
Submitted:
06 May 2023
Posted:
09 May 2023
You are already at the latest version
Abstract
The Epidermal Growth Factor Receptor (EGFR) overexpression or its mutation mediates the sustaining proliferative signaling in cancers. Human EGFR-targeting monoclonal antibody (mAb) therapy such as cetuximab has been approved for clinical use in patients with colorectal cancers and head and neck squamous cell carcinomas. A reliable preclinical mouse model is essential to further develop the mAb therapy against EGFR. However, a mAb against mouse EGFR (mEGFR) for flow cytometry has not been established. In this study, we developed a specific and sensitive mAb for mEGFR using the Cell-Based Immunization and Screening (CBIS) method. The established anti-mEGFR mAb, EMab-300 (rat IgG1, kappa) reacted with mEGFR-overexpressed Chinese hamster ovary-K1 (CHO/mEGFR) and endogenously mEGFR-expressed cell lines, including NMuMG (a mouse mammary gland epithelial cell) and Lewis lung carcinoma cells by flow cytometry. The kinetic analysis using flow cytometry indicated that the dissociation constant (KD) of EMab-300 for CHO/mEGFR and NMuMG was 4.3 × 10−8 M and 1.9 × 10−8 M, respectively. These results indicated that EMab-300 applies to the detection of mEGFR by flow cytometry, and is expected in the use of preclinical study.
Keywords:
mouse EGFR
; monoclonal antibody
; Cell-Based Immunization and Screening
; CBIS
1. Introduction
The Epidermal Growth Factor Receptor (EGFR) belongs to the ERBB family of receptor tyrosine kinases. EGFR is a type I transmembrane glycoprotein, which is composed of an extracellular ligand-binding domain and an intracellular tyrosine kinase domain [1]. Upon ligand binding to the extracellular domain of EGFR, the downstream signaling pathways, such as the mitogen-activated protein kinases, the phosphoinositide 3-kinase/Akt, the Janus kinase/signal transducer and activator of transcription, and the phospholipase C-γ/protein kinase C pathways are activated [2]. These pathways lead to the transcriptional activation of target genes, which are important for cell proliferation, survival, migration, invasion and cancer-initiating properties [2]. In many carcinomas, EGFR overexpression and its mutation are involved in sustaining proliferative signaling, which is an important hallmark of cancer [3,4]. Therefore, EGFR has been considered as an important target for cancer therapy.
The EGFR-targeted therapies have been used in the clinic, including monoclonal antibodies (mAbs) [5] and the tyrosine kinase inhibitors [6]. Cetuximab is a mouse/human chimeric mAb, that binds to the extracellular domain (domain III) of EGFR, which is important for the neutralization activity [7]. Cetuximab has been approved by the Food and Drug Administration for colorectal cancer [8] and head and neck squamous cell carcinoma (HNSCC) [9]. Although cetuximab has been used to treat patients with metastatic colorectal cancers, the use is limited to tumors harboring wild-type RAS [10]. The cetuximab treatment did not exhibit the benefit in patients with colorectal cancer harboring RAS mutations [10]. Moreover, the majority of HNSCC express EGFR; however, the benefit of therapy is limited to only 15-20% of HNSCC patients [11].
Preclinical mouse models have been developed for the establishment of cancer therapy. The earliest models were built through the transplantation of murine tumors into immunocompetent host mice [12]. Furthermore, tumors harvested from genetically modified mice can be transplanted and expanded into fully immunocompetent syngeneic hosts [13]. These syngeneic models were used in preclinical studies to evaluate not only small molecule chemotherapeutic drugs but also immunotherapies including immune checkpoint inhibitors [13]. Although several studies have shown that cetuximab can stimulate the innate or adaptive immune responses [14,15], the mechanisms have not been fully understood due to the lack of a preclinical model using anti-mouse EGFR (mEGFR) mAb. The model is also thought to be important for the evaluation of novel antibody-drug conjugates and prediction of the side effects.
The Cell-Based Immunization and Screening (CBIS) method includes the immunization of antigen-overexpressing cells and high-throughput hybridoma screening using flow cytometry. By the CBIS method, we have developed mAbs against human antigens, including human epidermal growth factor receptor 2 (HER2) [16], human epidermal growth factor receptor 3 (HER3) [17], epithelial cell adhesion molecule (EpCAM) [18,19], trophoblast cell surface antigen 2 (TROP2) [20,21], programmed cell death ligand 1 (PD-L1) [22], podoplanin (PDPN) [23,24,25,26,27,28,29,30,31,32,33,34], the cluster of differentiation 19 (CD19) [35], CD20 [36,37], CD44 [38,39,40,41], CD133 [42], killer cell lectin-like receptor G1 (KLRG1) [43], C-C motif chemokine receptor 9 (CCR9) [44], and T cell immunoreceptor with Ig and ITIM domains (TIGIT) [45] by immunization of antigen-overexpressing cells into mice. We also successfully developed mAbs against mouse antigens, including mouse CCR3 [46] and mouse CCR8 [47] by immunization them into rats. In this study, we developed novel anti-mEGFR mAbs by the CBIS method and evaluated its applications including flow cytometry.
2. Materials and methods
2.1. Preparation of plasmids
The expression plasmid of mEGFR (pCMV6-neo-mEGFR-Myc-DDK) was commercially available from OriGene Technologies, Inc. (Rockville, MD). The cDNA encoding mEGFR (NM_207655) was subcloned into pCAG-zeo_ssnPA and pCAG-zeo_MAP vectors, which are purchased from a pCAG-zeo vector (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan), with N-terminal PA tag [48,49,50] and MAP tag [51,52], respectively. The amino acid sequences of the tag system were as follows: PA tag, 12 amino acids (GVAMPGAEDDVV) and MAP tag, 12 amino acids (GDGMVPPGIEDK). The PA tag can be recognized by NZ-1 (an anti-human PDPN mAb) [48,49,50,53,54,55,56,57,58,59,60,61,62,63,64,65].
2.2. Antibodies
Alexa Fluor 488-conjugated anti-rat IgG was purchased from Cell Signaling Technology, Inc. (Danvers, MA).
2.3. Cell lines
P3X63Ag8U.1 (P3U1), Chinese hamster ovary (CHO)-K1, LN229, and NMuMG (a mouse mammary gland epithelial cell) were obtained from the American Type Culture Collection (ATCC; Manassas, VA). Lewis lung carcinoma was obtained from the Japanese Collection of Research Bioresources (JCRB; Osaka, Japan).
The pCAG-zeo_ssnPA-mEGFR and pCAG-zeo_MAP-mEGFR plasmids were transfected into LN229 and CHO-K1 cells, respectively. The stable transfectants were generated as described previously [40,41].
CHO-K1, mEGFR-overexpressed CHO-K1 (CHO/mEGFR), Lewis lung carcinoma, and P3U1 were cultured in an RPMI-1640 medium (Nacalai Tesque, Inc., Kyoto, Japan), with 10% fetal bovine serum (FBS; Thermo Fisher Scientific Inc.). A cocktail of 100 units/mL of penicillin, 100 μg/mL of streptomycin, and 0.25 μg/mL of amphotericin B (Nacalai Tesque, Inc.) was added to the medium. LN229, mEGFR-overexpressed LN229 (LN229/mEGFR), and NMuMG were cultured in DMEM (Nacalai Tesque, Inc.), supplemented as indicated above. For NMuMG cells, 10 μg/mL of insulin (Sigma-Aldrich Corp., St. Louis, MO, USA) was further added. All cells were cultured using a humidified incubator at 37°C, in an atmosphere of 5% CO2 and 95% air.
2.4. Development of hybridomas
A five-week-old Sprague–Dawley rat was purchased from CLEA Japan (Tokyo, Japan). To develop mAbs against mEGFR, we intraperitoneally immunized one rat with LN229/mEGFR (1 × 109 cells) plus Imject Alum (Thermo Fisher Scientific Inc.). After three additional injections in every week (1 × 109 cells/rat), a final booster injection (1 × 109 cells/rat) was performed two days before harvesting spleen cells. The hybridomas were produced, as described previously [46]. The hybridoma supernatants were screened using flow cytometry using CHO/mEGFR, CHO-K1, and NMuMG.
2.5. Purification of EMab-300
The cultured supernatant of EMab-300 hybridomas was applied to 1 mL of Ab-Capcher (ProteNova, Kagawa, Japan). Ab-capcher is a gel carrier in which the alkali-resistant antibody-binding protein Protein A-R28, developed by Protenova’s patented technology, was immobilized at multiple points at high density. After washing with phosphate-buffered saline (PBS), the antibodies were eluted with an IgG elution buffer (Thermo Fisher Scientific Inc.). Finally, the eluates were concentrated, and the elution buffer was replaced with PBS using Amicon Ultra (Merck KGaA, Darmstadt, Germany).
2.6. Flow cytometric analysis
CHO/mEGFR, CHO-K1, NMuMG, and Lewis lung carcinoma were harvested after a brief exposure to 1 mM ethylenediaminetetraacetic acid (EDTA, Nacalai Tesque, Inc.). The cells were treated with EMab-300 or blocking buffer (control) (0.1% BSA in PBS) for 30 min at 4°C, followed by treatment with Alexa Fluor 488-conjugated anti-rat IgG. The data were analyzed using the SA3800 Cell Analyzer and SA3800 software ver. 2.05 (Sony Corp., Tokyo, Japan).
2.7. Determination of dissociation constant (KD) by flow cytometry
The serially diluted EMab-300 was suspended with CHO/mEGFR and NMuMG cells for 30 min at 4°C. The cells were treated with 50 μL of Alexa Fluor 488-conjugated anti-rat IgG (1:200). The fluorescence data were collected, using the SA3800 Cell Analyzer. The KD was subsequently calculated by GraphPad PRISM 8 (GraphPad Software, Inc., La Jolla, CA).
3. Results
3.1. Development of anti-mEGFR mAbs by the CBIS method
To develop anti-mEGFR mAbs, one rat was immunized with LN229/mEGFR cells. (Figure 1A). The spleen was then excised from the rat, and splenocytes were fused with myeloma P3U1 cells (Figure 1B). The developed hybridomas were subsequently seeded into 96-well plates and cultivated for six days. The positive wells were screened by the selection of mEGFR-expressing cell-reactive and CHO-K1-non-reactive supernatants, using flow cytometry (Figure 1C). After the limiting dilution and several additional screenings, an anti-mEGFR mAbs, EMab-300 (rat IgG1, kappa) was finally established (Figure 1D).
3.2. Flow cytometric analysis using EMab-300
We conducted flow cytometry using EMab-300 against CHO/mEGFR, NMuMG, and Lewis lung carcinoma cell lines. EMab-300 recognized CHO/mEGFR cells dose-dependently at 10, 1, 0.1, and 0.01 μg/mL (Figure 2A). Parental CHO-K1 cells were not recognized even at 10 μg/mL of EMab-300 (Figure 2B).
Next, we investigated the reactivity of EMab-300 against endogenously mEGFR-expressing cell lines, NMuMG and Lewis lung carcinoma. EMab-300 reacted with NMuMG and Lewis lung carcinoma in a dose-dependent manner (Figure 2C,D.) These results suggested that EMab-300 specifically recognizes mEGFR, and is also useful for detecting endogenous mEGFR by flow cytometry.
3.3. Kinetic analysis of EMab-300 using flow cytometry
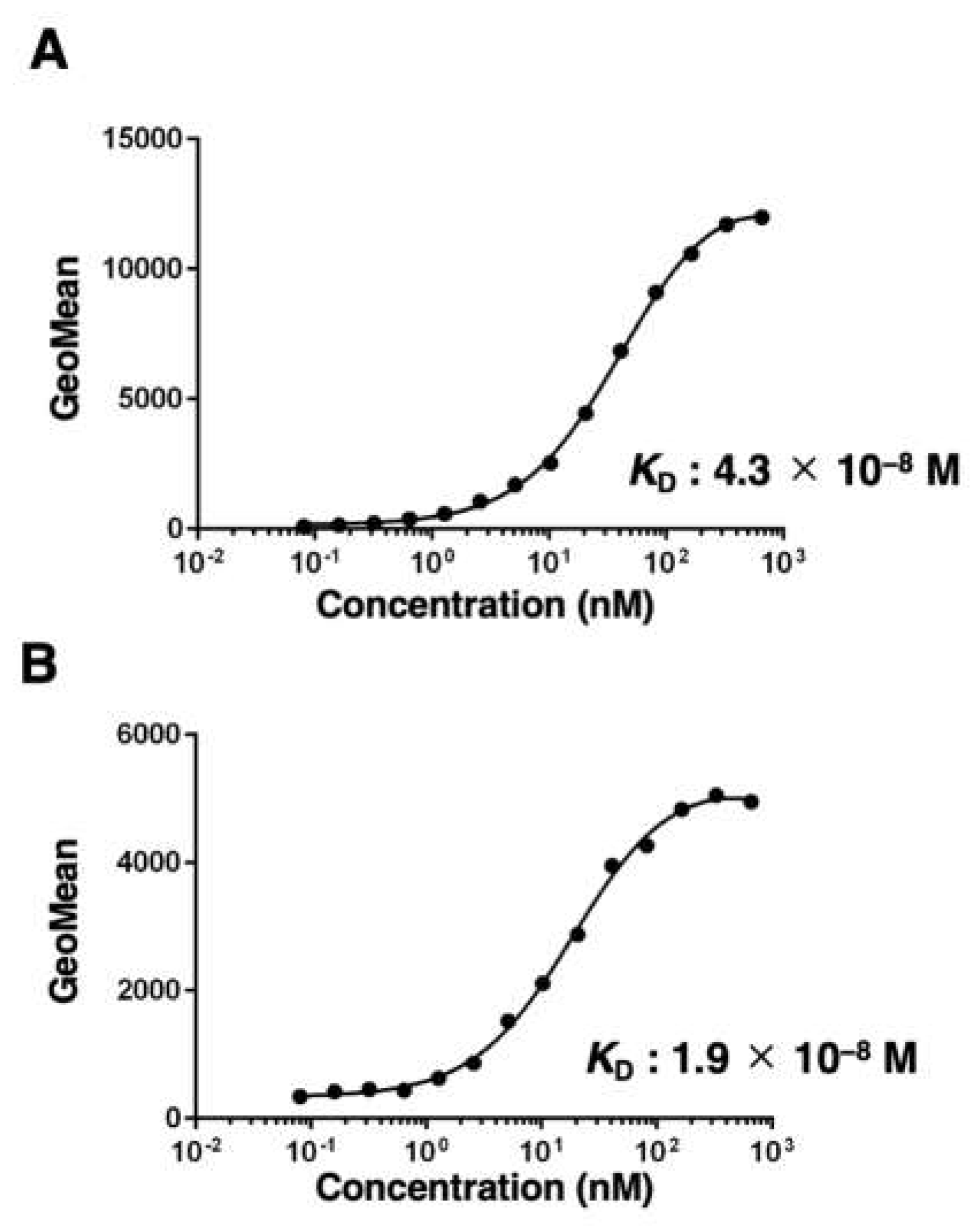
To determine the KD of EMab-300 with mEGFR-expressing cells, we conducted kinetic analyses by flow cytometry using CHO/mEGFR and NMuMG cells. The KD values of EMab-300 for CHO/mEGFR and NMuMG were determined as 4.3 × 10−8 M and 1.9 × 10−9 M, respectively (Figure 3A,B). These results indicate that EMab-300 possesses a moderate affinity for both CHO/mEGFR and NMuMG cells.
4. Discussion
Therapeutic strategies and modalities for cancer have been revolutionary developed. However, only about five percent of new cancer therapies are approved, and most fail due to the lack of efficacy [13]. The failures cost enormous finance and lose the quality of patient life. The failures also indicate that current preclinical methods are not sufficient to predict successful outcomes. Although EGFR is one of the important targets for cancer therapy, anti-mEGFR mAbs for preclinical study have not been developed. In this study, we developed a novel anti-mEGFR mAbs (EMab-300) using the CBIS method and showed the application to flow cytometry (Figure 2 and Figure 3). EMab-300 could contribute to the preclinical study to evaluate the antitumor effects and predict the side effects of the EGFR targeting therapy.
EMab-300 could react with NMuMG and Lewis lung carcinoma cells (Figure 2). NMuMG has been used in the study of epithelial-to-mesenchymal transition (EMT), which is induced by various cytokines, such as TGF-β [66] and transcriptional factors [67]. The activation of EMT program confers tumor cells the ability of migration, invasion, extravasation, and stemness [68]. Once the EMT-induced tumor cells reach to distant organs, these mesenchymal properties revert to epithelial properties via mesenchymal–epithelial transition (MET) in order to form a secondary tumor in distant organs [68]. The EMT-induced NMuMG cells can make spheres in the presence of EGF in vitro, and exhibit the tumorigenic potential in vivo [69]. Therefore, we can evaluate the neutralization activity of EMab-300 in the sphere formation assay in the future study.
Lewis lung carcinoma has been widely used as a syngenetic model [70]. The model was a successful preclinical model for the evaluation of a chemotherapeutic agent, navelbine, prior to its implementation in clinical trials [70]. Recently, syngenetic models are also important for the evaluation of combination therapy with immune checkpoint inhibitors [71,72]. Therefore, the conversion of EMab-300 (rat IgG1, kappa) to mouse IgG is essential for evaluating antitumor effects and developing antibody-drug conjugates or chimeric antigen receptor T cell therapies. We previously produced recombinant antibodies, which were converted into mouse IgG2a subclass from mouse IgG1. Furthermore, we produced defucosylated IgG2a mAbs using fucosyltransferase 8-deficient CHO-K1 cells to enhance the antibody-dependent cellular cytotoxicity activity [73,74,75,76,77,78,79,80]. The defucosylated mAbs showed an antitumor effect in xenograft models. Therefore, a class-switched and defucosylated type of EMab-300 could contribute to the treatment of syngenetic mouse tumors and spontaneous mice tumors in the future.
The identification of the epitope is also important to assess the property of mAbs. We have established anti-human EGFR mAbs using the CBIS method. Most of the mAbs recognized the conformational epitopes. Therefore, we have developed the RIEDL insertion for epitope mapping (REMAP) method [81,82,83,84] to identify the conformational epitope. We determined the conformational epitopes of anti-human EGFR mAbs (EMab-51 and EMab-134) [82,84] and anti-CD44 mAbs (C44Mab-5 and C44Mab-46) [81,83] using the REMAP method. Therefore, further investigations are required to determine the epitopes of EMab-300.
References
- Zandi, R.; Larsen, A.B.; Andersen, P.; Stockhausen, M.T.; Poulsen, H.S. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal 2007, 19, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Sequist, L.V.; Lin, J.J. Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management. Nat Rev Clin Oncol 2022, 19, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Tsao, L.C.; Force, J.; Hartman, Z.C. Mechanisms of Therapeutic Antitumor Monoclonal Antibodies. Cancer Res 2021, 81, 4641–4651. [Google Scholar] [CrossRef]
- Zubair, T.; Bandyopadhyay, D. Small Molecule EGFR Inhibitors as Anti-Cancer Agents: Discovery, Mechanisms of Action, and Opportunities. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007, 357, 2040–2048. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Ohishi, T.; Kaneko, M.K.; Yoshida, Y.; Takashima, A.; Kato, Y.; Kawada, M. Current Targeted Therapy for Metastatic Colorectal Cancer. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Bardelli, A.; Jänne, P.A. The road to resistance: EGFR mutation and cetuximab. Nat Med 2012, 18, 199–200. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; Merlino, G.; Van Dyke, T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.M.; Trivedi, S.; Concha-Benavente, F.; Gibson, S.P.; Reeder, C.; Ferrone, S.; Ferris, R.L. CD137 Stimulation Enhances Cetuximab-Induced Natural Killer: Dendritic Cell Priming of Antitumor T-Cell Immunity in Patients with Head and Neck Cancer. Clin Cancer Res 2017, 23, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Kubach, J.; Hubo, M.; Amendt, C.; Stroh, C.; Jonuleit, H. IgG1 anti-epidermal growth factor receptor antibodies induce CD8-dependent antitumor activity. Int J Cancer 2015, 136, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Itai, S.; Fujii, Y.; Kaneko, M.K.; Yamada, S.; Nakamura, T.; Yanaka, M.; Saidoh, N.; Chang, Y.W.; Handa, S.; Takahashi, M.; et al. H(2)Mab-77 is a Sensitive and Specific Anti-HER2 Monoclonal Antibody Against Breast Cancer. Monoclon Antib Immunodiagn Immunother 2017, 36, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Ohishi, T.; Takei, J.; Nakamura, T.; Nanamiya, R.; Hosono, H.; Tanaka, T.; Sano, M.; Harada, H.; Kawada, M.; et al. Anti-HER3 monoclonal antibody exerts antitumor activity in a mouse model of colorectal adenocarcinoma. Oncol Rep 2021, 46. [Google Scholar] [CrossRef]
- Li, G.; Suzuki, H.; Asano, T.; Tanaka, T.; Suzuki, H.; Kaneko, M.K.; Kato, Y. Development of a Novel Anti-EpCAM Monoclonal Antibody for Various Applications. Antibodies (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Ohishi, T.; Takei, J.; Sano, M.; Nakamura, T.; Hosono, H.; Yanaka, M.; Asano, T.; Sayama, Y.; Harada, H.; et al. Anti-EpCAM monoclonal antibody exerts antitumor activity against oral squamous cell carcinomas. Oncol Rep 2020, 44, 2517–2526. [Google Scholar] [CrossRef]
- Sayama, Y.; Kaneko, M.K.; Takei, J.; Hosono, H.; Sano, M.; Asano, T.; Kato, Y. Establishment of a novel anti-TROP2 monoclonal antibody TrMab-29 for immunohistochemical analysis. Biochem Biophys Rep 2021, 25, 100902. [Google Scholar] [CrossRef]
- Tanaka, T.; Ohishi, T.; Asano, T.; Takei, J.; Nanamiya, R.; Hosono, H.; Sano, M.; Harada, H.; Kawada, M.; Kaneko, M.K.; et al. An anti-TROP2 monoclonal antibody TrMab-6 exerts antitumor activity in breast cancer mouse xenograft models. Oncol Rep 2021, 46. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Itai, S.; Nakamura, T.; Yanaka, M.; Chang, Y.W.; Suzuki, H.; Kaneko, M.K.; Kato, Y. Monoclonal Antibody L(1)Mab-13 Detected Human PD-L1 in Lung Cancers. Monoclon Antib Immunodiagn Immunother 2018, 37, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Itai, S.; Nakamura, T.; Yanaka, M.; Saidoh, N.; Chang, Y.W.; Handa, S.; Harada, H.; Kagawa, Y.; Ichii, O.; et al. PMab-52: Specific and Sensitive Monoclonal Antibody Against Cat Podoplanin for Immunohistochemistry. Monoclon Antib Immunodiagn Immunother 2017, 36, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Kaneko, M.K.; Nakamura, T.; Itai, S.; Fukui, M.; Harada, H.; Yamada, S.; Kato, Y. Establishment of a Monoclonal Antibody PMab-231 for Tiger Podoplanin. Monoclon Antib Immunodiagn Immunother 2019, 38, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Takei, J.; Sayama, Y.; Yamada, S.; Kaneko, M.K.; Kato, Y. Development of an anti-bear podoplanin monoclonal antibody PMab-247 for immunohistochemical analysis. Biochem Biophys Rep 2019, 18, 100644. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Yamada, S.; Itai, S.; Nakamura, T.; Takei, J.; Sano, M.; Harada, H.; Fukui, M.; Kaneko, M.K.; Kato, Y. Establishment of a monoclonal antibody PMab-233 for immunohistochemical analysis against Tasmanian devil podoplanin. Biochem Biophys Rep 2019, 18, 100631. [Google Scholar] [CrossRef] [PubMed]
- Goto, N.; Suzuki, H.; Tanaka, T.; Asano, T.; Kaneko, M.K.; Kato, Y. Development of a Monoclonal Antibody PMab-292 Against Ferret Podoplanin. Monoclon Antib Immunodiagn Immunother 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Yamada, S.; Itai, S.; Sano, M.; Nakamura, T.; Yanaka, M.; Handa, S.; Mizuno, T.; Maeda, K.; Fukui, M.; et al. Establishment of Monoclonal Antibody PMab-202 Against Horse Podoplanin. Monoclon. Antib. Immunodiagn. Immunother. 2018, 37, 233–237. [Google Scholar] [CrossRef]
- Kato, Y.; Yamada, S.; Furusawa, Y.; Itai, S.; Nakamura, T.; Yanaka, M.; Sano, M.; Harada, H.; Fukui, M.; Kaneko, M.K. PMab-213: a monoclonal antibody for immunohistochemical analysis against pig podoplanin. Monoclon. Antib. Immunodiagn. Immunother. 2019, 38, 18–24. [Google Scholar] [CrossRef]
- Furusawa, Y.; Yamada, S.; Nakamura, T.; Sano, M.; Sayama, Y.; Itai, S.; Takei, J.; Harada, H.; Fukui, M.; Kaneko, M.K.; et al. PMab-235: A monoclonal antibody for immunohistochemical analysis against goat podoplanin. Heliyon 2019, 5, e02063. [Google Scholar] [CrossRef]
- Kato, Y.; Furusawa, Y.; Yamada, S.; Itai, S.; Takei, J.; Sano, M.; Kaneko, M.K. Establishment of a monoclonal antibody PMab-225 against alpaca podoplanin for immunohistochemical analyses. Biochem Biophys Rep 2019, 18, 100633. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Furusawa, Y.; Itai, S.; Takei, J.; Nakamura, T.; Sano, M.; Harada, H.; Yamada, S.; Kaneko, M.K. Establishment of an Anticetacean Podoplanin Monoclonal Antibody PMab-237 for Immunohistochemical Analysis. Monoclon. Antib. Immunodiagn. Immunother. 2019, 38, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Furusawa, Y.; Sano, M.; Takei, J.; Nakamura, T.; Yanaka, M.; Okamoto, S.; Handa, S.; Komatsu, Y.; Asano, T.; et al. Development of an Anti-Sheep Podoplanin Monoclonal Antibody PMab-256 for Immunohistochemical Analysis of Lymphatic Endothelial Cells. Monoclon Antib Immunodiagn Immunother 2020, 39, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Asano, T.; Sano, M.; Takei, J.; Hosono, H.; Nanamiya, R.; Nakamura, T.; Yanaka, M.; Harada, H.; Fukui, M.; et al. Development of Monoclonal Antibody PMab-269 Against California Sea Lion Podoplanin. Monoclon Antib Immunodiagn Immunother 2021, 40, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Kaneko, M.K.; Sayama, Y.; Asano, T.; Sano, M.; Yanaka, M.; Nakamura, T.; Okamoto, S.; Handa, S.; Komatsu, Y.; et al. Development of Novel Mouse Monoclonal Antibodies Against Human CD19. Monoclon Antib Immunodiagn Immunother 2020, 39, 45–50. [Google Scholar] [CrossRef]
- Furusawa, Y.; Kaneko, M.K.; Kato, Y. Establishment of C(20)Mab-11, a novel anti-CD20 monoclonal antibody, for the detection of B cells. Oncol Lett 2020, 20, 1961–1967. [Google Scholar] [CrossRef]
- Furusawa, Y.; Kaneko, M.K.; Kato, Y. Establishment of an Anti-CD20 Monoclonal Antibody (C(20)Mab-60) for Immunohistochemical Analyses. Monoclon Antib Immunodiagn Immunother 2020, 39, 112–116. [Google Scholar] [CrossRef]
- Tawara, M.; Suzuki, H.; Goto, N.; Tanaka, T.; Kaneko, M.K.; Kato, Y. A Novel Anti-CD44 Variant 9 Monoclonal Antibody C44Mab-1 was Developed for Immunohistochemical Analyses Against Colorectal Cancers Curr. Issues Mol. Biol. 2023, 45, 3658–3673. [Google Scholar] [CrossRef]
- Kudo, Y.; Suzuki, H.; Tanaka, T.; Kaneko, M.K.; Kato, Y. Development of a Novel Anti-CD44 variant 5 Monoclonal Antibody C44Mab-3 for Multiple Applications against Pancreatic Carcinomas. Antibodies 2023, 12, 31. [Google Scholar] [CrossRef]
- Ejima, R.; Suzuki, H.; Tanaka, T.; Asano, T.; Kaneko, M.K.; Kato, Y. Development of a Novel Anti-CD44 Variant 6 Monoclonal Antibody C(44)Mab-9 for Multiple Applications against Colorectal Carcinomas. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Yamada, S.; Itai, S.; Nakamura, T.; Yanaka, M.; Kaneko, M.K.; Kato, Y. Detection of high CD44 expression in oral cancers using the novel monoclonal antibody, C(44)Mab-5. Biochem Biophys Rep 2018, 14, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Itai, S.; Fujii, Y.; Nakamura, T.; Chang, Y.W.; Yanaka, M.; Saidoh, N.; Handa, S.; Suzuki, H.; Harada, H.; Yamada, S.; et al. Establishment of CMab-43, a Sensitive and Specific Anti-CD133 Monoclonal Antibody, for Immunohistochemistry. Monoclon Antib Immunodiagn Immunother 2017, 36, 231–235. [Google Scholar] [CrossRef]
- Asano, T.; Nanamiya, R.; Tanaka, T.; Kaneko, M.K.; Kato, Y. Development of Antihuman Killer Cell Lectin-Like Receptor Subfamily G Member 1 Monoclonal Antibodies for Flow Cytometry. Monoclon Antib Immunodiagn Immunother 2021, 40, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Nanamiya, R.; Takei, J.; Asano, T.; Tanaka, T.; Sano, M.; Nakamura, T.; Yanaka, M.; Hosono, H.; Kaneko, M.K.; Kato, Y. Development of Anti-Human CC Chemokine Receptor 9 Monoclonal Antibodies for Flow Cytometry. Monoclon Antib Immunodiagn Immunother 2021, 40, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Takei, J.; Asano, T.; Nanamiya, R.; Nakamura, T.; Yanaka, M.; Hosono, H.; Tanaka, T.; Sano, M.; Kaneko, M.K.; Harada, H.; et al. Development of Anti-human T Cell Immunoreceptor with Ig and ITIM Domains (TIGIT) Monoclonal Antibodies for Flow Cytometry. Monoclon Antib Immunodiagn Immunother 2021, 40, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Nanamiya, R.; Takei, J.; Nakamura, T.; Yanaka, M.; Hosono, H.; Tanaka, T.; Sano, M.; Kaneko, M.K.; Kato, Y. Development of Anti-Mouse CC Chemokine Receptor 3 Monoclonal Antibodies for Flow Cytometry. Monoclon Antib Immunodiagn Immunother 2021, 40, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nanamiya, R.; Takei, J.; Nakamura, T.; Yanaka, M.; Hosono, H.; Sano, M.; Asano, T.; Kaneko, M.K.; Kato, Y. Development of Anti-Mouse CC Chemokine Receptor 8 Monoclonal Antibodies for Flow Cytometry. Monoclon Antib Immunodiagn Immunother 2021, 40, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Oi, R.; Akashi, S.; Kaneko, M.K.; Kato, Y.; Nogi, T. Application of the NZ-1 Fab as a crystallization chaperone for PA tag-inserted target proteins. Protein Sci 2019, 28, 823–836. [Google Scholar] [CrossRef]
- Fujii, Y.; Matsunaga, Y.; Arimori, T.; Kitago, Y.; Ogasawara, S.; Kaneko, M.K.; Kato, Y.; Takagi, J. Tailored placement of a turn-forming PA tag into the structured domain of a protein to probe its conformational state. J Cell Sci 2016, 129, 1512–1522. [Google Scholar] [CrossRef]
- Fujii, Y.; Kaneko, M.; Neyazaki, M.; Nogi, T.; Kato, Y.; Takagi, J. PA tag: a versatile protein tagging system using a super high affinity antibody against a dodecapeptide derived from human podoplanin. Protein Expr Purif 2014, 95, 240–247. [Google Scholar] [CrossRef]
- Fujii, Y.; Kaneko, M.K.; Kato, Y. MAP Tag: A Novel Tagging System for Protein Purification and Detection. Monoclon Antib Immunodiagn Immunother 2016, 35, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Wakasa, A.; Kaneko, M.K.; Kato, Y.; Takagi, J.; Arimori, T. Site-specific epitope insertion into recombinant proteins using the MAP tag system. J Biochem 2020, 168, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kaneko, M.K.; Kuno, A.; Uchiyama, N.; Amano, K.; Chiba, Y.; Hasegawa, Y.; Hirabayashi, J.; Narimatsu, H.; Mishima, K.; et al. Inhibition of tumor cell-induced platelet aggregation using a novel anti-podoplanin antibody reacting with its platelet-aggregation-stimulating domain. Biochem Biophys Res Commun 2006, 349, 1301–1307. [Google Scholar] [CrossRef]
- Chalise, L.; Kato, A.; Ohno, M.; Maeda, S.; Yamamichi, A.; Kuramitsu, S.; Shiina, S.; Takahashi, H.; Ozone, S.; Yamaguchi, J.; et al. Efficacy of cancer-specific anti-podoplanin CAR-T cells and oncolytic herpes virus G47Delta combination therapy against glioblastoma. Mol Ther Oncolytics 2022, 26, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.; Waseda, M.; Ishii, T.; Kaneko, M.K.; Kato, Y.; Kaneko, S. Improved anti-solid tumor response by humanized anti-podoplanin chimeric antigen receptor transduced human cytotoxic T cells in an animal model. Genes Cells 2022, 27, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Tamura-Sakaguchi, R.; Aruga, R.; Hirose, M.; Ekimoto, T.; Miyake, T.; Hizukuri, Y.; Oi, R.; Kaneko, M.K.; Kato, Y.; Akiyama, Y.; et al. Moving toward generalizable NZ-1 labeling for 3D structure determination with optimized epitope-tag insertion. Acta Crystallogr D Struct Biol 2021, 77, 645–662. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.K.; Ohishi, T.; Nakamura, T.; Inoue, H.; Takei, J.; Sano, M.; Asano, T.; Sayama, Y.; Hosono, H.; Suzuki, H.; et al. Development of Core-Fucose-Deficient Humanized and Chimeric Anti-Human Podoplanin Antibodies. Monoclon Antib Immunodiagn Immunother 2020, 39, 167–174. [Google Scholar] [CrossRef]
- Abe, S.; Kaneko, M.K.; Tsuchihashi, Y.; Izumi, T.; Ogasawara, S.; Okada, N.; Sato, C.; Tobiume, M.; Otsuka, K.; Miyamoto, L.; et al. Antitumor effect of novel anti-podoplanin antibody NZ-12 against malignant pleural mesothelioma in an orthotopic xenograft model. Cancer Sci 2016, 107, 1198–1205. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Abe, S.; Ogasawara, S.; Fujii, Y.; Yamada, S.; Murata, T.; Uchida, H.; Tahara, H.; Nishioka, Y.; Kato, Y. Chimeric Anti-Human Podoplanin Antibody NZ-12 of Lambda Light Chain Exerts Higher Antibody-Dependent Cellular Cytotoxicity and Complement-Dependent Cytotoxicity Compared with NZ-8 of Kappa Light Chain. Monoclon Antib Immunodiagn Immunother 2017, 36, 25–29. [Google Scholar] [CrossRef]
- Ito, A.; Ohta, M.; Kato, Y.; Inada, S.; Kato, T.; Nakata, S.; Yatabe, Y.; Goto, M.; Kaneda, N.; Kurita, K.; et al. A Real-Time Near-Infrared Fluorescence Imaging Method for the Detection of Oral Cancers in Mice Using an Indocyanine Green-Labeled Podoplanin Antibody. Technol Cancer Res Treat 2018, 17, 1533033818767936. [Google Scholar] [CrossRef]
- Shiina, S.; Ohno, M.; Ohka, F.; Kuramitsu, S.; Yamamichi, A.; Kato, A.; Motomura, K.; Tanahashi, K.; Yamamoto, T.; Watanabe, R.; et al. CAR T Cells Targeting Podoplanin Reduce Orthotopic Glioblastomas in Mouse Brains. Cancer Immunol Res 2016, 4, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, T.; Yoneda, K.; Mori, M.; Kanayama, M.; Kuroda, K.; Kaneko, M.K.; Kato, Y.; Tanaka, F. Detection of Circulating Tumor Cells (CTCs) in Malignant Pleural Mesothelioma (MPM) with the “Universal” CTC-Chip and An Anti-Podoplanin Antibody NZ-1.2. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Nishinaga, Y.; Sato, K.; Yasui, H.; Taki, S.; Takahashi, K.; Shimizu, M.; Endo, R.; Koike, C.; Kuramoto, N.; Nakamura, S.; et al. Targeted Phototherapy for Malignant Pleural Mesothelioma: Near-Infrared Photoimmunotherapy Targeting Podoplanin. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kaneko, M.K.; Kunita, A.; Ito, H.; Kameyama, A.; Ogasawara, S.; Matsuura, N.; Hasegawa, Y.; Suzuki-Inoue, K.; Inoue, O.; et al. Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci 2008, 99, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Vaidyanathan, G.; Kaneko, M.K.; Mishima, K.; Srivastava, N.; Chandramohan, V.; Pegram, C.; Keir, S.T.; Kuan, C.T.; Bigner, D.D.; et al. Evaluation of anti-podoplanin rat monoclonal antibody NZ-1 for targeting malignant gliomas. Nucl Med Biol 2010, 37, 785–794. [Google Scholar] [CrossRef]
- Rasmussen, S.; Rapraeger, A. Altered structure of the hybrid cell surface proteoglycan of mammary epithelial cells in response to transforming growth factor-beta. J Cell Biol 1988, 107, 1959–1967. [Google Scholar] [CrossRef]
- Meyer-Schaller, N.; Cardner, M.; Diepenbruck, M.; Saxena, M.; Tiede, S.; Lüönd, F.; Ivanek, R.; Beerenwinkel, N.; Christofori, G. A Hierarchical Regulatory Landscape during the Multiple Stages of EMT. Dev Cell 2019, 48, 539–553. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Okita, Y.; Kimura, M.; Xie, R.; Chen, C.; Shen, L.T.; Kojima, Y.; Suzuki, H.; Muratani, M.; Saitoh, M.; Semba, K.; et al. The transcription factor MAFK induces EMT and malignant progression of triple-negative breast cancer cells through its target GPNMB. Sci Signal 2017, 10. [Google Scholar] [CrossRef]
- Kellar, A.; Egan, C.; Morris, D. Preclinical Murine Models for Lung Cancer: Clinical Trial Applications. Biomed Res Int 2015, 2015, 621324. [Google Scholar] [CrossRef]
- Xin, M.; Lin, D.; Yan, N.; Li, H.; Li, J.; Huang, Z. Oxaliplatin facilitates tumor-infiltration of T cells and natural-killer cells for enhanced tumor immunotherapy in lung cancer model. Anticancer Drugs 2022, 33, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Suzuki, H.; Ohishi, T.; Asano, T.; Tanaka, T.; Yanaka, M.; Nakamura, T.; Yoshikawa, T.; Kawada, M.; Kaneko, M.K.; et al. Antitumor activities of a defucosylated anti-EpCAM monoclonal antibody in colorectal carcinoma xenograft models. Int J Mol Med 2023, 51. [Google Scholar] [CrossRef] [PubMed]
- Nanamiya, R.; Takei, J.; Ohishi, T.; Asano, T.; Tanaka, T.; Sano, M.; Nakamura, T.; Yanaka, M.; Handa, S.; Tateyama, N.; et al. Defucosylated Anti-Epidermal Growth Factor Receptor Monoclonal Antibody (134-mG(2a)-f) Exerts Antitumor Activities in Mouse Xenograft Models of Canine Osteosarcoma. Monoclon Antib Immunodiagn Immunother 2022, 41, 1–7. [Google Scholar] [CrossRef]
- Kawabata, H.; Suzuki, H.; Ohishi, T.; Kawada, M.; Kaneko, M.K.; Kato, Y. A Defucosylated Mouse Anti-CD10 Monoclonal Antibody (31-mG(2a)-f) Exerts Antitumor Activity in a Mouse Xenograft Model of CD10-Overexpressed Tumors. Monoclon Antib Immunodiagn Immunother 2022, 41, 59–66. [Google Scholar] [CrossRef]
- Kawabata, H.; Ohishi, T.; Suzuki, H.; Asano, T.; Kawada, M.; Suzuki, H.; Kaneko, M.K.; Kato, Y. A Defucosylated Mouse Anti-CD10 Monoclonal Antibody (31-mG(2a)-f) Exerts Antitumor Activity in a Mouse Xenograft Model of Renal Cell Cancers. Monoclon Antib Immunodiagn Immunother 2022. [Google Scholar] [CrossRef]
- Asano, T.; Tanaka, T.; Suzuki, H.; Li, G.; Ohishi, T.; Kawada, M.; Yoshikawa, T.; Kaneko, M.K.; Kato, Y. A Defucosylated Anti-EpCAM Monoclonal Antibody (EpMab-37-mG(2a)-f) Exerts Antitumor Activity in Xenograft Model. Antibodies (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Tateyama, N.; Nanamiya, R.; Ohishi, T.; Takei, J.; Nakamura, T.; Yanaka, M.; Hosono, H.; Saito, M.; Asano, T.; Tanaka, T.; et al. Defucosylated Anti-Epidermal Growth Factor Receptor Monoclonal Antibody 134-mG(2a)-f Exerts Antitumor Activities in Mouse Xenograft Models of Dog Epidermal Growth Factor Receptor-Overexpressed Cells. Monoclon Antib Immunodiagn Immunother 2021, 40, 177–183. [Google Scholar] [CrossRef]
- Takei, J.; Ohishi, T.; Kaneko, M.K.; Harada, H.; Kawada, M.; Kato, Y. A defucosylated anti-PD-L1 monoclonal antibody 13-mG(2a)-f exerts antitumor effects in mouse xenograft models of oral squamous cell carcinoma. Biochem Biophys Rep 2020, 24, 100801. [Google Scholar] [CrossRef]
- Takei, J.; Kaneko, M.K.; Ohishi, T.; Hosono, H.; Nakamura, T.; Yanaka, M.; Sano, M.; Asano, T.; Sayama, Y.; Kawada, M.; et al. A defucosylated antiCD44 monoclonal antibody 5mG2af exerts antitumor effects in mouse xenograft models of oral squamous cell carcinoma. Oncol Rep 2020, 44, 1949–1960. [Google Scholar] [CrossRef]
- Asano, T.; Kaneko, M.K.; Takei, J.; Tateyama, N.; Kato, Y. Epitope Mapping of the Anti-CD44 Monoclonal Antibody (C44Mab-46) Using the REMAP Method. Monoclon Antib Immunodiagn Immunother 2021, 40, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Kaneko, M.K.; Aasano, T.; Kato, Y. Epitope Mapping of an Antihuman EGFR Monoclonal Antibody (EMab-134) Using the REMAP Method. Monoclon Antib Immunodiagn Immunother 2021, 40, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Kaneko, M.K.; Kato, Y. Development of a Novel Epitope Mapping System: RIEDL Insertion for Epitope Mapping Method. Monoclon Antib Immunodiagn Immunother 2021, 40, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Nanamiya, R.; Sano, M.; Asano, T.; Yanaka, M.; Nakamura, T.; Saito, M.; Tanaka, T.; Hosono, H.; Tateyama, N.; Kaneko, M.K.; et al. Epitope Mapping of an Anti-Human Epidermal Growth Factor Receptor Monoclonal Antibody (EMab-51) Using the RIEDL Insertion for Epitope Mapping Method. Monoclon Antib Immunodiagn Immunother 2021, 40, 149–155. [Google Scholar] [CrossRef]
Figure 1.
The production of anti-mEGFR mAb, EMab-300. (A) LN229/mEGFR cells were immunized into a Sprague–Dawley rat. (B) The spleen cells were fused with P3U1 cells. (C) To select anti-mEGFR mAb-producing hybridomas, the supernatants were screened by flow cytometry using CHO-K1 and CHO/mEGFR cells. (D) After limiting dilution, an anti-mEGFR mAb, EMab-300 was finally established.
Figure 1.
The production of anti-mEGFR mAb, EMab-300. (A) LN229/mEGFR cells were immunized into a Sprague–Dawley rat. (B) The spleen cells were fused with P3U1 cells. (C) To select anti-mEGFR mAb-producing hybridomas, the supernatants were screened by flow cytometry using CHO-K1 and CHO/mEGFR cells. (D) After limiting dilution, an anti-mEGFR mAb, EMab-300 was finally established.

Figure 2.
Flow cytometry to mEGFR-expressing cells using EMab-300. CHO/mEGFR (A), CHO-K1 (B), NMuMG (C), and Lewis lung carcinoma (D) cells were treated with 0.01–10 µg/mL of EMab-300, followed by treatment with anti-rat IgG conjugated with Alexa Fluor 488. The black line represents the negative control.
Figure 2.
Flow cytometry to mEGFR-expressing cells using EMab-300. CHO/mEGFR (A), CHO-K1 (B), NMuMG (C), and Lewis lung carcinoma (D) cells were treated with 0.01–10 µg/mL of EMab-300, followed by treatment with anti-rat IgG conjugated with Alexa Fluor 488. The black line represents the negative control.
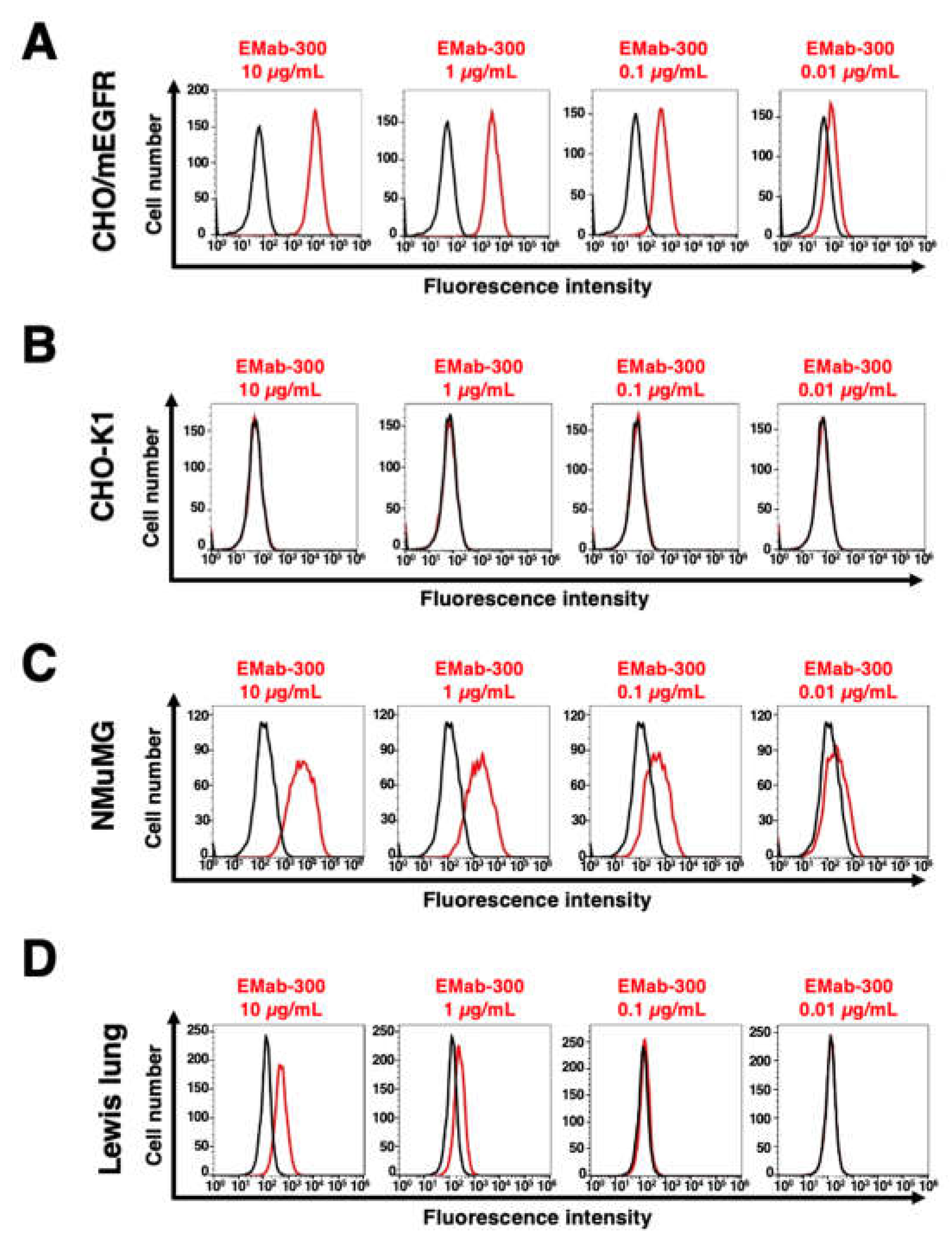
Figure 3.
The binding affinity of EMab-300. CHO/mEGFR (A) and NMuMG (B) cells were suspended in serially diluted EMab-300 as described in “Materials and methods”. The cells were treated with anti-rat IgG conjugated with Alexa Fluor 488. The fluorescence data were subsequently collected using the SA3800 Cell Analyzer, followed by the calculation of the dissociation constant (KD) by GraphPad PRISM 8.
Figure 3.
The binding affinity of EMab-300. CHO/mEGFR (A) and NMuMG (B) cells were suspended in serially diluted EMab-300 as described in “Materials and methods”. The cells were treated with anti-rat IgG conjugated with Alexa Fluor 488. The fluorescence data were subsequently collected using the SA3800 Cell Analyzer, followed by the calculation of the dissociation constant (KD) by GraphPad PRISM 8.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



