
Submitted:
10 May 2023
Posted:
10 May 2023
You are already at the latest version
Abstract
Antimicrobial resistance (AMR) is a major global concern; antibiotics and other regular treatment methods have failed to overcome the increasing number of infectious diseases. Bacteriophages (phages) are viruses that specifically target/ kill bacterial hosts without affecting other human microbiome. Phage therapy provides optimism in the current global healthcare scenario with a long history of its applications in humans that has now reached various clinical trials. Phages in clinical trials have specific requirements of being exclusively lytic, free from toxic genes with an enhanced host range that adds an advantage to this requisite. This review explains in detail the various phage engineering methods and their potential applications in therapy. To make phages more efficient, engineering has been attempted using techniques like conventional homologous recombination, Bacteriophage Recombineering of Electroporated DNA (BRED), clustered regularly interspaced short palindromic repeats (CRISPR)-Cas, CRISPY-BRED/Bacteriophage Recombineering with Infectious Particles (BRIP), chemically accelerated viral evolution (CAVE), and phage genome rebooting. Phages are administered in cocktail form in combination with antibiotics, vaccines, and purified proteins, such as endolysins. Thus, phage therapy is proving to be a better alternative for treating life-threatening infections, with more specificity and fewer detrimental consequences.
Keywords:
Antimicrobial Resistance
; Phage engineering
; endolysins
; phage therapy
1. Introduction
From ancient times till date, mankind has faced serious health concerns due to several bacterial infections, such as tuberculosis, typhoid, syphilis, diphtheria, and cholera etc. [1]. After their discovery since 1930s, antibiotics were the only easily available treatment for curing such infections and were quite effective for these prolonged illnesses; however, this shining era of antibiotics soon started to decline with the emergence of antibiotic resistance. Natural causes of antibiotic resistance include rapid mutations in the genes involved in antibiotic transport/ metabolism, horizontal transfer of mutated genes to other bacteria and selective pressures for the survival of these resistant bacteria due to uncontrolled and inappropriate use of antibiotics [2]. Approximately, 4.95 million deaths occurred due to bacterial antimicrobial resistance in 2019, with the maximum resistance caused by Escherichia coli (E. coli). In addition, Staphylococcus aureus (S. aureus), Pseudomonas aeruginosa (P. aeruginosa), Streptococcus pneumonia (S. pneumonia), Klebsiella pneumonia (K. pneumonia), Acinetobacter baumannii (A. baumannii), Mycobacterium tuberculosis (Mtb), Enterococcus faecalis (E. faecalis), Enterococcus faecium (E. faecium), and Streptococcus agalactiae (S. agalactiae ) (group B Streptococcus) are also responsible for higher mortality rates [3]. In 2016, the United Nations General Assembly graded the problem of AMR as the “greatest and most urgent global risk,” aiming to search for better alternatives for treating deadly bacterial infections [4]. In this search, probiotics, nanobiotics, antibody-antibiotic conjugates, vaccines, stem cell-based antimicrobial peptides, CRISPR-Cas editing machinery and phage therapy are the currently most widespread options to combat AMR. Among these, phage therapy is considered the most effective in the treatment of persistent bacterial infections that are globally prevalent [5]. Phages are self-replicating, highly specific to their host, and quite resistant to environmental changes such as pH and temperature, making them suitable candidates for combating ongoing AMR [6].
Phage therapy utilizes phages, which are the natural predators of bacteria that hijack their machinery to reproduce by the process commonly known as transduction. Phages are made up of two components: nucleic acid (DNA/RNA) encapsulated in a protein capsid that attaches to the bacterial surface via specific receptors, inserts their genetic material, and completes their life cycle either by lytic cycle (release of newer phage progeny by lysis of the bacterial cell) or by lysogenic cycle (integrating the phage genome into the bacterial genome) [7]. Felix d’Herelle was the first person to propose the idea of using phages therapeutically, with the clinical application of phages to treat four pediatric instances of bacterial diarrhea in 1919 at the Hôpital des Enfants-Malades in Paris [8]. However, with continued efforts in the early 20th century, d'Herelle advanced phage therapy by treating cholera, bubonic plague, and dysentery using a network of phage therapy facilities in Europe and India [9]. Phage therapy in India was first carried out in the Punjab region for the treatment of cholera, where the mortality rate was reduced by 90% in the experimental group compared to that in the control group [8].
Phages should possess specific characteristics that include a strict lytic lifecycle, absence of toxic genes, broader host range, good transduction and virulence potential [10]. Phages co-evolve with their bacterial hosts, hence; they possess a rich diversity of genetic elements in their compact genomes to fight their bacterial hosts, such as tail-fibre/ spike proteins for specific recognition of their hosts and the holin-endolysin machinery for cell lysis. For ex. in a particular genus like Mycobacterium, pathogenic (Mtb) and non-pathogenic (Mycobacterium smegmatis mc2155 (M.smeg)) species differ only in the composition of the sugar moieties present in their cell wall that imparts specificity in their phage binding [11]. In addition, phages and their respective hosts have co-evolved, also leading to the emergence of phage resistance, limiting their antimicrobial efficiency and host range [12]. Phages isolated from the natural environment may or may not meet the criteria for successful phage therapy hence; they require modifications for further use in phage therapy. Therefore, to overcome these limitations, genetic engineering of phages may provide better insight into their applications in phage therapy. Also, a rich library of diverse phages against a bacterial host can provide a better opportunity for the production of desired genetically engineered phages against a particular bacterial host species/ strain. Phage engineering can lead to the production of host range mutants generated by tail fibre mutations, exclusively lytic phages generated from lysogenic phages, phages with non-toxic genes generated by gene-deletions and diagnostic phages generated by incorporation of reporter genes. This review is divided into two sections; the first section explains in detail the various methods that have been successfully applied for the generation of genetically engineered phages and the second section explains the important applications of the genetically engineered phages till date.
2. Methods for Phage Genetic Engineering (PGE)
The various methods that are employed in phage genome engineering for better therapeutic outcomes are as follows:
2.1 Conventional homologous recombination (HR)
2.2 Bacteriophage Recombineering of Electroporated DNA (BRED)
2.3 CRISPR-CAS
2.4 CRISPY BRED and CRISPY BRIP
2.5 Chemically accelerated viral evolution (CAVE)
2.6 Rebooting of Phage Genome
2.1. Conventional homologous recombination (HR)
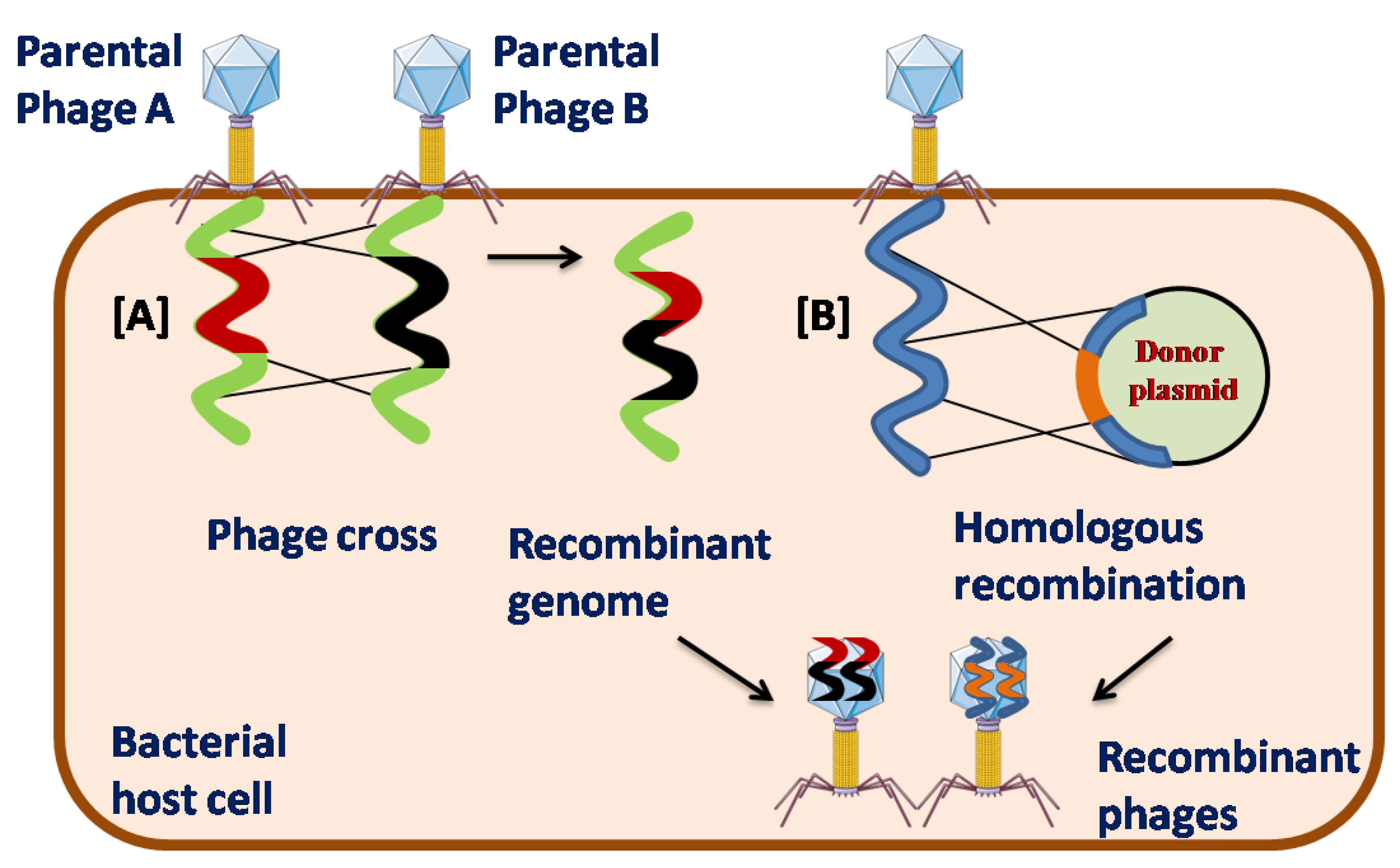
The most conventional method employed for PGE is homologous recombination (HR) of two wild-type parental phages inside the host cell. This process is also known as phage cross, in which bacterial host cells are co-infected with wild-type parental phages carrying different phenotypes. [13]. Although rare, once inside the host cell, the phage genome can either opt for its own or its bacterial host recombination system. Bacterial recombination machinery cannot carry out recombination between divergent genome sequences because phages with divergent genome sequences are likely to avoid the bacterial recombination machinery and preferably precede their own genome-encoded recombination functions. There are three major superfamilies of phage recombinases, which are divided into Rad51, Rad52, and Gp2.5 like proteins, among which Rad52 is the most divergent and the largest family of phage recombinases [14]. Modifications of the phage genome via HR usually do not occur at a specific position, limiting the use of this method. To overcome this shortcoming, HR between the phage genome and a plasmid carrying the desired mutation flanked by the corresponding sequence in the phage genome is required. In this method, a plasmid containing the desired mutation is first transformed into the host bacterium, followed by infection of the phage genome to be modified and mutants with insertions, deletions, or gene replacement can be generated [15]. The recombination frequency for some phages was approximately 5 × 10−3, which was much higher than that of the conventional phage crossing method. However, in general, the recombination frequency is relatively low, limiting its use [16]. Thus, HR is a long-drawn method that makes it difficult to screen recombinants with lower recombination frequencies [17].
Figure 1.
Schematic representation of [A] Phage Cross and [B] Homologous recombination (HR) in phage genetic engineering.
Figure 1.
Schematic representation of [A] Phage Cross and [B] Homologous recombination (HR) in phage genetic engineering.
2.2. Bacteriophage Recombineering of Electroporated DNA (BRED)
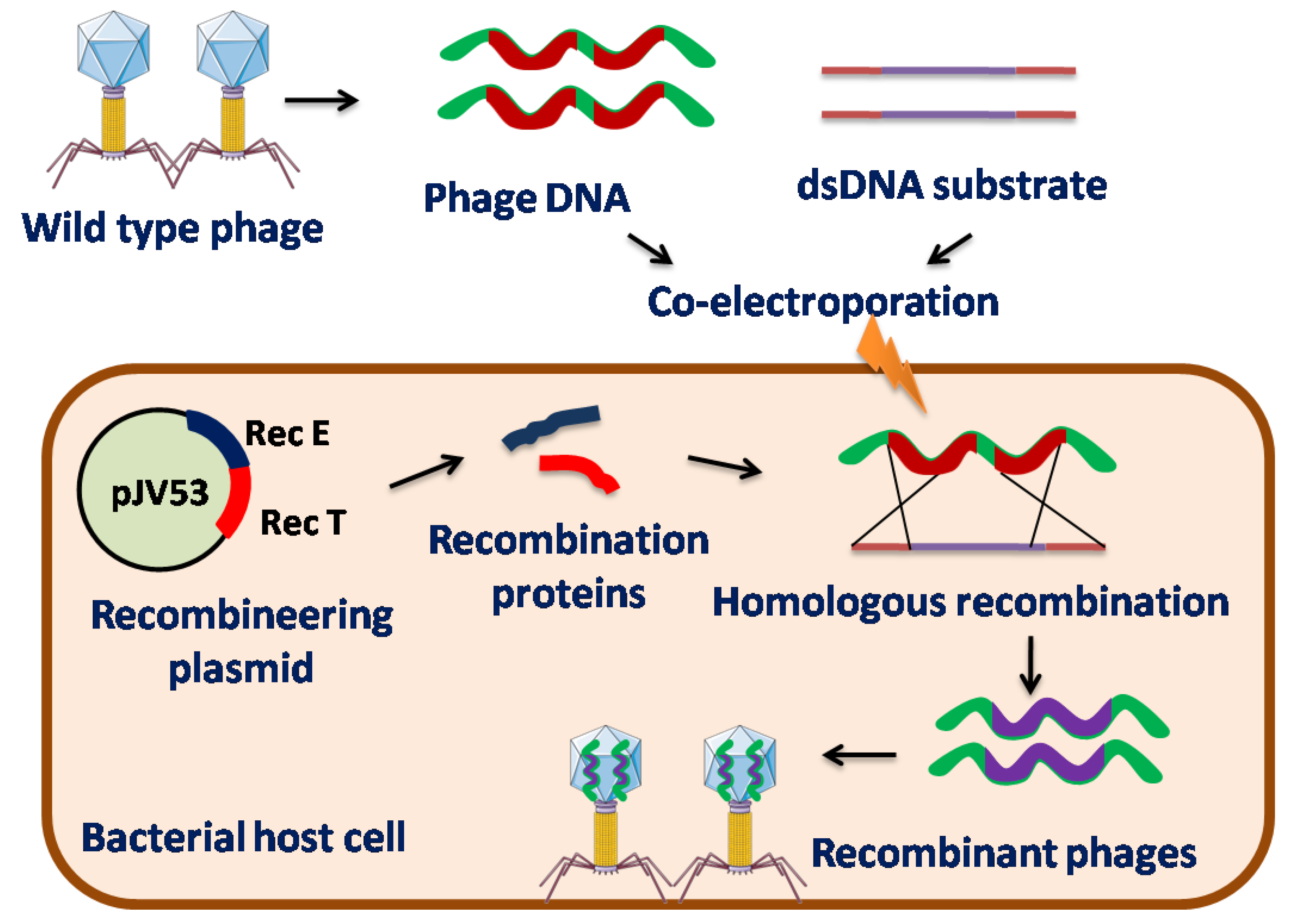
Recombineering is a genetic engineering technique which is based upon HR [18,19]. Initially, HR systems such as the red system of bacteriophage λ and the RecE/RecT system of the Rac prophage were used for genetic engineering. The red system of lambda phage primarily encodes three proteins, Exo, Beta, and Gam, whereas Rac-prophage encodes two proteins, RecE and RecT [20,21]. Rec E and Exo has 5’–3’ dsDNA exonuclease activity that cleaves one strand of dsDNA to generate a single-stranded DNA (ssDNA) substrate, whereas RecT and Beta are single-strand binding proteins (SSBs) that encourage the annealing of one strand of the DNA substrate to its recombination target in the phage genome. The λ phage Gam protein binds to the host E. coli RecBCD exonuclease complex and SbcD enzymes to inhibit their activities to prevent the degradation of the linear dsDNA substrate [22,23].
Recombineering requires co-electroporation of the substrate DNA and phage DNA template into a recombinant bacterial strain that expresses the phage recombineering proteins via inducible plasmid to promote HR [24,25,26]. Substrate DNA is designed on the basis of the required genome alterations to be made in phages [25] that are flanked by homologous sequences of the phage region where the mutation has to be incorporated which leads to HR between the phage genome and the substrate DNA. Recombination is thought to occur only after phage genome replication begins [25]. Plaques produced by phages contain both wild-type (non-recombinant) and mutant (recombinant) phages. Phage particles containing the desired mutation can then be retrieved by plating transformed cells, followed by screening these plaques using Polymerase Chain Reaction (PCR). Therefore, several rounds of plating and PCR are required to isolate recombinants [25,26]. This recombineering technique, when used in phages against Mycobacterium species, is known as BRED [23,27]. This technique was first devised by Marinelli et al. for mycobacteriophages and utilizes a recombination system encoded by mycobacteriophages with enhanced frequency of HR [28].
Homologs of RecE and RecT are rare among mycobacteriophages [29]. However, recombinant proteins from the mycobacteriophage Che9c, gp60, and gp61, which are distant relatives of RecE and RecT, respectively, have been identified and isolated [30]. BLAST analysis and biochemical characterization confirmed these functions. BLAST analysis depicted that Che9c gp60 protein shares 28% identity with λ RecE C-terminus, while Che9c gp61 shares 29% identity with λ RecT (contains a motif common to the family of ssDNA annealing proteins) [24,31,32]. Hence, these genes are introduced into an inducible plasmid (named pJV53) that is widely used to engineer mycobacteriophages. However, there is a need to identify a comparable recombineering system encoded by mycobacteriophages because the high GC content of E. coli-derived proteins does not produce or function properly in mycobacteria [31]. BRED has been applied to mycobacteriophages to construct chromosomal gene knockouts, gene deletions, base substitutions, heterologous gene insertions, and specific gene replacements [24,25,33]. Similar alterations can also be done in other phages using BRED.
BRED also has certain drawbacks such as the co-transformation of substrate DNA and phage DNA into the same cell is generally low. Hence, it is particularly challenging to apply this technique to gram-positive bacteria that exhibit low transformation efficiencies [17].
Figure 2.
Schematic representation of Bacteriophage Recombineering of Electroporated DNA (BRED) in phage genetic engineering.
Figure 2.
Schematic representation of Bacteriophage Recombineering of Electroporated DNA (BRED) in phage genetic engineering.

| Phage Strain | Engineered phage | Cluster | Mutated gene | Type of mutation | Bacterial strain | Observation |
|---|---|---|---|---|---|---|
| Adephagia | AdephagiaΔ41Δ43 | K1 | Repressor and Integrase | Deletion |
M.smeg mc2155 and Mtb H37Rv |
Lysogenic phage converted to lytic |
| Fionnbharth | FionharthΔ45Δ47 | K4 | Repressor and Integrase | Deletion |
M.smeg mc2155 and Mtb H37Rv |
Lysogenic phage converted to lytic |
| BPs | BPsΔHTH33 | G1 | Integrase | Deletion |
M.smeg mc2155 and Mtb H37Rv |
Lysogenic phage converted to lytic |
| D29 | D29:: Phsp60-egfp | A2 | Egfp gene | Insertion | mc2155 | Reporter phage |
| ZoeJ | ZoeJΔ45 | K2 | Repressor gene | Deletion |
M.smeg mc2155 and Mtb H37Rv |
Lysogenic phage converted to lytic |
2.3. CRISPR-CAS
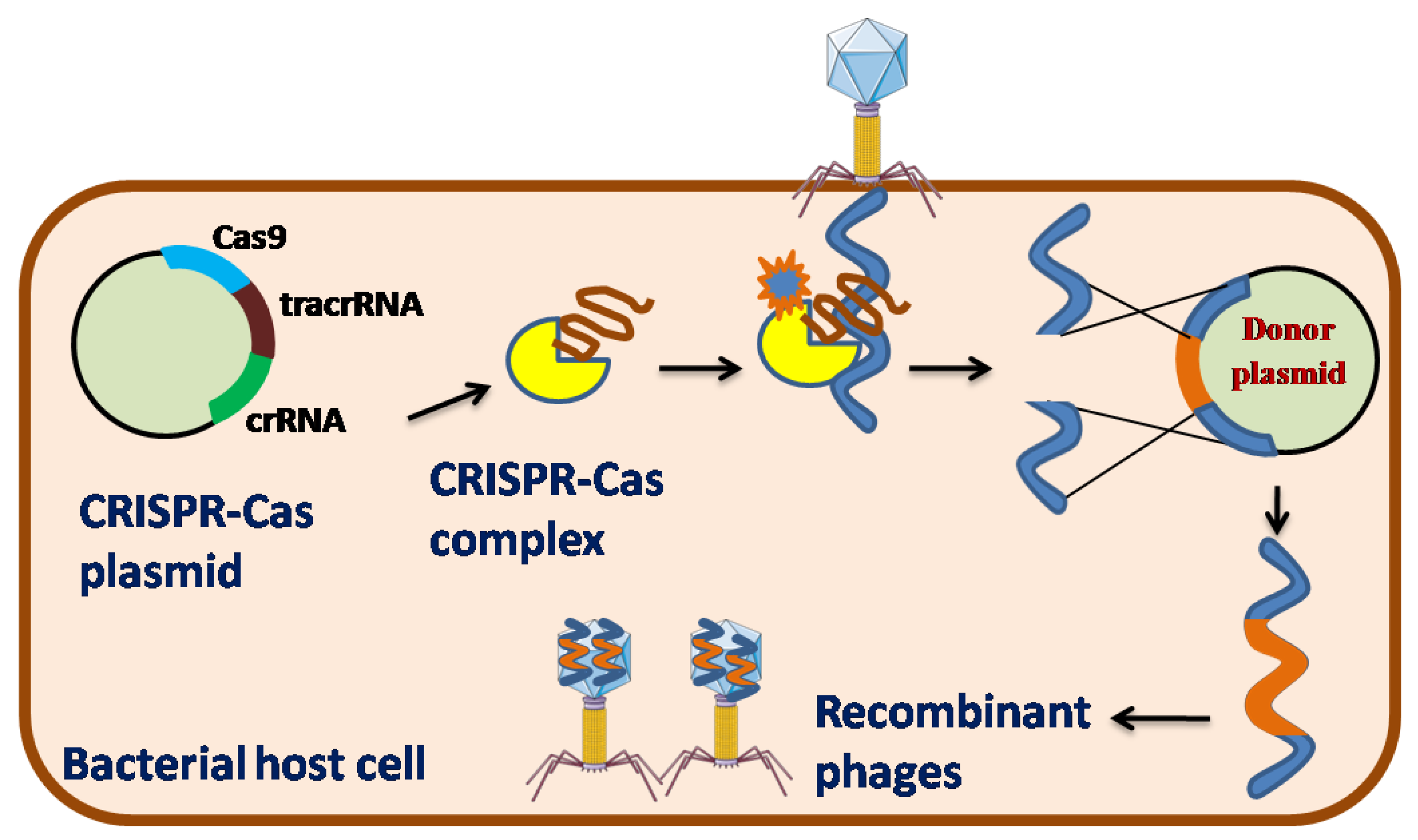
To counter the invasion of phages, bacteria have developed a specific immunity mechanism popularly known as the CRISPR-Cas system, in which the nucleic acids of any foreign organisms are targeted and cleaved with the help of distinct nucleases [34]. It is very similar to the pattern recognition receptors (PRRs) of the mammalian innate immune defense mechanism [35]. The CRISPR-Cas system adapts to a short stretch of the phage genome of approximately 30-40 nucleotides (called “spacers”) by binding and merging them into the CRISPR loci of the bacterial genome. Furthermore, these spacers are transcribed into CRISPR RNAs (crRNAs), which form effector complexes that combine with single or multiple Cas proteins. This effector complex further interferes with the infection of phage genome which is complementary to the crRNA by recognizing and cleaving its nucleic acid (called “protospacers”) and hence providing immunity to bacteria [36]. In light of all other phage engineering strategies, CRISPR-Cas systems in various bacteria have recently been utilized with remarkable effectiveness to aid phage genome engineering [37].
CRISPR-Cas has been classified into two classes based on the composition of cas genes. The Class 1 system is based on multiple subunits of the effector complex and has three types I, III, and IV, whereas the Class 2 system is based on a single subunit of the effector complex and has three types II, V, and VI [38]. Among the six types of CRISPR-Cas systems type I, II, and III have been efficiently applied for the engineering of phage genomes.
The type I-E system observed in E. coli is the most well-known instance of a Type I CRISPR-Cas system [39]. The endoribonuclease Cas6, also known as CasE or Cas6e, is essential for crRNA synthesis, as it detects and cleaves the precursor crRNA within each repetition. Cas6 and crRNA were further merged with Cas8 (large type component), Cas7 (six sets), CasB or Cse2 (two sets of small subtype subunits), and Cas5. In addition, a two to six nucleotide protospacer-adjacent motif (PAM) must be present on the non-complementary DNA strand. After recognizing the target DNA by Cascade (CRISPR-associated complex for antiviral defense), another protein helicase-nuclease, Cas3, cleaves the target DNA [40]. This type I-E system has been successfully utilized to generate recombinant phages for E. coli [41] and Vibrio cholera [42]. Two non-essential genes were deleted from phage T7, which infects E. coli using this approach. The wild-type T7 phage was infected with an E. coli strain carrying a plasmid containing homologous phage sequences flanking the gene to be deleted. This step generated both wild type and recombinant phages, which were further selected by propagating these phages onto the bacterial strain carrying plasmids encoding cascade, cas3, and spacer sequences, where the recombinant phages were automatically selected by cleaving the protospacer of wild-type phages, generating 38% and 42% recombinant phages for deleting two non-essential genes, respectively [41]. Using a similar approach, two deletions and one replacement were produced in the ICP1_2011_A phage infecting V. cholera, where approximately 50% of the recombinants were discovered, whereas a small deletion of approximately 33 nucleotides generated 100% recombinant phages [42].
The most commonly used CRISPR –Cas system in genome editing applications is CRISPR-Cas9 [43], which belongs to the type II –A CRISPR-Cas family typically found in Streptococcus thermophilus and Streptococcus pyogenes [36]. Here, a short trans-activating crRNA (tracrRNA), is necessary for crRNA production having a corresponding region to repeat-derived sequences of crRNA. In this system, the 5’end of crRNA are modified using some unspecified nucleases while the 3’end of crRNA is trimmed by RNase III, a host encoded nuclease when the complementary short trans-activating crRNA (tracrRNA) and progenitor crRNA binds with each other. This complex binds to Cas9, which recognizes and cleaves the targeted double-stranded DNA. Similar to the CRISPR type I system, CRISPR-Cas9 requires PAM and a corresponding crRNA with a protospacer in the seed region to induce productive interference [44]. Some non-essential genes have been identified in phage 2972 against four different S. thermophilus strains, each carrying their own type II CRISPR-Cas system. In addition, with the help of donor DNA and plasmids carrying CRISPR-Cas9, point mutations and single gene deletions (100% efficiency), two-nucleotide deletions (80% efficiency), and gene exchange were efficiently performed in phage 2972 using this method [37]. CRISPR-Cas9 has also been used to engineer P2, a lytic phage, against Lactococcus lactis. Several short nucleotide insertions, point mutations, and single-gene deletions were introduced into the P2 phage using plasmid-encoded donor DNA and CRISPR-Cas9 [45].
The type III-A system is well described in Staphylococcus epidermidis, showing a close similarity with the type I CRISPR-Cas system [46], which also depends on the Cas6 endonuclease for crRNA biogenesis at its 5’ end, while the 3’ end is modified by some non-cas nucleases. The effector complex is composed of Cas10 (large type subunit), Csm2 (small type subunit), Cas5, and Cas7 (numerous homologs), which along with crRNA and Csm6, target and cleave foreign DNA and RNA [47]. The type III system functions in a transcription-dependent manner; hence it cleaves the target molecule only after successful transcription. Therefore, it is difficult to engineer lysogenic phages using this system because the late genes of lysogenic phages are usually silent during this phase. However, lytic phages of S. epidermidis and S. aureus were efficiently engineered using the type III CRISPR-Cas system. Point mutations have been introduced in phage Andhra [48] against S. epidermidis and in phage ISP [49] against S. aureus with 100% mutant phages.
Therefore, owing to the diversity of CRISPR-Cas in various bacterial species, it can be used to modify various phages using both host-encoded and plasmid-encoded CRISPR constructs. Some off-target cleavage can be performed by CRISPR nucleases; however, it can be easily screened once a perfect recombinant phage is found [36]. One very important aspect of using CRISPR-Cas systems in phage engineering is its diverse role in different hosts, proving its utility in a longer run once optimized properly.
Figure 3.
Schematic representation of CRISPR-Cas Type II system in phage genetic engineering.
2.4. CRISPY BRED and CRISPY BRIP
Phage engineering is mostly based on either a phage-encoded recombination system or host-derived CRISPR-Cas machinery. The engineering of mycobacteriophages [25] has been carried out by BRED which is further implemented for phages of other bacterial species including E. coli [33], Klebsiella [50], and Salmonella [51], whereas CRISPR-Cas is readily used to engineer phages of E. coli [41], V. cholerae [42] and S. thermophilus [37]. When combined, these techniques can be used to efficiently carry out engineering known as CRISPY-BRED, where BRED or recombineering is used to carry out recombinations and CRISPR is used to select recombinant phages. CRISPY-BRED has been used to modify various mycobacteriophages such as Alma, BPs, BuzzLysyear, LadyBird, Miko, PhiFW1, Fionnbharth and Adephagia [52]. The CRISPR system is encoded by a plasmid derived from pIRL53 containing an sgRNA (single guide RNA of approximately 20 bp in length, which is positioned 5’ to the PAM sequence used to target the gene to be modified), the cas9 gene17 of S. thermophilus [53], an E. coli replication origin, and a kanamycin resistance gene. The integration of this cassette into mycobacterial chromosomes contains an attP-Int site [54]. BRED contains the parental phage DNA to be modified and an artificial DNA substrate (150-250 bp long) that is homologous to the target phage DNA sequence. They are co-electroporated in M. smeg cells, which expresses recombination genes from the mycobacteriophage Che9c. Once the target phage DNA sequence, artificial DNA substrate, recombination genes, and CRISPR cassette are inside the M. smeg cell, the guide RNA cleaves the target DNA, followed by the recombination of the artificial DNA substrate with the targeted phage sequence using recombination genes. After one round of the lytic cycle, the recombinant phages are recovered by plating on the M. smeg psgRNA strain. Plaques are detected by PCR in the presence of mutant alleles. CRISPY-BRED is advantageous over BRED, as the former usually obtains homogenous mutant phages in its primary plaque screening, whereas the latter has heterogeneity of both wild and mutant phages in primary plaque screening, which is further confirmed by secondary plaque screening. Therefore, CRISPY-BRED simplifies the screening procedure because the recombination efficiency is low in the case of BRED in M. smeg [52].
CRISPY-BRIP is another approach for phage engineering, which is similar to CRISPY-BRED in all aspects, except the co-electroporation of the target phage DNA sequence and the artificial DNA substrate. In CRISPY-BRIP, cells carrying the recombination genes are first electroporated with the artificial DNA substrate, followed by further infection with phage particles by natural route to release their DNA inside the host cell. This approach is useful for engineering phages with hosts that have low electroporation efficiency, such as gram-positive bacteria, particularly Mycobacterium sp. Compared to CRISPY-BRED, CRISPY-BRIP is less efficient than replacing the repressor gene (gene 47) of the mycobacteriophage Fionnbharth with the mutant variant gene (gene 52) of mycobacteriophage Fruitloop, which yielded only two recombinants out of fourteen plaques (14% efficiency), while similar replacement with CRISPY-BRED yielded twenty two recombinants out of twenty four plaques (~92% efficiency) [52].
Owing to the higher efficiency and easier screening of recombinants, CRISPY-BRED has been efficiently used to introduce deletions and replacements in various mycobacteriophages, with a decent number of viable mutants. PAM site choice in Cas9 of S. thermophilus is restricted to 5bp whose target is quite precise in case of deletion and replacement, but for targeted insertion and point mutation, PAM site choice is still restricted and requires other CRISPR-Cas systems with more specific PAM site choices. Recombineering remains a limitation for other phages and requires extensive screening of mutant phages. Therefore, CRISPY-BRED can be a better platform for producing engineered phages, whereas CRISPY-BRIP can be readily used for engineering phages possessing a larger genome size and is unable to transfect its host organism efficiently [52].
Table 2.
CRISPY-BRED engineered phages [52].
Table 2.
CRISPY-BRED engineered phages [52].
| Mycobacteriophage | Mutated Gene | Type of Mutation |
|---|---|---|
| Alma | Ori ncRNA (35) | Deletion |
| BPs HRM 10 | Integrase (32), Repressor (33) | Deletion |
| BuzzLysyear | 41,42,43 | Deletion |
| LadyBird | Ori ncRNA (34) | Deletion |
| Miko | repA (36) | Deletion |
| PhiFW1 | Capsid (14) | Deletion |
| Fionnbharth | Repressor (47) of Fionnbharth/F52mut3 of Fruitloop | Replacement |
| Fionnbharth | Integrase (45), 46,Repressor (47)/mCherry (Fluorescent protein) | Replacement |
| BPs HRM 10 | Integrase (32), Repressor (33)/ mCherry (Fluorescent protein) | Replacement |
| Adephagia | Integrase (41), 42, Repressor (43)/mCherry (Fluorescent protein) | Replacement |
2.5. Chemically accelerated viral evolution (CAVE)
Chemically accelerated viral evolution (CAVE) is based on a directed evolutionary strategy in which phages undergo a series of chemical mutagenesis cycles to evolve with better functional characteristics than the original wild type form [55]. Ethyl methanesulfonate (EMS), an alkylating substance, is used to perform chemical mutagenesis in various E. coli phages, producing effective variants of these phages in vitro [56]. Here, a random mutation is inserted within the phage genome by chemical mutagenesis, which generates progeny of mismatched mutated phages upon replication in its host bacteria. Furthermore, these mutated phages are exposed to high temperatures, resulting in variants with enhanced thermal stability. These variants are multiplied and subjected to 30 cycles. Next-generation sequencing of these variants depicted that during the initial cycle of mutagenesis, mutations were restricted to fewer mutations, which further increased and saturated with the latter cycle of mutagenesis. The stability of mutated phages at higher temperatures increased by approximately 63% compared to that of wild-type phages. Sequencing results demonstrated that all these mutations occurred within the coding regions of phages, especially in the structural and assembly protein subunits. In these mutations, the original amino acids were replaced with more hydrophobic amino acids than their corresponding wild-type counterparts. Except for changes in the structural genes of phages, CAVE did not cause any alterations in lytic activity or host range capability of these phages. CAVE can also be used to generate resilient phages at acidic pH, without causing any significant changes in the structural biology of phages [57].
Therefore, CAVE can prove to be an efficient tool for carrying out phage engineering with a directed evolutionary approach, as this strengthens the functional attributes of phages with a variety of criteria for phage selection [57]. CAVE has developed engineered phages with increased thermal stability at 60°C and acidic pH resistance. Furthermore, this technique can be employed for engineering phages with much better phenotypic characteristics, which are usually a constraint in wild-type phages and can be effectively used in phage therapy and other antimicrobial purposes.
Table 3.
CAVE induced mutations in various structural proteins of T3 and T7 phage [57].
Table 3.
CAVE induced mutations in various structural proteins of T3 and T7 phage [57].
| Bacteriophage | Gene no. | Gene name | Mutation rate |
|---|---|---|---|
| T3 | gp37 | Head-to-tail joining protein | 0.998 |
| T3 | gp45 | Internal virion B | 0.713 |
| T3 | gp48 | Tail fiber protein | 0.197 |
| T7 | gp47 | Tail tubular protein B | 0.951 |
| T7 | gp51 | Internal virion protein D | 0.935 |
| T7 | gp42 | Head-to-tail joining protein | 0.930 |
| T7 | gp57 | DNA maturation protein | 0.877 |
| T7 | gp17 | DNA binding protein | 0.838 |
| T7 | gp43 | Capsid assembly protein | 0.720 |
2.6. Rebooting of Phage Genome
The basic principle behind the rebooting approach is that the full-length phage genome containing the desired mutation is generated by artificial methods such as Gibson assembly or transformation-associated recombination (TAR). This synthetically generated phage genome is directly transformed into phage-specific host cells, which produce modified phages after the lysis of the host cell. This approach is helpful for certain bacteriophage gene products that are harmful to their host cells and was first applied to the phage genome of phiX174 (5386 bp). The phiX174 genome was assembled in vitro using artificial oligonucleotides by Polymerase Cycling Assembly (PCA) and further transformed into yeast artificial chromosomes by the transformation-associated recombination (TAR) method in vivo. These fragments are then liberated by digestion with restriction enzymes and finally rebooted into their respective host cell that is E. coli [58].
Similar modifications have been applied to T7 phages to modulate their host range, particularly in gram-negative bacteria. This shows that the tail fiber gene gp17 plays a significant role in determining the host range of T7 phages [59,60]. In addition, rebooting was successful in the Salmonella Myovirus FelixO1 strain [61], P. aeruginosa [62], and Klebsiella sp. [63]. Therefore, this method is more suitable for gram-negative bacteria because it has greater transformation efficiency than gram-positive bacteria.
For the rebooting of phages for gram-positive bacteria, L-form cell wall-deficient bacterial strains are used, which can easily uptake the genomic DNA of the phage to be modified. This technique has been used to reboot the gram-positive L-form of Listeria monocytogenes which has been applied to alter the host range of Listeria phages [64] in addition to generating reporter phages [65].
Phage rebooting can also be performed outside the host cell using a cell-free transcription and translation (TXTL) system. This method utilizes phages with a higher self-assembly capacity, such as T4, T7, and phiX174. These phage particles are further grown in test tubes along with cell extracts of E. coli using the TXTL system [66]. Cell-free systems of genetic engineering can overcome the hurdle of bacterial species with inefficient transformation capacity, and can be used to engineer their respective phages.
3. Applications of Phage Therapy
Phages assist in the treatment of bacterial infections at both the pre-infection (prophylaxis) and post-infection (therapy) stages. The findings that no major adverse side-effects of phage therapy have been documented till date, substantiallysupports the safety of bacteriophage preparations. Most of the applications of the phage therapy that have been employed clinically till date are most often personalized for a particular patient and there are perhaps no report that describes its characterization or usage on a population or mass level. Several attempts have been made to aid in the selection of particular conditions and methods so that the phage therapy experiments can be targeted most effectively in the future.Still,phages have been identified to have huge potential in therapy against several critical bacterial illnesses where antibiotics and (or) other treatment modalities are not very effective[67,68]. Phage therapy can be administered in several ways, some of which are listed below:
3.1 Phage Host Range Mutants
3.2 Phage endolysin derived enzybiotics
3.3 Phage vaccines
3.4 Engineered phages
3.5 Phage Cocktail/ Cocktail+Antibiotic
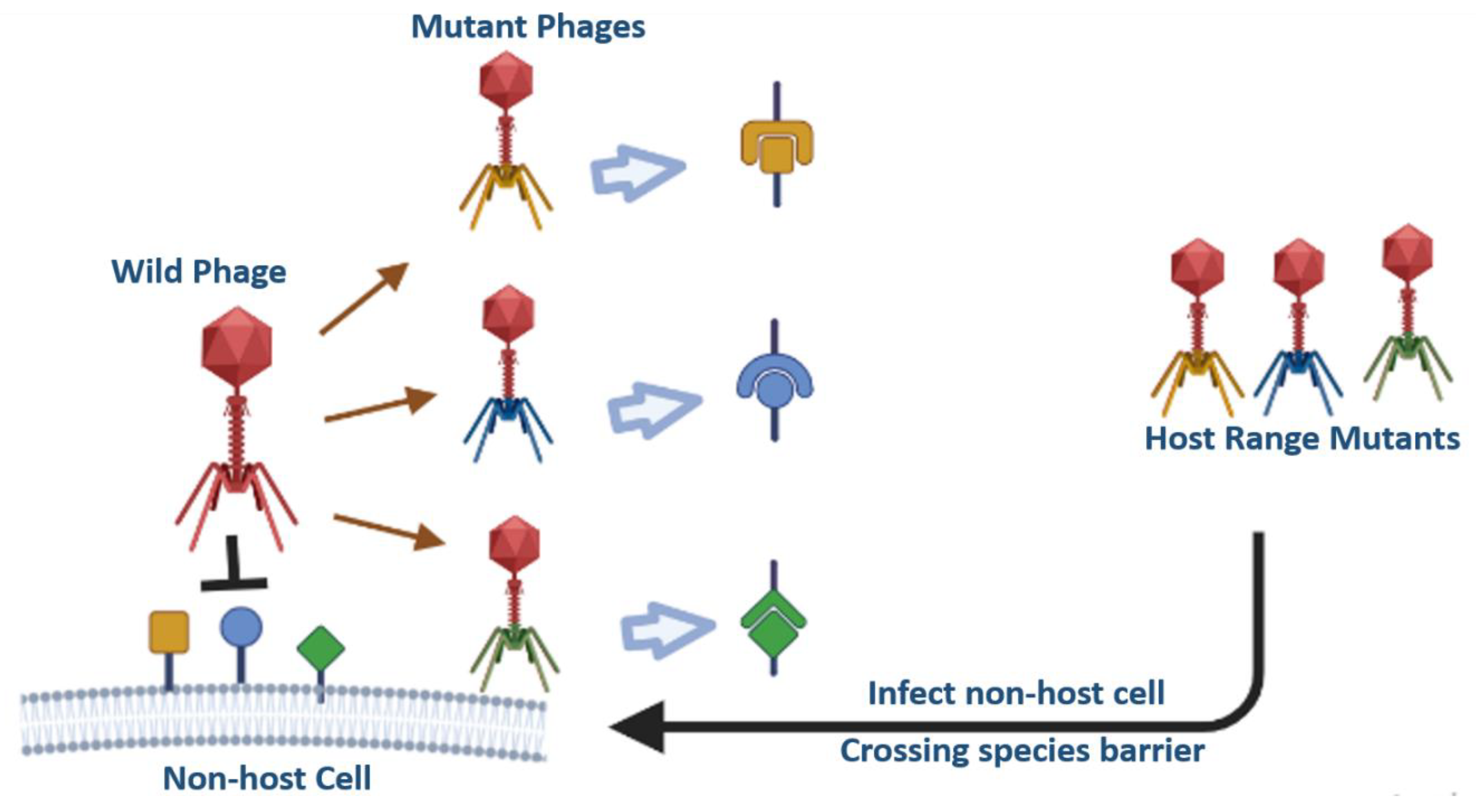
3.1. Phage Host Range Mutants (HRM)
Phages are very specific to their host, and the major drawback of phage therapy is their limited host range [69].It is nearly impossible to use a single phage type to target every strain of a species in a given genus.Through advancements in genetic engineering, receptor-binding proteins of phages can be swapped or modified to expand their host range in phage therapy. Changes in host specificity have been made by substituting receptor-binding protein genes across many strains, each of which targets a different host. For instance, switching the long-tail fiber genes of T2 and PPO1 phages leads to changes in the host of T2 fromE. coli-K12 to E. coli O157:H7 [70]. Additionally, the T2 phage's long tail fiber gene was also switched with the IP008 phage tail fibre, which increased T2 host range for additional E. coli strains [71]. By swapping heterologous receptor-binding genes between distant phages,Even a phage intended to infect E. coli bacteria could possibly be capable of infecting Klebsiella bacteria and vice versa [72]. Filamentous phages, such as fd and IKe, have minor pIII coat proteins that are responsible for their infectivity. The pIII coat protein of the fd phage which infects E. coli containing F pili was fused with the pIII coat protein (receptor-binding region) of the IKe phage that infects E. coli containing N or I pili that lead to widened host range of mutated fd phage as it can now infect either with N or even F pili [73]. The fd phage was also engineered to infect V. cholera by adding N-terminal 274 amino acids of the pIII gene of the filamentous phage CTXphi, which infects V. cholera through toxin-co-regulated pili [74].
Figure 4.
Schematic representation of Host Range Mutants (HRMs) by swapping the receptor binding domains.
Figure 4.
Schematic representation of Host Range Mutants (HRMs) by swapping the receptor binding domains.
3.2. Phage endolysin derived enzybiotics as potential therapeutics
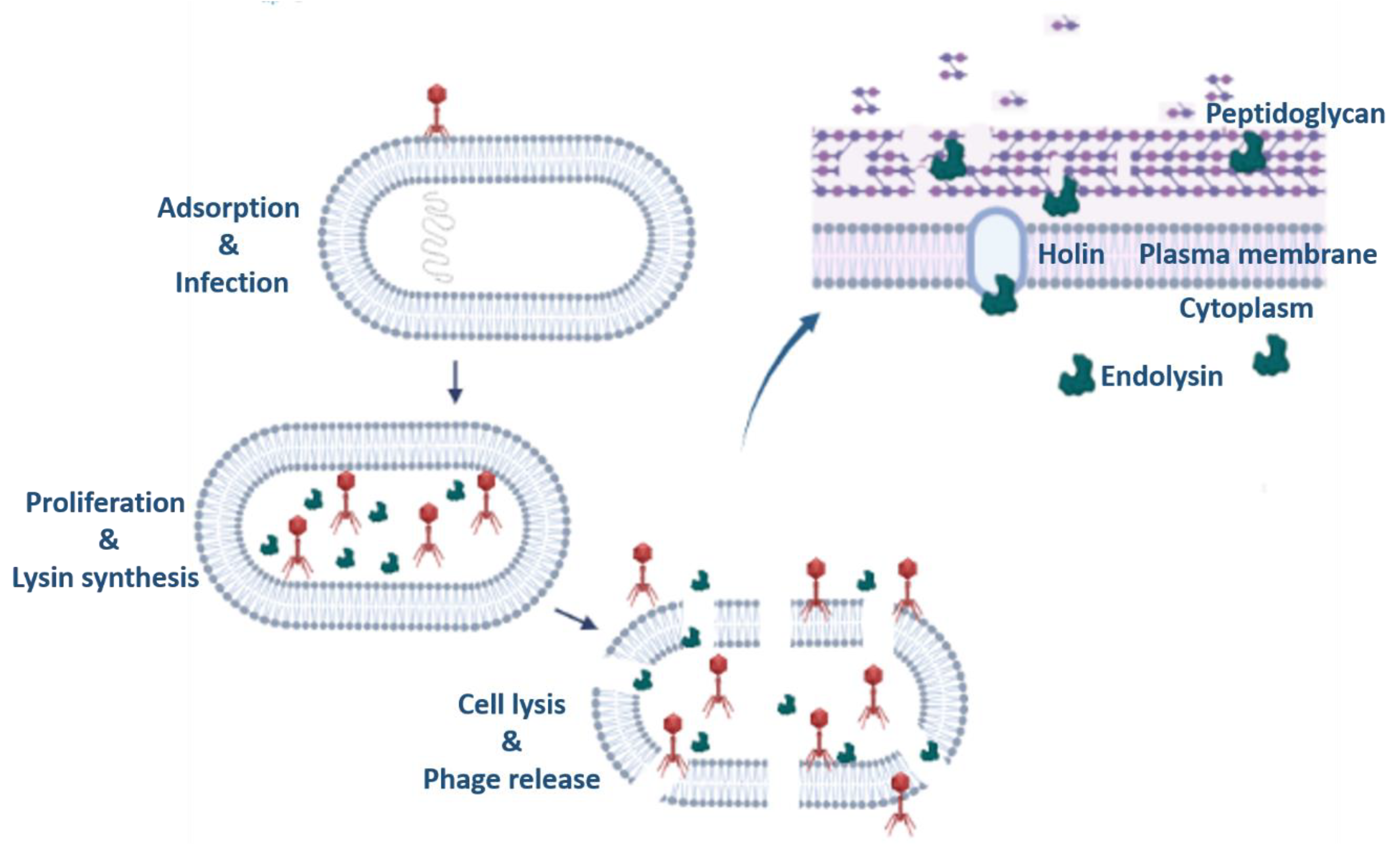
The term “enzybiotics” is derived from two words “enzyme” and “antibiotics” [75]. Enzybiotics are enzymes or in some cases non-enzymatic derivatives of phages which have been extensively utilized for their antibacterial and antimicrobial properties. The foundation of enzybiotic research is the hydrolytic enzyme class known as endolysins (or lysins), which are particularly successful in eradicating a variety of bacterial infections [76,77]. Endolysins also known as murein hydrolases, cleave the bacterial host cell wall towards the end stage of the lytic cycle. Endolysins require a group of proteins known as holins to make pores/ gaps in the cytoplasmic membrane from inside so that they can access the bacterial peptidoglycan since they lack their own signal sequences. Only when the holin’s concentration exceeds a predetermined level does this closely controlled chain of events begin. Endolysins may now access the peptidoglycan and break it down, which kills the bacterial cells [78].
Phage endolysins have been studied as potential medicines for the treatment of bacterial infections in both humans and animals ever since it was discovered that their exogenous application causes lysis of the host bacteria [79,80]. In 2001, Nelson et al. released the first study that demonstrated an endolysin's effectiveness in vivo. [75] As the phage and their hosts have co-evolved, there is far less risk of resistance to endolysins than to antibiotics [81]. Over the past 20 years, research on these enzymes have advanced from straightforward in vitro characterization to sophisticated protein engineering methods, including state-of-the-art pre-clinical and clinical testing [82,83]. Despite these advancements, there are still certain problems that need to be addressed with the systemic use of endolysins. They include immunogenicity and circulation half-life, as well as characteristics that target and penetrate particular cells and tissues [84,85].
Owing to the distinct cell wall topologies of these two important bacterial groupings, endolysins that target Gram-positive, Gram-negative, and mycobacteria frequently have different structures. Most endolysins are composed of a polypeptide chain that is divided into two parts: a catalytic domain (CD) at the protein's N-terminus and a cell wall-binding domain (CBD) at the C-terminus connected by a brief, bendable linker between these two components. [86,87].
Nelson et al. demonstrated in 2001 that the streptococcal phage lysin PlyC could both prevent and treat group. A streptococcal upper respiratory colonisation in mice (then known as C1 lysin). The test shows that lysin is a unique murein hydrolase that rapidly eliminates group A streptococci both in vitro and in vivo while having no impact on the other native microorganisms being studied. Using this broad approach, streptococci may be decreased or eradicated from carriers' or sick individuals' upper respiratory mucosal epithelium, hence reducing the associated disease [88].
3.2.1. Gram positive bacteria-targeting endolysins
Most Gram-positive bacteria have a continuous, somewhat thick cell wall that is mostly composed of peptidoglycan and ranges in thickness from 30 to 100 nm [89]. Single type of endolysin is produced by bacteriophages that attack gram-positive bacteria [90] that have evolved a modular structure that typically separates enzymatic activity and cell wall recognition into different domains and connects them with flexible linkers. The endolysin's cell wall-binding domain (CBD) enables it to make a specific, targeted connection to the target bacterial cell wall whereas its enzymatically active domains (EAD), which give its catalytic activity, often reside at its N-terminus [91,92].
The PlyG endolysin from the Bacillus anthracis -phage, a chemical significant with regard to biowarfare, has demonstrated remarkable results in addition to treating streptococcal infections. Mice that were intra-peritoneally injected with Bacillus cells and given buffer died after two hours, whereas mice that were given PlyG lived for 70–80% of the 72 hours of the experiment [93]. The treatment of staphylococcal infections in animal models, particularly Multiple Resistance Staphylococcus aureus (MRSA), has been the subject of numerous articles. In the first study from 2007, MRSA was eliminated from mouse nares using the MV-L endolysin from phage MR11. When the same enzyme was supplied intravenously at 0, 30 and 60 minutes following systemic MRSA infection, respectively, 100%, 100%, and 60% of the mice survived the test [94]. S. aureus frequently causes osteomyelitis, a bone infection that is difficult to treat because the bacteria are resistant to medications and persist inside the cells. To treat experimental osteomyelitis in rats, Karau et al. gave endolysin CF-301 intravenously one week after infection and the localised S. aureus bacteria in the bone were greatly reduced by this systemic injection. However, compared to the control, the impact was only about 0.48 log decrease [95].
3.2.2. Gram negative bacteria-targeting endolysins
Gram-negative bacteria are shielded from the environment by an outer membrane made of lipopolysaccharides that surrounds the peptidoglycan cell wall [96]. Gram-negative bacteria have thin cell walls that are between 20 and 30 nm in thickness [97]. While endolysins that attack Gram-negative bacteria are typically small, single-domain globular proteins (molecular mass between 15 and 20 kDa), they can also be found in other structural forms [98,99].
The concept of Artilysins is introduced to combat Gram-negative bacteria which is constructed by alteration of endolysin structure with peptides that destabilise lipopolysaccharide. Artilysins, are able to pass through the outer membrane and reach the peptidoglycan where they exert their action. In a study, novel artilysins (designated as AL-3AA, AL-9AA, and AL-15AA) are developed with antimicrobial-peptide SMAP29 fusion at the N-terminal of LysPA26 and utilised them. The findings demonstrated the significant bactericidal activity of artilysin AL-3AA; even 0.05 mg/mL AL-3AA could kill 5.81 log units of P. aeruginosa without EDTA in 60 minutes. AL-3AA significantly reduced P. aeruginosa biofilms and prevented the development of P. aeruginosa PAO1 biofilms. Additionally, it may have had broad-spectrum activity against K. pneumoniae and E. coli, two susceptible Gram-negative bacteria most often found in hospitals [100,101]. The Gram-positive Bacillus amyloliquefaciens phage IAM 1521 endolysin (Lys1521) is the most extensively researched example. Lys1521 (40 g/ml) externally applied to gram negative bacteria like E. coli W3110 and P. aeruginosa decreased the number of cells by 98% (1.90 log) and 99.78% (2.66 log) in 10 minutes, respectively [102]. The SPN9CC endolysin worked by cleaving the glycosidic linkages of peptidoglycan; demonstrated lytic activity by exogenous applications and exclusively had antibacterial action against Gram-negative bacteria. It's interesting to note that without EDTA, a permeabilizer of the outer membrane, the SPN9CC endolysin could still lyse intact Gram-negative bacteria [103]. LysAB54, a novel endolysin with limited homology to other well-known related endolysins from bacteriophage p54 was cloned, expressed and characterised. LysAB54 has demonstrated considerable bactericidal action in the absence of outer membrane permeabilizers against multidrug-resistant A. baumannii and other Gram-negative bacteria, such as P. aeruginosa, K. pneumoniae, and E. coli making it a promising therapy option for Gram-negative superbugs that are multidrug resistant [104].
3.2.3. Mycobacteriophage endolysin
Mycobacteriophages, viruses that infect mycobacterial hosts, face significant obstacles because of the unusual structure of the mycobacterial cell wall, which consists of a mycolic acid-rich mycobacterial outer membrane attached to an arabinogalactan layer connected to the peptidoglycan. The mycobacterial cell wall has a complex structure that makes it unique and sets it apart from both Gram-positive and Gram-negative bacteria [105]. Its exceptionally high lipid content (up to 60% of the cell wall is made of lipids) accounts for the highly hydrophobic cell surface properties, leading to a naturally impermeable cell wall, resistance to many antibacterial drugs, and exceptional inflammatory activity, playing a key role in virulence [106].
While the arabinogalactan-peptidoglycan complex is covalently attached to the outer membrane rich in mycolic acid, lysis must not only remove the mycolic acid layer but also penetrate it in order to split the peptidoglycan layer. Consequently, two different endolysins are produced by mycobacteriophages: Lysin A (LysA), which hydrolyzes peptidoglycan, and Lysin B (LysB), a novel mycolylarabinogalactan esterase, which cleaves the mycolylarabinogalactan bond to release free mycolic acid [107,108,109].
Mycobacteriophage Ms6 endolysins, Lysin384 and Lysin241 were found to inhibit the growth of mycobacterial species like M. smegmatis, M. aurum, and M. fortuitum when exogenously applied. In a different investigation, mycobacteriophage BTCU-1 lysin A and lysin B demonstrated alteration of the morphology of M. smegmatis and its increased capacity to eradicate intracellular M. smegmatis [110,111].
There is an advantage over using complete mycobacteriophages, as only a few numbers of phages have been demonstrated to infect tubercle bacilli thus far. Therefore, the large number of isolated mycobacteriophages represents a vast reservoir of various endolysins degrading enzymes that have potential for numerous therapeutic uses.
The applications of microbial recombinant enzymes have increased the therapeutic possibilities for humans, however challenges such as high immunogenicity, protein instability, brief half-lives, and low substrate affinity still need to be overcome [112]. Enzymes with higher activity and fewer side effects, as well as those that can be genetically changed, are all still being sought after. Endolysins have a lot of promise as potential alternatives or complements to traditional antibiotics. When endolysins are administered exogenously to some bacteria, cell lysis occurs quickly [113]. We can develop novel endolysins with higher stability, specificity, and lytic action, which increases the potential of endolysins. To successfully create and use endolysins, it is essential to thoroughly comprehend how their biochemical, biophysical, and bacteriolytic characteristics interact with one another.
Figure 5.
Schematic representation of the action of Phage derived endolysins in phage therapy.
3.3. Phage vaccines
Phages specifically infect their bacterial hostswithout affecting mammalian cells and have characteristics such as small size, highly ordered structure, multivalent surface antigens and repeated structures to trigger pathogen specific immune response in the mammalian host which are desirable traits for use as candidate phage based vaccines [114,115]Numerous vaccine frameworks have been developed so far using various phages, including filamentous phages [116], MS2 [117], phage λ [118], T4 [119] and others [120].
Adjuvants are immunological agents that regulate innate and adaptive immune responses. Phages have the potential to function as natural adjuvants because of their viral traits and the presence of CpG (the ligand for Toll-like receptor 9) in their genomes [121,122,123]. The basic idea behind using phages as antigen delivery vehicles is to assemble pathogenic antigens onto phage capsids, either in vivo or in vitro, to produce virus-like particles (VLPs) that cause a strong immune response without the need for external adjuvants [124]. Being a natural adjuvant they also enhance IgM and IgG responses, activate CD8+ and CD4+ T cells, facilitate B cell activation, and can also be engineered to activate dendritic cells (DCs) [123,125,126].
Researchers have observed compared to soluble counterparts, particulate surface-bound antigens elicit higher immunological responses [119]. For example, antigens presented on phage Qβ VLPs packed with CpG showed a much higher level of antigen-specific IgG antibody production than a straightforward combination of CpG-packaged Qβ VLPs and soluble antigen[126].Surface-bound particulate antigens also activate follicular dendritic cells (FDCs) by binding to natural IgM and fixing complement component 1q, followed by the selection of B cells during germinal center reactions. However, the soluble capsid protein was unable to trigger this humoral innate immune response and was ineffective in depositing FDCs [127]. Particulate antigens can also activate CD4+ and CD8+ T cells, as VLPs can be present by both class I and class II MHC [130]. Research has demonstrated that CD8+ T lymphocytes specific for p24 may be stimulated by HIV-1 p24 antigens packed on the T4 capsid in infected mice. However, soluble p24 triggers few or no CD8+ T cells with p24 specificity [128].Similarly, Yersinia pestis F1mutV antigen activates both type 1 and type 2 helper T cells in mice when produced on the T4 capsid, whereas preferentially, type 2 helper T cells are only activated by soluble F1mutV antigen[119].
It has been suggested that the highly confined epitope density promotes the cross-linking of B cell receptors to antigens, which facilitates B cell activation[129,130]. In a study using the Qβ phage capsid, a high-density model peptide (D2) caused increased titers of D2-specific IgG compared to medium- or low-density peptides [131]. One of the promising methods to increase vaccine effectiveness is antigen targeting the immune cells. The primary targeted immune cells are dendritic cells (DCs), which play a crucial role in linking innate and adaptive immune responses [132]. Engineered phages can target the DCs of mammalian cells by displaying a targeting molecule that is specific to DC, which boosts the immune response against delivered antigens. For example, researchers have engineered filamentous fd phages using pIII and pVIII capsid proteins to display a single-chain variable fragment (scFV) of an antibody against a DC-specific receptor (DEC-205) and ovalbumin peptide [123]. The resulting phages produce higher antibody titers than phages that merely display ovalbumin peptides and lack targeting molecules [133]. Most phages can connect the antigen and DC-targeting molecule to the same VLP because they contain many structural proteins that can be used for display. Similarly, two non-essential capsid proteins of phage T4Hoc is used to display an antigen and the Soc has been used to display a DC-targeting molecule (for instance, a monoclonal antibody against DEC-205) thus used to display two distinct foreign proteins[134,135].
Despite the numerous benefits of phages, no phage-based vaccines have yet reached the market. Several phage platform-based vaccine candidates are currently undergoing clinical trials [136,137] but the majority of these are currently only useful for basic research. The fact that most phages cannot adequately display the antigen at a high density could be one factor which is necessary to high levels of neutralising antibodies that are specific in their conformations[138]. Another drawback is the quick changes in a few essential amino acids in the epitopes soon after infection, leading to peptide vaccines based on only one or a few of these epitopes being less effective. In addition, it is not possible to employ phages to show antigens that need post-translational changes, such as glycosylation, which is essential for the structural and conformational integrity of proteins. This is because bacteria lack post-translational modification (PTM) mechanisms.Secondly, phages are naturally occurring protein nanoparticles that trigger immunological responses, which may limit their application in situations where multiple vaccinations are required [139].
However, recent advances in vaccine development and phage engineering have overcome some of these drawbacks, such as the fact that the T4 phage platform has demonstrated that the full-length antigen can be displayed at a high density. For instance, 360 copies of the 83 kDa protective antigen (PA) [124,134], 650 copies of plague F1mutV [119], 350 copies of the 90 kDa fatal factor (LF) [134] and 200 copies of tetrameric 129 kDa β-galactosidase [119] can all be individually observed on the surface of the T4 capsid. Like any other pathogen, the epitope areas of phage capsid proteins exhibit varying immunogenicities. The epitope that triggers the strongest immune response is referred to as the immunodominant epitope [140]. Thus, immunodominant epitopes can be disrupted by phage engineering to reduce their immunogenicity. The process of attaching polyethylene glycol (PEG), commonly known as "PEGylation," permits increased solubility and decreased renal clearance, which lengthens the time a substance remains in the bloodstream [141].
Figure 6.
Schematic representation of target delivery of phage vaccine to increase the immune response.
Figure 6.
Schematic representation of target delivery of phage vaccine to increase the immune response.

3.4. Engineered phages
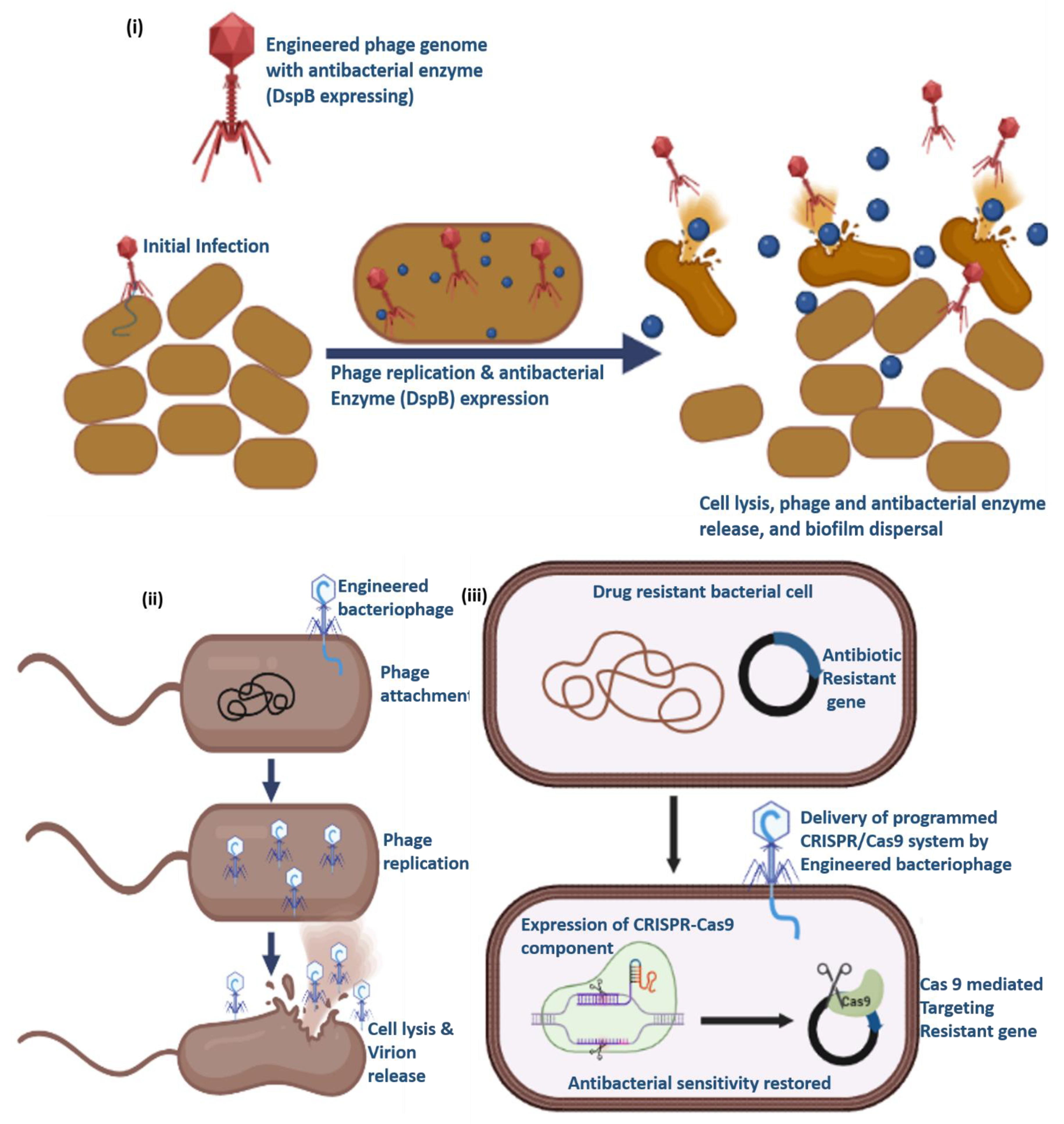
In addition to their inherent antimicrobial properties, phage activity can be readily increased by genetically modifying them to carry genes that can code for antimicrobial proteins and(or) antimicrobial substances. An excellent illustration is the use of a modified T7 phage to deliver biofilm-degrading enzyme dispersin B (DspB) [142]. The DspB gene of Actinobacillus actinomycetemcomitans was expressed by an engineered phage T7, which can be recognized by T7 RNA polymerase. As a result, it could considerably lower the the number of bacteria in a single-species E. coli biofilm than in the T7 phage control [142].Similarly, In order to prevent quorum sensing, the T7 phage was altered to express a lactonase enzyme which is crucial for the development of biofilms. Compared to the no-phage control, the resulting T7 phage decreased biofilm formation by 74.9% and 65.9% at 4 and 8 hrs after plating, respectively. However, after 4 and 8 hrs, the wild-type T7 phage decreased biofilm by only 23.8% and 31.7%, respectively [143].
Besides targeting biofilms, phages can also be used to cleave antibiotic resistance gene either by delivering a specific antibiotic drug or a programmed CRISPR-Cas system [144,145]. For instance, phagemids encoding the CRISPR-Cas9 system were packaged in the staphylococcal phage NM1 and designed to target the aph-3 kanamycin resistance gene [144]. Strong suppression of bacterial growth was observed when the recombinant ΦNM1phage was introduced into S. aureus RN4220 cells that had a kanamycin resistance gene. The ΦNM1phage, on the other hand, did not result in any appreciable suppression when combined with a non-targeting CRISPR-Cas system.
Artificial selection of useful traits to enhance the phage capability by phageengineering can be used to kill resistant bacteria more effectively than their wild type forms. For example, engineered mycobacteriophages that contained host range mutants and exclusively lytic phages have been successfully used therapeutically to treat human infections inrecent studies [146,147]. A 15-year-old patient with cystic fibrosis (homozygous for ∆F508) after receiving bilateral lung transplantation had non-tuberculous Mycobacterium (NTM) infections, including Mycobacterium abscessus, which was antibiotic-resistant. To investigate possible therapeutic phages, M. abscessus subsp. massiliense with a rough colony morphotype (named strain GD01) was isolated one month post-transplantation. The GD01 strain was then used to screen 1000 phages. A phage cocktail was designed, and the patient underwent a single topical test of the sternal wound and continued intravenous (IV) therapy with a three-phage cocktail. The patient improved clinically with healing of surgical wounds and skin lesions and the lung function improved with no side effects [146].
Table 4.
The components of the phage cocktail [146].
Table 4.
The components of the phage cocktail [146].
| Phages | Cluster/Subcluster | Used in PT | Mutant gene | GD01 Survivors |
|---|---|---|---|---|
| Muddy | Singleton | Wild | - | No |
| ZoeJ | K/K2 | Engineered (ZoeJ∆45) | Repressor gene | Yes |
| BPs | G/G1 | Engineered, mutant (BPs∆33HTH-HRM10) | Integrase | Yes |
Another successful clinical case was the treatment of Mycobacterium chelonae infection. Clinical manifestations of M. chelonae include localized skin or soft tissue infection, as well as extensive cutaneous disease. This rapidly proliferating non-tuberculous Mycobacterium infects immuno-compromised patients for a prolonged period. A 56-year-old man at a dermatology clinic complained of weight loss, sweats and new nodular lesions on his left upper limb. The patient was diagnosed with M. chelonae infection. The strain (GD153) of M. chelonae was used to investigate possible therapeutic phages that might be helpful in the treatment of illnesses. Only Muddy and Muddy_HRMGD04, variations with a wider host range, demonstrated successful results in infecting GD153. In addition to medication and surgical treatment, a single bacteriophage was administered intravenously to the patient. Patient's illness improved steadily and there were no signs of bacterial resistance to the phage [147].
The BRED technique has also been used to engineer phages from other strains such as Klebsiella[148] and Escherichia coli [149] and vaccine development is performed by engineering Shigella [150].In Klebsiella, recombineering was used to demonstrate that the multihost bacteriophage ΦK64-1 infects a different capsular strain of Klebsiella. Eleven of the genes in the bacteriophage ΦK64-1 had a sequence similar to that of tail fiber/spike or lyase. Eight of the 11 genes (S1-1, S1-2, S1-3, S2-1, S2-4, S2-5, S2-6, and S2-8) encoded capsule depolymerases, which allowed them to infect several Klebsiella capsular strains, including K1, K11, K21, K25, K30, K35, K64, and K69, as well as the novel capsular strains KN4 and KN5. The roles of these genes in phage infection were examined by deleting the gene followed by purifying mutant phages in a presence of hosts. Thus, Mutant phages for the capsule depolymerase gene (ΔS1-2, ΔS2-2, ΔS2-3, and ΔS2-6) lost their ability to infect specific Klebsiella capsular strains (KN4, K25, K35, and K30/K69) respectively. This implies that capsule depolymerase is necessary for the infection to propagate in a specific host [148].
In coliphages, BRED was initially employed to eliminate a copy of the mobile element IS1 (transposon), which has been shown to be active, from the P1vir genome. The results demonstrated that the engineered phage with deleted IS1 (specific copy of lS1) did not directly contribute to lytic replication and displayed normal plaque morphology, burst size, phage titer, and capacity for generalized transduction. Therefore, P1virΔIS is a tool for genome engineering that is devoid of IS contamination [149].
In Shigella, recombineering (Short homology recombination) was carried out to create Shigella vaccine strains with several gene deletions. The virulence gene, also known as icsA or virG, are major targets for attenuation since strains with this mutation are unable to spread to neighboring host cells and demonstrate avirulence in animal models. Recombineering is, therefore, employed to produce a number of isogenic strains from different Shigella serotypes by meticulously removing virulence-associated genes. These bacteria have been characterized for their attenuation in both in vivo and in vitro studies. As a result, prototype Shigella vaccine strains have been developed. Contrary to popular belief, evidence acquired from recent clinical studies have demonstrated that Shigella vaccine strains with virG mutations (SC602, WRSS1, and WRSd1) are highly immunogenic and safe when administered at low dosages [150].
Figure 7.
Schematic representation of phage engineering to target i) biofilms, ii) by producing exclusively lytic phages and iii) by targeting the antibiotic resistant gene to regain the sensitivity against antibiotics.
Figure 7.
Schematic representation of phage engineering to target i) biofilms, ii) by producing exclusively lytic phages and iii) by targeting the antibiotic resistant gene to regain the sensitivity against antibiotics.
3.5. Phage Cocktail/ Cocktail+Antibiotic
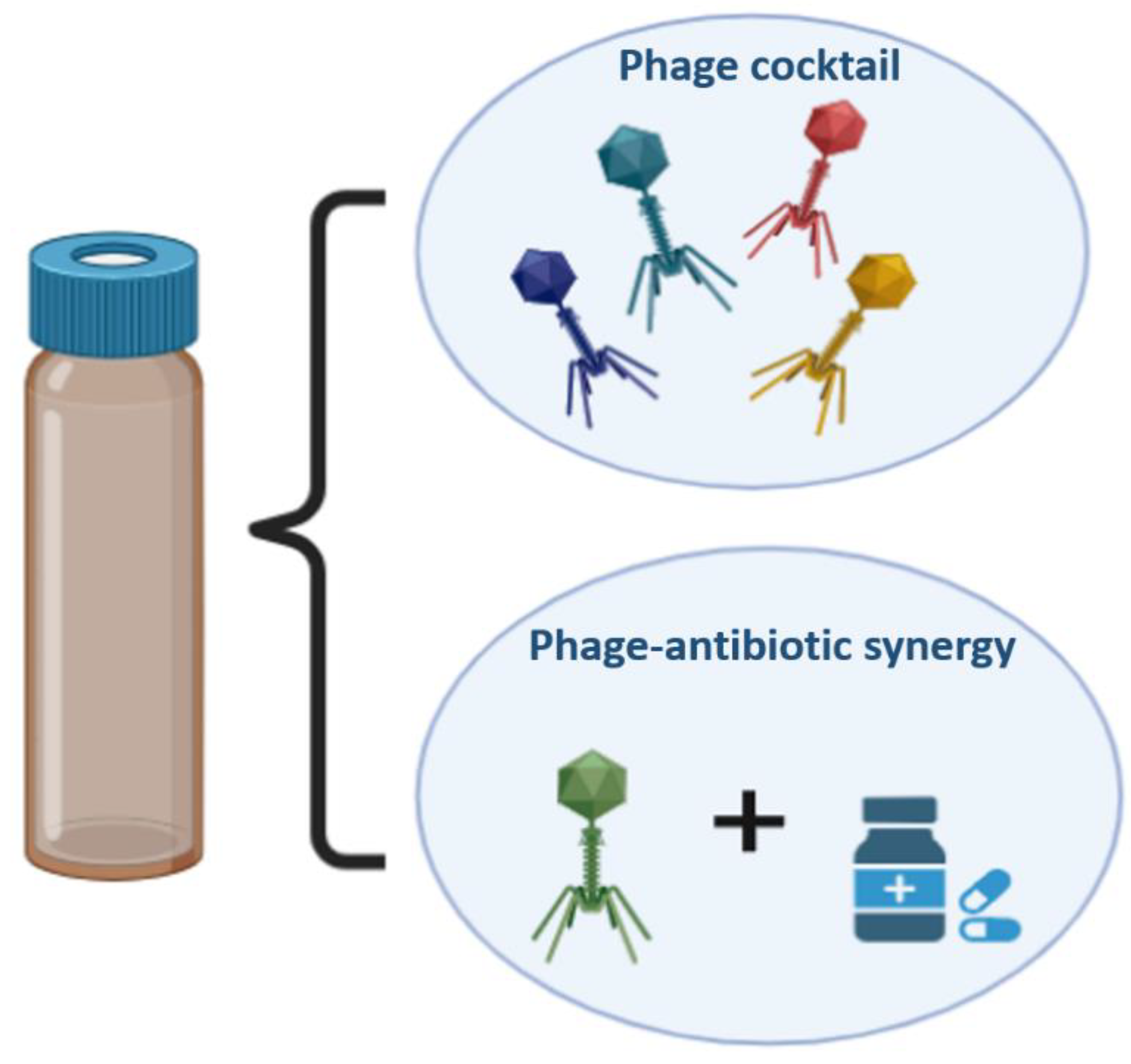
Phage treatment is most appealing sinceit can be applied to get rid of antibiotic-resistant bacteria[151]. There is no cross-resistance between antibiotics and phages because the processes by which phages and antibiotics eliminate the bacterial pathogens are fundamentally different from one another. Administration of a single phage may not be effective in eradicating the target bacteria and can lead to the emergence of resistance. However, this can be overcome by using a phage cocktail. A combination of six phages has been demonstrated to effectively treat sepsis in Galleria mellonella models and respiratory P. aeruginosa infections in mice [152]. In theory, phages can lyse both antibiotic-sensitive and -resistant bacteria with equal effectiveness. In addition, phages and antibiotics can be used in conjunction to treat bacterial infections [153]. Research has also demonstrated that the co-administration of phages and antibiotics restores antibiotic sensitivity [154]. In a case study, phage and ceftazidime antibiotic were administered directly to an aortic prosthetic graft patient that had been infected with P. aeruginosa. The infection was successfully treated and could potentially be eradicated [155]. According to Kirby's research, treating Staphylococcus aureus with gentamicin and phage SA5 together is more effective than using gentamicin or SA5 alone [156]. Similarly, when phage LUZ7 and streptomycin were administered together rather than individually, Pseudomonas aeruginosa titers were reduced [157]. Same results were observed in a mouse model for diabetes where S. aureus was infected on each mouse's hind paw [158]. When phage MR-10 and linezolid were used to treat the infections simultaneously, the most significant decrease in the bacterial titer was observed [158]. It has been demonstrated that the combined therapy employing ciprofloxacin and phage KPO1K2 can effectively halt the establishment of resistant variants in vitro, in addition to eradicating Klebsiella pneumoniae biofilms [159]. Researchers have discovered OMKO1, a lytic phage that attacks Pseudomonas aeruginosa by attaching to its outer membrane porin M (OprM) receptor. The P. aeruginosa antibiotic efflux pump contains an OprM channel. As a result of phage OMKO1 infection, OprM mutations that compromised its efflux function were selected to restore antibiotic sensitivity in P. aeruginosa. Recent research has discovered a phenomenon known as phage-antibiotic synergy (PAS), which causes host bacteria to produce more phages when treated with a phage plus sub-lethal dosages of specific antibiotics [160,161].
Figure 8.
Schematic representation of phage cocktail and also phage & antibiotic combination.
4. Phage therapy in clinical trials
Phage therapy is a promising treatment for persistent bacterial infections and is undergoing clinical trials. Spray-dried phage formulations have also been extensively tested for use in inhalational therapy of P. aeruginosa lung infections [155,162]. Even Phase I/II trials were completed in Belgium, France, and Switzerland and focused on P. aeruginosa associated with infected burn wounds [163]. To improve patient outcomes, an individualized phage cocktail was administered intravenously in a recent case study of infection caused by MDR-A. baumannii infections at the craniectomy site [164]. Phages were administered to patients with venous leg ulcers in a limited phase I safety study, and researchers have also reported safety and no adverse effects associated with phage delivery [67]. A trial targeting bacteriophage consumption as a prebiotic for gastrointestinal illnesses was completed in the US.
Table 5.
Clinical Trials of Phage therapy for various infectious diseases [165].
Table 5.
Clinical Trials of Phage therapy for various infectious diseases [165].
| Conditions | Intervention | Phases |
|---|---|---|
| Prosthetic joint infection/bacterial infections | Single phage/phages cocktail | Phase 1/phase 2 |
| Atopic dermatitis | Single phage/phages cocktail | Phase 1/phase 2 |
| Venous leg ulcers | phage cocktail | Phase 1 |
| Infectious disease/bacterial infections | Phage lytic enzymes | Phase 1/phase 2 |
| Wound infection | phages cocktail | Phase 1/phase 2 |
| Diabetes/diabetic foot | phages cocktail | Phase 1/phase 2 |
| Urinary tract infection, bacterial | Single phage/phages cocktail | Phase 1/phase 2 |
| Cystic fibrosis/lung infection | phage cocktail | Phase 1/phase 2 |
| Acute tonsillitis | Single phage | Phase 3 |
| Cystic fibrosis | Single phage | Phase 1/phase 2 |
| Prosthetic joint infection | Single phage/phages cocktail | Phase 1/phase 2 |
| Osteomyelitis/diabetic foot osteomyelitis | Single phage/phages cocktail | Phase 2 |
| Prosthetic joint infection | Single phage/phages cocktail | Phase 2/phase 3 |
| Necrotizing enterocolitis/microbial substitution | Fecal phages transfer | Early Phase 1 |
| E. coli infections/bloodstream infection | Phages cocktail | Phase 1 |
5. Conclusions
Phages have certain advantages and disadvantages that have been explored, and can be utilized to combat resistant bacterial strains. Wild phages cannot be directly applied for therapeutic purposes using phage therapy because they may not be able to meet the characteristics required for phage therapy. However, various advancements in genetic engineering, can be used to overcome some of these limitations. There are various methods for phage engineering, some of which are described in this review. These methods can be time-consuming, labor-intensive, and may require some modifications for an improved screening of recombinant phages. Still some engineered phages have shown effective results and have filled the gap between laboratory and therapeutic purposes. Phages and phage-derived proteins, such as endolysins and hydrolases, have been proven to be effective in combination with antibiotics to target resistant bacteria. Phages are highly specific and only target the host bacteria and do not affect the other useful gut microbiota, unlike the long-term use of antibiotics, which causes the destruction of the target host and other symbiotic bacteria. Currently, phage therapy is administered in combination with antibiotics or as a phage cocktail. Since antibiotics have a broad spectrum whereas "cocktail therapy," is personalized to have more effective results than individual administrations. Much effort is needed to increase the effectiveness of phage cocktails on a large scale, or to generalize phage cocktails. This raises the cost of production because the cocktail design depends on the patient’s health condition, medications, and duration of infection. There are further known risks associated with phage therapy, including the likelihood of virulence or antibiotic resistance genes being transferred to infected bacteria, which could result in concerns, including the establishment of extremely pathogenic strains.
The Antibacterial Resistance Leadership Group (ARLG) provided advice on possible clinical circumstances, laboratory evaluations, and pharmacokinetics that may need to be considered while using phage therapy. Administration route, dose, frequency, and duration are some of the factors thathave shown that an ideal methodology still needs to be explored. For any given method of administration or a particular illness type, there are currently insufficient data to offer a definite prescription on the best frequency and duration of phage therapy. However, the available data indicate that phages must be administered regularly to maximize their concentration at the infected site. The amount and time frame required may change depending on factors such as phage products, pathogens, the severity of the illness, and the site of infection.
Although clinical success demonstrates that the phage therapy is tolerable, in some cases, effective clinical functionality still needs to be defined carefully. Most recent human data generation has been performed on a single patient, with compassionate use based on the lack of well-controlled clinical trial data and complex regulatory frameworks. However, the use of naturally occurring phages has been limited. Fortunately, recent advances in phage genome engineering have helped overcome these limitations, including expanding the phage host range for improving phage treatment and impeding the immunodominant epitope of the phage capsid to remove the immune response against phages and, consequently, generate accurate and consistent variants against infectious diseases. It is challenging to draw firm conclusions on the effectiveness of phages alone because the majority of studies incorporate the concurrent use of antibiotics. However, the investigation of the pairing of phages and antibiotics is supported by the compassionate use of human data, and represents a potential route for near-term clinical advancement.
There is no one-size-fits-all solution available. Therefore, building models specifically for phages through collective efforts will undoubtedly improve health. Furthermore, we must never forget how crucial it is to maintain the highest levels of efficacy, quality, and safety, despite the difficulties posed by regulatory standards. It is crucial for researchers and healthcare professionals to discuss with essential regulatory agencies and to advance this field as quickly as possible. Consequently, animal studies and clinical cases continue to be the main focus of current research. Phage treatment has shown good efficacy and safety in these tests, but many issues still needs to be resolved before it can be used in healthcare situations.
Author Contributions
AK: GK, TN were involved in collecting and updating research and also wrote the manuscript, RKS and LKJ read the manuscript and provided suggestions for editing; AG conceptualized the idea, supervised, reviewed and edited the manuscript.
Funding
AK: GK: TN, received the financial assistance from BHU, DBT, CSIR respectively while LKJ and RKS received the financial assistance from ICMR, India.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yoshikawa, T.T. Antimicrobial resistance and aging: beginning of the end of the antibiotic era? Journal of the American Geriatrics Society 2002, 50, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a new metallo-β-lactamase gene, bla NDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrobial agents and chemotherapy 2009, 53, 5046–5054. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Tasak, N. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. The Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- United Nations. 2017. PRESS RELEASE: High-Level Meeting on Antimicrobial Resistance. 2016; Accessed Mar 29, 2017. 2016; 29.
- Kumar, M.; Sarma, D.K.; Shubham, S.; Kumawat, M.; Verma, V.; Nina, P.B.; Tiwari, R.R. Futuristic non-antibiotic therapies to combat antibiotic resistance: A review. Frontiers in microbiology 2021, 12, 609459. [Google Scholar] [CrossRef] [PubMed]
- Gibb, B.; Hyman, P.; Schneider, C.L. The many applications of engineered bacteriophages—An overview. Pharmaceuticals 2021, 14, 634. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World journal of gastrointestinal pharmacology and therapeutics 2017, 8, 162. [Google Scholar] [CrossRef] [PubMed]
- Chanishvili, N. Phage therapy—history from Twort and d'Herelle through Soviet experience to current approaches. Advances in virus research 2012, 83, 3–40. [Google Scholar] [PubMed]
- Sulakvelidze, A.; Alavidze, Z.; Morris Jr, J.G. Bacteriophage therapy. Antimicrobial agents and chemotherapy 2001, 45, 649–659. [Google Scholar] [CrossRef]
- Hyman, P. Phages for phage therapy: isolation, characterization, and host range breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef]
- Sho, M. Identification and characterization of nutrient transporters in Mycobacterium smegmatis. 2011. [Google Scholar]
- Meile, S.; Du, J.; Dunne, M.; Kilcher, S.; Loessner, M.J. Engineering therapeutic phages for enhanced antibacterial efficacy. Current opinion in virology 2022, 52, 182–191. [Google Scholar] [CrossRef]
- Lopes, A.; Amarir-Bouhram, J.; Faure, G.; Petit, M.A.; Guerois, R. Detection of novel recombinases in bacteriophage genomes unveils Rad52, Rad51 and Gp2. 5 remote homologs. Nucleic acids research 2010, 38, 3952–3962. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, M.; Hutinet, G.; Son, O.; Amarir-Bouhram, J.; Schbath, S.; Petit, M.A. Temperate phages acquire DNA from defective prophages by relaxed homologous recombination: the role of Rad52-like recombinases. PLoS genetics 2014, 10, e1004181. [Google Scholar] [CrossRef] [PubMed]
- Namura, M.; Hijikata, T.; Miyanaga, K.; Tanji, Y. Detection of Escherichia coli with fluorescent labeled phages that have a broad host range to E. coli in sewage water. Biotechnology progress 2008, 24, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Tanji, Y.; Furukawa, C.; Na, S.H.; Hijikata, T.; Miyanaga, K.; Unno, H. Escherichia coli detection by GFP-labeled lysozyme-inactivated T4 bacteriophage. Journal of biotechnology 2004, 114, 11–20. [Google Scholar] [CrossRef]
- Chen, Y.; Batra, H.; Dong, J.; Chen, C.; Rao, V.B.; Tao, P. Genetic engineering of bacteriophages against infectious diseases. Frontiers in microbiology 2019, 10, 954. [Google Scholar] [CrossRef]
- Yu, D.; Ellis, H.M.; Lee, E.C.; Jenkins, N.A.; Copeland, N.G.; Court, D.L. An efficient recombination system for chromosome engineering in Escherichia coli. Proceedings of the National Academy of Sciences 2000, 97, 5978–5983. [Google Scholar] [CrossRef]
- Ellis, H.M.; Yu, D.; DiTizio, T.; Court, D.L. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proceedings of the National Academy of Sciences 2001, 98, 6742–6746. [Google Scholar] [CrossRef]
- Zhang, Y.; Buchholz, F.; Muyrers, J.P.; Stewart, A.F. A new logic for DNA engineering using recombination in Escherichia coli. Nature genetics 1998, 20, 123–128. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Gottesman, M.E.; Gottesman, S.; Martin, G. Characterization of bacteriophage λ reverse as an Escherichia coli phage carrying a unique set of host-derived recombination functions. Journal of molecular biology 1974, 88, 471–487. [Google Scholar] [CrossRef]
- Kulkarni, S.K.; Stahl, F.W. Interaction between the sbcC gene of Escherichia coli and the gam gene of phage lambda. Genetics 1989, 123, 249–253. [Google Scholar] [CrossRef]
- Court, R.; Cook, N.; Saikrishnan, K.; Wigley, D. The crystal structure of λ-Gam protein suggests a model for RecBCD inhibition. Journal of molecular biology 2007, 371, 25–33. [Google Scholar] [CrossRef]
- Marinelli, L.J.; Hatfull, G.F.; Piuri, M. Recombineering: A powerful tool for modification of bacteriophage genomes. Bacteriophage 2012, 2, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.J.; Piuri, M.; Swigoňová, Z.; Balachandran, A.; Oldfield, L.M.; van Kessel, J.C.; Hatfull, G.F. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One 2008, 3, e3957. [Google Scholar] [CrossRef] [PubMed]
- Thomason, L.C.; Oppenheim, A.B.; Court, D. L. Modifying Bacteriophage λ with Recombineering. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions; 2009; pp. 239–251. [Google Scholar]
- Marsić, N.; Roje, S.; Stojiljković, I.; Salaj-Smic, E.; Trgovcević, Z. In vivo studies on the interaction of RecBCD enzyme and lambda Gam protein. Journal of bacteriology 1993, 175, 4738–4743. [Google Scholar] [CrossRef] [PubMed]
- Nafissi, N.; Slavcev, R. Bacteriophage recombination systems and biotechnical applications. Applied microbiology and biotechnology 2014, 98, 2841–2851. [Google Scholar] [CrossRef] [PubMed]
- Van Kessel, J.C.; Hatfull, G.F. Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: characterization of antimycobacterial drug targets. Molecular microbiology 2008, 67, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- van Kessel, J.C.; Hatfull, G.F. Mycobacterial recombineering. Chromosomal Mutagenesis 2008, 203–215. [Google Scholar]
- Van Kessel, J.C.; Hatfull, G.F. Recombineering in Mycobacterium tuberculosis. Nature methods 2007, 4, 147–152. [Google Scholar] [CrossRef]
- Marinelli, L.J.; Piuri, M.; Hatfull, G.F. Genetic manipulation of lytic bacteriophages with BRED: bacteriophage recombineering of electroporated DNA. Bacteriophages: Methods and Protocols, Volume IV 2019, 69–80. [Google Scholar]
- Fehér, T.; Karcagi, I.; Blattner, F.R.; Pósfai, G. Bacteriophage recombineering in the lytic state using the lambda red recombinases. Microbial biotechnology 2012, 5, 466–476. [Google Scholar] [CrossRef]
- Tao, P.; Wu, X.; Rao, V. Unexpected evolutionary benefit to phages imparted by bacterial CRISPR-Cas9. Science advances 2018, 4, eaar4134. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.; Vogel, P.; Alber, G.; Schaub, G.A. The innate immune system of mammals and insects. Trends in innate immunity 2008, 15, 21–44. [Google Scholar]
- Hatoum-Aslan, A. Phage genetic engineering using CRISPR–Cas systems. Viruses 2018, 10, 335. [Google Scholar] [CrossRef] [PubMed]
- Martel, B. , & Moineau, S. CRISPR-Cas: an efficient tool for genome engineering of virulent bacteriophages. Nucleic acids research 2014, 42, 9504–9513. [Google Scholar]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Current opinion in microbiology 2017, 37, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Brouns, S.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.; Snijders, A.P.; Van Der Oost, J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Barrangou, R.; Garneau, J.E.; Labonté, J.; Fremaux, C.; Boyaval, P.; Moineau, S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. Journal of bacteriology 2008, 190, 1390–1400. [Google Scholar] [CrossRef]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type IE CRISPR-Cas system. RNA biology 2014, 11, 42–44. [Google Scholar] [CrossRef]
- Box, A.M.; McGuffie, M.J.; O'Hara, B.J.; Seed, K.D. Functional analysis of bacteriophage immunity through a type IE CRISPR-Cas system in Vibrio cholerae and its application in bacteriophage genome engineering. Journal of bacteriology 2016, 198, 578–590. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Garneau, J.E.; Dupuis M, È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Lemay, M.L.; Tremblay, D.M.; Moineau, S. Genome engineering of virulent lactococcal phages using CRISPR-Cas9. ACS synthetic biology 2017, 6, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. science 2008, 322, 1843–1845. [Google Scholar] [CrossRef] [PubMed]
- Samai, P.; Pyenson, N.; Jiang, W.; Goldberg, G.W.; Hatoum-Aslan, A.; Marraffini, L.A. Co-transcriptional DNA and RNA cleavage during type III CRISPR-Cas immunity. Cell 2015, 161, 1164–1174. [Google Scholar] [CrossRef]
- Cater, K.; Dandu, V.S.; Bari, S.N.; Lackey, K.; Everett, G.F.; Hatoum-Aslan, A. A novel Staphylococcus podophage encodes a unique lysin with unusual modular design. MSphere 2017, 2, e00040–17. [Google Scholar] [CrossRef] [PubMed]
- Vandersteegen, K.; Mattheus, W.; Ceyssens, P.J.; Bilocq, F.; De Vos, D.; Pirnay, J.P.; Lavigne, R. Microbiological and molecular assessment of bacteriophage ISP for the control of Staphylococcus aureus. PLoS One 2011, 6, e24418. [Google Scholar] [CrossRef]
- Pan, Y.J.; Lin, T.L.; Chen, C.C.; Tsai, Y.T.; Cheng, Y.H.; Chen, Y.Y.; Wang, J.T. Klebsiella phage ΦK64-1 encodes multiple depolymerases for multiple host capsular types. Journal of virology 2017, 91, e02457–16. [Google Scholar] [CrossRef]
- Shin, H.; Lee, J.H.; Yoon, H.; Kang, D.H.; Ryu, S. Genomic investigation of lysogen formation and host lysis systems of the Salmonella temperate bacteriophage SPN9CC. Appl. Environ. Microbiol. 2014, 80, 374–384. [Google Scholar] [CrossRef]
- Wetzel, K.S.; Guerrero-Bustamante, C.A.; Dedrick, R.M.; Ko, C.C.; Freeman, K.G.; Aull, H.G.; Hatfull, G.F. CRISPY-BRED and CRISPY-BRIP: efficient bacteriophage engineering. Scientific Reports 2021, 11, 1–6. [Google Scholar] [CrossRef]
- Rock, J.M.; Hopkins, F.F.; Chavez, A.; Diallo, M.; Chase, M.R.; Gerrick, E.R.; Fortune, S.M. Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nature microbiology 2017, 2, 1–9. [Google Scholar] [CrossRef]
- Lee, M.H.; Pascopella, L.; Jacobs Jr, W.R.; Hatfull, G.F. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guérin. Proceedings of the National Academy of Sciences 1991, 88, 3111–3115. [Google Scholar] [CrossRef] [PubMed]
- Maheshri, N.; Koerber, J.T.; Kaspar, B.K.; Schaffer, D.V. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nature biotechnology 2006, 24, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Krieg, D.R. Ethyl methanesulfonate-induced reversion of bacteriophage T4rII mutants. Genetics 1963, 48, 561. [Google Scholar] [CrossRef] [PubMed]
- Favor, A.H.; Llanos, C.D.; Youngblut, M.D.; Bardales, J.A. Optimizing bacteriophage engineering through an accelerated evolution platform. Scientific Reports 2020, 10, 13981. [Google Scholar] [CrossRef] [PubMed]
- Jaschke, P.R.; Lieberman, E.K.; Rodriguez, J.; Sierra, A.; Endy, D. A fully decompressed synthetic bacteriophage øX174 genome assembled and archived in yeast. Virology 2012, 434, 278–284. [Google Scholar] [CrossRef]
- Chan, L.Y.; Kosuri, S.; Endy, D. Refactoring bacteriophage T7. Molecular systems biology 2005, 1, 2005–0018. [Google Scholar] [CrossRef] [PubMed]
- Calendar, R.L.; Abedon, S.T. (Eds.) The bacteriophages (Vol. 2); Oxford University Press on Demand, 2006. [Google Scholar]
- Kilcher, S.; Loessner, M.J. Engineering bacteriophages as versatile biologics. Trends in microbiology 2019, 27, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Monteiro, R.; Mil-Homens, D.; Fialho, A.; Lu, T.K.; Azeredo, J. Designing P. aeruginosa synthetic phages with reduced genomes. Scientific Reports 2021, 11, 2164. [Google Scholar] [CrossRef]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering modular viral scaffolds for targeted bacterial population editing. Cell systems 2015, 1, 187–196. [Google Scholar] [CrossRef]
- MDunne, M.; Rupf, B.; Tala, M.; Qabrati, X.; Ernst, P.; Shen, Y.; Kilcher, S. Reprogramming bacteriophage host range through structure-guided design of chimeric receptor binding proteins. Cell reports 2019, 29, 1336–1350. [Google Scholar]
- Meile, S.; Sarbach, A.; Du, J.; Schuppler, M.; Saez, C.; Loessner, M.J.; Kilcher, S. Engineered reporter phages for rapid bioluminescence-based detection and differentiation of viable Listeria cells. Applied and environmental microbiology 2020, 86, e00442–20. [Google Scholar] [CrossRef] [PubMed]
- Bundy, B.C.; Franciszkowicz, M.J.; Swartz, J.R. Escherichia coli-based cell-free synthesis of virus-like particles. Biotechnology and bioengineering 2008, 100, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, D.D.; Wolcott, R.D.; Kuskowski, M.A.; Wolcott, B.M.; Ward, L.S.; Sulakvelidze, A. Bacteriophage therapy of venous leg ulcers in humans: results of a phase I safety trial. Journal of wound care 2009, 18, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Gutiérrez, D.; García, P.; Rodríguez, A. The perfect bacteriophage for therapeutic applications—a quick guide. Antibiotics 2019, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, F.L.; Vlot, M.; de Jonge, P.A.; Dreesens, L.L.; Beaumont, H.J.; Lavigne, R.; Brouns, S.J. Targeting mechanisms of tailed bacteriophages. Nature Reviews Microbiology 2018, 16, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Yoichi, M.; Abe, M.; Miyanaga, K.; Unno, H.; Tanji, Y. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157: H7. Journal of biotechnology 2005, 115, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Mahichi, F.; Synnott, A.J.; Yamamichi, K.; Osada, T.; Tanji, Y. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS microbiology letters 2009, 295, 211–217. [Google Scholar] [CrossRef]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering modular viral scaffolds for targeted bacterial population editing. Cell systems 2015, 1, 187–196. [Google Scholar] [CrossRef]
- Marzari, R.; Sblattero, D.; Righi, M.; Bradbury, A. Extending filamentous phage host range by the grafting of a heterologous receptor binding domain. Gene 1997, 185, 27–33. [Google Scholar] [CrossRef]
- Heilpern, A.J.; Waldor, M.K. pIIICTX, a predicted CTXφ minor coat protein, can expand the host range of coliphage fd to include Vibrio cholerae. Journal of bacteriology 2003, 185, 1037–1044. [Google Scholar] [CrossRef]
- Nelson, D.; Loomis, L.; Fischetti, V.A. Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proceedings of the National Academy of Sciences 2001, 98, 4107–4112. [Google Scholar] [CrossRef] [PubMed]
- Danis-Wlodarczyk, K.M.; Wozniak, D.J.; Abedon, S.T. Treating bacterial infections with bacteriophage-based enzybiotics: in vitro, in vivo and clinical application. Antibiotics 2021, 10, 1497. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V.A. Development of phage lysins as novel therapeutics: a historical perspective. Viruses 2018, 10, 310. [Google Scholar] [CrossRef] [PubMed]
- Young, R.Y. Bacteriophage lysis: mechanism and regulation. Microbiological reviews 1992, 56, 430–481. [Google Scholar] [CrossRef] [PubMed]
- Loessner, M.J. Bacteriophage endolysins—current state of research and applications. Current opinion in microbiology 2005, 8, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.R.; March, J.B. Bacteriophages and biotechnology: vaccines, gene therapy and antibacterials. Trends in biotechnology 2006, 24, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.U.; Wang, W.; Sun, Q.; Shah, J.A.; Li, C.; Sun, Y.; Wang, S. Endolysin, a promising solution against antimicrobial resistance. Antibiotics 2021, 10, 1277. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Delgado-Martínez, J. Bacteriophages: protagonists of a post-antibiotic era. Antibiotics 2018, 7, 66. [Google Scholar] [CrossRef]
- Fischetti, V.A. Lysin therapy for Staphylococcus aureus and other bacterial pathogens. In Staphylococcus aureus: Microbiology, Pathology, Immunology, Therapy and Prophylaxis; 2017; pp. 529–540. [Google Scholar]
- Briers, Y.; Walmagh, M.; Van Puyenbroeck, V.; Cornelissen, A.; Cenens, W.; Aertsen, A.; Lavigne, R. Engineered endolysin-based “Artilysins” to combat multidrug-resistant gram-negative pathogens. MBio 2014, 5, e01379–14. [Google Scholar] [CrossRef]
- Pinto, A.M.; Silva, M.D.; Pastrana, L.M.; Bañobre-López, M.; Sillankorva, S. The clinical path to deliver encapsulated phages and lysins. FEMS Microbiology Reviews 2021, 45, fuab019. [Google Scholar] [CrossRef]
- Gerstmans, H.; Criel, B.; Briers, Y. Synthetic biology of modular endolysins. Biotechnology advances 2018, 36, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Low, L.Y.; Yang, C.; Perego, M.; Osterman, A.; Liddington, R. Role of net charge on catalytic domain and influence of cell wall binding domain on bactericidal activity, specificity, and host range of phage lysins. Journal of Biological Chemistry 2011, 286, 34391–34403. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Schuch, R.; Chahales, P.; Zhu, S.; Fischetti, V.A. PlyC: a multimeric bacteriophage lysin. Proceedings of the National Academy of Sciences 2006, 103, 10765–10770. [Google Scholar] [CrossRef] [PubMed]
- Rohde, M. The Gram-positive bacterial cell wall. Microbiology Spectrum 2019, 7, 7–3. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V.A. Bacteriophage endolysins: a novel anti-infective to control Gram-positive pathogens. International Journal of Medical Microbiology 2010, 300, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V.A. Bacteriophage lysins as effective antibacterials. Current opinion in microbiology 2008, 11, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Gondil, V.S.; Harjai, K.; Chhibber, S. Endolysins as emerging alternative therapeutic agents to counter drug-resistant infections. International journal of antimicrobial agents 2020, 55, 105844. [Google Scholar] [CrossRef]
- Schuch, R.; Nelson, D.; Fischetti, V.A. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 2002, 418, 884–889. [Google Scholar] [CrossRef]
- Gutiérrez, D.; Fernández, L.; Rodríguez, A.; García, P. Are phage lytic proteins the secret weapon to kill Staphylococcus aureus? MBio 2018, 9, e01923–17. [Google Scholar] [CrossRef]
- Masters, E.A.; Trombetta, R.P.; de Mesy Bentley, K.L.; Boyce, B.F.; Gill, A.L.; Gill, S.R.; Muthukrishnan, G. Evolving concepts in bone infection: redefining “biofilm”,“acute vs. chronic osteomyelitis”,“the immune proteome” and “local antibiotic therapy”. Bone research 2019, 7, 20. [Google Scholar] [CrossRef]
- Lüderitz, O.; Freudenberg, M.A.; Galanos, C.; Lehmann, V.; Rietschel, E.T.; Shaw, D.H. Lipopolysaccharides of gram-negative bacteria. In Current topics in membranes and transport; Academic Press, 1982; Volume 17, pp. 79–151. [Google Scholar]
- Pasquina-Lemonche, L.; Burns, J.; Turner, R.D.; Kumar, S.; Tank, R.; Mullin, N.; Hobbs, J.K. The architecture of the Gram-positive bacterial cell wall. Nature 2020, 582, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yu, J.; Wei, H. Engineered bacteriophage lysins as novel anti-infectives. Frontiers in microbiology 2014, 5, 542. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future microbiology 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [PubMed]
- Gerstmans, H.; Rodríguez-Rubio, L.; Lavigne, R.; Briers, Y. From endolysins to Artilysin® s: novel enzyme-based approaches to kill drug-resistant bacteria. Biochemical Society Transactions 2016, 44, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zheng, Y.; Dai, J.; Zhou, J.; Yu, R.; Zhang, C. Design SMAP29-LysPA26 as a Highly Efficient Artilysin against Pseudomonas aeruginosa with Bactericidal and Antibiofilm Activity. Microbiology Spectrum 2021, 9, e00546–21. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Tanji, Y.; Mizoguchi, K.; Soejima, A.; Orito, Y.; Unno, H. Antibacterial activity of Bacillus amyloliquefaciens phage endolysin without holin conjugation. Journal of bioscience and bioengineering 2001, 91, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Lai WC, B.; Chen, X.; Ho MK, Y.; Xia, J.; Leung SS, Y. Bacteriophage-derived endolysins to target gram-negative bacteria. International Journal of Pharmaceutics 2020, 589, 119833. [Google Scholar] [PubMed]
- Khan, F.M.; Gondil, V.S.; Li, C.; Jiang, M.; Li, J.; Yu, J.; Yang, H. A novel Acinetobacter baumannii bacteriophage endolysin LysAB54 with high antibacterial activity against multiple Gram-negative microbes. Frontiers in cellular and infection microbiology 2021, 11, 637313. [Google Scholar] [CrossRef]
- Chatterjee, D. The mycobacterial cell wall: structure, biosynthesis and sites of drug action. Current opinion in chemical biology 1997, 1, 579–588. [Google Scholar] [CrossRef]
- Abrahams, K.A.; Besra, G.S. Mycobacterial cell wall biosynthesis: a multifaceted antibiotic target. Parasitology 2018, 145, 116–133. [Google Scholar] [CrossRef]
- Catalão, M.J.; Pimentel, M. Mycobacteriophage lysis enzymes: targeting the mycobacterial cell envelope. Viruses 2018, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Payne, K.M.; Hatfull, G.F. Mycobacteriophage endolysins: diverse and modular enzymes with multiple catalytic activities. PLoS One 2012, 7, e34052. [Google Scholar] [CrossRef] [PubMed]
- Gigante, A.M.; Hampton, C.M.; Dillard, R.S.; Gil, F.; Catalão, M.J.; Moniz-Pereira, J.; Pimentel, M. The Ms6 mycolyl-arabinogalactan esterase LysB is essential for an efficient mycobacteriophage-induced lysis. Viruses 2017, 9, 343. [Google Scholar] [CrossRef] [PubMed]
- Catalao, M.J.; Milho, C.; Gil, F.; Moniz-Pereira, J.; Pimentel, M. A second endolysin gene is fully embedded in-frame with the lysA gene of mycobacteriophage Ms6. PLoS One 2011, 6, e20515. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.J.; Liu, C.C.; Jiang, S.J.; Soo, P.C.; Tu, M.H.; Lee, J.J.; Chang, K.C. Antimycobacterial activities of endolysins derived from a mycobacteriophage, BTCU-1. Molecules 2015, 20, 19277–19290. [Google Scholar] [CrossRef] [PubMed]
- Murray, E.; Draper, L.A.; Ross, R.P.; Hill, C. The advantages and challenges of using endolysins in a clinical setting. Viruses 2021, 13, 680. [Google Scholar] [CrossRef]
- Love, M.J.; Abeysekera, G.S.; Muscroft-Taylor, A.C.; Billington, C.; Dobson, R.C. On the catalytic mechanism of bacteriophage endolysins: Opportunities for engineering. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 2020, 1868, 140302. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nature Reviews Immunology 2010, 10, 787–796. [Google Scholar] [CrossRef]
- -Zepp, F. Principles of vaccine design—lessons from nature. Vaccine 2010, 28, C14–C24. [Google Scholar] [CrossRef]
- Henry, K.A.; Arbabi-Ghahroudi, M.; Scott, J.K. Beyond phage display: non-traditional applications of the filamentous bacteriophage as a vaccine carrier, therapeutic biologic, and bioconjugation scaffold. Frontiers in microbiology 2015, 6, 755. [Google Scholar] [CrossRef]
- Fu, Y.; Li, J. A novel delivery platform based on Bacteriophage MS2 virus-like particles. Virus Research 2016, 211, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, J.; Sheldon, K.; Slavcev, R.A. Bacteriophage lambda display systems: developments and applications. Applied microbiology and biotechnology 2014, 98, 2853–2866. [Google Scholar] [CrossRef] [PubMed]
- Tao, P.; Mahalingam, M.; Kirtley, M.L.; van Lier, C.J.; Sha, J.; Yeager, L.A.; Rao, V.B. Mutated and bacteriophage T4 nanoparticle arrayed F1-V immunogens from Yersinia pestis as next generation plague vaccines. PLoS pathogens 2013, 9, e1003495. [Google Scholar] [CrossRef] [PubMed]
- Tissot, A.C.; Renhofa, R.; Schmitz, N.; Cielens, I.; Meijerink, E.; Ose, V.; Jennings, G.T.; Saudan, P.; Pumpens, P.; Bachmann, M.F. Versatile virus-like particle carrier for epitope based vaccines. PloS one 2010, 5, e9809. [Google Scholar] [CrossRef] [PubMed]
- Zimecki MI CH A, Ł.; Weber-Dabrowska, B.; Lusiak-Szelachowska, M.; Mulczyk, M.; Boratynski, J.; Pozniak, G.; Górski, A. Bacteriophages provide regulatory signals in mitogen-induced murine splenocyte proliferation. Cellular and Molecular Biology Letters 2003, 8, 699–712. [Google Scholar]
- Kaur, T.; Nafissi, N.; Wasfi, O.; Sheldon, K.; Wettig, S.; Slavcev, R. Immunocompatibility of bacteriophages as nanomedicines. Journal of Nanotechnology 2012. [CrossRef]
- Sartorius, R.; D'Apice, L.; Trovato, M.; Cuccaro, F.; Costa, V.; De Leo, M.G.; De Berardinis, P. Antigen delivery by filamentous bacteriophage fd displaying an anti-DEC-205 single-chain variable fragment confers adjuvanticity by triggering a TLR 9-mediated immune response. EMBO molecular medicine 2015, 7, 973–988. [Google Scholar] [CrossRef]
- Shepardson, K.M.; Schwarz, B.; Larson, K.; Morton, R.V.; Avera, J.; McCoy, K.; Rynda-Apple, A. Induction of antiviral immune response through recognition of the repeating subunit pattern of viral capsids is toll-like receptor 2 dependent. MBio 2017, 8, e01356–17. [Google Scholar] [CrossRef]
- Mantegazza, A.R.; Magalhaes, J.G.; Amigorena, S.; Marks, M.S. Presentation of phagocytosed antigens by MHC class I and II. Traffic 2013, 14, 135–152. [Google Scholar] [CrossRef]
- Gomes, A.C.; Flace, A.; Saudan, P.; Zabel, F.; Cabral-Miranda, G.; Turabi, A.E.; Bachmann, M.F. Adjusted particle size eliminates the need of linkage of antigen and adjuvants for appropriated T cell responses in virus-like particle-based vaccines. Frontiers in immunology 2017, 8, 226. [Google Scholar] [CrossRef]
- Link, A.; Zabel, F.; Schnetzler, Y.; Titz, A.; Brombacher, F.; Bachmann, M.F. Innate immunity mediates follicular transport of particulate but not soluble protein antigen. The Journal of Immunology 2012, 188, 3724–3733. [Google Scholar] [CrossRef]
- Sathaliyawala, T.; Rao, M.; Maclean, D.M.; Birx, D.L.; Alving, C.R.; Rao, V.B. Assembly of human immunodeficiency virus (HIV) antigens on bacteriophage T4: a novel in vitro approach to construct multicomponent HIV vaccines. Journal of virology 2006, 80, 7688–7698. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Rohrer, U.H.; Kündig, T.M.; Bürki, K.; Hengartner, H.; Zinkernagel, R.M. The influence of antigen organization on B cell responsiveness. Science 1993, 262, 1448–1451. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Zinkernagel, R.M. The influence of virus structure on antibody responses and virus serotype formation. Immunology today 1996, 17, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Jegerlehner, A.; Storni, T.; Lipowsky, G.; Schmid, M.; Pumpens, P.; Bachmann, M.F. Regulation of IgG antibody responses by epitope density and CD21-mediated costimulation. European journal of immunology 2002, 32, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, R.; Bettua, C.; D'Apice, L.; Caivano, A.; Trovato, M.; Russo, D.; De Berardinis, P. Vaccination with filamentous bacteriophages targeting DEC-205 induces DC maturation and potent anti-tumor T-cell responses in the absence of adjuvants. European journal of immunology 2011, 41, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Shivachandra, S.B.; Zhang, Z.; Rao, V.B. Assembly of the small outer capsid protein, Soc, on bacteriophage T4: a novel system for high density display of multiple large anthrax toxins and foreign proteins on phage capsid. Journal of molecular biology 2007, 370, 1006–1019. [Google Scholar] [CrossRef]
- Shivachandra, S.B.; Li, Q.; Peachman, K.K.; Matyas, G.R.; Leppla, S.H.; Alving, C.R.; Rao, V.B. Multicomponent anthrax toxin display and delivery using bacteriophage T4. Vaccine 2007, 25, 1225–1235. [Google Scholar] [CrossRef]
- Low, J.G.; Lee, L.S.; Ooi, E.E.; Ethirajulu, K.; Yeo, P.; Matter, A.; Novotny-Diermayr, V. Safety and immunogenicity of a virus-like particle pandemic influenza A (H1N1) 2009 vaccine: results from a double-blinded, randomized Phase I clinical trial in healthy Asian volunteers. Vaccine 2014, 32, 5041–5048. [Google Scholar] [CrossRef]
- Huang, X.; Wang, X.; Zhang, J.; Xia, N.; Zhao, Q. Escherichia coli-derived virus-like particles in vaccine development. npj Vaccines 2017, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Joshi, M.D.; Singhania, S.; et al. Peptide vaccine: progress and challenges. Vaccines 2014, 2, 515–36. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowska, K.; Miernikiewicz, P.; Piotrowicz, A.; Hodyra, K.; Owczarek, B.; Lecion, D.; Górski, A. Immunogenicity studies of proteins forming the T4 phage head surface. Journal of virology 2014, 88, 12551–12557. [Google Scholar] [CrossRef] [PubMed]
- Akram, A.; Inman, R.D. Immunodominance: a pivotal principle in host response to viral infections. Clinical immunology 2012, 143, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. Sciences, H.; Sciences, M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Delivery Rev 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.K.; Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proceedings of the National Academy of Sciences 2007, 104, 11197–11202. [Google Scholar] [CrossRef] [PubMed]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of biofilm formation by T7 bacteriophages producing quorum-quenching enzymes. Applied and environmental microbiology 2014, 80, 5340–5348. [Google Scholar] [CrossRef] [PubMed]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Marraffini, L.A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nature biotechnology 2014, 32, 1146–1150. [Google Scholar] [CrossRef]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nature biotechnology 2014, 32, 1141–1145. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Spencer, H. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nature medicine 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Little, J.S.; Dedrick, R.M.; Freeman, K.G.; Cristinziano, M.; Smith, B.E.; Benson, C.A.; Hatfull, G.F. Bacteriophage treatment of disseminated cutaneous Mycobacterium chelonae infection. Nature communications 2022, 13, 2313. [Google Scholar] [CrossRef]
- Pan, Y.J.; Lin, T.L.; Chen, C.C.; Tsai, Y.T.; Cheng, Y.H.; Chen, Y.Y.; Wang, J.T. Klebsiella phage ΦK64-1 encodes multiple depolymerases for multiple host capsular types. Journal of virology 2017, 91, e02457–16. [Google Scholar] [CrossRef] [PubMed]
- Fehér, T.; Karcagi, I.; Blattner, F.R.; Pósfai, G. Bacteriophage recombineering in the lytic state using the lambda red recombinases. Microbial biotechnology 2012, 5, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Ranallo, R.T.; Barnoy, S.; Thakkar, S.; Urick, T.; Venkatesan, M.M. Developing live Shigella vaccines using λ Red recombineering. FEMS Immunology & Medical Microbiology 2006, 47, 462–469. [Google Scholar]
- Viertel, T.M.; Ritter, K.; Horz, H.P. Viruses versus bacteria—novel approaches to phage therapy as a tool against multidrug-resistant pathogens. Journal of Antimicrobial Chemotherapy 2014, 69, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Forti, F.; Roach, D.R.; Cafora, M.; Pasini, M.E.; Horner, D.S.; Fiscarelli, E.V.; Ghisotti, D. Design of a broad-range bacteriophage cocktail that reduces Pseudomonas aeruginosa biofilms and treats acute infections in two animal models. Antimicrobial agents and chemotherapy 2018, 62, e02573–17. [Google Scholar] [CrossRef] [PubMed]
- Torres-Barceló, C.; Hochberg, M.E. Evolutionary rationale for phages as complements of antibiotics. Trends in microbiology 2016, 24, 249–256. [Google Scholar] [CrossRef]
- Liu, C.; Hong, Q.; Chang RY, K.; Kwok PC, L.; Chan, H.K. Phage–Antibiotic therapy as a promising strategy to combat multidrug-resistant infections and to enhance antimicrobial efficiency. Antibiotics 2022, 11, 570. [Google Scholar] [CrossRef]
- Chan, B.K.; Turner, P.E.; Kim, S.; Mojibian, H.R.; Elefteriades, J.A.; Narayan, D. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evolution, medicine, and public health 2018, 2018, 60–66. [Google Scholar] [CrossRef]
- Kirby, A.E. Synergistic action of gentamicin and bacteriophage in a continuous culture population of Staphylococcus aureus. Plos one 2012, 7, e51017. [Google Scholar] [CrossRef]
- Torres-Barceló, C.; Arias-Sánchez, F.I.; Vasse, M.; Ramsayer, J.; Kaltz, O.; Hochberg, M.E. A window of opportunity to control the bacterial pathogen Pseudomonas aeruginosa combining antibiotics and phages. PloS one 2014, 9, e106628. [Google Scholar] [CrossRef] [PubMed]
- Chhibber, S.; Kaur, T.; Kaur, S. Co-therapy using lytic bacteriophage and linezolid: effective treatment in eliminating methicillin resistant Staphylococcus aureus (MRSA) from diabetic foot infections. PloS one 2013, 8, e56022. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Harjai, K.; Chhibber, S. Characterization of a T7-like lytic bacteriophage of Klebsiella pneumoniae B5055: a potential therapeutic agent. Current microbiology 2009, 59, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Tétart, F.; Trojet, S.N.; Prère, M.F.; Krisch, H.M. Phage-antibiotic synergy (PAS): β-lactam and quinolone antibiotics stimulate virulent phage growth. Plos one 2007, 2, e799. [Google Scholar] [CrossRef] [PubMed]
- Kamal, F.; Dennis, J.J. Burkholderia cepacia complex phage-antibiotic synergy (PAS): antibiotics stimulate lytic phage activity. Applied and environmental microbiology 2015, 81, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.Y.; Wong, J.; Mathai, A.; Morales, S.; Kutter, E.; Britton, W.; Chan, H.K. Production of highly stable spray dried phage formulations for treatment of Pseudomonas aeruginosa lung infection. European Journal of Pharmaceutics and Biopharmaceutics 2017, 121, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sybesma, W.; Rohde, C.; Bardy, P.; Pirnay, J.P.; Cooper, I.; Kurtbӧke, D.İ.; Expert Round Table on Acceptance and Re-Implementation of Bacteriophage Therapy. Silk route to the acceptance and re-implementation of bacteriophage therapy—part II. Antibiotics 2018, 7, 35. [Google Scholar]
- Furfaro, L.L.; Payne, M.S.; Chang, B.J. Bacteriophage therapy: clinical trials and regulatory hurdles. Frontiers in cellular and infection microbiology 2018, 8, 376. [Google Scholar] [CrossRef]
- Furfaro, L.L.; Payne, M.S.; Chang, B.J. Bacteriophage therapy: clinical trials and regulatory hurdles. Frontiers in cellular and infection microbiology 2018, 8, 376. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



