
Submitted:
08 May 2023
Posted:
09 May 2023
You are already at the latest version
Abstract
Acute lymphoblastic leukemia (ALL) is a blood cancer that primarily affects children but also adults. It is due to the malignant proliferation of lymphoid precursor cells that invade the bone marrow and can spread to extramedullary sites. ALL is divided into B cell (85%) and T cell lineages (10 to 15%); rare cases are associated with the natural killer (NK) cell lineage (<1%). To date, the survival rate in children with ALL is excellent while in adults continues to be poor. Despite the therapeutic progress, there are subsets of patients that still have high relapse rates after chemotherapy or hematopoietic stem cell transplantation (HSCT) and an unsatisfactory cure rate. Hence, the identification of more effective and safer therapy choices represents a primary issue. In this review, we will discuss novel therapeutic options including bispecific antibodies, antibody-drug conjugates,chimeric antigen receptor (CAR)-based therapies, and other promising treatments for both pediatric and adult patients.
Keywords:
ALL
; immunotherapy
; antibody-drug conjugate
; CAR-based therapies
; targeted therapies
1. Introduction
ALL is a hematologic malignancy characterized by the uncontrolled proliferation of early lymphoid precursors thatreplace the normal hematopoietic cells of the bone marrow[1,2,3].
The central nervous system (CNS) and testes are the most common sites of precursors’ extra-medullary spread[4], although theoretically any organ or tissue could be infiltrated. The involvement of skin, kidneys, and ovaries has also been extensively described[5,6].
ALL is divided into tumors of B-lineage, T-lineage, and uncommon variants of NK cell lineage which are morphologically indistinguishable. According to the 2016 revision to the World Health Organization (WHO), the classification of major subtypes of ALL includes 4 distinct entities: B-ALL/LBL not otherwise specified (NOS), B-ALL/LBL with recurrent genetic abnormalities, T-ALL/LBL, and NK-ALL/LBL[7], as shown in Table 1.
Few environmental and/or genetic factors have been associated with an increased risk of ALL. Among these, ionizing radiation, pesticide exposure, childhood infections[8,9,10], and genetic conditions such as Down syndrome or ataxia telangiectasia are included [11,12,13,14].
Its incidence varies among people of different ages, sex, and race[15,16,17]. Age-specific incidence curve for ALL has a bimodal distribution with peak incidences in children aged between 1 and 4, and adults aged 55 or above[18]. Males develop it more than females with a ratio of 1.2:1[19].
Globally, the estimated annual incidence of ALL is 1 to 5 cases/100.000 population, and more than two-thirds of cases of ALL are B cell phenotype[16,20,21,22]. Italy, USA, Switzerland, and Costa Rica are the countries with the highest ALL incidence [16].In the USA about 6,660 new cases and 1,560 deaths (including both children and adults) are estimated in 2022 [19].
The outcome is more disappointing in adults (5-year overall survival (OS) <45%) than in children (5-year survival rate of over 90%) [23,24,25] and this is related to multiple factors such as a higher incidence of poor prognostic markers, a lower incidence of favourable subtypes and traditional chemotherapy regimens[26].
Although there has been a substantial improvement in OS over time, there is still a gap in the availability of leukemia treatments between countries. This is partly due to the different socio-economic status; low-income countries are less likely to use available treatments and this may contribute to poor survival[27].
Therefore, there is a joint effort to find more accessible solutions and to develop promising therapeutic strategies aiming to mantain remission, improve survival, and control the toxicities associated with chemotherapy regimens.
2. Different biological characteristics in pediatric and adults ALL patients
Childhood and adult leukemias are biologically distinct and diverge in their molecular landscape but also in their cellular origin[1].Even if the exact causes of ALL are not yet understood, it has been demonstrated that in children it is the result of a multistep process associated with the acquisition of genetic alterations in lymphoid progenitors during in-utero development[28,29]. Chromosome aneuploidy, structural alterations and rearrangements, copy number variations (CNVs), and sequence mutations all contribute to leukemogenesis[30].
Disease cytogenetic abnormalities have a different prognostic impact between age categories. Usually, adult patients have a higher white blood cell count, an increased frequency of T-lineage ALL, and a decreased incidence of hyperdiploidy than children[31,32]. It was also demonstrated an increase in the presence of unfavorable genetic anomalies with increasing age (incidence up to 53% over 55 years), such as the Philadelphia chromosome[33]. In contrast, genetic alterations, such as hyperdiploid karyotype, frequently seen in pediatric ALL patients, are related to a favorable outcome[34].
The gap in outcome between children and adults is due to the differences in disease biology and treatment tolerance and also to the intensified chemotherapy regimens used in children that permit improved response rates and prolonged survival[35]. Fortunately, the management of ALL in adult patients has significantly improved thanks to the administration of pediatric-inspired regimens or even unmodified pediatric protocols (adults up to 60 years old), so chemotherapy intensity has increased[36].
3. Evolution of ALL treatment applications
Treatment for ALL is divided into four different phases: remission induction, consolidation, intensification, and long-term maintenance. CNS prophylaxis is given at the proper intervals during the treatment. Allogeneic HCT is optional after consolidation.
Standard frontline chemotherapy is used for induction therapy while targeted drug therapy, alone or combined with chemotherapy, is employed for all phases.
The achievement of the current treatment modalities is the result of changes that happened in a temporal space that started in the 70s when the older strategies were applied[37]. At that time, cranial radiotherapy (CRT) to prevent CNS relapse was used for all patients[38,39]. However, intensive and prolonged therapy for ALL was considered responsible for detrimental effects on intellectual and learning abilities[40,41,42]. As a result, some years later, CRT intensity has been reduced and intrathecal therapy and high doses of systemic chemotherapy substituted the previous method[43,44,45,46,47].
Then, conventional chemotherapy has been optimized, raising the chance of cure in the highest-risk patients while minimizing long-term adverse events in those with the lowest risk[48,49,50,51].
However, in childhood ALL, CNS-directed prophylaxis remains an obliged choice. Indeed, the high possibility of infiltration of the CNS, by massive numbers of leukemic cells, puts patients at a higher chance of CNS relapse leading to severe morbidity and mortality[52,53].
Until now, little information is known on where leukemia cells reside in the CNS and about their interactions with cellular components of the CNS microenvironment, which could induce their quiescence and survival. Certain studies suggested that some B-Cell Precursor (BCP)-ALL cells would be able to survive in particular CNS niches for a very long time as extramedullary minimal residue disease and could be responsible for CNS relapse[54,55,56].
Fernandez-Sevilla et al. proposed that the choroid plexus (CP), asecretory tissue responsible for producing cerebrospinal fluid, constitutes a sanctuary for paediatric BCP-ALL cells. Inside it, interactions between BCP-ALL cells and microenvironment cell components promote their survival and chemoresistance[57].
Allo-HCT used as a consolidation therapy, contributes to the considerable improvement in the prognosis of patients with ALL, but not without complications [complexity and graft versus host disease (GVHD)]. Access to allo-HCT is usually reserved for patients with high-risk characteristics or relapsing disease[58,59,60,61]. Until recently, for those patients as well as for older adults, the treatment options were extremely limited.
4. Immunotherapy for ALL
Finding the best treatment for ALL is an ongoing challenge leading to the continuous development of new therapeutic approaches. Among these, immunotherapy stands out, exploiting the patient’s immune system to target cancer cells, improving survival, and reducing the toxicity of chemotherapy. Major immunotherapies include the use of bispecific antibodies, CART or CARNK cells, and antibody-drug conjugates, which are showing important results primarily in the treatment of B-ALL. CART or CARNK cells and antibody therapy hold promise also for the treatment of T-ALL.
4.1. Bispecific antibodies (BsAbs)
BsAbs are antibodies engineered to contain two different fragment-binding antigens (Fabs) regions that allow for the concurrent targeting of two antigens. One of the main mechanisms of action of BsAbs is to recruit and activate effector cells (i.e., T cells) against target cells (i.e., tumor cells). These antibodies, unlike conventional ones, induce enhanced T-cell activation. BsAb-activated T cells exert their cytotoxic action on tumor cells by producing proteins such as perforin and granzymes. Perforin is responsible for pore formation in cell membranes, facilitating the entry of granzymes and thus, enabling their delivery into the cytosol to initiate apoptosis[62,63,64].
Blinatumomab is a bispecific Tcell-engaging (BiTE) antibody. BiTe antibodies lack the Fc fragment and are composed of the fusion of two different single-chain variable fragments (scFvs); one scFv binds the CD3 expressed on effector T cells and the other binds a tumor-associated antigen. Blinatumomab by simultaneously binding CD19 on B-ALL cells and CD3 on T cells, can mediate a direct cross-link between T cells and tumor cells[65,66], resulting in a targeted and highly effective tumor cell killing [67].
Figure 1.
Blinatumomab immunotherapy for the treatment of CD19-positive Bcell precursor ALL. (A) Representative scheme depicting the structure of Blinatumomab. Blinatumomab is comprised of the fusion of the scFv of CD3 fused with a linker to the scFv of CD19. (B) Representative scheme depicting the mechanism of action of Blinatumomab. Blinatumomab can specifically bind both the CD3 expressed on T cells and the CD19 expressed on B-ALL cells, cross-linking the T cells and B-ALL cells and mediating cell lysis. Note: Figure created with BioRender.com.
Figure 1.
Blinatumomab immunotherapy for the treatment of CD19-positive Bcell precursor ALL. (A) Representative scheme depicting the structure of Blinatumomab. Blinatumomab is comprised of the fusion of the scFv of CD3 fused with a linker to the scFv of CD19. (B) Representative scheme depicting the mechanism of action of Blinatumomab. Blinatumomab can specifically bind both the CD3 expressed on T cells and the CD19 expressed on B-ALL cells, cross-linking the T cells and B-ALL cells and mediating cell lysis. Note: Figure created with BioRender.com.
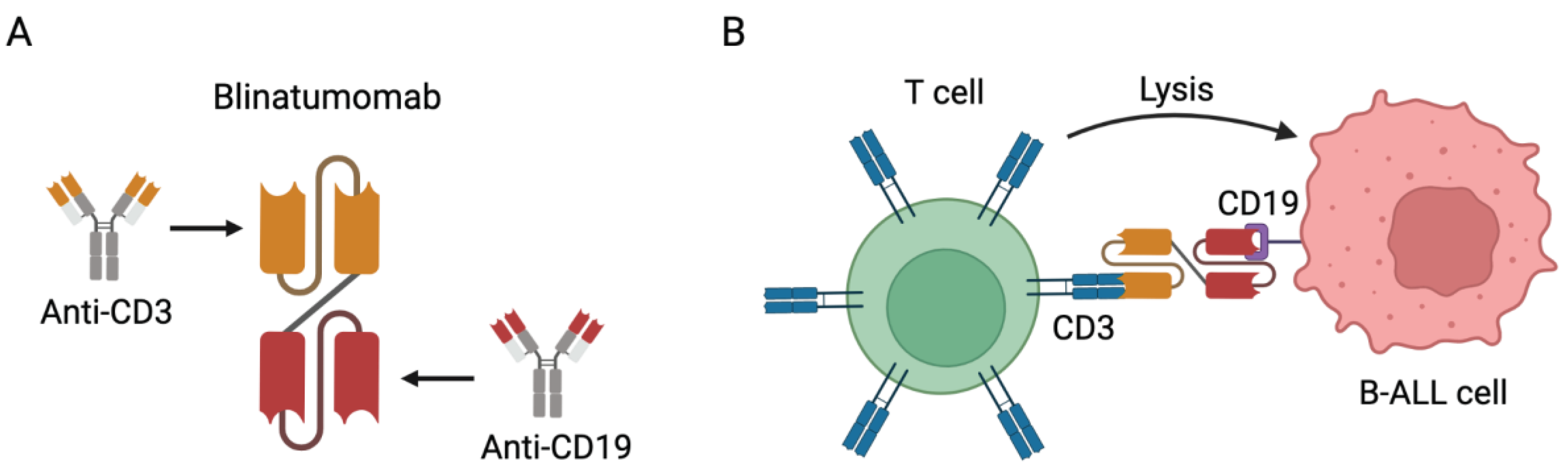
It is the first FDA-approved BiTE antibody to treat minimal residual disease (MRD)-positive BCP-ALL, as well as for relapsed or refractory (R/R) ALL, both in adult [68,69] and pediatric patients[70]. According to European Medicines Agency (EMA), the use of blinatumomab in the pediatric population is limited to children aged one or older with Philadelphia chromosome-negative CD19 positive Bcell precursor ALL (CD19+R/R Ph-negative BCP-ALL), which is refractory or in relapse after receiving at least two prior therapies or in relapse after receiving prior HSCT[71]. Cytokine release syndrome (CRS) and neurotoxicity are reported as the main adverse events, most of which can be rapidly resolved.
Blinatumomab in adult patients
The first single-arm study on the use of blinatumomab involved 189 adults with R/R Ph-negative B-ALL. Complete remission/complete remission with partial hematologic recovery (CR/CRh) was achieved in 43% (81/189) of patients within 2 cycles of blinatumomab with a median OS of 6.1 months[69]. The positive data produced has accelerated, at the end of 2014, FDA approval for the use of blinatumomab in adults with R/R Ph-negative B-ALL. The greater efficacy of blinatumomab compared to conventional chemotherapy was then confirmed in the TOWER study. In this randomized, open-label, multicenter phase III trial, 271 out of 495 patients with R/R B-ALL received blinatumomab and 134 patients received standard chemotherapy (2:1 ratio). Patients who received blinatumomab had a better CR rate (34% vs 16%, p<0.001), greater MRD negativity (76% vs 48%), and a longer median OS (7.7 vs 4.0 months;p=0.001) than those treated with chemotherapy.
The efficacy of blinatumomab was tested also in the setting of MRD[72]. In the BLAST study, it was used as monotherapy in adults with MRD-positive B-ALL. For this study, 116 patients were enrolled and, of the 113 evaluable patients, 88 (78%) had a complete MRD response after the first treatment cycle. The median OS was 36.5 months and the relapse-free survival (RFS) was 54% after 18 months of follow-up[73].
Blinatumomab has been evaluated also in patients with Ph-positive B-ALL previously treated with tyrosine kinase inhibitor (TKI)-based therapy (ALCANTARA study). Sixteen patients out of 45 enrolled achieved a CR/CRh rate of 36% with 88% complete MRD[74].
Furthermore, the Foà et al. study assessed a chemotherapy-free induction and consolidation first-line treatment with dasatinib and blinatumomab for Ph+ ALL in adults. More specifically, the 63 adult patients enrolled in the study underwent treatment with dasatinib plus glucocorticoids, followed by 2 cycles of blinatumomab[75,76]. Ninety-eight percent of the patients achieved CR and 60% had a molecular response (MR) that further increased up to 81% after the fourth cycle of blinatumomab. At a median follow-up of 18 months, the OS was 95% and RFS was 88%. Finally, 24 patients received HSCT and the transplantation-related mortality was considerably lower (4%) than that of other previous studies. Induction with TKIs is showing promising results also in the phase 3 PhALLCON study(NCT03589326), where a comparison of first-line ponatinib (PON) vs imatinib (IM) with reduced-intensity chemotherapy (CT) has been done in patients (pts) with newly diagnosed Philadelphia chromosome–positive (Ph+) ALL.
Blinatumomab in pediatric patients
The blinatumomab investigation in the pediatric population has been encouraging even from the first small case series performed in patients with relapsed ALL after HSCT[77,78]. Since then, worldwide research in paediatric use of blinatumomab exponentially increased(Figure 2)[77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103].
Two randomized controlled trials have recently published their results. Data from the first trial (NCT02101853) are reported by Brown and colleagues. They described the experience of the Children’s Oncology Group that conducted a randomized phase 3 clinical trial in the USA, Australia, and New Zealand[99]. Two-hundred and eight children, adolescents, and young adults (aged 1 to 30 years) with B-ALL first relapse were eligible. They received a 4-week reinduction chemotherapy course followed by blinatumomab as post-reinduction consolidation or conventional chemotherapy; HSCT followed both treatments. Improved survival has been detected by substituting intensive chemotherapy with blinatumomab in consolidation therapy. The blinatumomab group showed improved DFS (54.4% vs 39%), OS (71.3% vs 58.4%), and MRD clearance (75% vs 32%) with lower toxicities[99].
Furthermore, results of the trial NCT02393859 were published by Locatelli and colleagues, who showed data obtained using blinatumomab as consolidation therapy instead of chemotherapy before allogeneic HSCT for patients (aged >28 days up to 18 years) with high-risk first-relapse B-ALL. A total of 108 patients were included in this study. Enrollment was terminated early because the prespecified criterion to declare benefit in favor of blinatumomab was met. After a median follow-up time of 22.4 months, the adverse events in the blinatumomab arm were significantly fewer than in the consolidation chemotherapy group (31.5% and 57.4%). The death occurred in 8 blinatumomab-treated patients and 16 chemotherapy-treated ones (14.8% vs 29.6%). The OS hazard ratio from a stratified Cox proportional hazard model was 0.43 (95% CI, 0.18-1.01). CR was seen in 44 of 49 patients (90%) who were treated with blinatumomab and in 26 of 48 patients (54%) treated with chemotherapy. Treatment with 1 cycle of blinatumomab before allogeneic HSCT resulted in an improved event-free survival at a median of 22.4 months of follow-up over intensive multidrug chemotherapy[100].
Moreover, in January 2022, Locatelli and colleagues reported the final analysis data of an open-label, single-arm, extended-access international study (RIALTO) conducted across 16 specialized hospitals in Europe and the USA and confirmed the results published in 2020 on the safety and efficacy of blinatumomab. Among 110 patients with CD19 positive R/R BCP ALL (aged >28 days up to 18 years) enrolled in the study, 69 achieved CR within the first 2 treatment cycles; most of them (73.5%) received an allogeneic HSCT and had better OS compared with those who did not (1-year OS probability: 87% vs 29%). Authors referred also to a very low incidence of severe CRS (grades 3–4) and grade 3 neurotoxicities arising with blinatumomab treatment. Only 2 patients developed severe grade ≥3 treatment-related CRS and only 6 experienced severe grade ≥3 neurologic toxicity. These events resolved quickly (CRS median resolution time, 6.5 days; neurologic toxicity median resolution time, 2.0 days). No adverse events with fatal outcomes were associated with blinatumomab treatment[101].
Recent retrospective studies have also provided evidence supporting the anti-leukemic activity of blinatumomab in pediatric R/R. Among 113 children who received blinatumomab treatment, via an expanded access program (EAP), thirty-eight of 72 patients in the R/R group achieved a hematological response within 2 cycles of blinatumomab. Fifty percent of these patients underwent a transplant without bridging myelosuppressive therapy. In patients with evaluable MRD (n = 36), 83% (n = 30) achieved an MRD response. Taken together, these results demonstrated that the real-world effectiveness of blinatumomab in this cohort of patients was similar to that demonstrated in clinical studies[102]. In addition, the Italian real–life multicenter retrospective study on 39 R /R BCP-ALL pediatric patients (0-21 years old) treated in seven National Cooperative Pediatric Oncology Group (AIEOP) centers represents a further demonstration of blinatumomab efficacy (CR rate 46% in 13 patients) and good tolerability (34.8% grade ≥3 AE rates, no CRS and no associated toxic deaths)[103].
4.2. Antibody-drug conjugates (ADC)
ADCs are obtained by combining a mAb with a cytotoxic drug using various linkers that determine how and when the drug is detached from the mAb[104]. Following the binding of the surface antigen, ADCs are internalized into the endosomes and then released in the lysosomes. Finally, when ADCs are delivered to the nucleus, they induce cell death[105,106]. It has also been shown that non-internalized ADCs directed against the tumor microenvironment (TME) components can efficiently liberate their drug in the extracellular space and mediate a potent therapeutic activity (bystander killing effect) [107](Figure 3).
CD22 is expressed in around 90% of B-ALL cases and can be considered an ideal B cell target for immunoconjugate treatment. Inotuzumab ozogamicin (InO) is a humanized CD22 monoclonal antibody conjugated to calicheamicin [108], approved for R/R ALL adult patients’ treatment. Its approval was supported by the results of the global, open-label, phase 3 randomized INO-VATE study (NCT01564784) that investigated the efficacy and safety of InO vs. chemotherapy in R/R ALL patients with low, moderate, or high disease burden. Patients treated with InO had improved CR rates and improved median OS than those treated with chemotherapy (CR rates: 81% vs 29%; OS: 7.7 vs 6.7 months) [108].
Moreover, DeAngelo et al. carried out a post hoc analysis of INO-VATE to evaluate the long-term efficacy and safety profile of InO also in adult patients R/R -ALL with high baseline disease burden. They reported comparable clinically meaningful benefits to those observed in subgroups with low and moderate disease burden[109]. However, patients receiving a starting dose of 1.8 mg/m2/cycle (in 3 divided doses) of InO had an increased risk of sinusoidal obstruction syndrome (SOS), especially following HSCT, compared with those who received standard chemotherapy (13% vs <1%) [108]. Therefore, other groups are evaluating how to prevent SOS. One of the strategies (NCT03677596) is based on the use of a lower dose of InO (1.2 mg/m2/cycle). Initial results showed that half of the patients (11/22) achieved remission, and more than 70% of them achieved MRD negativity[110]. Additional studies recommend ursodiol prophylaxis, limited use of InO at no more than two cycles before HCT, and no dual alkylating agents such as thiotepa and melphalan and hepatotoxic agents[111].
Despite the good results of InO treatment in adult ALL, there is still little information on its safety and efficacy in childhood ALL. However, available data from European and American compassionate use programs demonstrated a favorable benefit-risk profile of InO in children with R/R BCP-ALL. To provide more comprehensive data on InO effectiveness and tolerability, phase II prospective studies on pediatric patients with R/R ALL are currently underway in the United States (NCT02981628/ AALL1621) and in Europe (EudraCT 2016– 000227–71). Results of the Children’s Oncology Group trial AALL1621 on InO treatment in children and adolescents with R/R B-ALL were presented at Asco 2022. In cycle 1, patients received a starting dose of InO of 0.8 mg/m2 intravenously on day 1 and 0.5 mg/m2 on days 8 and 15 of a 28-day cycle with response evaluation at day 28. Dose-limiting toxicities and SOS were continuously checked. Nineteen out of 48 patients had CR and 9 CRi after cycle 1. Twenty-one patients received HSCT after InO, of whom 6 developed grade 3 SOS. InO was effective with high response rates and MRD< 0.01% in two-thirds of responders even if SOS after HSCT and prolonged cytopenias were notable[112].
Furthermore, the phase 1 trial (ITCC-059), registered as EudraCT 2016–000227–71, investigated the recommended phase 2 dose of InO in children with multiple R/R ALL. Twenty-five patients aged 1 to 18 years were enrolled in the study and, among these, 23 were included in the dose escalation analysis (3 doses of InO per course). Severe adverse events were observed in 23 patients and hepatic SOS was evidenced in 2 patients following chemotherapy. OR after 1 course was achieved in 20 of 25 patients, of whom 84% reached CR-MRD negative. The OR at 12 months was 40%. Recommended phase 2 dose (RP2D) of InO was established at 1.8 mg/m2 per course for adults[113].
Such results demonstrate the efficacy of these antibody constructs and support new design approaches based on the synergistic potential of either or both agents with low-intensity chemotherapy to further improve outcomes, especially in older patients. The first data obtained indicate that the use of InO in combination with low-intensity chemotherapy (mini-hyper-CVD) with or without blinatumomab confers better outcomes than standard intensive chemotherapy (hyper-CVAD) as first-line therapy in adult patients with ALL [114,115,116,117,118].
4.3. Chimeric antigen receptor (CAR)-engineered immune cell therapy in ALL
In this section, we will review one of the most promising immunotherapy approaches for ALL, consisting of the genetic modification of immune cells such as T-cells and also NK cells with chimeric antigen receptors (CARs). It’s known that the immune system plays a crucial role in tumor growth control. The tumor, however, may escape detection by the immune system, and its growth and progression are controlled by TME. Hypoxia, also as a consequence of ischemia or nutrient deprivation are only some of the ways used by TME to destabilize immune cells. Hypoxia can shape the type and function of immune cell infiltration in the TME by polarizing tumor-associated macrophages (TMA) toward anti-inflammatory M2 macrophages and cytokines[119,120,121,122]. Furthermore, immune cell dysfunction is mediated by a series of factors including the changes in signal transduction molecules, loss of TSA, stimulation of CTLA4 on T cells, and secretion of some soluble molecules by tumor or non-tumor cells in the TME, other than by the presence of some immunosuppressive cells in TME[123,124]. In this context, the engineering of CART cells has become the new frontier of immunotherapy in the treatment of hematological malignancies, even if it has important adverse events limiting its success. CRS and neurotoxicity together to on-target off-tumor effects and GVHD are only some of the restrictions linked to a broader application of CART cell therapy in hematological disease treatment. To overcome these limitations, other immune effector cells that may be modified with CARs and used in immunotherapy are being studied. The scientific focus has recently shifted to NK cells, whose particular molecular peculiarities make them suitable for an “off-the-shelf” allogeneic therapy. First, it’s possible to produce big NK cells quantities from several sources and this, together with a minimal risk of toxicity or GVHD permitted by the HLA-I dominant-negative regulatory function on NK killing activity, and a minor cost of production makes CARNK cell therapy the future of immunotherapy for leukemia.
4.3.1. CART Cells
CART cell therapy is based on the genetic engineering of a patient’s T cells to induce the expression of a chimeric receptor able to recognize a marker expressed on tumor cells leading to cancer cell elimination. T cells are sampled from the patient’s peripheral blood and transduced with viral vectors encoding the desired genes. Genetically engineered cells are then expanded in vitro before re-infusion into the patient’s blood (Figure 4).
Currently, CART cells can be categorized into four generations based on the organization of their intracellular signaling domain, with fifth-generation CARs similar to the second-generation, but with an intracellular domain of a cytokine receptor[125,126,127,128,129,130].
CD19 is an ideal target antigen for CART cell therapy and encouraging results have been reported in the treatment of several types of B cell malignancies[131]. In 2013, for the first time, CD19-directed CART (CART 19) cells have been successfully used in two children with chemotherapy-resistant ALL, and, despite the presence of severe CRS and B-aplasia, CR was achieved in both patients[132]. Since that, several investigations have been carried out to better understand the CR rate and the durability of the CART cell therapy effect, and, early reports demonstrated the potential benefits of CAR T cells in R/R B-ALL[133,134,135,136,137,138].
In 2018, FDA approved the anti-CD19 CART cell therapy tisagenlecleucel (CTL019) for R/R B-ALL based on the results of the ELIANA multicenter study (ClinicalTrials.gov number, NCT02435849) that showed high response rates in patients up to 25 years of age. Although transient high-grade toxic effects occurred, an overall remission rate of 81% among 75 patients at 3 months of follow-up after a single infusion of tisagenlecleucel has been reported[133].
In the same year, Park et al. used a CD19 CART construct with a CD28 costimulatory domain (19-28z) in a phase 1 trial on 53 adults with relapsed B-cell ALL. They hypothesized that the safety and long-term efficacy of 19-28z CART cells may be associated with the clinical characteristics of the patients, disease characteristics, the treatment regimen, and the kinetics of T-cell expansion. They reported that 14 out of 53 patients developed severe CRS and 1 patient died. CR was achieved in 83% of the patients and MRD response was observed in 67%. The median OS was 12.9 months. 19-28z CART cell therapy (median follow-up of 29 months) showed a favorable long-term remission rate in patients with a low disease burden, who had significantly longer event-free survival and OS with a markedly lower incidence of toxic effects than did those with a high disease burden[135].
KTE-X19, another anti-CD19 CART cell therapy, already approved for non-Hodgkin lymphoma, has also been studied for the treatment of B-ALL. Data from the ZUMA-3 trial, which has been conducted on adult patients with R/R B-ALL, showed a high response rate and tolerable safety of KTE-X19. Fifty-four patients were enrolled in phase 2 of the trial and 45 of them received a single infusion of the CD19-directed product (2×106, 1×106, or 0.5×106 cells per kg) after lymphodepleting chemotherapy. Severe CRS and neurotoxicity, which occurred respectively in 31% and 38% of the patients, were successfully managed. CR was achieved in 52% of patients within 3 months[139]. Furthermore, at the 2021 American Society of Clinical Oncology (ASCO) Annual Meeting, Shah, et al. presented the results of the phase 2 portion of this trial reporting that the CR/CRi rate was 71%. After a median follow-up of 16.4 months, KTE-X19 showed compelling clinical benefit in heavily pretreated adults with R/R B-ALL, with the median OS not yet reached for responding pts and a manageable safety profile[140]. Finally, in October 2021, KTE-X19 was approved as the first CART cell therapy for adults with R/R B-ALL.
Interestingly, CART cells can migrate to CNS or testes and thus, can be considered a good therapeutic choice also for the treatment of CNS-relapsed leukemia[141,142,143]. In 2015, Rheingold et al. demonstrated that CTL019 was detectable in cerebrospinal fluid in 46 out of 47 treated patients with B-ALL, indicating the ability of this therapy to cross the blood-brain barrier [144]. To date, only a few results are available on the efficacy of CART cell therapy in patients with R/R B ALL and active CNS disease [136,145,146]. However, data from the CHP959 trial (NC01626495), carried out on 65 patients with CNS involvement showed no significant differences in relapse-free survival or neurological toxicities between patients with active CNS disease and those without it, before CD19 CART cell infusion[147]. Other studies confirmed the efficacy of this treatment in patients with multiply relapsed or refractory extramedullary leukemia [148,149].
The value of CART cells is undeniable in the treatment of B-ALL, but it is necessary to understand how to minimize toxicities such as CRS, immune effector cell associated neurotoxicity syndrome (ICANS), and B-cell aplasia related to it, particularly in adult patients. CRS is a systemic inflammatory response that is often associated with CART cell therapy within 1-4 days after the infusion and can progress to multiple organ dysfunction. Learning how to recognize early CRS is a fundamental step to treating it promptly and preserving life-threatening consequences. Of note, higher grade CRS has been associated with higher disease burden and may be effectively treated with the anti-interleukin-6 receptor antibody tocilizumab that, however, could limit the efficacy of the immunotherapy [150,151]. In addition, a relationship between CART dose and CRS occurrence has been highlighted[152]. Therefore, the adoption of a fractionated dosing scheme might be a good strategy to retain high response rates with acceptable tolerability. Meaningful advancement was shown by Frey’s group which demonstrated that fractionation of CTL019 dosing treatment can help manage CRS toxicity and maintain efficacy in adults with R/R ALL [153]. ICANS is also associated with CART cell therapies and it seems due to both activated CART and endogenous T lymphocytes and the cytokines secreted by them [154,155,156]. It can occur in association with or following CRS and its management continues to evolve and constitutes an area of ongoing research. In addition, B cell aplasia represents another CART cell-related toxicity linked to CD19 CART cell therapy for B-ALL[157]. Hypogammaglobulinemia and agammaglobulinemia caused by Bcell aplasia expose patients to an increased risk of infections that needs to be promptly managed to avoid lethal consequences[157,158]. To this aim, immunoglobulin replacement may help to prevent serious bacterial infections[133,159,160,161,162]. It has been demonstrated that increasing serum IgG levels may result in protection against infections [157]. Moreover, antimicrobial and antifungal prophylaxis is also recommended in the prevention of infections in patients treated with CD19-redirected CART cell therapy [163,164,165]. In case of viral infections such as herpes simplex virus and varicella zoster virus reactivation, following CD19 redirected CART therapy, antiviral prophylaxis should be considered. Already in the past, researchers were interested in studying the possible impacts of immunotherapy in leukemic patients with viral infections such as HIV, hepatitis B, and C. Until the past few years, in the presence of viral infections, patients were not considered for immunotherapy treatment that could worsen the infection.
However, during the COVID-19 pandemic, studies on the interaction between the immune system and acute respiratory syndrome coronavirus 2 (Sars-Cov-2) indicated that the coronavirus can promote PD-L1 expression, towards which several immunotherapy drugs are directed. Therefore, in the case of coronavirus infection, in the early phases, an immunotherapy regimen could have positive effects in counteracting the virus, by stimulating the patient’s immune system against it. Instead, in severe Sars-Cov-2, the use of immunotherapies could represent a risk for the inflammatory storm associated with a hyperactive inflammatory response. Recently, a prolonged severe Sars-Cov-2 infection in a BCMA-redirected CART cell therapy recipient was described. Despite convalescent plasma therapy and antiviral prophylaxis with the agent remdesivir, the patient experienced a massive lung infection and died from infection-related complications[166].
4.3.2. CARNK cell therapy
As depicted in Figure 5, NK cell activity is controlled by multiple inhibitory or stimulating receptor-ligand interactions depending on a health condition or disease [167].
NK cells are heterogenous and distinct cell subsets mediating specialized functions. The tissue of origin of NK cells is the bone marrow in which IL15 and a to lesser extent IL2 play a pivotal role in NK cell development and differentiation[168,169]. Among NK cells, two cell subsets emerge in terms of cell function. The first subset is composed of the classic cytotoxic cells while the second subset is composed of NK cells with regulatory functions. Both NK cell subsets are defined according to the intensity of cell surface expression of CD56 and CD16. The previous includes CD56lowCD16highcells while the latter is characterized by CD56highCD16low/negcells. The CD56lowCD16high are professional killer cells. NK cell cytotoxicity is regulated by a balance between activatory and inhibitory molecules termed natural cytotoxic receptors and killer inhibitory receptors (KIRs) respectively (missing-cell hypothesis); Figure 5[170]. The role of NK cells in the host defense against solid tumors is unclear. However, there is evidence that NK cells may play a minimal direct role in counteracting epithelial cancers, but, they can cooperate with T cells in controlling tumor progression. For example, NK cells are barely found in the TME, and even if they are found may not be associated with improved survival[171,172,173]. However, pre-clinical and clinical studies have shown that NK cells play a pivotal role in the immune response against leukaemia in allogeneic, HLA-matched, and unmatched settings. Unlike T cells, they do not mediate GVHD. As a consequence, NK cells are interesting effector cells for cell-based immunotherapy for leaukemias[174,175].
NK cells can be obtained from several sources including donor or autologous peripheral blood mononuclear cells (PBMC), umbilical cord (UCB), cell lines (NK-92), pluripotent stem cells (PSC) from human embryonic stem cells (hESC), and induced pluripotent stem cells (iPSC). All these sources of NK cells can, subsequently, be engineered with a CAR, expanded, and infused into the patient.
Figure 6.
Novel CARNK cell therapy.Representative scheme depicting the process of CARNK cell generation for clinical use. Various cellularsources are utilized for the isolation or differentiation of NK cells.The NK cells are then engineered to express a CAR on their surface and expanded in culture.Following ex vivoexpansion, the CARNK cells are infused into the patient and are re-directed to target and destroy cancer cells. Note: Figure created with BioRender.com.
Figure 6.
Novel CARNK cell therapy.Representative scheme depicting the process of CARNK cell generation for clinical use. Various cellularsources are utilized for the isolation or differentiation of NK cells.The NK cells are then engineered to express a CAR on their surface and expanded in culture.Following ex vivoexpansion, the CARNK cells are infused into the patient and are re-directed to target and destroy cancer cells. Note: Figure created with BioRender.com.
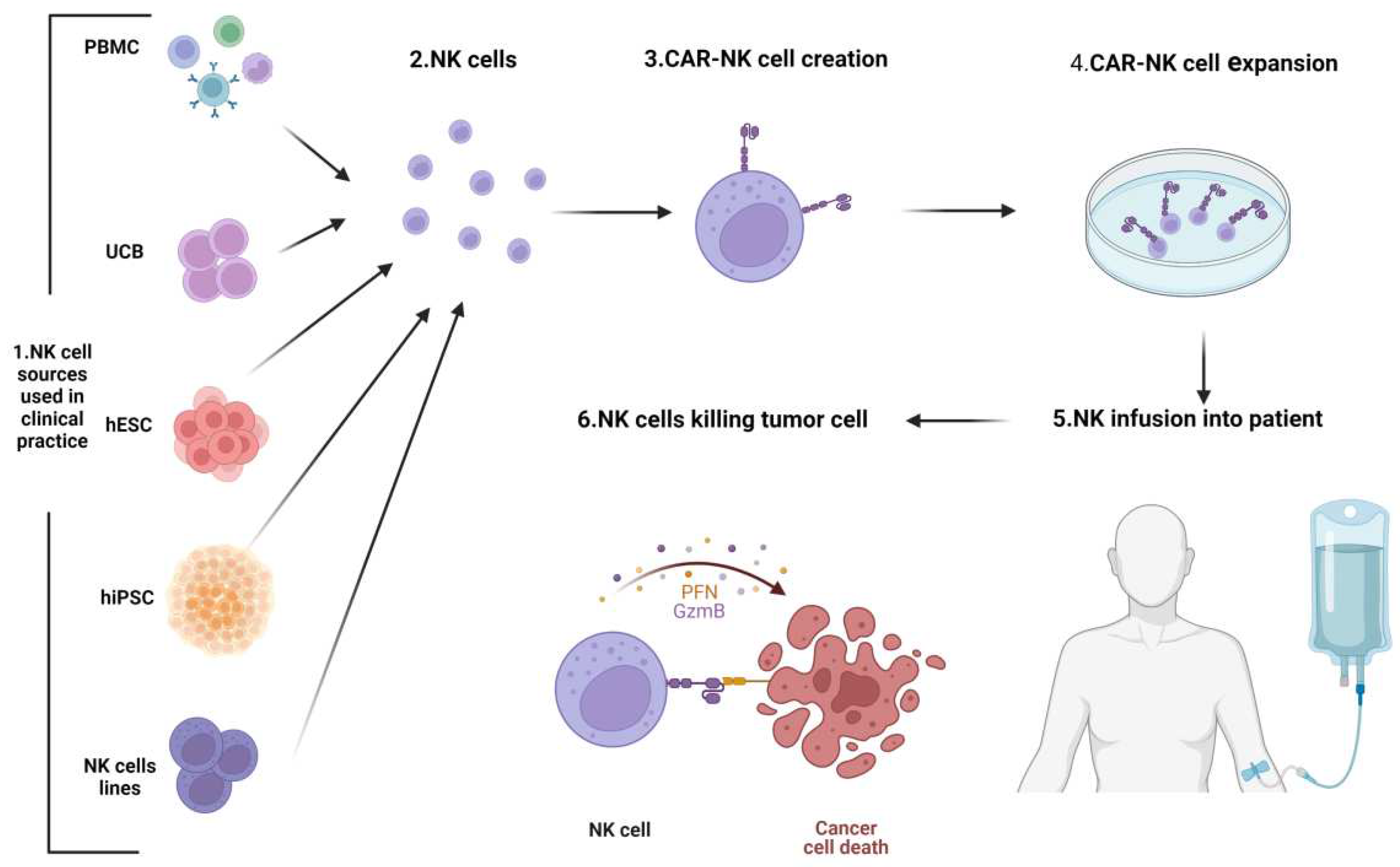
Analyzing and comparing the efficacy of methods for NK cell engineering, it has been shown that NK cells can be quickly isolated from peripheral blood (PB) however, they are difficult to engineer due to low transduction efficiency combined with poor expansion. Instead, NK-92 cells demonstrate a strong anti-tumor activity [176] that makes them a good option for engineering, even if they need to be irradiated before use to prevent lethal effects. To overcome the limitations of long-term storage decreasing the cytotoxic capabilities, a good choiceis given by UCB-derived NK cells. Indeed, these cells may undergo cryopreservation with minimal alterations, and despite their relatively immature nature, exhibit high proliferative capabilities and work effectively for in vivo studies compared to PB-derived NK cells[176]. Also manufacturing iPSC-NK cells may be considered a good alternative. IPSC-NK cells are quick to obtain, safe, and show high cytotoxic activity against tumor cells. To further improve the efficacy of CARNK cells, several gene editing strategies to enhance their potential, their persistence, and homing are being studied [177].
NK cell-mediated immunotherapy is based on increased NK cell activation via blocking inhibitory interactions, expanding NK cell populations, and improving overall function. Although it is still in the experimental phase, its potential is amply suggested by longer survival and reduced relapse together with fewer adverse effects than CART cell therapies[178].
Initial studies on CARNK cells have been initiated by CART cell constructs since NK cells and T cells share some costimulator domains such as 4-1BB. However, other co-stimulatory domains, more specific for NK cell signaling, are being investigated. In particular, NKG2D and CD244 (2B4) are the two costimulatory molecules through which, NK cells raise their cytotoxic capability and cytokine production [179]. Early results from ongoing clinical trials are encouraging and demonstrate that NK cells provide a safer and more advantageous CAR-engineering platform than that T cells[180]. This permits us to hypothesize that a large number of patients can be treated on demand with this new immunotherapy. However, to date, only a few clinical studies of CARNK cell immunotherapy for ALL patients are going on. This may be partially due to the modest outcomes obtained from the use of first-generation CARNK cells [181]. Among the most promising clinical trials, there is NCT05020678, a single-arm, open-label, multicenter, phase 1 study that is ongoing to evaluate the safety and tolerability of an experimental intravenous allogeneic CARNK cell targeting CD19 (NKX019) in adult patients (n=60) with relapsed/refractory NHL, CLL or B-ALL. NCT05563545, a single-arm clinical study, is instead recruiting cases with recurrent or refractory CD19 positive ALL to evaluate the safety, dose, tolerance, and pharmacokinetic characteristics of CARNK-CD19 (SNC103) and also define the effectiveness, the immunogenicity of the product, and the correlation between the changes of cytokines after infusion and CRS, ICANS. Furthermore, in NCT04796688, patients with CD19+ R/R ALL are being recruited to be treated with fludarabine + cyclophosphamide + CARNK-CD19 cells and evaluate the safety and efficacy of universal CAR-modified AT19 cells. Instead, in NCT04796675, NK cells derived from healthy donor cord blood (CB) have been engineered with an anti-CD19 CAR to test their safety and efficacy in patients with CD19+ Bcell malignancies. Also in the active, but still not recruiting NCT03056339 clinical study, CB-derived NK cells are being used. The purpose of the study is to learn if iC9/CAR.19/IL15-transduced CB-NK cells infusion, after fludarabine and cyclophosphamide or mesna chemotherapy, improves the disease in patients with R/R B-cell leukemia. Another goal of the study is to find the highest tolerable dose of CARNK cells to give to patients and evaluate the safety of this treatment. Among the several NK lines, the NK-92 cell line has been successfully modified to express CARs recognizing antigens expressed on tumor cells and may be considered an ideal source for cell-based immunotherapy. Previous phase 1 clinical study has demonstrated that NK-92 cells can be irradiated at very high doses with minimal toxicity in patients with refractory hematologic tumors, who had relapsed after autologous hematopoietic cell transplantation [182]. The clinical trial NCT02892695 started in 2016, is one of the first clinical trials with engineered NK-92 cells for CAR therapy. Ten patients with leukemia (including ALL) or lymphoma have been enrolled to evaluate the safety and optimal dose of CARNK PCAR-119 used as bridge immunotherapy before receiving stem cell transplantation. In the ongoing NCT02727803 phase II study, CAR-engineered NK-92 cells are used in patients (estimated enrollment of 100 patients with myelodysplastic syndrome, leukemia, lymphoma, or multiple myeloma) that have received cord blood transplantation.
The primary objective of the study is to define the progression-free survival (PFS) time and then evaluate OS time for treatment-related mortality (TRM) and adverse events (GVHD/infection). In addition to CD19, another promising target for CARNK cell therapy is CD7. In NCT02742727, the NK-92 cell line has been engineered to express an anti-CD7 attached to TCRzeta, CD28, and 4-1BB signaling domains and to be infused in patients with CD7-positive R/R leukemia and lymphoma to evaluate its safety and effectiveness. The NCT02890758 clinical trial is underway to investigate the number of NK cells from non-HLA matched donors (this kind of infusion is still experimental and not approved by the FDA) that can be safely infused into patients with hematologic tumors. After receiving the NK cells, patients may also be given ALT803, a drug that keeps NK cells alive, promotes their expansion, and supports their cancer-fighting characteristics. Furthermore, to enhance the therapeutic utility of NK-92 cells for the treatment of B-ALL, Oelsner et al. engineered NK-92 cells with an FMS-like tyrosine kinase 3 (FLT3)-specific CAR containing a composite CD28-CD3ζ signaling domain. Their results suggest that FLT3-specific CAR NK cells exhibit high and selective cytotoxic activity against established and primary B-ALL cells in vitro and, in a NOD/SCID IL2Rγ-null mouse xenograft model of B-ALL, a remarkable inhibition of disease progression is observed thus demonstrating high antileukemic activity in vivo [183].
5. New treatments under investigation
Future research directions aim to minimize chemotherapy and HSCT and to improve the life expectancy of patients with ALL, especially the older ones and those with ALL resistant to available treatments. The question is: what real advances in ALL therapy can we expect in the next future? The answer will come from the use of combination therapies capable of reducing drug resistance and improving drug efficacy to obtain a better and longer outcome. Therefore, several studies are currently ongoing to evaluate different drug-combination uses in relapse and frontline treatment settings.
In Ph+ ALL, the combination of a new TKI named ponatinib with blinatumomab is showing promising efficacy with a deep and durable response and less need for both chemotherapy and HCST in the first remission. Results of the phase 2 monocentric study presented at ASH 2021 by Short et al. evidenced that the combination of these two agents had synergistic effects on apoptosis. While ponatinib inhibits BCR-ABL kinases, blinatumomab promotes an antitumor response against CD19-expressing B cells[184].
This chemotherapy-free combination of ponatinib and blinatumomab resulted in safe and effective in both newly diagnosed (ND) and R/R Ph+ ALL patients. Particularly favorable outcomes (estimated 2-year EFS and OS 95%) were reported for the ND cohort that was not transplanted in first remission, suggesting that this regimen may serve as an effective transplant-sparing therapy in these patients[184]. Good results were obtained by combining ponatinib also with lower-intensity chemotherapy (hyper-CVAD) as an initial treatment for adult patients (age ≥ 18-75 years) with ND-Ph+ ALL (NCT01424982). A stable long-term remission has been shown in 70% of patients[185]. Other encouraging data were presented by a Spanish group (PETHEMA), that carried out a phase 2 PONALFIL trial, in which ponatinib (30 mg/d) was combined with an induction/consolidation chemotherapy followed by HSCT to treat adults with ND-Ph+ ALL[186]. In comparison to a more conventional therapeutic approach, this combination therapy showed good clinical activity and a favorable toxicity profile. CR was achieved in 100% of patients (30/30), 14 (47%) of whom obtained CMR and 5 (17%) MMR.
With regards to CART cell therapy, given the success of tisagenlecleucel (Kymriah), approved for use in children and young adults up to the age of 25 years, and more recently of brexucabtagene autoleucel (Tecartus), approved for all adult patients with R/R B-ALL, future works are focusing on designing new CAR structures with improved anti-tumor efficacy and a better safety profile.
Among multiple strategies, there is one of the phase 1 ALLCAR19 study (NCT02935257), which evaluated the effectiveness of a novel CD19 CAR that uses non-mobilized autologous leukapheresis (CAT-41BBzCAR, also known as AUTO1, another name: obecabtagene autoleucel, obe-cel) in adult patients with R/R B-ALL. Updated data showed a tolerable safety profile of AUTO1 in adult patients with R/R B-ALL despite the high disease burden[187,188]. Furthermore, another phase 1b/2 trial (FELIX trial – NCT04404660) aims to find out the best balance between the safety and efficacy of this CAR construct. Obe-cel has a lower affinity for CD19 than similar CART cell products and this helps to avoid CARTcell over-activation and exhaustion so the T cells can stay active for a longer period[189]. The first results by Roddie et al. demonstrated that this therapy has a good safety profile and high remission rates and it might offer a durable treatment option for these patients [188].
To overcome frequent relapses (10-20% of patients) following CD19 CART therapy, a CD22 CART cell therapy has been developed. Pan et al. demonstrated that CD22 CART cell therapy was highly effective in inducing remission in R/R B-ALL patients who failed from previous CD19 CART cell therapy and can be also considered a good bridge for subsequent transplantation to achieve durable remission [190]. Another useful strategy seems to be the development of dual-target CARs by simultaneously targeting CD19 and a second antigen (CD22 or CD20). As shown by Dai et al., this approach is feasible, safe, and able to induce remission in adult patients with R/R B-ALL [191].
Progresses in targeted immunotherapies for B-ALL generated big expectations also for T-ALL therapy. Glucocorticoids (GC) represent central components of T-ALL therapy, and the early response to GC-based therapy is an important predictor of long-term outcomes[192]. Nevertheless, relapse continues to represent a challenge in the clinical management of T-ALL. At the moment, nelarabine, a purine deoxyguanosine analog that inhibits DNA synthesis, remains the only drug for treating the relapse of T-ALL (response rates of over 50% in children and 36% in adults)[193,194,195]. Therefore, there is a need to develop efficient methods of augmenting the response to GC and overcoming resistance to steroid treatment. Most of the ongoing preclinical studies involve novel drugs able to enhance the results of glucocorticoid therapy and that could potentially be included in the induction phase in newly diagnosed ALL patients to prevent relapse and provide better outcomes.
6. Conclusions
Immunotherapy revolutionized the treatment of ALL permitting the achievement of remarkably effective and durable clinical responses. However, there is still a significant subset of patients that do not benefit from it. Therefore, research is now focusing on understanding the mechanisms of immune evasion employed by leukemia cells for developing novel therapeutic strategies. These tools, together with single-cell techniques, including massive DNA-RNA sequencing and mass cytometry, will help investigators understand which patients will benefit the most from immunotherapeutic approaches.
Author Contributions
All authors contributed to drafting and revising the article. All authors read and approved the final manuscript.
Funding
Not applicable
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Sandra Gessani for the helpful discussion and Nunziatina Cherubini for the administrative assistance. GS is supported by the Italian Association for Cancer Research (AIRC), Investigator Grant (IG) 2020-24440, and National Operational Program (PON) “TITAN” Ministry of the University and Research (MUR)-EU.
Conflicts of Interest
The author declares no conflict of interest.
References
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017 76 2017, 7, e577–e577. [Google Scholar] [CrossRef]
- Berg, S.L.; Steuber, P.; Poplack, D.G. Clinical manifestations of acute lymphoblastic leukemia. In: Hematology, Basic Principles, and Practice; 2000;
- Del Principe, M.I.; Buzzatti, E.; Piciocchi, A.; Forghieri, F.; Bonifacio, M.; Lessi, F.; Imbergamo, S.; Orciuolo, E.; Rossi, G.; Fracchiolla, N.; et al. Clinical significance of occult central nervous system disease in adult acute lymphoblastic leukemia. A multicenter report from the Campus ALL Network. Haematologica 2021, 106, 39–45. [Google Scholar] [CrossRef]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. 2015, 373, 1541–1552. [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Spector, L.G.; Ross, J.A.; Robison, L.L.; Bhatia, S. Epidemiology and etiology. Child. Leuk. Second Ed. 2006, 48–66. [Google Scholar] [CrossRef]
- Sehgal, S.; Mujtaba, S.; Gupta, D.; Aggarwal, R.; Marwaha, R.K. High incidence of Epstein Barr virus infection in childhood acute lymphocytic lukemia: A preliminary study. Indian J. Pathol. Microbiol. 2010, 53, 63. [Google Scholar] [CrossRef]
- Geriniere, L.; Bastion, Y.; Dumontet, C.; Salles, G.; Espinouse, D.; Coiffier, B. Heterogeneity of acute lymphoblastic leukemia in HFV-seropositive patients. Ann. Oncol. 1994, 5, 437–440. [Google Scholar] [CrossRef]
- Chessells, J.M.; Harrison, G.; Richards, S.M.; Bailey, C.C.; Hill, F.G.H.; Gibson, B.E.; Hann, I.M.; Bailey, C.C.; Barton, C.; Broadbent, V.; et al. Down’s syndrome and acute lymphoblastic leukaemia: clinical features and response to treatment. Arch. Dis. Child. 2001, 85, 321–325. [Google Scholar] [CrossRef]
- Dördelmann, M.; Schrappe, M.; Reiter, A.; Zimmermann, M.; Graf, N.; Schott, G.; Lampert, F.; Harbott, J.; Niemeyer, C.; Ritter, J.; et al. Down’s syndrome in childhood acute lymphoblastic leukemia: clinical characteristics and treatment outcome in four consecutive BFM trials. Leuk. 1998 125 1998, 12, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Bielorai, B.; Fisher, T.; Waldman, D.; Lerenthal, Y.; Nissenkorn, A.; Tohami, T.; Marek, D.; Amariglio, N.; Toren, A. Acute Lymphoblastic Leukemia in Early Childhood as the Presenting Sign of Ataxia-Telangiectasia Variant. Pediatric hematology and oncology 2013, 30, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Toledano, S.R.; Lange, B.J. Ataxia-telangiectasia and acute lymphoblastic leukemia. Cancer 1980, 45, 1675–8. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.S.; Bhatia, S.; Robison, L.L.; Yang, J.J. Genomics of racial and ethnic disparities in childhood acute lymphoblastic leukemia. Cancer 2014, 120, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Dores, G.M.; Devesa, S.S.; Curtis, R.E.; Linet, M.S.; Morton, L.M. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood 2012, 119, 34–43. [Google Scholar] [CrossRef]
- Feng, Q.; De Smith, A.J.; Vergara-Lluri, M.; Muskens, I.S.; McKean-Cowdin, R.; Kogan, S.; Brynes, R.; Wiemels, J.L. Trends in Acute Lymphoblastic Leukemia Incidence in the United States by Race/Ethnicity From 2000 to 2016. Am. J. Epidemiol. 2021, 190, 519–527. [Google Scholar] [CrossRef]
- Hallböök, H.; Gustafsson, G.; Smedmyr, B.; Söderhäll, S.; Heyman, M. Treatment outcome in young adults and children >10 years of age with acute lymphoblastic leukemia in Sweden. Cancer 2006, 107, 1551–1561. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA. Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Redaelli, A.; Laskin, B.L.; Stephens, J.M.; Botteman, M.F.; Pashos, C.L. A systematic literature review of the clinical and epidemiological burden of acute lymphoblastic leukaemia (ALL). Eur. J. Cancer Care (Engl). 2005, 14, 53–62. [Google Scholar] [CrossRef]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.S.; Bannon, F.; Ahn, J. V.; Johnson, C.J.; Bonaventure, A.; et al. Global surveillance of cancer survival 1995-2009: Analysis of individual data for 25 676 887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet 2015, 385, 977–1010. [Google Scholar] [CrossRef]
- Dong, Y.; Shi, O.; Zeng, Q.; Lu, X.; Wang, W.; Li, Y.; Wang, Q.; Wang, Q.; Wang, Q. Leukemia incidence trends at the global, regional, and national level between 1990 and 2017. Exp. Hematol. Oncol. 2020, 9, 1–11. [Google Scholar] [CrossRef]
- Bassan, R.; Hoelzer, D. Modern therapy of acute lymphoblastic leukemia. J. Clin. Oncol. 2011, 29, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Pulte, D.; Gondos, A.; Brenner, H. Improvement in survival in younger patients with acute lymphoblastic leukemia from the 1980s to the early 21st century. Blood 2009, 113, 1408–1411. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Pei, D.; Campana, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Coustan-Smith, E.; Jeha, S.; et al. Improved prognosis for older adolescents with acute lymphoblastic leukemia. J. Clin. Oncol. 2011, 29, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; O’Brien, S.; Konopleva, M.; Kantarjian, H. New insights into the pathophysiology and therapy of adult acute lymphoblastic leukemia. Cancer 2015, 121, 2517–2528. [Google Scholar] [CrossRef]
- Bonaventure, A.; Harewood, R.; Stiller, C.A.; Gatta, G.; Clavel, J.; Stefan, D.C.; Carreira, H.; Spika, D.; Marcos-Gragera, R.; Peris-Bonet, R.; et al. Worldwide comparison of survival from childhood leukaemia for 1995–2009, by subtype, age, and sex (CONCORD-2): a population-based study of individual data for 89 828 children from 198 registries in 53 countries. Lancet Haematol. 2017, 4, e202–e217. [Google Scholar] [CrossRef]
- Ghosn, E.; Yoshimoto, M.; Nakauchi, H.; Weissman, I.L.; Herzenberg, L.A. Hematopoietic stem cell-independent hematopoiesis and the origins of innate-like B lymphocytes. Dev. 2019, 146. [Google Scholar] [CrossRef]
- Greaves, M.F.; Maia, A.T.; Wiemels, J.L.; Ford, A.M. Leukemia in twins: lessons in natural history. Blood 2003, 102, 2321–2333. [Google Scholar] [CrossRef]
- Shin, S.Y.; Lee, H.; Lee, S.T.; Choi, J.R.; Jung, C.W.; Koo, H.H.; Kim, S.H. Recurrent somatic mutations and low germline predisposition mutations in Korean ALL patients. Sci. Reports 2021 111 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Mancini, M.; Scappaticci, D.; Cimino, G.; Nanni, M.; Derme, V.; Elia, L.; Tafuri, A.; Vignetti, M.; Vitale, A.; Cuneo, A.; et al. A comprehensive genetic classification of adult acute lymphoblastic leukemia (ALL): analysis of the GIMEMA 0496 protocol. Blood 2005, 105, 3434–3441. [Google Scholar] [CrossRef]
- Cytogenetic Abnormalities in Adult Acute Lymphoblastic Leukemia: Correlations with Hematologic Findings and Outcome. A Collaborative Study of the Groupe Français de Cytogέnέtique Hέmatologique: By The Groupe FranGais de Cytogέnέtique Hέmatologique (partic). Blood 1996, 87, 3135–3142. [Google Scholar] [CrossRef]
- Advani, A.S.; Hunger, S.P.; Burnett, A.K. Acute Leukemia in Adolescents and Young Adults. Semin. Oncol. 2009, 36, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Litzow, M.R. Antigen-based immunotherapy for the treatment of acute lymphoblastic leukemia: the emerging role of blinatumomab. ImmunoTargets Ther. 2014, 3, 79–89. [Google Scholar] [CrossRef]
- Stary, J.; Zimmermann, M.; Campbell, M.; Castillo, L.; Dibar, E.; Donska, S.; Gonzalez, A.; Izraeli, S.; Janic, D.; Jazbec, J.; et al. Intensive chemotherapy for childhood acute lymphoblastic leukemia: Results of the randomized intercontinental trial ALL IC-BFM 2002. J. Clin. Oncol. 2014, 32, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Beldjord, K.; (GRAALL), on behalf of the G. for R. on A.A.L.L.; Chevret, S.; (GRAALL), on behalf of the G. for R. on A.A.L.L.; Asnafi, V.; (GRAALL), on behalf of the G. for R. on A.A.L.L.; Huguet, F.; (GRAALL), on behalf of the G. for R. on A.A.L.L.; Boulland, M.-L.; (GRAALL), on behalf of the G. for R. on A.A.L.L.; et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood 2014, 123, 3739–3749. [CrossRef]
- Pinkel, D. Five-Year Follow-Up of Total Therapy of Childhood Lymphocytic Leukemia. JAMA 1971, 216, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Aur, R.J.; Simone, J.; Hustu, H.O.; Walters, T.; Borella, L.; Pratt, C.; Pinkel, D. Central Nervous System Therapy and Combination Chemotherapy of Childhood Lymphocytic Leukemia. Blood 1971, 37, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Bleyer, W.A.; Poplack, D.G. Prophylaxis and treatment of leukemia in the central nervous system and other sanctuaries. Semin. Oncol. 1985, 12, 131–48. [Google Scholar]
- Anderson, F.S.; Kunin-Batson, A.S. Neurocognitive late effects of chemotherapy in children: The past 10 years of research on brain structure and function. Pediatr. Blood Cancer 2009, 52, 159–164. [Google Scholar] [CrossRef]
- Kadan-Lottick, N.S.; Zeltzer, L.K.; Liu, Q.; Yasui, Y.; Ellenberg, L.; Gioia, G.; Robison, L.L.; Krull, K.R. Neurocognitive Functioning in Adult Survivors of Childhood Non-Central Nervous System Cancers. JNCI J. Natl. Cancer Inst. 2010, 102, 881–893. [Google Scholar] [CrossRef]
- Bleyer, W.A. Neurologic sequelae of methotrexate and ionizing radiation: a new classification. Cancer Treat. Rep. 1981, 65 Suppl 1, 89–98. [Google Scholar]
- Oeffinger, K.C.; Mertens, A.C.; Sklar, C.A.; Kawashima, T.; Hudson, M.M.; Meadows, A.T.; Friedman, D.L.; Marina, N.; Hobbie, W.; Kadan-Lottick, N.S.; et al. Chronic Health Conditions in Adult Survivors of Childhood Cancer. N. Engl. J. Med. 2006, 355, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Cheng, C.; Leung, W.; Rai, S.N.; Rivera, G.K.; Sandlund, J.T.; Ribeiro, R.C.; Relling, M. V.; Kun, L.E.; Evans, W.E.; et al. Extended Follow-up of Long-Term Survivors of Childhood Acute Lymphoblastic Leukemia. 2003, 349, 640–649. [CrossRef]
- Hijiya, N.; Hudson, M.M.; Lensing, S.; Zacher, M.; Onciu, M.; Behm, F.G.; Razzouk, B.I.; Ribeiro, R.C.; Rubnitz, J.E.; Sandlund, J.T.; et al. Cumulative Incidence of Secondary Neoplasms as a First Event After Childhood Acute Lymphoblastic Leukemia. JAMA 2007, 297, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Geenen, M.M.; Cardous-Ubbink, M.C.; Kremer, L.C.M.; Van Den Bos, C.; Van Der Pal, H.J.H.; Heinen, R.C.; Jaspers, M.W.M.; Koning, C.C.E.; Oldenburger, F.; Langeveld, N.E.; et al. Medical Assessment of Adverse Health Outcomes in Long-term Survivors of Childhood Cancer. JAMA 2007, 297, 2705–2715. [Google Scholar] [CrossRef] [PubMed]
- Waber, D.P.; Turek, J.; Catania, L.; Stevenson, K.; Robaey, P.; Romero, I.; Adams, H.; Alyman, C.; Jandet-Brunet, C.; Neuberg, D.S.; et al. Neuropsychological outcomes from a randomized trial of triple intrathecal chemotherapy compared with 18 Gy cranial radiation as CNS treatment in acute lymphoblastic leukemia: Findings from Dana-Farber Cancer Institute ALL Consortium Protocol 95-01. J. Clin. Oncol. 2007, 25, 4914–4921. [Google Scholar] [CrossRef] [PubMed]
- Schrappe, M.; Reiter, A.; Ludwig, W.-D.; Harbott, J.; Zimmermann, M.; Hiddemann, W.; Niemeyer, C.; Henze, G.; Feldges, A.; Zintl, F.; et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. Blood 2000, 95, 3310–3322. [Google Scholar] [CrossRef] [PubMed]
- Silverman, L.B.; Gelber, R.D.; Dalton, V.K.; Asselin, B.L.; Barr, R.D.; Clavell, L.A.; Hurwitz, C.A.; Moghrabi, A.; Samson, Y.; Schorin, M.A.; et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood 2001, 97, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Lukenbill, J.; Advani, A.S. The treatment of adolescents and young adults with acute lymphoblastic leukemia. Curr. Hematol. Malig. Rep. 2013, 8, 91–97. [Google Scholar] [CrossRef]
- Rowe, J.M.; Buck, G.; Burnett, A.K.; Chopra, R.; Wiernik, P.H.; Richards, S.M.; Lazarus, H.M.; Franklin, I.M.; Litzow, M.R.; Ciobanu, N.; et al. Induction therapy for adults with acute lymphoblastic leukemia: results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood 2005, 106, 3760–3767. [Google Scholar] [CrossRef]
- Williams, M.T.S.; Yousafzai, Y.M.; Elder, A.; Rehe, K.; Bomken, S.; Frishman-Levy, L.; Tavor, S.; Sinclair, P.; Dormon, K.; Masic, D.; et al. The ability to cross the blood–cerebrospinal fluid barrier is a generic property of acute lymphoblastic leukemia blasts. Blood 2016, 127, 1998–2006. [Google Scholar] [CrossRef]
- Krishnan, S.; Wade, R.; Moorman, A. V.; Mitchell, C.; Kinsey, S.E.; Eden, T.O.B.; Parker, C.; Vora, A.; Richards, S.; Saha, V. Temporal changes in the incidence and pattern of central nervous system relapses in children with acute lymphoblastic leukaemia treated on four consecutive Medical Research Council trials, 1985–2001. Leuk. 2010 242 2009, 24, 450–459. [Google Scholar] [CrossRef]
- Akers, S.M.; Rellick, S.L.; Fortney, J.E.; Gibson, L.F. Cellular elements of the subarachnoid space promote ALL survival during chemotherapy. Leuk. Res. 2011, 35, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Gaynes, J.S.; Jonart, L.M.; Zamora, E.A.; Naumann, J.A.; Gossai, N.P.; Gordon, P.M. The central nervous system microenvironment influences the leukemia transcriptome and enhances leukemia chemo-resistance. Haematologica 2017, 102, e136–e139. [Google Scholar] [CrossRef] [PubMed]
- Jonart, L.M.; Ebadi, M.; Basile, P.; Johnson, K.; Makori, J.; Gordon, P.M. Disrupting the leukemia niche in the central nervous system attenuates leukemia chemoresistance. Haematologica 2020, 105, 2130–2140. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sevilla, L.M.; Valencia, J.; Flores-Villalobos, M.A.; Gonzalez-Murillo, Á.; Sacedón, R.; Jiménez, E.; Ramírez, M.; Varas, A.; Vicente, Á. The choroid plexus stroma constitutes a sanctuary for paediatric B-cell precursor acute lymphoblastic leukaemia in the central nervous system. J. Pathol. 2020, 252, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Schrappe, M.; Bernardo, M.E.; Rutella, S. How I treat relapsed childhood acute lymphoblastic leukemia. Blood 2012, 120, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, A.H.; Richards, S.M.; Lazarus, H.M.; Tallman, M.S.; Buck, G.; Fielding, A.K.; Burnett, A.K.; Chopra, R.; Wiernik, P.H.; Foroni, L.; et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/. Blood 2008, 111, 1827–1833. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.J.; Group, on behalf of the D.-B.H.C.; van der Holt, B.; Group, on behalf of the D.-B.H.C.; Verhoef, G.E.G.; Group, on behalf of the D.-B.H.C.; van ’t Veer, M.B.; Group, on behalf of the D.-B.H.C.; van Oers, M.H.J.; Group, on behalf of the D.-B.H.C.; et al. Myeloablative allogeneic versus autologous stem cell transplantation in adult patients with acute lymphoblastic leukemia in first remission: a prospective sibling donor versus no-donor comparison. Blood 2009, 113, 1375–1382. [CrossRef]
- Jamieson, C.H.M.; Amylon, M.D.; Wong, R.M.; Blume, K.G. Allogeneic hematopoietic cell transplantation for patients with high-risk acute lymphoblastic leukemia in first or second complete remission using fractionated total-body irradiation and high-dose etoposide: A 15-year experience. Exp. Hematol. 2003, 31, 981–986. [Google Scholar] [CrossRef]
- Gruen, M.; Bommert, K.; Bargou, R.C. T-cell-mediated lysis of B cells induced by a CD19xCD3 bispecific single-chain antibody is perforin dependent and death receptor independent. Cancer Immunol. Immunother. 2004, 53, 625–632. [Google Scholar] [CrossRef]
- Thiery, J.; Keefe, D.; Boulant, S.; Boucrot, E.; Walch, M.; Martinvalet, D.; Goping, I.S.; Bleackley, R.C.; Kirchhausen, T.; Lieberman, J. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat. Immunol. 2011 128 2011, 12, 770–777. [Google Scholar] [CrossRef]
- Trapani, J.A. Target cell apoptosis induced by cytotoxic T cells and natural killer cells involves synergy between the pore-forming protein, perforin, and the serine protease, granzyme B. Aust. N. Z. J. Med. 1995, 25, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Brischwein, K.; Schlereth, B.; Guller, B.; Steiger, C.; Wolf, A.; Lutterbuese, R.; Offner, S.; Locher, M.; Urbig, T.; Raum, T.; et al. MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 2006, 43, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Nagorsen, D.; Baeuerle, P.A. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp. Cell Res. 2011, 317, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.; Hofmeister, R.; Brischwein, K.; Brandl, C.; Crommer, S.; Bargou, R.; Itin, C.; Prang, N.; Baeuerle, P.A. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int. J. Cancer 2005, 115, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Chow, V.; Weissman, A.; Tulpule, S.; Aldoss, I.; Akhtari, M. Clinical use of blinatumomab for B-cell acute lymphoblastic leukemia in adults. Ther. Clin. Risk Manag. 2016, 12, 1301–1310. [Google Scholar] [CrossRef]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.A.; et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015, 16, 57–66. [Google Scholar] [CrossRef]
- Shukla, N.; Sulis, M.L. Blinatumomab for Treatment of Children With High-risk Relapsed B-Cell Acute Lymphoblastic Leukemia. JAMA 2021, 325, 830–832. [Google Scholar] [CrossRef]
- Queudeville, M.; Ebinger, M. Blinatumomab in Pediatric Acute Lymphoblastic Leukemia—From Salvage to First Line Therapy (A Systematic Review). J. Clin. Med. 2021, Vol. 10, Page 2544 2021, 10, 2544. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef]
- Martinelli, G.; Boissel, N.; Chevallier, P.; Ottmann, O.; Gökbuget, N.; Topp, M.S.; Fielding, A.K.; Rambaldi, A.; Ritchie, E.K.; Papayannidis, C.; et al. Complete hematologic and molecular response in adult patients with relapsed/refractory philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia following treatment with blinatumomab: Results from a phase II, single-arm, multicenter study. J. Clin. Oncol. 2017, 35, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Foà, R.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, M.-C.; Canichella, M.; Viero, P.; Ferrara, F.; Lunghi, M.; et al. Dasatinib–Blinatumomab for Ph-Positive Acute Lymphoblastic Leukemia in Adults. N. Engl. J. Med. 2020, 383, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, S.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Ferrara, F.; Lunghi, M.; Fabbiano, F.; Bonifacio, M.; Fracchiolla, N.; et al. S1617 A DASATINIB-BLINATUMOMAB COMBINATION FOR THE FRONT-LINE TREATMENT OF ADULT PH+ ALL PATIENTS. PRELIMINARY RESULTS OF THE GIMEMA LAL2116 D-ALBA TRIAL; ON BEHALF OF GIMEMA ACUTE LEUKEMIA WORKING PARTY. HemaSphere 2019, 3, 746. [Google Scholar] [CrossRef]
- Handgretinger, R.; Zugmaier, G.; Henze, G.; Kreyenberg, H.; Lang, P.; Von Stackelberg, A. Complete remission after blinatumomab-induced donor T-cell activation in three pediatric patients with post-transplant relapsed acute lymphoblastic leukemia. Leuk. 2011 251 2010, 25, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, P.; Lang, P.; Zugmaier, G.; Ebinger, M.; Kreyenberg, H.; Witte, K.E.; Feucht, J.; Pfeiffer, M.; Teltschik, H.M.; Kyzirakos, C.; et al. Pediatric posttransplant relapsed/refractory B-precursor acute lymphoblastic leukemia shows durable remission by therapy with the T-cell engaging bispecific antibody blinatumomab. Haematologica 2014, 99, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Von Stackelberg, A.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; Trippett, T.M.; Rizzari, C.; Bader, P.; O’brien, M.M.; Brethon, B.; Bhojwani, D.; et al. Phase I/Phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J. Clin. Oncol. 2016, 34, 4381–4389. [Google Scholar] [CrossRef] [PubMed]
- Mejstríková, E.; Hrusak, O.; Borowitz, M.J.; Whitlock, J.A.; Brethon, B.; Trippett, T.M.; Zugmaier, G.; Gore, L.; Von Stackelberg, A.; Locatelli, F. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017 712 2017, 7, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, A.; zur Stadt, U.; Winkler, B.; Müller, I.; Escherich, G. Lineage switch under blinatumomab treatment of relapsed common acute lymphoblastic leukemia without MLL rearrangement. Pediatr. Blood Cancer 2017, 64, e26594. [Google Scholar] [CrossRef]
- Wadhwa, A.; Kutny, M.A.; Xavier, A.C. Blinatumomab activity in a patient with Down syndrome B-precursor acute lymphoblastic leukemia. Pediatr. Blood Cancer 2018, 65, e26824. [Google Scholar] [CrossRef]
- Gore, L.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; O’Brien, M.M.; Bader, P.; Bhojwani, D.; Schlegel, P.G.; Tuglus, C.A.; von Stackelberg, A. Survival after blinatumomab treatment in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia. Blood Cancer J. 2018 89 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Wölfl, M.; Rasche, M.; Eyrich, M.; Schmid, R.; Reinhardt, D.; Schlegel, P.G. Spontaneous reversion of a lineage switch following an initial blinatumomab-induced ALL-to-AML switch in MLL-rearranged infant ALL. Blood Adv. 2018, 2, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Elitzur, S.; Arad-Cohen, N.; Barzilai-Birenboim, S.; Ben-Harush, M.; Bielorai, B.; Elhasid, R.; Feuerstein, T.; Gilad, G.; Gural, A.; Kharit, M.; et al. Blinatumomab as a bridge to further therapy in cases of overwhelming toxicity in pediatric B-cell precursor acute lymphoblastic leukemia: Report from the Israeli Study Group of Childhood Leukemia. Pediatr. Blood Cancer 2019, 66, e27898. [Google Scholar] [CrossRef] [PubMed]
- Keating, A.K.; Gossai, N.; Phillips, C.L.; Maloney, K.; Campbell, K.; Doan, A.; Bhojwani, D.; Burke, M.J.; Verneris, M.R. Reducing minimal residual disease with blinatumomab prior to HCT for pediatric patients with acute lymphoblastic leukemia. Blood Adv. 2019, 3, 1926–1929. [Google Scholar] [CrossRef] [PubMed]
- Mouttet, B.; Vinti, L.; Ancliff, P.; Bodmer, N.; Brethon, B.; Cario, G.; Chen-Santel, C.; Elitzur, S.; Hazar, V.; Kunz, J.; et al. Durable remissions in TCF3-HLF positive acute lymphoblastic leukemia with blinatumomab and stem cell transplantation. Haematologica 2019, 104, e244–e247. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Gore, L.; et al. A Randomized Phase 3 Trial of Blinatumomab Vs. Chemotherapy As Post-Reinduction Therapy in High and Intermediate Risk (HR/IR) First Relapse of B-Acute Lymphoblastic Leukemia (B-ALL) in Children and Adolescents/Young Adults (AYAs) Demonstrates Superior Eff. Blood 2019, 134, LBA–1. [Google Scholar] [CrossRef]
- Locatelli, F.; Whitlock, J.A.; Peters, C.; Chen-Santel, C.; Chia, V.; Dennis, R.M.; Heym, K.M.; Katz, A.J.; Kelsh, M.A.; Sposto, R.; et al. Blinatumomab versus historical standard therapy in pediatric patients with relapsed/refractory Ph-negative B-cell precursor acute lymphoblastic leukemia. Leuk. 2020 349 2020, 34, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Zugmaier, G.; Mergen, N.; Bader, P.; Jeha, S.; Schlegel, P.G.; Bourquin, J.P.; Handgretinger, R.; Brethon, B.; Rossig, C.; et al. Blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia: results of the RIALTO trial, an expanded access study. Blood Cancer J. 2020 107 2020, 10, 1–5. [Google Scholar] [CrossRef]
- Clesham, K.; Rao, V.; Bartram, J.; Ancliff, P.; Ghorashian, S.; O’Connor, D.; Pavasovic, V.; Rao, A.; Samarasinghe, S.; Cummins, M.; et al. Blinatumomab for infant acute lymphoblastic leukemia. Blood 2020, 135, 1501–1504. [Google Scholar] [CrossRef]
- Ampatzidou; Kattamis, A. ; Baka, M.; Paterakis, G.; Anastasiou, T.; Tzanoudaki, M.; Kaisari, A.; Avgerinou, G.; Doganis, D.; Papadakis, V.; et al. Insights from the greek experience of the use of blinatumomab in pediatric relapsed and refractory acute lymphoblastic leukemia patients. Neoplasma 2020, 67, 1424–1430. [Google Scholar] [CrossRef]
- Horibe, K.; Morris, J.D.; Tuglus, C.A.; Dos Santos, C.; Kalabus, J.; Anderson, A.; Goto, H.; Ogawa, C. A phase 1b study of blinatumomab in Japanese children with relapsed/refractory B-cell precursor acute lymphoblastic leukemia. Int. J. Hematol. 2020, 112, 223–233. [Google Scholar] [CrossRef]
- Mikhailova, E.; Illarionova, O.; Shelikhova, L.; Zerkalenkova, E.; Molostova, O.; Olshanskaya, Y.; Novichkova, G.; Maschan, A.; Maschan, M.; Popov, A. Immunophenotypic changes in leukemic blasts in children with relapsed/refractory B-cell precursor acute lymphoblastic leukemia after treatment with CD19-directed chimeric antigen receptor (CAR)- expressing T cells. Haematologica 2022, 107, 970–974. [Google Scholar] [CrossRef]
- Contreras, C.F.; Higham, C.S.; Behnert, A.; Kim, K.; Stieglitz, E.; Tasian, S.K. Clinical utilization of blinatumomab and inotuzumab immunotherapy in children with relapsed or refractory B-acute lymphoblastic leukemia. Pediatr. Blood Cancer 2021, 68, e28718. [Google Scholar] [CrossRef] [PubMed]
- Brethon, B.; Lainey, E.; Caye-Eude, A.; Grain, A.; Fenneteau, O.; Yakouben, K.; Roupret-Serzec, J.; Le Mouel, L.; Cavé, H.; Baruchel, A. Case Report: Targeting 2 Antigens as a Promising Strategy in Mixed Phenotype Acute Leukemia: Combination of Blinatumomab With Gemtuzumab Ozogamicin in an Infant With a KMT2A-Rearranged Leukemia. Front. Oncol. 2021, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Queudeville, M.; Schlegel, P.; Heinz, A.T.; Lenz, T.; Döring, M.; Holzer, U.; Hartmann, U.; Kreyenberg, H.; von Stackelberg, A.; Schrappe, M.; et al. Blinatumomab in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia. Eur. J. Haematol. 2021, 106, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Sutton, R.; Pozza, L.D.; Khaw, S.L.; Fraser, C.; Revesz, T.; Chamberlain, J.; Mitchell, R.; Trahair, T.N.; Bateman, C.M.; Venn, N.C.; et al. Outcomes for Australian children with relapsed/refractory acute lymphoblastic leukaemia treated with blinatumomab. Pediatr. Blood Cancer 2021, 68, e28922. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.E.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Bernhardt, M.B.; et al. Effect of Postreinduction Therapy Consolidation With Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults With First Relapse of B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 833–842. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A.; et al. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children With High-risk First-Relapse B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 843–854. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Mergen, N.; Bader, P.; Jeha, S.; Schlegel, P.G.; Bourquin, J.P.; Handgretinger, R.; Brethon, B.; Rössig, C.; et al. Blinatumomab in pediatric relapsed/refractory B-cell acute lymphoblastic leukemia: RIALTO expanded access study final analysis. Blood Adv. 2022, 6, 1004–1014. [Google Scholar] [CrossRef]
- Locatelli, F.; Maschan, A.; Boissel, N.; Strocchio, L.; Alam, N.; Pezzani, I.; Brescianini, A.; Kreuzbauer, G.; Baruchel, A. Pediatric patients with acute lymphoblastic leukemia treated with blinatumomab in a real-world setting: Results from the NEUF study. Pediatr. Blood Cancer 2022, 69, e29562. [Google Scholar] [CrossRef]
- Beneduce, G.; De Matteo, A.; Stellato, P.; Testi, A.M.; Bertorello, N.; Colombini, A.; Putti, M.C.; Rizzari, C.; Cesaro, S.; Cellini, M.; et al. Blinatumomab in Children and Adolescents with Relapsed/Refractory B Cell Precursor Acute Lymphoblastic Leukemia: A Real-Life Multicenter Retrospective Study in Seven AIEOP (Associazione Italiana di Ematologia e Oncologia Pediatrica) Centers. Cancers 2022, Vol. 14, Page 426 2022, 14, 426. [Google Scholar] [CrossRef]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody–drug conjugates: Recent advances in linker chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-drug conjugates: A comprehensive review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Kalim, M.; Chen, J.; Wang, S.; Lin, C.; Ullah, S.; Liang, K.; Ding, Q.; Chen, S.; Zhan, J.B. Intracellular trafficking of new anticancer therapeutics: antibody–drug conjugates. Drug Des. Devel. Ther. 2017, 11, 2265–2276. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br. J. Cancer 2017 11712 2017, 117, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Advani, A.S.; Marks, D.I.; Stelljes, M.; Liedtke, M.; Stock, W.; Gökbuget, N.; Jabbour, E.; Merchant, A.; Wang, T.; et al. Inotuzumab ozogamicin for relapsed/refractory acute lymphoblastic leukemia: outcomes by disease burden. Blood Cancer J. 2020 108 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Özcan, M.; Cassaday, R.D.; Singh, P.; Zarzycka, E.; Zhang, X.; Nègre, E.; Vandendries, E.; Altuntas, F. The Efficacy and Safety of Low-Dose Inotuzumab Ozogamicin in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia: Interim Results of a Phase 4 Study. Blood 2021, 138, 1208–1208. [Google Scholar] [CrossRef]
- Kebriaei, P.; Cutler, C.; De Lima, M.; Giralt, S.; Lee, S.J.; Marks, D.; Merchant, A.; Stock, W.; Van Besien, K.; Stelljes, M. Management of important adverse events associated with inotuzumab ozogamicin: expert panel review. Bone Marrow Transplant. 2017 534 2018, 53, 449–456. [Google Scholar] [CrossRef]
- O’Brien, M.M.; Ji, L.; Shah, N.N.; Rheingold, S.R.; Bhojwani, D.; Yuan, C.M.; Xu, X.; Yi, J.S.; Harris, A.C.; Brown, P.A.; et al. Phase II Trial of Inotuzumab Ozogamicin in Children and Adolescents With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia: Children’s Oncology Group Protocol AALL1621. J. Clin. Oncol. 2022, 40, 956–967. [Google Scholar] [CrossRef]
- Brivio, E.; Locatelli, F.; Lopez-Yurda, M.; Malone, A.; Díaz-de-Heredia, C.; Bielorai, B.; Rossig, C.; van der Velden, V.H.J.; Ammerlaan, A.C.J.; Thano, A.; et al. A phase 1 study of inotuzumab ozogamicin in pediatric relapsed/refractory acute lymphoblastic leukemia (ITCC-059 study). Blood 2021, 137, 1582–1590. [Google Scholar] [CrossRef]
- Jabbour, E.; Sasaki, K.; Ravandi, F.; Huang, X.; Short, N.J.; Khouri, M.; Kebriaei, P.; Burger, J.; Khoury, J.; Jorgensen, J.; et al. Chemoimmunotherapy with inotuzumab ozogamicin combined with mini-hyper-CVD, with or without blinatumomab, is highly effective in patients with Philadelphia chromosome–negative acute lymphoblastic leukemia in first salvage. Cancer 2018, 124, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.J.; Sasaki, K.; Ravandi, F.; Short, N.J.; Garcia-Manero, G.; Daver, N.; Kadia, T.; Konopleva, M.; Jain, N.; Cortes, J.; et al. Inotuzumab ozogamicin in combination with low-intensity chemotherapy (mini-HCVD) with or without blinatumomab versus standard intensive chemotherapy (HCVAD) as frontline therapy for older patients with Philadelphia chromosome-negative acute lymphoblastic. Cancer 2019, 125, 2579–2586. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Kantarjian, H.M.; Ravandi, F.; Short, N.J.; Kebriaei, P.; Huang, X.; Rytting, M.E.; Jain, N.; Konopleva, M.Y.; Garcia-Manero, G.; et al. Sequential Combination of Inotuzumab Ozogamicin (InO) with Low-Intensity Chemotherapy (Mini-hyper-CVD) with or without Blinatumomab Is Highly Effective in Patients (pts) with Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia (ALL) in First Rel. Blood 2019, 134, 3806–3806. [Google Scholar] [CrossRef]
- Kantarjian, H.; Ravandi, F.; Short, N.J.; Huang, X.; Jain, N.; Sasaki, K.; Daver, N.; Pemmaraju, N.; Khoury, J.D.; Jorgensen, J.; et al. Inotuzumab ozogamicin in combination with low-intensity chemotherapy for older patients with Philadelphia chromosome-negative acute lymphoblastic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2018, 19, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Richard-Carpentier, G.; Kantarjian, H.M.; Short, N.J.; Ravandi, F.; Ferrajoli, A.; Schroeder, H.M.; Garcia-Manero, G.; Montalban Bravo, G.; Cortes, J.E.; Kwari, M.; et al. Updated Results from the Phase II Study of Hyper-CVAD in Sequential Combination with Blinatumomab in Newly Diagnosed Adults with B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood 2019, 134, 3807–3807. [Google Scholar] [CrossRef]
- Henze, A.T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Invest. 2016, 126, 3672–3679. [Google Scholar] [CrossRef]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef]
- Gravante, G.; Ong, S.L.; Metcalfe, M.S.; Sorge, R.; Sconocchia, G.; Orlando, G.; Lloyd, D.M.; Dennison, A.R. Cytokine Response to Ischemia/Reperfusion Injury in an Ex Vivo Perfused Porcine Liver Model. Transplant. Proc. 2009, 41, 1107–1112. [Google Scholar] [CrossRef]
- Ohno, S.; Ohno, Y.; Suzuki, N.; Kamei, T.; Koike, K.; Inagawa, H.; Kohchi, C.; Soma, G.I.; Inoue, M. Correlation of Histological Localization of Tumor-associated Macrophages with Clinicopathological Features in Endometrial Cancer. Anticancer Res. 2004, 24, 3335–3342. [Google Scholar]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, Vol. 9, Page 992 2020, 9, 992. [Google Scholar] [CrossRef]
- Khalaf, K.; Hana, D.; Chou, J.T.T.; Singh, C.; Mackiewicz, A.; Kaczmarek, M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front. Immunol. 2021, 12, 1764. [Google Scholar] [CrossRef] [PubMed]
- Duell, J.; Lurati, S.; Dittrich, M.; Bedke, T.; Pule, M.; Einsele, H.; Rossig, C.; Topp, M.S. First Generation Chimeric Antigen Receptor Display Functional Defects In Key Signal Pathways Upon Antigen Stimulation. Blood 2010, 116, 2088. [Google Scholar] [CrossRef]
- Van Der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015 147 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell–mediated Tumor Eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: the fourth generation of CARs. 2015, 15, 1145–1154. [CrossRef]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: next-generation CAR T cells. Br. J. Cancer 2018 1201 2018, 120, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef]
- Miller, B.C.; Maus, M. V. CD19-Targeted CAR T Cells: A New Tool in the Fight against B Cell Malignancies. Oncol. Res. Treat. 2015, 38, 683–690. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Stetler-Stevenson, M.; Yuan, C.M.; Shah, N.N.; Delbrook, C.; Yates, B.; Zhang, H.; Zhang, L.; Kochenderfer, J.N.; Rosenberg, S.A.; et al. Long-Term Outcomes Following CD19 CAR T Cell Therapy for B-ALL Are Superior in Patients Receiving a Fludarabine/Cyclophosphamide Preparative Regimen and Post-CAR Hematopoietic Stem Cell Transplantation. Blood 2016, 128, 218–218. [Google Scholar] [CrossRef]
- Shah, B.D.; Bishop, M.R.; Oluwole, O.O.; Logan, A.C.; Baer, M.R.; Donnellan, W.B.; O’Dwyer, K.M.; Holmes, H.; Arellano, M.L.; Ghobadi, A.; et al. KTE-X19 anti-CD19 CAR T-cell therapy in adult relapsed/refractory acute lymphoblastic leukemia: ZUMA-3 phase 1 results. Blood 2021, 138, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.; Boissel, N.; Cassaday, R.D.; Forcade, E.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. Phase 2 results of the ZUMA-3 study evaluating KTE-X19, an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy, in adult patients (pts) with relapsed/refractory B-cell acute lymphoblastic leukemia (R/R B-ALL). 2021, 39, 7002–7002. [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Talekar, M.K.; Maude, S.L.; Hucks, G.E.; Motley, L.S.; Callahan, C.; White, C.M.; Baniewicz, D.; Barrett, D.M.; Rheingold, S.R.; Lacey, S.F.; et al. Effect of chimeric antigen receptor-modified T (CAR-T) cells on responses in children with non-CNS extramedullary relapse of CD19+ acute lymphoblastic leukemia (ALL). 2017, 35, 10507–10507. [CrossRef]
- Chen, X.; Wang, Y.; Ruan, M.; Li, J.; Zhong, M.; Li, Z.; Liu, F.; Wang, S.; Chen, Y.; Liu, L.; et al. Treatment of Testicular Relapse of B-cell Acute Lymphoblastic Leukemia With CD19-specific Chimeric Antigen Receptor T Cells. Clin. Lymphoma, Myeloma Leuk. 2020, 20, 366–370. [Google Scholar] [CrossRef]
- Rheingold, S.R.; Chen, L.N.; Maude, S.L.; Aplenc, R.; Barker, C.; Barrett, D.M.; Callahan, C.; Cebry, K.; Kulikovskaya, I.; Lacey, S.F.; et al. Efficient Trafficking of Chimeric Antigen Receptor (CAR)-Modified T Cells to CSF and Induction of Durable CNS Remissions in Children with CNS/Combined Relapsed/Refractory ALL. Blood 2015, 126, 3769–3769. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Rheingold, S.R.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Barker, C.S.; Callahan, C.; Frey, N. V.; Nazimuddin, F.; et al. Sustained remissions with CD19-specific chimeric antigen receptor (CAR)-modified T cells in children with relapsed/refractory ALL. 2016, 34, 3011–3011. [CrossRef]
- Newman, H.; Leahy, A.E.B.; Li, Y.; Liu, H.; Myers, R.M.; DiNofia, A.M.; Dolan, J.G.; Callahan, C.; Devine, K.J.; Wray, L.; et al. CD19-targeted chimeric antigen receptor (CAR) T cells in CNS relapsed acute lymphoblastic leukemia (ALL). 2020, 38, 10511–10511. [CrossRef]
- Zhang, X.; Lu, X.A.; Yang, J.; Zhang, G.; Li, J.; Song, L.; Su, Y.; Shi, Y.; Zhang, M.; He, J.; et al. Efficacy and safety of anti-CD19 CAR T-cell therapy in 110 patients with B-cell acute lymphoblastic leukemia with high-risk features. Blood Adv. 2020, 4, 2325–2338. [Google Scholar] [CrossRef]
- Rubinstein, J.D.; Krupski, C.; Nelson, A.S.; O’Brien, M.M.; Davies, S.M.; Phillips, C.L. Chimeric Antigen Receptor T Cell Therapy in Patients with Multiply Relapsed or Refractory Extramedullary Leukemia. Biol. Blood Marrow Transplant. 2020, 26, e280–e285. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M. V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Frey, N. V.; Porter, D.L. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematology 2016, 2016, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Frey, N. V.; Shaw, P.A.; Hexner, E.O.; Pequignot, E.; Gill, S.; Luger, S.M.; Mangan, J.K.; Loren, A.W.; Perl, A.E.; Maude, S.L.; et al. Optimizing chimeric antigen receptor T-cell therapy for adults with acute lymphoblastic leukemia. J. Clin. Oncol. 2020, 38, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Taraseviciute, A.; Turtle, C.J. Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs 2018, 32, 1091–1101. [Google Scholar] [CrossRef]
- Neelapu, S.S. Managing the toxicities of CAR T-cell therapy. Hematol. Oncol. 2019, 37, 48–52. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef]
- Arnold, D.E.; Maude, S.L.; Callahan, C.A.; DiNofia, A.M.; Grupp, S.A.; Heimall, J.R. Subcutaneous immunoglobulin replacement following CD19-specific chimeric antigen receptor T-cell therapy for B-cell acute lymphoblastic leukemia in pediatric patients. Pediatr. Blood Cancer 2020, 67, e28092. [Google Scholar] [CrossRef]
- Doan, A.; Pulsipher, M.A. Hypogammaglobulinemia due to CAR T-cell therapy. Pediatr. Blood Cancer 2018, 65, e26914. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N. V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.N.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Perez, E.E.; Orange, J.S.; Bonilla, F.; Chinen, J.; Chinn, I.K.; Dorsey, M.; El-Gamal, Y.; Harville, T.O.; Hossny, E.; Mazer, B.; et al. Update on the use of immunoglobulin in human disease: A review of evidence. J. Allergy Clin. Immunol. 2017, 139, S1–S46. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and biological correlates of neurotoxicity associated with car t-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef]
- Hill, J.A.; Li, D.; Hay, K.A.; Green, M.L.; Cherian, S.; Chen, X.; Riddell, S.R.; Maloney, D.G.; Boeckh, M.; Turtle, C.J. Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood 2018, 131, 121–130. [Google Scholar] [CrossRef]
- Kansagra, A.J.; Frey, N. V.; Bar, M.; Laetsch, T.W.; Carpenter, P.A.; Savani, B.N.; Heslop, H.E.; Bollard, C.M.; Komanduri, K. V.; Gastineau, D.A.; et al. Clinical Utilization of Chimeric Antigen Receptor T Cells in B Cell Acute Lymphoblastic Leukemia: An Expert Opinion from the European Society for Blood and Marrow Transplantation and the American Society for Blood and Marrow Transplantation. Biol. Blood Marrow Transplant. 2019, 25, e76–e85. [Google Scholar] [CrossRef]
- Hensley, M.K.; Bain, W.G.; Jacobs, J.; Nambulli, S.; Parikh, U.; Cillo, A.; Staines, B.; Heaps, A.; Sobolewski, M.D.; Rennick, L.J.; et al. Intractable Coronavirus Disease 2019 (COVID-19) and Prolonged Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Replication in a Chimeric Antigen Receptor-Modified T-Cell Therapy Recipient: A Case Study. Clin. Infect. Dis. 2021, 73, e815–e821. [Google Scholar] [CrossRef]
- Biassoni, R. Natural killer cell receptors. Adv. Exp. Med. Biol. 2008, 640, 35–52. [Google Scholar] [CrossRef]
- Mrózek, E.; Anderson, P.; Caligiuri, M. Role of Interleukin-15 in the Development of Human CD56+ Natural Killer Cells From CD34+ Hematopoietic Progenitor Cells. Blood 1996, 87, 2632–2640. [Google Scholar] [CrossRef]
- Sconocchia, G.; Fujiwara, H.; Rezvani, K.; Keyvanfar, K.; El Ouriaghli, F.; Grube, M.; Melenhorst, J.; Hensel, N.; Barrett, A.J. G-CSF-mobilized CD34+ cells cultured in interleukin-2 and stem cell factor generate a phenotypically novel monocyte. J. Leukoc. Biol. 2004, 76, 1214–1219. [Google Scholar] [CrossRef]
- Coppola, A.; Arriga, R.; Lauro, D.; del Principe, M.I.; Buccisano, F.; Maurillo, L.; Palomba, P.; Venditti, A.; Sconocchia, G. NK cell inflammation in the clinical outcome of colorectal carcinoma. Front. Med. 2015, 2, 33. [Google Scholar] [CrossRef]
- Sconocchia, G.; Arriga, R.; Tornillo, L.; Terracciano, L.; Ferrone, S.; Spagnoli, G.C. Melanoma cells inhibit NK cell functions. Cancer Res. 2012, 72, 5428–5429. [Google Scholar] [CrossRef]
- Sconocchia, G.; Zlobec, I.; Lugli, A.; Calabrese, D.; Iezzi, G.; Karamitopoulou, E.; Patsouris, E.S.; Peros, G.; Horcic, M.; Tornillo, L.; et al. Tumor infiltration by FcγRIII (CD16)+ myeloid cells is associated with improved survival in patients with colorectal carcinoma. Int. J. Cancer 2011, 128, 2663–2672. [Google Scholar] [CrossRef]
- Sconocchia, G.; Eppenberger, S.; Spagnoli, G.C.; Tornillo, L.; Droeser, R.; Caratelli, S.; Ferrelli, F.; Coppola, A.; Arriga, R.; Lauro, D.; et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. Oncoimmunology 2014, 3, 1–6. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Casucci, M.; Volpi, I.; Tosti, A.; Perruccio, K.; Urbani, E.; Negrin, R.S.; Martelli, M.F.; Velardi, A. Role of Natural Killer Cell Alloreactivity in HLA-Mismatched Hematopoietic Stem Cell Transplantation. Blood 1999, 94, 333–339. [Google Scholar] [CrossRef]
- Sconocchia, G.; Lau, M.; Provenzano, M.; Rezvani, K.; Wongsena, W.; Fujiwara, H.; Hensel, N.; Melenhorst, J.; Li, J.; Ferrone, S.; et al. The antileukemia effect of HLA-matched NK and NK-T cells in chronic myelogenous leukemia involves NKG2D–target-cell interactions. Blood 2005, 106, 3666–3672. [Google Scholar] [CrossRef]
- Schmidt, P.; Raftery, M.J.; Pecher, G. Engineering NK Cells for CAR Therapy—Recent Advances in Gene Transfer Methodology. Front. Immunol. 2021, 11, 3404. [Google Scholar] [CrossRef]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin. Immunopathol. 2018 411 2018, 41, 59–68. [Google Scholar] [CrossRef]
- Pan, K.; Farrukh, H.; Chittepu, V.C.S.R.; Xu, H.; Pan, C. xian; Zhu, Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 2022, 41, 1–21. [Google Scholar] [CrossRef]
- Chang, Y.H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef]
- Klingemann, H. Are natural killer cells superior CAR drivers? 2014, 3. [CrossRef]
- Rafei, H.; Daher, M.; Rezvani, K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: leveraging the power of innate immunity. Br. J. Haematol. 2021, 193, 216–230. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; DenHollander, N.; Gupta, V.; Wang, X.-H.; Chaboureau, A.; Viswanathan, S.; Keating, A.; Williams, B.A.; et al. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef]
- Oelsner, S.; Waldmann, A.; Billmeier, A.; Röder, J.; Lindner, A.; Ullrich, E.; Marschalek, R.; Dotti, G.; Jung, G.; Große-Hovest, L.; et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int. J. Cancer 2019, 145, 1935–1945. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.; Konopleva, M.; Desikan, S.P.P.; Jain, N.; Ravandi, F.; Huang, X.; Wierda, W.G.; Borthakur, G.; Sasaki, K.; et al. Updated Results of a Phase II Study of Ponatinib and Blinatumomab for Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Blood 2021, 138, 2298–2298. [Google Scholar] [CrossRef]
- Jabbour, E.; Short, N.J.; Ravandi, F.; Huang, X.; Daver, N.; DiNardo, C.D.; Konopleva, M.; Pemmaraju, N.; Wierda, W.; Garcia-Manero, G.; et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: long-term follow-up of a single-centre, phase 2 study. Lancet Haematol. 2018, 5, e618–e627. [Google Scholar] [CrossRef]
- Ribera, J.-M.; Garcia, O.; Ribera, J.; Montesinos, P.; Cano, I.; Martínez, P.; Esteve, J.; Esteban, D.; García-Fortes, M.; Alonso, N.; et al. Ponatinib and Chemotherapy in Adults with De Novo Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Final Results of Ponalfil Clinical Trial. Blood 2021, 138, 1230–1230. [Google Scholar] [CrossRef]
- Roddie, C.; O’Reilly, M.A.; Marzolini, M.A. V; Wood, L.; Dias, J.; Cadinanos Garai, A.; Bosshard, L.; Abbasian, M.; Lowdell, M.W.; Wheeler, G.; et al. ALLCAR19: Updated Data Using AUTO1, a Novel Fast-Off Rate CD19 CAR in Relapsed/Refractory B-Cell Acute Lymphoblastic Leukaemia and Other B-Cell Malignancies. Blood 2020, 136, 3–4. [Google Scholar] [CrossRef]
- Roddie, C.; Dias, J.; O’Reilly, M.A.; Abbasian, M.; Cadinanos-Garai, A.; Vispute, K.; Bosshard-Carter, L.; Mitsikakou, M.; Mehra, V.; Roddy, H.; et al. Durable responses and low toxicity after fast off-rate cd19 chimeric antigen receptor-t therapy in adults with relapsed or refractory b-cell acute lymphoblastic leukemia. J. Clin. Oncol. 2021, 39, 3352–3364. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019 259 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Pan, J.; Niu, Q.; Deng, B.; Liu, S.; Wu, T.; Gao, Z.; Liu, Z.; Zhang, Y.; Qu, X.; Zhang, Y.; et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leuk. 2019 3312 2019, 33, 2854–2866. [Google Scholar] [CrossRef]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Gao, J.; Liu, W.J. Prognostic value of the response to prednisone for children with acute lymphoblastic leukemia: a meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7858–7866. [Google Scholar] [CrossRef]
- Berg, S.L.; Blaney, S.M.; Devidas, M.; Lampkin, T.A.; Murgo, A.; Bernstein, M.; Billett, A.; Kurtzberg, J.; Reaman, G.; Gaynon, P.; et al. Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: A report from the children’s oncology group. J. Clin. Oncol. 2005, 23, 3376–3382. [Google Scholar] [CrossRef]
- Dunsmore, K.P.; Winter, S.S.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Children’s oncology group AALL0434: A phase III randomized clinical trial testing nelarabine in newly diagnosed t-cell acute lymphoblastic leukemia. J. Clin. Oncol. 2020, 38, 3282–3293. [Google Scholar] [CrossRef]
- Gökbuget, N.; Basara, N.; Baurmann, H.; Beck, J.; Brüggemann, M.; Diedrich, H.; Güldenzoph, B.; Hartung, G.; Horst, H.A.; Hüttmann, A.; et al. High single-drug activity of nelarabine in relapsed T-lymphoblastic leukemia/lymphoma offers curative option with subsequent stem cell transplantation. Blood 2011, 118, 3504–3511. [Google Scholar] [CrossRef]
- Chun-fung, S.; Wan, T.M.; Mohan, A.A.M.; Qiu, Y.; Jiao, A. Bortezomib Is Effective in Treating T-ALL, Inducting G2/M Cell Cycle Arrest and WEE1 Downregulation. Blood 2021, 138, 4360–4360. [Google Scholar] [CrossRef]
- Zheng, R.; Li, M.; Wang, S.; Liu, Y. Advances of target therapy on NOTCH1 signaling pathway in T-cell acute lymphoblastic leukemia. Exp. Hematol. Oncol. 2020, 9, 1–9. [Google Scholar] [CrossRef]
- Jaramillo, S.; Hennemann, H.; Horak, P.; Teleanu, V.; Heilig, C.E.; Hutter, B.; Stenzinger, A.; Glimm, H.; Goeppert, B.; Müller-Tidow, C.; et al. Ruxolitinib is effective in the treatment of a patient with refractory T-ALL. eJHaem 2021, 2, 139–142. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Jabbour, E.; Short, N.J.; Rausch, C.R.; Savoy, J.M.; Bose, P.; Yilmaz, M.; Jain, N.; Borthakur, G.; Ohanian, M.; et al. Clinical Experience With Venetoclax Combined With Chemotherapy for Relapsed or Refractory T-Cell Acute Lymphoblastic Leukemia. Clin. Lymphoma, Myeloma Leuk. 2020, 20, 212–218. [Google Scholar] [CrossRef]
- Bride, K.L.; Vincent, T.L.; Im, S.Y.; Aplenc, R.; Barrett, D.M.; Carroll, W.L.; Carson, R.; Dai, Y.; Devidas, M.; Dunsmore, K.P.; et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood 2018, 131, 995–999. [Google Scholar] [CrossRef]
Figure 2.
Blinatumomab in paediatric ALL. Clinical trials of blinatumomab are reported by year, country, and population of paediatric patients with ALL (on the left) and are visualized on the map (on the right).
Figure 2.
Blinatumomab in paediatric ALL. Clinical trials of blinatumomab are reported by year, country, and population of paediatric patients with ALL (on the left) and are visualized on the map (on the right).
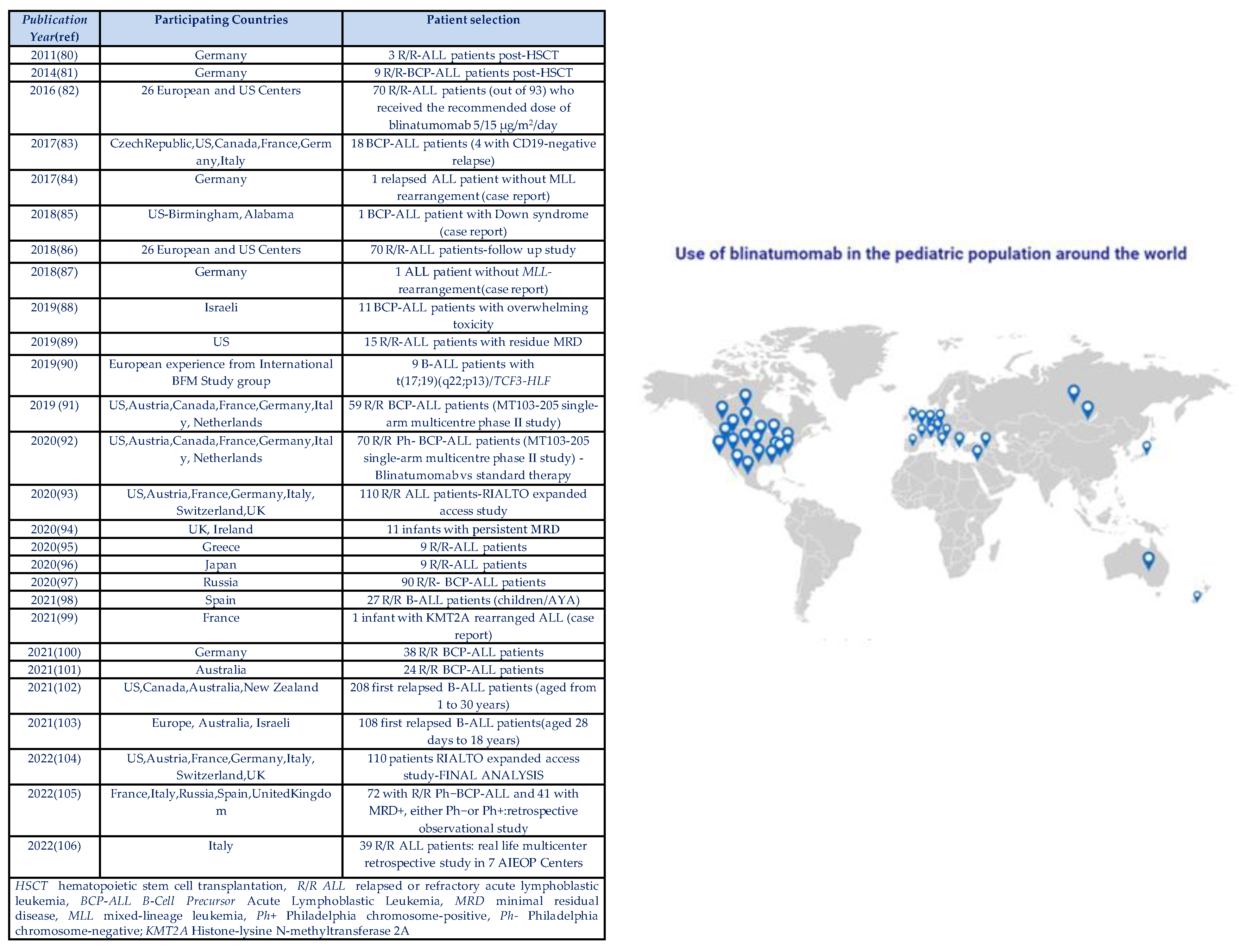
Figure 3.
Mechanism of action of ADC. The principal steps of the mechanism of action are as follows:1) the ADC binds to a tumor-cell specific antigen,2)the ADC-antigen becomes a coated pit vesicle and undergoes endocytosis, 3) the ADC is degraded by lysosomal proteases, 4) the drug is released into the cytoplasm, and 5)the drug disrupts the DNA of the targeted cell leading to cell death. Note: Figure created with BioRender.com.
Figure 3.
Mechanism of action of ADC. The principal steps of the mechanism of action are as follows:1) the ADC binds to a tumor-cell specific antigen,2)the ADC-antigen becomes a coated pit vesicle and undergoes endocytosis, 3) the ADC is degraded by lysosomal proteases, 4) the drug is released into the cytoplasm, and 5)the drug disrupts the DNA of the targeted cell leading to cell death. Note: Figure created with BioRender.com.
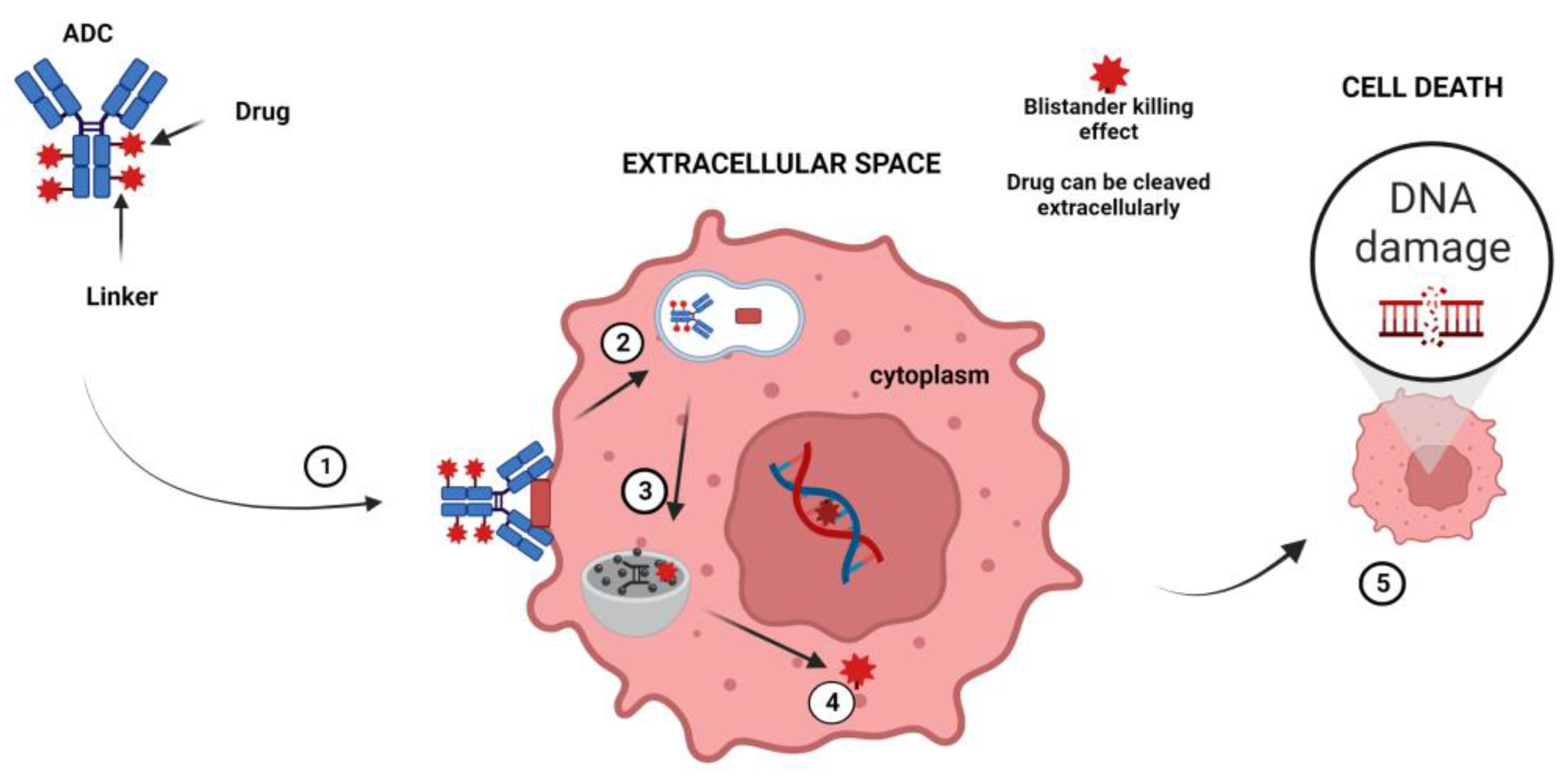
Figure 4.
CART cellgeneration and mechanism of action.The process of CART cell therapy includes the following steps: 1) the patient’s T cells are collected by leukapheresis, 2)a viral vector delivers a gene encoding the CAR into the T cells, 3) the T cells start expressing the CAR on their surface, 4)the CART cells are expanded and then infused back into the patient’s blood, 5)the CART cells can attack and destroy cancer cells. Note: Figure created with BioRender.com.
Figure 4.
CART cellgeneration and mechanism of action.The process of CART cell therapy includes the following steps: 1) the patient’s T cells are collected by leukapheresis, 2)a viral vector delivers a gene encoding the CAR into the T cells, 3) the T cells start expressing the CAR on their surface, 4)the CART cells are expanded and then infused back into the patient’s blood, 5)the CART cells can attack and destroy cancer cells. Note: Figure created with BioRender.com.
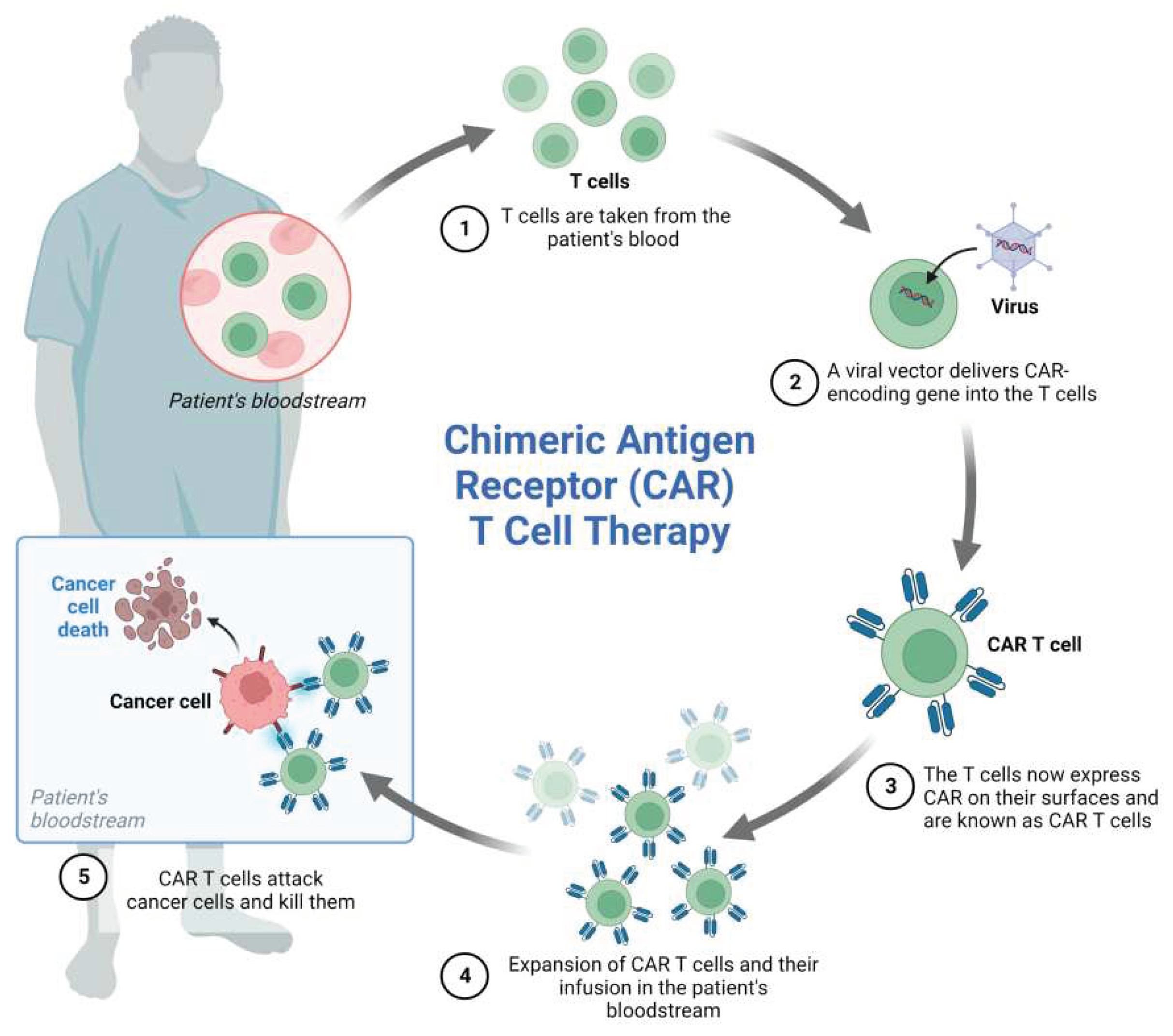
Figure 5.
NK cell activity.The engagement of NK cell inhibitory or activating receptor signaling decides the outcome of the immune synapse. Note: Figure created with BioRender.com.
Figure 5.
NK cell activity.The engagement of NK cell inhibitory or activating receptor signaling decides the outcome of the immune synapse. Note: Figure created with BioRender.com.
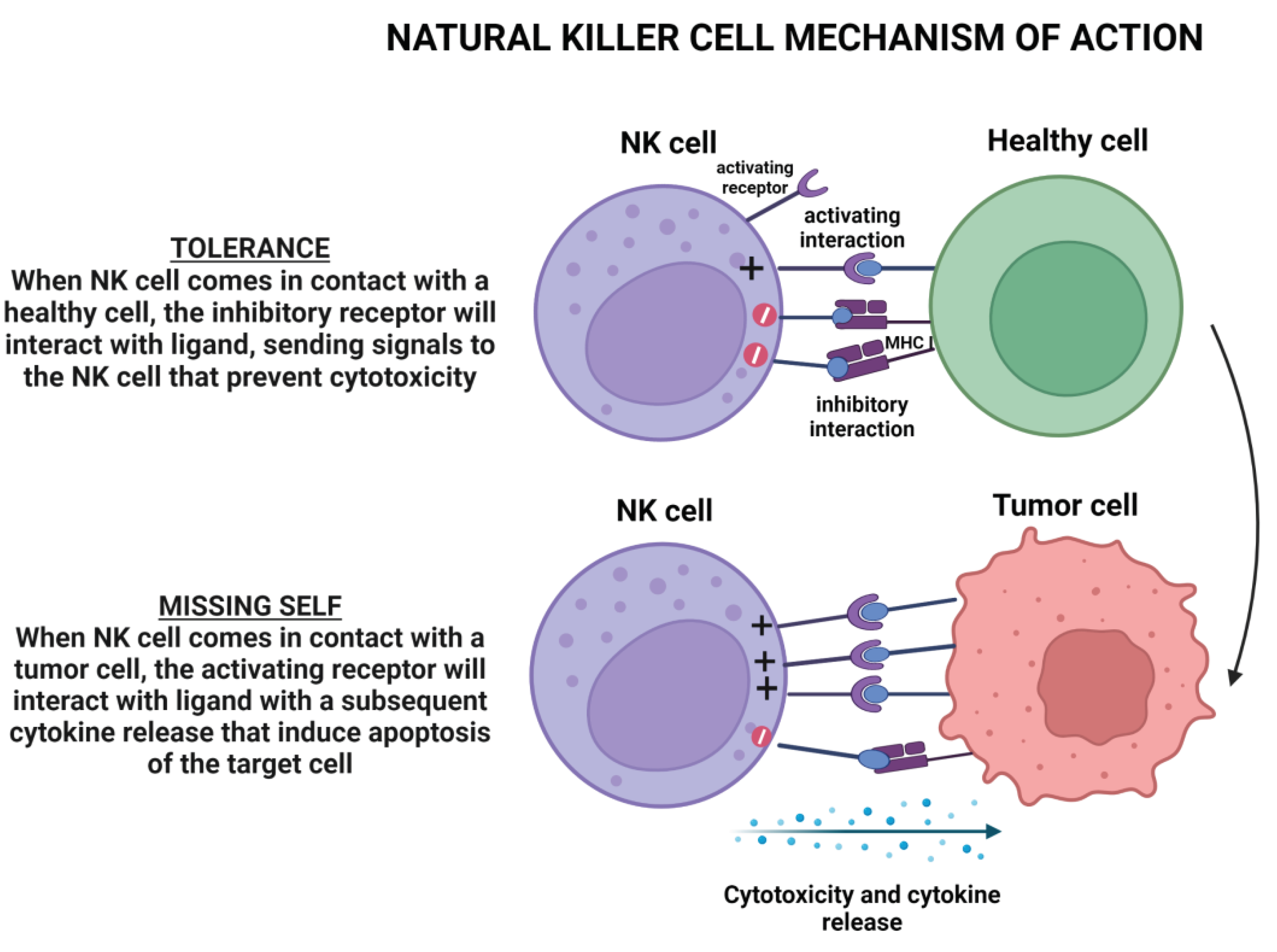

Table 1.
WHO classification of acute lymphoblastic leukemia.
| B-lymphoblastic leukemia/lymphoma |
|---|
| B-lymphoblastic leukemia/lymphoma, NOS |
| B-lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities |
| B-lymphoblastic leukemia/lymphoma with t(9;22)(q34.1;q11.2);BCR-ABL1 |
| B-lymphoblastic leukemia/lymphoma with t(v;11q23.3);KMT2Arearranged |
| B-lymphoblastic leukemia/lymphoma with t(12;21)(p13.2;q22.1);ETV6-RUNX1 |
| B-lymphoblastic leukemia/lymphoma with hyperdiploidy |
| B-lymphoblastic leukemia/lymphoma with hypodiploidy |
| B-lymphoblastic leukemia/lymphoma with t(5;14)(q31.1;q32.3)IL3-IGH |
| B-lymphoblastic leukemia/lymphoma with t(1;19)(q23;p13.3);TCF3-PBX1 |
| Provisional entity:B-lymphoblastic leukemia/lymphoma with translocations involving tyrosine kinases or cytokine receptors (“ BCR-ABL1–like“) |
| Provisional entity:B-lymphoblastic leukemia/lymphoma with intrachromosomal amplification of chromosome 21 (iAMP21) |
| T-lymphoblastic leukemia/lymphoma(can only be differentiated from B-ALL/LBL based on IHC and/or flow cytometry). |
| Provisional entity: Early T-cell precursor lymphoblastic leukemia |
| Provisional entity: NK cell lymphoblastic leukemia/lymphoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



