
Submitted:
08 May 2023
Posted:
09 May 2023
You are already at the latest version
Abstract
The circadian clock regulates daily changes in behavioral, endocrine and metabolic activities in mammals. Circadian rhythms in cellular physiology are significantly affected by aging. In particular, we previously found that aging has profound impact on daily rhythms in mitochondrial functions, leading to increased oxidative stress. However, this is unlikely due to malfunction of molecular clocks in the suprachiasmatic nucleus or peripheral tissues, as robust clock oscillations are observed therein in old animals. Nonetheless, aging induces changes in gene expression levels and rhythms in various peripheral and probably central tissues. In this article, we review findings on the roles of the circadian clock and aging in regulating mitochondrial rhythms and redox homeostasis. Chronic sterile inflammation is implicated in mitochondrial dysfunction and increased oxidative stress during aging. In particular, upregulation of CD38 by inflammatory response can disturb mitochondrial functions in multiple ways.
Keywords:
circadian rhythms
; aging
; suprachiasmatic nucleus
; peripheral clock
; inflammatory response
1. Introduction
Intrinsic circadian oscillations are present in certain nuclei of the central nervous system (e.g., suprachiasmatic nucleus (SCN)) and most peripheral tissues in mammals (1). At the molecular level, circadian oscillations are driven by core clock proteins that serve as transcription factors (TFs). In particular, the clock proteins CLOCK and BMAL1 dimerize to transcriptionally activate other core clock genes such as Per1/2 and Cry1/2, whose protein products antagonize the actions of CLOCK/BMAL1, forming a negative feedback loop (1). CLOCK/BMAL1 also activate the transcription of clock genes Nr1d1/2, whose protein products inhibit Bmal1 transcription, forming another feedback loop (1). In addition to regulating their own transcription to sustain circadian oscillation, clock proteins also exert broad control over other genes. As a result, about 10% of genes expressed in peripheral tissues are rhythmic, and the rhytmic genes are involved in nearly all aspects of cellular functions (e.g., metabolism, immune defense, and cell cycle regulation) (2, 3). Clock functions are tissue-specific, and gene expression rhythms vary greatly across tissues. Indeed, genomic binding of clock proteins are tissue-specific (4, 5). Those sites are found within open chromatin regions established by tissue-specific TFs (e.g., pionneer TFs) and some ubiquitously expressed TFs (6).
Core clock genes typically harbor mutiple cis-elements for clock proteins themselves, a strong control thought to ensure robust and resilient clock oscillation across cell types (6). However, circadian rhythms are not regulated by the intrinsic tissue clock alone. Extrinsic cues, including those derived from the SCN (neural, humoral, and behavioral cues such as body temperature) and communicating signals from other tissues, also regulate gene expression rhythms (7). Previous studies emphasize the influence of extrinsic cues on instrinsic clocks per se. Indeed, extrinsic cues can enage TFs (e.g., CREB and SRF) that regulate transcription of clock gene (1). Given that TFs typically have many genomic binding sites, they could also influence other genes besides clock genes. Indeed, in the absence of rhythmic cues from the SCN and other tissues, the intrinsic clock can only sustain the rhythms of a limited set of genes (e.g., core clock genes and some clock-controlled genes). Those results indicate that clock proteins often collaborate with other TFs in rhythm regulation (6). The collaboration can occur within the same enhancer co-bound by clock proteins and other TFs; it also can be achieved through looping of distinct enhancers bound by clock proteins and other TFs respectively to promoters of their common target genes. Such combinatorial control permits plasticity in circadian rhythm regulation by allowing TFs other than clock proteins to control rhythms (6). For example, high fat diet reprograms liver gene expression via PPPAγ and SREBP, but leaves the liver clock intact (8, 9). Lung adenocarcinoma in young mice releases cytokines to reprogram liver gene expression without disturbing the liver clock (10). Interestingly, aging also leads to extensive reprogramming of daily gene expression in peripheral tissues of mouse and man, while clock oscillations remain normal therein (11-14).
Mitochondria are the hub of metabolism and a significant source of reactive oxygen species (ROS). Their dysfunction is a hallmark of aging (15). Previously, we found robust daily rhythms of mitochondrial functions in livers of young mice, which are regulated by the clock (16). Moreover, we found that mitochondrial rhythms are disrupted in livers of old mice, while normal clock oscillation persists (16). Those results indicate that mitochondrial functions are regulated not only by the clock but also by age-related mechanisms. In particular, aging disrupts mitochondrial redox homeostasis and elevates oxidative stress. In this article, we first provided an overview of ROS production and antioxidant defense. We then focused on clock roles in regulating mitochondrial functions, including redox homeostasis. Finally, we discussed mechanisms that dysregulate mitochondrial functions during aging, and the consequences of age-related increase in oxidative stress to circadian timing functions.
2. ROS and Oxidative Stress
ROS is an umbrella term for free radicals (species with at least one free electron. E.g., the superoxide anion O2-·) and non-radical species derived from oxygen (e.g., H2O2) (17). Electron leakage from protein redox centers can produces O2- (18). Mitochondrial dehydrogenases and respiratory complexes of the electron transport chain (ETC) are all O2·- production sites (19). O2·- can also be directly produced by enzymes such as NADPH oxidases (NOXs), cytochrome P450 enzymes (CYPs), and xanthine oxidases (18). O2·- can react with nitric oxide (NO) to form peroxynitrite (ONOO-), and oxidants derived from nitric oxide are called reactive nitrogen species (RNS). O2·- can be dismutated to H2O2 spontaneously or by superoxide dismutase (SOD) enzymes. H2O2 is also enzymatically produced by oxidases. H2O2 can oxidize the sulfhydryl (-SH) group of cysteine in proteins into sulfenic acid (-SOH). The cysteine -SH group can also be converted into –SNO (S-nitrosylation) by NO. Both -SOH and -SNO can react with glutathione, either spontaneously or by enzymatic actions (S-glutathionylation); deglutathionylation is carried out by specialized enzymes (20). S-nitrosylation and S-glutathionylation are though to protect proteins from further oxidative and/or nitrosative modification. Indeed, the sulfenic acid group can be further oxidized by H2O2 into sulfinic acid (-SO2H) and sulfonic acid (-SO3H). While the former can be reversed by sulfiredoxin (SRX) (21), the latter form is not reversible. Importantly, H2O2 leads to production of the hydroxyl radical OH·-, a powerful oxidant that damages proteins, lipids, DNAs and sugars. OH·- can directly oxidize various protein residues. OH·- and free radicals derived from fatty acids damage lipids through lipid peroxidation reactions. The lipid peroxidation products (e.g., 4-HNE) are also highly reactive toward proteins and other biomolecules. Oxidization of proteins by OH·- and lipid peroxidation products leads to protein carbonylations that are not readily reversed.
Molecular damages by ROS/RNS result in oxidative stress (22). While ROS and RNS as well as oxidative stress can play signaling roles (see below), oxidative damages also disturb cellular functions. Dysfunctional biomolecules thus need to be eliminated, a task carried out by the proteasome and via autophagy. Hyperoxidized, S-glutathionylated and carbonylated proteins can be degraded by the 20S proteasome in an ATP- and ubiquitin-independent manner (23). Some damaged proteins and organelles (e.g. mitochondria) are degraded through autophagy (23). RNS plays complex roles in mitophagy (24). We focus below on the biology of ROS.
3. Redox Homeostasis: Maintaining the Balance between ROS Production and Antioxidant Defense
Besides eliminating oxidative damages via proteasome and autophagy, cells are endowed with antioxidant defense systems to reduce ROS production and oxidative stress. They include the glutathione (GSH), glutaredoxin (GRX), and thioredoxin (TRX) systems. While all systems use NADPH as the ultimate reducing power, each can have specialized functions (22). For example, peroxinredoxin 6 (PRDX6), glutathione S-transferases (GSTs), and glutathione peroxidase 4 (GPX4) can restrain damages by lipid peroxidation. GSTs and GRXs are mainly responsible for protein S-glutathionylation and its reversal, respectively. GPXs and PRDXs mitigate oxidative stress by catalyzing H2O2 removal. In particular, PRDX3 plays a prominent role in eliminating H2O2 in mitochondria. A cysteine residue is found at the catalytic center of PRDXs, and its sulfhydryl group is oxidized by H2O2 to sulfenic acid. Upon oxidization of typical 2-Cys PRDXs (PRDX1-4), intermolecular disulfide bonds are formed, which are resolved by TRX (25, 26). The dimer/monomer ratio of PRDX can be used as an indicator of oxidative stress. Sulfenic acid at PRDX active site can be further oxidized by H2O2 into sulfinic acid (antagonized by SRX) and even sulfonic acid. However, under physiological conditions, hyperoxidized PRDXs are typically of low abundance in cells and tissues, and they can be degraded by the 20S proteasome (27, 28).
4. ROS and Oxidative Stress Play Signaling Roles
ROS play signaling roles in physiology (17, 29). For example, O2·- can release Fe2+ from Fe-S (iron-sulfur) clusters in proteins to affect metabolism (29). ROS can also activate uncoupling proteins (UCPs) in mitochondrial inner membrane to dissipate the H+ electrochemical gradient for heat production instead of ATP synthesis (30, 31). ROS production by the ETC is associated with mitochondrial energetics and varies by ATP supply and demand (32, 33). A high electrochemical potential of H+ (the protonmotive force, pmf) promotes ROS production by the ETC (33). Buildup of pmf is favored under nutrient-rich conditions (e.g., using glucose or pyruvate as substrate), due to abundant NADH and FADH2 supply to the ETC. Increased ROS, in synergy with other factors such as mitochondrial [Ca2+] elevation, can trigger transient opening of the mitochondrial permeability transition pore (mPTP) (34) to decrease pmf and halt ATP production (32). Thus, ROS plays a signaling role to maintain ATP homeostasis (32). The molecular identity of mPTP and how ROS, Ca2+ and H+ regulate its opening are being elucidated (32, 35-37). Interestingly, transient mPTP opening triggers a burst of O2·- production, known as “ROS-induced ROS release” (38) or “mitoflash”(39). Mitoflash frequency signals basal ROS and oxidative stress levels.
Besides the roles in metabolism, ROS also regulate signal transduction. This role is carried out mainly by H2O2, which can oxidize redox-sensitive cysteines in target proteins. For example, AMPK, the metabolic regulator activated by glucose shortage and low energy charge (40), is subject to redox-based regulation. H2O2 can oxidize cysteines in AMPKα to hinder its activation by upstream kinases (41). PTEN, which dephosphorylates phosphatidylinositol 3,4,5-triphosphate (PIP3) to inhibit PI3K signaling, is also subject to redox-based regulation. Like AMPK, PTEN activity is inhibited when H2O2 oxidizes its active site cysteine, turning the sulfhydryl group (-SH) into sulfenic acid (-SOH). The –SOH group reacts with the -SH group of another cysteine in PTEN to form an intramolecular disulfide bond, thus inactivating PTEN (42). Protein tyrosine kinases (PTKs) are activated by cytokines, growth factors (e.g., insulin), and T- and B-cell receptor stimulation; protein tyrosine phosphatases (PTPs) negatively regulate PTK signaling (43). During PTK activation, O2·- is produced by NOXs and converted to H2O2; PTKs phosphorylates and inactivates PRDXs to allow localized H2O2 accumulation (44, 45). H2O2 oxidizes the active site cysteine to inactivate PTP, thus prolonging PTK signaling. PTP oxidization leads to the intramolecular formation of a disulfide (e.g., SHP2) or a sulfenyl-amide bond (e.g., PTP1B). Oxidation and inactivation of AMPK, PTEN, and PTPs by H2O2 are transient, and they can be reversed by reducing agents such as TRX and GSH (46).
5. ROS and Oxidative Stress Regulate Gene Transcription
Besides modulating signal transduction, ROS and oxidative stress also regulate transcription. A well-known example is their activation of the KEAP1-NRF2 system for antioxidant defense to maintain redox homeostasis. From a mechanistic point of view, antioxidant defense is related to xenobiotic detoxification, which consists of three phases (47). Phase I enzymes (e.g., CYPs) can activate xenobiotics to produce electrophilic products, which are conjugated with reducing agents by phase II enzymes (e.g., GSTs); conjugation products are excreted through transporters encoded by phase III genes. Some phase I (e.g., ALDH2) and II (e.g., GST) enzymes can detoxify lipid peroxidation products. NRF2 is a TF that can upregulate phase II genes. Normally, KEAP1 complexes with NRF2 at a 2:1 ratio and promotes NRF2 ubiquitination and degradation by proteasome. During xenobiotic detoxification, electrophilic products from phase I reactions can oxidize cysteine residues in KEAP1, leading to formation of disulfide bonds between KEAP1 dimer (48). The conformational changes in KEAP1 liberate NRF2. Evading degradation, NRF2 accumulates and moves into the nucleus to upregulate phase II genes (48). CYPs are also a ROS source. Similar to electrophiles, H2O2 oxidizes cysteine residues in KEAP1 to enable NRF2 for transcription regulation (49). Thus, the KEAP1-NRF2 system is employed as a negative feedback mechanism to defend against both electrophiles and ROS oxidants. ROS and oxidative stress also regulate other TFs, such as HIF-1α for adaptions to hypoxia (50). ROS also facilitates inflammatory responses and NF-κB activation (17). As discussed later, inflammatory TFs including NF-κB are most probably involved in age-related reprogramming of daily gene expression.
6. Reciprocal Regulation between the Circadian Clock and Redox Homeostasis
Given the broad control over cellular functions by the circadian clock, it is not a surprise that daily changes in redox regulation are observed in various tissues (51). The liver plays essential roles in metabolism and is an important model of peripheral clocks. Microarray studies revealed that some rhythmic genes in mouse liver are involved in xenobiotic detoxification (52, 53). Those include phase I (e.g., CYPs), II (e.g., GST), and III (e.g., ABC transporters) genes. In addition, xenobiotic receptors (e.g., Car) and Alas1 (for biosynthesis of heme, the prosthetic group of CYPs) are also rhythmically expressed. The circadian clock also regulates antioxidant defense genes Aldh2 and Nqo1 (54). The pentose phosphate pathway (PPP) is the major cellular source of NADPH. The circadian clock indirectly regulates PPP genes and NADPH production via PPARδ (55, 56). The clock also regulates NRF2 to control GSH-mediated antioxidant defense (57). Finally, the clock regulates autophagy (58, 59), which is known to reduce oxidative stress. The ULK1/2 kinases not only promote autophagy (peaking during late daytime in the mouse liver) but also promote NADPH production by the PPP (60). Those results highlight the critical roles of the circadian clock in coordinating redox regulation across the day.
In a reciprocal manner, redox states also affect the clock. For example, the redox states of NAD(H) and NADP(H) influence clock proteins’ DNA binding (61), and perturbing the PPP can alter clock dynamics (62). NRF2 can regulate clock genes Cry2 and Nr1d1 (62, 63). Another link between redox states and the circadian clock is redox regulation of ion channel functions in the SCN to influences its neural output (64).
Reciprocal regulation between the clock and redox states most probably has adaptive values. For most animals, a feeding/fasting cycle co-varies with their sleep/wake cycle across the day. Many liver functions (e.g., bile production) are related to feeding and exhibit robust daily changes. Clock-controlled induction of xenobiotic detoxification genes in anticipation of feeding could be beneficial for organismal health (53). During the glucose-rich feeding phase, increased ROS production may facilitate insulin signaling to regulate liver metabolism. On the other hand, during the fasting phase, glucose shortage can activate AMPK to promote autophagy and fatty acid oxidation (FAO) as alternative energy source, a metabolic adaptation that favors oxidative stress reduction (see next section). Thus, circadian redox homeostasis is manifested as a daily rhythm of redox states, which is an integral part of daily changes in metabolism and physiology. Clock gene mutations significantly disturb redox homeostasis. In particular, deficiencies within the positive limb of the clock (i.e., Clock, Bmal1, and Npas2) decrease the expression of antioxidant defense genes and increase oxidative stress in young mice (16, 54, 57, 65). A premature aging phenotype is observed in Bmal1 knockout mice (66, 67).
Intriguingly, metabolic and redox oscillations are found in red blood cells devoid of nuclear transcription and cells lacking functional molecular clocks (27, 68). Nonetheless, an intact clock sustains more robust and widespread circadian oscillations at molecular and cellular levels (69).
7. Mitochondrial Functions Are Rhythmic and Regulated by the Circadian Clock in Young Mice
Except for the 13 OXPHOS subunits encoded in mtDNA, all other mitochondrial proteins, including those involved in mtDNA replication, transcription, and mitochondrial protein translation, are encoded by the nuclear genome (70). Some of those genes appear under clock control (71, 72). We and others found daily changes in the expression of OXPHOS genes encoded not only by the nuclear genome but also by mtDNA in livers of young mice (16, 73). The daily changes in OXPHOS protein composition, along with other mitochondrial rhythms (see below), are probably optimized for daily changes in nutrient supply and energy demand that are associated with the feeding-fasting cycle. Depending on the feeding state, mitochondria switch fuel choice between pyruvate and fatty acid (74). That mitochondrial energetics and fuel usage vary over the day is clearly evidenced by daily changes in oxygen consumption rate and the respiratory exchange ratio in mice and men (73, 75, 76). Mitochondrial respiratory activities in vitro also change across the day, but the peak phases differ by substrates, consistent with daily changes in fuel usage (73, 77). Some mechanisms of mitochondrial fuel selection are known. For example, reversible phosphorylation of pyruvate dehydrogenase (PDH) controls mitochondrial pyruvate use. PDH is inactivated upon phosphorylation by PDH kinases and re-activated by a Ca2+-activated phosphatase (78). Ca2+ influx promotes pyruvate oxidation (78), and is limited by MICU1 (regulating Ca2+ influx via MCU) and OPA1 present at cristae junction (79). We found that phosphorylation of PDH-E1α in livers of young mice is increased at daytime, indicating that mitochondrial pyruvate oxidation is reduced during fasting (16). Mitochondria undergo structural changes in response to nutrient availability (80). Under nutrient-poor situations, OPA1 promotes fusion of the mitochondrial inner membrane and intracristal assembly of OXPHOS complexes and supercomplexes to facilitate ATP production (81, 82). We found daily changes in mitochondrial OPA1 abundance in mouse liver, with higher levels at daytime (16), consistent with the role of OPA1 in promoting FAO (83).
OPA1 not only promotes FAO but also curtail oxidative stress (84). ROS production rate by mitochondria in vitro is low during FAO (85). We found that mitochondrial oxidative stress in vivo, judged by the degree of PRDX3 dimerization in young mouse liver, is decreased during daytime, with a nadir at ZT10 (16). From a metabolic point of view, mitochondrial dehydrogenases and respiratory complexes are differentially employed when different respiratory substrates are used, so ROS production rate would vary by substrate, as indeed observed in vitro (85). Low ROS production rate during FAO could be accounted for by several mechanisms. First of all, FAO produces NADH and FADH2 at an equal molar ratio. Compared to NADH, FADH2 derived from FAO donates its electrons (via ETF) to the ETC downstream of ROS production sites in complex I, thus favors less ROS production (86). Other mechanisms exist to restrain oxidative stress during FAO. For example, during FAO in liver, TCA cycle metabolites are depleted by gluconeogenesis, and acetyl-CoAs from FAO are diverted to ketogenesis. As a result, less FADH2, NADH and ROS are made by TCA cycle in liver during FAO. Moreover, a limited NADH supply to the ETC during FAO would raise the NAD+/NADH ratio, thus activating sirtuins (87). Notably, the mitochondrial sirtuin SIRT3 plays a role in reducing oxidative stress. SIRT3 deacetylates various proteins (86), such as ETC proteins, FAO and antioxidant defense enzymes (e.g., IDH2 and SOD2). SIRT3 increases efficiency of ETC electron transfer to reduce ROS production, and also promotes antioxidant defense (86). We found that acetylation levels of SIRT3 targets change over the day, reaching lowest levels near ZT10, concurrent with the nadir of mitochondrial oxidative stress in livers of young mice (16). Overall, the daily rhythm in mitochondrial redox states is closely integrated with daily metabolic changes in mouse liver, such that low oxidative stress level is associated with FAO during fasting.
Consistent with clock control of mitochondrial functions (Figure 1) (88), most mitochondrial rhythms we found in livers of young mice, including the redox rhythm, are disrupted by the Clock△19 mutation (16). The circadian clock also controls a redox rhythm at the whole cell level (89).
8. Mitochondrial Rhythms in Mouse Liver Are Disrupted by aging Despite Normal Circadian Clock Oscillation
Intriguingly, we found that the mitochondrial rhythms evident in young mice are disrupted by aging, due mainly to rhythm damping by age-related changes during daytime, especially at ZT10 (16). For example, mtDNA transcripts in livers of old mice are much less abundant at ZT10, the peak time in young mice. Meanwhile, SIRT3 target acetylation in old mice is increased at ZT10, the nadir of corresponding rhythms in young mice. Aging abolishes the mitochondrial redox rhythm, owing to a prominent increase of oxidative stress in old mice at ZT10, the nadir of PRDX3 dimerization in young mice. The age-related disruption of mitochondrial rhythms, however, is not associated with overt clock defects. We found that clock gene rhythms remain normal with age in mouse liver (16), consistent with other studies on mouse peripheral tissues (11-13) and the SCN (90-92). Those results indicate that aging disrupts mitochondrial rhythms through molecular mechanism(s) downstream of the circadian clock. Indeed, deep sequencing studies revealed that, while robust clock oscillations are preserved in peripheral tissues, aging induces extensive changes in gene levels and rhythms therein (11-13). Age-related global changes in SCN gene expression await future studies. Some genes related to mitochondrial functions in various tissues, including the few we identified in mouse liver (16), are expected to be affected by aging. Such genes remain to be fully characterized.
9. Mechanism(s) for Age-Related Dysregulation of Mitochondrial Rhythms
The mechanisms for age-related reprogramming of daily gene expression are under current investigation (Figure 2). Gene expression is known to be controlled by both the circadian clock and other TFs, either independently or in combination (6). Conceivably, some TFs whose activities are altered by aging take part in reprogramming daily gene expression. Plausible candidates are TFs (e.g., NF-κB, IRFs and STATs) activated by chronic inflammation, a hallmark of aging (93). Indeed, inflammatory response genes are enriched in tissues of old mice (11, 13). Unlike the strong immune response that disrupts the molecular clock after a LPS challenge (94), age-related sterile inflammation is of low grade, thus may not significantly disturb the molecular clock (95), at least at the early stage of aging examined (11-13, 16).
One notable gene target of inflammatory signaling is CD38, a NADase whose up-regulation contributes to the age-related decline in NAD+ levels in mouse tissues (96). Low NAD+ levels lead to insufficient sirtuin activation that has pleotropic effects (97). For example, SIRT6 restrains NF-κB signaling, and its deficiency leads to premature aging. SIRT1 deficiency promotes P53 activation and cellular senescence. SIRT1 also deacetylates PGC1α to activate the transcription of genes involved in mitochondrial functions (98). Insufficient SIRT1 activation during aging may disturb mitochondrial rhythms. Decreased SIRT3 activation may be particularly relevant to the disruption of mitochondrial redox rhythms during aging, as evidenced by the increase in both SIRT3 target acetylation and mitochondrial oxidative stress in livers of old mice (16). SIRT3 also promotes mitobiogenesis via deacetylating TFAM, a protein essential for mtDNA maintenance and transcription (99). Decreased SIRT3 activation probably contributes to the reduction of mtDNA transcripts during aging (16).
Increased oxidative stress is implicated in age-related dysregulation of SCN neural activity, which impairs circadian timing functions at the system level (100, 101). Age-related disturbance of redox homeostasis could also impact signaling pathways in various tissues. For example, AMPK and mTOR are alternatively activated across the day in young mice, in part due to influence by the redox rhythm. Dysregulation of AMPK and mTOR during aging could induce changes in metabolism and gene expression (102). Finally, ROS causes damages to DNA, which activate ATM to regulate the DNA damage response (DDR) of DNA repair, cell cycle arrest and/or apoptosis (103). ATM is also activated by ROS and promote NADPH production and autophagy to lower oxidative stress (103). ATM activation in young animals is expected to be rhythmic across the day. However, during aging, loss of redox homeostasis and chronic increase in oxidative stress could favor persistent ATM activation, a condition under which ATM and the DDR promote SASP (senescence-associated secretory phenotype) in cells to exacerbate systemic inflammation (104). Such a vicious cycle may eventually lead to the accumulation of DNA damages and loss of proteostasis in various tissues, which can aggravate cellular dysfunctions and age-related diseases.
10. Summary and Perspective
From the perspective of circadian rhythms, mitochondrial functions are regulated by both the clock and age-related mechanisms. In young animals, mitochondrial functions and redox states are rhythmic and integrated with daily changes in metabolic activities. Mitochondrial rhythms are disrupted during aging, when redox homeostasis is disturbed due to unbalanced ROS production and antioxidant defense, resulting in a net increase in oxidative stress. Age-related disruption of mitochondrial rhythms is associated with global changes in gene expression levels and rhythms in spite of intact tissue clocks. Such reprogramming of daily gene expression is a manifestation of plasticity in circadian rhythm control, and involves TFs activated during aging (e.g., inflammatory TFs).
Maintaining proper circadian rhythms at the cellular and organismal levels is beneficial to health. For example, time-restricted feeding (tRF) enable robust daily rhythms to prevent metabolic diseases in young mice and men (105). The tRF factor contributes to the lifespan-extending effect of caloric restriction (CR) in mice (13). It is of interest to examine the effect of CR on mitochondrial rhythms during aging. Finally, the phases of mitochondrial rhythms should be taken into account when administering daily pharmaceutical interventions aimed for healthy aging (102).
Acknowledgments
This work is supported by the National Natural Science Foundation of China (NSFC31971091).
References
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Mure, L.S.; et al. Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 2018, 359, eaao0318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef] [PubMed]
- Beytebiere, J.R.; et al. R.; et al. Tissue-specific BMAL1 cistromes reveal that rhythmic transcription is associated with rhythmic enhancer-enhancer interactions. Genes. Dev. 2019, 33, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; et al. GENE REGULATION. Discrete functions of nuclear receptor Rev-erbalpha couple metabolism to the clock. Science 2015, 348, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Beytebiere, J.R.; Greenwell, B.J.; Sahasrabudhe, A.; Menet, J.S. Clock-controlled rhythmic transcription: Is the clock enough and how does it work? Transcription 2019, 10, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Koronowski, K.B.; Sassone-Corsi, P. Communicating clocks shape circadian homeostasis. Science 2021, 371, eabd0951. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Mahan, K.L.; et al. Reprogramming of the circadian clock by nutritional challenge. Cell 2013, 155, 1464–1478. [Google Scholar] [CrossRef]
- Guan, D.; et al. Diet-Induced Circadian Enhancer Remodeling Synchronizes Opposing Hepatic Lipid Metabolic Processes. Cell 2018, 174, 831–842. [Google Scholar] [CrossRef]
- Masri, S.; et al. Lung Adenocarcinoma Distally Rewires Hepatic Circadian Homeostasis. Cell 2016, 165, 896–909. [Google Scholar] [CrossRef]
- Solanas, G.; et al. Aged Stem Cells Reprogram Their Daily Rhythmic Functions to Adapt to Stress. Cell 2017, 170, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; et al. Circadian Reprogramming in the Liver Identifies Metabolic Pathways of Aging. Cell 2017, 170, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Rodriguez, V.; et al. Circadian alignment of early onset caloric restriction promotes longevity in male C57BL/6J mice. Science 2022, 376, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Talamanca, L.; Gobet, C.; Naef, F. Sex-dimorphic and age-dependent organization of 24-hour gene expression rhythms in humans. Science 2023, 379, 478–483. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Gong, C.; et al. The daily rhythms of mitochondrial gene expression and oxidative stress regulation are altered by aging in the mouse liver. Chronobiol. Int. 2015, 32, 1254–1263. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2033, 552 Pt 2, 335–344. [Google Scholar] [CrossRef]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef]
- Belcastro, E.; et al. Regulation of protein function by S-nitrosation and S-glutathionylation: Processes and targets in cardiovascular pathophysiology. Biol. Chem. 2017, 398, 1267–1293. [Google Scholar] [CrossRef]
- Jeong, W.; Bae, S.H.; Toledano, M.B.; Rhee, S.G. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic. Biol. Med. 2012, 53, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Montagna, C.; Cirotti, C.; Rizza, S.; Filomeni, G. When S-Nitrosylation Gets to Mitochondria: From Signaling to Age-Related Diseases. Antioxid. Redox Signal 2020, 32, 884–905. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.S.; Yoon, H.J.; Kim, J.Y.; Woo, H.A.; Rhee, S.G. Circadian rhythm of hyperoxidized peroxiredoxin II is determined by hemoglobin autoxidation and the 20S proteasome in red blood cells. Proc. Natl. Acad. Sci. USA 2014, 111, 12043–12048. [Google Scholar] [CrossRef]
- Kil, I.S.; et al. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell 2012, 46, 584–594. [Google Scholar] [CrossRef]
- Sies, H.; et al. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Echtay, K.S.; Murphy, M.P.; Smith, R.A.; Talbot, D.A.; Brand, M.D. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J. Biol. Chem. 2002, 277, 47129–47135. [Google Scholar] [CrossRef]
- Chouchani, E.T.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; et al. Mitochondrial flashes regulate ATP homeostasis in the heart. Elife 2017, 6, e23908. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Ramzan, R.; Wen, L.; Vogt, S. New extension of the Mitchell Theory for oxidative phosphorylation in mitochondria of living organisms. Biochim. Biophys. Acta 2017, 1800, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Carraro, M.; Lippe, G. The mitochondrial permeability transition: Recent progress and open questions. FEBS J. 2022, 289, 7051–7074. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; et al. Protons Trigger Mitochondrial Flashes. Biophys. J. 2016, 111, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; et al. Synergistic triggering of superoxide flashes by mitochondrial Ca2+ uniport and basal reactive oxygen species elevation. J. Biol. Chem. 2013, 288, 4602–4612. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Feng, G.; et al. . Mitoflash biogenesis and its role in the autoregulation of mitochondrial proton electrochemical potential. J. Gen. Physiol. 2019, 151, 727–737. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef]
- Shao, D.; et al. . A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; et al. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.A.; et al. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Schwertassek, U.; et al. Reactivation of oxidized PTP1B and PTEN by thioredoxin 1. FEBS J. 2014, 281, 3545–3558. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, C.Y.; Kong, A.N. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 2005, 28, 249–268. [Google Scholar] [CrossRef]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox Signal 2010, 13, 1665–1678. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef]
- Hardeland, R.; Coto-Montes, A.; Poeggeler, B. Circadian rhythms, oxidative stress, and antioxidative defense mechanisms. Chronobiol. Int. 2003, 20, 921–962. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002, 109, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Gachon, F.; Olela, F.F.; Schaad, O.; Descombes, P.; Schibler, U. The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell Metab. 2006, 4, 25–36. [Google Scholar] [CrossRef]
- Musiek, E.S.; et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Invest. 2013, 123, 5389–5400. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; et al. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 2013, 502, 550–554. [Google Scholar] [CrossRef]
- Pekovic-Vaughan, V.; et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes. Dev. 2014, 28, 548–560. [Google Scholar] [CrossRef]
- Ma, D.; Panda, S.; Lin, J.D. Temporal orchestration of circadian autophagy rhythm by C/EBPbeta. EMBO J. 2011, 30, 4642–4651. [Google Scholar] [CrossRef]
- Sulli, G.; et al. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018, 553, 351–355. [Google Scholar] [CrossRef]
- Li, T.Y.; et al. ULK1/2 Constitute a Bifurcate Node Controlling Glucose Metabolic Fluxes in Addition to Autophagy. Mol. Cell 2016, 62, 359–370. [Google Scholar] [CrossRef]
- Rutter, J.; Reick, M.; Wu, L.C.; McKnight, S.L. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 2001, 293, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Rey, G.; et al. The Pentose Phosphate Pathway Regulates the Circadian Clock. Cell Metab. 2016, 24, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Wible, R.S.; et al. NRF2 regulates core and stabilizing circadian clock loops, coupling redox and timekeeping in Mus musculus. Elife 2018, 7, e31656. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.A.; et al. Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science 2012, 337, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Q.; et al. Diurnal variation of hepatic antioxidant gene expression in mice. PLoS ONE 2012, 7, e44237. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.A.; Weaver, D.R. Disrupting the circadian clock: Gene-specific effects on aging, cancer, and other phenotypes. Aging 2011, 3, 479–493. [Google Scholar] [CrossRef]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes. Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef]
- O'Neill, J.S.; Reddy, A.B. Circadian clocks in human red blood cells. Nature 2011, 469, 498–503. [Google Scholar] [CrossRef]
- Greco, C.M.; et al. Integration of feeding behavior by the liver circadian clock reveals network dependency of metabolic rhythms. Sci. Adv. 2021, 7, eabi7828. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef]
- Rey, G.; et al. Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol. 2011, 9, e1000595. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, B.M.; et al. Disrupted circadian oscillations in type 2 diabetes are linked to altered rhythmic mitochondrial metabolism in skeletal muscle. Sci. Adv. 2021, 7, eabi9654. [Google Scholar] [CrossRef] [PubMed]
- Neufeld-Cohen, A.; et al. Circadian control of oscillations in mitochondrial rate-limiting enzymes and nutrient utilization by PERIOD proteins. Proc. Natl. Acad. Sci. USA 2016, 113, E1673–E1682. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Chaix, A.; Lin, T.; Le, H.D.; Chang, M.W.; Panda, S. Time-Restricted Feeding Prevents Obesity and Metabolic Syndrome in Mice Lacking a Circadian Clock. Cell Metab. 2019, 29, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Zitting, K.M.; et al. Human Resting Energy Expenditure Varies with Circadian Phase. Curr. Biol. 2018, 28, 3685–3690. [Google Scholar] [CrossRef] [PubMed]
- Peek, C.B.; et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 2013, 342, 1243417. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef]
- Gottschalk, B.; Madreiter-Sokolowski, C.T.; Graier, W.F. Cristae junction as a fundamental switchboard for mitochondrial ion signaling and bioenergetics. Cell Calcium, 1025. [Google Scholar]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef]
- Patten, D.A.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Cabrera, R.; et al. Opa1 relies on cristae preservation and ATP synthase to curtail reactive oxygen species accumulation in mitochondria. Redox Biol. 2021, 41, 101944. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Guarente, L. The SirT3 divining rod points to oxidative stress. Mol. Cell 2011, 42, 561–568. [Google Scholar] [CrossRef]
- Canto, C.; et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Mezhnina, V.; Ebeigbe, O.P.; Poe, A.; Kondratov, R.V. Circadian Control of Mitochondria in Reactive Oxygen Species Homeostasis. Antioxid. Redox Signal 2022, 37, 647–663. [Google Scholar] [CrossRef]
- Khapre, R.V.; Kondratova, A.A.; Susova, O.; Kondratov, R.V. Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle 2011, 10, 4162–4169. [Google Scholar] [CrossRef]
- Farajnia, S.; et al. Evidence for neuronal desynchrony in the aged suprachiasmatic nucleus clock. J. Neurosci. 2012, 32, 5891–5899. [Google Scholar] [CrossRef]
- Nakamura, T.J.; et al. Age-related decline in circadian output. J. Neurosci. 2011, 31, 10201–10205. [Google Scholar] [CrossRef]
- Yamazaki, S.; et al. Effects of aging on central and peripheral mammalian clocks. Proc. Natl. Acad. Sci. USA 2002, 99, 10801–10806. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.K.; et al. Requirement for NF-kappaB in maintenance of molecular and behavioral circadian rhythms in mice. Genes. Dev. 2018, 32, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.C.; et al. NAD(+) Controls Circadian Reprogramming through PER2 Nuclear Translocation to Counter Aging. Mol. Cell 2020, 78, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Pereira, J.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann. N. Y Acad. Sci. 2008, 1147, 321–334. [Google Scholar] [CrossRef]
- Liu, H.; Li, S.; Liu, X.; Chen, Y.; Deng, H. SIRT3 Overexpression Inhibits Growth of Kidney Tumor Cells and Enhances Mitochondrial Biogenesis. J. Proteome Res. 2018, 17, 3143–3152. [Google Scholar] [CrossRef]
- Farajnia, S.; Deboer, T.; Rohling, J.H.; Meijer, J.H.; Michel, S. Aging of the suprachiasmatic clock. Neuroscientist 2014, 20, 44–55. [Google Scholar] [CrossRef]
- Allen, C.N.; Nitabach, M.N.; Colwell, C.S. Membrane Currents, Gene Expression, and Circadian Clocks. Cold Spring Harb. Perspect. Biol. 2017, 9, a027714. [Google Scholar] [CrossRef]
- Acosta-Rodriguez, V.A.; Rijo-Ferreira, F.; Green, C.B.; Takahashi, J.S. Importance of circadian timing for aging and longevity. Nat. Commun. 2021, 12, 2862. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 796–814. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Chaix, A.; Manoogian, E.N.C.; Melkani, G.C.; Panda, S. Time-Restricted Eating to Prevent and Manage Chronic Metabolic Diseases. Annu. Rev. Nutr. 2019, 39, 291–315. [Google Scholar] [CrossRef]
Figure 1.
Mitochondrial function are regulated by the circadian clock and other TFs in young animals. Such combinatorial regulation coordinates robust daily changes in mitochondrial protein composition, structural dynamics, energtics and metabolism. There is a clear mitochondrial redox rhythm that is integrated with daily rhythms in metabolism.
Figure 1.
Mitochondrial function are regulated by the circadian clock and other TFs in young animals. Such combinatorial regulation coordinates robust daily changes in mitochondrial protein composition, structural dynamics, energtics and metabolism. There is a clear mitochondrial redox rhythm that is integrated with daily rhythms in metabolism.
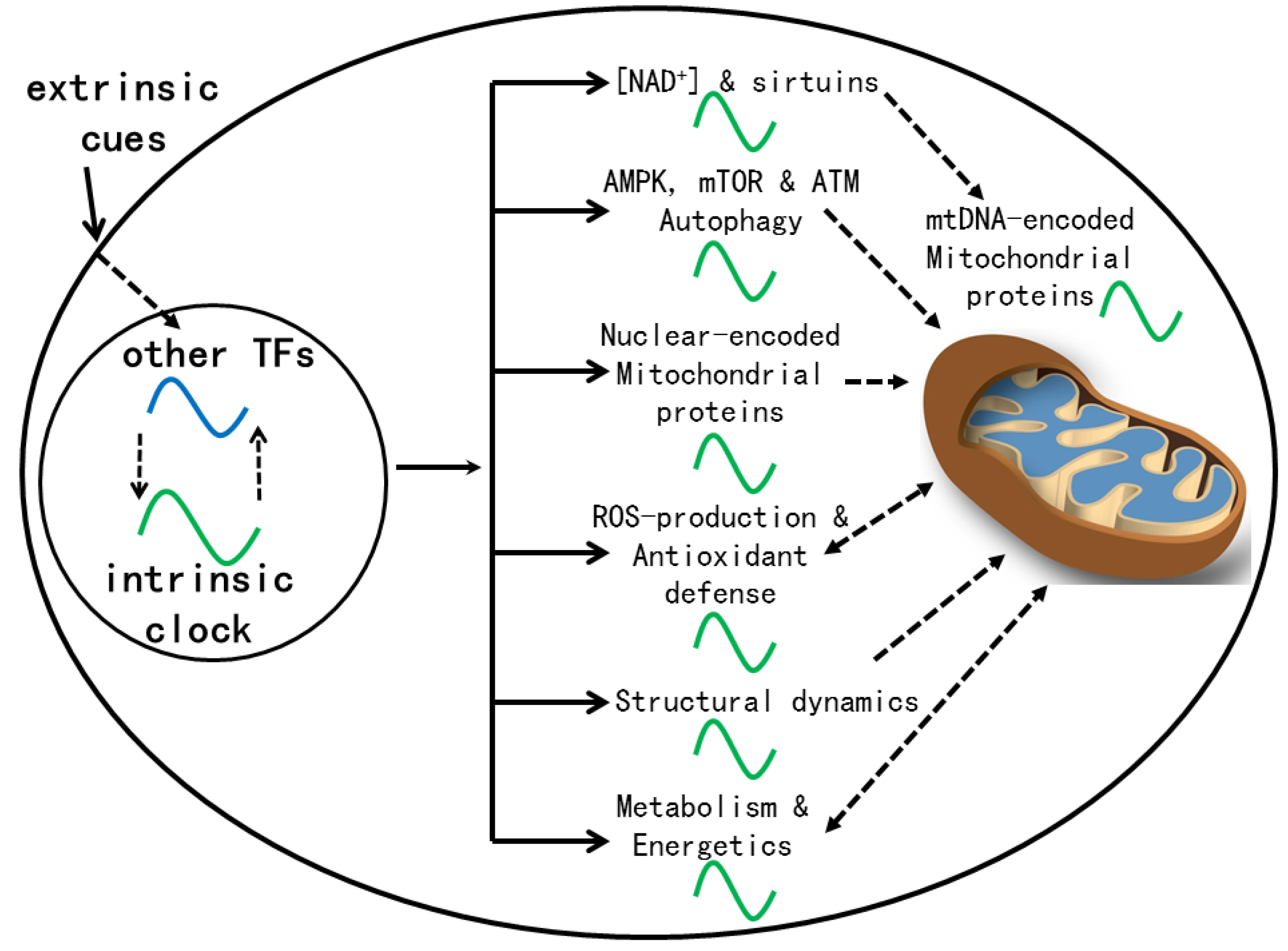
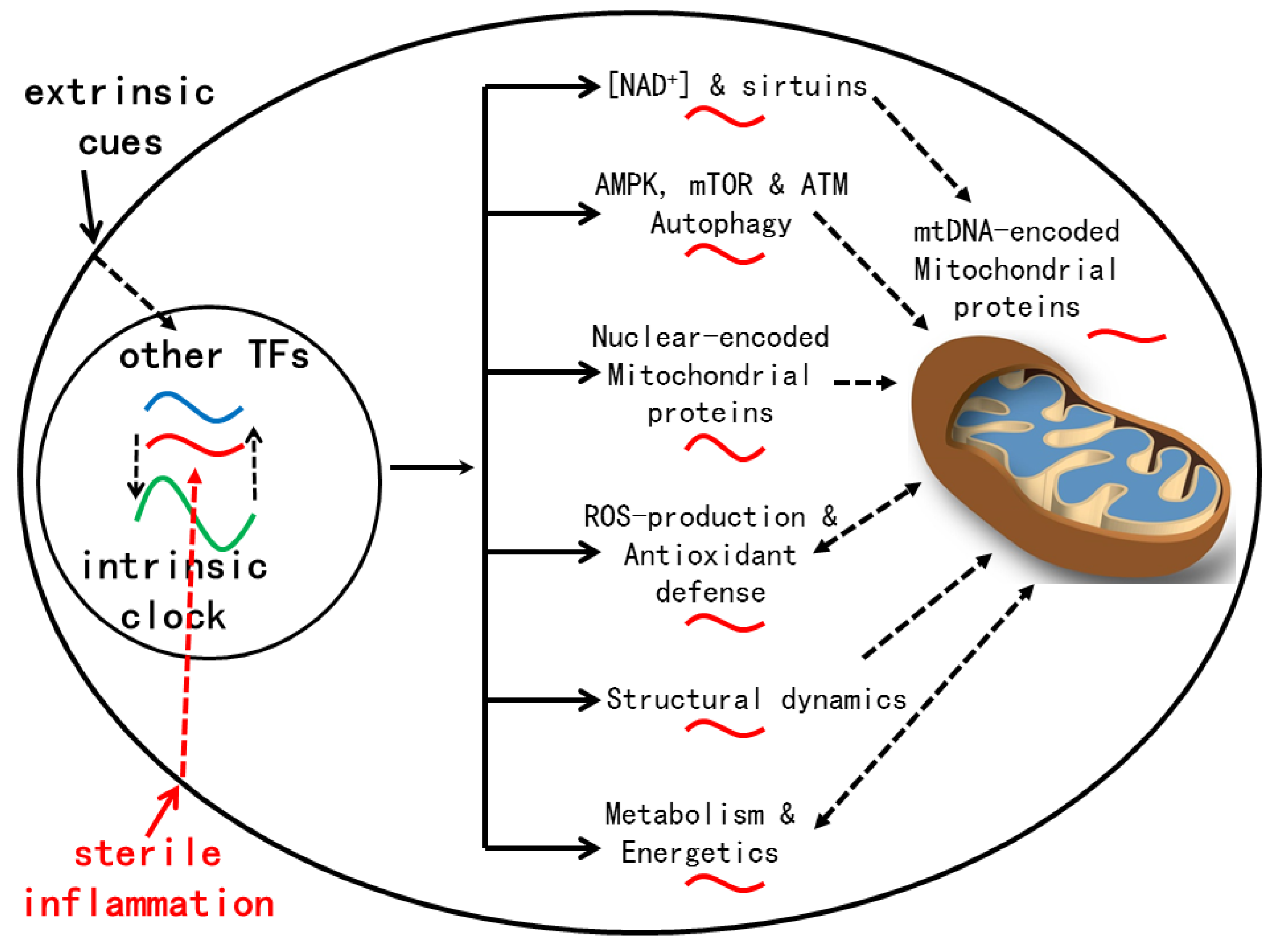
Figure 2.
During aging, age-related activation of certain TFs such as inflammatory TFs leads to the reprogramming of daily gene expression that has profound effects on mitochondrial physiology, leading to the disruption of most mitochondrial rhythms that are evident in young animals. Such dysregulation of mitochondrial functions is associated with metabolic changes and disturbed redox homeostasis.
Figure 2.
During aging, age-related activation of certain TFs such as inflammatory TFs leads to the reprogramming of daily gene expression that has profound effects on mitochondrial physiology, leading to the disruption of most mitochondrial rhythms that are evident in young animals. Such dysregulation of mitochondrial functions is associated with metabolic changes and disturbed redox homeostasis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



