Submitted:
08 May 2023
Posted:
09 May 2023
You are already at the latest version
Abstract
Alcohol effects on hepatic lipid metabolism through various mechanisms, leading synergistically to an accumulation of fatty acids (FA) and triglycerides. Obesity, as well as, the dietary fat [saturated fatty acids (FA) versus poly-unsaturated fatty acids (PUFA)] may modulate the hepatic fat. Alcohol inhibits adenosine monophosphate activated kinase (AMPK). AMPK activates peroxisome proliferators activated receptor a (PPARα) and leads to a decreased activation of sterol regulatory element binding protein 1c (SRABP1c).
The inhibition of AMPK, and thus of PPARα results in an inhibition of FAs oxidation. This ß-oxidation is further reduced due to mitochondrial damage induced through cytochrome P4502E1 (CYP2E1) driven oxidative stress. Furthermore, the synthesis of FAs is stimulated through an activation of SHREP1. In addition, alcohol consumption leads to a reduced production of adiponectin in adipocytes due to oxidative stress and to an increased mobilization of FAs from adipose tissue and from the gut as chylomicrons.
On the other side, the secretion of FAs via very low density lipoproteins (VLDL) from the liver is inhibited by alcohol. Alcohol also affects signal pathways such as early growth response 1 (Egr-1) associated with the expression of tumour necrosis factor (TNF), and the mammalian target of rapamycin (mTOR) a key regulator of autophagy. Both have influence the pathogenesis of alcoholic fatty liver.
Alcohol-induced gut dysbiosis is contributing to the severity of ALD by increasing metabolism of ethanol in the gut and promoting intestinal dysfunction. Moreover, pathogen associated molecular paterns (PAMPS) via specific Toll-like receptors (TLR) bacterial overgrowth is leading to the translocation of bacteria. Endotoxins, and toxic ethanol metabolites enter the enterohepatic circulation reaching the liver, inducing the activation of the nuclear factor kappa-B (NFB) pathway.
Pro-inflammatory cytokines released in the process contribute to inflammation and fibrosis. In addition, cellular apoptosis is inhibited in the favor of necrosis.
Keywords:
alcoholic fatty liver
; adenosine monophosphate activated kinase
; sterol regulatory element binding protein 1c
; peroxisome proliferator activated receptor
; oxidative stress
; cytochrome P450 2E1
1. Introduction
Alcohols are a group of hydrocarbons containing one or more hydroxyl (-OH) groups. Ethanol (C2H5OH, ethyl alcohol) is the main psychoactive ingredient in alcoholic beverages. Under usual conditions, beverages produced by fermentation have an alcohol concentration of maximum 14%. By distillation, the fermented mixture has is a pure condensate. Alcohol is a sedative with hypnotic effects. Alcohol intoxication may result in amnesic syndrome, long-term dependence and in a variety of physical and mental disorders [1. 2].
A volume of beverage alcohol ( e.g. a glass of wine, a can of beer, or a mixed drink containing distilled spirits) that contains approximately the same amounts (in grams) of ethanol regardless of the type of beverage is termed standard drink. The term is often used to educate alcohol users about the similar effects associated with consuming different alcoholic beverages served in standard-sized glasses or containers (e.g. the effects of one glass of beer are equal to those of one glass of wine). In the United Kingdom, the term ''unit" is employed, where one unit of an alcoholic beverage contains approximately 8-9 grams of ethanol; in North American literature, ''a drink" contains about 12 grams of ethanol. In other countries, the amounts of alcohol chosen to approximate a standard drink may be greater or less, depending on local customs and beverage packaging.
Alcohol-related mental and behavioral disorders are classified as psychoactive substance use disorders in International Classification of Diseases (ICD-10). Other organ-induced damage is represented by cardiomyopathy, alcoholic hepatitis, pancreatitis, and injury produced to other systems. The physiological health consequences of alcohol consumption are closely correlated with the pattern of drinking. The first pattern of drinking is moderate alcohol consumption (in the USA defined as no more than one drink/day for women, and no more than two drinks/day for men). The second pattern of drinking which could be harmful to health is the high-risk drinking. Organ toxicity is mostly associated with the second pattern of drinking [1].
Fat accumulation in the liver is an early and frequent response of the liver towards chronic alcohol consumption. Over 90% of regular alcohol consumers over a longer period of time develop fatty liver (FL) [1,2]. In addition, FL also occurs in binge drinkers. These are individuals who consume large amounts of alcohol within hours [2]. This effect has been shown in animal experiments. Already 12 hours after the application of a high dose of alcohol to mice, a 10-fold increase in hepatic triglycerides has been observed [3].
Histologically, FL is characterized by the deposition of fat droplets in the cytoplasm of hepatocytes. FL is defined as a liver in which more than 5% of hepatocytes reflect fat deposition. Hepatic fat accumulation after alcohol consumption occurs primarily around the central vein, from where it extends to the centre of hepatic lobule and, finally, towards the portal area. In alcoholics, steatosis is predominantly a macro-vesicular and rarely micro-vesicular. Chemically, the deposition of triglycerides in fat droplets of hepatocytes is very characteristic [4].
Following alcohol abstinence, alcoholic FL disappears fast [5,6,7]. Steatosis is not associated with any symptoms. This is why alcoholic FL has been classified as a side effect of alcohol consumption and as a harmless precondition of alcohol-induced liver damage. However, under certain conditions alcoholic fatty liver may progress to inflammation, fibrosis, and cirrhosis [1]. Data emphasized the fact that free fatty acids (FFA) play a pathological role in this process [2]. Therefore, it is of major significance to understand the effect of alcohol on fatty acid metabolism and the mechanisms by which alcohol induces FL.
Under physiological condition, fatty acids from circulation enter into the hepatocytes. The neo-synthesis of fatty acids on one side, as well as, the degradation of fatty acids and their secretion on the other side keep a certain balance. If one of these factors changes, it will be compensated by another mechanism. For example an increased availability and an increased uptake of fatty acids into the hepatocyte are followed by an increased ß-oxidation to acetyl-CoA which can then be utilized in the citric cycle for energy generation. Acetyl-CoA can also be used for the generation and secretion of ketone-bodies to other organs, for example to muscle cells and the brain, where the ketone bodies are used for energy production. If no energy is required, fatty acids are esterified to triglycerides, stored in fat droplets and then oxidized when needed or secreted in the form of very low-density lipoproteins (VLDL) into the circulation [8,9,10].
Therefore, under physiological conditions, intercellular lipid concentrations are observed only for a short period of time. Alcohol, however, disturbs the homeostasis of lipid metabolism by various mechanisms and, as a consequence, fat is accumulated in the liver (Fig.1).
This review describes metabolic and molecular factors inducing alcoholic FL. For more details it is referred to the following review articles [1,2,8,9,10,11,12].
The types of alcoholic drinks impact liver damage by adding new converges. There is an association between the drink, its specific taste, scent and these congeners. The specific taste of the drink has an influence on the consumer preference and consumption of alcohol. Alcohol congener’s interaction with the human organism follows different pathways in comparison to ethanol concentration [10]. Product flavor is an important component in product selection, primarily endorsing preference for sweet e-liquid flavor (e.g., fruit) in women and young adolescents, compared with preferences reported by older adults [10]
2. Metabolism of Ethanol and Ethanol as a Source of Energy
The physiological burning value of pure alcohol is 30 kJ/g, which is approximately 7 kcal/g ethanol. This means for example that 500 mL beer with 5% volume of alcohol content have approximately 140 kcal. Furthermore, each beverage has, in addition to its ethanol content, also calories from other components such as sugar as a source of energy [13].
Ethanol cannot be stored in the body. Ethanol is primarily to over 90% metabolized in the liver and is oxidized via alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH). These enzymes are in cytoplasm of hepatocytes. Chronic alcohol consumption induces cytochrome P450 2E1 in the endoplasmic reticulum of the hepatocytes. This so called microsomal ethanol oxidizing system (MEOS) also generates acetaldehyde (AA) [14]. Only a small percentage of ethanol is oxidized through catalase in the peroxisomes of the hepatocyte.
Regardless of the metabolic pathway, AA is further metabolized to acetate in the mitochondria by aldehyde dehydrogenases (ALDH). After binding to Coenzyme A acetate is further synthesized to acetyl-CoA through acetyl Co-A-synthetase. Both ADH and ALDH need nicotinamide-adenine dinucleotide (NAD+) as a co-enzyme, which is then transferred into its reduced form nicotinamide-adenine dinucleotide-hydrogen (NADH). Thus, oxidation of ethanol leads to an excess of NADH and acetyl Co-A [1,15].
Acetyl Co-A can be generated from carbohydrates such as glucose, but not the other way around. In an excess of acetyl Co-A, it is used either to generate energy in the citric cycle, or for the synthesis of fatty acids. NADH, however, which also exists in excess, inhibits the citric cycle. This is the reason why acetyl Co-A from ethanol oxidation is predominantly used for the synthesis of fatty acids (FA). FAs are esterified with glycerol to triglycerides and stored as fat drops in the cytosol of the hepatocyte [1,15].
Steatosis is also promoted by excess dietary lipids and can be attenuated by their replacement with medium-chain triglycerides [16]
3. Inhibition of Fatty Acid Oxidation
Due to the fact that ethanol metabolism produces NADH and decreases the NAD+/NADH ratio, other metabolic pathways leading to hepatic steatosis are activated [15]. The result is a change in the cellular redox potential with an inhibition of the oxidative degradation of fat in the hepatocytes, since many enzymes of fat acid oxidation require NAD+ and are inhibited by higher NADH levels [10].
For a long time, the redox change of NAD+/NADH ratios was seen as a major cause for FL. Such a metabolic change, however, may not explain completely the fast increase in hepatic fat by acute alcohol consumption. In addition, experimental data show that the change in NAD+/NADH ratio cannot explain hepatic fatty liver induced by alcohol alone since animal studies have shown that ethanol may lead to hepatic fat deposits even after normalization of the NAD+/NADH ratio [17].
The pathogenesis of alcoholic FL is a multifactorial process, requiring in addition to metabolic changes also various signal transduction pathways which are changed and which affect not only the degradation of fat and fatty acids, but also other components of lipid metabolism. Alcohol leads to a depolarization of mitochondria. The consequences of mitochondrial depolarization leads to reduced energy generated from adenosine triphosphate (ATP). In addition, this depolarization leads to a decreased influx of fatty acids into the mitochondria with a consecutively reduced ß-oxidation [18].
Alcohol has an effect on the transcriptional factor peroxisome proliferation-activated receptor α (PPAR α) [19]. PPAR α is a nuclear receptor interacting with the retinoid x receptor. The protein complex is activated through a binding of fatty acids and leads to an increased expression of genes responsible for the transport and oxidation of fatty acids. Animal experiments and cell cultures have shown that alcohol application leads to a decreased binding of PPAR α to their promotor regions and, consecutively, to a decreased expression of coded protein.
Acetaldehyde deactivates PPAR α, possibly through a covalent binding or the production of protein adducts which inhibit the binding to the promotor elements [19]. Also, alcohol directly deactivates PPAR α through CYP2E1-mediated oxidative stress. Other factors which inhibit PPAR a are NADH/NAD or via the inhibition of adiponectin, AMPK and zinc levels. The importance of the inhibitory effect of alcohol on PPAR α for the generation of FL has been shown in animal experiments where the application of PPAR α agonists inhibits alcohol-mediated FL [20].
4. Enhancement of De-Novo Lipogenesis
Acute and chronic alcohol consumption increases the synthesis of fatty acid in the hepatocytes. This alcohol effect results from an increased expression of enzymes involved in lipogenesis, such as fatty acid synthase (FASN), acyl Co-A carboxylase (ACC) and ATP citrate lyase (ACL), or stearyl- CoA desaturase. These enzymes are coded through genes which are regulated through the transcription factor sterol-regulatory element binding protein-1 (SREBP-1) [21]. Alcohol induces in tissue cultures, in vitro, and in the animal model an increased SREBP-1 level [21]. Serine-arginine-rich protein kinase 2 (SRPK2), a key kinase controlling alternative splicing, is activated in hepatocytes in response to alcohol, in mice with chronic-plus-binge alcohol feeding, and in patients with ALD. Such induction activates sterol regulatory element binding transcription factor 1 (SREBP-1) and promotes lipogenesis in ALD.
Fibroblast growth factor 21 (FGF21) is upregulated in alcohol induction of SRPK2. This results in steatosis, lipotoxicity, and inflammation. Mechanistically, SRPK2 is required for alcohol-mediated impairment of serine-arginine splicing factor 10 (SRSF10), which generates exon 7 inclusion in lipin 1, and triggers concurrent induction of lipogenic regulators—lipin 1β and SREBP-1. FGF21 suppresses alcohol-induced SRPK2 accumulation through mTORC1 inhibition-dependent degradation of SRPK2 [10,21].
This effect of alcohol is mediated through an alcohol-mediated inhibition of AMPK or primarily by acetaldehyde [19,22]. In this context, it is important to note that increased concentrations of malonyl CoA are produced. malonyl CoA inhibits allosterically carnitin-palmitoyl-transferase 1 (CPT-1). CPT-1 is a key factor in the uptake of fatty acids and fatty acids-Acyl-CoA from the plasma into the mitochondria. Thus, allosterically the oxidation of fatty acids is inhibited through the alcohol-induced lipogenesis and malonyl-CoA, which is a further mechanism by which alcohol affects fat degradation in the liver negatively.
In addition to an inhibition of AMPK by acetaldehyde SREBP1c is also activated by lipopolysaccharides, 2-arachidonoylglycerol (2-AG), complement, endoplasmic reticulum stress, reduced adiponectin, NAD-dependent protein deacetylase sirtuin 1 (SIRT1), signal transducers and activator of transcriptor 3 (STAT3) and H3K9 (trimethylation of lysine 9 of histone 3) methylation [2,21,22].
5. The Role of Cytochrome P4502E1
It has been shown in cell cultures of human hepatocytes that the induction of CYP2E1 correlated significantly with the degree of hepatic steatosis [23]. Furthermore, hepatic fat content was found to be significantly reduced in CYP2E1 knockout mice following alcohol administration when compared to wild type animals [24,25,26].
Cytochrome P4502E1 may play an important role in the pathogenesis of alcoholic fatty liver [27,28,29]. The NADPH oxidase activity of CYP2E1 can stimulate NADPH transport into the mitochondria. In addition, ROS generated by CYP2E1 may damage mitochondria leading to an inhibition of ß-oxidation [28]. Other mechanisms of CYP2E1 mediated hepatic steatosis may include an impairment of protein kinase B activity by CYP2E1 - mediated oxidative stress [29] and a suppression of PPAR a by oxidative stress [25]. The non-competitive CYP2E1 inhibitor, clomethiazole, attenuates acute ethanol-induced hepatic steatosis by suppressing oxidative stress, an adiponectin decline and an activation of autophagy [30]. In patients with alcoholic fatty liver, clomethiazole administered for 1 week improved hepatic steatosis only [31].
6. Cytochrome P4502A5 and Nicotine
Cytochrome P4502A5 (CYP2A5) is also induced by ethanol and this induction is regulated by nuclear factor-erythroid 2-related factor 2 (NRF2) [32]. CYP2A5 knockout mice develop more severe hepatic steatosis that wild type animals [32]. A complex cascade through PPARa is responsible for this effect. Interestingly, alcohol and tobacco are often misused together. Recent data have shown that alcoholic fatty liver was enhanced by nicotine in cyp2a5+ mice, but not knock out mice. Nicotine is metabolized through CYP2A5 and both ethanol as well as nicotine induces CYP2A5. The mechanisms by which nicotine enhances alcoholic fatty liver is mediated through CYP2A5 induction enhanced nicotine metabolism and ROS generation [33].
7. Effect of Alcohol on Fat Tissue
A further mechanism by which alcohol contributes to fatty liver is its effect on fat tissue. Fat tissue is an important source of energy. Food calories are stored as fat in adipocytes. In the fasting state or under exercise conditions, adipocytes liberate the stored fat. Animal data show that chronic alcohol consumption increases lipolysis in fat tissue. An increased degradation of triglycerides to free fatty acids, which are secreted into the circulation. As a consequence, the mass of body fat decreases. On the other hand, fatty acids secreted from fat tissue are taken up by the liver and is esterified with glycerol to triglycerides. Thus, a shift of triglycerides in adipocytes to triglycerides in hepatocytes occurs, which increases hepatic fat deposition in the liver [34].
It is interesting to note that alcoholics with fatty liver have a significantly lower body weight as compared to control persons. They also have a lower body mass index (BMI) and lower body fat. Alcohol consumption results in a decreased production of adiponectin, which is generated by adipocytes. Ethanol-induced oxidative stress via CYP2E1 disrupts adiponectin secretion from adipocytes [35]. Adiponectin is secreted from adipocytes into the circulation and its serum concentration is significantly reduced in heavy drinkers. Carnitine palmitoyl transferase 1 (CPT1) is the enzyme responsanle for fatty acid oxidation, as the key target of adipokines. The physiological effect of adiponectin includes an increase in CPT1 activity, as well as, the oxidation of fatty acids, while the activity of acetyl CoA carboxylase and fatty acid synthetase, two key enzymes of the de-novo lipogenesis, is inhibited [36,37,38]. In animal experiments, alcohol-induced hepatic FL could be significantly inhibited by the administration of recombinant adiponectin [38].
8. Disturbed Lipid Transport
Stored lipids or triglycerides can be exported into the circulation from the hepatocytes in the form of very low-density lipoproteins (VLDL) only. Before secretion stored triglycerides in the fat droplets are split and free fatty acids (FFA) are generated. These fatty acids are then loaded with Apolipoprotein B100 in the endoplasmic reticulum together with phospholipids and cholesterol and finally synthesized to VLDL particles [39].
PPAR α regulates not only genes involved in fat oxidation, but also regulates the expression of key factors in fat transport such as Apolipoprotein B, triglyceride transfer protein and fatty acid binding protein (FABP). By inhibiting PPAR activity, alcohol leads to disturbances of VLDL generation and secretion with the consequence of intercellular accumulation of fatty acids [40].
9. Effect of Alcohol on Various Intracellular Signal Pathways
9.1. AMP Activated Protein Kinase (AMPK)
Alcohol inhibits AMP-activated protein kinase (AMPK) [17]. AMPK is an enzyme that protects cells against ATP deficiency, or energy deficiency. AMPK is regulated through AMP and ATP concentrations in the cells. AMP is a degradation product of ATP and therefore an excellent indicator for energy deficiency. AMPK regulates energy deficiency through a shutdown of energy-effective biosynthesis. For example, AMPK inhibits enzymes responsible for cholesterol and fatty acid synthesis through phosphorylation, such as the Acetyl CoA carboxylase (ACC). ACC is a key enzyme of lipid synthesis. ACC catalyses the carboxylation of acetyl CoA to malonyl CoA, the initial product for de novo lipogenesis. At the same time, malonyl CoA inhibits fat burning allosterically. Additionally, oxidation of fatty acids is stimulated through a diminished malonyl CoA concentration due to increased AMPK activity. This process as well as further ATP-generating catabolic processes such as the citric cycle or glycolysis, are activated directly through AMPK as a counter-regulation in the presence of energy-deficiency. Fat burning is inhibited and lipogenesis stimulated through the inhibitory effect of alcohol on AMPK, independent of the real energy need of the cell. This effect of alcohol is due to the stimulation of SREBP1-levels. Alcohol-induced hepatic fat accumulation can be significantly inhibited through the application of an AMPK-activator like metformin [22]
9.2. Early Growth Response 1 (Egr-1)
Alcohol affects hepatic lipid accumulation through a further transcription factor, namely early growth response 1 (Egr-1) [41]. Egr-1 regulates the expression of genes, which are induced by cellular stress (alcohol causes cellular stress by various mechanisms). Egr-1 increases the expression of tumour necrosis factor α (TNFα). TNF-α is lipogenic, through activation of SHREP1, which leads to an increased de novo lipogenesis.
Alcohol also activates Kupffer cells to release pro-inflammatory cytokines such as TNF-α that promote fat accumulation in the liver. Another activation of Kupffer cells may occur through pathogen-associated molecular patterns (PAMP)s from alcohol induced gut bacterial overgrowth and dysbiosis. The importance of Egr-1 for alcohol-induced fatty liver could be clearly shown in animal experiments. Egr-1-deficient mice do not show an increase in TNF-α expression and hepatic fat accumulation is prevented after alcohol administration [41].
10. The Mammalian Target of Rapamycin (mTOR) of Autophagy
Autophagy is a genetically determined evolutionary conserved process by which cells degrade their own cellular constituents. Lipophagy is a special form of autophagy and plays an important role in the initiation of fat burning. During lipophagy, the fat droplets have to be cracked and triglycerides have to be liberated. For this process, fat droplets are included in autophagosomes. Autophagosomes have a double membrane and transport their load to liposomes and merge with them. Fat is digested through liposomal enzymes. The fatty acids can be oxidized via ß-oxidation in the mitochondria. The alcohol affects autophagy [42,43]
The mTOR signal pathway is a key regulator of autophagy. Studies have shown different effect of alcohol on mTOR activation and autophagy. Long-time application of alcohol inhibits autophagy while short-term application results in an increased autophagy. During acute alcohol exposition or in an acute phase of alcoholic liver damage, the inhibitory effect of hepatic steatosis is induced. On the other hand, the alcohol effect of ß-oxidation is inhibited. Thus, an accumulation of toxic free FFAs in the cytoplasm of hepatocytes could be induced through the induction of lipophagy [34,40,44].
11. Endotoxin and Presepsin
Chronic alcohol consumption damages almost all tissues in the human body, which may additionally enhance fatty liver, for example effects on the glucose metabolism, the endocrine pancreas or effects on the intestinal barrier function. Intestinal CYP2E1 may be a mediator of alcohol-induced intestinal leakiness [45] with the uptake of endotoxins resulting in fatty liver. The gut microbiota which is changed by chronic alcohol consumption may also play an important role in hepatic steatosis and alcoholic liver disease [46,47].
11.1. Alcohol and Endotoxin
ALD is associated with elevated plasma endotoxin levels in alcoholic liver [48,49,50]. The endotoxemia results in an increase level of lipopolysaccharides (LPS) [51]. Endotoxemia increases intestinal permeability [52].
Ethanol effects glycosylation of epithelial mucins, which alters the protective mucus layer and may cause a change in adherent bacterial species [53]. The effect of alcohol on intestinal permeability is in part due to bacterial ethanol metabolism to acetaldehyde [53] .
Patients with alcoholic liver disease have an overgrowth of Candida sp. compared to non-alcoholic controls [54,55,56,57,58]. Moreover, 2 weeks of abstinence from alcohol reduces the proportion of Candida albicans in patients with alcohol use disease. In addition , mice fed a chronic diet supplemented with alcohol have an increase in Meyerozyma guillermondii [59] .
11.2. Alcohol and Presepsin
A very useful biomarker in monitoring LPS is presepsin. The presepsin molecule is characterized by rapid kinetics: activation time is only 2 hours following a bacterial or fungal event, with a peak concentration after 3 hours. Presepsin and resistin are better markers for bacterial infection in patients with decompensated liver cirrhosis [60].
Presepsin secretion sCD14-ST is a 13k Da fragment derived from cleavage of CD14, a glycoprotein of 55k Dalton anchored to the membrane of monocytes, macrophages and polymorphic neutrophils. CD14 acts as a receptor for LPS complexes and the specific LPS binding protein (LBP). It can bind to peptidoglycans and other surface structures present in both Gram- positive and Gram-negative bacteria. Once bound to the LPS-LBP complex, it activates the intracellular inflammatory response of the TLR4/MD2-complex, triggering the host’s inflammatory cascade against the infectious pathogenic agent. Phagocytosis and activity of plasma proteases (lysosomal enzymes, cathepsin D) result in the formation of the fragments CD14 subtype, in particular the 13 kDa fragment of sCD14-ST [60].
12. The Intestinal Microbiome
The intestinal microbiome is now recognized as playing a central role in the proper functioning of the intestine. The GI microbiome consists of bacteria, fungi, and viruses .The major portion of the microbiome is located in the ileum and proximal colon. The multiple effects of the gut microbiota influence physiological function [61].
Gut microbial imbalance are alterations in the small intestinal flora, changes in the ratio of useful to harmful bacteria and the translocation of colonic bacteria producing dysbiosis. The dysbiosis increases intestinal permeability to endotoxin and has a key role in the metabolic changes leading to pathogenesis. This results in decreased production of long-chain fatty acids. In experimental mouse model, ethanol feeding, down-regulated in thee liver, let-7a and let-7b. The treatment of human hepatic stellate cells (HSCs) with LPS and TGF-β similarly decreased the expressions of let-7a and let-7b. Conversely, overexpression of let-7a and let-7b suppressed the myofibroblastic activation of cultured human HSCs induced by LPS and TGF-β, as evidenced by repressed ACTA2 (α-actin 2), COL1A1 (collagen 1A1), TIMP1 (TIMP metallopeptidase inhibitor 1), and FN1 (fibronectin 1) [62].
ALD is associated with elevated endotoxin levels in patients and rodent models of alcohol consumption [62,63,64]. The LPS, binds to TLR4. The TLR4- deficient mice develop less liver injury following alcohol exposure as compared to wildtype mice, suggesting a role for bacterial metabolom in the pathogenesis of liver injury from alcohol [65]. Increased endotoxemia produces intestinal permeability [66].
Finally, alcohol induced changes in the microbiome enhances hepatic steatosis.
Alcohol affects lipid metabolism through various mechanisms, leading synergistically to an accumulation of fatty acids and triglycerides. Overweight and obesity as well as the quality of dietary fat (saturated FA versus PUFA) may modulate the quantity and quality of hepatic fat. Alcohol inhibits adenosine monophosphate activated kinase (AMPK). AMPK activates PPARa and leads to a decreased activation of SRABP1c. Ethanol toxicity is enhanced by the presence of copper or iron, which promote lipid peroxidation. Impairment of mitochondrial function is an early step in the ethanol-induced toxicity.
In Figure 1 we present a schmatic sossible scenario of the mechanism of alcohol-induced toxic effect on liver cell.
13. Inflamasome
The proinflammatory cytokines are activated by alcohol signalling for inflammation via multi-faceted innate immunity-activating alarmin. Ethanol signals via Tall Receptors (TLR2, TLR4, TLR9), and chemokine receptor (CXCR4) to activate inflammation. It also induces the release of interleukines (IL-1α, IL-1β, IL-8, IL-6), TNF -α, interferon (IFN)-γ,as well as, chemolines (CXCL11 and CXCL12) [65,66].
Ethanol enhance the inflammation in the liver by activating hepatic stellate cells (HSCs). HSCs reside in the perisinusoidal space between sinusoidal endothelial cells and hepatocytes. HSCs and Kupffer cells integrate cytokine-mediated inflammatory responses in the sinusoids [67,68,69] . NF-κβ can also induce the antiapoptotic genes (TRAF-1 and TRAF-2) influencing the appearence and the growth of HCC. Increased TNF-α production has been shown to deregulate the tight junction in GI tract, causing disruption of the intestinal barrier. The change induced by acetaldehyde and ROS is related to folate metabolism . ALD patients have been found with polymorphisms in the methylene tetrahydrofolate reductase gene, leading to an alteration in folate metabolism and HCC development. Ethanol effects glycosylation of epithelial mucins, which may cause a change in adherent bacterial species [70] . Germ-free mice studies suggest that the effect of alcohol on intestinal permeability is in part due to ethanol metabolism to acetaldehyde [71] . Other bacterial metabolites also effect gut barrier disruption [72] . The changes in bacterial variety of the host induced by ALD brings with it an important modify in the bile acids composition and their concentration. The bacterial dehydroxylation, results in an increase in Deoxycholic Acid synthesis. Accumulation of bile acids and activation of farnesoid X receptor (FXR) induce the expression of the bile salt export pump, organic solute transporter alpha, and beta.
Alcohol administration to rats resulted in a decrease in the Lactobacillus sp., Pediococcus sp., Leuconostoc sp., and Lactococcus sp. [73]. Alcohol-fed mice also developed an increase in Proteobacteria sp.and Actinobacteria sp. [74] . Alcohoic individuals present a decrease in the abundance of Bacteroidetes together with an increase in Enterobacteriaceae and Proteobacteria (75) as well as decreased fungal species richness [76,77,78]. Two weeks of abstinence from alcohol reduces the proportion of Candida albicans in patients with alcohol use disease [79].
Examination of the stools from alcoholic patients have shown a correlation between the levels of long chain fatty acids (LCFA) and the numbers of Lactobacilli in the stools. The Lactobacilli in the intestinal microbiome (IM) metabolize saturated LCFAs which promote the growth of the bacteria. Dietary fat composition influences the IM and modulates liver injury. In a murine model administration of a diet rich in unsaturated fats resulted in a decrease in Bacteroidetes and an increase in Proteobacteria and Actinobacteria, resulting in a more severe ALD. In addition, there were metabolomic changes -a decrease in long chain fatty acids (LCFA)s, medium-chain fatty acids (MCFA)s, and ahort chain fatty acids (SCFA)s. Supplementation of saturated LCFAs maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice (80). Thus dietary lipids have a role in dysbiosis, leading to the development of ALD, enhancing the severity of the disease or reducing the inflammation [81,82,83,84,85,86,87].
14. The Significance of Alcohol Consumption to Liver Toxicity
The alcohol role in the etiology of liver disease has established several codes for categories of liver diseases, primarily caused by alcohol. For example, ICD-10(7) recognizes several forms of alcoholic liver disease (ICD-10, K70), sometimes considered stages that range from reversible alcoholic steatosis (K70.0) and alcoholic hepatitis (K70.1), to alcoholic fibrosis and sclerosis of the liver (K70.2), and further to severe and irreversible stages such as alcoholic liver cirrhosis (K70.3) and alcoholic hepatic failure (K70.4). Alcohol consumption, in particular heavy use over time, has been central in the etiology and development of these diseases [88,89,90]. However, liver diseases are multifactorial, and alcohol use may play a role in the progression of all types of cirrhosis, [91]. Even one drink per day may have an effect on the incidence of liver cirrhosis [92]. Scientific review of liver related diseases including cirrhosis, it is important to include both alcoholic and non-alcoholic-induced liver injury when examining the impact of alcohol use [93,94,95,96,97,98].
In the metanalysis performed by us, alcohol consumption showed a complex association with hepatic steatosis with substantial differences by ethnicity and sex. Low alcohol consumption was beneficial in Japan with good epidemiological evidence. There was no association in other countries. However, heterogeneity was large in countries other than Japan [95. 96].
Becker U et al., explained the lower risk for alcohol-induced cirrhosis in wine drinkers [101]. The authors studied prospectively 30630 participants from the Copenhagen area. The following parameters were noted: weekly intake of beer, wine, and spirits. The participants data (sex, age, body mass index, smoking habits, and education was noted . The first noxpital admission or death, due to alcohol-induced cirrhosis obtained from the National Hospital Discharge Register. The individuals who drank more than 5 drinks per day had a higher risk for developing cirrhosis compared with non- or light drinkers. Also individuals who drank no wine, individuals drinking 16% to 30% wine of the alcohol total intake had a higher risk of developing cirrhosis compared to the people drinking 51% of developing cirrhosis. The authors suggest that wine drinkers are at a lower risk than beer and spirits[101]
14.2. Biological Mechanisms That Contributed to Alcohol-Related Research
Many scientists had a specific interest to understand the role of polyphenols in wine, especially flavonoids, that stimulated research into their potential benefits to human health. One of the main properties of polyphenols is their antioxidant activity which enables them to attenuate the development of atherosclerosis, inflammatory diseases, and cancer. Wine contains trans-resveratrol that may be the most effective anticancer polyphenol present in red wine as consumed by healthy human subjects [102]. Yhe group of clinical chemists tested the absorptive efficiency of three of these constituents (trans-resveratrol, [+]-catechin and quercetin) when given orally to healthy human subjects in three different media. The absorption of these polyphenols was equivalent in aqueous and alcoholic matrices, but at peak concentrations was inadequate to permit circulating concentrations consistent with biologic activity. The author concluded that literature reporting powerful in vitro anticancer and anti-inflammatory effects of the free polyphenols is irrelevant, given that the polyphenols are absorbed as conjugates [103]. Moreover, specific methods to determine the possible tent of white and red wines with cork have been establish in biochemical laboratory [104]. The importance of polyphenols was also studied in people with different medical condition [105].
However, after 30 years of dedicated research yjere is a lack of pharmacological, human evidence to confirm the poly)phenols' biological actions. This is since in each type of grapes the quantity and quality of polyphenols differ. Moreover, the fermentattion process differ from one winery to the other. Future research will eventually clarify the positive activities of the compounds. There is a clear recommendation of responsibly drinking moderate amounts of wine with meals.
15. Conclusions
The metabolism of alcohol is involved in alcoholic fatty liver disease leading to tissue changes. There are ethnic differences, sex, age, and environmental differences. Alcohol -induced toxic metabolites interfere with the mitochondrial regulation pathways is important in understanding ALD pathogenesis. In addition, alcohol has the capacity to disturb gut microbiota, favoring the expansion of endotoxin-producing bacteria and intestinal permeability leading to inflammation and fibrogenesis. Different biomarkers are used to define the severity of the disease and its possible repair.
Author Contribution
Conceptualization, H.K.S., B.M., M.G.N; methodology, H.K.S., M.G.N; resources, none; data curation, M.N, H.K.S.; writing, original draft preparation, H.K.S., B.M., M.G.N; review and editing, M.G.N., H.K.S.; visualization. All authors have read and agreed to the published version of the manuscript.” All authors have contributed substantially to the work reported.
Acknowledgements
The authors want to thank Ms. Laura Ehrhard for typing the manuscript.
Conflict of Interest
None
Abbreviations
| 2-AG | 2-arachidonoyl-glycerol |
| AA | acetaldehyde |
| Acetyl | acetyl-Coenzyme A |
| ACC | CoA acyl Co-A carboxylase |
| ACE-2 | angiotensin-converting enzyme 2 |
| ACL | ATP citrate lyase |
| ACTA-2 | α-actin 2 |
| ADH | alcohol dehydrogenase |
| AH | alcoholic hepatitis |
| AIH | autoimmune hepatitis |
| ALD | alcoholic liver disease |
| ALI | acute liver injury |
| ALP | alkaline phosphatase |
| ALT | alanine aminotransferase (glutamic-pyruvic transaminase, GPT) |
| AMA | anti-mitochondrial antibody |
| AMPK | adenosine monophosphate activated kinase |
| ANA | antinuclear antibody |
| AST | aspartate aminotransferase (glutamic-oxalic- transaminase, GOT) |
| ASH | alcoholic steatohepatitis |
| ATP | adenosine triphosphate |
| BMI | body mass index |
| CCK | caspase cleaved cytokeratin (8 & 18)- M30-M 65 |
| C2H5OH | ethyl alcohol, ethanol |
| CoA | coenzyme A |
| COL1A1 | collagen 1A1 |
| CPT-1 | carnitin-palmitoyl-transferase1 |
| CXCL | chemokine |
| CYP2E1 | cytochrome P4502E1 |
| DAMPs | danger-associated molecular patterns |
| DNA | deoxyribonucleic acid |
| EGF | epithelial growth factor |
| Egr-1 | early growth response 1 |
| FA | fatty acids |
| FABP | fatty acid binding protein |
| FASN | fatty acid synthetase |
| FDA | Food and Drug Administration |
| FFA | free fatty acid |
| FL | fatty liver |
| FN1 | fibronectin 1 |
| GGT | γ-glutamyl-transferase |
| GSH | glutathione |
| H3K9 | tri-methylation of lysine 9 of histone 3 |
| HER | hydroxyethyl radical |
| HGF | hepatocyte growth factor |
| ICD | International Classification of Diseases |
| IFN | interferon |
| IL | interleukine |
| LPB | LPS binding protein |
| LPS | lipopolysaccharides |
| KC | Kuppfer cells |
| MAA | malon-dialdehyde |
| MAFLD | metabolic associated fatty liver disease, |
| MEOS | microsomal ethanol oxidizing system |
| Mean ± SD | mean ± standard deviation |
| mTOR | mammalian target of rapamycin |
| NAD+ | nicotin-amide-adenine dinucleotide |
| NADH | nicotin-amide-adenine dinucleotide-hydrogen |
| NADP+ | nicotinamide adenine dinucleotide phosphate, |
| NADPH | reduced nicotinamide adenine dinucleotide phosphate |
| NRF2 | nuclear factor-erythroid 2-related factor 2 |
| PAMP | pathogen-associated molecular patterns |
| PhET | phosphatidyl ethanol |
| PPAR alpha: peroxisome proliferator | activated receptor alpha |
| PRRs | pattern-recognition receptors |
| PUFA | poly-unsaturated fatty acids |
| ROS | reactive oxygen specie |
| SIRT1 | sirtuin 1 |
| SRPK2 | serine-arginine-rich protein kinase 2 (SRPK2) . |
| SOCS | suppressor of cytokine signaling |
| SRABP1c | sterol element binding protein 1c |
| SREBP-1 | sterol-regulatory element binding protein-1 |
| STAT3 | signal transducers and activator of transcriptor 3 |
| TGF-β | transforming growth factor-β |
| TIMP1 | TIMP metallopeptidase inhibitor |
| TLR4 | toll-like receptor 4 |
| TNF a | tumour necrosis factor a |
| VLDL | very low-density lipoproteins |
References
- Seitz, H.K.; Mueller, S. Alcoholic liver disease. In Clinical Hepatology. Principle and practice of hepatobiliary diseases; Springer, Heidelberg: New York, NY, USA, 2010; pp. 1111–1151. [Google Scholar]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; et al. Alcoholic liver disease. Nat Rev Dis Primers. 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Hege, M.; Jung, F.; Sellmann, C.; Jin, C.; Ziegenhardt, D.; Hellerbrand, C.; Bergheim, I. An iso-α-acid-rich extract from hops ( Humulus lupulus ) attenuates acute alcohol-induced liver steatosis in mice. Nutrition 2018, 45, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.; de la, M. Pathological spectrum of alcoholic liver disease. In Alcoholic liver disease. Pathology and Pathogenesis; 2nd ed.; Edward Arnold: London, UK, 1995; pp. 41–70. [Google Scholar]
- Simonis, B.; Reimann, F.; Waldherr, R. Sonographic course of alcoholic fatty liver by inter-observer and digital evaluation of liver echogenicity. Zeitschr Gastroenterol. 2007, 45, 689–696. [Google Scholar]
- Thiele, M.; Rausch, V.; Fluhr, G.; Piecha, F.; Mueller, J.; Straub, B.; Lupsor-Platon, M.; Ledinghen, V.; Seitz, H.K.; Detlefsen, S.; et al. s01-3controlled attenuation parameter for the assessment of alcoholic hepatic steatosis: biopsy-controlled diagnostic accuracy and role of detoxification. Alcohol Alcohol. 2017, 52, i4–i30. [Google Scholar] [CrossRef]
- Hohmann, N.; Schröder, F.; Moreira, B.; Teng, H.; Burhenne, J.; Bruckner, T.; Mueller, S.; E Haefeli, W.; Seitz, H.K. Clomethiazole inhibits cytochrome P450 2E1 and improves alcoholic liver disease. Gut 2021, 71, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Baraona, E.; Lieber, C.S. Effects of ethanol on lipid metabolism. J. Lipid Res. 1979, 20, 289–315. [Google Scholar] [CrossRef]
- Purohit, V.; Gao, B.; Song, B. Molecular Mechanisms of Alcoholic Fatty Liver. Alcohol. Clin. Exp. Res. 2009, 33, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Malnick, S.D.H.; Alin, P.; Somin, M.; Neuman, M.G. Fatty Liver Disease-Alcoholic and Non-Alcoholic: Similar but Different. Int. J. Mol. Sci. 2022, 23, 16226. [Google Scholar] [CrossRef]
- Lívero, F.A.; Acco, A. Molecular basis of alcoholic fatty liver disease: From incidence to treatment. Hepatol. Res. 2015, 46, 111–123. [Google Scholar] [CrossRef]
- Völzke, H. Multi-causality in fatty liver disease: Is there a rationale to distinguish between alcoholic and non-alcoholic origin. World J. Gastroenterol. 2012, 18, 3492–3501. [Google Scholar] [CrossRef]
- Seitz, H.K. Do alcoholic calories count? Front. Alcohol. 2014, 2, 52–58. [Google Scholar]
- Lieber, C.S.; Rubin, E.; DeCarli, L.M. Hepatic microsomal ethanol oxidizing system (MEOS): Differentiation from alcohol dehydrogenase and NADPH oxidase. Biochem. Biophys. Res. Commun. 1970, 40, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Mueller, S. Metabolism of alcohol and its consequences. In Metabolism of drugs and other xenobiotics; Wiley-VCH GmbH: Weinheim, Germany, 2012; pp. 493–516. [Google Scholar]
- Lieber, C.S.; Lefèvre, A.; Spritz, N.; Feinman, L.; DeCarli, L.M. Difference in Hepatic Metabolism of Long- and Medium-Chain Fatty Acids: The Role of Fatty Acid Chain Length in the Production of the Alcoholic Fatty Liver*. J. Clin. Investig. 1967, 46, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Pan, J.; Qu, N.; Lei, Y.; Han, J.; Zhang, J.; Han, D. The AMPK pathway in fatty liver disease. Front. Physiol. 2022, 13, 970292. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Ramshesh, V.K.; Rehman, H.; Liu, Q.; Theruvath, T.P.; Krishnasamy, Y.; Lemasters, J.J. Acute Ethanol Causes Hepatic Mitochondrial Depolarization in Mice: Role of Ethanol Metabolism. PLoS ONE 2014, 9, e91308. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Pinaire, J.; Fischer, M.; Dorris, R.; Crabb, D.W. The Transcriptional and DNA Binding Activity of Peroxisome Proliferator-activated Receptor α Is Inhibited by Ethanol Metabolism. J. Biol. Chem. 2001, 276, 68–75. [Google Scholar] [CrossRef]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol Induces Fatty Acid Synthesis Pathways by Activation of Sterol Regulatory Element-binding Protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef]
- You, M.; Matsumoto, M.; Pacold, C.M.; Cho, W.K.; Crabb, D.W. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004, 127, 1798–1808. [Google Scholar] [CrossRef]
- Mahli, A.; Thasler, W.E.; Müller, M.; et al. Identification of cytochrome P4502E1 as critical mediator of alcohol effects on steatotic hepatocytes. Oncotarget 2015, 6, 41464–41478. [Google Scholar] [CrossRef]
- Lu, Y.; Wu, D.; Wang, X.; Ward, S.C.; Cederbaum, A.I. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free. Radic. Biol. Med. 2010, 49, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhuge, J.; Wang, X.; Bai, J.; Cederbaum, A.I. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology 2008, 47, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.-H.; Yun, J.-W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.-J. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free. Radic. Biol. Med. 2013, 65, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Harjumäki, R.; Pridgeon, C.S.; Ingelman-Sundberg, M. CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload. Int. J. Mol. Sci. 2021, 22, 8221. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Intoductional serial review: Alcohol, oxidative stress and cell injury. Free Radic Biol Med 2001, 31, 1524–1526. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Zhang, C.L.; Zhao, N.; et al. Impairment of Akt activity by CPY2E1-mediated oxidative stress is involved in chronic ethanol-induced fatty liver. Redox Biol. 2018, 14, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Zhang, C.-L.; Zhao, X.-L.; Xie, K.-Q.; Zeng, T. Inhibition of cytochrome P4502E1 by chlormethiazole attenuated acute ethanol-induced fatty liver. Chem. Interactions 2014, 222, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, N.; Schröder, F.; Moreira, B.; Teng, H.; Burhenne, J.; Bruckner, T.; Mueller, S.; E Haefeli, W.; Seitz, H.K. Effect of Clomethiazole Vs. Clorazepate on Hepatic Fat and Serum Transaminase Activities in Alcohol-Associated Liver Disease: Results from a Randomized, Controlled Phase II Clinical Trial. Alcohol Alcoholism 2023, 58, 134–141. [Google Scholar] [CrossRef]
- Chen, X.; Ward, S.C.; Cederbaum, A.I.; Xiong, H.; Lu, Y. Alcoholic fatty liver is enhanced in CYP2A5 knockout mice: The role of the PPARα-FGF21 axis. Toxicology 2017, 379, 12–21. [Google Scholar] [CrossRef]
- Chen, X.; Owoseni, E.; Salamat, J.; Cederbaum, A.I.; Lu, Y. Nicotine enhances alcoholic fatty liver in mice: Role of CYP2A5. Arch. Biochem. Biophys. 2018, 657, 65–73. [Google Scholar] [CrossRef]
- Neuman, M.G.; Seitz, H.K.; Teschke, R.; Malnick, S.; Johnson-Davis, K.L.; Cohen, L.B.; German, A.; Hohmann, N.; Moreira, B.; Moussa, G.; et al. Molecular, Viral and Clinical Features of Alcohol- and Non-Alcohol-Induced Liver Injury. Curr. Issues Mol. Biol. 2022, 44, 1294–1315. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Sebastian, B.M.; Axhemi, A.; Chen, X.; Hillian, A.D.; Jacobsen, D.W.; Nagy, L.E. Ethanol-Induced Oxidative Stress via the CYP2E1 Pathway Disrupts Adiponectin Secretion from Adipocytes. Alcohol. Clin. Exp. Res. 2011, 36, 214–222. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Rogers, C.Q. Adinopectin: A key adipokine in alcoholic fatty liver. Exp Biol Med 2009, 234, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.Q.; Ajmo, J.M.; You, M. Adiponectin and alcoholic fatty liver disease. IUBMB Life 2008, 60, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Wang, Y.; Keshaw, H.; et al. The fat-derived hormone adiponectin alleviates alcoholic and non-alcoholic fatty liver diseases in mice. J Clin Invest. 2001, 112, 91–100. [Google Scholar] [CrossRef]
- Zhong, W.; Zhao, Y.; Tang, Y.; Wei, X.; Shi, X.; Sun, W.; Sun, X.; Yin, X.; Sun, X.; Kim, S.; et al. Chronic Alcohol Exposure Stimulates Adipose Tissue Lipolysis in Mice: Role of Reverse Triglyceride Transport in the Pathogenesis of Alcoholic Steatosis. Am. J. Pathol. 2012, 180, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Rasineni, K.; Ganesan, M.; Feng, D.; McVicker, B.L.; McNiven, M.A.; Osna, N.A.; Mott, J.L.; Casey, C.A.; Kharbanda, K.K. Structure, Function and Metabolism of Hepatic and Adipose Tissue Lipid Droplets: Implications in Alcoholic Liver Disease. Curr. Mol. Pharmacol. 2017, 10, 237–248. [Google Scholar] [CrossRef]
- McMullen, M.R.; Pritchard, M.T.; Wang, Q.; Millward, C.A.; Croniger, C.M.; Nagy, L.E. Early Growth Response-1 Transcription Factor Is Essential for Ethanol-Induced Fatty Liver Injury in Mice. Gastroenterology 2005, 128, 2066–2076. [Google Scholar] [CrossRef]
- Ding, W. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef]
- Dolganiuc, A.; Thomes, P.G.; Ding, W.; Lemasters, J.J.; Donohue, T.M. Autophagy in Alcohol-Induced Liver Diseases. Alcohol. Clin. Exp. Res. 2012, 36, 1301–1308. [Google Scholar] [CrossRef]
- Rasineni, K.; Donohue, T.M., Jr.; Thomes, P.G.; et al. Ethanol-induced steatosis involves impairment of lipophagy, associated with reduced Dynamin2 activity. Hepatol Commun. 2017, 1, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Voigt, R.M.; Keshavarzian, A. Intestinal CYP2E1: A mediator of alcohol-induced gut leakiness. Redox Biol. 2014, 3, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.P.; Suk, K.T.; Kim, D.J. Significance of gut microbiota in alcoholic and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2021, 27, 6161–6179. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.L.; Brach, T.; Kern, T.; Bak, E.G.; Nielsen, T.; Arumugam, M. The Role of the Bacterial Microbiota in Alcoholic and Non-alcoholic Fatty Liver Disease. In The Human Gut-Liver-Axis in Health and Disease; 2019; pp. 89–104.
- Parlesak, A.; Schäfer, C.; Schütz, T.; Bode, J.C.; Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Mathurin, P.; Deng, Q.; Keshavarzian, A.; Choudhary, S.; Holmes, E.W.; Tsukamoto, H. Exacerbation of Alcoholic Liver Injury by Enteral Endotoxin in Rats. Hepatology 2000, 32, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.; Kugler, V.; Bode, J.C. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J. Hepatol. 1987, 4, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef]
- Ferrier, L.; Bérard, F.; Debrauwer, L.; Chabo, C.; Langella, P.; Buéno, L.; Fioramonti, J. Impairment of the Intestinal Barrier by Ethanol Involves Enteric Microflora and Mast Cell Activation in Rodents. Am. J. Pathol. 2006, 168, 1148–1154. [Google Scholar] [CrossRef]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A.; Voigt, R.M.; et al. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef]
- Yang, A.-M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef]
- Lang, S.; Duan, Y.; Liu, J.; Ventura-Cots, M.; Lucey, M.R.; Padilla, F.J.B.; Mathurin, P.; Louvet, A.; Garcia-Tsao, G.; Verna, E.C.; et al. 246 – Intestinal Fungal Dysbiosis and Systemic Immune Response to Fungi in Patients with Alcoholic Hepatitis. Gastroenterology 2019, 156. [Google Scholar] [CrossRef]
- Chu, H.; Duan, Y.; Lang, S.; Jiang, L.; Wang, Y.; Llorente, C.; Liu, J.; Mogavero, S.; Bosques-Padilla, F.; Abraldes, J.G.; et al. The Candida albicans exotoxin candidalysin promotes alcohol-associated liver disease. J. Hepatol. 2020, 72, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Lang, S.; Zeng, S.; Duan, Y.; Zhang, X.; Wang, Y.; Bondareva, M.; Kruglov, A.; Fouts, D.E.; Stärkel, P.; et al. Dynamic Changes of the Fungal Microbiome in Alcohol Use Disorder. Front. Physiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, K.; Sun, L.; Cheng, B.; Qiao, S.; Dai, H.; Shi, W.; Ma, J.; Liu, H. Therapeutic manipulation of gut microbiota by polysaccharides of Wolfiporia cocos reveals the contribution of the gut fungi-induced PGE2 to alcoholic hepatic steatosis. Gut Microbes 2020, 12, 1830693. [Google Scholar] [CrossRef] [PubMed]
- Couch, R.D.; Dailey, A.; Zaidi, F.; Navarro, K.; Forsyth, C.B.; Mutlu, E.; Engen, P.A.; Keshavarzian, A. Alcohol Induced Alterations to the Human Fecal VOC Metabolome. PLoS ONE 2015, 10, e0119362. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.; Grigoras, C.; Bugariu, A.; Nicoara-Farcau, O.; Stefanescu, H.; Benea, A.; Hadade, A.; Margarit, S.; Sparchez, Z.; Tantau, M.; et al. Are presepsin and resistin better markers for bacterial infection in patients with decompensated liver cirrhosis? Dig. Liver Dis. 2019, 51, 1685–1691. [Google Scholar] [CrossRef]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- McDaniel K, Huang L, Sato K, Wu N, Annable T, Zhou T, Ramos-Lorenzo S, Wan Y, Huang Q, Francis H, Glaser S, Tsukamoto, H. , Alpini G, Meng, F. The let-7/Lin28 axis regulates activation of hepatic stellate cells in alcoholic liver injury. J Biol Chem. 2017, 292, 11336–11347. [Google Scholar] [CrossRef]
- Nanji, A.; Khettry, U.; Sadrzadeh, S.M.; Yamanaka, T. Severity of liver injury in experimental alcoholic liver disease. Correlation with plasma endotoxin, prostaglandin E2, leukotriene B4, and thromboxane B2. Am. J. Pathol. 1993, 142, 367–373. [Google Scholar]
- Hritz, I.; Mandrekar, P.; Velayudham, A.; Catalano, D.; Dolganiuc, A.; Kodys, K.; Kurt-Jones, E.; Szabo, G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008, 48, 1224–1231. [Google Scholar] [CrossRef]
- Francis, H.; McDaniel, K.; Han, Y.; Liu, X.; Kennedy, L.; Yang, F.; McCarra, J.; Zhou, T.; Glaser, S.; Venter, J.; et al. Regulation of the Extrinsic Apoptotic Pathway by MicroRNA-21 in Alcoholic Liver Injury. J. Biol. Chem. 2014, 289, 27526–27539. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Fields, J.Z.; Vaeth, J.; Holmes, E.W. The differing effects of acute and chronic alcohol on gastric and intestinal permeability. Am. J. Pathol. 1994, 89, 2205–2211. [Google Scholar]
- Wu, N.; McDaniel, K.; Zhou, T.; Ramos-Lorenzo, S.; Wu, C.; Huang, L.; Chen, D.; Annable, T.; Francis, H.; Glaser, S.; Alpini, G.; Meng, F. Knockout of microRNA-21 attenuates alcoholic hepatitis through the VHL/NF-κB signaling pathway in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2018, 315, G385–G398. [Google Scholar] [CrossRef] [PubMed]
- Slevin, E.; Baiocchi, L.; Wu, N.; Ekser, B.; Sato, K.; Lin, E.; Ceci, L.; Chen, L.; Lorenzo, S.R.; Xu, W.; Kyritsi, K.; Meadows, V.; Zhou, T.; Kundu, D.; Han, Y.; Kennedy, L.; Glaser, S.; Francis, H.; Alpini, G.; Meng, F. Kupffer Cells: Inflammation Pathways and Cell-Cell Interactions in Alcohol-Associated Liver Disease. Am J Pathol. 2020, 190, 2185–2193. [Google Scholar] [CrossRef] [PubMed]
- Elamin, E.; Masclee, A.; Dekker, J.; Jonkers, D.M. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr. Rev. 2013, 71, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Grewal, R.K.; Mahmood, A. The effects of ethanol administration on brush border membrane glycolipids in rat intestine. Alcohol 2010, 44, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Stärkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2010, 53, 96–105. [Google Scholar] [CrossRef]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; et al. Metagenomic analyses of alohol induced pathogenic alterations in the intestinal microbiome and the Effect of Lactobacillus rhamnosus GG treatment. PLoS ONE 2013, 8, 4–13. [Google Scholar] [CrossRef]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The Gastrointestinal microbiome: Alcohol effects on the composition of intestinal microbiota. Alcohol Res. 2015, 37, 223–236. [Google Scholar]
- Chu, H.; Duan, Y.; Lang, S.; Jiang, L.; Wang, Y.; Llorente, C.; Liu, J.; Mogavero, S.; Bosques-Padilla, F.; Abraldes, J.G.; et al. The Candida albicans exotoxin candidalysin promotes alcohol-associated liver disease. J. Hepatol. 2020, 72, 391–400. [Google Scholar] [CrossRef]
- Sun, S.; Wang, K.; Sun, L.; Cheng, B.; Qiao, S.; Dai, H.; Shi, W.; Ma, J.; Liu, H. Therapeutic manipulation of gut microbiota by polysaccharides of Wolfiporia cocos reveals the contribution of the gut fungi-induced PGE2 to alcoholic hepatic steatosis. Gut Microbes 2020, 12, 1830693. [Google Scholar] [CrossRef] [PubMed]
- Llopis, M.; Cassard, A.M.; Wrzosek, L.; Boschat, L.; Bruneau, A.; Ferrere, G.; Puchois, V.; Martin, J.C.; Lepage, P.; Le Roy, T.; et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 2016, 65, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Torralba, M.; Tan, J.; Embree, M.; Zengler, K.; Stärkel, P.; van Pijkeren, J.-P.; DePew, J.; Loomba, R.; Ho, S.B.; et al. Supplementation of Saturated Long-Chain Fatty Acids Maintains Intestinal Eubiosis and Reduces Ethanol-induced Liver Injury in Mice. Gastroenterology 2015, 148, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Petrosino, J.; Ajami, N.; Feng, W.; Wang, Y.; Liu, Y.; Beier, J.I.; Barve, S.S.; Yin, X.; Wei, X.; et al. Saturated and Unsaturated Dietary Fats Differentially Modulate Ethanol-Induced Changes in Gut Microbiome and Metabolome in a Mouse Model of Alcoholic Liver Disease. Am. J. Pathol. 2016, 186, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E.; Torralba, M.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J. Hepatol. 2012, 56, 1283–1292. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Petrosino, J.; Ajami, N.; Feng, W.; Wang, Y.; Liu, Y. , et al. Saturated and Unsaturated Dietary Fats Differentially Modulate Ethanol-Induced Changes in Gut Microbiome and Metabolome in a Mouse Model of Alcoholic Liver Disease. Am. J. Pathol. 2016, 186, 765–776. [Google Scholar] [CrossRef]
- Xu, W.; Kyritsi, K.; Meadows, V.; Zhou, T.; Kundu, D.; Han, Y.; Kennedy, L.; Glaser, S.; Francis, H.; Alpini, G.; Meng, F. Kupffer Cells: Inflammation Pathways and Cell-Cell Interactions in Alcohol-Associated Liver Disease. Am J Pathol. 2020, 190, 2185–2193. [Google Scholar]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Solovieva, N.V.; Leikhter, S.N.; Shidakova, N.A.; Lebedeva, O.V.; Sidorov, P.I.; Bazhukova, T.A.; Soloviev, A.G.; Barve, S.S.; McClain, C.J.; et al. Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: A pilot study. Alcohol 2008, 42, 675–682. [Google Scholar] [CrossRef]
- Stadlbauer, V.; Mookerjee, R.P.; Hodges, S.; Wright, G.A.; Davies, N.A.; Jalan, R. Effect of probiotic treatment on deranged neutrophil function and cytokine responses in patients with compensated alcoholic cirrhosis. J. Hepatol. 2008, 48, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Slevin, E.; Baiocchi, L.; Wu, N.; Ekser, B.; Sato, K.; Lin, E.; Ceci, L.; Chen, L.; Lorenzo, S.R.; Xu, W.; et al. Kupffer Cells. Am. J. Pathol. 2020, 190, 2185–2193. [Google Scholar] [CrossRef] [PubMed]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Tapper, E.B.; Parikh, N.D. Mortality due to cirrhosis and liver cancer in the United States, 1999-2016: Observational study. BMJ 2018, 362, k2817. [Google Scholar] [CrossRef] [PubMed]
- O'Shea, R.S.; Dasarathy, S.; McCullough, A.J. Alcoholic liver disease. Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Hepatology 2010, 51, 307–328. [Google Scholar] [CrossRef]
- Menon, K.V.; Gores, G.J.; Shah, V.H. Pathogenesis, diagnosis, and treatment of alcoholic liver disease. Mayo Clin Proc 2001, 76, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- EASL clinical practical guidelines: Management of alcoholic liver disease. J Hepatol 2012, 57, 399–420. [CrossRef]
- Schwarzinger, M.; Baillot, S.; Yazdanpanah, Y.; et al. Alcohol use disorders and the burden of chronic hepatitis C in France, 2008-2013: A nationwide retrospective cohort study. J Hepatol 2017, 67, 454–461. [Google Scholar] [CrossRef]
- Roerecke, M.; Nanau, R.; Rehm, J.; Neuman, M. Ethnicity matters: A Systematic Review and Meta-Analysis of the Non-Linear Relationship Between Alcohol Consumption and Prevalence and Incidence of Hepatic Steatosis. EBioMedicine 2016, 8, 317–330. [Google Scholar] [CrossRef]
- Rehm, J.; Taylor, B.; Mohapatra, S.; et al. Alcohol as a risk factor for liver cirrhosis - a systematic review and meta-analysis. Drug Alcohol Rev 2010, 29, 437–445. [Google Scholar] [CrossRef]
- Askgaard, G.; 97Gronbaek, M.; Kjaer, M.S.; et al. Alcohol drinking pattern and risk of alcoholic liver cirrhosis: A prospective cohort study. J Hepatol 2015, 62, 106. [Google Scholar] [CrossRef] [PubMed]
- Klatsky AL, Friedman GD, Armstrong MA; et al. Wine, liquor, beer, and mortality. Am J Epidemiol 2003, 158, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Stampfer, M.J.; Colditz, G.A.; Giovannucci, E.L.; Manson, J.E.; Kawachi, I.; Hunter, D.J.; Hankinson, S.E.; Hennekens, C.H.; Rosner, B.; et al. Alcohol Consumption and Mortality among Women. New Engl. J. Med. 1995, 332, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Becker U, Deis A, Sorensen TI; et al. Prediction of risk of liver disease by alcohol intake, sex, and age: A prospective population study. Hepatology 1996, 23, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Becker, U.; Grønbaek, M.; Johansen, D.; Sørensen, T.I.A. Lower risk for alcohol-induced cirrhosis in wine drinkers. Hepatology 2002, 35, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Soleas, G.J.; Grass, L.; Josephy, P.; Goldberg, D.M.; Diamandis, E.P. A comparison of the anticarcinogenic properties of four red wine polyphenols. Clin. Biochem. 2002, 35, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.M.; Yan, J.; Soleas, G.J. Absorption of three wine-related polyphenols in three different matrices by healthy subjects. Clin. Biochem. 2003, 36, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Soleas, G.J.; Yan, J.; Seaver, T.; Goldberg, D.M. Method for the Gas Chromatographic Assay with Mass Selective Detection of Trichloro Compounds in Corks and Wines Applied To Elucidate the Potential Cause of Cork Taint. J. Agric. Food Chem. 2002, 50, 1032–1039. [Google Scholar] [CrossRef]
- Arranz, S.; Chiva-Blanch, G.; Valderas-Martínez, P.; Medina-Remón, A.; Lamuela-Raventós, R.M.; Estruch, R. Wine, Beer, Alcohol and Polyphenols on Cardiovascular Disease and Cancer. Nutrients 2012, 4, 759–781. [Google Scholar] [CrossRef]
- Visioli, F.; Panaite, S.-A.; Tomé-Carneiro, J. Wine’s Phenolic Compounds and Health: A Pythagorean View. Molecules 2020, 25, 4105. [Google Scholar] [CrossRef]
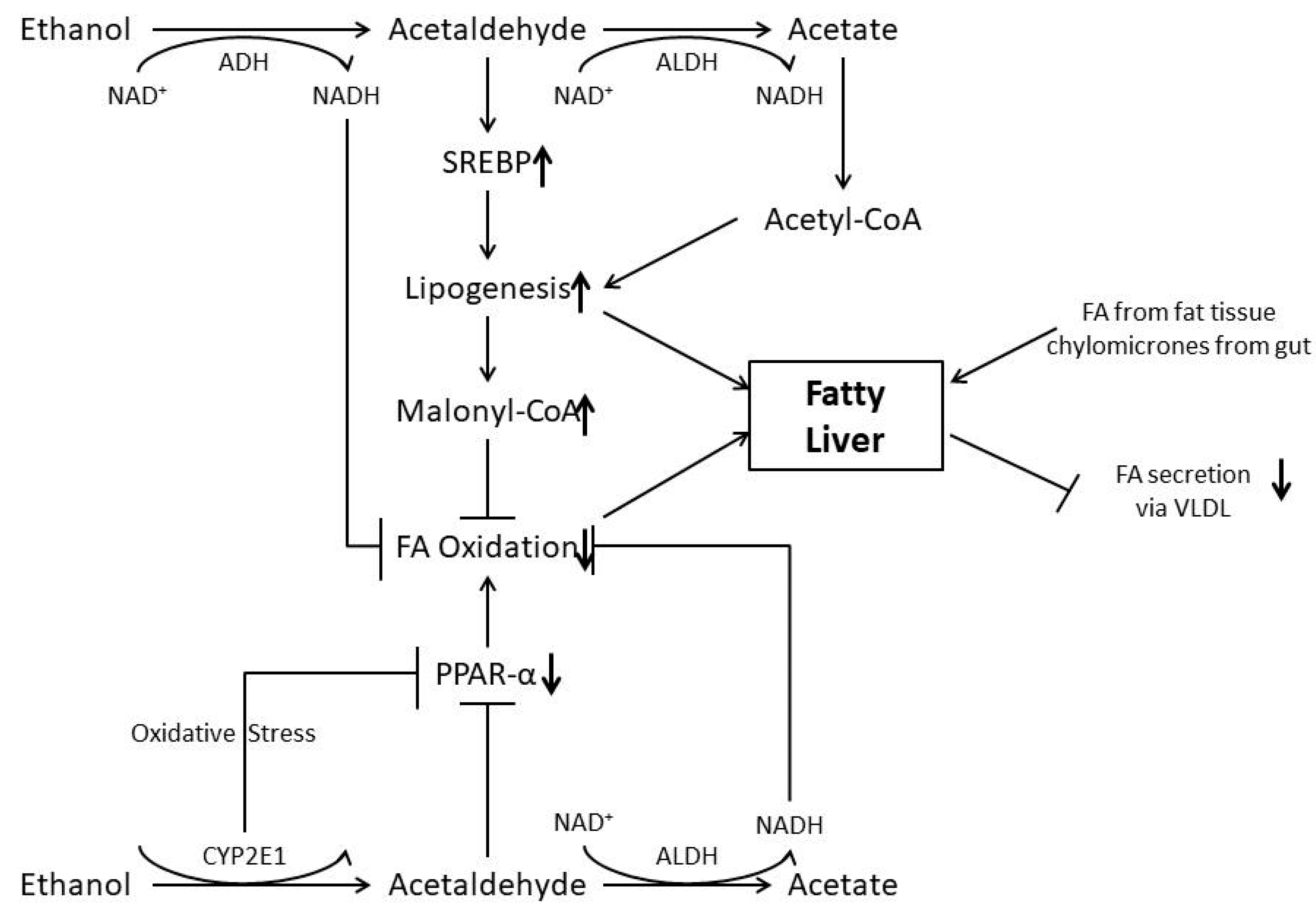
Figure 1.
Mechanisms involved in the pathogenesis of alcoholic fatty liver. Ethanol via the CYP 2E1 is oxidize; Acetaldehyde in the presence of acetaldehyde dehydrogenase generate NADH and acetated which inhibit FA oxidation. Acetaldehyde stimulate SREBP and inhibit PPARα. While increased SREBP stimulates lipogenesis, decreased PPARα inhibit FA oxidation, so does malonyl-CoA addition. Oxidative stress generated by ethanol oxidation through cytochrome P4502E1 also inhibits PPARα and may damage mitochondria with a decrease in NADH- and acetaldehyde oxidation. In addition, FA and chylomicrons are delivered from fat tissue and from the gut, respectively. Finally, FA secretion as VLDL from the hepatocyte is inhibited by alcohol. NAD (nicotine adenine dinucleotide); NADH (nicotine adenine dinucleotide) in reduced form; ADH (alcohol dehydrogenase); ALDH (acetaldehyde dehydrogenase); FA (fatty acid) SREBP1c; PPARa; CYP2E1 (cytochrome P4502E1); VLDL (very low density lipoprotein) .
Figure 1.
Mechanisms involved in the pathogenesis of alcoholic fatty liver. Ethanol via the CYP 2E1 is oxidize; Acetaldehyde in the presence of acetaldehyde dehydrogenase generate NADH and acetated which inhibit FA oxidation. Acetaldehyde stimulate SREBP and inhibit PPARα. While increased SREBP stimulates lipogenesis, decreased PPARα inhibit FA oxidation, so does malonyl-CoA addition. Oxidative stress generated by ethanol oxidation through cytochrome P4502E1 also inhibits PPARα and may damage mitochondria with a decrease in NADH- and acetaldehyde oxidation. In addition, FA and chylomicrons are delivered from fat tissue and from the gut, respectively. Finally, FA secretion as VLDL from the hepatocyte is inhibited by alcohol. NAD (nicotine adenine dinucleotide); NADH (nicotine adenine dinucleotide) in reduced form; ADH (alcohol dehydrogenase); ALDH (acetaldehyde dehydrogenase); FA (fatty acid) SREBP1c; PPARa; CYP2E1 (cytochrome P4502E1); VLDL (very low density lipoprotein) .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



