
Submitted:
23 September 2023
Posted:
25 September 2023
You are already at the latest version
Abstract
Prostate cancer (PCa) is a major cause of cancer-related mortality worldwide, with a rising incidence observed over the years. The androgen receptor (AR) signaling pathway plays a pivotal role in male development and maintaining masculine characteristics. Dysregulation of AR signaling in prostate cancer can lead to disease progression and resistance to standard therapies. Understanding the intricate regulation and function of AR in both healthy and diseased states is crucial for developing effective treatment strategies. This review comprehensively explores the role of androgen receptors in prostate cancer susceptibility, disease progression, and treatment response by analyzing recent literature. An extensive search of peer-reviewed publications in major databases, including PubMed, Scopus, and Web of Science, was conducted using specific keywords related to androgen receptor, prostate cancer, disease progression, and treatment resistance. Relevant conference abstracts and clinical trial reports were also included. The review presents an overview of the role of androgen receptors in prostate cancer initiation, progression, and treatment resistance. It highlights emerging biomarkers associated with AR signaling dysregulation and their potential utility for early detection and personalized treatment approaches. Additionally, recent advances in targeting the AR pathway for novel therapeutic strategies to improve patient outcomes and overcome treatment resistance in advanced prostate cancer are discussed. The findings contribute to a comprehensive understanding of the AR signaling pathway in prostate cancer and offer insights into its multifaceted role in disease development and treatment response. They may pave the way for innovative therapeutic interventions and precision medicine approaches based on specific AR signaling profiles, enhancing patient care and reducing the burden of this lethal disease.
Keywords:
Prostate cancer
; androgen receptor
; AR signaling pathway
; disease progression
; therapeutic re-sistance
; personalized treatment
; androgen deprivation therapy
; AR-targeted therapies
1. Introduction
Uncontrolled cellular proliferation in the body is a hallmark of the disease known as cancer [1]. Prostate cancer (PCa) is the type of cancer that affects the prostate gland, a tiny organ with a walnut-like shape in the male reproductive system [2]. It is among men’s most commonly diagnosed cancers, and it remains the reason for cancer-related deaths globally [3]. The frequency of PCa has risen over the past few years, with an estimated incidence rate increase of 1 in every 52 males aged 50 to 59 years developing the disease in their lifetime [4,5]. Age increases the risk of PCa, with men over fifty being diagnosed with the majority of cases. More than 1.4 million new cases of prostate cancer were found globally in 2020 [6]. Men from Europe, Latin America, the Caribbean, and Northern America, have high incidence rates but low mortality compared to those from Africa and Asia which have shown lower incidence and higher PCa mortality rates [7]. A lot of factors could be responsible for the disparities in incidence and mortality of PCa according to geographic and racial distribution [8]. The incidence of PCa varies widely across different populations and regions, with higher incidence rates and low mortality being observed in Western countries compared to Asian and African populations [6]. However, this in contrast to African countries including Nigeria, that has low incidence but higher mortality. This disparities could be due to environment, variations in genetic factors and improved access to health care and increased awareness of the disease [8]. Despite the low incidence rate which could be attributed to poor information system and data management, it is the most common cause of cancer related death in Nigerian men.
To gain a better understanding of the natural history and true prevalence of PCa in Nigeria and Africa as a whole, more extensive studies are needed. One method for looking for early indications of PCa is the Prostate Specific Antigen (PSA) test. Over the years PSA test has been used to diagnose PCa although not very reliable because it is not specific. PSA levels can rise as a result of prostate cancer. However, many non-cancerous illnesses can also raise PSA levels. Therefore, it cannot offer accurate diagnostic data regarding the prostate’s health. A digital rectal exam is an additional screening procedure that is frequently performed in conjunction to a PSA test. PCa may be indicated by high PSA levels, but many other conditions, such as an enlarging or inflamed prostate, can also cause elevated PSA levels. As a result, research has concentrated on finding precise and more trustworthy biomarkers for PCa early detection. Technological advancements have made new and inventive techniques for detecting and treating PCa possible. The advancement of complex imaging methods, such as magnetic resonance imaging (MRI), has increased the precision of diagnosis and enabled earlier disease detection [9]. It is crucial to remember that the prevalence of PCa is predicted to rise as a result of both population aging and economic growth. Three known risk factors for PCa are getting older, being black, and having a family history of the condition.
The complex process of cancer progression is influenced by numerous genetic and molecular abnormalities; people with tumors have been shown to have more genetic mutations [10]. PCa, among other types of cancer, develops and progresses in part due to the androgen receptor (AR), a transcription factor [11]. In PCa, AR is expressed more frequently, and its activity is crucial for the growth and survival of prostate cancer cells. Numerous studies have demonstrated that inhibiting AR can successfully stop prostate cancer cells from growing [12,13]. PCa patients frequently receive androgen deprivation therapy (ADT), which lowers the levels of androgens like testosterone that bind to and activate AR [14]. However, many PCa cells eventually become resistant to ADT, and leading to castration resistant prostate cancer (CRPC) is often associated with increased AR activity (Crawford et al., 2018). Recent research has focused on developing new strategies to target AR in PCa, including the development of AR antagonists that can bind to and inhibit the activity of AR, and the inhibition of AR signaling pathways [15]. Additionally, studies have shown that AR can interact with other signaling pathways, such as the PI3K/Akt pathway, and targeting these interactions may also be a promising strategy for treating PCa. In this review, we summarized the current understanding of the role of AR in PCa susceptibility, diagnosis and treatment to highlight potential therapeutic strategies for targeting AR in PCa.
2. Methodology and Search Strategy
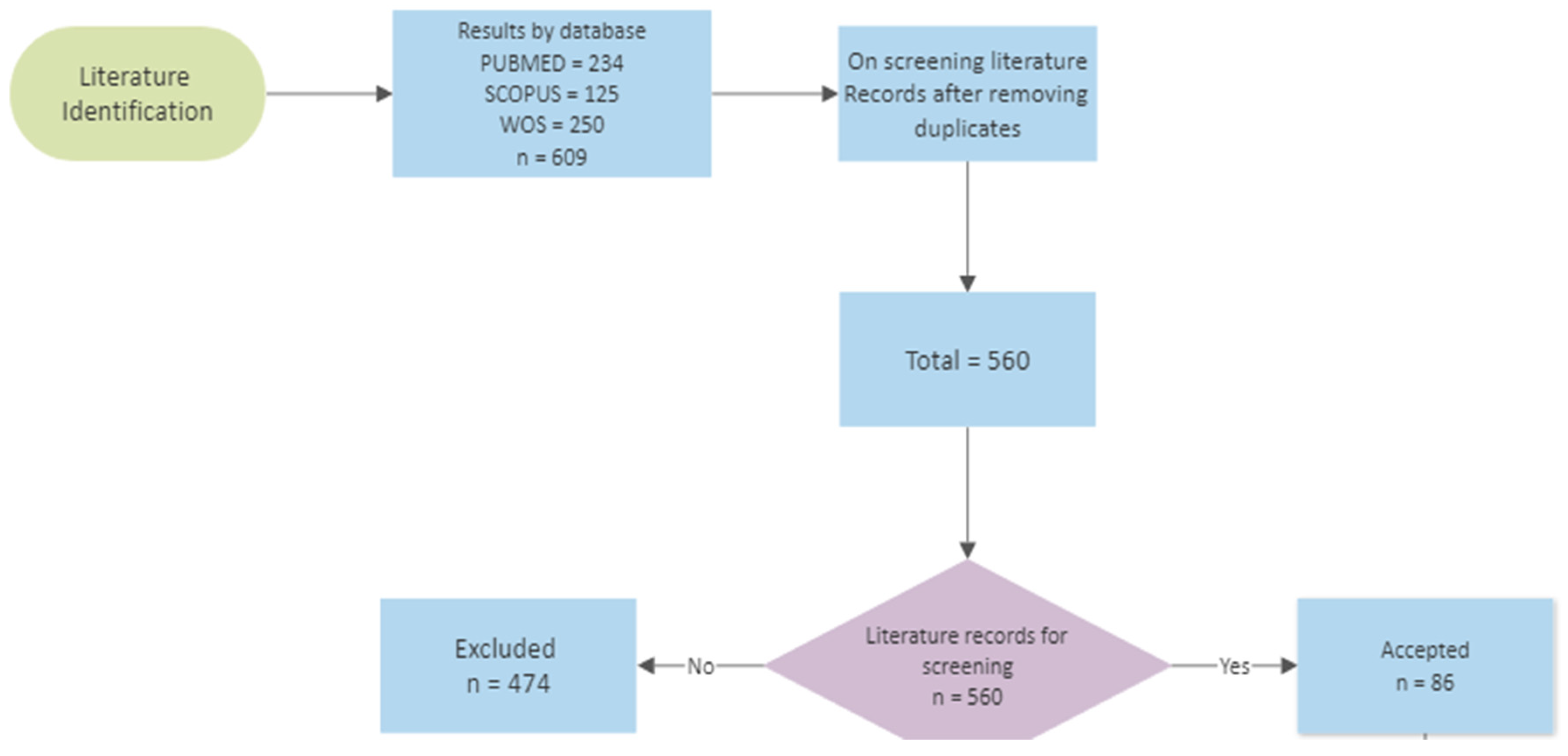
We conducted a comprehensive systematic search of relevant literature to gather articles for this review. Our search was executed across multiple reputable databases, including PubMed, Scopus, WOS, and Google Scholar. The search strategy involved using a combination of targeted search terms. Specifically, we employed the search terms "androgen receptors" or "androgen deprivation therapy" and "prostate cancer susceptibility" or "prostate cancer progression" or "prostate cancer treatment" to ensure a comprehensive retrieval of articles up until February 2023. It’s worth noting that the search terms were slightly adapted to match the unique features of each database. The retrieved articles underwent a rigorous screening process, which was carried out independently by two reviewers (Reviewer A and Reviewer B). Initially, titles and abstracts were screened to assess their relevance. Subsequently, the full-text versions of the selected articles were scrutinized in detail (see Figure 1 for a flowchart depicting the selection process). In cases where discrepancies arose between the two reviewers, a third reviewer (Reviewer C) was consulted to resolve any disagreements through consensus discussions. To be included in this review, articles were required to meet specific criteria. They had to provide quantitative insights into the functions of androgen receptors in prostate cancer. Additionally, the studies needed to have been conducted on samples collected within the context of prostate cancer susceptibility, progression, or treatment. Conversely, articles falling under the categories of methods, non-English publications, and studies involving animal models were excluded from consideration. Notably, thesis and dissertation works adhering to our inclusion criteria were included in our analysis. To facilitate our presentation and comparison of findings, we categorized the collected samples into distinct groups: (1) assessments related to prostate cancer susceptibility, (2) investigations into prostate cancer progression, and (3) studies focusing on prostate cancer treatment. Moreover, we ensured to give the basic biology of androgen and androgen receptors signalling pathways. Furthermore, the research publications pertaining to androgen receptors in prostate cancer was visually represented through the creation of a flow chart using data visualization tools like cloud smartdraw and Biorender plartforms.
3. Results and Discussion
The culmination of our systematic review underscores the pivotal role of androgen receptors (AR) in the susceptibility, progression, and treatment of prostate cancer. A comprehensive analysis of the literature reveals a multitude of studies that converge on the significance of AR in shaping the dynamics of this prevalent malignancy.
Figure 1.
Study flow chart.
3.1. Brief History of Androgen Receptor (AR)
The testis and adrenal glands produce androgens, a group of steroid hormones that have a role in puberty’s secondary sexual characteristics as well as the development of the male sex in a healthy foetus [16]. Androgens are a type of steroid hormone that is derived from cholesterol with a 19-carbon structure. Moreover, androgens are the precursors for the production of female sex hormones, called estrogens, which are formed from androgens through various enzymatic processes such as hydroxylation, elimination, and aromatization, with the help of an enzyme called aromatase. Androgens serve a crucial part in the survival and growth of organs of the male reproductive system, including the prostate, through the regulation of gene expression levels. They do this largely through AR, which is a transcription factor that is ligand-dependent [17]. The earliest proof of androgen receptors came from research on the effects of androgens on the reproductive system conducted in the 1930s and 1940s. Researchers began investigating the androgens’ mode of action in the 1950s and 1960s. In 1958, a group at the University of Illinois under the direction of Paul Zamboni discovered that androgens encourage the development of the prostate gland in rats. As a result, it was postulated that androgens exert their effects via interacting with certain cell receptors. Androgen receptors were first identified from rat prostate tissue in 1968 by a team at the University of Chicago under the direction of Elwood Jensen. The receptors were discovered to be androgen-specific and to have a strong affinity for testosterone [18]. This finding opened the door for more investigation and offered compelling evidence for the existence of AR.
The AR is found in a variety of target tissues, and its activity and levels change by the initiation of certain cellular processes (e.g., malignant transformation, sexual development) [19]. Three researchers independently identified and characterised the AR in the late 1960s: Ian Mainwaring, Nicholas Bruchovsky, and Shutsung Liao [20]. Through a chemical screen, the first anti-androgen, cyproterone, was unintentionally found. Cyproterone acetate (CPA), which displayed improved anti-androgenic activity, was created by adding an acetate group later on [21]. The need for non-steroidal anti-androgens was sparked by the limits of CPA, particularly its link to a severe sexual dysfunction that is comparable to surgical castration [22].
3.2. AR Family Members
Three receptors make up the AR family: the androgen receptor (AR), the glucocorticoid receptor (GR), and the progesterone receptor (PR). These receptors, which are members of the nuclear receptor superfamily, control how genes are expressed in response to the binding of hormones [23]. A transcription factor known as androgen receptor (AR) controls how genes are expressed in response to androgens like testosterone and dihydrotestosterone (DHT). The most bioactive AR ligand, DHT, is mostly produced from circulating testosterone that is intracellularly transformed into its more active metabolite. It is activated by androgens, its cognate ligands [25,24]. The Progesterone Receptor (PR) is a transcription factor belonging to the nuclear receptors superfamily. It can activate and inhibit the transcription of its target genes and is activated by the hormone progesterone. The PR molecule has domains for ligand binding, DNA binding, and transcription activation. In the reproductive system of females which includes the ovaries, uterus, and mammary glands, PR is necessary for ovulation, implantation of the fertilised egg, and the upkeep of pregnancy [26]. In the ovary, PR is only expressed in the granulosa cells of preovulatory follicles [27]. Furthermore, PR determines immune function, bone density, mammary gland development, and the onset and progression of breast and ovarian cancer. Breast cancer can be classified as either estrogen-receptor-positive (ER+), progesterone-receptor-positive (PR+), or ER+/PR+. Mammary gland growth is directly tied to the actions of progesterone and oestrogen on breast tissue. As a result, the activation of these receptors in breast tissue can boost cell proliferation in the mammary gland by raising the proportion of breast cells in the G2/M phase, which raises the risk of breast cancer [28]. Glucocorticoid receptor (GR), which is activated by the hormone cortisol, mediates the effects of glucocorticoids in cells. It belongs to the nuclear receptor (NR) family of intracellular receptors together with the oestrogen receptor (ER), PR, AR, and the mineralocorticoid receptor (MR) that lacks a recognised ligand [29]. The hypothalamic-pituitary-adrenal (HPA) axis, the body’s stress response system, is controlled by the GR, which is also important in controlling inflammation, immunological response, and glucose metabolism. Additionally, GR contributes to the onset and progression of numerous illnesses, such as autoimmune diseases, rheumatoid arthritis, and asthma [30]. The AR, GR, and, PR play a role in many physiological processes, including bone density, immunological function, sexual differentiation, and reproductive function. Several diseases, including cancer and autoimmune disorders, have been linked to the onset and progression of deregulation of these receptors.
3.3. AR Structure
Androgens exert their effects through the binding to Androgen receptor (AR) [31,32]. AR belongs to the superfamily of nuclear hormone and steroid receptors, including glucocorticoids, mineralocorticoids, progesterone, oestrogens, and vitamin D. Steroid receptors, including the AR, have three functional domains: an NH2-terminal domain (NTD) that contains the transcriptional activation function 1 (AF-1), a central DNA-binding domain (DBD) linked to hinge region, and a COOH-terminal ligand-binding domain (LBD), which is linked to the DBD by a hinge region and contains the transcriptional activation function 2 (AF-2) [33,34]. The AR gene is situated on Xq11-12 and creates a protein that weighs 110 kDa and has 920 amino acids [35]. The AR gene consists of eight exons, with exon 1 encoding the NTD, exons 2-3 encoding the DBD, exon 4 encoding the HR, and exons 5-8 encoding the LBD as shown in (see Figure 2). This ligand-dependent transcription factor controls the expression of genes that are involved in the growth and differentiation of the prostate gland [36]. Family members differ in the amino-terminal domain and the hinge region that joins the core DBD to the C-terminal ligand-binding domain [37].
3.4.1. The NH2-terminal domain
About half of the receptor’s 919 amino acid core sequence is taken up by the NTD, which ranges from amino acids 1 to 559 [38]. The AR-NTD differs most from other members of the steroid receptor family in terms of amino acid variability, sharing less than 15% of its amino acid sequence with those of the other steroid receptor-NTDs. It produces the AF-1, which has the Tau1 and Tau5 transcriptional activation units (Tau). When the AR-LBD is removed, the Tau5 area (amino acids 360–528) exhibits constitutive transcription of the AR-NTD without the need for ligands, whereas the Tau1 region (amino acids 141-338) is necessary for ligand-dependent transactivation of the AR [35]. Tau-5 is a signal-dependent transactivation site, in contrast to Tau-1, and is activated by signaling events from the protein kinase C related kinase (PRK-1). Other steroid receptor-NTDs do not have the three distinct homo-polymeric amino acid repeats found in the AR-NTD. There are three types of repeats: poly-glutamine (poly-Q), poly-proline (poly-P), and poly-glycine (poly-G). The poly-P tract is 9 residues long and begins at amino acid 327. The poly-glycine tract is 24 residues long and begins at amino acid 449. The poly-Q tract is found at amino acid 59 and has a usual range of 17–29 residues. Although the particular relevance of these three repetitions is unknown, the poly-Q tract has been the subject of intense study to understand its involvement in AR activity. It has been demonstrated that the length of the poly-Q tract and AR transcriptional activity are inversely correlate. The AR-poly-Q tract’s length may also affect how directly AR interacts with its co-regulatory proteins, which control AR-mediated transcription. Shortening the poly-Q tract of AR to 17 amino acids or less, as was described earlier, has been linked to an increased risk of prostate cancer. The AR-NTD is appealing for AR-specific protein interactions due to its distinctive sequences and characteristics, which may be crucial for guiding AR-specific responses. Finding novel protein partners that interact with the AR-NTD may help to clarify the process by which cells are able to respond to androgenic ligands in an AR-specific manner. In the reverse yeast two-hybrid system (RTA), our group has discovered a number of novel AR-NTD interacting proteins by using the N-terminus of AR as bait. An example of these protein is the TATA binding protein Associated Factor 1 (TAF1).
3.4.2. The Central DNA-binding domain and hinge region
The DBD and hinge region of the AR are respectively comprised of amino acids 560-623 and 624-676. These areas perform a variety of tasks, such as dimerization of active AR molecules, nuclear localization of activated receptors, and binding to DNA at consensus sequences in the promoter/enhancer region of AR-regulated genes [39]. Moreover, the DBD of AR interacts with potential transcriptional co-regulators as well as proteins that make up the basic transcriptional apparatus. It is important for the dimerization of AR and the binding of dimerized AR to certain DNA patterns. The cysteine residues in this domain, which promote the development of two zinc finger motifs, contribute to these DBD activities [35]. Two conserved zinc finger motifs in the DBD of the AR and other steroid receptors interact with DNA regulatory regions. These DNA sequences in the promoters of androgen-regulated genes are referred to as androgen response elements (ARE) for AR. Inverted palindromic sequences with two half-sites and a 3-nucleotide spacer (5’-GGA/TACAnnnTGTTCT-3’) make up the ARE. Whereas the second zinc finger of the DBD stabilizes receptor-DNA connections, the first NH2-terminal zinc finger of the DBD is in charge of detecting ARE sequences and selectively binding to AREs in the main groove of DNA. The AR-second DBD’s zinc finger may have an impact on how well the receptor binds to AR-specific ARE. Nuclear localization sequence (NLS) (amino acids 613-633) found in the hinge region of the AR directs the activated receptor to the nucleus. The bipartite NLS is made up of two basic amino acid clusters spaced apart by ten amino acids. Because of the disruption caused by Lys-to-Ala mutations of these residues, the hinge region’s lysine residues (K630, 632, and 633) that are acetylated during receptor activation are thought to be crucial for nuclear translocation [40].
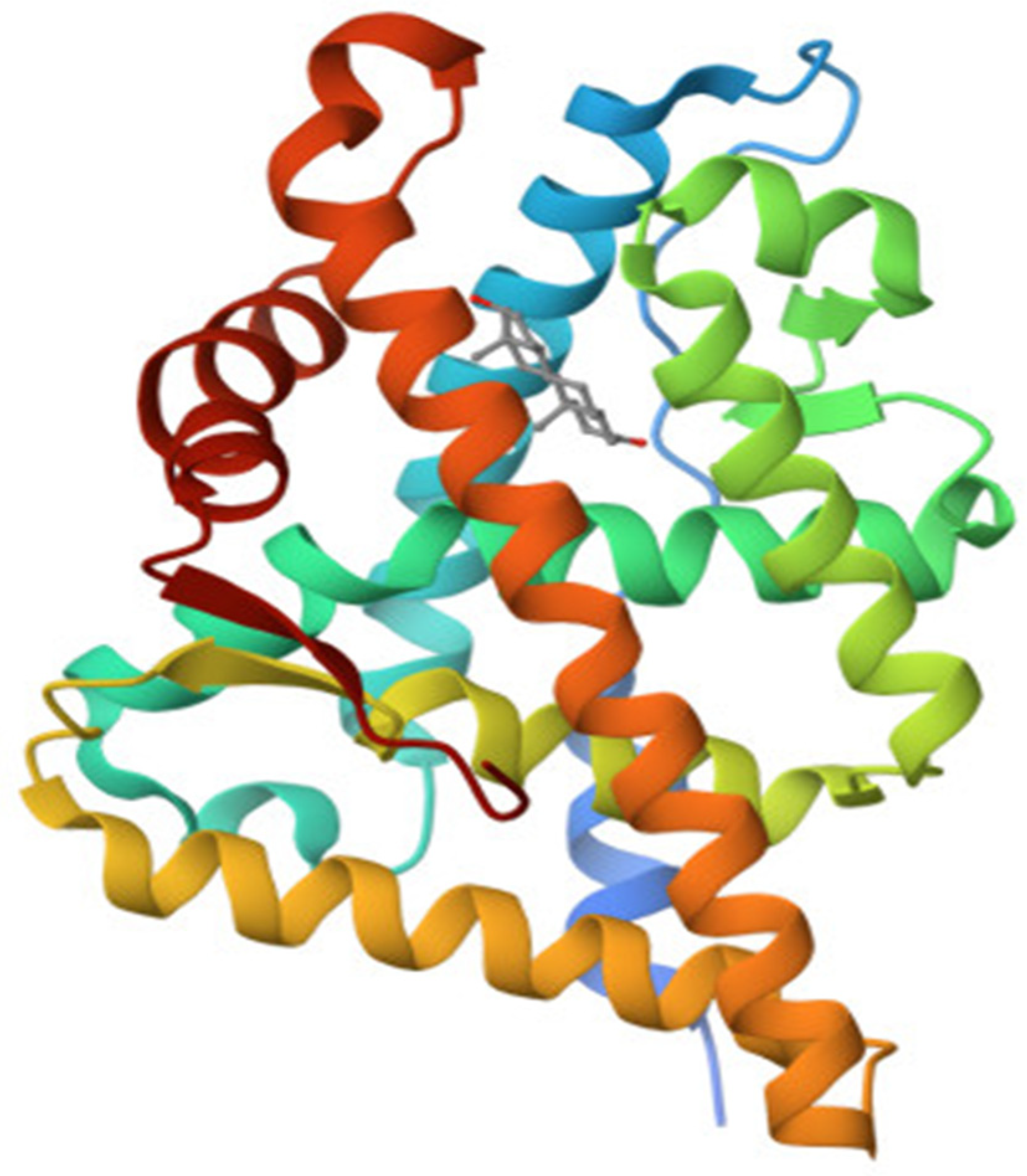
Figure 3.
Structure of AR bound to DHT.
3.4.3. The Ligand Binding Domain
The ligand binding domain (LBD) of AR is a region within the AR protein that is responsible for binding to androgens, which are hormones that play a key role in the development and maintenance of male characteristics [41]. The LBD is located at the C-terminus of the AR protein and is composed of several structural elements, including 12 alpha-helices and several beta-strands. The LBD of AR, which consists of amino acids 616-919, includes a hydrophobic pocket that accepts androgenic ligands such as DHT and testosterone. The LBD is well conserved among different species such as human, rat, and mouse, with degrees of homology ranging from 20-55% with LBDs of other members of the steroid receptor family. When an androgen hormone binds to the LBD of the AR, it causes a conformational change in the receptor, which allows it to translocate to the nucleus of the cell and bind to androgen response elements (AREs) on DNA. This binding leads to the activation of target genes involved in the regulation of a wide range of physiological processes, including male sexual development, muscle growth, and bone density. The AR-LBD is particularly critical for prostate cancer because it is the main target of current androgen deprivation therapies. Despite the availability of potent androgen antagonists in clinics, mutations in the AR-LBD can result in the improper activation of AR by non-androgenic substances, leading to ligand-binding promiscuity. Over 30% of prostate cancers possess AR mutations, and several AR variants have been discovered that lack receptor specificity in the absence of traditional ligands. The majority of mutations in the AR affect the ligand binding pocket and are found in three primary regions of the LBD, specifically amino acids 670-678, 710-730, and 874-910 [37]. The most frequently observed variants in tumors are T877A, T877S, and H874Y. The T877A mutation is particularly well-known as it is found in the LNCaP human PCa cell line, as well as cases of advanced prostate cancer. Overall, these mutations make the receptor more sensitive to adrenal androgens or other steroid hormones compared to the wild type AR. This may be due to the recruitment of various co-activators, which enable the AR to bind other steroid ligands, and allow antagonists to act as agonists to activate the AR in an androgen-depleted environment [36].
4. Physiological Role of AR and Signal Tranduction
The androgen receptor (AR) plays a vital role in the development and maintenance of male characteristics, including the development of male reproductive organs, the growth of muscle and bone mass, and the deepening of the voice during puberty. However, its role is not limited to males as the AR is also present and active in females, although to a lesser degree. In addition to its role in sexual development, the AR has been shown to play a role in the growth and function of other tissues, such as the skin, hair follicles, and brain. It has also been implicated in the development of certain diseases, including prostate cancer, and is the target of antiandrogen therapy in the treatment of prostate cancer. Furthermore, the AR has been found to be involved in the regulation of energy metabolism and the immune system, suggesting that it may play a broader role in overall health and disease beyond its role in sexual development and function.
The androgen receptor (AR) signaling pathway is a complex process that involves multiple steps and regulatory factors. The ligand-free AR is cytoplasmic in location, bound by a number of cochaperones such as HSP90 and HSP70/HSC70, which keep the protein in a ligand-binding conformation and prevent it from degradation. When androgens bind to the AR, the receptor undergoes a conformational change, leading to its translocation to the nucleus. AR changes conformation when activated and promotes NTD/LBD interactions and the binding to AR coregulators, for the best transcriptional response [[43]. The AR subsequently attaches to motor and transport proteins, such as dynein and importin-α/-β, which recognize the nuclear localization signal of the AR and facilitate the AR complex’s translocation to the nucleus. Once in the nucleus, the AR dimerizes with another AR molecule and binds to specific regions of DNA known as androgen response elements (AREs), which are located in the promoters of androgen-responsive genes. The binding of the AR to AREs initiates a cascade of events that result in the transcription of androgen-responsive genes. This process is regulated by several co-regulators, including co-activators and co-repressors, which can modulate the activity of the AR by either enhancing or inhibiting its transcriptional activity. AR signaling is activated by binding of androgens, such as testosterone and dihydrotestosterone, to the receptor. This triggers a cascade of events leading to the regulation of target gene expression and subsequent cellular responses [44,45,31]. However, AR signaling is also subject to crosstalk with other signaling pathways, which can modulate its activity and contribute to prostate cancer progression especially observed in castration-resistant prostate cancer cases and androgen-independent prostate cancer cases [44]. Also, Genes involved in preventing apoptosis and promoting cellular proliferation are subject to transcriptional activity modulation by the AR [44].
4.2. AR Ligand—Androgens
Androgens are steroid hormones that exert their effects through the Androgen receptor (AR). They include testosterone and dihydrotestosterone (DHT), dehydroepiandrosterone (DHEA), and androstenedione, among others. Testosterone and DHT are the two most significant androgens involved with prostate cancer. Each androgen, which functions via the androgen receptor (AR), has a distinct function during the development of male sexual differentiation. Both androgens interact differently with the androgen receptor. In comparison to DHT, testosterone has a two-fold reduced affinity for the androgen receptor. Androgens are synthesized from cholesterol in the human body. The process of turning cholesterol into testosterone occurs in a number of distinct steps, the first of which is the transfer of cholesterol from the outer to inner mitochondral membrane by the steroidogenic acute regulatory protein and the subsequent side chain cleavage of cholesterol by the enzyme P450 side-chain-cleavage. This conversion, which results in the creation of pregnenolone, is a rate-limiting step in the biosynthesis of testosterone. Several enzymes, such as 17-hydroxylase, 3-hydroxysteroid dehydrogenase, and 3-hydroxysteroid dehydrogenase, are needed for subsequent processes (Figure 4).
In males, the testes’ Leydig cells serve as the body’s main source of androgens like testosterone [46]. These cells respond to the pituitary gland’s luteinizing hormone (LH), which stimulates them to produce and secrete testosterone. However, the adrenal cortex, which is part of the adrenal gland located on top of the kidneys, also contributes to the production of androgens in males. The adrenal cortex produces a precursor hormone called dehydroepiandrosterone (DHEA), which may be transform to testosterone in peripheral tissues such as testes [46]. The hypothalamic/pituitary/gonadal axis then directs androgens to diffusely penetrate the cytoplasm of target organs like the prostate and skin via the bloodstream. Androgens are then transported via the bloodstream to target organs like the prostate and skin, where they diffusely penetrate the cytoplasm, under the direction of the hypothalamic-pituitary-gonadal axis. Hydrophobic molecules, which are not soluble in water, include androgens like testosterone and dihydrotestosterone (DHT). They attach to particular transport proteins, such as albumin and sex hormone-binding globulin (SHBG), in order to move through the bloodstream [38]. The majority of circulating androgens (approximately 98%) are bound to these transport proteins, leaving only a small percentage (approximately 2%) in the free, unbound state. This bound fraction of androgens is reversible and can be released from the protein binding sites as needed to exert their biological effects in target tissues. The binding of androgens to transport proteins is an important mechanism for regulating their distribution and availability in the body, and alterations in the levels of these transport proteins can have significant effects on androgen bioactivity.
Testosterone and its metabolite DHT play important roles in the development and differentiation of male reproductive structures. The Wolffian duct-derived structures such as the epididymides, vasa deferentia, seminal vesicles, and ejaculatory ducts, are all androgen-sensitive tissues that respond to testosterone during fetal development and at puberty [47,48]. However, the effects of testosterone and DHT are not the same in all androgen-sensitive tissues. The androgen receptor interacts differently with each androgen, with DHT binding to the receptor with higher affinity than testosterone [38]. For example, the prostate gland, scrotum, urethra, and penis are all androgen-sensitive tissues that respond to DHT, not testosterone.
4.3. Prostate Cancer Susceptibility and AR
While lifestyle and environmental factors play a role in its development, genetics also plays a significant role in PCa susceptibility [49]. Genetics plays key role in the development of PCa and has potential implications for its prevention, diagnosis, and treatment. Age, ethnic background, and family history are the three main factors that increase the chance of developing prostate cancer [50]. Current epidemiological studies indicate that the development of prostate cancer is strongly associated with hereditary variables, making it one of the most prevalent heritable malignancies. When compared to families without a history of PCa, those with heritable ties to men who have been diagnosed with the disease have a 2-3-fold increased risk of developing PCa. The estimated percentage of hereditary factors contributing to PCa is approximately 5-15% [51]. Genetic mutations or alterations can occur in several genes that play a role in PCa development [8]. Mutation of mismatch repair genes (MMR, MLH1, MSH2, MSH6, and PMS2) and homologous recombination genes (BRCA1/2, ATM, PALB2, and CHEK2) are currently the most consistently associated genes with PCa susceptibility. Research have verified their link to a higher incidence of PC alongside other genes such as HOXB13, BRP1, and NSB1 although recommended for further research. HOXB13 mutation is related to high risk of PCa in a more recent study [51]. Additionally, studies have identified other genetic variations that are associated with an increased risk of PCa, such as mutations in the BRCA1 and BRCA2 genes. Mutations in this gene have been associated with an increased risk of prostate cancer. Another gene known to play a role in prostate cancer susceptibility is the BRCA1 and BRCA2 genes [52]. These genes are commonly associated with breast and ovarian cancer, but have also been linked to an increased risk of prostate cancer. Other genes, such as the RNASEL gene and the MSR1 gene, have also been implicated in PCa development [53].
The well-known androgen receptor (AR) gene, which regulates the activity of hormones that promote prostate cell growth is at the center of prostate cancer progression. The androgen receptor protein is made according to instructions from the AR gene [54]. . Studies have shown that genetic variations in the AR gene are associated with an increased risk of PCa. These variations can alter the function of AR and lead to an increased sensitivity to androgens, which can promote the development of PCa. Studies of polymorphisms in androgen-related genes have shown that the variation in the androgen receptor impacts the risk of prostate cancer [55]. A polymorphism sequence of CAG repeats encoding polyglutamine can be found in the first exon of AR, which codes for the N-terminal domain important for transactivational regulation. There are a variety of racial or ethnic group discrepancies as a result of the mean repeat number of CAGs in white people being longer than in African-Americans [56]. Longer CAG repeats are linked to androgen insensitivity syndrome, while shorter CAG repeats are correlated with enhanced AR transcriptional activity. Men with shorter CAG repeats had a slightly higher chance of developing prostate cancer, according to epidemiological studies on prostate cancer and CAG repeat length in the AR gene. The understanding of the genetic basis of PCa has led to the development of genetic testing for individuals at high risk for the disease [57]. This testing can help identify individuals who may have an inherited risk for prostate cancer, and enable them to take steps to reduce their risk, such as increased screening and lifestyle modifications.
4.4. Role of Androgen Receptor in PCa
AR is a critical player in the development and progression of PCa (Shaffer et al., 2004). In healthy prostate tissue, AR signalling is tightly regulated, and androgens stimulate the growth and differentiation of prostate epithelial cells. However, in PCa, AR signalling becomes dysregulated, and the cancer cells become more dependent on androgen signalling for their growth and survival. This is why ADT, which reduces the levels of androgens in the body, is often used to treat PCa. AR is expressed in prostate cells, and androgen binds to it, triggering a cascade of cellular events that lead to cell growth, proliferation, and survival. Androgen acts on prostate normal or cancer cells through its receptor AR (Zhang et al., 2022). The biochemical activity of androgen is started by testosterone or DHT attaching to its specific receptor, the AR, activating it (Figure 5).
When unattached from its ligand, AR is found in the cytoplasm as a component of an inactive complex that also contains the heat shock proteins HSP40, HSP70, and HSP90 and other chaperone proteins to protect the receptor against degradation [58]. These proteins play a crucial function in maintaining an accessible AR shape and preventing premature AR degradation. The AR undergoes a conformational change when it binds to a particular ligand, creating a more compact and stable form of the receptor. When AR is activated, it separates from heat shock proteins and moves to the nucleus, engaging in homodimer interactions with DNA androgen response elements [35]. A second dimerization interface is needed to bind to an AR response element since AR employs the DBD to bind to ARE elements. The protein-protein and protein-DNA connections that allow for this unexpected structure are preserved, and some AR-specific dimerization contacts are responsible for the AR specificity of AR response elements. In order to control the gene-specific expression that affects the development and survival of target cells, the active DNA-bound AR can entice co-regulator proteins and fundamental transcriptional machinery [59].
4.5. Prostate Cancer Progression to a Castration-Resistant State
The androgen receptor (AR) is a critical driver of prostate cancer progression, and its role in the development of metastatic castration-resistant prostate cancer (mCRPC) is well established. . Androgen deprivation therapy (ADT) is a common treatment for advanced prostate cancer. ADT works by reducing levels of androgens (male hormones), which are the primary fuel for prostate cancer growth. However, despite initial response to ADT, prostate cancer cells eventually develop mechanisms to bypass the need for androgens and continue to grow in the absence of these hormones. This is known as castration-resistant prostate cancer (CRPC). In CRPC, the AR signaling pathway becomes hyperactive and plays a critical role in driving prostate cancer progression. This hyperactivation of the AR can occur through a variety of mechanisms, including: Amplification or overexpression of the AR gene, Mutation of the AR gene, Ligand-independent activation of the AR (i.e., activation of the receptor in the absence of androgens). The hyperactive AR signaling pathway drives several key aspects of mCRPC, including: Increased cell proliferation and survival, Resistance to apoptosis (programmed cell death), Increased cell migration and invasion, leading to metastasis, Increased expression of genes associated with stemness, which may contribute to the development of treatment resistance.
PCa relies on androgens for growth, and early-stage disease can be managed with radical prostatectomy or radiation ablation of the prostate gland [60]. However, once cancer cells have spread outside of the prostate capsule, the disease is much more difficult to treat. When surgery is no longer an option, androgen withdrawal is usually used to treat prostate cancer patients with advanced disease, which affects about one-third of patients. By eliminating hormones through androgen ablation therapy, the growth-promoting effects of androgens are prevented, resulting in cancer cell apoptosis and ultimately tumour regression. However, this approach is not a complete cure, and the average overall survival time is less than 2-3 years. Bypassing the requirement for androgenic growth signals, PCa cells eventually develop castration resistance, which causes uncontrolled growth and elevated PSA levels, even when there are small amounts of androgen. This phenomenon’s mechanisms are intricate and still not fully understood. As androgens are crucial for maintaining prostate cells, a key issue is how prostate cells continue to survive after androgen deprivation therapy. Recent discoveries by several researchers suggest that AR is essential for the growth of androgen-resistant PCa. Over 80% of locally advanced castration-resistant prostate tumors have been found to have high levels of nuclear AR, and in bone metastases, the amount present is often higher than in primary tumors. In most cases, some sort of inappropriate activation of AR is associated with recurrent growth of prostate cancers. Studies have showed that the AR gene is the only gene consistently up-regulated during tumor progression in various experimental models of androgen-resistant prostate cancer [61]. The molecular mechanisms by which progression shifts from androgen dependence to castration-resistant state can be divided into two pathways: those that bypass the AR and those that operate through the receptor. These pathways are not mutually exclusive and often coexist in castration-resistant prostate cancer.
Figure 6.
Androgen-dependent vs. castration-resistant PCa progression.

Although AR is involved in the primary pathway, the unfettered pathway avoids it [62]. Castration resistance can be reached through either one of two pathways: one that avoids AR or one that uses the receptor. Apoptotic genes like PTEN and Bcl-2 are downregulated in the first pathway, increasing cell survival. In the second pathway, PCa cells are able to survive via AR due to dysregulated cytokines or growth factors, anomalies in receptor amplitude or genetics, autocrine generation of active androgens, and changes in co-activator expression In the second pathway, unregulated growth factors or cytokines, abnormalities in receptor amplitude or genetics, autocrine synthesis of active androgens, altered co-activator expression, and expression of alternatively spliced AR variants all contribute to PCa cells’ survival via AR. These processes result in an AR being stimulated in a way that is binding site-reduced or independent. Targeting the AR signaling pathway has become a cornerstone of treatment for mCRPC. This includes the use of second-generation anti-androgens, such as enzalutamide and apalutamide, which block the AR from binding to androgens, as well as the use of androgen synthesis inhibitors, such as abiraterone acetate, which reduce the production of androgens.
4.6. Crosstalk between AR and other pathways
The androgen receptor (AR) regulates the transcription of a wide range of genes involved in cell proliferation, differentiation, and survival. However, the AR signaling pathway is not independent and often interacts with other signaling pathways, which can modulate its activity and contribute to the development of prostate cancer. Several signaling pathways have been implicated in crosstalk with AR signaling, including the PI3K/AKT pathway, Wnt/β-catenin pathway, MAPK/ERK pathway, Hedgehog pathway, and Notch pathway. Activation of these pathways can promote or inhibit AR signaling, depending on the context and cellular environment. Once active, AR regulates a number of regulatory elements to change the metabolism of PCa cells. Importantly, the AR-driven metabolic program depends on activation of the mTOR signaling pathway [63]. An enzyme called phosphatidylinositol 3-kinase (PI3K) is essential for cellular processes like cell division, growth, and proliferation. However, PI3K is frequently altered in cancer. Due of the reciprocal feedback loops that AR and PI3K signaling use, Qi et al. discovered that the suppression of both can be synergistic. Inhibiting AR in conjunction with PI3K or mTOR pathway led to a rise in apoptosis and cell cycle arrest in CRPC cells, which in turn reduced cell proliferation [64]. The PI3K/AKT/mTOR pathway, the glucocorticoid receptor (GR) pathway, and alternative proteins can all be used to activate alternative signaling pathways, restore downstream signaling, and provide resistance to AR-targeted drugs in PCa [44].
The cell-cycle regulation system heavily relies on the PI3K/AKT/mTOR pathway. PIP2 (phosphatidylinositol-(4,5)-bisphosphate) is converted by PI3K into PIP3 (phosphatidylinositol(3,4,5)-triphosphate), which attracts proteins with pleckstrin homology domains to the cell membrane, including AKT kinase. This causes phosphorylation, structural changes, and activation of AKT. AKT that is activated moves to the cell nucleus and engages downstream pathways, such as mTOR, that are involved in angiogenesis, growth, migration, cell division, and survival. Instead, PTEN removes a phosphate from PIP3 to create PIP2, acting as a negative regulator of the PI3K signaling pathway. As a result, increased PI3K/AKT/mTOR downstream signaling activation is caused by PTEN inactivation. Between 20 and 40 percent of primary PCa and 50 percent of metastatic castration resistant prostate cancer (mCRPC) have changed PI3K/AKT/mTOR pathways. PTEN inactivation by bi-allelic loss and hotspot mutations commonly results in hyperactivity of the PI3K/AKT/mTOR pathway; if not, it is caused by PIK3CA/B mutations, amplifications, and activating fusions or AKT activating mutations. It has been shown in vitro that the PI3K/AKT/mTOR pathway and AR signaling cross-regulate each other through mutual feedback. After PTEN deletion, if the PI3K/AKT/mTOR pathway is upregulated, the AR signaling is downregulated, and vice versa, so that inhibiting one pathway stimulates the other. These findings provide a compelling preclinical case for combining treatments that block PI3K/AKT/mTOR and AR signaling [65].
Crosstalk between AR and CDK/pRb promotes cell cycle progression and this has been found to be a very promising therapeutic strategy in PCa especially with combined inhibition of AR and CDK4/6. AR controls the G1-S phase transition of the cell cycle. Thus, boosting CDK activity and causing phosphorylation to cause the inactivation of pRb [64]. In yeast and mammalian two-hybrid tests, it has been demonstrated that β-catenin directly interacts with AR. The LBD of AR and the armadillo repeats in β-catenin were shown to contain the interaction sites. Using an AR deletion construct as a lure, β-catenin was identified as an AR interacting protein from a library of gonadotropin-releasing hormone neuronal cells. In the absence of androgen, β-catenin was largely located in the cytoplasm, whereas in the presence of DHT, it completely co-localized with AR in the nucleus. Other liganded receptors, such as progesterone, glucocorticoids, or estrogen alpha, were unable to translocate β-catenin into the nucleus, indicating that the action was unique to AR. Additionally, it was demonstrated that agonist-bound AR was required for translocation by the inability of AR antagonists such bicalutamide and hydroxyflutamide to move β-catenin. The time course experiments revealed that the co-translocation occurred with similar kinetics and was independent of the GSK3, p42/44 ERK/MAPK, and phosphatidylinositol 3-kinase (PI3K) pathways. Inhibitors of these pathways had no effect on the co-translocation. The transcriptional effects of the p160 coactivator transcriptional mediators/intermediary factor 2 (TIF2) and NTD were modified by β-catenin upon interaction with LBD. NTD and TIF2 were not necessary for the binding, but they were cooperative. The reduction of AR-mediated transcription was caused by the redistribution of cytoplasmic β-catenin to the cell membrane as a result of E-cadherin expression in E-cadherin null PCa cells. According to this finding, the absence of E-cadherin can increase the cellular levels of β-catenin in PCa cells, which directly contributes to a more malignant and invasive tumor phenotype by enhancing AR activity during PCa formation and progression [66].
The activation of a number of signaling molecules and pathways, including but not limited to PI3K, WNT/-catenin, SRC, IL-6/STAT3, and MAPK, always follows genomic changes in PCa. These signals support PCa growth, create genotype-phenotype connection, and help receive inputs from the tumor microenvironment. Additionally, active signals cause epithelial mesenchymal transition (EMT), cancer stem cell (CSC)-like characteristics/stemness, and neuroendocrine differentiation (NED), which alter PC behavior [67]. In gene editing mouse models, Wnt/β-catenin signaling activation in the prostate has oncogenic roles in maintaining CRPC proliferation, encouraging EMT and NED, and converting stem cell-like properties to PC cells (Yeh et al., 2019). It has been demonstrated that Wnt/β-catenin and AR signaling interact. In order to activate AR signaling and advance CRPC, β-catenin forms complexes with and enhances AR. Additionally, β-catenin increases CRPC by acting as a coactivator with mutant AR (W741C and T877A). AR is recruited to the promoter regions of Myc, cyclin D1, and PSA by Wnt/β-catenin. On the other hand, AR overexpression boosts Wnt/β-catenin signaling’s transcriptional activity. Whether in LNCaP cells that express AR or PC3 cells that do not, AR has the ability to induce β-catenin translocation into the nucleus. SOX9 transcriptional factor activation may be used to achieve Wnt/β-catenin-AR feedback signaling [67]. AR controls the G1-S phase transition of the cell cycle, boosting CDK activity and causing phosphorylation to cause the inactivation of pRb. Combining AR with CDK4/6 inhibition has also been demonstrated to be a therapeutic approach in prostate cancer due to the interaction of AR with CDK/pRb in encouraging cell cycle progression [64].
AR signaling and SPOP dysregulation
Emerging evidence suggests that mutations in the speckle-type POZ protein (SPOP) gene can play a critical role in prostate cancer development and progression [68]. The SPOP is an E3 ubiquitin ligase that plays a key role in mediating the proteasomal degradation of several substrate proteins, including the AR. SPOP has emerged as a significant regulator of AR signaling, as it targets AR for ubiquitination and subsequent degradation, thereby influencing AR protein stability and transcriptional activity. SPOP mutations have been identified in a subset of prostate cancer cases, with recurrent hotspot mutations mainly occurring in the substrate-binding MATH domain [69]. These mutations have context-dependent effects on AR signaling and are associated with distinct clinical outcomes. In some cases, SPOP mutations lead to aberrant AR signaling, resulting in increased AR protein stability, enhanced transcriptional activity, and persistent androgen-dependent growth even in castration-resistant prostate cancer (CRPC) settings. On the other hand, certain SPOP mutations can impair AR binding, leading to reduced AR protein stability and decreased AR signaling, which may affect treatment response and patient outcomes. The crosstalk between androgen receptor (AR) signaling and SPOP dysregulation in prostate cancer is a complex and important aspect of disease pathogenesis. SPOP mutations or dysregulation can influence AR signaling through various mechanisms, leading to altered AR activity and subsequent effects on prostate cancer development and progression [70]. Here are some ways in which AR signaling and SPOP dysregulation can interact in prostate cancer:
AR Protein Stability and Degradation: Wild-type SPOP functions as an E3 ubiquitin ligase that promotes the proteasomal degradation of AR. It recognizes specific substrates on the AR protein, leading to its ubiquitination and subsequent degradation. SPOP mutations in the substrate-binding MATH domain can disrupt its ability to interact with AR, impairing AR ubiquitination, and resulting in increased AR protein stability [71]. As a consequence, the dysregulated AR becomes more resistant to degradation and retains higher transcriptional activity, promoting aberrant cell growth and survival in prostate cancer.
AR-ARV Interaction: In addition to full-length AR, SPOP dysregulation can also affect the stability of AR variants (ARVs), which are truncated forms of AR that lack the ligand-binding domain. ARVs have constitutive transcriptional activity and are associated with castration-resistant prostate cancer (CRPC). Certain SPOP mutations have been reported to preferentially target ARVs for degradation, leading to increased ARV stability and transcriptional activity [72]. This dysregulation can contribute to the development of CRPC and resistance to androgen-deprivation therapy.
Impact on Other Signaling Pathways: Dysregulated SPOP may impact other signaling pathways that intersect with AR signaling, leading to alterations in the AR response. For example, SPOP has been shown to interact with the phosphoinositide 3-kinase (PI3K) pathway, which can influence AR activity and cell proliferation in prostate cancer. Crosstalk with other pathways may further modulate AR signaling and contribute to disease progression and treatment resistance.
SPOP-AR Signaling Feedback Loop: The dysregulation of SPOP and AR signaling can lead to a feedback loop that perpetuates oncogenic signaling. Dysregulated AR activity resulting from SPOP mutations can drive the expression of genes involved in tumorigenesis and treatment resistance [73]. In turn, these altered gene expression patterns may further impact SPOP function and dysregulate its interactions with other cellular proteins, including the AR. Understanding the crosstalk between AR signaling and SPOP dysregulation is critical for developing targeted therapies in prostate cancer. Targeting the AR signaling axis or restoring SPOP function may hold therapeutic promise for treating SPOP-mutated prostate cancers or those with dysregulated AR signaling.
4.7. Targeting AR in Prostate cancer
ADT is a popular remedy for advanced PCa because it prevents androgen production or action. Although the majority of advanced prostate tumors respond to ADT, resistance to the drug eventually develops in most sick people, causing the disease to proceed to the circumcision PCa phenotype (CRPC), which is always fatal [5]. In the past, it was believed that resistance to ADT was unrelated to the AR axis, but in the past 20 years, it has been evident that, even after castration, AR signaling is still essential for tumor growth. Abiraterone, enzalutamide, darolutamide, and apalutamide are a few examples of novel hormonal drugs with increased anticancer activity that have been developed as a result of this discovery [75]. This includes LHRH analogs, which have already been reviewed and are almost entirely provided through injection. The metabolic syndrome, diabetes, elevated cardiovascular risk, and cognitive and sexual symptoms are just a few of the negative effects of these treatments. Other approaches that preserve clinical benefit while having fewer side effects are therefore required. Relugolix, an orally accessible, nonpeptide LHRH antagonist, has recently been created. It competitively binds to and blocks the anterior pituitary gland’s LHRH receptor. Relugolix reduces LH secretion and release as well as LHRH binding to the LHRH receptor, which lowers the secretion of testosterone from Leydig cells in the testes.
Recent research has focused on developing new strategies to target AR in PCa [76,77]. These include the development of AR antagonists that can bind to and inhibit the activity of AR, and interference with AR signal transduction. More recently, novel AR-targeted therapies, such as second-generation anti-androgens and androgen receptor signaling inhibitors (ARSIs), have been developed that target the AR directly or it’s downstream signaling pathways. These therapies are being actively researched in clinical trials because they have shown promise in improving outcomes for patients with advanced PCa.
4.7.1. Emerging Therapies Targeting AR
Prostate cancers need various forms of treatment since they might behave in various ways. Indeed, ADT has served as the primary form of therapy for advanced PCa for many years [76]. Androgens are male hormones, such as testosterone, which can stimulate the growth of PCa cells. By reducing the levels of androgens in the body, ADT can help slow the growth of PCa and reduce burden [62]. There are different types of ADT, but the most common approach is to use medications called luteinizing hormone-releasing hormone (LHRH) sympathomimetic or adversaries that can inhibit yield of testosterone by the testicles. Another approach is to use medications called anti-androgens, which block the effects of androgens on PCa cells. Depending on the stage and severity of the PCa, ADT may be used alone or in conjunction with other therapies like radiation therapy or chemotherapy. Despite the fact that ADT can be helpful in symptom improvement and slowing the spread of PCa, all patients eventually develop deadly metastatic CRPC [62,78]. As a result, second-generation hormonal treatments have substantially improved the prognosis for those with advanced PCa. Precision medicine is the subject of recent research. Every patient will receive treatment that is specifically tailored to their needs from the outset. Precision medicine must cover a lot of ground because there are so many different forms of prostate cancer. It’s critical to keep in mind that not everyone benefits equally from developing medicines. Also, some of these medicines are still awaiting FDA approval. There are several emerging therapies for PCa including: Immunotherapy: A form of therapy called immunotherapy aids the immune system’s recognition and destruction of cancerous cells [79]. Several immunotherapy drugs, such as checkpoint inhibitors, CAR-T cells, and cancer vaccines, are being developed and tested in clinical trials for prostate cancer. Targeted therapies: Targeted therapies are drugs that specifically target cancer cells based on their genetic mutations or other specific characteristics. For prostate cancer, several targeted therapies are being developed, including drugs that target the androgen receptor pathway and drugs that target specific enzymes and proteins involved in prostate cancer growth. Radiopharmaceuticals: Drugs called radiopharmaceuticals can be used to target and eliminate cancer cells because they includes a radioactive materials. Several radiopharmaceuticals, such as radium-223 and lutetium-177, are being developed and tested for prostate cancer. Gene therapy: Gene therapy is a type of treatment that involves inserting or altering genes in a person’s cells to treat or prevent disease. For prostate cancer, several gene therapies are being developed, including therapies that target the androgen receptor pathway and therapies that use viruses to deliver therapeutic genes to cancer cells. Nanoparticle-based therapies: Nanoparticles are tiny particles that can be used to deliver drugs directly to cancer cells. Several nanoparticle-based therapies, such as liposomes and polymer nanoparticles, are being developed and tested for prostate cancer. It’s crucial to keep in mind that many of these treatments remain in the phase of development and research and might not be widely accessible over many years. We would look at some of this emerging therapies based on those related AR.
4.7.2. Androgen Deprivation Therapy (ADT)
ADT is a typical prostate treatment option that lowers androgens, such as testosterone, which bind to and activate AR. However, many of cancerous cells eventually are immune to ADT, and the development of castration resistant PCa (CRPC) is often implicated with increased AR activity. ADT can substantially lower the amount of available androgens in men with PCa, which in turn lowers AR’s transcriptional activity [40]. AR itself experiences numerous changes as the course of treatment progresses in response to these changes. Prostate cancer that is resistant to treatment and no longer relies on androgens for growth and survival is known as castration-resistant prostate cancer. By using androgen ablation therapy, CRPC is targeted. There are five different types of probable development mechanisms for CRPC. AR mutations, higher concentrations of co-activators, or enhanced synthesis of the potent androgen dihydrotestosterone all increase sensitivity to low levels of androgen in the blood in the hypersensitive pathway. The AR and androgen continue to be essential for the growth of tumors that use this mechanism. All of these ultimately lead to androgen-independent AR activation, which is the fundamental reason for anti-androgen therapy resistance. AR can be stimulated regardless of the existence of other hormones and medications, such as progesterone, flutamide, bicalutamide, and enzalutamide, and sustain its transcription activity to promote PCa carcinogenesis. The gene amplification and some acquire genetic abnormalities can cause this. The existing treatments for advanced prostate cancer comprise inhibitors of androgen synthesis that suppress the production of testosterone and/or DHT, as well as inhibitors of AR that prevent the binding of ligands at the LBD [80]. Nevertheless, LBD-specific therapies may become ineffective due to AR genetic variations and discrepancies. Because they both play crucial part in its gene transcription action and are less susceptible to AR alternative splicing than the LBD, the DBD and NTD have therefore emerged as new targets for inhibition [74]. By inhibiting DBD and NTD, it may be possible to enhance men’s longevity, living conditions, and acquired resistance to current therapies will all be improved.
4.7.3. Androgen Receptor Signaling Inhibitors (ARSIs)
Androgen receptor signaling inhibitors are quite a type of medication utilized for treating PCa. Because AR signaling promotes tumor growth, medicines known as AR signaling inhibitors (ARSIs), which work to block AR signaling, have long been used in clinical settings. The majority of individuals treated with ARSIs develop a castration-resistant (CRPC) phenotype, which is unfortunate because the therapeutic efficacy of ARSIs is transient [34]. These drugs work by blocking the activity of androgen receptors, which are proteins found in prostate cells that bind to androgens and stimulate the growth and spread of prostate cancer. These drugs can be used alone or in combination with other treatments, such as chemotherapy or radiation therapy, to treat prostate cancer. They can also be used to manage the symptoms of advanced prostate cancer. There are several types of androgen receptor signaling inhibitors, including: Anti-androgens: These drugs impair androgen from forming complex to its receptors, preventing the cancer cells from receiving the signal to grow and divide. Examples include bicalutamide, flutamide, and nilutamide. GnRH agonists: These drugs reduce the production of androgens by the testes by inhibiting the release of gonadotropin-releasing hormone (GnRH) from the hypothalamus. This causes reduction in testosterone levels in the body, which can slow the growth of prostate cancer. Examples include leuprolide and goserelin. CYP17 inhibitors: These drugs block an enzyme called CYP17, which is involved in the production of androgens in the adrenal gland and in the prostate cancer cells themselves. Examples include abiraterone acetate. Androgen receptor antagonists: These drugs block the activity of androgen receptors directly, preventing them from stimulating the growth of prostate cancer cells. Examples include enzalutamide and apalutamide.
The biotransformation of androgens in the body is the foundation that drives the creation of medications for PCa [59]. Androgens are synthesized from cholesterol, which is converted into pregnenolone and then into various hormones, including DHEA, androstenedione, and testosterone [81]. Enzymes involved in androgen synthesis include CYP11A1, 3β-HSD, and CYP17A1. DHEA biogenesis from cholesterol is crucial in the treatment of PCa individuals who may have rising androgen levels and overactive AR because DHEA is a crucial forerunner in androgen biosynthetic pathways in the prostate. Abiraterone, enzalutamide, apalutamide, and darolutamide are drugs recommended by the NCCN for PCa treatment and act on the androgen axis by targeting androgen synthesis and metabolism pathways [81]. These medications, renowned as ARSI, are used to block pathways, restore DHEA levels in the body, and impede AR signaling, which are the main causes of ARSI resistance.
Abiraterone is a CYP17A1 enzyme-selective, irrevocable blocker that has received FDA approval that efficiently prevents androgen synthesis in the testis, adrenal gland, and PCa. Abiraterone is administered with prednisone to reduce adverse reactions caused by the inhibition of corticosterone and 11-deoxycorticosterone. According to medical findings, those with metastatic CRPC who take abiraterone and prednisone together have longer overall survival times and disease-free survival times. However, alterations in androgen synthesis and metabolic pathways eventually lead to the development of rebellion to abiraterone. The de novo synthesis of androgens is activated by CYP17A1 increased expression and mutation, which also raises the level of DHEA in the tumor. Abiraterone rebellion is brought on by the aberrant expression of 3-HSDs and AKR1C3, which boost the "backdoor pathway" and "alternative pathway" of androgen formation, respectively, while reducing the metabolic activity of the active form of androgens by the glucuronidation pathway. Adrenocorticotropic hormone (ACTH), pregnenolone, and other hormones upstream of P450c17 build up as a result of abiraterone’s inhibitory activity of enzyme activity, which encourages the improvement of the "backdoor pathway" of androgen synthesis and also activates mutated AR. Moreover, the addition of exogenous glucocorticoids to reduce the adverse effects of abiraterone is also thought to activate mutant AR, causing drug resistance. Furthermore, the development of truncated androgen receptor variants (such as AR-V7), as well as the activation of the PI3K/AKT/mTOR and ErbB2 pathways, also contribute to the resistance of abiraterone.
Apalutamide is a new generation AR inhibitor approved for nmCRPC with improved CNS safety compared to enzalutamide. Its mechanism involves inhibiting AR nuclear translocation and binding to AREs. Apalutamide resistance is caused by AR mutations, splicing variants, and PI3K pathway activation. Darolutamide is another novel oral non-steroidal AR inhibitor that inhibits AR function and cell growth in PCa without crossing the BBB, resulting in fewer CNS side effects. Studies on darolutamide resistance are limited, but there is evidence of cross-resistance with other AR inhibitors and significant inhibition of AR-mutated variants. Enzalutamide is an FDA-approved second-generation androgen receptor antagonist used to treat CRPC and non-metastatic CRPC. The drug works by preventing the nuclear translocation of stimulated AR, which prevents it from being localized to AREs, and inducing apoptosis and inhibiting CRPC cell proliferation. Despite its effectiveness, patients may develop resistance to enzalutamide. Enzalutamide resistance may be due to various mechanisms, such as changes in the quantity or structure of AR, over-activation of GR, activation of the Wnt pathway and Warburg effect, autophagy inhibition, and microRNA-mediated gene expression activation, leading to neuroendocrine trans-differentiation of CRPC cells. The majority of these AR-targeted therapies target the LBD. The NTD contains AF-1, which is required for AR transcriptional activity. The first androgen receptor amino-terminal domain inhibitor is EPI-001 [36]. EPI-001, an innovation of Marianne Sadar and Raymond Andersen, a group of stereoisomers and analogues with a special interest in the clinical development of anitens for the treatment of CRPC. EPI-001 is an AR antagonist that inhibits protein-protein interactions required for AR transcriptional activity by binding covalently to the AR’s NTD [82]. A The C-terminal ligand-binding domain (LBD) of the androgen receptor (AR) binds to all currently used anti-androgens, which then compete for androgen binding and activation. EPI-001 type compounds may be helpful in the management of advanced PCa that is resistant to traditional anti-androgens like enzalutamide due to their distinct mode of action.
4.7.4. Combinational Therapies
There is a proven substantial correlation between the PI3K/AKT/mTOR and AR signaling in PCa [62]. Combination therapy targeting these pathways has shown promising results in preclinical and clinical studies [83,84]. One potential combination therapy for PCa is the use of AR inhibitors such as enzalutamide or abiraterone acetate, in combination with PI3K inhibitors such as buparlisib or idelalisib. The combination of these drugs has been shown to have a synergistic effect in preclinical studies, resulting in decreased proliferation of cancer cells and increased apoptosis. Table 1 lists some of these dual-targeting combination therapies. In a preclinical study, found that combining an AKT inhibitor with an anti-androgen prolonged the stabilization of the disease in a CRPC model [85]. The AKT inhibitor in the androgen-sensitive and castration-resistant phases of the LNCaP mouse xenograft, AZD5363 proved anticancer activity. It also successfully decreased propagation and induced apoptosis in PCa cell lines expressing AR [86]. But after about 30 days of treatment, increasing PSA levels signaled AZD5363 resistance. It was discovered that AZD5363 enhanced AR transcriptional activity, AR binding to androgen response elements, and AR-dependent gene expression, including PSA and NKX3.1 [85]. Combining AZD5363 with the antiandrogen bicalutamide prevented these side effects and prolonged the inhibition of tumor growth and stabilized PSA in CRPC in vivo (Table 1).
Additionally, in vitro experiments revealed a synergism reduction in cell prepon-derance and trigger of apoptosis Synergistic inhibition of cell proliferation and induction of apoptosis were also observed in vitro [86]. Another approach is to target both AR and PI3K pathways with a single drug. For example, a novel drug called AZD5363 has been developed that inhibits both PI3K and AKT, a downstream effector of PI3K, as well as AR signaling. This drug has shown promising results in preclinical studies, and is currently being evaluated in clinical trials. It is important to note that combination therapy targeting AR and PI3K pathways may also have increased toxicity compared to single-agent therapy. Thus, careful patient selection and monitoring are essential to ensure the safety and efficacy of these treatments. Overall, combinational therapies for prostate cancer offer a promising approach to improving patient outcomes. However, further research is needed to determine the optimal combination of therapies and identify biomarkers that can predict treatment response and guide personalized treatment strategies.
5. Conclusions and future directions
Androgen receptors (AR) play a pivotal role in prostate cancer (PCa) susceptibility, progression, and treatment. Targeting AR has emerged as a promising strategy against this complex malignancy. Integrating diagnostic approaches like AR, SPOP and PSA measurements could enhance PCa detection accuracy. Further research is vital to uncover the intricate mechanisms by which AR influences PCa initiation and progression, leading to innovative early detection strategies. Overcoming resistance to androgen deprivation therapy requires novel tactics to effectively target AR in advanced PCa stages. Identifying genetic variations impacting AR activity, developing innovative AR-targeted therapies, and optimizing combination pharmaceuticals are essential. Future investigations should focus on elucidating AR’s molecular mechanisms, circumventing resistance, exploring the genetic landscape, optimizing combination therapies, discovering predictive biomarkers, translating findings into practice, and tailoring treatments based on patient profiles. These endeavors hold the potential to transform PCa diagnosis and treatment approaches by deepening our understanding of AR’s intricate involvement.
Author Contributions
Conceptualization, O.O.O. and S.Z.; Literature search, E.C.A; validation, C.C.E.; writing—original draft preparation, S.Z.; writing—review and editing, O.O.O.; supervision, O.O.O. All authors have read and agreed to the published version of the manuscript. All authors made substantial contribution to the work and as well approved the final version for publication.
Funding
This research received no external funding.
Data Availability Statement
Data sharing is not applicable to this article
Conflicts of Interest
Authors declare that no conflict of interest exist.
References
- Chu, J.J. & Mehrzad, R. (2023) The biology of cancer. In: The Link Between Obesity and Cancer. Elsevier. pp. 35–45. [CrossRef]
- Ha, H., Kwon, H., Lim, T., Jang, J., Park, S.-K. & Byun, Y. (2021) Inhibitors of prostate-specific membrane antigen in the di-agnosis and therapy of metastatic prostate cancer – a review of patent literature. Expert Opinion on Therapeutic Patents. 31 (6), 525–547. [CrossRef] [PubMed]
- Ellinger, J.; Alajati, A.; Kubatka, P.; Giordano, F.A.; Ritter, M.; Costigliola, V.; Golubnitschaja, O. Prostate cancer treatment costs increase more rapidly than for any other cancer—how to reverse the trend? EPMA J. 2022, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef]
- Wang, L.; Lu, B.; He, M.; Wang, Y.; Wang, Z.; Du, L. Prostate Cancer Incidence and Mortality: Global Status and Temporal Trends in 89 Countries From 2000 to 2019. Front. Public Heal. 2022, 10, 811044. [Google Scholar] [CrossRef]
- Osadchuk, L.V.; Osadchuk, A.V. Role of CAG and GGC Polymorphism of the Androgen Receptor Gene in Male Fertility. Russ. J. Genet. 2022, 58, 247–264. [Google Scholar] [CrossRef]
- Badal, S.; Aiken, W.; Morrison, B.; Valentine, H.; Bryan, S.; Gachii, A.; Ragin, C. Disparities in prostate cancer incidence and mortality rates: Solvable or not? Prostate 2020, 80, 3–16. [Google Scholar] [CrossRef]
- Hu, L.; Fu, C.; Song, X.; Grimm, R.; von Busch, H.; Benkert, T.; Kamen, A.; Lou, B.; Huisman, H.; Tong, A.; et al. Automated deep-learning system in the assessment of MRI-visible prostate cancer: comparison of advanced zoomed diffusion-weighted imaging and conventional technique. Cancer Imaging 2023, 23, 6. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, G.; Zhu, S.; Dai, J.; Chen, J.; Zhang, M.; Ni, Y.; Zhang, H.; Shen, P.; Zhao, X.; et al. Circulating tumour DNA reveals genetic traits of patients with intraductal carcinoma of the prostate. BJU Int. 2022, 129, 345–355. [Google Scholar] [CrossRef]
- He, Y.; Hooker, E.; Yu, E.-J.; Wu, H.; Cunha, G.R.; Sun, Z. An Indispensable Role of Androgen Receptor in Wnt Responsive Cells During Prostate Development, Maturation, and Regeneration. STEM CELLS 2018, 36, 891–902. [Google Scholar] [CrossRef]
- Manzar, N.; Ganguly, P.; Khan, U.K.; Ateeq, B. Transcription networks rewire gene repertoire to coordinate cellular reprograming in prostate cancer. Semin. Cancer Biol. 2023, 89, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Perez, M.P.; Perez-Navarro, E.; Alonso-Gordoa, T.; Conteduca, V.; Font, A.; Vázquez-Estévez, S.; González-Del-Alba, A.; Wetterskog, D.; Antonarakis, E.S.; Mellado, B.; et al. A correlative biomarker study and integrative prognostic model in chemotherapy-naïve metastatic castration-resistant prostate cancer treated with enzalutamide. Prostate 2023, 83, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.D. (2022) Triplet therapy for prostate cancer. The Lancet. 399 (1 0336), 1670–1671. [CrossRef]
- Crawford, E.D.; Schellhammer, P.F.; McLeod, D.G.; Moul, J.W.; Higano, C.S.; Shore, N.; Denis, L.; Iversen, P.; Eisenberger, M.A.; Labrie, F. Androgen Receptor Targeted Treatments of Prostate Cancer: 35 Years of Progress with Antiandrogens. J. Urol. 2018, 200, 956–966. [Google Scholar] [CrossRef]
- Weidemann, W.; Hanke, H. Cardiovascular Effects of Androgens. Cardiovasc. Drug Rev. 2002, 20, 175–198. [Google Scholar] [CrossRef]
- Levine, P.M.; Garabedian, M.J.; Kirshenbaum, K. Targeting the Androgen Receptor with Steroid Conjugates. J. Med. Chem. 2014, 57, 8224–8237. [Google Scholar] [CrossRef]
- Mendelsohn, M.E.; Karas, R.H.; Yamamoto, Y.; Brady, M.P.; Lu, Z.P.; Maziasz, P.J.; Liu, C.T.; Pint, B.A.; More, K.L.; Meyer, H.M.; et al. Molecular and Cellular Basis of Cardiovascular Gender Differences. Science 2005, 308, 1583–1587. [Google Scholar] [CrossRef]
- Chang, C.; Saltzman, A.; Yeh, S.; Young, W.; Keller, E.; Lee, H.-J.; Wang, C.; Mizokami, A. Androgen Receptor: An Overview. Crit. Rev. Eukaryot. Gene Expr. 1995, 5, 97–125. [Google Scholar] [CrossRef]
- Mainwaring, W.I.P. A SOLUBLE ANDROGEN RECEPTOR IN THE CYTOPLASM OF RAT PROSTATE. J. Endocrinol. 1969, 45, 531–541. [Google Scholar] [CrossRef]
- Neumann, F. The antiandrogen cyproterone acetate: discovery, chemistry, basic pharmacology, clinical use and tool in basic research*. Exp. Clin. Endocrinol. Diabetes 1994, 102, 1–32. [Google Scholar] [CrossRef]
- Davies, A.H.; Zoubeidi, A. Targeting androgen receptor signaling: a historical perspective. Endocrine-Related Cancer 2021, 28, T11–T18. [Google Scholar] [CrossRef] [PubMed]
- Willems, A.; De Gendt, K.; Deboel, L.; Swinnen, J.V.; Verhoeven, G. The development of an inducible androgen receptor knockout model in mouse to study the post-meiotic effects of androgens on germ cell development. Spermatogenesis 2011, 1, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Senapati, D.; Kumari, S.; Heemers, H.V. Androgen receptor co-regulation in prostate cancer. Asian J. Urol. 2020, 7, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Wetendorf, M.; DeMayo, F.J. The progesterone receptor regulates implantation, decidualization, and glandular development via a complex paracrine signaling network. Mol. Cell. Endocrinol. 2012, 357, 108–118. [Google Scholar] [CrossRef]
- Akison, L.; Robker, R. The Critical Roles of Progesterone Receptor (PGR) in Ovulation, Oocyte Developmental Competence and Oviductal Transport in Mammalian Reproduction. Reprod. Domest. Anim. 2012, 47, 288–296. [Google Scholar] [CrossRef]
- Cable, J.K. & Grider, M.H. (2023) Physiology, Progesterone. In: StatPearls. Treasure Island (FL), StatPearls Publishing. p. Available online: http://www.ncbi.nlm.nih.gov/books/NBK558960/.
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef]
- Nicolaides, N.C., Chrousos, G. & Kino, T. (2000) Glucocorticoid Receptor. In: K.R. Feingold, B. Anawalt, M.R. Blackman, A. Boyce, G. Chrousos, et al. (eds.). Endotext. South Dartmouth (MA), MDText.com, Inc. p. Available online: http://www.ncbi.nlm.nih.gov/books/NBK279171/.
- Aurilio, G.; Cimadamore, A.; Mazzucchelli, R.; Lopez-Beltran, A.; Verri, E.; Scarpelli, M.; Massari, F.; Cheng, L.; Santoni, M.; Montironi, R. Androgen Receptor Signaling Pathway in Prostate Cancer: From Genetics to Clinical Applications. Cells 2020, 9, 2653. [Google Scholar] [CrossRef]
- Vidula, N.; Yau, C.; Wolf, D.; Rugo, H.S. Androgen receptor gene expression in primary breast cancer. npj Breast Cancer 2019, 5, 1–7. [Google Scholar] [CrossRef]
- Tindall, D.J.; Lonergan, P.E. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [CrossRef]
- Jamroze, A.; Chatta, G.; Tang, D.G. Androgen receptor (AR) heterogeneity in prostate cancer and therapy resistance. Cancer Lett. 2021, 518, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Messner, E.A.; Steele, T.M.; Tsamouri, M.M.; Hejazi, N.; Gao, A.C.; Mudryj, M.; Ghosh, P.M. The Androgen Receptor in Prostate Cancer: Effect of Structure, Ligands and Spliced Variants on Therapy. Biomedicines 2020, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Crona, D.J.; Whang, Y.E. Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers 2017, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, P.L.; Jivan, A.; Dollins, D.E.; Claessens, F.; Gewirth, D.T. Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. 2004, 101, 4758–4763. [Google Scholar] [CrossRef]
- Alemany, M. The Roles of Androgens in Humans: Biology, Metabolic Regulation and Health. Int. J. Mol. Sci. 2022, 23, 11952. [Google Scholar] [CrossRef]
- Shaffer, P.L.; McDonnell, D.P.; Gewirth, D.T. Characterization of Transcriptional Activation and DNA-Binding Functions in the Hinge Region of the Vitamin D Receptor. Biochemistry 2005, 44, 2678–2685. [Google Scholar] [CrossRef]
- Ban, F.; Leblanc, E.; Cavga, A.D.; Huang, C.-C.F.; Flory, M.R.; Zhang, F.; Chang, M.E.K.; Morin, H.; Lallous, N.; Singh, K.; et al. Development of an Androgen Receptor Inhibitor Targeting the N-Terminal Domain of Androgen Receptor for Treatment of Castration Resistant Prostate Cancer. Cancers 2021, 13, 3488. [Google Scholar] [CrossRef]
- El Kharraz, S.; Dubois, V.; E van Royen, M.; Houtsmuller, A.B.; Pavlova, E.; Atanassova, N.; Nguyen, T.; Voet, A.; Eerlings, R.; Handle, F.; et al. The androgen receptor depends on ligand-binding domain dimerization for transcriptional activation. Embo Rep. 2021, 22, e52764. [Google Scholar] [CrossRef]
- Estébanez-Perpiñá, E.; Arnold, L.A.; Nguyen, P.; Rodrigues, E.D.; Mar, E.; Bateman, R.; Pallai, P.; Shokat, K.M.; Baxter, J.D.; Guy, R.K.; et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Natl. Acad. Sci. 2007, 104, 16074–16079. [Google Scholar] [CrossRef]
- Westaby, D.; Maza, M.d.L.D.F.d.L.; Paschalis, A.; Jimenez-Vacas, J.M.; Welti, J.; de Bono, J.; Sharp, A. A New Old Target: Androgen Receptor Signaling and Advanced Prostate Cancer. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 131–153. [Google Scholar] [CrossRef]
- Jacob, A.; Raj, R.; Allison, D.B.; Myint, Z.W. Androgen Receptor Signaling in Prostate Cancer and Therapeutic Strategies. Cancers 2021, 13, 5417. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P., Yetiskul, E. & Jialal, I. (2021) Physiology, male reproductive system. In: StatPearls [Internet]. StatPearls Publish-ing. p.
- Carpenter, V.; Saleh, T.; Lee, S.M.; Murray, G.; Reed, J.; Souers, A.; Faber, A.C.; Harada, H.; Gewirtz, D.A. Androgen-deprivation induced senescence in prostate cancer cells is permissive for the development of castration-resistance but susceptible to senolytic therapy. Biochem. Pharmacol. 2021, 193, 114765. [Google Scholar] [CrossRef]
- Molina, P.E. (2013) Chapter 8. Male Reproductive System. In: Endocrine Physiology. 4th edition. New York, NY, The McGraw-Hill Companies. p. accessmedicine.mhmedical.com/content.aspx?aid=57307916.
- Mbemi, A.; Khanna, S.; Njiki, S.; Yedjou, C.G.; Tchounwou, P.B. Impact of Gene–Environment Interactions on Cancer Development. Int. J. Environ. Res. Public Heal. 2020, 17, 8089. [Google Scholar] [CrossRef]
- Oladoyinbo, C.A., Akinbule, O.O., Bolajoko, O.O., Aheto, J.M., Faruk, M., Bassey, I.E., Odedina, F., Ogunlana, O.O., Ogun-sanya, M., Suleiman, A.M., Obialor, S. & Gali, R. (2020) Risk Factors for Prostate Cancer in West African Men: The Prostate Cancer Transatlantic Consortium (CaPTC) Cohort Study. Cancer Health Disparities. 4. Available online: https://www.companyofscientists.com.
- Vietri, M.T.; D’elia, G.; Caliendo, G.; Resse, M.; Casamassimi, A.; Passariello, L.; Albanese, L.; Cioffi, M.; Molinari, A.M. Hereditary Prostate Cancer: Genes Related, Target Therapy and Prevention. Int. J. Mol. Sci. 2021, 22, 3753. [Google Scholar] [CrossRef] [PubMed]
- Foley, G.R.; Marthick, J.R.; Ostrander, E.A.; Stanford, J.L.; Dickinson, J.L.; FitzGerald, L.M. Association of a novel BRCA2 mutation with prostate cancer risk further supports germline genetic testing. Eur. J. Cancer 2023, 180, 155–157. [Google Scholar] [CrossRef]
- Johnson, J.R. , Woods-Burnham, L. A. ( 2021) Genetic contributions to prostate cancer disparities in men of West African descent. Frontiers in oncology. 11, 770500.
- A Rey, R. The Role of Androgen Signaling in Male Sexual Development at Puberty. Endocrinology 2021, 162. [Google Scholar] [CrossRef]
- Song, S.H.; Kim, E.; Jung, Y.J.; Kim, H.-M.; Park, M.S.; Kim, J.K.; Lee, H.; Oh, J.J.; Lee, S.; Hong, S.K.; et al. Polygenic risk score for tumor aggressiveness and early-onset prostate cancer in Asians. Sci. Rep. 2023, 13, 1–6. [Google Scholar] [CrossRef]
- Hsing, A.W., Gao, Y.-T., Wu, G., Wang, X., Deng, J., Chen, Y.-L., Sesterhenn, I.A., Mostofi, F.K., Benichou, J. & Chang, C. (2000) Polymorphic CAG and GGN repeat lengths in the androgen receptor gene and prostate cancer risk: a popula-tion-based case-control study in China. Cancer research. 60 (18), 5111–5116.
- Allemailem, K.S., Almatroudi, A., Alrumaihi, F., Makki Almansour, N., Aldakheel, F.M., Rather, R.A., Afroze, D. & Rah, B. (2021) Single nucleotide polymorphisms (SNPs) in prostate cancer: its implications in diagnostics and therapeutics. Ameri-can Journal of Translational Research. 13 (4), 3868–3889.
- Ratajczak, W.; Lubkowski, M.; Lubkowska, A. Heat Shock Proteins in Benign Prostatic Hyperplasia and Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 897. [Google Scholar] [CrossRef]
- Takayama, K. (2018) The Biological Role of Androgen Receptor in Prostate Cancer Progression. In: M. Estrada (ed.). Advanc-es in Testosterone Action. IntechOpen. p. [CrossRef]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Men’s Heal. 2019, 37, 288–295. [Google Scholar] [CrossRef]
- Student, S.; Hejmo, T.; Poterała-Hejmo, A.; Leśniak, A.; Bułdak, R. Anti-androgen hormonal therapy for cancer and other diseases. Eur. J. Pharmacol. 2020, 866, 172783. [Google Scholar] [CrossRef]
- Tortorella, E.; Giantulli, S.; Sciarra, A.; Silvestri, I. AR and PI3K/AKT in Prostate Cancer: A Tale of Two Interconnected Pathways. Int. J. Mol. Sci. 2023, 24, 2046. [Google Scholar] [CrossRef]
- Gonthier, K.; Poluri, R.T.K.; Audet-Walsh. Functional genomic studies reveal the androgen receptor as a master regulator of cellular energy metabolism in prostate cancer. J. Steroid Biochem. Mol. Biol. 2019, 191, 105367. [Google Scholar] [CrossRef] [PubMed]
- Michmerhuizen, A.R.; Spratt, D.E.; Pierce, L.J.; Speers, C.W. ARe we there yet? Understanding androgen receptor signaling in breast cancer. npj Breast Cancer 2020, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Pisano, C.; Tucci, M.; Di Stefano, R.F.; Turco, F.; Scagliotti, G.V.; Di Maio, M.; Buttigliero, C. Interactions between androgen receptor signaling and other molecular pathways in prostate cancer progression: Current and future clinical implications. Crit. Rev. Oncol. 2021, 157, 103185. [Google Scholar] [CrossRef] [PubMed]
- Khurana, N.; Sikka, S.C. Interplay Between SOX9, Wnt/β-Catenin and Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2066. [Google Scholar] [CrossRef]
- Tong, D. Unravelling the molecular mechanisms of prostate cancer evolution from genotype to phenotype. Crit. Rev. Oncol. 2021, 163, 103370. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.; Burleson, M. SPOP and cancer: a systematic review. Am J Cancer Res 2020, 10, 704–726. [Google Scholar]
- Wang, Z.; Song, Y.; Ye, M.; Dai, X.; Zhu, X.; Wei, W. The diverse roles of SPOP in prostate cancer and kidney cancer. Nat. Rev. Urol. 2020, 17, 339–350. [Google Scholar] [CrossRef]
- Kwon, J.E.; La, M.; Oh, K.H.; Oh, Y.M.; Kim, G.R.; Seol, J.H.; Baek, S.H.; Chiba, T.; Tanaka, K.; Bang, O.S.; et al. BTB Domain-containing Speckle-type POZ Protein (SPOP) Serves as an Adaptor of Daxx for Ubiquitination by Cul3-based Ubiquitin Ligase. J. Biol. Chem. 2006, 281, 12664–12672. [Google Scholar] [CrossRef]
- Hernandez-Munoz I, Lund AH, Van Der Stoop P, Boutsma E, Muijrers I, Verhoeven E, Nusinow DA, Panning B, Marahrens Y & Van Lohuizen M (2005) Stable X chromosome inactivation involves the PRC1 polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc Natl Acad Sci USA 102, 7635–7640.
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef]
- Bernasocchi, T.; Theurillat, J.-P.P. SPOP-mutant prostate cancer: Translating fundamental biology into patient care. Cancer Lett. 2021, 529, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Severson, T.; Qiu, X.; Alshalalfa, M.; Sjöström, M.; Quigley, D.; Bergman, A.; Long, H.; Feng, F.; Freedman, M.L.; Zwart, W.; et al. Androgen receptor reprogramming demarcates prognostic, context-dependent gene sets in primary and metastatic prostate cancer. Clin. Epigenetics 2022, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.; Marozzi, M.; Trisciuoglio, D.; Rotili, D.; Mai, A.; Rizzi, F. Simultaneous administration of EZH2 and BET inhibitors inhibits proliferation and clonogenic ability of metastatic prostate cancer cells. J. Enzym. Inhib. Med. Chem. 2023, 38, 2163242. [Google Scholar] [CrossRef] [PubMed]
- Westaby, D.; Maza, M.d.L.D.F.d.L.; Paschalis, A.; Jimenez-Vacas, J.M.; Welti, J.; de Bono, J.; Sharp, A. A New Old Target: Androgen Receptor Signaling and Advanced Prostate Cancer. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 131–153. [Google Scholar] [CrossRef] [PubMed]
- Ekenwaneze CC, Zakari S, Amadi EC, Ogunlana OO. Recent Advances in Immunotherapy for Prostate Cancer Treatment. Presented at: Annual Conference of the Nigerian Society for Microbiology - NSM-OTA; July 2023; Covenant University, Ota, Ogun State, Nigeria. Available online: https://www.researchgate.net/publication/372915334_Recent_Advances_in_Immunotherapy_for_Prostate_Cancer_Treatment.
- Iheagwam FN, Iheagwam OT, Odiba JK, Ogunlana OO, Chinedu SN. Cancer and Glucose Metabolism: A Review on Warburg Mechanisms. TJNPR [Internet]. 2022 May 1 [cited 2023 Aug 6];6(5):661-7. Available online: https://tjnpr.org/index.php/home/article/view/42.
- Alabi, B.R.; Liu, S.; Stoyanova, T. Current and emerging therapies for neuroendocrine prostate cancer. Pharmacol. Ther. 2022, 238, 108255. [Google Scholar] [CrossRef]
- El Kharraz, S.; Dubois, V.; E van Royen, M.; Houtsmuller, A.B.; Pavlova, E.; Atanassova, N.; Nguyen, T.; Voet, A.; Eerlings, R.; Handle, F.; et al. The androgen receptor depends on ligand-binding domain dimerization for transcriptional activation. Embo Rep. 2021, 22, e52764. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, Y.; Xing, Z.; Sah, R.K.; Hu, J.; Hu, H. Androgen Metabolism and Response in Prostate Cancer Anti-Androgen Therapy Resistance. Int. J. Mol. Sci. 2022, 23, 13521. [Google Scholar] [CrossRef]
- Narayanan, R. Therapeutic targeting of the androgen receptor (AR) and AR variants in prostate cancer. Asian J. Urol. 2020, 7, 271–283. [Google Scholar] [CrossRef]
- Crook JM, Iweala E, Popoola A, Jibrin P, Faruk M, Sowumi A, Fatiregun O, Blaise N, Oladoyinbo C, Okoye I, Salako AA, Omonisi A, Bassey IE, Adeniji K, Asura NH, Kaninjing E, Kukoyi O, Fathi P, Enuka R, Toye O, CaPTC Investigators, Odedina F. Vitamin C prediction of cancer diagnosis in men of African ancestry in Nigeria, Cameroon, and the United States: An analysis of the CaPTC prostate cancer study [abstract]. In: Proceedings of the 15th AACR Conference on the Science of Cancer Health Disparities in Racial/Ethnic Minorities and the Medically Underserved; 2022 Sep 16-19; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Epidemiol Biomarkers Prev 2022;31(1 Suppl):Abstract nr C068.
- Otero J, Iweala E, Popoola A, Jibrin P, Faruk M, Sowumi A, Fatiregun O, Blaise N, Oladoyinbo C, Okoye I, Salako AA, Omonisi A, Bassey IE, Adenji K, Asura NH, Kaninjing E, Kukoyi O, Fathi P, Enuka R, Toye O, Crook J, Odedina F. Influence of access to care on cancer diagnoses in the CaPTC Prostate Cancer Familial Cohort Study of African Ancestry in Nigeria, Cameroon, and the United States [abstract]. In: Proceedings of the 15th AACR Conference on the Science of Cancer Health Disparities in Racial/Ethnic Minorities and the Medically Underserved; 2022 Sep 16-19; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Epidemiol Biomarkers Prev 2022;31(1 Suppl):Abstract nr B072.
- Thomas, C.; Lamoureux, F.; Crafter, C.; Davies, B.R.; Beraldi, E.; Fazli, L.; Kim, S.; Thaper, D.; Gleave, M.E.; Zoubeidi, A. Synergistic Targeting of PI3K/AKT Pathway and Androgen Receptor Axis Significantly Delays Castration-Resistant Prostate Cancer ProgressionIn Vivo. Mol. Cancer Ther. 2013, 12, 2342–2355. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.; Kim, S.; Cordonnier, T.; Crafter, C.; Davies, B.R.; Fazli, L.; Gleave, M.E.; Zoubeidi, A. Combination AZD5363 with Enzalutamide Significantly Delays Enzalutamide-resistant Prostate Cancer in Preclinical Models. Eur. Urol. 2015, 67, 986–990. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Androgen receptor gene and protein.
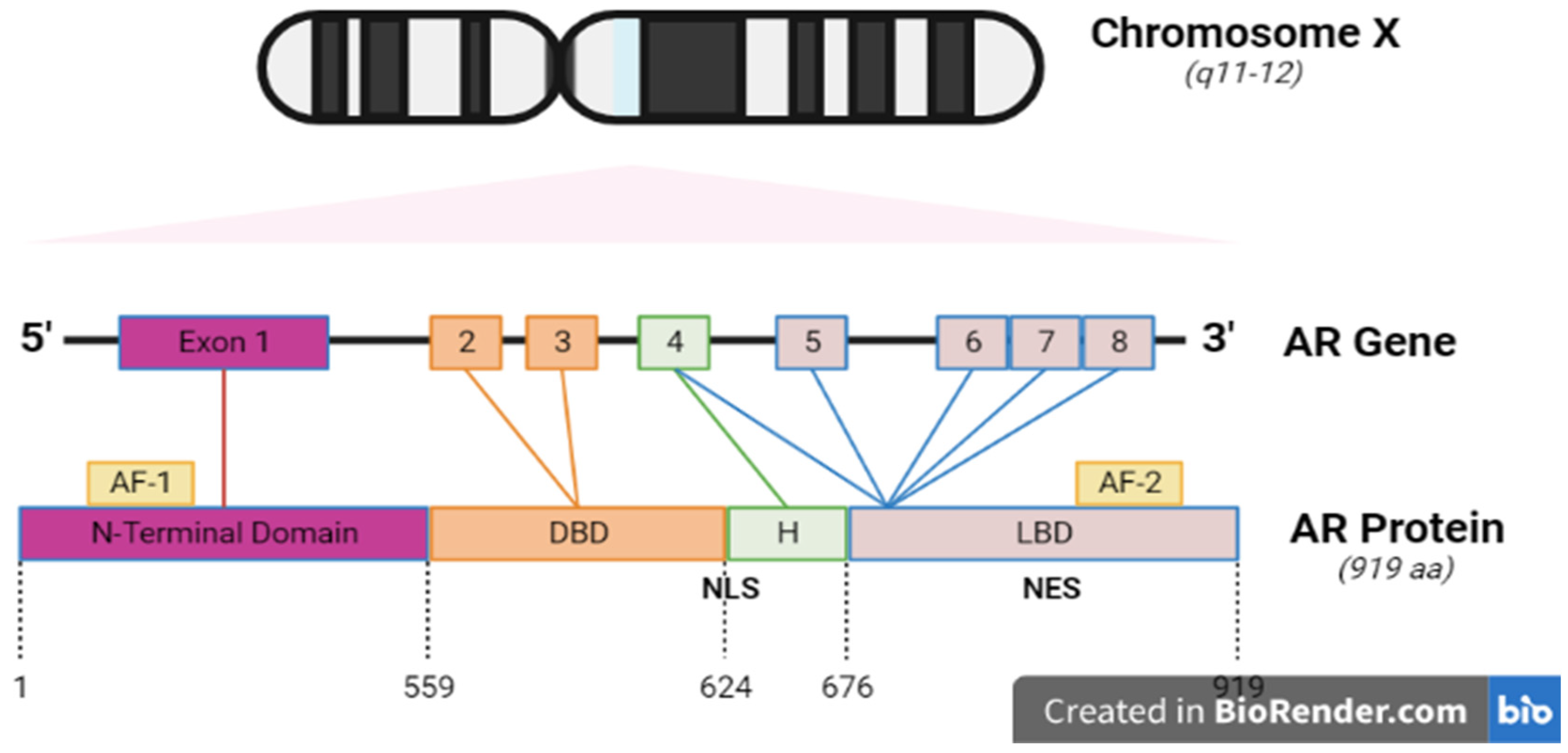
Figure 4.
Androgen biosynthesis.
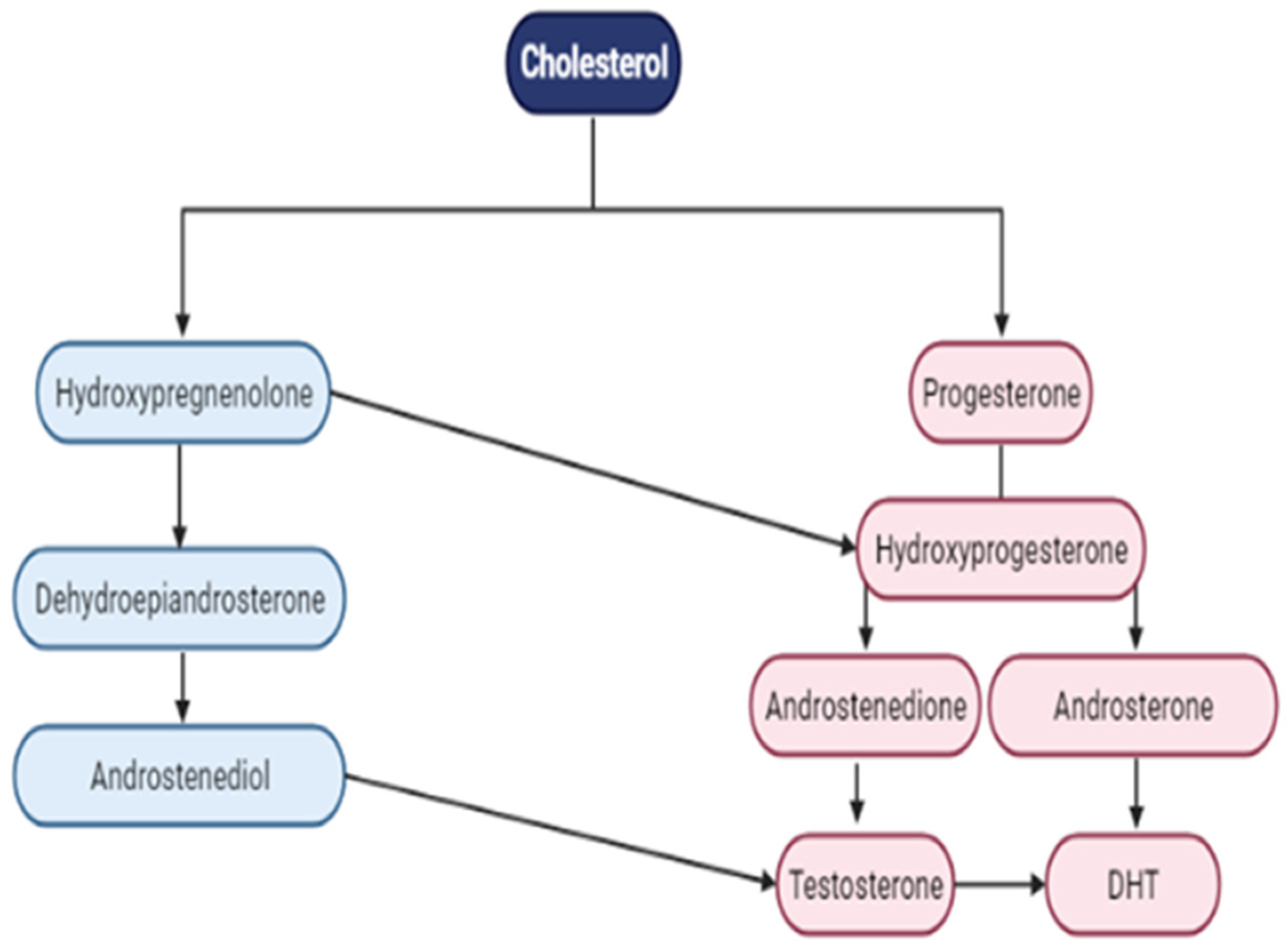
Figure 5.
Androgen signaling pathway.
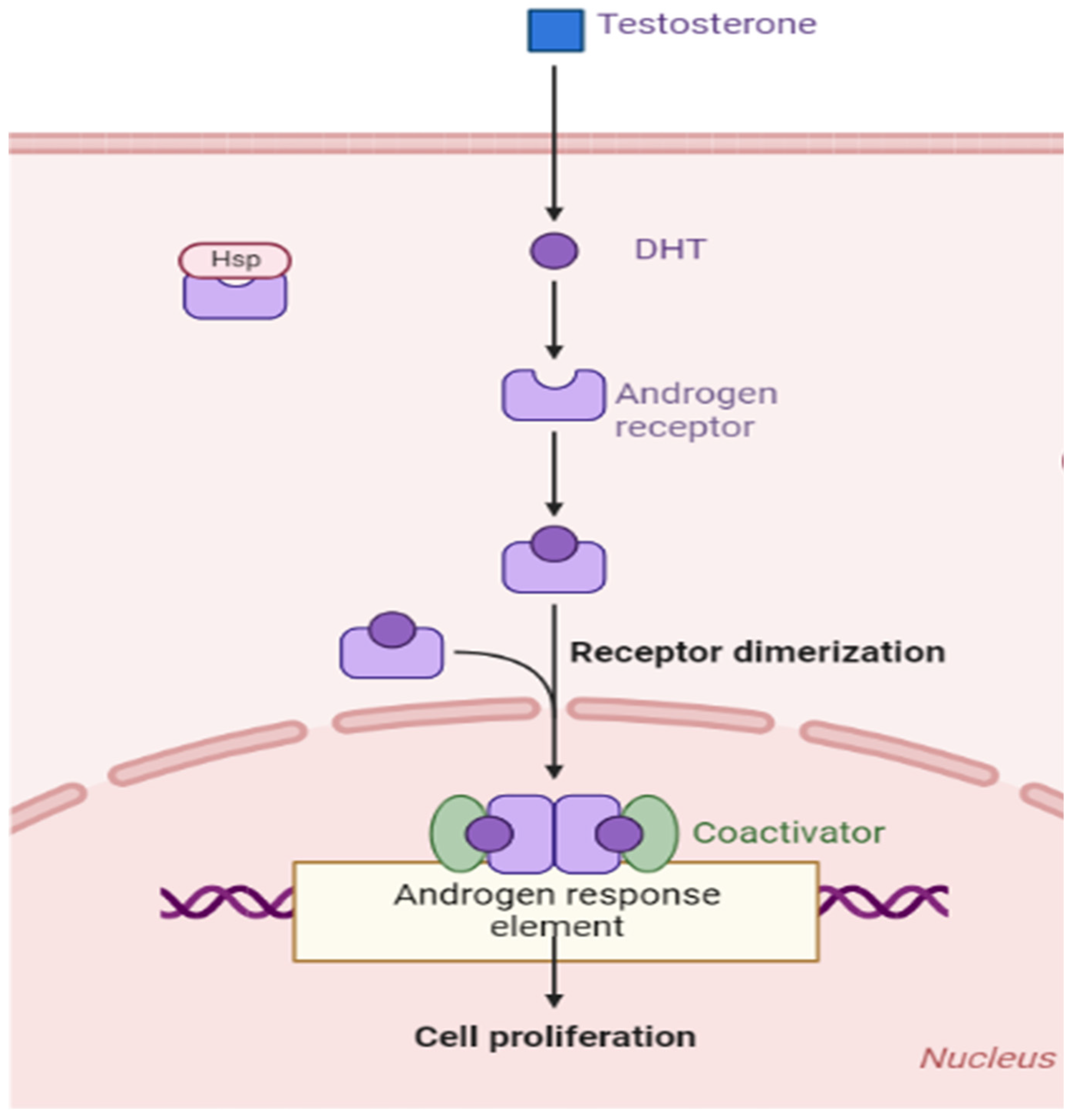

Table 1.
Combination therapies targeting AR and PI3K/AKT/mTOR pathways.
| Target | Agent | Phase | Administration | Condition | I.D on Clinicaltrial.gov |
|---|---|---|---|---|---|
| AKT | AZD5363 | III | Docetaxel | mCRPC | NCT05348577 |
| MK2206 | II | Bicalutamide | High-Risk of Progression | NCT01251861 | |
| Capiversertib | II | Abiraterone acetate | High Risk Localized PCa | NCT05593497 | |
| PI3K | AZD8186 | I | Docetaxel | mPCa with PTEN Mut | NCT03218826 |
| GSK2636771 | I | Enzalutamide | PTEN(-) mCRPC Mut | NCT02215096 | |
| mTOR | Sepanisertib | II | Monotherapy | CRPC | NCT02091531 |
| Everolimus | I | + standard radiation therapy | PCa with rising PSA | NCT01548807 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



