
Submitted:
11 May 2023
Posted:
11 May 2023
You are already at the latest version
Abstract
Metagenomics have opened our eyes to the otherwise enigmatic microbial consortia that relies on different human body niches that impact on disease pathogenesis. This work originally aims to re-analyse a public shotgun metagenomics dataset to glean insights on the microbial species that may partake in stomach cancer pathogenesis. However, a random selection of sequenced reads reveals poor percent identities (79 to 94%) of the microbial identification obtained in a BLAST search. This surprising result suggests possible stomach acid induced degradation or modification of the sequenced read that reduced the effectiveness of the metagenomics approach in microbial identification. Further analysis of the dataset highlights that poor percent identities did not arise due to highly fragmented nature of the genetic material where there is a good abundance of sequenced reads in the 100 to 1300 base pair category. More intriguingly, attempts to obtain a correlation between percent identity and read length did not reveal a correlation which meant that stomach acid did not modify all types of nucleotides. More likely, stomach acid only modified low abundance nucleotide in this case, which point to the probability of identifying a microbe in the modified gene fragment in totally unpredictable ways. Overall, data reported in this work suggests caution in the interpretation of results of shotgun metagenomics studies of stomach cancer microbiota where stomach acid likely degraded the genetic material of microbes that may result in mis-identification.
Keywords:
degraded metagenome
; shotgun metagenomics
; stomach acid
; microbial identification
; percent identity
Profiling the genetic and genomic sequences of the consortia of microorganisms that inhabit a particular body niche is one approach for understanding the microbial basis of many human diseases. Current understanding in microbiome studies posit that there exists a unique ensemble of microbes for each niche in the human body that differs in composition and relative abundance due to various environmental and nutritional factors. [1,2] Hence, various methods for sequencing the genetic and genomic repertoire of a particular microbiome have come into vogue as the dominant methodology for understanding the composition of microbiome. One such approach is shotgun metagenomics.[3,4]
In shotgun metagenomics, the genetic and genomic materials from all microbial cells in the sample is isolated after a DNA extraction step. This genetic material is subsequently fragmented into shorter fragments with varied read length via either the use of restriction endonucleases or bead milling. Sanger sequencing is then employed to sequence the different fragments, and this constitutes the reads of the metagenome, which can be aligned to the reference genomes of different microbial species in a BLAST search for identification.
This work sought to use the metagenomics approach to identify the stomach microbiota of stomach cancer patients for identifying possible correlations between relative abundance of particular microbes and pathogenesis and phenotype of the stomach cancer disease. To this end, a stomach cancer metagenomics dataset prepared by the shotgun metagenomics approach was downloaded from Mgnify database that links to the larger European Nucleotide Archive database. The dataset accession number is MGYS00005768. Using an in-house MATLAB metagenomics read analysis software, the much larger original dataset was randomly sampled to output a smaller and more manageable 5000 reads dataset for analysis. The length of each read was analysed and histogram analysis was employed to characterise the read length characteristics. Subsequently, the sequence of each metagenomic read was subjected to BLAST analysis to determine which microbial species the read originated from.
What emerges from the analysis described above was fairly low percent identity of the reads which could yield a microbial identification tag. These percent identities ranged between 79% to 94% (Figure 1), which fall short of the 97% percent identity that is internationally recognised to be minimum for microbial identification via the genetic profiling approach. Such a low percent identity of sequenced reads reveal possible degradation or modification of the genetic material in the microbial species due to the high acid stress of the stomach. Noting that this is a single patient sample to which we are analysing, the phenomenon of acid induced modification or degradation of the genetic material of microbe in the stomach may be more or less severe depending on the amount of acid production in each patient.
Analysing the read length distribution of this stomach shotgun metagenomics dataset reveals a cross-segment of read lengths ranging from 100 base pairs to 8800 base pairs (Figure 2). Most of the reads clustered between 100 and 1300 base pairs, which theoretically show yield good identification as the read is sufficiently long to encode genetic diversity present in different microbial species. Thus, the issue of low percent identities of the sequence reads could not be attributed to a prevalence of short sequenced reads in this metagenomics dataset. Rather, it is mutations and chemical modifications of the nucleotides in the genetic material of the different microbial species in a non species-specific way that resulted in the low percent identities of the profiled sequenced reads.
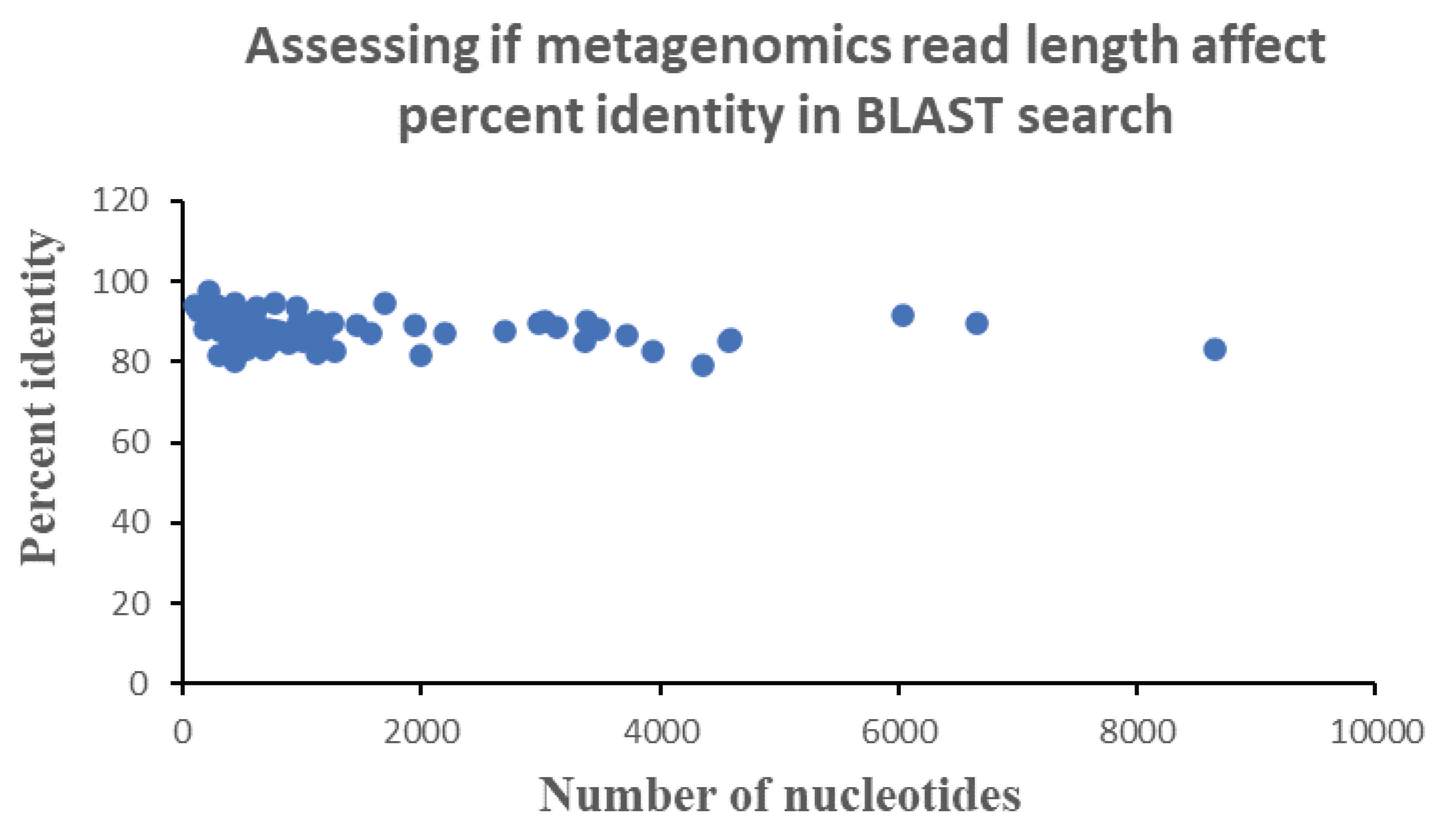
To understand the possibility of how read length may modulate the percent identities of the sequenced read, the correlation or dependence of percent identity of each sequenced read was plotted against their read length (Figure 3). Results show that there is no dependence of percent identities on read length. This suggests that the effect of acid induced degradation of the genetic material is not across all types of nucleotides. In particular, stomach acid likely exerts its effect on specific type of nucleotide. Note here that changing or modifying a nucleotide due to acid stress may result in sequencing error which can tilt the species identification process one way or another, with an overall effect that cannot be readily predicted due to the under-determinate nature of the problem. This explains why we did not obtain a downward sloping curve during a plot of percent identity and read length (number of nucleotides).
Overall, attempt to profile the microbial species in the stomach microbiome of stomach cancer patients reveal a poor percent identity of sequenced reads that hamper microbial identification. Using different metrics to help determine the cause of the low percent identities reveal that the problem is not likely due to a high level fragmentation of the genetic material into very short reads (i.e., 50 to 75 base pairs) given that there is a preponderance of 100 to 1300 base pairs reads in the shotgun metagenomics dataset. In addition, correlating percent identities with read length did not reveal a correlation, which suggests that stomach acid may not modified all types of nucleotides in the gene fragment. Rather, the acid induced modification only occurs on low abundance nucleotide in this sample, which suggests possible patient specificity in the type of degraded metagenome that emerges in the stomach environment of stomach cancer patients.
Funding
No funding was used in this work.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Niche Specific Microbiota-Dependent and Independent Bone Loss around Dental Implants and Teeth - O. Heyman, Y. Niche Specific Microbiota-Dependent and Independent Bone Loss around Dental Implants and Teeth - O. Heyman, Y. Horev, N. Koren, O. Barel, I. Aizenbud, Y. Aizenbud, M. Brandwein, L. Shapira, A.H. Hovav, A. Wilensky, 2020. https://journals.sagepub.com/doi/abs/10.1177/0022034520920577.
- Deines, P.; Hammerschmidt, K.; Bosch TC, G. Exploring the Niche Concept in a Simple Metaorganism. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, T. J. An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Histogram distribution of percentage identity of microbial species identified based on a BLAST search of shotgun metagenomic reads. Results indicate relatively low percentage identities of most reads which meant that the stomach metagenome may be degraded by high levels of stomach acids and is unable to yield microbial identification.
Figure 1.
Histogram distribution of percentage identity of microbial species identified based on a BLAST search of shotgun metagenomic reads. Results indicate relatively low percentage identities of most reads which meant that the stomach metagenome may be degraded by high levels of stomach acids and is unable to yield microbial identification.
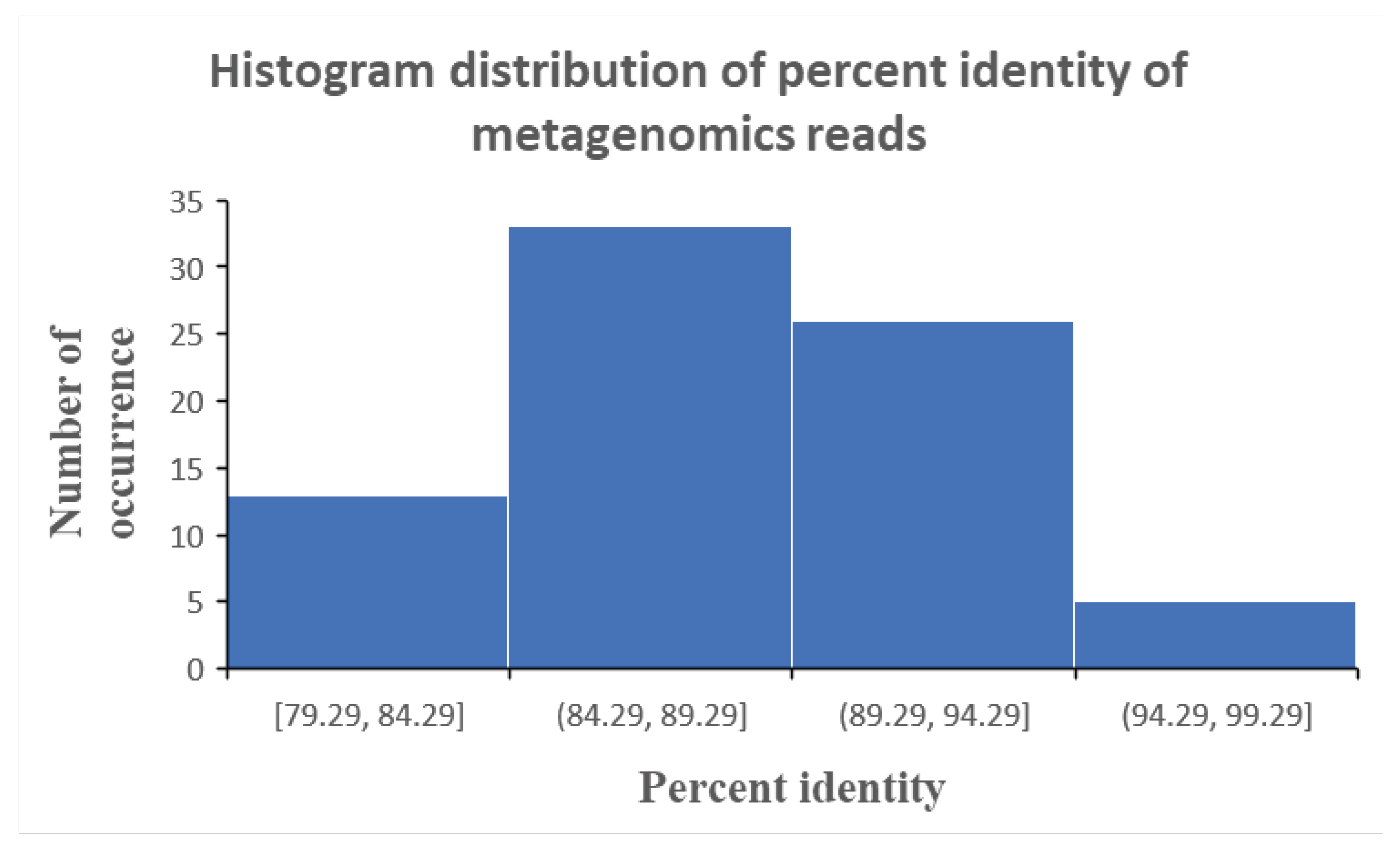
Figure 2.
Histogram distribution of read length of shotgun metagenomics assay on stomach cancer patient showing that majority of reads clustered between 100 and 1300 base pairs.
Figure 2.
Histogram distribution of read length of shotgun metagenomics assay on stomach cancer patient showing that majority of reads clustered between 100 and 1300 base pairs.
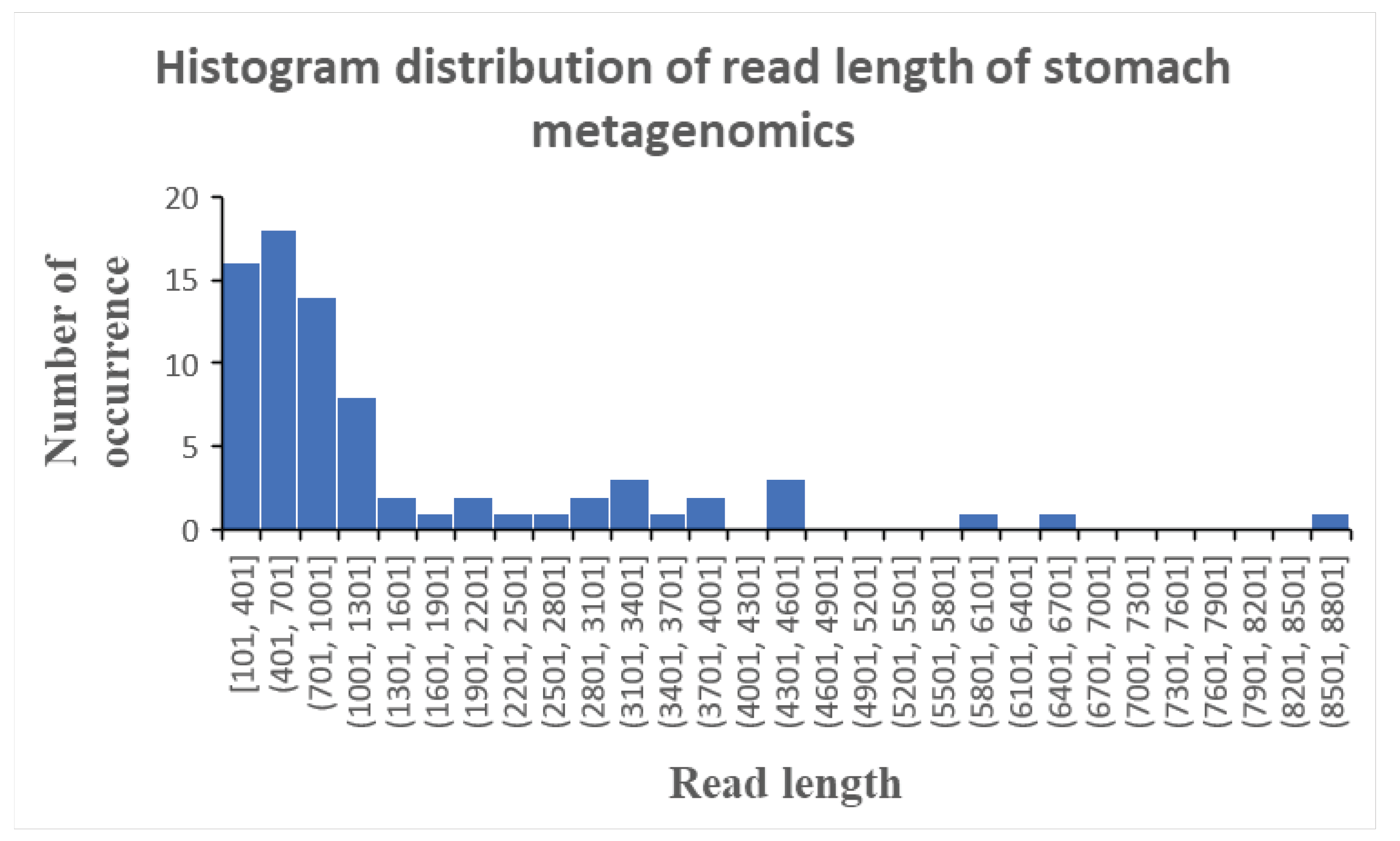
Figure 3.
Effect of metagenomic read length on the percentage identity of microbial species identified using reads from the shotgun metagenomics assay.
Figure 3.
Effect of metagenomic read length on the percentage identity of microbial species identified using reads from the shotgun metagenomics assay.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



