
Submitted:
08 May 2023
Posted:
09 May 2023
You are already at the latest version
Abstract
A conjugate compound 5, (E)-6-hydroxy-2-oxo-2H-chromen-7-yl 3-(4-hydroxy-3-methoxyphenyl)acrylate, of 6,7-hydroxycoumarin (esculetin) 3 and (E)-3-(4-hydroxy-3-methoxyphenyl)- acrylic acid (ferulic acid) 1 was prepared in 61% yield by the esterification reaction of a protected ferulic acid 2a with esculetin 3 in the presence of triethylamine in dichloromethane for 3 h, followed by deprotection using 3M HCl. The structure of compound 5 was confirmed by 1H, 13C NMR spectroscopy, mass-spectrometry and elemental analysis.
Keywords:
coumarin
; esculetin
; ferulic acid
; esterification
; antioxidant
1. Introduction
Many coumarin-based derivatives are important structural scaffolds for the synthesis of potential biologically active compounds with different pharmacological applications [1]. They continue to be designed and synthesized [2] because of remarkable biological properties, including anticancer [3], anticonvulsant [4] antimicrobial [5], and antiviral [6] activities. Coumarins having intramolecular charge transfer character have also been investigated for fluorescence sensors [7,8]. Among them, 6,7-dihydroxycoumarin (esculetin) 3 displayed various biological activities such as anticancer [9,10], free radical scavenging [11], anti-inflammatory [12], anti-arthritic [13], and hepatoprotective [14]. And, (E)-3-(4-hydroxy-3-methoxyphenyl)acrylic acid (ferulic acid) 1 is widely present in seeds, nuts, leaves, and fruits. It has many pharmacological effects including antioxidant [15], anticancer [16], neuroprotective [17], and anti-metabolic syndrome [18]. However, there have been no reports on the synthesis of hybrid compounds made of 1 and 3. We report herein the synthesis of a new hybrid compound 5 of potential biological interest, (E)-6-hydroxy-2-oxo-2H-chromen-7-yl 3-(4-hydroxy-3-methoxyphenyl)acrylate.
2. Results
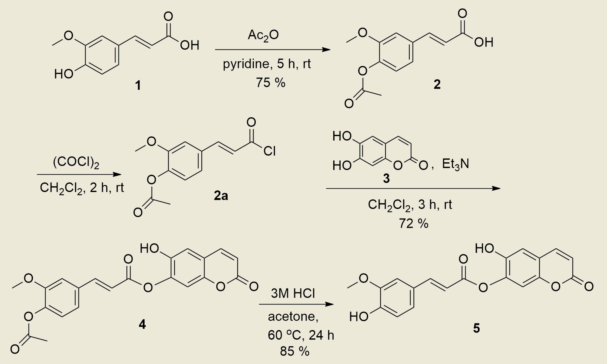
The new compound 5 was prepared as shown in Scheme 1. The hydroxy group of starting material 1 was first protected with acetic anhydride and pyridine according to the previously reported procedure [19] to give 2, (E)-3-(4-acetoxy-3-methoxyphenyl)acrylic acid. After 2 was activated with oxalyl chloride including DMF, the resultant 2a was allowed to react with 3 in dichloromethane at room temperature for 3 h in the presence of triethylamine to afford an esterified product 4, (E)-6-hydroxy-2-oxo-2H-chromen-7-yl 3-(4-acetoxy-3-methoxyphenyl)acrylate in 72% yield. The deprotection of acetyl group of 4 was achieved by use of 3M HCl solution in acetone at room temperature for 24 h to give a conjugate compound 5 in 85% yield.
The 1H NMR spectrum of 4 showed the expected pattern with two sharp singlets at δ 3.82 and 2.24 ppm attributed to methoxy and acetyl protons, respectively, and two doublets at δ 7.81 and 6.93 ppm (J = 16.0 Hz) due to trans vinyl protons in ferulic acid moiety. It also showed two doublets at δ 7.89 and 6.24 ppm (J = 9.5 Hz) due to cis vinyl protons of esculetin moiety, and the aromatic protons were shown as two singlets at δ 7.48, 6.86 and three doublets at δ 7.59 (d, J = 1.7 Hz), 7.36 (dd, J = 8.2, 1.7 Hz) and 7.13 ppm (d, J = 8.2 Hz). A sharp singlet at δ 10.93 of low field was shown for a hydroxy proton of esculetin moiety (Supplementary Materials). In the 13C NMR spectrum, compound 4 displayed three peaks δ 168.8, 165.0, 160.7 ppm for the two carbonyl and newly formed an ester carbon, including sixteen peaks for aromatic and vinyl carbons at δ 153.6, 153.2, 151.7, 146.4, 144.5, 141.9, 136.1, 133.3, 123.8, 122.5 (2C), 117.7, 112.9, 112.8, 111.4, 104.0 ppm, and two peaks for two methyl carbons at δ 56.6, 20.9 ppm. The mass spectrum showed m/z = 395 (M+-1) corresponding the molecular formula, C21H16O8, and elemental analysis also provided satisfactory results.
Compound 5 was confirmed by the absence of signals such as acetyl protons at δ 2.24 ppm in the 1H NMR, and a carbonyl carbon at δ 168.8 ppm in the 13C NMR spectrum, compared to the spectra of compound 4. Two singlets due to two hydroxy groups were shown at δ 10.88 and 9.64 ppm in the 1H NMR spectrum. The mass spectrum provided m/z = 353 (M+-1) corresponding the molecular formula, C19H14O7, and elemental analysis gave satisfactory results. The preliminary biological test of DPPH free radical scavenging activity [20,21] for 4 and 5 as antioxidant exhibited SC50 values of 40.4 and 2.36 μg/mL, respectively, compared to 1 (2.58 μg/mL) and 3 (0.82 μg/mL) with ascorbic acid (1.65 μg/mL) as positive control.
In conclusion, a new compound 5 of biological importance was effectively prepared in 61% yield by the esterification reaction of a protected ferulic acid 2a with esculetin 3 in the presence of triethylamine in dichloromethane for 3 h, followed by deprotection of acetyl group using 3M HCl in acetone.
3. Materials and Methods
3.1. General Information
Ferulic acid, esculetin, oxalic chloride, acetic anhydride, triethylamine, 1,1-diphenyl-2-picryhydrazyl (DPPH), ascorbic acid, and the dry organic solvents were purchased from Sigma-Aldrich (St. Louis, MO, USA) and TCI (Tokyo, Japan). Melting point was determined on Kofler apparatus. Thin-layer chromatography (TLC) was used to monitor reactions and performed using aluminum sheets precoated with silica gel 60 (HF254, Merck, Waltham, MA, USA), and visualized with UV radiation (Fisher Scientific, Waltham, MA, USA). The 1H, and 13C NMR spectrum were recorded in deuterated DMSO with TMS as the standard on a JEOL JNM-ECZ600R 500 FT-NMR (Tokyo, Japan). The mass spectrum was obtained with AGILENT1100 LCMS (Santa Clara, CA, USA) under electrospray ionization (ESI) conditions. Absorbance for the compounds was measured using SpectraMax Paradigm multi-mode microplate reader (San Jose, CA, USA).
3.2. Synthesis of (E)-6-Hydroxy-2-Oxo-2H-Chromen-7-yl 3-(4-Acetoxy-3-Methoxyphenyl)Acrylate (4)
To a stirred solution of 2 (1.0 g, 4.23 mmol) in dry dichloromethane (20 mL) containing few drops of DMF was added oxalyl chloride (1.07 g, 8.45 mmol), and stirred at room temperature for 2 h. After evaporation of the solution, the mixture was diluted with dichloromethane (20 mL), and added 3 (0.75 g, 4.23 mmol) and triethylamine (1.19 mL, 8.50 mmol). The resulting solution was stirred at room temperature for 3 h with monitoring. When the reaction was completed, the mixture was washed with 0.1M HCl solution (10 mL), water (10 mL), and extracted with dichloromethane (2 x 15 mL). The organic extracts were dried over MgSO4, filtered, and concentrated to dryness. The crude product was purified by column chromatography (eluent: ethyl acetate/n-hexane = 1/1, v/v) and recrystallized from ethanol to give white solid of 4 in 72% yield (1.20 g). Mp 212–213 °C; TLC Rf = 0.48 (dichloromethane/MeOH = 90/10). 1H NMR (500 MHz, DMSO-d6) (ppm) δ 10.93 (s, 1H), 7.89 (d, J = 9.5 Hz, 1H), 7.81 (d, J = 16.0 Hz, 1H), 7.59 (d, J = 1.7 Hz, 1H), 7.48 (s, 1H), 7.36 (dd, J = 8.2, 1.7 Hz, 1H), 7.13 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 16.0 Hz, 1H), 6.86 (s, 1H), 6.24 (d, J = 9.5 Hz, 1H), 3.81 (s, 3H), 2.24 (s, 3H). 13C NMR (126 MHz, DMSO-d6) (ppm) δ 168.8, 165.0, 160.7, 153.6, 153.2, 151.7, 146.4, 144.5, 141.9, 136.1, 133.3, 123.8, 122.5 (2C), 116.1, 113.6, 112.8, 112.1, 111.3, 104.0, 56.3. MS (ESI) m/z = 395 (M+-1). Anal. calcd. for C21H16O8, %: C, 63.64; H, 4.07. Found, %: C, 63.88; H, 4.20.
3.3. Synthesis of (E)-6-Hydroxy-2-Oxo-2H-Chromen-7-yl 3-(4-Hydroxy-3-Methoxyphenyl)Acrylate (5)
A solution of 4 (1.0 g, 2.82 mmol) in acetone (15 mL) containing 3M HCl (1 mL) was heated at 60 °C with stirring for 24 h. After reaction was completed, the mixture was added to saturated aqueous sodium bicarbonate (10 mL) and extracted with ethyl acetate (2 x 15 mL). The organic extracts were dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (eluent: dichloromethane/MeOH = 95/5, v/v) and recrystallized from ethanol to give white solid of 5 in 85% yield (0.84 g). Mp 232–233 °C; TLC Rf = 0.38 (dichloromethane/MeOH = 90/10). 1H NMR (500 MHz, DMSO-d6) (ppm) δ 10.88 (s, 1H), 9.64 (s, 1H), 7.88 (d, J = 9.5 Hz, 1H), 7.71 (d, J = 15.9 Hz, 1H), 7.45 (s, 1H), 7.39 (d, J = 1.6 Hz, 1H), 7.18 (dd, J = 8.2, 1.7 Hz, 1H), 6.85 (s, 1H), 6.79 (d, J = 8.2 Hz, 1H), 6.69 (d, J = 15.9 Hz, 1H), 6.24 (d, J = 9.5 Hz, 1H), 3.80 (s, 3H). 13C NMR (126 MHz, DMSO-d6) (ppm) δ 165.3, 160.7, 153.7, 153.1, 150.3, 148.5, 147.6, 144.5, 136.3, 125.9, 124.1, 122.5, 116.1, 113.6, 112.8, 112.1, 111.3, 104.0, 56.6. MS (ESI) m/z = 353 (M+-1). Anal. calcd. for C19H14O7, %: C, 64.41; H, 3.98. Found, %: C, 64.30; H, 4.09. 3.4. DPPH radical scavenging assay for the compounds.
Each sample was dissolved in methanol at various concentrations ranging from 0 to 100 μg/mL. Then, 50 μL of the sample solution was mixed with 450 μL of a DPPH solution (400 μM) and incubated for 30 minutes at 4 °C. The absorbance was measured at 517 nm using a microplate reader (SpectraMax Paradigm). The SC50, which is the minimum concentration (μg/mL) required to scavenge 50% of the DPPH radicals, was calculated based on the measured absorbance. Ascorbic acid was used as a positive control.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1-S6: 1H NMR, 13C NMR, and Mass spectra of compound 4 and 5.
Author Contributions
Conceptualization, methodology, investigation, writing—original draft preparation, writing—review and editing, Y.-H.S.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
This work was supported by Mokwon University.
Conflicts of Interest
The author declares no conflict of interest.
References
- Flores-Morales, V.; Villasana-Ruíz, A.P.; Garza-Veloz, I.; González-Delgado, S.; Martinez-Fierro, M.L. Therapeutic effects of coumarins with different substitution patterns. Molecules 2023, 28, 2413. [Google Scholar] [CrossRef] [PubMed]
- Lončarić, M.; Gašo-Sokač, D.; Jokič, S.; Molnar, M. Recent advances in the synthesis of coumarin derivatives from different starting materials. Biomolecules 2020, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, N.; Kumbhar, A.A.; Pokharel, Y.R.; Yadav, P.N. Anticancer potentials of coumarin and its derivatives. Mini Rev. Med. Chem. 2021, 21, 2996–3029. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Budagumpi, S.; Balappa Somappa, S. Synthetic and natural coumarins as potent anticonvulsant agents: A review with structure-activity relationship. J. Clin. Pharm. Ther. 2022, 47, 915–931. [Google Scholar] [CrossRef]
- Cheke, R.S.; Patel, H.M.; Ansari, I.A.; Ambhore, J.P.; Shinde, S.D.; Kadri, A.; Snoussi, M.; Adnan, M.; Kharkar, P.S.; Pasupuleti, V.R.; Deshmukh, P.K. Molecular insights into coumarin analogues as antimicrobial agents: Recent developments in drug discovery. Antibiotics 2022, 11, 566. [Google Scholar] [CrossRef]
- Li, Z.; Kong, D.; Liu, Y.; Li, M. Pharmacological perspectives and molecular mechanisms of coumarin derivatives against virus disease. Genes Dis. 2021, 9, 80–94. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, T.Z.; Zhao, Y.L.; Fan, D.W.; Xia, Y. J.; Zhang, P. Synthesis, crystal structure, photo- and electro-luminescence of 3-(4-(anthracen-10-yl)phenyl)-7-(N,N’-diethylamino)coumarin. Synth. Met. 2010, 160, 1642–1647. [Google Scholar] [CrossRef]
- Voutsadaki, S.; Tsikalas, G.K.; Klontzas, E.; Froudakis, G.E.; Katerinopoulos, H.E. A ‘turn-on” coumarin-based fluorescent sensor with high selectivity for mercury ions in aqueous media. Chem. Commun. 2010, 46, 3292–3294. [Google Scholar] [CrossRef]
- Arora, R.; Sawney, S.; Saini, V.; Steffi, C.; Tiwari, M.; Saluja, D. Esculetin induces antiproliferative and apoptotic response in pancreatic cancer cells by directly binding to KEAP1. Mol. Cancer. 2016, 15, 64. [Google Scholar] [CrossRef]
- Wang, G.; Lu, M.; Yao, Y.; Wang, J.; Li, J. Esculetin exerts antitumor effect on human gastric cancer cells through IGF-1/PI3K/Akt signaling pathway. Eur. J. Pharmacol. 2017, 814, 207–215. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, K.A.; Zhang, R.; Piao, M.J.; Ko, D.O.; Wang, Z.H.; Chae, S.W.; Kang, S.S.; Lee, K.H.; Kang, H.K.; et al. Protective effect of esculetin against oxidative stress-induced cell damage via scavenging reactive oxygen species. Acta Pharmacol. Sin. 2008, 29, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, T.; Huang, C.J.; Yen, T.L.; Hsia, C.W.; Sheu, J.R.; Bhavan, P.S.; Huang, W.C.; Hsieh, C.Y.; Hsia, C.H. Activation of Nrf2 by esculetin mitigates inflammatory responses through suppression of NF-κB signaling cascade in RAW 264.7 Cells. Molecules 2022, 27, 5143. [Google Scholar] [CrossRef] [PubMed]
- Rzodkiewicz, P.; Gasińska, E.; Gajewski, M.; Bujalska-Zadrożny, M.; Szukiewicz, D.; Maśliński, S. Esculetin reduces leukotriene B4 level in plasma of rats with adjuvant induced arthritis. Reumatologia 2016, 54, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Choi, R.Y.; Ham, J.R.; Lee, M.K. Esculetin prevents non-alcoholic fatty liver in diabetic mice fed high-fat diet. Chem. Biol. Interact. 2016, 260, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Sudheer, A.R.; Menon, V.P. Ferulic acid: Therapeutic potential through its antioxidant property. J. Clini. Biochem. Nutr. 2007, 40, 92–100. [Google Scholar] [CrossRef]
- Singh Tuli, H.; Kumar, A.; Ramniwas, S.; Coudhary, R.; Aggarwal, D.; Kumar, M.; Sharma, U.; Chaturvedi Parashar, N.; Haque, S.; Sak, K. Ferulic Acid: A Natural Phenol That Inhibits Neoplastic Events through Modulation of Oncogenic Signaling. Molecules 2022, 27, 7653. [Google Scholar] [CrossRef]
- Di Giacomo, S.; Percaccio, E.; Gullì, M.; Romano, A.; Vitalone, A.; Mazzanti, G.; Gaetani, S.; Di Sotto, A. Recent advances in the neuroprotective properties of ferulic acid in Alzheimer’s disease: A narrative review. Nutrients 2022, 14, 3709. [Google Scholar] [CrossRef]
- Li, Y.; Sair, A.T.; Zhao, W.; Li, T.; Liu, R.H. Ferulic acid mediates metabolic syndrome via the regulation of hepatic glucose and lipid metabolisms and the insulin/IGF-1 receptor/PI3K/AKT pathway in palmitiate-treated HepG2 cells. Agric. Food Chem. 2022, 70, 14706–14717. [Google Scholar] [CrossRef]
- Shirai, A.; Kajiura, M.; Omasa, T. Synergistic photobactericidal activity based on ultraviolet-A irradiation and ferulic acid derivatives. Photochem. Photobiol. 2015, 91, 1422–1428. [Google Scholar] [CrossRef]
- Um, J.N.; Mim, J.W.; Joo, K.S.; Kang, H.C. Antioxidant, anti-wrinkle activity and whitening effect of fermented mixture extracts of angelica gigas, paeonia lactiflora, rehmannia chinensis and cnidium officinale. Korean J. Medicinal Crop Sci. 2017, 25, 152–159. [Google Scholar] [CrossRef]
- Blois, M.S. Antioxidant determinations of by the use of a stable free radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of the target compound 5.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



