reactor FIA for tracing MTX in pharmaceutical formulations. (A) The system is shown in its initial condition, ready for sample injection, derivatization, and enrichment. Position (B) shows the back-flush elution and measurement procedure; six port high pressure switching valve (S.V.).
Figure 4.
Schematic diagram of an on-line-SPEn connected to a packed reactor FIA for tracing MTX in pharmaceutical formulations. (A) The system is shown in its initial condition, ready for sample injection, derivatization, and enrichment. Position (B) shows the back-flush elution and measurement procedure; six port high pressure switching valve (S.V.).
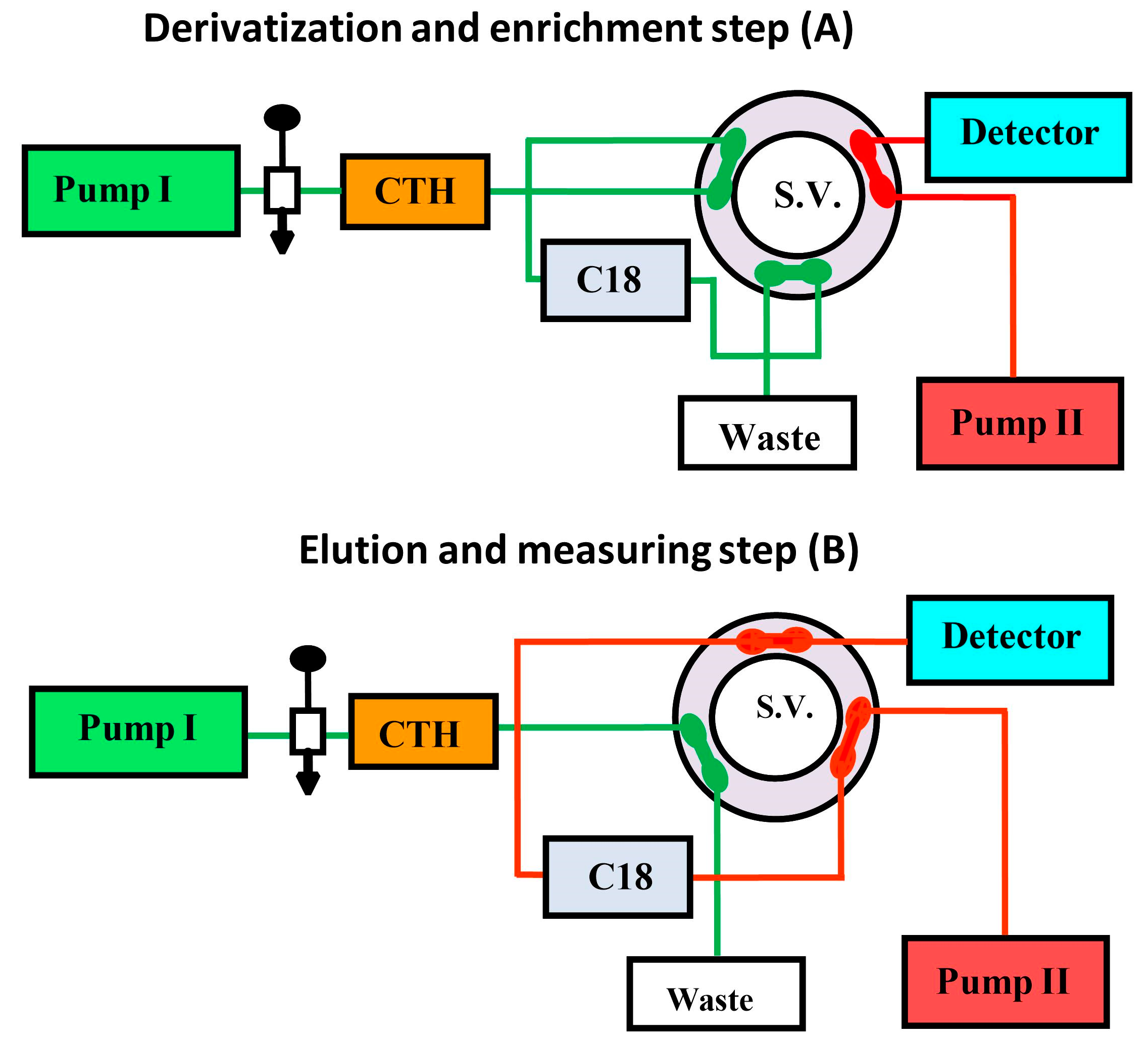
Figure 5.
Schematic diagram of online pre-column oxidation combined with column switching with an RP-C18 analytical column for sample enrichment and separation by HPLC to analyze MTX in pharmaceutical formulations.
Figure 5.
Schematic diagram of online pre-column oxidation combined with column switching with an RP-C18 analytical column for sample enrichment and separation by HPLC to analyze MTX in pharmaceutical formulations.
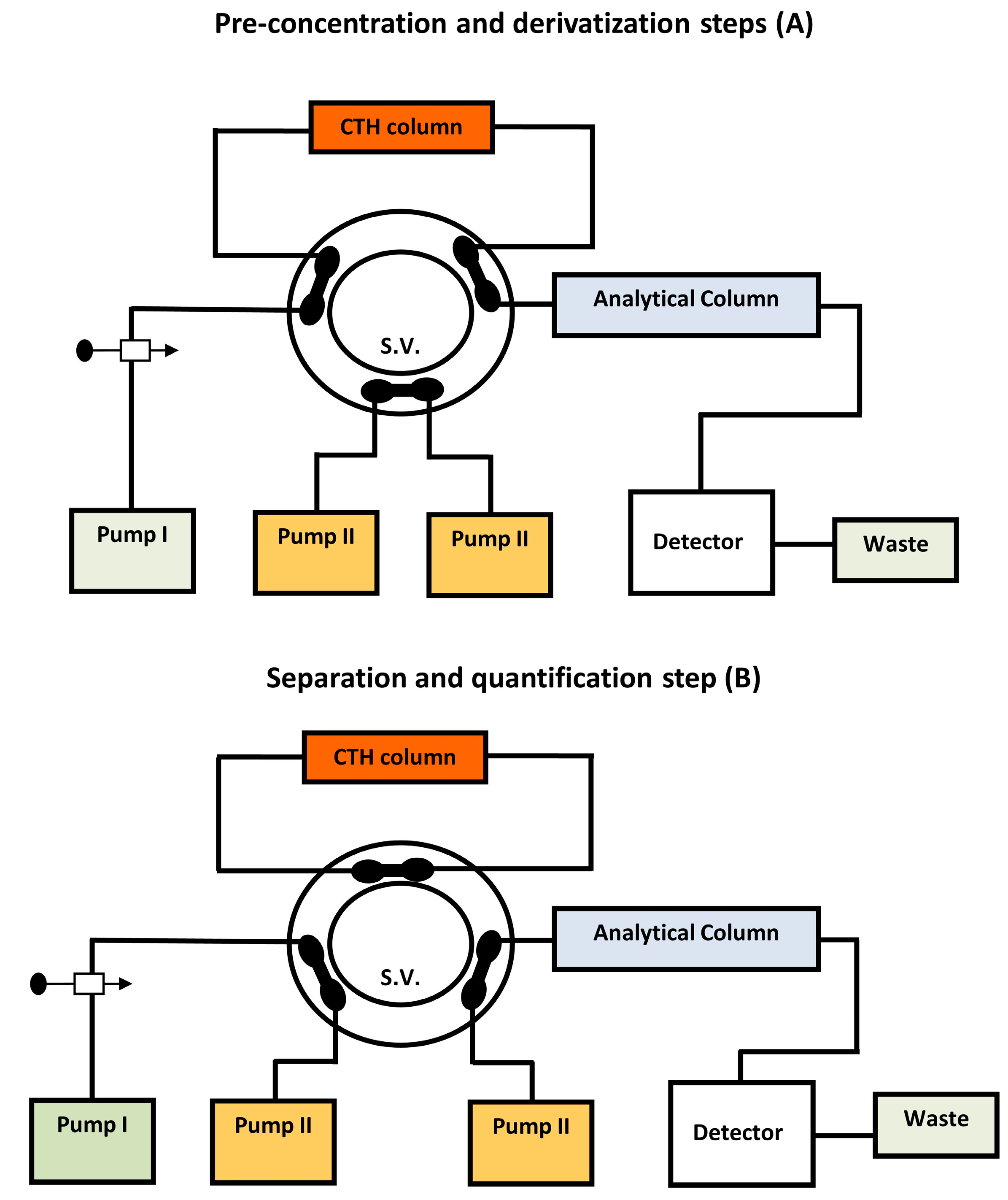
Figure 6.
Schematic diagram of the on-line pre-column derivatization and automated SPE utilizing packed oxidant and PC-RP-pre-column for the analysis of MTX in plasma.
Figure 6.
Schematic diagram of the on-line pre-column derivatization and automated SPE utilizing packed oxidant and PC-RP-pre-column for the analysis of MTX in plasma.
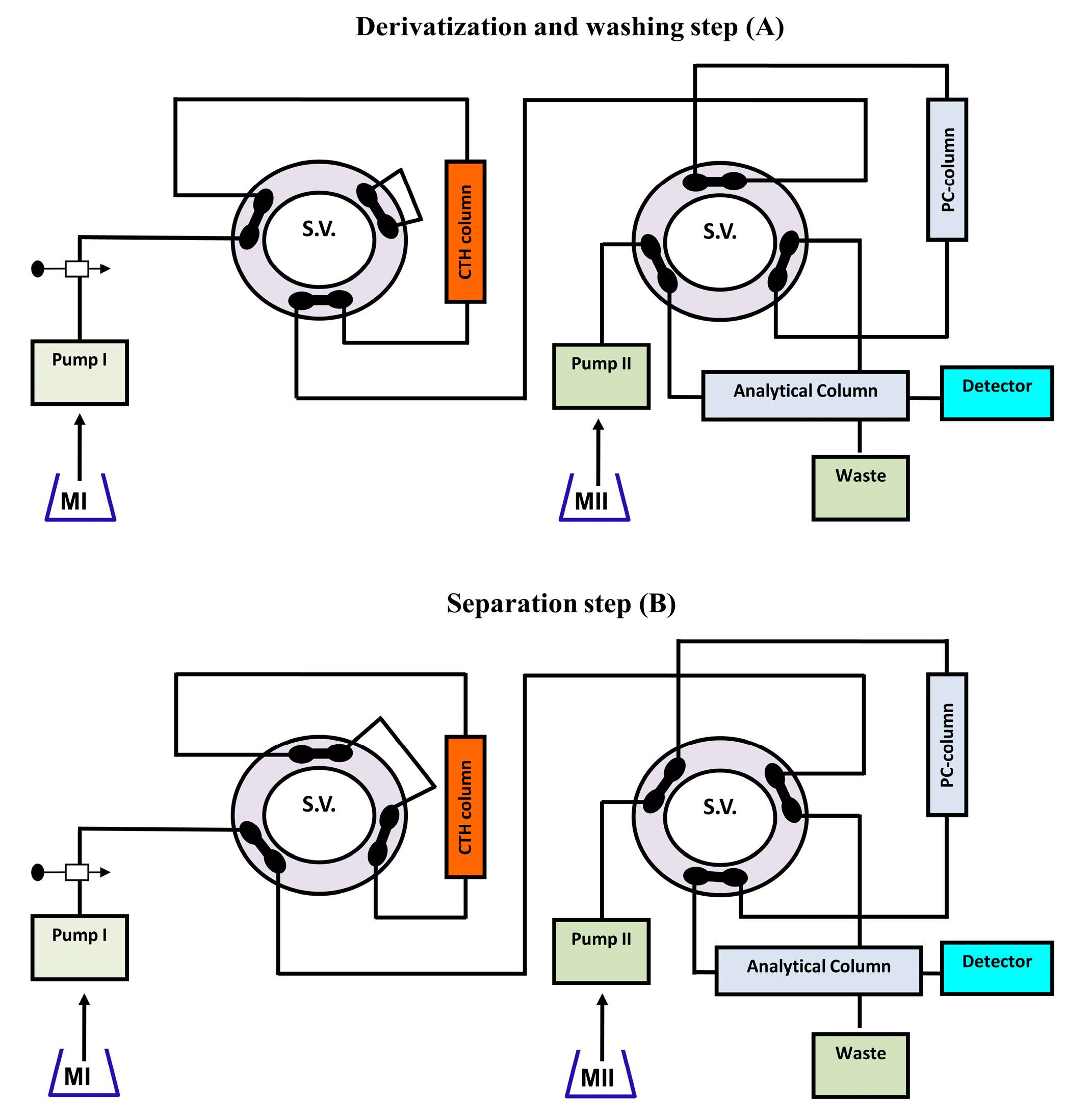
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



