
Submitted:
10 May 2023
Posted:
10 May 2023
You are already at the latest version
Abstract
Oxidative stress (OS) is involved in the pathogenesis of retinal neurodegenerative diseases like age-related macular degeneration (AMD) and diabetic retinopathy (DR) and an important target of therapeutic treatments. New therapeutics are tested in vivo despite limits in transferability and ethical concerns. Retina cultures using human tissue can deliver critical information and significantly reduce the number of animal experiments along with increased transferability. We cultured up to 32 retina samples derived from one eye, analyzed models’ quality, induced OS, and tested efficiency of antioxidative therapeutics. Bovine, porcine, rat, and human retinae were cultured in different experimental settings for 3-14 d. OS was induced by high-glucose or hydrogen peroxide (H2O2) and treated by Scutellarin, pigment epithelium-derived factor (PEDF), and/or granulocyte macrophage-colony stimulating factor (GM-CSF). Tissue morphology, cell viability, inflammation, and glutathione level were determined. Retina samples showed only moderate necrosis (23.83±5.05 increased to 27.00±1.66 AU PI-staining over 14 d) after 14 days in culture. OS was successfully induced (reduced ATP content of 288.3±59.9 vs. 435.7±166.8 nM ATP in controls); antioxidants reduced OS-induced apoptosis (from 124.20±51.09 to 60.80±319.66 cells/image after scutellarin-treatment). Enhanced mammalian animal and human retina cultures allow reliable, highly transferable research on OS-triggered age-related diseases and pre-clinical testing during drug development.
Keywords:
retina organ culture
; neuroretinal degenerative disease
; oxidative stress
; antioxidant
; age-related macular degeneration (AMD)
; diabetic retinopathy (DR)
; Scutellarin
; PEDF
; GM-CSF
; 4R
1. Introduction
Aging is seen as a disruption of homeostasis and affects all organs [1]. One key factor is oxidative stress (OS) induced by an imbalance of reactive oxygen species (ROS) and a weakened antioxidant cellular defense, though, in normal aging, the excess of ROS can be still tackled [2]. Nevertheless, this imbalance together with mitochondrial dysfunction, damaged protein accumulation, epigenetic alterations, telomere shortening, and abnormal intracellular signaling result in the frailty syndrome, which is defined by a lowered physiological reserve and reduced resistance to stressors [3]. In accelerated aging, ROS production cannot anymore be controlled and age-related diseases like cardiovascular diseases, metabolic disorders, cancer, and neurodegeneration are the consequence [1,2,3]. Particularly in industrialized countries, health care improved and consequently life expectancy increased [4,5]. Thus, the prevalence of age-related diseases is increasing significantly and becomes a severe burden for the quality of life of patients, health care and socio-economic systems (as analyzed, e.g., by Bae et al. for Korea [6]).
Age-related macular degeneration (AMD) and diabetic retinopathy (DR) belong to this group of OS-triggered, age-related disorders [7,8,9]. AMD is the major cause of blindness in elderly patients in industrialized countries and DR is the main reason of severe vision loss in the working age population [10]. In contrast to an imbalance of angiogenic and anti-angiogenic factors as seen in neovascular AMD (nAMD), the avascular form of AMD (aAMD) is mainly triggered by inflammatory processes and increased OS [11]. Bruch’s membrane and retinal pigment epithelial (RPE) cells become dysfunctional, and metabolic waste products (“drusen”) accumulate [11]. This breakdown of the retinal homeostasis leads to RPE cell death, followed by photoreceptor and finally, retinal neuron cell death cumulating in geographic atrophy (GA), the late stage of aAMD [11]. In DR, badly controlled hyperglycemia leads to microangiopathy, vessel occlusion, and leakage of the blood-retina barrier causing hemorrhages, edema, and diminished retinal neuronal function; finally, it results in retinal neurodegeneration and cell death [12]. It has to be noted that the brain and the eyes are particularly prone for OS and the development of associated diseases due to their particularly high oxygen consumption but, on the other hand, restraint antioxidant defense capacities [2,13]. A better understanding of underlying pathomechanisms and treatment approaches for these diseases is highly demanded.
Novel treatments have to be tested in vivo in animals to get approved by regulatory authorities; however, ethical issues and limits in transferability to patients make the development of alternative models necessary [14]. Cell culture is limited to preliminary testing due to missing complexity and transferability of the systems [14,15]. Organ cultures can be performed using human tissue and perfectly combine the methodological simplicity of in vitro studies with higher biological complexity as of in vivo models [16]. Additionally, they save animals and are ethically preferable to in vivo projects (4R principles: refine, reduce, replace, responsible). For example, retina organ culture allows for analysis of pathomechanisms including OS, and for testing different treatments or doses of molecules on multiple samples from the same individual animal/human donor eye. It was first described in 1926 by Strangeways and Fells who cultured 64-72 h young chick embryonic eyes up to 32 d in an extract from 64-72 h young chick embryos mixed in a balanced salt solution [17]. However, compared to the culture of fetal or newborn tissue that contains potent immature, retinal progenitor cells, the culture of adult, mature retinae remains challenging independent from the quality of the starting material and due to special demands in culture conditions. Murali et al. reported cell culture of isolated neural retinal cells (from 31-89 years old human donors) for up to 8 weeks as suspension culture [18]. However, culture of the complex retinal tissue structure (from adult donors) can be maintained only for 7 to 14 d; missing vascular perfusion, RPE, connectivity to the brain via the optic nerve, and the continuous high demand of nutrients and oxygen are fostering degeneration by inflammation and OS [19]. Moreover, except one report [20], retinal function, i.e., electric activity can be recorded only up to 8 h post-mortem [19,21,22]. Finally, the culture of aged and diseased human retinae - the tissue of interest in research of OS and ageing and useful to analyze pathomechanisms and to test novel treatment approaches in disease models, is even more challenging; OS-induced damage and other imbalances of normal retinal homeostasis are rendering maintenance of the tissue in culture additionally difficult. Despite these challenges, important insights into retinal physiology [23,24] and pathology [25,26] have been gained, and novel treatment options [27,28] been evaluated in retina organ culture.
We are working on the enhancement of adult/aged retina organ culture. It is used as a preclinical test system for novel treatments against retinal neurodegeneration like the cell-based non-viral ex vivo gene therapy approach to treat AMD. To recover a cell-protective, antioxidant retinal environment, RPE or iris pigment epithelial (IPE) cells are transfected using the non-viral Sleeping Beauty (SB100X) transposon system with the genes coding for PEDF and GM-CSF [29,30]. PEDF is a cell protective protein with multiple beneficial functions as evaluated, e.g., in models of osteosarcoma [31] and photoreceptor and amacrine cell survival [32]. PEDF, ubiquitously expressed by RPE cells, is a molecule that acts anti-angiogenic, anti-tumorigenic, and neurotrophic. It promotes survival and function of neurons, protects from oxidative damage, and inhibits apoptosis by activation of the NF-κB signaling pathway [33]. GM-CSF is a multi-faceted factor expressed in RPE cells [34] responsible for proliferation, differentiation, and maturation of myeloid cells and adaptive immune responses to inflammation and infection [35] especially in the central nervous system [36]. It counteracts oxidative damage by inhibiting apoptosis via the SRC-dependent STAT3 (signal transducer and activator of transcription 3) pathway, decreased BAD (Bcl-2 agonist of cell death) and increased BCL-2 (B-cell lymphoma 2) expression, and GM-CSF upregulates the production of neurotrophic factors [37]. Alongside, the phytochemical Scutellarin, found in Scutellaria Barbata, S. lateriflora and S. baicalensis, is tested as antioxidant nutrient in DR [38]. The flavonoid is used in Chinese medicine with confirmed efficiency in, e.g., cardiovascular diseases and diabetic complications [39] due to its function as a potent radical scavenger, inhibitor of induced nitric oxide synthase (iNOS) expression, lipid peroxidation, and cell death [39].
In the present work, different retina organ culture models were evaluated, and their advantages and drawbacks as preclinical test system discussed. Techniques were continuously improved and OS-triggered degeneration provoked by high-glucose or H2O2 treatment. The antioxidant and cell protective effects of Scutellarin, PEDF, and GM-CSF were analyzed in retina culture confirming the suitability of the model as pre-clinical test system in general and for OS-triggered retinal neurodegeneration particularly.
2. Materials and Methods
2.1. Animals and cells
Globes from adult cattle and pig derived from local slaughterhouses 1 to 6 h from slaughter, transported on ice, were used for Models 1-3 and 5a. Adult rat retinae from 8 weeks aged Brown Norway rats (Charles River, L'Arbresle Cedex, France) were used for Model 4 immediately after sacrifice. In the universities’ animal facility (University of Geneva, Geneva, Switzerland), animals had access to food and water ad libitum. Euthanasia was performed under general anesthesia using ketamin (Ketalar®, Pfizer, Zurich, Switzerland; 100 mg/kg) and Xylazine (Rompun®, Bayer, Leverkusen, Germany; 10 mg/kg) diluted in NaCl (Merck, Darmstadt, Germany), at room temperature (RT), intra-peritoneally (i.p.) injected; then, Thiopental (Inresa, Freiburg, Germany; 150 mg/kg) diluted in NaCl was i.p. injected. Human donor eyes were purchased from the Lions Gift of Sight Eye Bank (Saint Paul, MN, USA) 2–8 d post-mortem.
2.2. Tissue isolation and culture conditions
2.2.1. Bovine Retina-RPE culture (Model 1)
The eyes were washed 3 min with 70% Ethanol (EtOH) (Bichsel, Interlaken, Switzerland) followed by ice cold sterile phosphate buffer saline (PBS, Merck, Darmstadt, Germany) containing 0.1% EtOH. The anterior segment was cut approximately 3.5 mm posterior to the limbus and the posterior segment was flattened by a radial incision of the sclera using microscissors (Fine Science Tools, Heidelberg, Germany). With the help of forceps (Fine Science Tools), the vitreous was carefully removed and the globe was sectioned into four pieces. Using a cell scraper (Milian, Boswil, Switzerland), the retina-retinal pigment epithelium (RPE)-choroid complex was separated from the underlying sclera and placed on a nitrocellulose membrane (pore size 0.45 µm, Schleicher & Schuell, Dassel, Germany) with the choroid facing downwards.
The retina-RPE-choroid complex was then sectioned into 5 x 5 mm samples using a surgical blade (Swann-Morton, Sheffield, UK). Nine samples were placed on customized, sterile, perforated, high-grade stainless-steel grid support benches in 12-well culture plates (Vitaris AG, Baar, Switzerland) and the wells were filled with serum-free DMEM (GIBCO, Eggenstein, Germany) supplemented with 2.8 mM L-glutamine (GIBCO), 673 U/mL penicillin and 673 µg/ml streptomycin (Merck) (n=36 from 4 retinas). All wells were filled with medium up to a level just below the upper, retina-vitreous interface of the explant. Culture medium was changed every 24 h for a total culture duration of 4 d.
2.2.2. Dynamic porcine Retina culture (Model 2)
Enucleated eyes were cleaned from rests of muscle tissue using forceps and microscissors (Fine Science Tools) and put into a beaker (Schott, Jena, Germany) filled with PBS (Merck). Then, globes were disinfected for 2 min in 70% EtOH (Bichsel) before they were transferred to a beaker (Schott) filled with sterile Ringer solution (B.Braun, Melsungen, Germany). For RPE-retina complex isolation, eyes were placed on a mull compress (Lohman & Rauscher, Rengsdorf, Germany) on a ceramic tile. Using loupe-glasses (magnification 2.5x, Starmed, Grafing, Germany), the bulb was opened by a radial cut with a scalpel no. 11 (VWR, Dietikon, Switzerland) within the pars plana to ensure that retina and RPE stay bound. Using forceps, the anterior part of the globe was lifted, the vitreous grasped using Colibri forceps, both separated from the posterior part and discarded. Always the nasal orientated quarter of each retina (part of the visual streak and thus showing a high rod density) was cut from the rest of the posterior part using forceps and microscissors (Fine Science Tools); drying of the tissue was avoided by rinsing with Ringer solution. Under tension, the tissue was fixed on styrofoam with three canulae (Beckton & Dickinson, Franklin Lakes, NJ, USA). At the ora serrata, the RPE was detached from the capsule, held by Colibri forceps. Between capsule and RPE by dissecting present vessels, a pocket has been carefully prepared without lesioning RPE and retina. The white ring of the MinuSheet® system (Minucells and Minutissue Vertriebs GmbH, Bad Abbach, Germany) [40,41] was pushed into the pocket while the black ring was placed onto the tissue and the ring system gently closed using blunt forceps to avoid lesioning of the tissue. Excess tissue was cut, and the sample placed in the MinuSheet® chamber for perfusion culture. One chamber was filled with 6 samples, which were vertically oriented to the flow of medium, which was freshly prepared and stored for max. 3 d at 4°C. During perfusion, the ice-cooled medium flew with a fix flowrate of 56.5 µm/h through semi-permeable silicon tubes into the airtight closed Minuth-chamber passing a peristaltic pump (Fisher Scientific, Reinach, Switzerland). The flow rate was chosen to create a fluid pressure in the range of normal in vivo intraocular pressure (10-21 mmHg). The chamber and at least 10 cm of the tubes were placed on a 42°C warmed heating plate to guarantee an inner-chamber temperature of 37°C; for experiments at 21°C inner chamber temperature, perfusion took place at RT. Finally, the system was protected by a heat-conserving plastic cover. Used medium left the perfusion chamber after passing the tissue and was collected in a glass bottle (Schott). The maximum culture duration was 3 d. Used media were DMEM (Biochrome, Cambridge, UK) or Ames’ medium (8.9 g/L, Merck) supplemented with 10 mL Amphotericin B (AMIMED, Allschwil, Switzerland), and 8 mL Penicilin/Streptomycin (Merck) for 1 L medium; Ames’ was additionally supplemented with 0.538 g NaCl (Merck), 0.634 g NaHCO₃ (Merck), and 5.957 g HEPES (Merck).
2.2.3. Static porcine Retina culture (Model 3)
Before rinsing the globes in ice-cold PBS (Merck), followed by disinfection in EtOH 70% for 2 min and rinsing in sterile ice-cold PBS, remaining muscle tissue and adnexa were removed with surgical scissors (Fine Science Tools). A scleral incision was made using a surgical blade around 3 mm posterior to the limbus and extended circumferentially with surgical scissors to dissect the anterior segment; the vitreous was kept in place to hydrate the retina and avoid RPE detachment. Next, the globe was cut into five equal pieces by cuts from the limbus to the optic nerve using scissors (Fine Science Tools). The samples were placed with the vitreous downwards onto a glass Petri dish; the retina was gently dissected from overlaying RPE, choroid, and sclera. Remaining RPE cells were washed off using BSS Plus (Alcon, Zug, Switzerland). A sterile, 15 x 15 mm small nitrocellulose membrane was placed on the photoreceptor layer, the tissue was turned and the vitreous removed. The tissue samples were put on customized, sterile, perforated, high-grade stainless-steel grid support benches (0.8 mm) in 12-well culture plates with the photoreceptors directed downwards; culture wells contained only 1.5 mL medium allowing the retinae having air contact. Retinae were cultured for 1 to 4 d at 21°C, 5% CO2, and 95% humidity. Medium was not exchanged but freshly added if the level decreased below the photoreceptor layer due to evaporation. The medium used was Ames’ (8.9 g/L, Merck) supplemented with 2 mM NaCl (Merck), 7.5 mM NaHCO₃ (Merck), 25 mM HEPES (Merck), 1% Amphotericin B, and 0.8% Penicillin/Streptomycin.
2.2.4. Rat Retina culture (Model 4)
Due to the smaller size of the rat eye, tissue preparation was performed under semi-sterile conditions using a dissection microscope under a laboratory hood based on protocols from Bull et al., Kaempf et al. and Leroy-Ciocanea [38,42,43]. Immediately after sacrifice of the animals and enucleation, globes were washed in PBS (Merck) followed by disinfection for 30 s in Betadine® (Mundipharma, Frankfurt, Germany) before being again rinsed in sterile PBS. A scleral incision was made posterior to the limbus using a 25G needle (Beckton & Dickinson, Franklin Lakes, NJ, USA) and extended circumferentially using Vannas scissors (Fine Science Tools) to dissect the anterior from the posterior segment. As explained in Model 3, the vitreous was kept maintaining the retina hydrated and to avoid detachment of the RPE layer. Using Vannas scissors, the retina was then cut into two halves by two cuts towards the optic nerve head. The samples were transferred to a glass Petri dish with the vitreous directed downwards and, using a small steel sooth spoon, RPE, choroid, and sclera were removed. A sterile, 10 x 10 mm small nitrocellulose membrane was placed on the photoreceptor layer of the retina, the complex turned and placed on customized, sterile, perforated, high-grade stainless-steel grid support benches (0.8 mm) in 12-well culture plates with the photoreceptors directed down-wards; to allow air contact, only 1.5 mL medium were added to culture wells. The tissue was cultured at 21°C, 5% CO2, and 95% humidity for 9-13 d.
Different medium conditions were tested to improve the culture system, but in any case, half of the medium was exchanged every other day. First, Ames’ medium as described above was used; secondly, Neurobasal-A medium (ThermoFisher Scientific, Waltham, MA, USA) supplemented with the neurotrophic factors 2% B-27 and 1% N-2 (ThermoFisher Scientific) was tested. Third, Ames’ medium supplemented with 2% B-27 and 1% N-2 was added to culture wells; all media were supplemented with 10 mL Amphotericin B, and 8 mL Penicillin/Streptomycin in 1 L medium.
2.2.5. Semi-long porcine and human Retina culture (Model 5a and 5b)
Porcine and human retinae were similarly treated for culture. As aforementioned, the globes were rinsed in PBS (10 mL, Merck), disinfected in Betadine® (10 mL, 2 min) and a second time washed in sterile PBS (10 mL, 2 min) after removal of remaining muscles and adnexa with surgical scissors. A flat scalpel no. 11 (VWR) was used to make a small incision around 2 mm below the limbus and extended with curved micro-scissors (Fine Science Tools). Cornea, iris, lens, and vitreous were removed using forceps before four cuts towards the optic nerve were made into the posterior part using scalpel no. 10 (VWR) to flatten the tissue. The retina was detached from the RPE by flushing the tissue with sterile PBS and allowed to “slide” into a glass Petri dish (90 mm) filled with 10 mL sterile PBS with the photoreceptors directed downwards. A 6 mm small biopsy punch (Stiefel, Brantford, UK) was used to cut up to 32 samples per retina; the punch had to be positioned perpendicular to the retina and twisted a few times. The retina remained in the punch and like that could be easily transferred to a culture insert (0.4 µm permeable, transparent PET membrane, Corning, Corning, NY, USA) prefilled with 1 mL sterile PBS. Then, the retina detached itself from the punch and settled down to the base by gravity when immersed in PBS; if necessary, the punch was moved up and down slowly to allow detachment. PBS was carefully removed with a pipette without touching the tissue before transferring the insert to a 12-well plate prefilled with 1 mL Ames’ medium and culturing at 21°C and 5% CO2 for 14 d; medium was exchanged every day. Explants were cultured in serum-free Ames’ medium (pH=7.4) supplemented with growth factors 1% N2 and 1% B27 along with 1% antibiotics (Penicillin/Streptomycin) and 1% fungicide (Amphotericin B).
All models 1-5 are summarized and juxtaposed in Table 5.
2.2.6. Induction of OS by high glucose conditions and Scutellarin treatment
In Model 3, high glucose conditions were induced to mimic diabetic conditions and treated with different concentrations of Scutellarin. Commercially available Ames’ medium contains 6 mM D-glucose (1.081 g), which was increased by addition of 24 mM D-glucose (Merck) to a final concentration of 30 mM to reach high-glucose levels. Scutellarin (Xie Qifan, Chengdu, China) was added in concentrations of 1, 10 or 100 µM. Other culture conditions were maintained.
2.2.7. Induction of OS by H2O2 incubation and PEDF/GM-CSF treatment
OS was induced on retinae (Model 4) by adding 300 μM H2O2 (Merck) for 3 h on day 3. To test their neuroprotective and anti-oxidative effect, 500 ng PEDF (BioProductsMD, Middletown, MD, USA), 500 ng GM-CSF (Peprotech, London, UK) or both were added to the medium for 3 days; due to the short half-life time of the proteins, the medium was exchanged every day with freshly added proteins from day 1 to day 3. Cultures were terminated on day 13.
2.3. Brightfield microscopy of flatmount preparations and degeneration score
For Model 3, samples of one retina were fixed in one block but separated by the nitrocellulose membrane in freshly prepared 4% PFA (Applichem, Darmstadt, Germany) for 12 h at RT. Next, retinae were rinsed in PBS (Merck), dehydrated through a series of increasing EtOH concentrations and Xylol (Reactolab, Servion, Switzerland) (Table 1) before being embedded in Paraffin (Paraplast, Leica Biosystems, Muttenz, Switzerland) and stored at RT until use. Sections were analyzed for tissue integrity by bright field (BF) microscopy (Leica DM4000, Leica, Heerbrugg, Switzerland). One sample of the freshly isolated retina was mounted without fixation on a slide for BF microscopy.
For Models 5a and 5b, tissue processing was enhanced to preserve retinal laminar structure and to minimize damage during transfer from the culture insert to microscope slide (Superfrost Plus, VWR). First, the slide was cleaned with EtOH and dried for 5 min before two 5.5 mm small strips of double-sided scotch were put on both ends of the slide. A 15 x 15 mm large coverslip (ThermoFisher Scientific) was placed on top of the scotch tape. Then, 1 mL sterile PBS was pipetted on the slide. The membrane of the culture insert was cut out with a scalpel no. 11 without touching the tissue and placed in the drop of PBS. With help of forceps, the retina slipped in the PBS. The PBS was aspirated and replaced by a drop of mounting medium (ProLong Gold Antifade Reagent, ThermoFisher Scientific); a large cover slip (40 x 60 mm, ThermoFisher Scientific) was gently placed on the top and the sample dried for 15 min. Samples were evaluated by BF microscopy at a magnification of 100x using (Leica DM4000); five images were taken per sample with one micrograph from each peripheral side and one from the center. To quantify degeneration, a score shown in Table 2 from 0 to 3 was created.
2.4. Hemalum and Eosin (H&E) staining
After culture termination, samples were fixed in 4% PFA; for Model 1, every sample was fixed individually, for Model 2 to 4, all samples of one retina were fixed in one block, for Model 5a/b, two samples were fixed and embedded in one block separated by nitrocellulose membrane. Except for Model 5a/b (2-4 h, RT), samples were fixed for 12 h at RT. In Model 5a/b, tissue preservation was optimized by placing samples in a cassette cushioned with filter paper on both sides and a mesh on one side to prevent the movement of the tissue in the cassette. After rinsing in PBS (Merck) or distilled water, tissues were dehydrated by an increasing EtOH and Xylol series (Table 1). Then, samples were embedded in paraffin and cut into 5-6 μm thin slices. Before being stained with H&E (VWR), samples were deparaffined and rehydrated; finally, the tissue was mounted (Table 1). Two images per sample at 100x, 200x and 400x magnification were recorded.
2.5. Cell death and viability determination
2.5.1. CytoTox GloTM assay
CytoTox GloTM assay Model 5a, b. Retina viability was assessed by the CytoTox-GloTM assay (Promega, Fitchburg, MA, USA), which is based on the release of a protease by damaged cells with compromised membrane integrity. Measurements were done in triplicates using the FLUOstar Omega spectrophotometer (BMG Labtech, Ortenberg, Germany) (setting: gain: 3600; positioning delay: 0.1; measurement start: 0.0; measurement interval time: 1.0; time to normal: 0.0). Luminescence was directly used to compare cell viability at different days in standardized 6 mm punched samples and not normalized to a cell count. Supernatant was collected 24 h after medium change on days 3, 6, 9, 11 and 14. Fifty microliter of CytoTox-Glo reagent was added to 100 µL culture medium and incubated in the dark for 15 min at RT on an orbital shaker. A standard curve using ARPE-19 cells was prepared (10’000, 5’000, 2’500, 1’250, 625, and 313 cells).
CytoTox GloTM assay (after H2O2 treatment). The same assay was used to determine viability of retinal tissue after H2O2-induced OS. Here, samples were weighed, and luminescence correlated to the weight of the tissue. Retinae were cut into 2 halves using scalpel no. 10. With the help of Colibri forceps (Fine Science Tools), each half was transferred to a 1.5 mL tube (Milian) and weighed. Next, 50 μL of CytoTox-Glo reagent was added to each well, gently mixed and incubated in the dark on an orbital shaker at RT for 15 min, before luminescence was measured. Subsequently, according to manufacturer’s instructions, lysis reagent was added and luminescence was again measured after incubation in the dark. After 9 d of culture and the loss of the supportive nitrocellulose membrane, the tissue did not need a homogenization step; careful mixing with the assay reagent’s and shaking during incubation was sufficient.
2.5.2. CellTiter Glo® viability assay
The CellTiter-Glo® cell assay (Promega) detects cellular ATP, and thus determines the amount of metabolic active cells visualized by luminescence. Using a dermatological Biopsy Punch of 5 mm diameter (Stiefel), size-standardized samples from the different times of culture and experimental conditions were prepared. Samples were homogenized by up and down pipetting in 500 μL normal glucose medium. Ten microliters of one sample were diluted 1:1 in Trypan Blue (Fisher Scientific) and counted in a Neubauer chamber to estimate the cell number of all samples. Two hundred-fifty thousand cells in 100 μL normal glucose medium per sample were incubated in a 96-well plate. The assay was performed in triplicates according to manufacture instructions. Briefly, 100 μl CellTiter-Glo® reagent was mixed with the cells on an orbital shaker for 2 min. The plate was incubated 10 min at RT and luminescence recorded using the FLUOstar® spectrophotometer; the gain was adjusted for each measurement and an ATP standard curve of serial ten-fold dilutions (1 nM to 10 μM) was generated.
2.5.3. Propidium Iodide (PI) staining
Necrosis was determined in samples processed as described in section 2.3 in duplicates per analysis day by PI staining. Forty microliters of the 2 mg/mL PI stock solution (Merck) were diluted in 4 mL sterile PBS to a final working concentration of 0.01%. To maintain retinae’s orientation, PI staining solution was added to the culture insert carefully from the side. Samples were incubated in the dark at 21°C for 10 min. Next, tissues were rinsed twice with 1 mL of sterile PBS for 5 min before being mounted on a glass slide (section 2.3.) and microscopic pictures immediately taken at 100x magnification using a fluorescence microscope; five images were taken from every sample with one image from every peripheral side and one from the center.
2.5.4. TUNEL assay
The In Situ Cell Death Detection Kit® Fluorescein (Merck) was used to determine apoptosis in paraffin embedded retinal cross sections; broken DNA strands in apoptotic cells are detected by incorporation of fluorescein (FITC)-labelled nucleotides polymerized by the terminal deoxynucleotidyl transferase. Apoptotic cells were then visualized by fluorescence microscopy (Leica DM4000) and counted manually on images (1 micrograph/sample) of 200x magnification using ImageJ (software version 2.3.0/1.53q, National Institute of Health, Bethesda, MD, USA). The assay was performed according to manufacturers’ instructions.
2.6. GSH assay
The luminescent-based GSH-Glo™ assay (Promega) quantifies glutathione (GSH) in cells or tissues to assess cellular OS. Retinae were prepared as described in section 2.5.1 and luminescence correlated to the weight of the tissue. Each half was transferred to a 1.5 mL tube prefilled with 50 μL PBS. The assay was performed according to the manufacturer's instructions. Briefly, 50 μL of the cell suspension was transferred to a 96-well plate and 50 μL of GSH-Glo™ Reagent 2X were added to each well. After mixing the solution, it was incubated on an orbital shaker in the dark at RT for 30 min. Subsequently, 100 μL of the Luciferin Detection Reagent were added to each well, mixed and incubated in the dark on an orbital shaker at RT for 15 min. Luminescence was measured using the FLUOstar Omega spectrophotometer applying same settings as described in section 2.5.1. A standard curve of 5 to 0.005 mM glutathione was prepared.
2.7. Immunohistology
Single and double immunofluorescence staining were performed on paraffin embedded retinal sections processed as described in section 2.4. and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) according to the protocol described in Table 3. Table 4 lists used primary and secondary antibodies. After mounting, micrographs were recorded at 100x, 200x and 400x magnification using the Leica DM4000 fluorescence microscope taking three images from every sample (right side, left side, center).
2.8. Statistics
Descriptive statistics were calculated for all data (mean, SD, SEM, min, max, range). A D’Agostino & Pearson normality test recognized parametric and non-parametric data. Differences between groups were calculated using the parametric t-test (2 samples), ANOVA with Tukey’s multiple comparison test (≥3 samples; in case of missing values, a mixed-effects model was applied), non-parametric Mann-Whitney (2 samples) or Kruskal-Wallis with Dunn’s post-test (≥3 samples) depending on the nature of the data. Time courses were analyzed applying a paired ANOVA. If not noted otherwise, data are shown as mean±SD. All analyses were made using the GraphPad Prism® software version 9.3.1 (471) (GraphPad Software, LLC, San Diego, CA, USA).
3. Results
3.1. Model 1: Bovine, static, 4 days
Model 1 used bovine eyes and delivered first insight into the feasibility of ex vivo cultured retinae especially when cultured in conjunction choroid and RPE layers, and inflammatory reactions. H&E-stained sections confirmed that tissue integrity including photoreceptor outer segments (POS) was maintained for 4 d (Figure 1a). Nevertheless, signs of degeneration were visible, i.e., holes and cell loss in the retinal ganglion cell (RGC) and inner nuclear (INL) layer, and the connection between RPE and POS got lost. The protein kinase C (PKC) selectively stains bipolar cells indicating cell loss (reduced staining) or inflammation (increased staining); in the present model, the PKC expression (green) did not decline over time (Figure 1b, bottom) compared to sections stained directly after isolation (Figure 1b, top). The second inflammatory marker, the glial fibrillary acidic protein (GFAP), identifies “activated” Müller cells; shown pictures demonstrated a low positive GFAP signal at 4 d of culture (Figure 1c, bottom) compared to retinae stained directly after isolation (Figure 1c, top).
3.2. Model 2: porcine, dynamic, 3 days
Model 2 cultured porcine retinae and tested a perfusion culture. The aim of the study was to evaluate potential advantages of a perfusion culture, to optimize media and to compare the benefit of the isolation of the RPE-retina complex vs. the neural retina alone. To that goal, cell counts of retinal layers performed in H&E-stained retinal cross sections from retinae cultured 1 d with or without RPE were compared to retinae stained directly after isolation (“fresh”). Cultured neural retinae showed comparable cell counts as to directly processed tissue, while cell counts were significantly reduced in RPE-retina cultures in the outer nuclear (ONL), INL and RGC layers (Figure 2a,b). H&E-stained cross sections shown in Figure 2c confirmed better-preserved structure of samples isolated and cultured without RPE compared to RPE-retinae cultures.
Figure 3 reports the results of the comparison of Ames’ medium and DMEM in neural retinae cultured for 1 d. In the ONL, cell counts did not differ significantly, but showed a trend to lowered cell numbers in DMEM (Figure 3a,b). In the INL and RGC layers, this difference is significant with reduced cell counts after culture in DMEM. The result is supported by better tissue preservation shown in H&E-stained cross sections from retinae cultured in Ames’ medium (Figure 3c).
Next, culture temperatures 37°C vs. 21°C were compared. Cell counts in the ONL decreased from “21°C”, to “fresh”, to “37°C” samples. Also in the INL, highest numbers were counted in the “21°C” group, followed by almost similar counts in the groups “fresh” and “37°C” (Figure 4a,d). In the RGC, no differences between the groups were detected. Generally, the differences were not significant. However, a TUNEL assay revealed significant differences in apoptosis due to culture temperature (Figure 4b, d). The number of apoptotic cells was significantly lowered after culture at 21°C compared to 37°C-cultures and even compared to freshly prepared sections. Representative H&E-stained sections confirmed overall increased degeneration in retinae cultured at 37°C demonstrated by large holes and cell loss. Nevertheless, holes were also found in the inner plexiform (IPL) and nerve fiber (NFL) layers of retinae cultured at 21°C (Figure 4c).
3.3. Model 3: porcine, static, 4 days
Culture conditions established in Model 2 were adapted to static retina culture in Model 3. Figure 5a-e shows representative images of different stages of degeneration at different time points, quantified in Figure 5f. Degeneration increased over time, but stayed in average below highest score of ‘3’. The mean score was 0.80±0.45 at 1 d, increased to 2.0±0.82 at 2 d to 2.13±0.64 at 3 d to 2.33±0.82 at 4 d; differences were significant from 1 vs. 3 d (p=0.0496) and from 1 vs. 4 d (p=0.0234).
3.4. Model 4: rat, static, 9-13 days
Optimization of culture conditions could significantly slow down degeneration in Models 2 and 3; nevertheless, culture duration was still short. Thus, in Model 4, the effect of using fresh tissue, isolated immediately after animals’ sacrifice was evaluated. Tissue preservation was additionally improved by medium supplementation with growth factors N-2 and B-27. The use of fresh tissue enabled robust culture prolongation until 9 d. Retinae were cultured up to 13 d; however, after 9 d, degeneration became prominent, often not allowing reliable tissue analysis. H&E-stained sections demonstrated tissue preservation from day 1-9 and a non-significant, acceptable number of holes and cell loss (Figure 6a-d). POS were partly preserved until 9 d. At later time points, in many samples either thickening of the tissue let assume inflammatory reactions (Figure 6e) or laminar structure of retinae was lost, INL and ONL were no longer distinguishable, thinning of the tissue and cell lost were observed (Figure 6f).
3.5. Model 5 and 5b: porcine and human, static, 14 days
Knowledge from Model 4 was applied to porcine eyes. Significant improvement of tissue isolation and processing also enabled the transfer of the model to retina culture from human donor eyes cultured 6-8 d post-mortem. It was possible to preserve laminar structure and distinguishable retinal layers for 14 d in both porcine and human retinae as shown in H&E-stained tissue sections (Figure 7a, b). From day 1-14, main retinal layers (RGC, IPL, INL, OPL, ONL) were distinguishable and did not show significant thinning (indicating cell loss) or thickening (indicating inflammation). POS were hardly visible in H&E-stained sections. Retinal thickness was determined in micrographs from H&E-stained sections (Figure 7c). Porcine samples had a thickness from 54.74±12.81 µm (1 d) to 65.59±11.60 µm (day 6) to 46.42±10.48 µm (14 d) without significant differences (p=0.4882); thickness increased slightly at 6 d of culture, but declined back to base values at 14 d. Also, thickness in human retinae did not differ significantly over time (p=0.6126) with 51.09±15.28 µm (1 d) to 66.58±19.16 µm (6 d) to 53.08±3.08 µm (14 d).
Tissue quality was also analyzed by determining degeneration (score), viability (CytoTox-GlowTM), and necrosis (PI staining) (Figure 8). Cell viability correlated inversely to measured luminescence; thus, the lower luminescence the higher viability. The black curve (pRetina) illustrated high luminescence at 3 d that continuously decreased over time, indicating a decrease in cell death (Figure 8a). Viability in hRetina remained high over the whole culture period (Figure 8a, grey curve). Necrosis only slightly increased over time for both species (Figure 8b). Figure 8c shows the degeneration score over 14 d for pRetina; the score increased over time but generally remained low with a maximal score of 2.13. Degeneration in hRetinae was even lower with a maximal score of 1.60 at 14 d.
Immunohistochemical staining analyzed cell loss using cell type specific antibodies (rhodopsin for POS, vimentin for Müller cells) and detected potential inflammatory reactions using anti-GFAP and anti-Iba-1 antibodies. Vimentin staining (red) confirmed normal Müller cell morphology for the whole culture period (Figure 9g-l). Rhodopsin staining (green) detected POS for 14 d in both species. GFAP staining (red) increased at 6 d in pRetinae and hRetinae but decreased even below base values at 14 d (Figure 9a-f). Iba-1 staining (green) remained low at every time point in both species (Figure 9g-l).
3.6. Antioxidant Scutellarin to treat OS in high-glucose retina
Models 3 and 4 were used to analyze disease conditions linked to OS, i.e., high glucose conditions and incubation with H2O2 to test antioxidative treatment options. pRetinae (Model 3) were cultured in normal glucose (NG, 6 mM D-glucose) and high glucose (HG, 30 mM D-glucose) conditions to provoke cellular damage as seen in patients suffering from DR; Scutellarin was added, and its effect tested in HG conditions. Cell viability was determined by cells’ capability to produce ATP in retinae cultured in HG and with 0, 1, 10, or 100 µM Scutellarin added to the cell culture medium. At 24 h, viability increased after medium supplementation with of 1 µM (998.9±202.5 nM ATP) and 10 µM Scutellarin (783.6±164.6 nM ATP), while the addition of 100 µM Scutellarin did not increase viability (360.1±152.8 nM ATP) compared to NG (500.3±17.01 nM ATP) and HG (560.1±138.7 nM ATP) conditions (Figure 10a). Viability after treatment with 1 µM Scutellarin differed significantly to treatment with 100 µM Scutellarin (p=0.0053). After 4 d of culture at HG conditions (Figure 10b), its detrimental effect became more obvious by decreased ATP level of 288.3±59.9 nM compared to 435.7±166.8 nM ATP in NG. Scutellarin (1 µM: 515.9±172.0 nM ATP; 10 µM: 420.5±201.9 nM ATP) increased viability at 4 d to levels comparable to NG; 100 µM Scutellarin did not increase viability (325.7±154.7 nM ATP). The antioxidative effect of low and medium doses of Scutellarin was illustrated in H&E-stained cross sections (Figure 10c); only after treatment with 1-10 µM Scutellarin, POS were preserved. Nevertheless, degeneration, i.e., holes and cell loss, was still visible.
Apoptosis was determined at 1 d and 4 d (Figure 11). At 1 d, apoptosis was highest in HG conditions (69.60±27.70 cells/image) and decreased even below NG conditions (46.2±24.41 cells/image) after addition of Scutellarin. The beneficial effect was similar for all doses: 1 µM = 32.25±7.50; 10 µM = 41.60±18.06; 100 µM = 37.75±16.09 cells/image. The difference between the freshly fixed control and HG was significant (p=0.0050). At 4 d, apoptosis generally increased with similar differences between the groups. HG had the highest values (124.20±51.09 cells/image), followed by NG (71.83±35.38 cells/image). Apoptosis further decreased by Scutellarin to 64.80±30.68 cells/image (1 µM), 75.20±321.19 cells/image (10 µM), and 60.80±319.66 cells/image (100 µM). The difference between the fresh control (2.75±3.40 cells/image) and HG was significant (p=0.0017). Micrographs visualized results at 4 d (Figure 11c).
3.6. OS reduction and cell protection by PEDF & GM-CSF in H2O2-treated retina
OS induced by age-related cellular alterations plays a key role in aAMD pathogenesis. Thus, a reduction of OS might slow down or halt retinal degeneration. For this purpose, RPE or IPE cells are transfected using the SB100X transposon system with the genes coding for the PEDF and GM-CSF proteins. To test the proteins’ cell protective effect, rRetinae cultured for up to 13 d (Model 4) were treated with one or both proteins for the first 3 d of culture; on day 3, OS was induced by H2O2. OS levels of treated retinae were determined by a glutathione (GSH) assay; cell-protective effects were quantified by a viability assay (Figure 14a, c). After H2O2-treatment, the tissue was extremely fragile, such that not all measured samples delivered technically valid results. Therefore, the rate of valid/invalid results was analyzed as additional parameter for OS and the anti-oxidative effect of PEDF/GM-CSF, respectively (Figure 14b, d). Cell viability (Figure 12a) in freshly fixed retinae (control) was 31.03±19.50%. In contrast, tissue incubated with H2O2 was highly damaged and no valid results could be measured indicating a low viability. Treatment with PEDF and GM-CSF increased viability significantly to 45.44±9.02% (PEDF) and 50.40±7.65% (GM-CSF), when “normal” tissue (no H2O2 incubation) was treated with the proteins. After H2O2-incubation, viabilities of 52.80±18.24% (PH = PEDF+H2O2) and 60.07±13.26% (PGH = PEDF+GM-CSF+H2O2) were measured. No valid results were measured after treatment with GM-CSF only (GH = GM-CSF+H2O2). The highest percentages of valid viability measurements were seen in non-H2O2-incubated groups with 100% (fresh), 100% (PEDF), and 71% (GM-CSF) (Figure 12b). No valid results (0%) could be received in H2O2-incubated and GH-treated samples. The treatment with PEDF or PEDF+GM-CSF increased the number of valid results to 25% (PH) and 38% (PGH). Lowest GSH levels were measured in samples that were not incubated with H2O2 (fresh: 0.01±0.00 µg/10 mg tissue, PEDF: 0.01±0.00 µg/10 mg tissue, GM-CSF: 0.02±0.00 µg/10 mg tissue) (Figure 12c). GSH level increased after H2O2 treatment to 0.16±0.00 µg/10 mg tissue; when additionally treated with PEDF and/or GM-CSF, levels further risen (except the PH group with 0.13±0.07 µg/10 mg tissue) to 0.23±0.19 µg/10 mg tissue (GH) and 0.221±030 µg/10 mg tissue (PGH). Tissue protection, i.e., lowered OS, was reflected by percentages of valid GSH results in GM-CSF- (83.3%), and PEDF-GM-CSF-treated retinae (100%) after H2O2 incubation (Figure 12d). If treated with PEDF alone, the percentage of valid results did not differ to H2O2-incubated, untreated retinae (66.7%). The lowest numbers of valid results were seen in “normal” tissue: one of three for fresh tissue, one of five for tissues treated with PEDF and GM-CSF, respectively.
4. Discussion
ROS, continuously produced especially by mitochondria [44,45], are important signaling molecules in exerting beneficial effects to calorie, glucose limitation, and physical exercise (mitohormesis concept) [46]. However, excess of ROS induced by mitochondrial dysfunction and a downregulation of antioxidant enzymes leads to OS and cell damage and plays a key role in normal aging and age-related diseases [46,47]. ROS like, e.g., the superoxide anion (O2•−), H2O2, or the hydroxyl ion (OH-) damage proteins, nucleic acids and lipids leading to cancerogenesis, cellular dysfunction and death [47]. The body developed a three-level antioxidative defense system against cell damage: First, superoxide dismutase (SOD) and related enzymes remove H2O2 and lipid hydroperoxides. Secondly, thioredoxin, glutamate-cysteine ligase and the glutathione synthetase generate GSH, glutathione and thioredoxin reductases to reduce glutathione and thioredoxin disulfide. Finally, the NF-κB pathway and others are activated to eliminate oxidized macromolecules, repair damage, and inhibit apoptosis, e.g., by regulating the expression of Bcl-2 proteins [46,47]. In age, increased ROS level and decreased antioxidant defense results in the frailty syndrome characterized by a reduced physiological reserve and increased susceptibility to stressors; when this weakened balance is disrupted, age-related diseases evolve [3].
AMD is a typical age-related disease and as long-term complication of diabetes, DR can also be numbered among this group; untreated, both lead to retinal neurodegeneration and blindness [5]. As an organ with a particular high oxygen consumption and processing light to generate vision, the retina is constantly producing ROS and is thus particularly prone to OS [48]. A key process in the pathogenesis of aAMD is the interplay of inflammation and OS that leads to dysfunction of the RPE. Mitochondrial damage, ATP depletion, inflammation, and RIPK-3-mediated membrane damage lead to apoptotic and necrotic cell death [48]. Oxidatively-modified lipids, 89 proteins including the peroxiredoxin 1 enzyme from the antioxidant defense system, and trace elements accumulate at the interface of Bruch’s membrane and the RPE and form drusen [49,50]. As a consequence, the RPE cannot anymore support function and survival of photoreceptors which also undergo cell death [11,48]. The diabetic retina is harmed by lipid peroxidation, protein oxidation, and DNA damage, endogenous levels of antioxidant defense enzymes are diminished, while pro-apoptotic proteins are increased, and levels of neurotrophic factors are decreased [51]. Prevalence of AMD and DR are increasing due to aging populations and increasing rates of diabetes [52] and will become a severe burden for patients and health care systems [6]. In industrialized countries, they are already the first cause of blindness in elderly patients (AMD) and the working-age population (DR) [10]. Thus, there is an urgent need for innovative, reliable models for a better understanding of the pathogenesis of OS-triggered diseases and the development of novel therapies. Such models are ideally i) simple to generate and thus easily reproducible, ii) available at low-cost, iii) enable up-scaling, iv) ethically favorable, v) biologically complex, and vi) transferable to patients.
Cell culture systems fulfill many of the criteria; however, they cannot evaluate complex biological interactions and are limitedly transferable to patients. The Ala Octa® affair demonstrated issues with in vitro studies: Perfluorooctane (Ala Octa®) was a perfluorocarbon liquid used in retinal surgery as transient tamponade and cytotoxicity has been tested – accordingly to regulatory standards - on mouse fibroblasts. Pastor et al. could demonstrate that as a result of insufficient testing, cytotoxicity has been underestimated and led to severe ocular complications up to blindness in many patients [15]. In vivo studies are more suitable to detect potential local and systemic adverse effects. However, research institutions, pharmacological industry, regulatory and governmental bodies are motivated to reduce animal experiments in biomedical research to a minimum following the 4R principles [14]. Moreover, many animal models are only limitedly transferable to patients. Pennesi et al. reviewed AMD animal models [52], of which most are working with mice while theses have no macula and are night-active with very different retinal anatomy and cellular composition compared to human. Often, the animals are of young age [53] being useful to investigate distinct disease features, but insufficient to prove efficiency and safety of novel treatments against age-related maculopathies. Studies with larger animals and species that have a macula are possible; however, the larger the animal the higher the costs and the more ethical questions arise. Organ cultures offer an alternative. They are relatively easy to generate, available at low costs compared to in vivo studies, allow study of intercellular interactions of complex tissues, and are ethically favorable. Moreover, the use of human donor tissue promises transferability to patients and offers the evaluation of diseased or aged organs. Retina culture was reported first by Strangeways and Fells [17] who analyzed ocular development for 32 d in cultured chick embryonic eyes. The culture of adult retinae is more challenging: recent research reported a suspension culture of retinal cells for 8 weeks [18] but except single reports, organotypic culture with preserved tissue structure can be maintained for 7-14 d [19,54]. Thus, the retina culture is an excellent system to analyze organ development, disease pathogenesis, intoxications, novel drug candidates and treatment approaches, and particularly OS, but needs enhancement [19,54].
In the present study, five retina culture models comparing different isolation and tissue processing methods, media, temperature, mammalian species, and post-mortem times were presented (summarized in Table 5). Briefly, the use of porcine or bovine eyes provided by food industry is ethically preferable compared to laboratory rat tissue. By applying enhanced isolation and tissue processing techniques, culture of retinae with prolonged post-mortem time is not inferior to fresh rat tissue regarding tissue morphology, degeneration, cell viability, and death. Ames’ culture medium, optimized by supplementation with growth factors N-2 and B-27 and retina incubation at 21°C revealed superior culture conditions. The implementation of an enhanced isolation and tissue processing technique (“minimal-touch technique”) allowed transfer of the culture system from animal to human donor retinae, which are most fragile due to a post-mortem time of several days. Culture of the neural retina together with the choroid and/or RPE cell layers seemed to be cell protective; however, the complex isolation techniques offset the benefit.

Table 5.
Comparison of presented retina culture models. The table summarizes the set-up of the presented Models 1 to 5. Photographs illustrate the different culture systems. a) Minuth chamber with a retina clamped into the culture ring. b) In the multi-well plate are pRetinas cultured on nitrocellulose membranes on customized stainless-steel grids. c) The smaller rat retina is cut into halfs before cultured on nitrocellulose membranes and customized grids. d) The panel shows the punch, how it is cutting tissue, and the culture insert before use and in culture.
Table 5.
Comparison of presented retina culture models. The table summarizes the set-up of the presented Models 1 to 5. Photographs illustrate the different culture systems. a) Minuth chamber with a retina clamped into the culture ring. b) In the multi-well plate are pRetinas cultured on nitrocellulose membranes on customized stainless-steel grids. c) The smaller rat retina is cut into halfs before cultured on nitrocellulose membranes and customized grids. d) The panel shows the punch, how it is cutting tissue, and the culture insert before use and in culture.
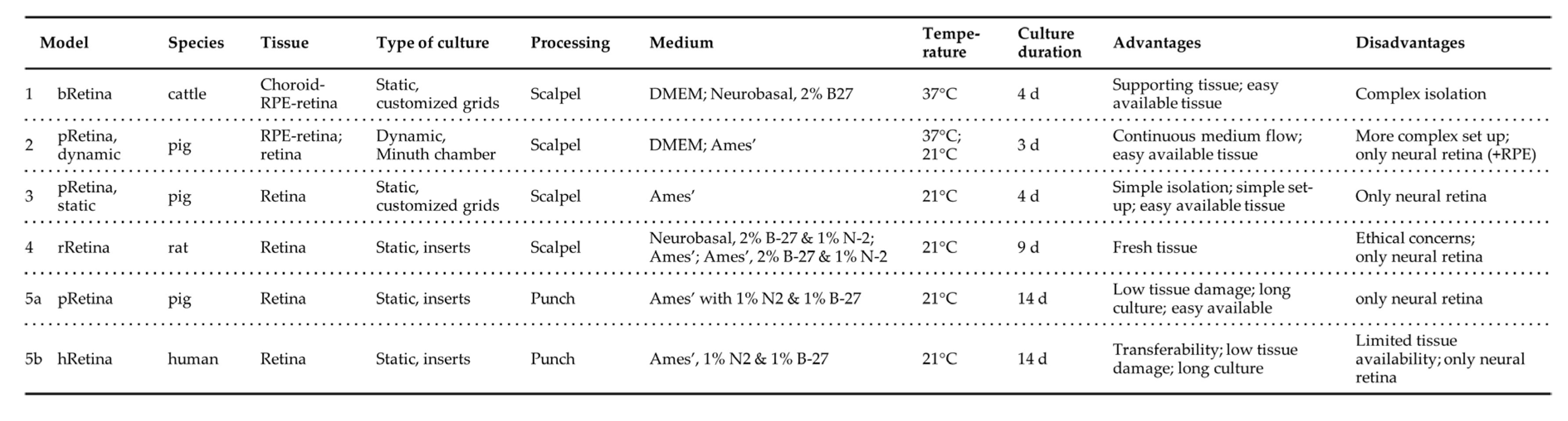
Various species are used for retina culture of which pig and cattle are frequently used since they are easily available. Kuehn et al. evaluated hypoxia-induced retinal degeneration following cobalt-chloride treatment in porcine retinae (8 d culture; Neurobasal medium supplemented with glutamine, B-27 and N-2; photoreceptors facing the multi-well inserts, 4-6 samples/retina; isolation using a 6 mm punch) [25]. General tissue quality was very good enabling analysis of dose-dependent effects of the cobalt-chloride treatment. Similarly, Models 2, 3, and 5a used porcine tissue and confirmed excellent tissue quality that enabled retina culture up to 14 d (Model 5a). Model 1 used cattle as starting material, as did Peynshaert et al. (2 d culture; isolation and culture of neural retina and vitreous, Neurobasal medium supplemented with glutamine and B-27; 3 explants/eye,) [23]. Results demonstrated excellent tissue quality in terms of tissue architecture, cell composition, nerve fiber layer and functionality comparable to data from Model 1. Schnichels et al. isolated young rat retinae immediately after sacrifice (7 d culture; R16-complete medium; retina of one eye cultured as one sample) [55]. Retinal architecture was maintained for 7 d and tissue thickness decreased only moderately. In line with this data, Model 4 demonstrated a retarded degeneration and prolonged culture duration of up to 9-13 d when using freshly isolated rat retinae (2 samples/eye). It is assumed that the short post-mortem time and not the species was responsible for the positive influence on cell viability in both studies. Yet, since Model 4 requested the sacrifice of one animal for 4 samples it is ethically questionable and not scalable.
The perfusion culture presented in Model 2 reported first by Kobuch et al. [56] guaranteed constant nutrient and oxygen delivery. The culture of the choroid-RPE-retina complex in a perfusion chamber allowed tissue preservation up to 10 d with POS showing first signs of degeneration after 1 d, apoptotic and necrotic cell death was seen after 4 d, while in control samples (static culture) tissue structure was disrupted after 4 d (10 d culture; DMEM, 15% porcine serum, 2.5% HEPES-buffer; 1 sample/retina) [56]. Similarly, Model 2 reported a decelerated degeneration compared to Model 1; however, compared to Model 3 and 5 no significant advantage could be observed; it is assumed that daily medium change (in static culture) delivers sufficient nutrients and oxygen for optimal tissue preservation. Thus, considering the simpler set up and easier up-scaling of static cultures, this system seems preferable.
Model 1 and 2 cultured the neural retina with supportive choroid and/or RPE tissue since Kobuch et al. reported a benefit of this technique on retina conservation [56]; also Peynshaert et al. reported low levels of inflammation, no significant changes in tissue thickness, and stable populations of bipolar cells after culture of the neural retina with supportive tissue (vitreous) [23]. Indeed, especially POS were better preserved when cultured together with RPE cells (and the choroid); however, enlarged analyses performed with Model 2 did not confirm the prominent differences between the culture of the neural retina alone or with (choroid-)RPE tissue. The higher complexity of the isolation technique seemed to abolish the positive effect of the supportive tissue layers, i.e., not all samples could be put in culture or showed damages due to isolation.
It is known that hypothermia causes immunosuppression and slows down metabolism and, thus, protects organs from damage due to hypoxia, ischemia, or trauma as shown by Narayanamurthy et al. who presented a cooling device that protected the rat brain from hypoxic-ischemic encephalopathy [57]. An hypothermia-induced metabolism deceleration, significantly inhibited the development of OS and its consequences as shown by Choi et al. in a model of renal ischemia-reperfusion injury [58]. Also various ocular conditions like allergic conjunctivitis or retinal hypoxia and ischemia are treated by hypothermia [59,60]. Due to the high metabolic demand and metabolic turnover of the retina, the idea to rescue cultured retinal tissue by hypothermia was already evaluated by Ames and Gurian in 1963: They showed that rabbit retinae recovered more rapidly at 30°C than at 37°C after glucose and oxygen deprivation [61]. Recently, Mueller-Buehl et al. reported a OS damage-reducing effect of hypothermia on H2O2-induced damage on porcine RGCs [28]. Accordingly, Model 2 cultured retinae in hypothermic conditions (21°C) compared to 37°C-cultured samples and revealed significant tissue preservation by higher cell counts and a lowered rate of apoptosis. It has to be noted that pharmacokinetics analyses have to be analyzed carefully due to the slowed metabolism.
While in Model 1 retina samples were cultured in DMEM, in Model 2, Ames’ medium was introduced and used in all following systems. Results of Model 2 demonstrated a significant benefit of Ames’ medium that has been especially developed for the culture of retinal tissue [62]. Many other groups are using Neurobasal medium for its protective function for neurons and report positive results [23,25]. However, particularly Model 5 confirmed that a medium that imitates the fluid that “bathes the retina in vivo” [62] is ideal to support this complex tissue that is composed of multiple neural and non-neural cell types. It has to be noted, that in Model 4 the medium has been optimized by addition of growth factors B-27 and N2 enabling the two-week long culture among other factors.
The experimental series in high glucose conditions and after H2O2 treatment demonstrated the benefit of the retina culture models in preclinical testing. Model 3 cultured porcine retinae in static culture using Ames’ medium at 21°C. Tissue degeneration could be kept at a low level for 4 d during which the detrimental effect of OS induced by “high glucose” and the benefit of antioxidant Scutellarin treatment could be determined. It has to be noted that high doses of Scutellarin might negate the positive effect, but more detailed analyses have to be done to confirm this tendency. Model 4 has been implemented to prolong the culture and its avail was evaluated by analyzing the effect of PEDF and/or GM-CSF on cell viability and OS after H2O2-treatment. Rat retinae cultured immediately after sacrifice, allowed cultures up to 13 d and confirmed cell viability rates in freshly fixed retinae of around 31%, what has been also measured in IPE cells immediately after isolation [63]. After H2O2-treatment, reduced GSH levels were expected as reported by Wang et al. in ARPE-19 cells [64] and by Zheng et al. in retinae from 18-months old hydroquinone fed mice [65]. In contrast, results showed lowest GSH concentrations in controls, while GSH levels in H2O2-treated samples were increased. It is assumed that while already improved compared to Models 1-3, tissue quality in Model 4 was not good enough to reliably measure GSH levels. This is supported by the low percentage of samples that delivered methodological valid results.
Thus, Model 5 has been developed to i) generally increase tissue quality and prolong culture duration, ii) implement a procedure that allows up-scaling of testing series, and iii) allow the transfer to human donor tissue with longer post-mortem time. Porcine retinae were cultured in static conditions at 21°C, in Ames’ medium supplemented with growth factors. Moreover, isolation and tissue processing techniques were improved; the novel “minimal-touch technique” includes sample preparation by 6 mm punches up to 32 samples/retina, special tissue handling and processing as detailed in section 2. It enabled significantly improved retina preservation shown by low degeneration scores at 14 d, high levels of viability, low apoptosis and inflammation. The results allowed transfer to human retinae with similar results and comparable to data reported by other groups like Jüttner et al. [66] and Schnichels et al. [67]. Ongoing experiments repeat the analyses of Model 4 evaluating the protective effect of PEDF and GM-CSF in OS to confirm superiority of Model 5. Future enhancements will implement a co-culture of the neural retina with iPS-derived RPE cells to keep simplicity of the model while adding supportive RPE tissue. Electroretinogram examinations will be added to the panel of analyses and OS-related damage will be evaluated in more detail by analyzing SODs, lipid peroxidation products, and DNA-oxidation products. This will be of particular interest by using human donor eyes from AMD or DR patients.
5. Conclusions
Five retina organ culture models from different mammalian species including human were evaluated for preclinical testing of OS and antioxidant intraocular therapy approaches were presented. Quality of starting material, tissue handling techniques, and culture conditions including hypothermia were assessed. Static culture of the neural retina in Ames’ medium on transwell-inserts with the photoreceptors facing the insert at 21°C resulted in optimal retina preservation even in human donor retina samples with a post-mortem time of up to 8 d (Model 5b). Further improvements are in development to allow more detailed analysis of OS-related damage. Today, the combination of Models 5a (porcine) and b (human) enables two weeks-long retina culture in large experimental series for a better understanding of pathogenesis of age-related, OS-triggered retinopathies and for extended drug candidate testing.
Author Contributions
Conceptualization, G.T., M.K.; methodology, G.T, M.K.; validation, G.T.; formal analysis, S.J., M.M., C.L., L.S., M.K.; resources, G.T.; data curation, M.K., S.J., M.M., C.L., L.S.; writing—original draft preparation, M.K.; writing—review and editing, M.M., C.L., L.S., N.H., T.B., EC, A.K., B.P., S.J., G.T.; visualization, M.K.; supervision, G.T., S.J., M.K; project administration, G.T., M.K.; funding acquisition, G.T. and M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by a grant from the Interdisciplinary Centre for Clinical Research „BIOMAT“ within the faculty of Medicine at the RWTH Aachen University (Model 1 and 2), by the Swiss National Science Foundation (grant no. 189341; Model 5a and b) and the ARVO Foundation for Eye Research (grant entitled “iPS-derived RPE cell & human retina co-culture for toxicity and potency assays in intraocular medicine.”, approved on July 13, 2021; Model 5b).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Commission cantonale d’éthique de la recherche of the canton Geneva, Switzerland (2016-01726, 11/24/2016)” for studies involving human material. The animal study was approved by the Département de la sécurité, de l’emploi et de la santé, Direction générale de la santé, Domaine de l’expérimentation animale of the canton Geneva, Switzerland (GE/116/1, 07/18/2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study and is archived by the Lions Gift of Sight Eye Bank (Saint Paul, MN, USA).
Data Availability Statement
The data presented in this study are openly available on the Zenodo repository [doi], [date of upload].
Acknowledgments
A special thanks goes to Dr. Stefanie Kaempf and the technicians from the Department of Ophthalmology at the University Hospital RWTH Aachen, and to Hajer Zedira for her excellent M.sc. work, Alain Conti and Gregg Sealy for their support and excellent technical assistance (Dept. of Ophthalmology, HUG & UNIGE, Geneva, Switzerland).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kumar, V.; Bishayee, K.; Park, S.; Lee, U.; Kim, J. Oxidative stress in cerebrovascular disease and associated diseases. Front. Endocrinol. (Lausanne) 2023, 14, 1124419. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, R.F.; Nagano, S. Crosstalk between Oxidative Stress and Aging in Neurodegeneration Disorders. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Fantini, C.; Corinaldesi, C.; Lenzi, A.; Migliaccio, S.; Crescioli, C. Vitamin D as a Shield against Aging. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Doblhammer, G.; Rau, R.; Vaupel, J.W. Ageing populations: the challenges ahead. Lancet 2009, 374, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Kai, J.-Y.; Xu, Y.; Li, D.-L.; Zhou, M.; Wang, P.; Pan, C.-W. Impact of major age-related eye disorders on health-related quality of life assessed by EQ-5D: a systematic review and meta-analysis. Graefes Arch. Clin. Exp. Ophthalmol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Bae, C.-Y.; Kim, B.-S.; Cho, K.-H.; Kim, I.-H.; Kim, J.-H.; Kim, J.-H. 10-year follow-up study on medical expenses and medical care use according to biological age: National Health Insurance Service Health Screening Cohort (NHIS-HealS 2002~2019). PLoS ONE 2023, 18, e0282466. [Google Scholar] [CrossRef]
- Tolentino, M.J.; Tolentino, A.J. Investigational drugs in clinical trials for macular degeneration. Expert Opin. Investig. Drugs 2022, 31, 1067–1085. [Google Scholar] [CrossRef]
- Li, Q.; Wang, M.; Li, X.; Shao, Y. Aging and diabetic retinopathy: Inherently intertwined pathophysiological processes. Exp. Gerontol. 2023, 112138. [Google Scholar] [CrossRef]
- Cabral de Guimaraes, T.A.; Daich Varela, M.; Georgiou, M.; Michaelides, M. Treatments for dry age-related macular degeneration: therapeutic avenues, clinical trials and future directions. Br. J. Ophthalmol. 2022, 106, 297–304. [Google Scholar] [CrossRef]
- Steinmetz, J.D.; Bourne, R.R.A.; Briant, P.S.; Flaxman, S.R.; Taylor, H.R.B.; Jonas, J.B.; Abdoli, A.A.; Abrha, W.A.; Abualhasan, A.; Abu-Gharbieh, E.G.; et al. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: the Right to Sight: an analysis for the Global Burden of Disease Study. The Lancet Global Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Deng, Y.; Qiao, L.; Du, M.; Qu, C.; Wan, L.; Li, J.; Huang, L. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 2022, 9, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Mounirou, B.A.M.; Adam, N.D.; Yakoura, A.K.H.; Aminou, M.S.M.; Liu, Y.T.; Tan, L.Y. Diabetic Retinopathy: An Overview of Treatments. Indian J. Endocrinol. Metab. 2022, 26, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Tisi, A.; Feligioni, M.; Passacantando, M.; Ciancaglini, M.; Maccarone, R. The Impact of Oxidative Stress on Blood-Retinal Barrier Physiology in Age-Related Macular Degeneration. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Kiani, A.K.; Pheby, D.; Henehan, G.; Brown, R.; Sieving, P.; Sykora, P.; Marks, R.; Falsini, B.; Capodicasa, N.; Miertus, S.; et al. Ethical considerations regarding animal experimentation. J. Prev. Med. Hyg. 2022, 63, E255–E266. [Google Scholar] [CrossRef] [PubMed]
- Pastor, J.C.; Coco, R.M.; Fernandez-Bueno, I.; Alonso-Alonso, M.L.; Medina, J.; Sanz-Arranz, A.; Rull, F.; Gayoso, M.J.; Dueñas, A.; Garcia-Gutierrez, M.T.; et al. ACUTE RETINAL DAMAGE AFTER USING A TOXIC PERFLUORO-OCTANE FOR VITREO-RETINAL SURGERY. Retina (Philadelphia, Pa.) 2017, 37, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.; Fietz, A.; Tsai, T.; Joachim, S.C.; Schnichels, S. Organ Cultures for Retinal Diseases. Front. Neurosci. 2020, 14, 583392. [Google Scholar] [CrossRef] [PubMed]
- Strangeways, T.; Fell, B. Experimental studies on the differentiation of embryonic tissues growing in vivo and in vitro.—II. The development of the isolated early embryonic eye of the fowl when cultivated in vitro. Proc. R Soc. B. 1926, 100, 273–281. [Google Scholar]
- Murali, A.; Ramlogan-Steel, C.A.; Steel, J.C.; Layton, C.J. Characterisation and validation of the 8-fold quadrant dissected human retinal explant culture model for pre-clinical toxicology investigation. Toxicol. In Vitro 2020, 63, 104716. [Google Scholar] [CrossRef]
- Schnichels, S.; Paquet-Durand, F.; Löscher, M.; Tsai, T.; Hurst, J.; Joachim, S.C.; Klettner, A. Retina in a dish: Cell cultures, retinal explants and animal models for common diseases of the retina.
- Koizumi, A.; Zeck, G.; Ben, Y.; Masland, R.H.; Jakobs, T.C. Organotypic culture of physiologically functional adult mammalian retinas. PLoS ONE 2007, 2, e221. [Google Scholar] [CrossRef]
- Ingensiep, C.; Schaffrath, K.; Denecke, B.; Walter, P.; Johnen, S. A multielectrode array-based hypoxia model for the analysis of electrical activity in murine retinae. J. Neurosci. Res. 2021, 99, 2172–2187. [Google Scholar] [CrossRef]
- Ingensiep, C.; Schaffrath, K.; Walter, P.; Johnen, S. Effects of Hydrostatic Pressure on Electrical Retinal Activity in a Multielectrode Array-Based ex vivo Glaucoma Acute Model. Front. Neurosci. 2022, 16, 831392. [Google Scholar] [CrossRef] [PubMed]
- Peynshaert, K.; Devoldere, J.; Forster, V.; Picaud, S.; Vanhove, C.; Smedt, S.C. de; Remaut, K. Toward smart design of retinal drug carriers: a novel bovine retinal explant model to study the barrier role of the vitreoretinal interface. Drug Deliv. 2017, 24, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, S.; Hurst, J.; Rensinghoff, F.; Tsai, T.; Grauthoff, S.; Satgunarajah, Y.; Dick, H.B.; Schnichels, S.; Joachim, S.C. Degenerative effects of cobalt-chloride treatment on neurons and microglia in a porcine retina organ culture model. Exp. Eye Res. 2017, 155, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Calbiague, V.M.; Vielma, A.H.; Cadiz, B.; Paquet-Durand, F.; Schmachtenberg, O. Physiological assessment of high glucose neurotoxicity in mouse and rat retinal explants. J. Comp. Neurol. 2020, 528, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Masri, R.A.; Lee, S.C.S.; Madigan, M.C.; Grünert, U. Particle-Mediated Gene Transfection and Organotypic Culture of Postmortem Human Retina. Transl. Vis. Sci. Technol. 2019, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Buehl, A.M.; Doepper, H.; Grauthoff, S.; Kiebler, T.; Peters, L.; Hurst, J.; Kuehn, S.; Bartz-Schmidt, K.U.; Dick, H.B.; Joachim, S.C.; et al. Oxidative stress-induced retinal damage is prevented by mild hypothermia in an ex vivo model of cultivated porcine retinas. Clin. Experiment. Ophthalmol. 2020, 48, 666–681. [Google Scholar] [CrossRef]
- Thumann, G.; Harmening, N.; Prat-Souteyrand, C.; Marie, C.; Pastor, M.; Sebe, A.; Miskey, C.; Hurst, L.D.; Diarra, S.; Kropp, M.; et al. Engineering of PEDF-Expressing Primary Pigment Epithelial Cells by the SB Transposon System Delivered by pFAR4 Plasmids. Mol. Ther. Nucleic Acids 2017, 6, 302–314. [Google Scholar] [CrossRef]
- Bascuas, T.; Zedira, H.; Kropp, M.; Harmening, N.; Asrih, M.; Prat-Souteyrand, C.; Tian, S.; Thumann, G. Human Retinal Pigment Epithelial Cells Overexpressing the Neuroprotective Proteins PEDF and GM-CSF to Treat Degeneration of the Neural Retina. CGT 2022, 21, 168–183basc. [Google Scholar] [CrossRef]
- Broadhead, M.L.; Dass, C.R.; Choong, P.F.M. Systemically administered PEDF against primary and secondary tumours in a clinically relevant osteosarcoma model. Br. J. Cancer 2011, 105, 1503–1511. [Google Scholar] [CrossRef]
- Michelis, G.; German, O.L.; Villasmil, R.; Soto, T.; Rotstein, P.N.; Politi, L.; Becerra, S.P. Pigment Epithelium-derived Factor (PEDF) and Derived Peptides Promote Survival and Differentiation of Photoreceptors and Induce Neurite-outgrowth in Amacrine Neurons. Journal of Neurochemistry 2021. [Google Scholar] [CrossRef]
- Craword, S.E.; Fitchev, P.; Veliceasa, D.; Volpert, O.V. The many facets of PEDF in drug discovery and disease: a diamond in the rough or split personality disorder? Expert Opin. Drug Discov. 2013, 8, 769–792. [Google Scholar] [CrossRef] [PubMed]
- Crane, I.J.; Wallace, C.A.; Forrester, J.V. Regulation of granulocyte-macrophage colony-stimulating factor in human retinal pigment epithelial cells by IL-1beta and IFN-gamma. Cell. Immunol. 2001, 209, 132–139. [Google Scholar] [CrossRef]
- Hamilton, J.A. GM-CSF in inflammation. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Chitu, V.; Biundo, F.; Stanley, E.R. Colony stimulating factors in the nervous system. Semin. Immunol. 2021, 54, 101511. [Google Scholar] [CrossRef]
- Schallenberg, M.; Charalambous, P.; Thanos, S. GM-CSF protects rat photoreceptors from death by activating the SRC-dependent signalling and elevating anti-apoptotic factors and neurotrophins. Graefes Arch. Clin. Exp. Ophthalmol. 2012, 250, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Leroy-Ciocanea, C. Establishment of an in vitro organotypic retinopathy model to test the neuroprotective impact of scutellarin. M.Sc.; Sorbonne Universités, Université Pierre et Marie Curie Paris, Paris, 2016.
- Wang, L.; Ma, Q. Clinical benefits and pharmacology of scutellarin: A comprehensive review. Pharmacol. Ther. 2018, 190, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Minuth, W.W.; Rudolph, U. A compatible support system for cell culture in biomedical research. Cytotechnology 1990, 4, 181–189. [Google Scholar] [CrossRef]
- Framme, C.; Kobuch, K.; Eckert, E.; Monzer, J.; Roininen, E. RPE in Perfusion Tissue Culture and Its Response to Laser Application: Preliminary Report. Ophthalmologica 2002, 216, 320–328. [Google Scholar] [CrossRef]
- Bull, N.; Johnson, T.; Martin, K.R. Organotypic explant culture of adult rat retina for in vitro investigations of neurodegeneration, neuroprotection and cell transplantation. Research Square 2011. Pre print. [Google Scholar] [CrossRef]
- Kaempf, S.; Walter, P.; Salz, A.K.; Thumann, G. Novel organotypic culture model of adult mammalian neurosensory retina in co-culture with retinal pigment epithelium. J. Neurosci. Methods 2008, 173, 47–58. [Google Scholar] [CrossRef]
- Zedira, H. Optimization of an in vitro retinal organotypic culture model of retinal degeneration to assess the neuroprotective effect of the recombinant proteins PEDF and GM-CSF; M.Sc., UNIGE, Université Grenoble Alpes, Geneva, Lyon, 2019.
- Mohit, M. Up-scaling and humanization of retinal organotypic culture as a model for intraocular efficiency and toxicity analyses; M.Sc.; University of Geneva, Geneva, 2023.
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon Olga, L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Jiang, S.; Gericke, A. Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.G.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. PNAS 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [PubMed]
- Bergen, A.A.; Arya, S.; Koster, C.; Pilgrim, M.G.; Wiatrek-Moumoulidis, D.; van der Spek, P.J.; Hauck, S.M.; Boon, C.J.F.; Emri, E.; Stewart, A.J.; et al. On the origin of proteins in human drusen: The meet, greet and stick hypothesis. Prog. Retin. Eye Res. 2019, 70, 55–84. [Google Scholar] [CrossRef] [PubMed]
- Al-Dosari, D.I.; Ahmed, M.M.; Al-Rejaie, S.S.; Alhomida, A.S.; Ola, M.S. Flavonoid Naringenin Attenuates Oxidative Stress, Apoptosis and Improves Neurotrophic Effects in the Diabetic Rat Retina. Nutrients 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Kropp, M.; Golubnitschaja, O.; Mazurakova, A.; Koklesova, L.; Sargheini, N.; Vo, T.-T.K.S.; Clerck, E. de; Polivka Jr., J.; Potuznik, P.; Polivka, J.; et al. Diabetic retinopathy as the leading cause of blindness and early predictor of cascading complications – Risks and mitigation. EMPA journal 2023. [Google Scholar] [CrossRef]
- Pennesi, M.E.; Neuringer, M.; Courtney, R.J. Animal models of age related macular degeneration. Mol. Aspects Med. 2012, 33, 487–509. [Google Scholar] [CrossRef]
- Murali, A.; Ramlogan-Steel, C.A.; Andrzejewski, S.; Steel, J.C.; Layton, C.J. Retinal explant culture: A platform to investigate human neuro-retina. Clin. Experiment. Ophthalmol. 2019, 47, 274–285. [Google Scholar] [CrossRef]
- Schnichels, S.; Dorfi, T.; Schultheiss, M.; Arango-Gonzalez, B.; Bartz-Schmidt, K.-U.; Januschowski, K.; Spitzer, M.S.; Ziemssen, F. Ex-vivo-examination of ultrastructural changes in organotypic retina culture using near-infrared imaging and optical coherence tomography. Exp. Eye Res. 2016, 147, 31–36. [Google Scholar] [CrossRef]
- Kobuch, K.; Herrmann, W.A.; Framme, C.; Sachs, H.G.; Gabel, V.-P.; Hillenkamp, J. Maintenance of adult porcine retina and retinal pigment epithelium in perfusion culture: characterisation of an organotypic in vitro model. Exp. Eye Res. 2008, 86, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Narayanamurthy, R.; Armstrong, E.A.; Yang, J.-L.J.; Yager, J.Y.; Unsworth, L.D. Administration of Selective Brain Hypothermia Using a Simple Cooling Device in Neonatal Rats. J. Neurosci. Methods 2023, 109838. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.E.; Jeong, J.Y.; Choi, H.; Chang, Y.K.; Ahn, M.S.; Ham, Y.R.; Na, K.R.; Lee, K.W. ERK phosphorylation plays an important role in the protection afforded by hypothermia against renal ischemia-reperfusion injury. Surgery 2017, 161, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Xi, L. Research Progress of the Application of Hypothermia in the Eye. Oxid. Med. Cell. Longev. 2020, 2020, 3897168. [Google Scholar] [CrossRef] [PubMed]
- Bilkhu, P.S.; Wolffsohn, J.S.; Naroo, S.A.; Robertson, L.; Kennedy, R. Effectiveness of nonpharmacologic treatments for acute seasonal allergic conjunctivitis. Ophthalmology 2014, 121, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Ames; III and B. S. Gurian. EFFECTS OF GLUCOSE AND OXYGEN DEPRIVATION ON FUNCTION OF ISOLATED MAMMALIAN RETINA. Neurophysiol. 1963, 26, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Ames III, A.; Nesbett, F.B. In vitro retina as an experimental model of the central nervous system. J Neurochem. 1981, 37, 867–877. [Google Scholar] [CrossRef]
- Kropp, M.; Harmening, N.; Bascuas, T.; Johnen, S.; Clerck, E. de; Fernández, V.; Ronchetti, M.; Cadossi, R.; Zanini, C.; Scherman, D.; et al. GMP-Grade Manufacturing and Quality Control of a Non-Virally Engineered Advanced Therapy Medicinal Product for Personalized Treatment of Age-Related Macular Degeneration. Biomedicines 2022, 10, 2777. [Google Scholar] [CrossRef]
- Wang, Q.; He, F.; Wu, L. NLRX1 increases human retinal pigment epithelial autophagy and reduces H2O2-induced oxidative stress and inflammation by suppressing FUNDC1 phosphorylation and NLRP3 activation. Allergol. Immunopathol. (Madr) 2023, 51, 177–186. [Google Scholar] [CrossRef]
- Zheng, H.-L.; Li, M.-T.; Zhou, T.; Wang, Y.-Y.; Shang, E.-X.; Hua, Y.-Q.; Duan, J.-A.; Zhu, Y. Protective effects of Lycium barbarum L. berry extracts against oxidative stress-induced damage of the retina of aging mouse and ARPE-19 cells. Food Funct. 2023, 14, 399–412. [Google Scholar] [CrossRef]
- Jüttner, J.; Szabo, A.; Gross-Scherf, B.; Morikawa, R.K.; Rompani, S.B.; Hantz, P.; Szikra, T.; Esposti, F.; Cowan, C.S.; Bharioke, A.; et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nature Neuroscience 2019, 22, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Schnichels, S.; Kiebler, T.; Hurst, J.; Maliha, A.M.; Löscher, M.; Dick, H.B.; Bartz-Schmidt, K.-U.; Joachim, S.C. Retinal Organ Cultures as Alternative Research Models. Altern. Lab. Anim. 2019, 47, 19–29. [Google Scholar] [CrossRef]
Figure 1.
Morphology, functional & inflammatory status of bovine retinae cultured with choroid and RPE. a) H&E-stained sections of the choroid-RPE-retina complex processed directly after isolation (0 d, left) and at 4 d in culture (right), both showed good tissue preservation. INL and RGC layer showed reduced cell numbers and holes at 4 d in culture. b) PKC expression in bipolar cells (green) was stable during ex vivo culture (bottom, 4 d) compared to sections stained directly after isolation (top, 0 d). c) GFAP expression was low in retinae at 4 d in culture (bottom, 4 d) compared to samples stained directly after isolation (top, 0 d). White arrows indicate areas of weak GFAP expression. Nuclei are counterstained using DAPI. NFL: nerve fiber layer; IPL: inter plexiform layer; OPL: outer plexiform layer; ONL: outer nuclear layer; PR: photoreceptors; BV: blood vessel.
Figure 1.
Morphology, functional & inflammatory status of bovine retinae cultured with choroid and RPE. a) H&E-stained sections of the choroid-RPE-retina complex processed directly after isolation (0 d, left) and at 4 d in culture (right), both showed good tissue preservation. INL and RGC layer showed reduced cell numbers and holes at 4 d in culture. b) PKC expression in bipolar cells (green) was stable during ex vivo culture (bottom, 4 d) compared to sections stained directly after isolation (top, 0 d). c) GFAP expression was low in retinae at 4 d in culture (bottom, 4 d) compared to samples stained directly after isolation (top, 0 d). White arrows indicate areas of weak GFAP expression. Nuclei are counterstained using DAPI. NFL: nerve fiber layer; IPL: inter plexiform layer; OPL: outer plexiform layer; ONL: outer nuclear layer; PR: photoreceptors; BV: blood vessel.
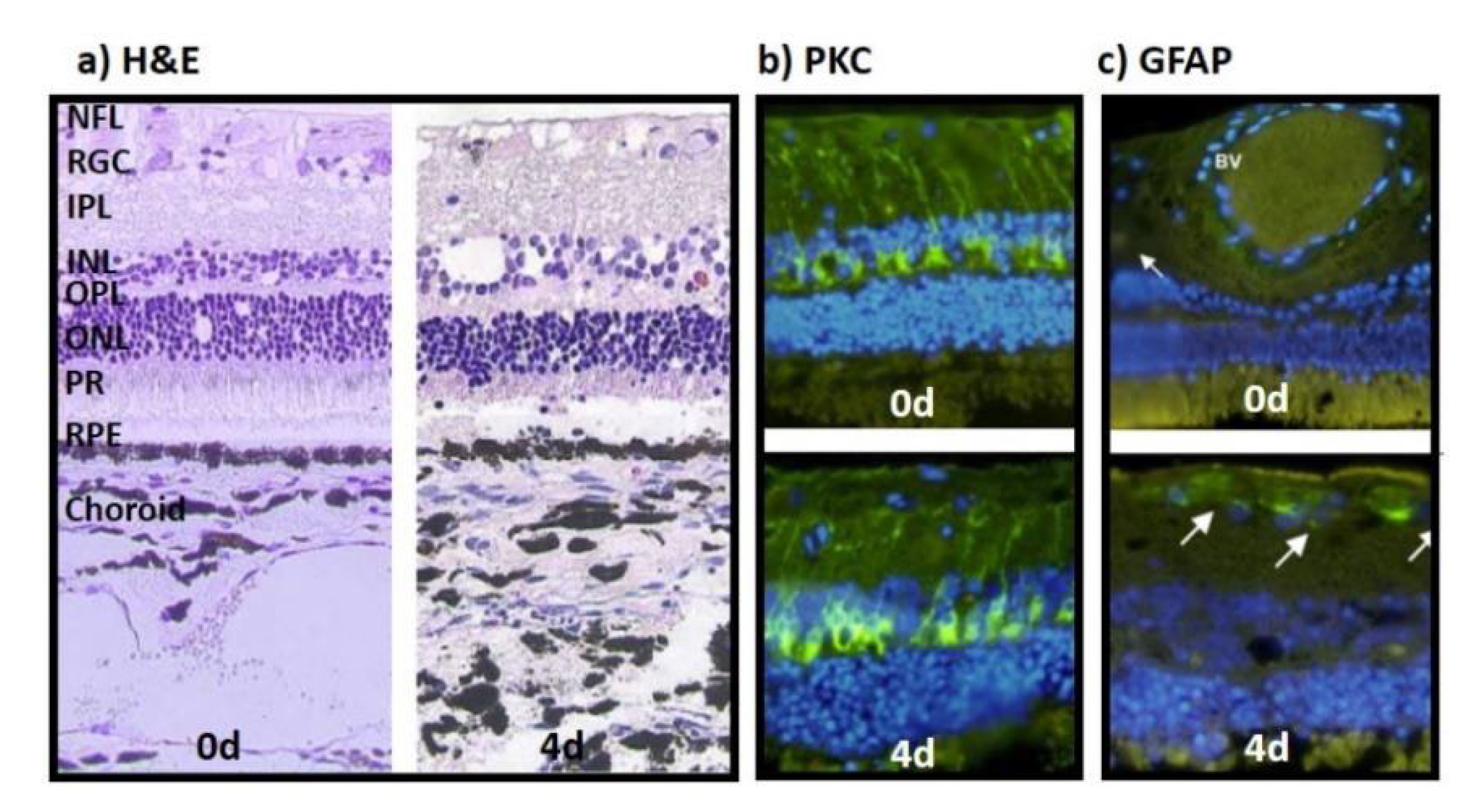
Figure 2.
Cell counts and morphology of porcine retinae cultured with & without RPE. a) Illustrated are cell counts in the ONL, INL, and RGC layers, determined in H&E-stained sections determined at 1 d from tissue samples isolated with (+RPE) and without (-RPE) RPE compared to sections directly stained after isolation without RPE (fresh -RPE). While cell counts of cultured neural retinae were similar to freshly processed tissue, cell numbers were significantly reduced in ONL, INL and RGC layers of cultures RPE-retina complexes. b) Individual values, n-numbers, and p-values of cell count in ONL, INL, and RGC layers. c) Representative H&E-stained sections illustrate tissue degeneration in retinae cultured with/without RPE. Overall, the “+RPE” culture decreased in thickness and revealed holes and cell loss. Magnification: 400x; scale bar: 50 µm.
Figure 2.
Cell counts and morphology of porcine retinae cultured with & without RPE. a) Illustrated are cell counts in the ONL, INL, and RGC layers, determined in H&E-stained sections determined at 1 d from tissue samples isolated with (+RPE) and without (-RPE) RPE compared to sections directly stained after isolation without RPE (fresh -RPE). While cell counts of cultured neural retinae were similar to freshly processed tissue, cell numbers were significantly reduced in ONL, INL and RGC layers of cultures RPE-retina complexes. b) Individual values, n-numbers, and p-values of cell count in ONL, INL, and RGC layers. c) Representative H&E-stained sections illustrate tissue degeneration in retinae cultured with/without RPE. Overall, the “+RPE” culture decreased in thickness and revealed holes and cell loss. Magnification: 400x; scale bar: 50 µm.
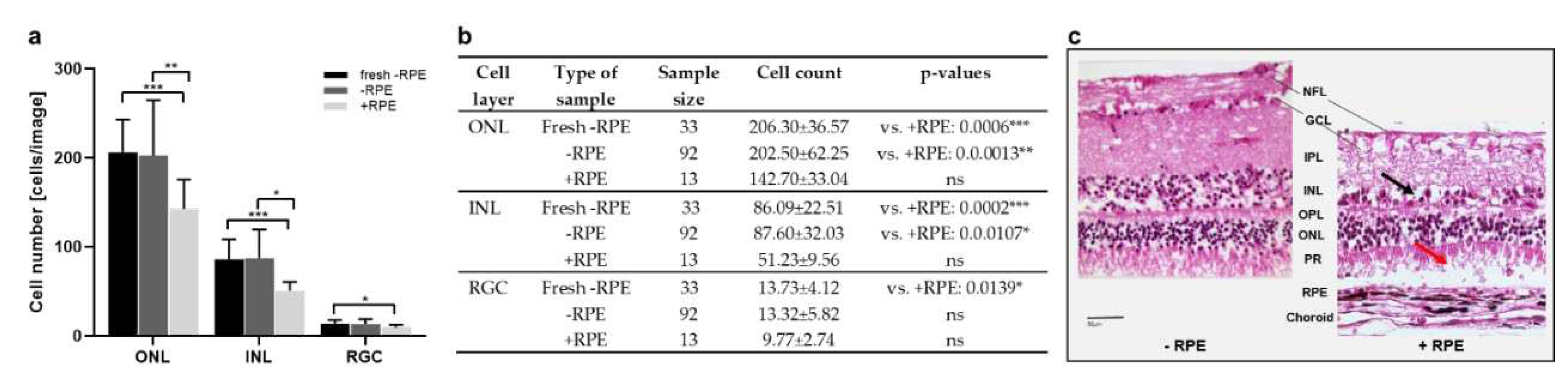
Figure 3.
Cell counts & morphology of porcine retinae cultured in Ames’ medium or DMEM. a) Illustrated are cell counts in the ONL, INL and RGC layers, counted in H&E-stained sections at 1d. The cell counts showed slightly reduced cell numbers in the ONL of tissue cultured in DMEM; in the INL and RGC layers this reduction was significant. b) Individual values, n-numbers, and p-values of cell count in ONL, INL, and RGC layers after culture in Ames’ medium vs. DMEM. c) H&E-stained sections illustrate representative retinae cultured 1 d in Ames’ or DMEM medium. Cross sections demonstrated signs of degeneration in DMEM-cultured retinae with large holes in the RGC (blue arrow), holes and cell loss in the INL (black arrow) and less structured, degenerated POS (red arrow). Fresh = stained directly after isolation. Magnification: 400x; scale bar: 50 µm.
Figure 3.
Cell counts & morphology of porcine retinae cultured in Ames’ medium or DMEM. a) Illustrated are cell counts in the ONL, INL and RGC layers, counted in H&E-stained sections at 1d. The cell counts showed slightly reduced cell numbers in the ONL of tissue cultured in DMEM; in the INL and RGC layers this reduction was significant. b) Individual values, n-numbers, and p-values of cell count in ONL, INL, and RGC layers after culture in Ames’ medium vs. DMEM. c) H&E-stained sections illustrate representative retinae cultured 1 d in Ames’ or DMEM medium. Cross sections demonstrated signs of degeneration in DMEM-cultured retinae with large holes in the RGC (blue arrow), holes and cell loss in the INL (black arrow) and less structured, degenerated POS (red arrow). Fresh = stained directly after isolation. Magnification: 400x; scale bar: 50 µm.
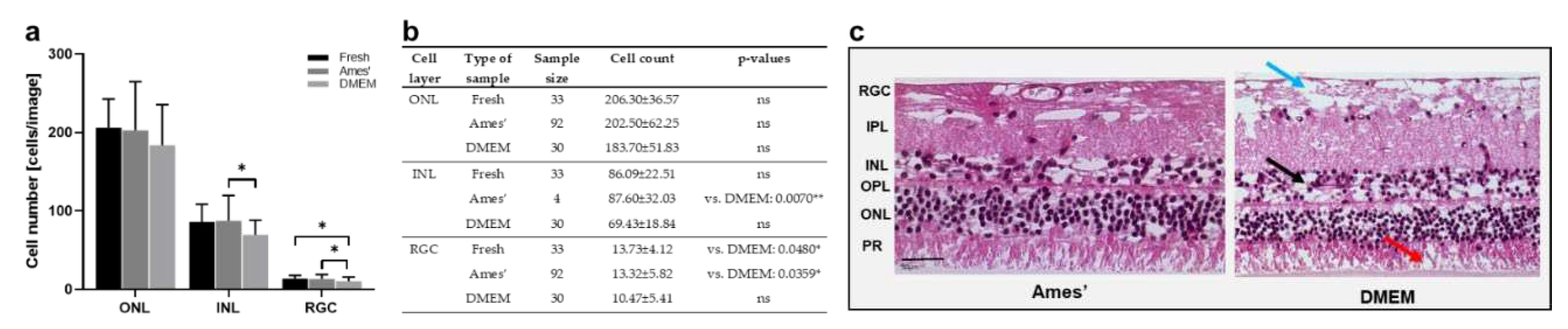
Figure 4.
Cell count and apoptosis in porcine retinae cultured at 37°C vs. 21°C. a) Increased cell counts were detected in the ONL of retinae cultured at 21°C for 1 d. Also in the INL, highest cell counts were found in 21°C-cultures. There was no important difference in cell counts in the RGC. b) The rate of apoptosis declined significantly in ONL, INL, and RGC layers of cultures incubated at 21°C. c) H&E-stained sections show representative examples of retinae cultured at 37°C or 21°C. Significant degeneration was seen after culture at 37°C with holes mainly in the INL and a significant loss of tissue integrity and POS. Also at 21°C, holes were found in the NFL and IPL (asterisks) layers. d) Individual values, n-numbers, and p-values of cell count and apoptosis analysis in ONL, INL, and RGC layers. Magnification: 400x; scale bar: 50 µm.
Figure 4.
Cell count and apoptosis in porcine retinae cultured at 37°C vs. 21°C. a) Increased cell counts were detected in the ONL of retinae cultured at 21°C for 1 d. Also in the INL, highest cell counts were found in 21°C-cultures. There was no important difference in cell counts in the RGC. b) The rate of apoptosis declined significantly in ONL, INL, and RGC layers of cultures incubated at 21°C. c) H&E-stained sections show representative examples of retinae cultured at 37°C or 21°C. Significant degeneration was seen after culture at 37°C with holes mainly in the INL and a significant loss of tissue integrity and POS. Also at 21°C, holes were found in the NFL and IPL (asterisks) layers. d) Individual values, n-numbers, and p-values of cell count and apoptosis analysis in ONL, INL, and RGC layers. Magnification: 400x; scale bar: 50 µm.
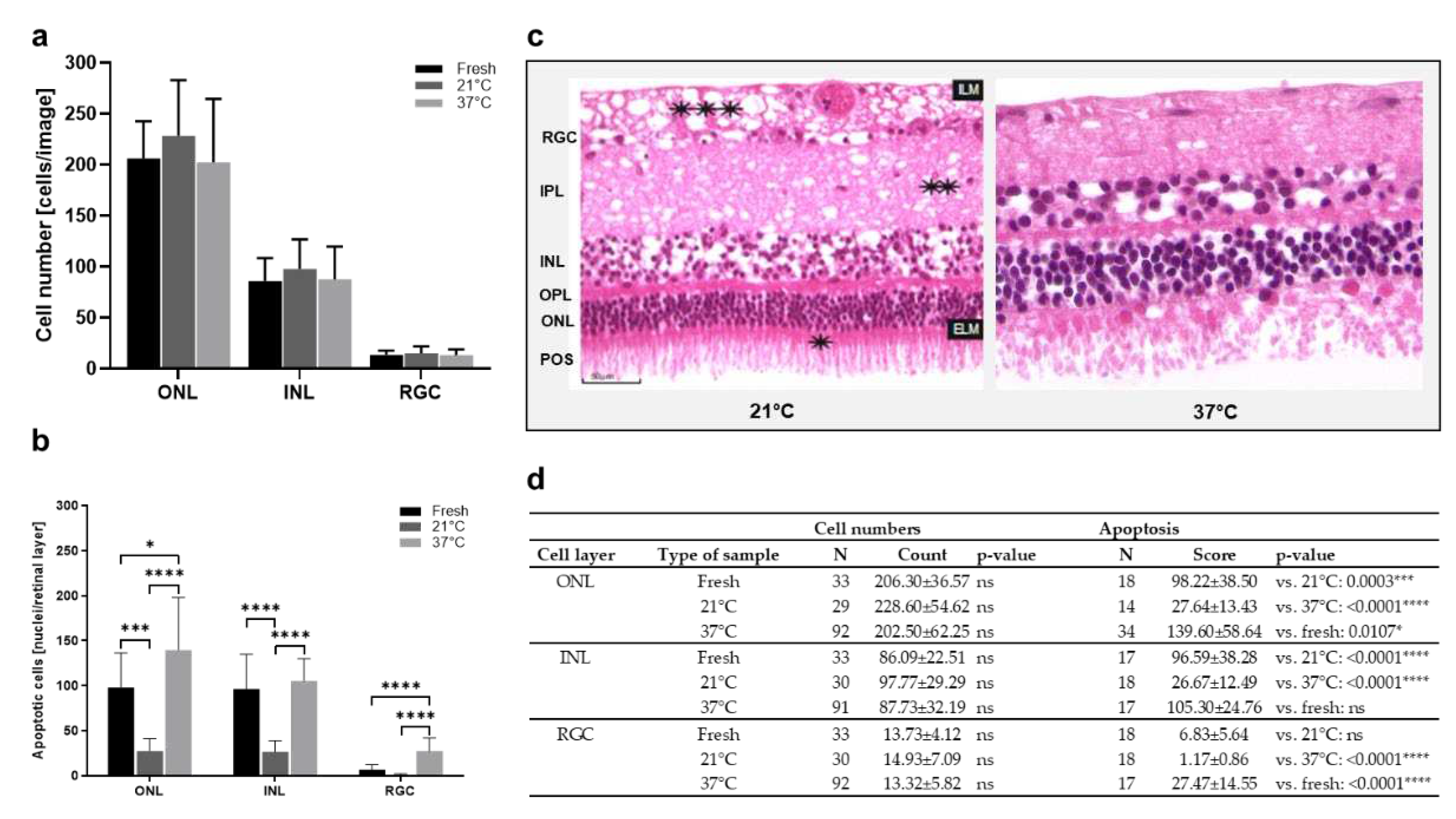
Figure 5.
Degeneration score in static porcine retina culture for 4 d. a) After isolation, retinae showed an integer structure without holes or clefts. b) At 1 d of culture, the tissue was still of good quality, but first brighter areas let assume cell loss (white arrow). Tissue borders were particularly exposed to degeneration (black arrow). c) At 2 days, degeneration became significant by bright spots, demonstrating lower cell density, i.e., cell loss (white arrows). d) Degeneration further increased at 3 d and bright areas progressed to holes (red arrows). e) After 4 d of culture, multiple holes and clefts demonstrated progressed tissue degeneration (red arrows); nevertheless, it must be noted that tissue assembly was still intact. f) Degeneration was quantified by the degeneration score. Though degeneration progressed over time, it never reached the highest score of ‘3’, i.e., a destruction of the tissue assembly. Significant differences were found between 1 vs. 3 and vs. 4 d of culture (p=0.0496 and p=0.0234, respectively). N= 5-8. Magnification: 200x; scale bar: 200 µm [41].
Figure 5.
Degeneration score in static porcine retina culture for 4 d. a) After isolation, retinae showed an integer structure without holes or clefts. b) At 1 d of culture, the tissue was still of good quality, but first brighter areas let assume cell loss (white arrow). Tissue borders were particularly exposed to degeneration (black arrow). c) At 2 days, degeneration became significant by bright spots, demonstrating lower cell density, i.e., cell loss (white arrows). d) Degeneration further increased at 3 d and bright areas progressed to holes (red arrows). e) After 4 d of culture, multiple holes and clefts demonstrated progressed tissue degeneration (red arrows); nevertheless, it must be noted that tissue assembly was still intact. f) Degeneration was quantified by the degeneration score. Though degeneration progressed over time, it never reached the highest score of ‘3’, i.e., a destruction of the tissue assembly. Significant differences were found between 1 vs. 3 and vs. 4 d of culture (p=0.0496 and p=0.0234, respectively). N= 5-8. Magnification: 200x; scale bar: 200 µm [41].
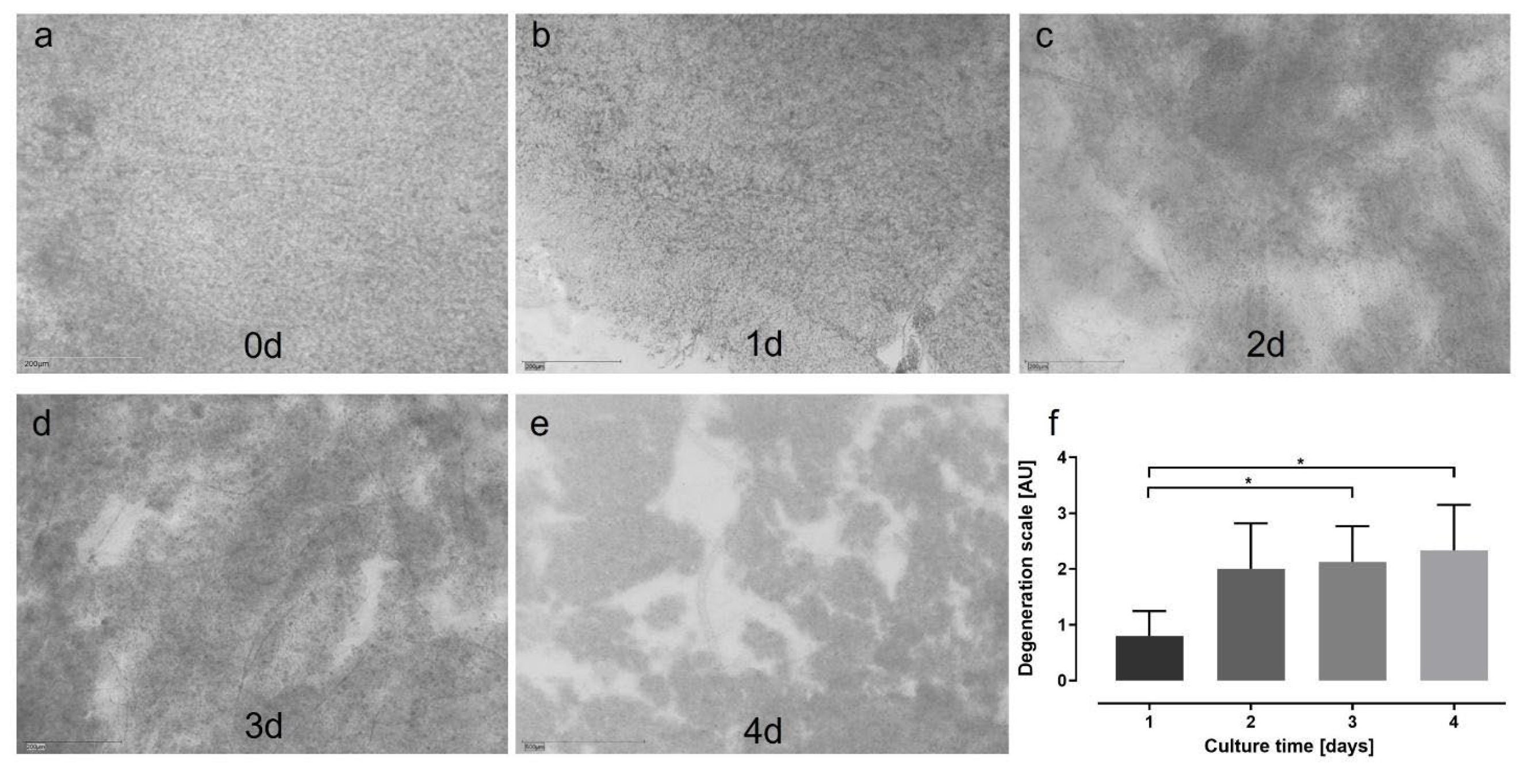
Figure 6.
Morphology of H&E-stained rat retina in static 12 d-long culture. a) Retinae stained directly after isolation showed the typical laminar structure without signs of degeneration; POS were present. b) At 4 d, tissue quality was comparable to directly stained retinae without significant signs of degeneration and preserved laminar structure. c) POS showed signs of degeneration after 6 d of culture. However, other retinal layers were preserved despite some holes indicating cell loss (red arrow). d) Until 9 d of culture, tissue integrity was maintained without significant signs of degeneration except the loss of POS. e+f) At 12 d of culture, degeneration became significant. Either retinae thickened indicating inflammatory processes (black arrow in (e)) or cell loss was significant and retinal layers, particularly INL and ONL, were no longer distinguishable (f). Clefts in (d) and (e) (white arrows) were due to tissue processing. N= 3. Magnification: 100x; scale bar: 100 µm [44].
Figure 6.
Morphology of H&E-stained rat retina in static 12 d-long culture. a) Retinae stained directly after isolation showed the typical laminar structure without signs of degeneration; POS were present. b) At 4 d, tissue quality was comparable to directly stained retinae without significant signs of degeneration and preserved laminar structure. c) POS showed signs of degeneration after 6 d of culture. However, other retinal layers were preserved despite some holes indicating cell loss (red arrow). d) Until 9 d of culture, tissue integrity was maintained without significant signs of degeneration except the loss of POS. e+f) At 12 d of culture, degeneration became significant. Either retinae thickened indicating inflammatory processes (black arrow in (e)) or cell loss was significant and retinal layers, particularly INL and ONL, were no longer distinguishable (f). Clefts in (d) and (e) (white arrows) were due to tissue processing. N= 3. Magnification: 100x; scale bar: 100 µm [44].
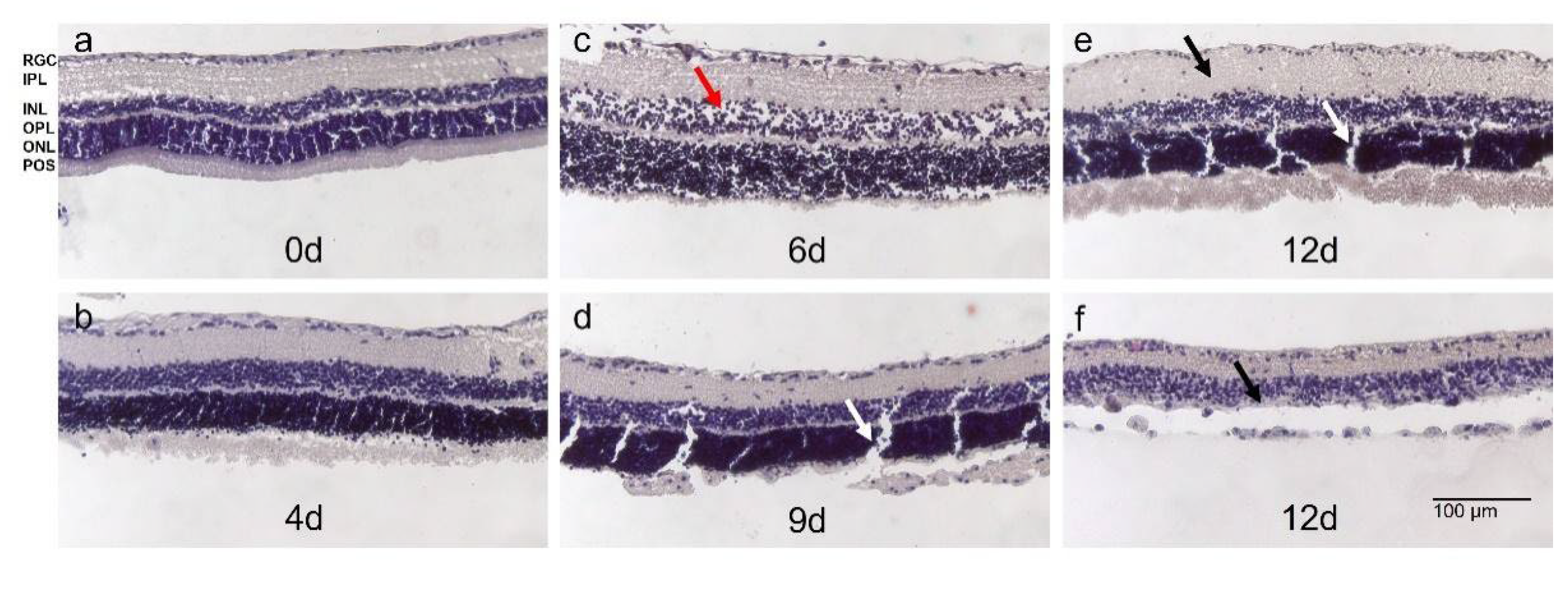
Figure 7.
Morphology and thickness of H&E-stained porcine and human retinae after static culture for 14 d. Sample isolation and tissue processing was improved (section 2.2.5) allowing for a 14 d-long culture. Shown are H&E-stained retinal sections from day 1 to day 14. a) The presented sections illustrate the quality of retinae at the time point of isolation, at 6 and 14 d. On day 1, layers were distinguishable, no signs of degeneration were seen (i). Also at 6 d, retinae look normal, though little cell loss might have occurred in the INL and ONL (ii). POS were present. After 14 d of culture, POS are no longer visible in H&E-stained sections, but laminar retinal structure is still intact (iii). N=16. b) The presented sections illustrate the quality of retinae at the time point of isolation, at 6 and 14 d. On day one, all layers were distinguishable, no signs of degeneration were seen (iv). On day 6, retinae look normal without significant signs of degeneration, though little cell loss might have occurred in the INL and ONL (black arrow) (v). POS were not visible. After 14 d of culture, laminar retinal structure is still intact (vi). N= 8. c) Retinal thickness was determined in porcine and human retinae cultured for 14 d using micrographs of H&E-stained samples. It did not differ significantly over 14 d; an ANOVA excluded significant changes between time points for both porcine (pRetina) (p=0.4882) and human (hRetina) tissue (p=0.6126). Magnification: 400x; scale bar: 50 µm [45].
Figure 7.
Morphology and thickness of H&E-stained porcine and human retinae after static culture for 14 d. Sample isolation and tissue processing was improved (section 2.2.5) allowing for a 14 d-long culture. Shown are H&E-stained retinal sections from day 1 to day 14. a) The presented sections illustrate the quality of retinae at the time point of isolation, at 6 and 14 d. On day 1, layers were distinguishable, no signs of degeneration were seen (i). Also at 6 d, retinae look normal, though little cell loss might have occurred in the INL and ONL (ii). POS were present. After 14 d of culture, POS are no longer visible in H&E-stained sections, but laminar retinal structure is still intact (iii). N=16. b) The presented sections illustrate the quality of retinae at the time point of isolation, at 6 and 14 d. On day one, all layers were distinguishable, no signs of degeneration were seen (iv). On day 6, retinae look normal without significant signs of degeneration, though little cell loss might have occurred in the INL and ONL (black arrow) (v). POS were not visible. After 14 d of culture, laminar retinal structure is still intact (vi). N= 8. c) Retinal thickness was determined in porcine and human retinae cultured for 14 d using micrographs of H&E-stained samples. It did not differ significantly over 14 d; an ANOVA excluded significant changes between time points for both porcine (pRetina) (p=0.4882) and human (hRetina) tissue (p=0.6126). Magnification: 400x; scale bar: 50 µm [45].
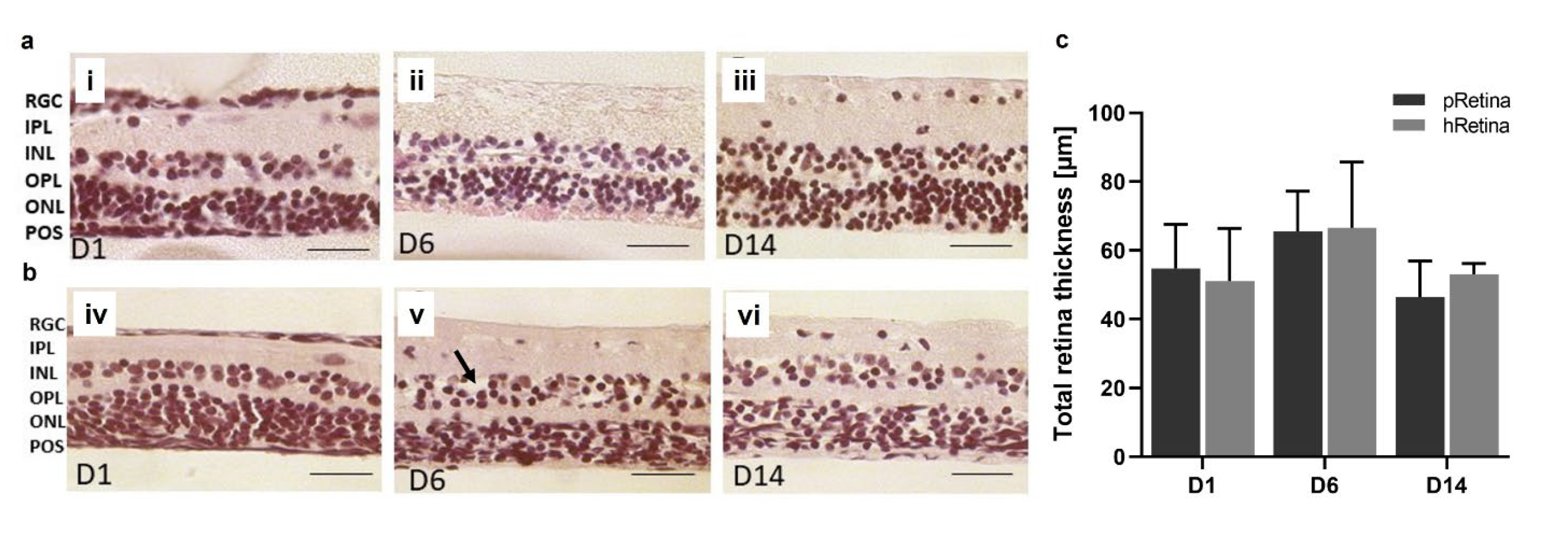
Figure 8.
Cell viability, death, & degeneration in static culture of p- and hRetina for 14 d. a) The curves show luminescence measured in culture media of p- and hRetina over 14 d. While luminescence remained low in hRetinae, it was high at the beginning in pRetinae followed by a decrease. Luminescence inversely correlates to viability indicating a high cell viability (or low cell death rate) for both species except within the first week in pRetinae. b) Necrosis was determined by PI-staining revealing low and stable rates of necrosis for both species from day 1-14. c) Degeneration in cultured pRetinae increased over time but remained moderate with a maximal increase to 2.13 (p<0.0001). d) In hRetina, degeneration remained stably low with scores from 1.03 to 1.60 (p=0.0290). e) Individual values of viability, death and degeneration analyses [45].
Figure 8.
Cell viability, death, & degeneration in static culture of p- and hRetina for 14 d. a) The curves show luminescence measured in culture media of p- and hRetina over 14 d. While luminescence remained low in hRetinae, it was high at the beginning in pRetinae followed by a decrease. Luminescence inversely correlates to viability indicating a high cell viability (or low cell death rate) for both species except within the first week in pRetinae. b) Necrosis was determined by PI-staining revealing low and stable rates of necrosis for both species from day 1-14. c) Degeneration in cultured pRetinae increased over time but remained moderate with a maximal increase to 2.13 (p<0.0001). d) In hRetina, degeneration remained stably low with scores from 1.03 to 1.60 (p=0.0290). e) Individual values of viability, death and degeneration analyses [45].
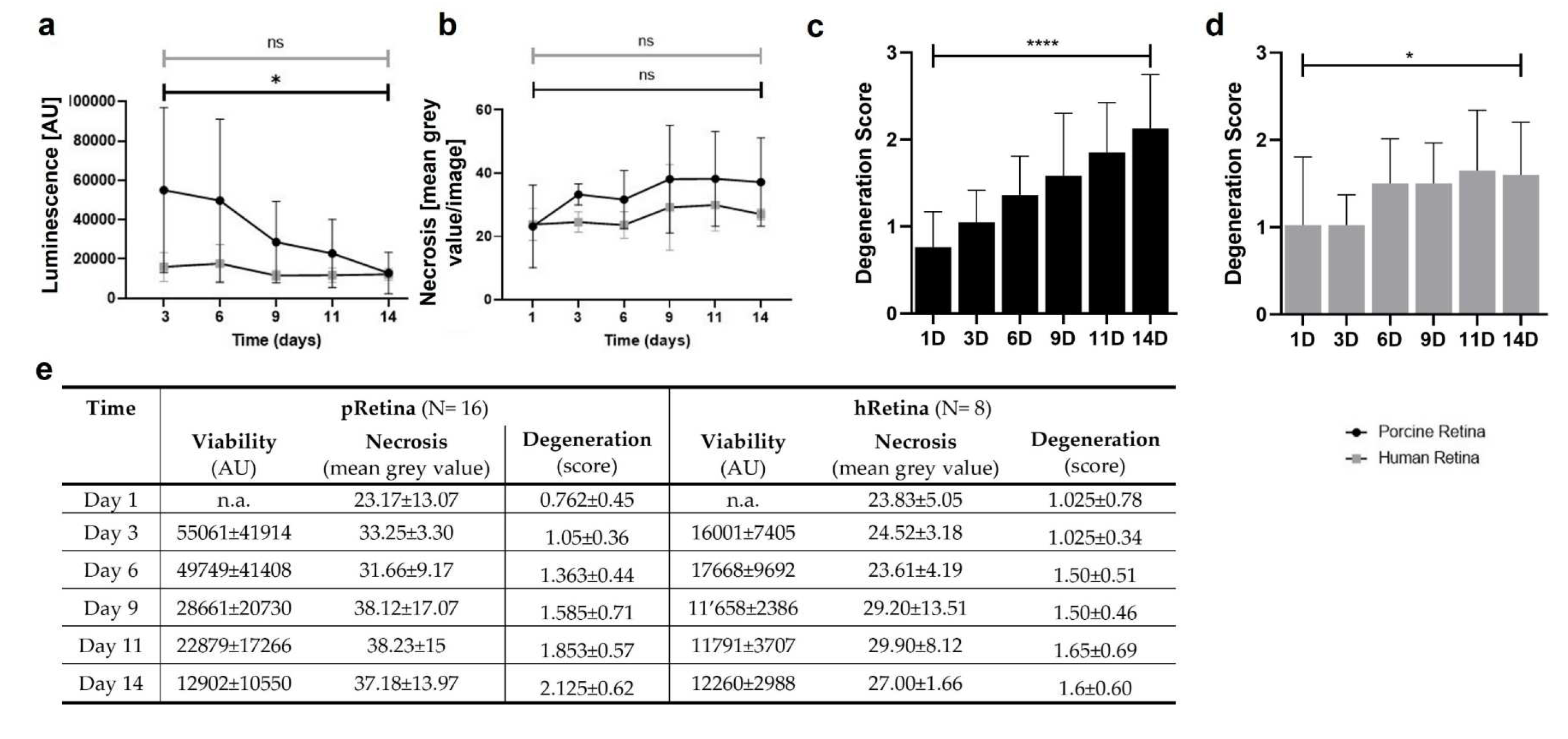
Figure 9.
Immunohistochemical staining of porcine and human retinae from 1-14 d of culture. a-f) Anti-GFAP-anti-rhodopsin double staining confirmed POS preservation up to day 14 (green). GFAP staining revealed inflammatory reactions in Müller cells at 6 d of culture that decreased in the second week of culture below base staining (red). The pattern was similar for pRetinae and hRetinae. g-l) Vimentin staining proved stable Müller cell presence and morphology for 14 d (red). Microglia activation, shown by Iba-1+ cell staining (green) remained low in both species at every time point. N= 16 (pRetinae); N=8 (hRetinae). Magnification: 200x. scale bar: 100 µm [45].
Figure 9.
Immunohistochemical staining of porcine and human retinae from 1-14 d of culture. a-f) Anti-GFAP-anti-rhodopsin double staining confirmed POS preservation up to day 14 (green). GFAP staining revealed inflammatory reactions in Müller cells at 6 d of culture that decreased in the second week of culture below base staining (red). The pattern was similar for pRetinae and hRetinae. g-l) Vimentin staining proved stable Müller cell presence and morphology for 14 d (red). Microglia activation, shown by Iba-1+ cell staining (green) remained low in both species at every time point. N= 16 (pRetinae); N=8 (hRetinae). Magnification: 200x. scale bar: 100 µm [45].
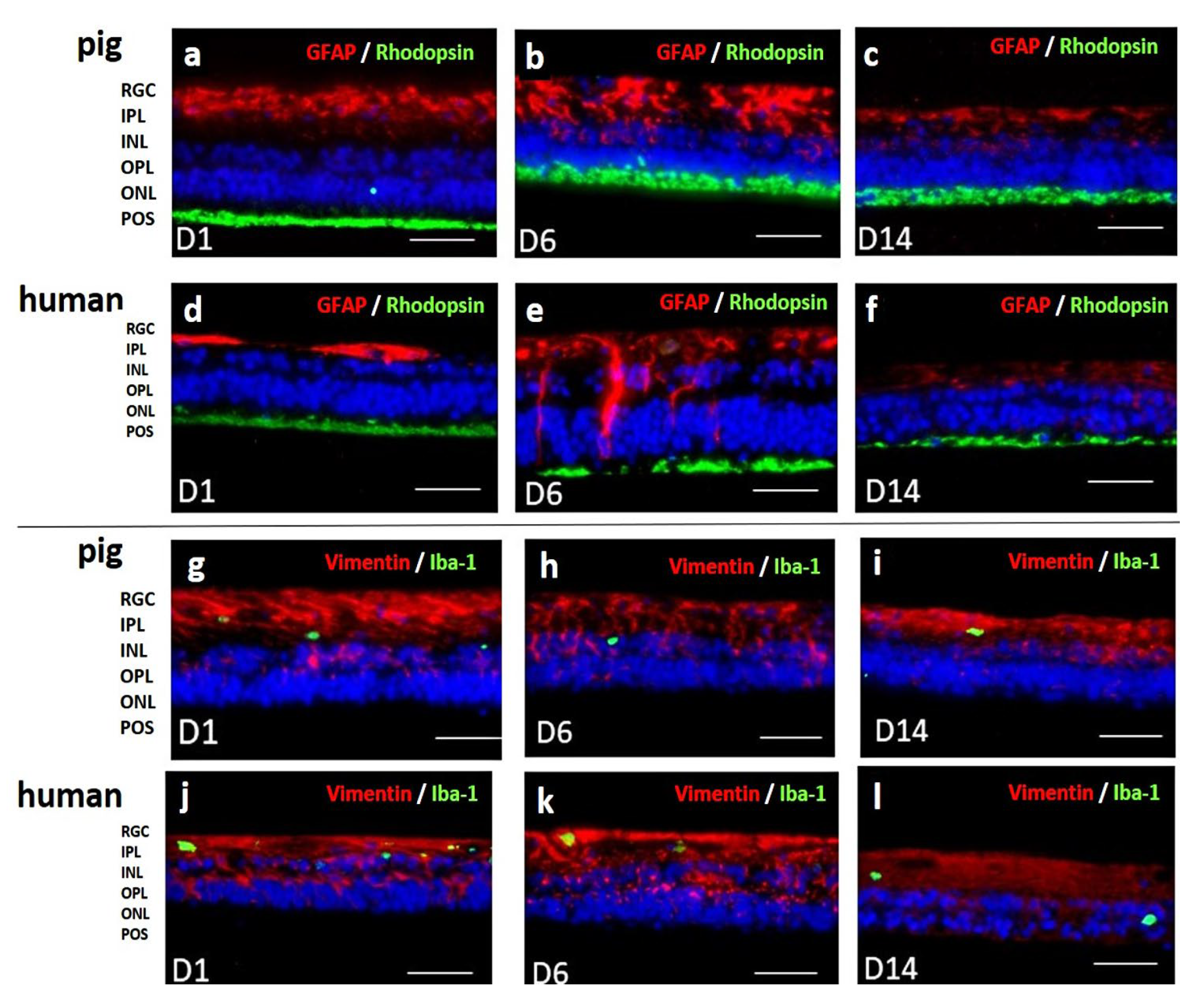
Figure 10.
Anti-oxidative cell protection by Scutellarin in HG. pRetinae were cultured for 4 d in static conditions (Model 3) in NG (6 mM) or HG conditions (30 mM) and treated with Scutellarin (1, 10 or 100 µM). a) Viability determined at 1 d (D1) revealed comparable ATP level in both glucose conditions, but significantly increased viability after treatment with low (1 µM) or medium (10 µM) doses of Scutellarin, while high doses (100 µM) had a detrimental effect. b) At 4 d (D4), viability decreased in HG, but increased after addition of low and medium doses of Scutellarin to levels of NG. c) H&E staining of retinal sections fixed at 4 d of culture (plus one control fixed immediately after isolation: “fresh”), revealed a lowered degeneration in POS of Scutellarin-treated samples if administered in low to medium doses. NG = normal glucose; HG = high glucose; HG1 = high glucose + 1 µM Scutellarin; HG10 = high glucose + 10 µM Scutellarin; HG100 = high glucose + 100 µM Scutellarin. N=4. Magnification: 200x. scale bar: 200 µm [38].
Figure 10.
Anti-oxidative cell protection by Scutellarin in HG. pRetinae were cultured for 4 d in static conditions (Model 3) in NG (6 mM) or HG conditions (30 mM) and treated with Scutellarin (1, 10 or 100 µM). a) Viability determined at 1 d (D1) revealed comparable ATP level in both glucose conditions, but significantly increased viability after treatment with low (1 µM) or medium (10 µM) doses of Scutellarin, while high doses (100 µM) had a detrimental effect. b) At 4 d (D4), viability decreased in HG, but increased after addition of low and medium doses of Scutellarin to levels of NG. c) H&E staining of retinal sections fixed at 4 d of culture (plus one control fixed immediately after isolation: “fresh”), revealed a lowered degeneration in POS of Scutellarin-treated samples if administered in low to medium doses. NG = normal glucose; HG = high glucose; HG1 = high glucose + 1 µM Scutellarin; HG10 = high glucose + 10 µM Scutellarin; HG100 = high glucose + 100 µM Scutellarin. N=4. Magnification: 200x. scale bar: 200 µm [38].
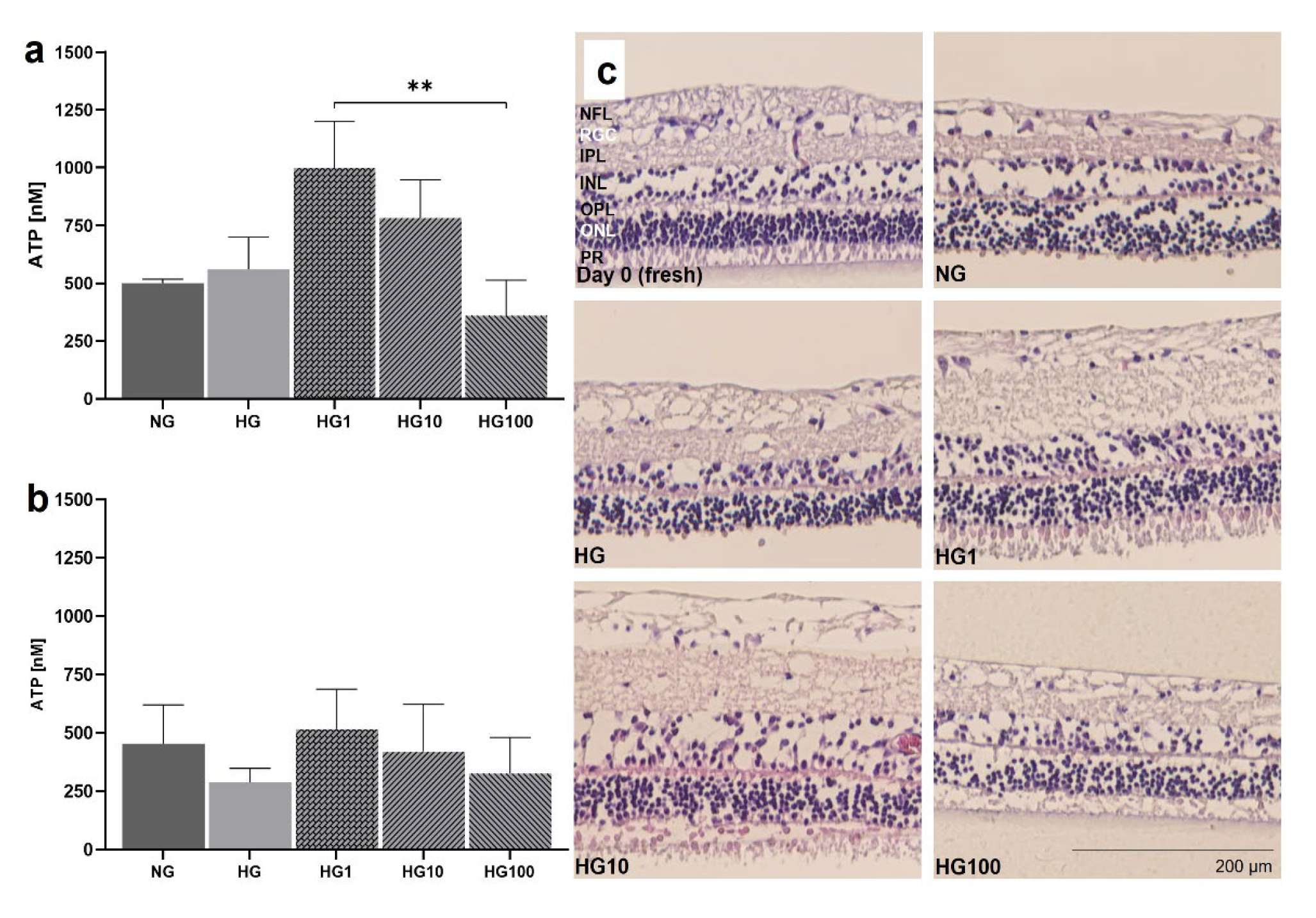
Figure 11.
Apoptosis at 1d and 4 d in HG and after treatment with the antioxidant Scutellarin. Apoptosis was measured by a TUNEL assay in retinal cross sections cultured in NG (6 mM) or HG (30 mM) treated with 1, 10, or 100 µM Scutellarin. a) The number of apoptotic cells at 1 d increased in HG cultures compared to NG samples but decreased after Scutellarin treatment, independently from the dose. b) At 4 d, the rate of apoptosis was generally increased compared to 1 d with similar differences between groups. The highest number of apoptotic cells was counted in HG, followed by NG. In all Scutellarin groups (HG1, HG10, HG100), apoptosis declined below NG values. c) Micrographs show representative results at 4 d with increased numbers of apoptotic cells in HG, which could be decreased by Scutellarin (HG1, HG10, HG100). N=4-6. Magnification: 200x. scale bar: 100 µm. NG = normal glucose; HG = high glucose; HG1 = high glucose + 1 µM Scutellarin; HG10 = high glucose + 10 µM Scutellarin; HG100 = 100 µM Scutellarin. Data are shown as mean±SD [38].
Figure 11.
Apoptosis at 1d and 4 d in HG and after treatment with the antioxidant Scutellarin. Apoptosis was measured by a TUNEL assay in retinal cross sections cultured in NG (6 mM) or HG (30 mM) treated with 1, 10, or 100 µM Scutellarin. a) The number of apoptotic cells at 1 d increased in HG cultures compared to NG samples but decreased after Scutellarin treatment, independently from the dose. b) At 4 d, the rate of apoptosis was generally increased compared to 1 d with similar differences between groups. The highest number of apoptotic cells was counted in HG, followed by NG. In all Scutellarin groups (HG1, HG10, HG100), apoptosis declined below NG values. c) Micrographs show representative results at 4 d with increased numbers of apoptotic cells in HG, which could be decreased by Scutellarin (HG1, HG10, HG100). N=4-6. Magnification: 200x. scale bar: 100 µm. NG = normal glucose; HG = high glucose; HG1 = high glucose + 1 µM Scutellarin; HG10 = high glucose + 10 µM Scutellarin; HG100 = 100 µM Scutellarin. Data are shown as mean±SD [38].
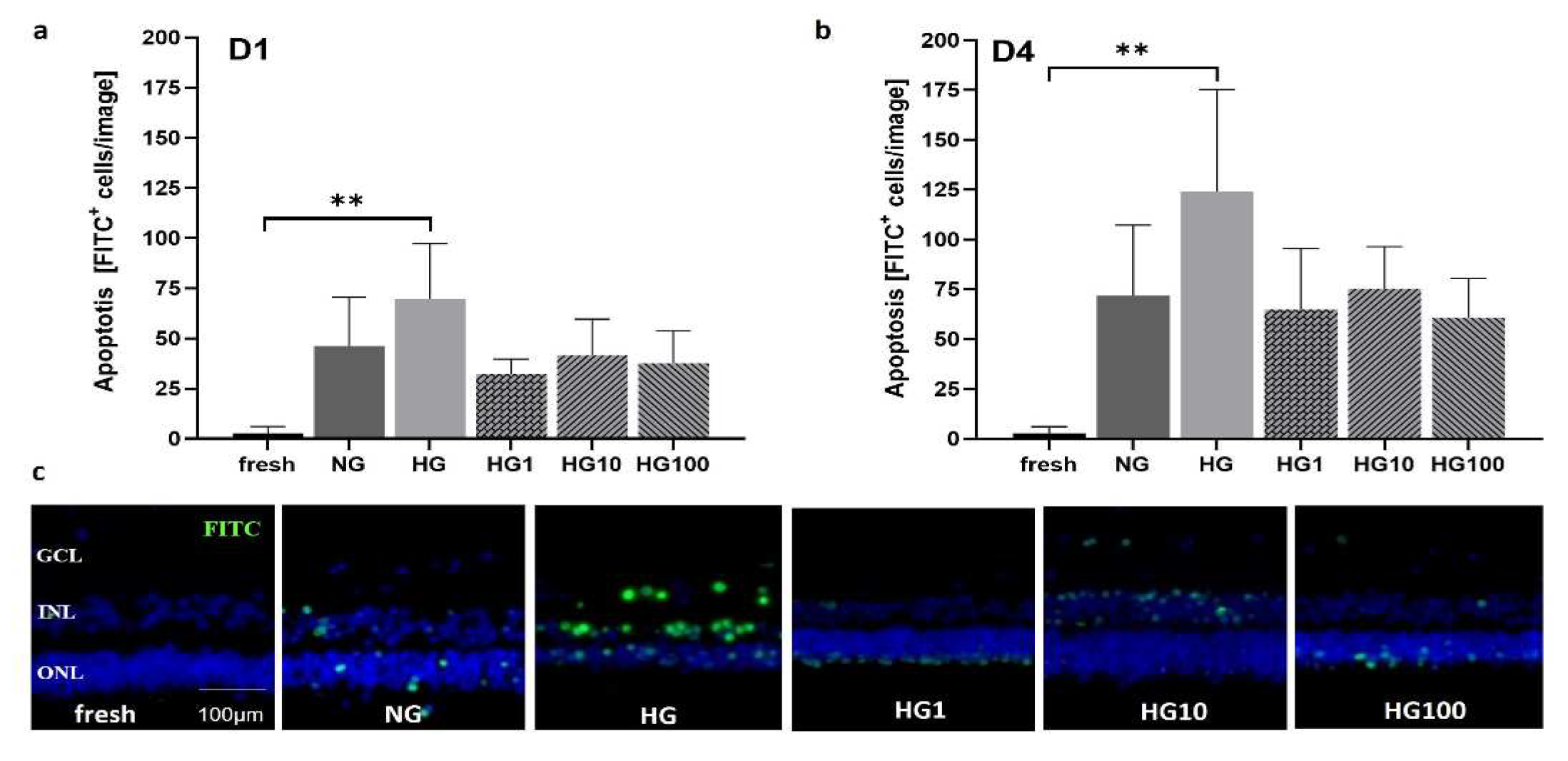
Figure 12.
OS in rat retinae incubated with H2O2 and treated with PEDF and/or GM-CSF. Retinae were treated for 3 d with PEDF and/or GM-CSF before incubation with H2O2 on day 3. At 4 d, viability and GSH levels were analyzed. Many samples were significantly degenerated and did not deliver technically valid results; thus, the number of valid results was analyzed as an additional parameter. a) Highest viabilities were detected in tissue treated with both proteins (after H2O2-incubation), followed by the PH group, GM-CSF and PEDF. Untreated, “normal” tissue without H2O2-incubation, had second lowest level. In groups H2O2 and GH, no valid results were measured. b) The number of valid viability measurements of data shown in (a) was increased in samples not H2O2-incubated and PEDF/GM-CSF-treated. The number of valid results decreased in PH and PGH groups, and no valid results were received in groups H2O2 and GH. c) Lowest GSH levels were measured in the groups fresh, PEDF, and GM-CSF. Medium levels were detected in groups H2O2 and PH, while highest GSH levels were determined in groups GH and PGH. d) The percentages of valid GSH assay results of data shown in (c) were highest in groups PH, GH, and PGH, followed by the H2O2. The lowest percentages were seen in groups fresh, PEDF, and GM-CSF. N=3-8. PH = PEDF+H2O2; GH = GM-CSF+H2O2; PGH = PEDF+GM-CSF. A chi-square test for trend was performed showing significant differences for the viability analysis (p<0.0001) and for the GSH assay (p=0.0069).
Figure 12.
OS in rat retinae incubated with H2O2 and treated with PEDF and/or GM-CSF. Retinae were treated for 3 d with PEDF and/or GM-CSF before incubation with H2O2 on day 3. At 4 d, viability and GSH levels were analyzed. Many samples were significantly degenerated and did not deliver technically valid results; thus, the number of valid results was analyzed as an additional parameter. a) Highest viabilities were detected in tissue treated with both proteins (after H2O2-incubation), followed by the PH group, GM-CSF and PEDF. Untreated, “normal” tissue without H2O2-incubation, had second lowest level. In groups H2O2 and GH, no valid results were measured. b) The number of valid viability measurements of data shown in (a) was increased in samples not H2O2-incubated and PEDF/GM-CSF-treated. The number of valid results decreased in PH and PGH groups, and no valid results were received in groups H2O2 and GH. c) Lowest GSH levels were measured in the groups fresh, PEDF, and GM-CSF. Medium levels were detected in groups H2O2 and PH, while highest GSH levels were determined in groups GH and PGH. d) The percentages of valid GSH assay results of data shown in (c) were highest in groups PH, GH, and PGH, followed by the H2O2. The lowest percentages were seen in groups fresh, PEDF, and GM-CSF. N=3-8. PH = PEDF+H2O2; GH = GM-CSF+H2O2; PGH = PEDF+GM-CSF. A chi-square test for trend was performed showing significant differences for the viability analysis (p<0.0001) and for the GSH assay (p=0.0069).
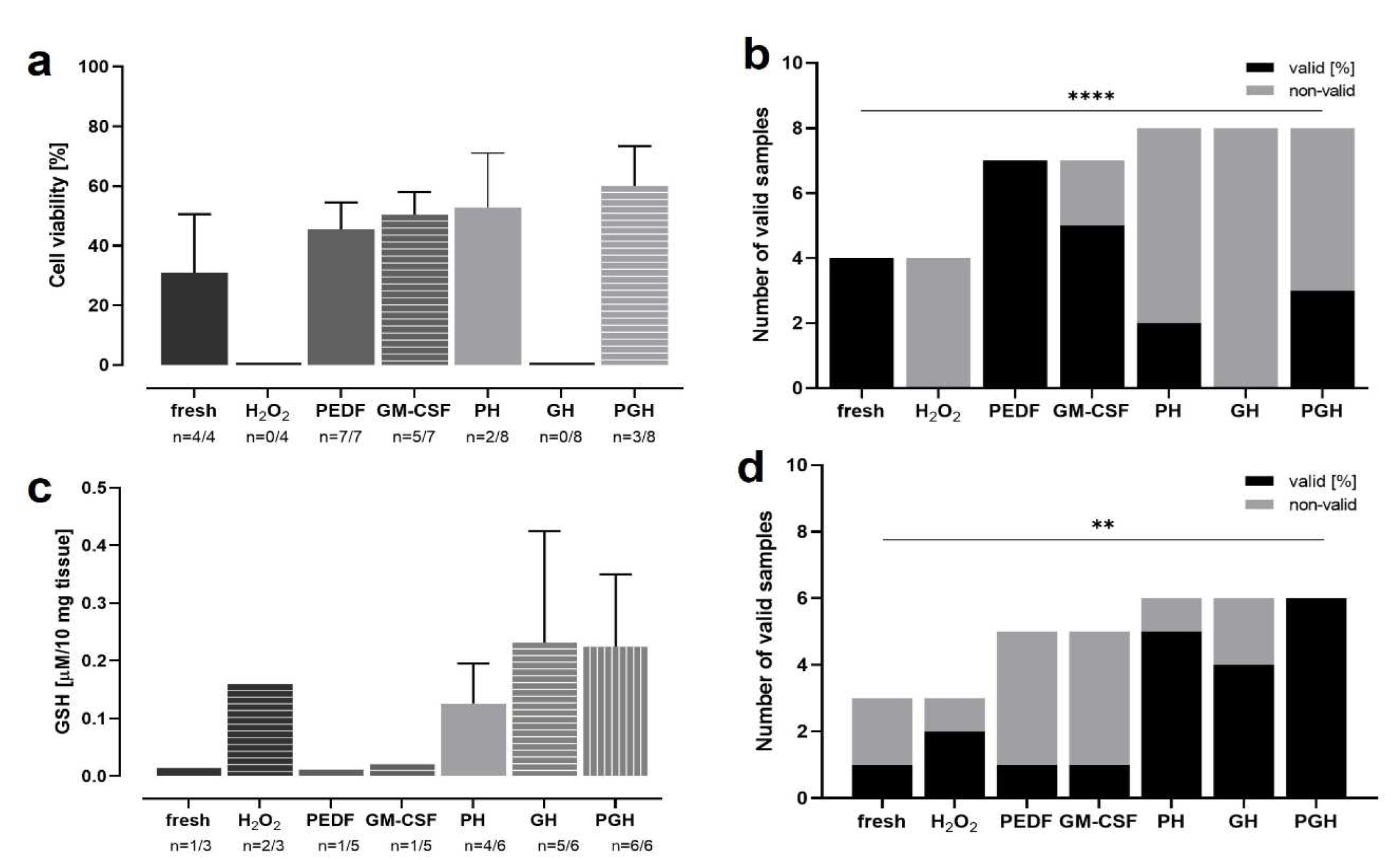
Table 1.
Hemalum and Eosin (H&E) staining protocol. The Table lists the staining protocol including the generally performed decreasing EtOH rehydration and increasing EtOH dehydration series.
Table 1.
Hemalum and Eosin (H&E) staining protocol. The Table lists the staining protocol including the generally performed decreasing EtOH rehydration and increasing EtOH dehydration series.
| Process | Reagents | Time | Temperature |
|---|---|---|---|
| Deparaffinization | Xylol | 3x3 min | RT |
| Rehydration | EtOH 2x 100%, 96%, 90%, 80%, 70%, 50%, dd H2O | 30 sec each 3 min |
RT |
| Staining | Hemalum Tap H2O Acid alcohol dd H2O Eosin dd H2O |
4 min 3x2 min 10 Sec 2x1 min 3 min 1 min |
RT |
| Dehydration | 50%, 70%, 80%, 90%, 96% 2x100% EtOH | 30 sec each 2x2 min |
RT |
| Mounting | Mounting medium | 1 min | RT |
Table 2.
Degeneration score. The score has been created to quantify degeneration in BF micrographs from flat mount preparations of retinal samples.
Table 2.
Degeneration score. The score has been created to quantify degeneration in BF micrographs from flat mount preparations of retinal samples.
| Grade | Tissue quality |
|---|---|
| 0 | Healthy tissue, integral. No damage |
| 1 | Irregular borders, small holes |
| 2 | Big holes, patches of cell loss |
| 3 | Big holes, major cell loss |
Table 3.
Immunofluorescence staining protocol.
| Process | Reagents | Time | Temperature |
|---|---|---|---|
| Deparaffinization | Xylol | 3x3 min | RT |
| Rehydration | EtOH 100%, 96%, 90%, 80%, 70%, 50%, dd H2O | 3 min each 3x5 min |
RT |
| Demasking | Citrate buffer, 0.1 M pH 6 (Boiling) cooling |
15 min 30 min |
95oC-100oC |
| Washing | PBS | 3x10 min | RT |
| Blocking | PBS-BSA 3% | 2 h | 370C |
| Staining | Primary antibody (PBS-BSA 1%) | overnight | 40C |
| Washing | PBS | 3x10 min | RT |
| Staining | Secondary antibody (PBS) | 30 min | 370C |
| Washing | PBS | 3x10 min | RT |
| Mounting | Mounting medium with DAPI | 1 min | RT |
Table 4.
Primary and secondary antibody used for immunofluorescence staining. GFAP: Glial fibrillary acidic protein, IBA-1: Ionized calcium-binding adapter molecule 1.
Table 4.
Primary and secondary antibody used for immunofluorescence staining. GFAP: Glial fibrillary acidic protein, IBA-1: Ionized calcium-binding adapter molecule 1.
| Specificity | Cells labelled | Host | Conjugate | Clonality | Company | ID | Dilution |
|---|---|---|---|---|---|---|---|
| Primary antibodies | |||||||
| Protein kinase C | Bipolar cells | Chicken | Unconjugated | Polyclonal | Abcam* ab14078 | n.a. | 1:50 |
| Rhodopsin | Rods | Rabbit | Unconjugated | Polyclonal | Novus Biological$ NLS1052 | AB_2178795 | 1:700 |
| GFAP | Astrocytes | Mouse | Unconjugated | Monoclonal | Millipore† MAB360 | AB_11212597 | 1:300 |
| Iba-1 | Microglia | Rabbit | Unconjugated | Polyclonal | Fujifilm Wako Pure Chemical Corporation§ 01-1874 | AB_2314666 | 1:750 |
| Vimentin | Müller cells | Mouse | Unconjugated | Monoclonal | Merck† MAB3400 | AB_94843 | 1:120 |
| Secondary antibodies | |||||||
| Mouse | NA | Donkey | AlexaFlour 647 | NA | Abcam ab150107 | AB_2890037 | 1:250 |
| Rabbit | NA | Donkey | AlexaFlour 488 | NA | Jackson# 711546152 | AB_2340619 | 1:100 |
*Cambridge, UK; $Zug, Switzerland; †Darmstadt, Germany; §Osaka, Japan; #Ely, UK.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



