
Submitted:
10 May 2023
Posted:
11 May 2023
You are already at the latest version
Abstract
The recently discovered Jingmenvirus group includes viruses with a segmented genome, RNA of a positive polarity, and several proteins with distant homology to the proteins of the members of the genus Flavivirus. Some Jingmenvirus group members, namely Alongshan virus (ALSV) and Jingmen tick virus, are reported to be tick-borne human pathogens, causing a wide variety of symptoms. ALSV is widely distributed in Eurasia, yet there is no reliable assay for its detection. Here, we describe a qPCR system for the detection of ALSV. Our data show that this system can detect as low as 104 copies of ALSV in the probe. It shows no amplification with common tick-borne viruses circulating in Eurasia, Yanggou tick virus—another member of the Jingmenvirus group—or some known members of the genus Flavivirus. The qPCR system was tested have no non-specific signal for Ixodes ricinus, I. persulcatus, Dermacentor reticulatus, D. marginatus, Haemaphysalis concinna, and H. japonica ticks. Overall, the qPCR system described here can be used for reliable and quantitative ALSV detection.
Keywords:
Jingmenvirus group
; Alongshan virus
; qPCR
; Flavivirus
; Yanggou tick virus
; tick-borne viruses
1. Introduction
Viruses of the genus Flavivirus (family Flaviviridae) are small, enveloped viruses with a non-segmented single strand RNA genome of a positive polarity. The genome encodes a polyprotein that is co- and post-translationally cleaved by viral and cellular proteases into ten proteins [1]. Many flaviviruses are arthropod-borne viruses (arboviruses) and are transmitted by mosquitoes and ticks [1]. Some members of the genus Flavivirus are well-known human pathogens, such as West Nile virus [2], Dengue virus [3], Zika virus [4], and Tick-borne encephalitis virus (TBEV) [5].
Recently the number of newly described viruses has increased dramatically, with several novel virus groups being discovered [6,7,8]. The Jingmenvirus (JMV) group is one of those novel virus groups. The genome of the JMV group members is segmented, and it has proteins with homology to genus Flavivirus polymerase [9] and helicase [9,10], and little homology to envelope protein [9,11]. JMV members include Jingmen tick virus (JMTV) [9], Yanggou tick virus (YGTV) [12], Guaico Culex virus [13], Alongshan virus (ALSV) [14,15], and several other viruses [16,17]. The geographic distribution of the JMV group is very wide, encompassing Europe, Asia, America, and Africa [9,12,13,14,15,18,19,20]. JMV group members are considered to be arboviruses. At least two members of the JMV group, JMTV and ALSV, are considered tick-borne human pathogens [15,21], while there is little information on the pathogenicity of other members of the JMV group.
JMTV was discovered in China and found to be highly prevalent in the tick samples from Hubei province. Moreover, a screening of cattle sera using an immunofluorescence assay and RT-PCR revealed JMTV-positive samples from Hubei and Zhejiang, China [9]. A follow-up study revealed four human cases in China by high throughput sequencing of skin biopsies and blood samples. Eight more cases of human JMTV were identified retrospectively in the same study. All patients had a history of a tick bite, and three of them were co-infected with Rickettsia [21]. Reported manifestations of JMTV infection were fever, headache, and malaise (typical for febrile illnesses), an itch or painful eschar after a tick bite, as well as lymphadenopathy [21]. Since the discovery of JMTV, it was detected in Europe, Turkey, Russia, Kenya, Japan, and Brazil [22].
ALSV was discovered also in China during surveillance for tick-borne diseases from a patient with a febrile illness [15]. A follow-up epidemiological investigation in China confirmed the detection of ALSV for 86 patients. The majority of patients (95%) had a clear history of tick bites before the onset of disease, and no evidence of other tick-borne pathogens were found. The most common symptoms in the patient assessed were headache and fever. Other symptoms include fatigue, depression, coma, poor appetite, nausea, myalgia or arthralgia, and rash. The symptoms resolved after six to eight days of treatment, no permanent clinical complications nor death occurred [15]. ALSV has been detected mostly in Ixodes ricinus and I. persulcatus ticks [12] and is currently considered a tick-borne arbovirus. Later ALSV was detected in the sheep and cattle in ALSV-endemic areas, revealing its potential veterinary importance [23,24].
ALSV has been detected in Eurasia, including France [25], Germany [24], Finland [19], Switzerland [20], China [15], and several regions across Russia [12,14,26]. In Russia alone, ALSV is circulating across territories where more than ten million people live. Currently, there is no systematic ALSV surveillance in Russia, so there is the possibility that ALSV might be more widely distributed than the data demonstrate [12]. There are various tick-borne pathogens, such TBEV, Omsk hemorrhagic fever virus (OHFV), Powassan virus (POWV), and Louping ill virus (LIV), circulating in Russia. Recently, another member of the JMV group, YGTV, was detected in different tick species in the territory where ALSV is circulating [26]. TBEV was also found in the same location as ALSV and YGTV, indicating the possibility of coinfection between classical members of the genus Flavivirus and the JMV group [26]. It should be noted, that clinical manifestation of the ALSV is similar to the TBEV infection [15], making differentiation between two viruses in the sympatric areas important for patient diagnosis, prognosis and treatment.
Considering all the above mentioned factors, there is a requirement for a tool designed for a quick and specific detection of ALSV. The current approaches for ALSV detection in ticks mostly include RT-PCR assays, followed by result confirmation with sequencing [12,26,27,28]. Sometimes, high throughput sequencing is used [28]. In two studies, a serological screening based on the purified VP2 protein, as well as RT-qPCR, were used [23,24]. However, such assays were never proved to not provide false-positive results with a wide array of co-circulating viruses. Here, we present a sensitive and specific RT-qPCR assay, which can be used to detect virus both in ticks and in human serum.
2. Materials and Methods
3.1. Virus-containing materials, serum and ticks
The JMV group members, the ALSV strains Miass519 and Miass527 [14] and YGTV strains Plast-T22438 and Bredy-T22181 [26], described previously were used in the current work. Several members of the genus Flavivirus from the institute collection were used: TBEV (strains Sofjin, EK-328, Absettarov), LIV strain S1, POWV strain Pow-24, West Nile virus (WNV) strain SHUA-3, Japanese encephalitis virus (JEV) strain Gagar, and Dengue-4 strain Cambodja. Kemerovo virus (KEMV) strain 21/10 (Sedoreoviridae, Orbivirus) from the institute collection was used. The full virus list, the system where the virus was replicated, and the amount of virus amount in plaque-forming units per ml (where possible) are presented in Table 1.
Poliovirus strain Sabin (104 copies/μl) from the institute collection was used as the internal control in the study (see below).
To test the specificity of the assay, pools of the various species of field-collected ticks were used. Tick species, pool composition, collection sites, and collection dates are presented in Table 2. Before RNA isolation, ticks were homogenized using the TissueLyser II (QIAGEN, Germany) in 0.9% saline solution (FSASI Chumakov FSC R&D IBP RAS, Moscow, Russia). The volume of the solution added was dependent on the tick’s species and the number of ticks in the pool: for each Ixodes spp. and Haemaphysalis spp. tick, 150 µL of solution was added; for each Dermacentor spp. tick, 200 µL of solution was added.
Additionally, ten negative human sera samples received previously during clinical trials of Tick-E-Vac vaccine [31], and a sheep serum (Gibco, New Zealand) were used in the study.
3.2. RNA isolation
Total RNA was isolated from the probes using TRI-reagent LS (Sigma-Aldrich, St. Louis, MO, USA), according to the manufacturer’s instructions. Briefly, 375 μl of the TRI-reagent LS was added to 125 μl of the sample, and then 100 μl of the chloroform was added. After that, the mix was shaken up by hand for 2 minutes, left at room temperature for 15 minutes, and centrifuged at 12000 g for 15 minutes using the Eppendorf 5424 centrifuge (Eppendorf). After that, the water phase (on top) was transferred in the fresh tube with the addition of 250 μl of water; the tube was gently mixed, left at the room temperature for 10 minutes, and centrifuged at 12000 g for 8 minutes using the Eppendorf 5424 centrifuge (Eppendorf). After that, the supernatant was discarded, 1 ml of the 80% ethanol was added, and the samples were centrifuged at 7500 g for 5 minutes. Then, ethanol was discarded from the probe and the precipitate was dried in the thermostat at 37°C for 15 minutes. After the procedure, RNA was dissolved in the water and it was either used instantaneously, for a downstream application, or was stored at −70°C.
3.3. Preparation of the standard RNA samples
Standard RNA samples were prepared as follows. First, a 1381 nt fragment of the segment 2 of strain Miass527 was amplified with oligonucleotides (Table 3), with the T7 promoter merging to the forward primer. The obtained PCR product was gel purified using the QIAGEN gel extraction kit (QIAGEN, Germany). The purified PCR products were used for in vitro transcription using T7 RNA polymerase (Sibenzime, Russia), according to the manufacturer’s instructions. The obtained RNA was purified in the sucrose density gradient (5-20%) using Optima L-90K Ultracentrifuge with SW-40 rotor at 40,000 rpm for 4h under 4°C. The resultant density gradient fractions were screened for their RNA amount using agarose gel electrophoresis, and the RNA was precipitated from the fraction with the highest amount of RNA as described in Section 3.2.
The amount of obtained pure RNA was measured using NanoDrop OneC (ThermoFischer Scientific). Using the formulae displayed in Figure 1, the RNA molecule quantity was calculated, ten-fold dilutions of the RNA in water were prepared, and RNA was stored at-70°C for the downstream applications.
3.3. Preparation of porcine embryo kidney total RNA
To isolate the total RNA of the porcine embryo kidney (PEK), 750 μl of TRI-reagent LS (Sigma-Aldrich, St. Louis, MO, USA) was added to the one-day PEK cells’ monolayer growing on the 25 cm2 cell culture flask (Corning), with the cell supernatant being discarded before the procedure. The TRI-reagent LS was then used to lyse the PEK cells; the obtained mixture was used to proceed with the RNA isolation protocol described in Section 3.2.
Purified RNA was dissolved in 50 μl of water, the RNA concentration was measured using NanoDrop OneC (ThermoFischer Scientific), and it was subsequently aliquoted to 250 ng/μl and stored at −70°C.
3.4. Reverse transcription and qPCR
Prior to the RNA isolation procedure followed for qPCR, 1 μg of the PEK cells RNA (see Section 3.3) was added to each sample to normalize the amount of RNA between probes. Additionally, 2 μl of the poliovirus strain Sabin (104 copies/μl) was added to each probe as an internal control. The total RNA was isolated from the probes using TRI-reagent LS (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions (see above).
Reverse transcription was carried out from the total RNA using MMLV Reverse Transcriptase (Evrogen JSC, Moscow, Russia) according to the manufacturer’s instructions. Briefly, RNA was dissolved in 9 μl of water and 2 μl of Miass_gly_3R (5 pmol/μl) oligonucleotide was added (Table 4). The mix was incubated for 2 minutes at 70°C and then placed on ice for 2 min. After that, 4 μl of 5x reaction buffer (Evrogen JSC, Moscow, Russia), 2 μl of DTT (Evrogen JSC, Moscow, Russia), 2 μl of 10mM dNTP (Evrogen JSC, Moscow, Russia), and 1 μl of MMLV Revertase (Evrogen JSC, Moscow, Russia) was added in the reaction mix, and the mix was incubated at 42°C for 1 hour. Reverse transcription for the internal control was conducted using the same approach, with a specific PVR1 oligonucleotide being used (Table S1).
qPCR was performed using the R-412 qPCR reaction kit (Syntol, Moscow, Russia) according to the manufacturer’s instructions. Briefly, a mix containing 2,5 μl 2,5 mM dNTP, 2,5 μl of 10x buffer, 2,5 μl 25 mM MgCl2 , 2 μl of forward and reverse oligonucleotides, 1 μl of qPCR probe, 0,25 μl of SynTaq polymerase and 10,25 μl of H2O was prepared (all reagents by Syntol, Moscow, Russia). Then, 2 μl of sample was added and probes were placed in C1000 Thermal Cycler (Bio-Rad, Hercules, CA, USA). Fluorescence detection was carried out by the CFX96 Real-Time System (Bio-Rad, Hercules, CA, USA). For ALSV detection, Miass_gly_3F and Miass_gly_3R oligonucleotides with Miass_gly3_PROBE probe (Table 4) were used. For the internal poliovirus control qPCR, PVL1 and PVR1 oligonucleotides with PVP1 probe were used (Table S1).The exact amplification cycles for ALSV and the internal poliovirus control are presented in Table S3 – S4.
The obtained amplification data were analyzed using Bio-Rad CFX Manager v.3.1 (Bio-Rad, Hercules, CA, USA), using the “Single Threshold” Cq Determination mode, “Baseline subtracted Curve Fit”, and “Apply Fluorescence Drift Correction” baseline settings, and other settings were used as default.
3. Results
Previously, Miass_gly_3F/Miass_gly_3R oligonucleotide pair (Table 4) was first designed to detect the ALSV strain Miass527 in the IRE/CTVM19 cell culture [14]. It was targeting the VP1a putative envelope protein [11] and amplified a sequence of 333 nt in length. Subsequently, this pair showed good results in screening ticks for the presence of ALSV, allowing us to discover more than 40 different ALSV isolates in ticks from various parts of Russia [12,14,26]. Here we further improved the detection system by designing the oligonucleotide probe Miass_gly3_PROBE (Table 4). This allowed for a less expensive and labor-intensive system, whilst at the same time preserving its specificity and sensitivity, as well as allowing a quantification of ALSV in the sample. Additionally, system was supplemented with a poliovirus internal control qPCR to prevent false-negative results due to mistakes during RNA isolation and reverse transcription procedures.
We tested the sensitivity of the assay by preparing standard RNA samples of the fragment of segment 2 (Section 3.3) and using ten-fold RNA dilutions in a qPCR. The quantification cycle (Cq) for the standard samples ranged from 29.7 (104 copies of RNA) to 16.4 (108 copies of RNA). The lower amount of RNA in the probe showed no amplification. The linear dependence between the amount of RNA copies and Cq had a R2 value of 0.997 (Figure 2). This indicates that this system may be used in conjunction with standards to calculate the virus load in the sample.
Since 104 copies of RNA was the lowest amount able to provide a signal, in further work a sample was considered “positive” if the calculated amount of the virus cDNA in the sample was higher than 104. Otherwise, amplification was considered to be non-specific.
We tested the specificity of the qPCR system to several possible targets. All samples mentioned below were tested to amplify internal control poliovirus RNA that was added to the sample before isolation. First, we tested the system with two strains of the ALSV: Miass519 and Miass527. qPCR system was able to successfully detect both viruses in the samples (Table 5). To test the ability of the system to detect ALSV in the serum samples, we used human and sheep sera. Both sera were tested with and without spiking them with 25 μl of the ALSV strain Miass519. We were able to successfully detect ALSV in the spiked serums (Table 5). In ten samples of non-spiked human sera, as well as in the unspiked sheep serum, no amplification was detected.
Additionally, we used a panel of the tick-borne flaviviruses, including three most common genotypes of TBEV, POWV, OHFV, and LIV. We report no detectable amplification of those viruses by our system.
The second panel included several other viruses, including highly relevant mosquito-borne flaviviruses: Japanese encephalitis virus, Dengue 4 virus, and West Nile virus. Additionally, we used two strains of YGTV, a close relative of the ALSV and Kemerovo virus, a tick-borne orbivirus isolated in Russia from I. persulcatus ticks. We report no detectable amplification of those viruses by our system. It should be noted, we detected no amplification in viruses replicated in PEK cells, Vero cell and mouse brain (Table 1). Since these samples contain also a host RNA, and we detected no amplification, we can conclude that no amplification would be detected in pure PEK cells, Vero cell and mouse brain as well.
One of the most predominant procedures used in epidemiological studies is testing collected ticks for the presence of the virus. Thus, we tested our system specificity against two of the most common tick species in Russia, Ixodes ricinus and I. persulcatus, as well as several other ticks that can be found in Russian territories: Dermacentor reticulatus, D. marginatus, Haemaphysalis concinna, and H. japonica. The ticks used in the experiment were studied in pools of two to six specimens in the sample and were previously tested for the absence of the flaviviruses, ALSV, and YGTV using RT-PCR. No detectable amplification was detected in all tick samples
Overall, no ALSV-free probes produced amplification signal higher than 104 copies threshold that we established earlier for this system (Table 5). This shows that qPCR system, together with standard RNA, does not give false-positive reactions in any systems tested in the article. At the same time, qPCR system successfully detected ALSV in two virus strains from the laboratory collection, as well as in serums spiked with ASLV.
4. Discussion
ALSV is spread across Europe [19,20,25], China [15,23], and throughout several regions in Russia [12,14,26]. Throughout Russian territory, ALSV is actively co-circulating with TBEV in the Republic of Karelia [26,32], Chelyabinsk Region [12,26] and the Republic of Tuva [12,33], which is leading to the possibility of co-infections. Currently, little is known about the biology of ALSV, as both the host range and pathogenic potential of the virus are not precisely known [15]. In order to estimate the epidemiological importance of ALSV, we need to accurately measure the presence of the virus in ticks, cattle, and humans.
This situation creates a necessity to develop a quick and easy system for the detection of ALSV. Currently, in the available scientific papers, ALSV is mostly surveyed using RT-PCR, followed by Sanger sequencing [12,26,27,28]. In one study, immunofluorescence assays and SYBR Green RT-qPCR were used. Interestingly, the prevalence of ALSV reported by the ELISA method was much lower than that reported by RT-qPCR [23]. Researchers believe that this situation is the result of collecting serum from young animals (less than one year old) in the beginning epidemic season of ticks [23]. However, there are no data on the specificity and sensitivity of the abovementioned assays, so it is possible that this is a result of the higher sensitivity of RT-qPCR or its low specificity. Overall, this highlights the necessity for the assays with proven specificity, especially if research is conducted in co-circulation areas.
It is also important to note, that many RT-PCR and RT-qPCR systems available today are mostly use oligonucleotides specific to the virus polymerase or helicase [15,23,24]. These proteins are very conservative, with high homology not only among the JMV group, but genus Flavivirus as well. This leads to the situation, where cross-reactions within JMV group and genus Flavivirus are possible. For example in some cases, genus Flavivirus polymerase-specific oligonucleotides [34], with very limited homology to JMV group, are able to amplify ALSV and YGTV [14]. Here we used a different approach by using oligonucleotides targeting VP1a protein, with very limited homology to genus Flavivirus and lower homology within JMV group.
During testing, our qPCR system amplified two different strains of ALSV, and did not amplify YGTV, three most common genotypes of the TBEV, and several other flaviviruses (both tick-borne and mosquito-borne) that can cause coinfection in Russia. Our system also produced no amplification signals in ALSV-free I. ricinus, I. persulcatus, D. reticulatus, D. marginatus, H. concinna, and H. japonica, as well as in ALSV-free sheep and human seraThese data highlights that system can be used for differential detection of the ALSV in various different systems. JMV group members are often isolated in the I. ricinus and Hyalomma anatolicum derived cell cultures [12,14,26], with many more tick-derived cell cultures availiable [29,30]. We can assume that qPCR system would also give no non-specific amplification tick cell lines derived from I. ricinus, I. persulcatus, D. reticulatus, D. marginatus, H. concinna, and H. japonica ticks, since system reported no amplification signal in ticks that cultures derive from.
Overall, the qPCR system presented here can detect ALSV in ticks, the blood serum of humans and animals, and a variety of common cell cultures, making it a useful tool for human diagnostics, epidemiological studies of ticks, and laboratory experiments involving ALSV. One additional benefit of our system is that with an amplicon length of 333 nt, Sanger sequencing can be used to confirm a positive result if necessary. Our system is designed to have a poliovirus internal control qPCR, as well as PEK cells RNA for the normalization of the RNA amount in the samples. It was designed this way based on the availability of these exact reagents in our lab. However they can be easily replaced with RNA from other sources if needed since qPCR assay shown high specificity.
For TBEV, virus loads in the serum are not associated with the patients’ clinical parameters, such as duration of the first phase of the disease, duration of the asymptomatic interval, TBE severity, and clinical presentation [35]. However, higher virus loads in plasma predicted a development the development of symptoms for a WNV infection [36,37]. Currently, there is no data on the importance of a virus load for ALSV infection course, but that data can be obtained using our system.
TBEV, as a member of the genus Flavivirus, shares some homology with JMV members [9,10,11], ALSV in particular. TBEV testing systems were designed before the discovery of the JMV group, so the specificity of those systems has not been tested on ALSV yet. According to the sanitary rules in Russia, when a potential patient brings tick to a laboratory, if the tick is positive for TBEV RNA, a specific immunoglobulin is administered [5]. However, in case of false-positive identification of ALSV as TBEV, immunoglobulin prescription would be useless.
Taking into account ALSV and TBEV co-circulation and homology, in order to obtain accurate epidemiological data on both ALSV and TBEV, reliable systems for the detection of both viruses are needed. Our work describes a specific system for ALSV detection. However, further research is required in order to test the specificity and, if necessary, improve the existing TBEV detection systems.
Supplementary Materials
Table S1: Oligonucleotides used for internal control Poliovirus RT-qPCR; Table S2: Amplification cycle for ALSV qPCR; Table S3: Amplification cycle for Poliovirus qPCR.
Author Contributions
Conceptualization, A.G.L. and G.G.K.; methodology, A.G.L.; validation, A.G.L.; investigation, A.G.L., E.V.O., I.S.K and A.E.P.; resources, A.G.L. and G.G.K; writing—original draft preparation, A.G.L.; writing—review and editing, A.G.L., I.S.K and G.G.K.; supervision, G.G.K.; project administration, G.G.K.; funding acquisition, A.G.L. and G.G.K All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, Grant no. 21-74-00083.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lindenbach, B.D.; Thiel, H.-J.; Rice, C.M. Flaviviridae: The Viruses and Their Replication. In Fields Virology, 5th Edition.; Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven Publishers: Philadelphia, 2007; pp. 1101–1151; ISBN 0781760607. [Google Scholar]
- Rossi, S.L.; Ross, T.M.; Evans, J.D. West Nile virus. Clin. Lab. Med. 2010, 30, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Whitehorn, J.; Simmons, C.P. The pathogenesis of dengue. Vaccine 2011, 29, 7221–8. [Google Scholar] [CrossRef] [PubMed]
- Wikan, N.; Smith, D.R. Zika virus: History of a newly emerging arbovirus. Lancet Infect. Dis. 2016, 16, e119–e126. [Google Scholar] [CrossRef] [PubMed]
- Ruzek, D.; Av, T.; Borde, J.; Chrdle, A.; Eyer, L.; Karganova, G.; Kholodilov, I.; Kozlovskaya, L.; Matveev, A.; Miller, A.D.; Osolodkin, D.I.; Överby, A.K.; Tikunova, N.; Tkachev, S.; Zajkowska, J. Tick-borne encephalitis in Europe and Russia : Review of pathogenesis, clinical features, therapy, and vaccines. Antiviral Res. 2019, 164, 23–51. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shi, M.; Tian, J.; Lin, X.; Kang, Y.; Chen, L.; Qin, X.; Xu, J.; Holmes, E.C.; Zhang, Y. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.; Tian, J.; Chen, L.; Chen, X.; Li, C.; Qin, X.; Li, J.; Cao, J.; Eden, J.; Buchmann, J.; Wang, W.; Xu, J.; Holmes, E.C.; Zhang, Y. Redefining the invertebrate RNA virosphere. Nature 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, S.; Ka, S.; Zirkel, F.; Donath, A.; Petersen, M.; Liu, S.; Zhou, X.; Drosten, C.; Misof, B.; Junglen, S. Viromics of extant insect orders unveil the evolution of the flavi-like superfamily. Virus 2021, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.-C.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Gao, D.-Y.; He, J.-R.; Wang, J.-B.; Li, C.-X.; Kang, Y.-J.; Yu, B.; Zhou, D.-J.; Xu, J.; Plyusnin, A.; Holmes, E.C.; Zhang, Y.-Z. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl. Acad. Sci. 2014, 111, 6744–6749. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Li, Z.; Wang, M.; Cui, S. Crystal structure of the NS3-like helicase from Alongshan virus. IUCrJ. 2020, 7, 375–382. [Google Scholar] [CrossRef]
- Domains, M.; Garry, C.E.; Garry, R.F. Proteomics Computational Analyses Suggest That the Envelope Glycoproteins of Segmented Jingmen Flavi-Like Viruses are Class II Viral Fusion Proteins. Viruses 2020, 12. [Google Scholar]
- Kholodilov, I.S.; Belova, O.A.; Morozkin, E.S.; Litov, A.G.; Ivannikova, A.Y.; Makenov, M.T.; Shchetinin, A.M.; Aibulatov, S.V.; Bazarova, G.K.; Bell-sakyi, L.; Bespyatova, L.A.; Bugmyrin, S.V.; Chernetsov, N.; Chernokhaeva, L.L.; Gmyl, L.V.; Khaisarova, A.N.; Khalin, A.V.; Klimentov, A.S.; Kovalchuk, I.V.; Luchinina, S.V.; Medvedev, S.G.; Nafeev, A.A.; Oorzhak, N.D.; Panjukova, E.V.; Polienko, A.E.; Purmak, K.A.; Romanenko, E.N.; Rozhdestvenskiy, E.N.; Saryglar, A.A.; Shamsutdinov, A.F.; Karganova, G.G. Geographical and Tick-Dependent Distribution of Flavi-Like Alongshan and Yanggou Tick Viruses in Russia. Viruses 2021, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ladner, J.T.; Wiley, M.R.; Beitzel, B.; Kramer, L.D.; Tesh, R.B.; Palacios, G.; Ladner, J.T.; Wiley, M.R.; Beitzel, B.; Auguste, A.J.; Ii, A.P.D.; Lindquist, M.E.; Sibley, S.D.; Kota, K.P.; Fetterer, D.; Eastwood, G.; Kimmel, D.; Prieto, K.; Guzman, H.; Aliota, M.T.; Reyes, D.; Brueggemann, E.E.; John, L.S.; Hyeroba, D.; Lauck, M. A Multicomponent Animal Virus Isolated from Mosquitoes. Cell Host Microbe 2016, 20, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Kholodilov, I.S.; Litov, A.G.; Klimentov, A.S.; Belova, O.A.; Polienko, A.E.; Nikitin, N.A.; Shchetinin, A.M.; Ivannikova, A.Y.; Bell-sakyi, L.; Yakovlev, A.S.; Bugmyrin, S.V.; Bespyatova, L.A.; Gmyl, L.V.; Luchinina, S.V.; Gmyl, A.P.; Gushchin, V.A.; Karganova, G.G. Isolation and Characterisation of Alongshan Virus in Russia. Viruses 2020, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-D.; Wang, B.; Wei, F.; Han, S.-Z.; Zhang, L.; Yang, Z.-T.; Yan, Y.; Lv, X.-L.; Li, L.; Wang, S.-C.; Song, M.-X.; Zhang, H.-J.; Huang, S.-J.; Chen, J.; Huang, F.-Q.; Li, S.; Liu, H.-H.; Hong, J.; Jin, Y.-L.; Wang, W.; Zhou, J.-Y.; Liu, Q. A New Segmented Virus Associated with Human Febrile Illness in China. N. Engl. J. Med. 2019, 380, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.; Kuwata, R.; Kimura, T.; Shimoda, H.; Fujita, R.; Faizah, A.N.; Kai, I.; Matsumura, R.; Kuroda, Y.; Watanabe, S.; Kuniyoshi, S.; Yamauchi, T.; Watanabe, M.; Higa, Y.; Hayashi, T.; Shinomiya, H.; Maeda, K.; Kasai, S.; Sawabe, K.; Isawa, H. Detection of Jingmenviruses in Japan with Evidence of Vertical Transmission in Ticks. Viruses 2021, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.; Vasilakis, N.; Tian, J.; Li, C.; Chen, L.; Eastwood, G.; Diao, X.; Chen, M.-H.; Chen, X.; Qin, X.-C.; Widen, S.G.; Wood, T.G.; Tesh, R.B.; Xu, J.; Holmes, E.C.; Zhanga, Y.-Z. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J. Virol. 2016, 90, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.C.; Maruyama, S.R.; de Miranda-Santos, I.K.F.; Palacios, G.; Ladner, J.T. Complete Coding Genome Sequence for Mogiana Tick Virus, a Jingmenvirus Isolated from Ticks in Brazil. Genome Announc. 2017, 5, 17–18. [Google Scholar] [CrossRef]
- Kuivanen, S.; Levanov, L.; Kareinen, L.; Sironen, T.; Jääskeläinen, A.J.; Plyusnin, I.; Zakham, F. Detection of novel tick-borne pathogen, Alongshan virus, in Ixodes ricinus ticks, south-eastern Finland. Eurosurveillance 2019, 24. [Google Scholar] [CrossRef]
- Stefanie, Stegmüller; Fraefel, C.; Kubacki, J. 20. Stefanie Stegmüller; Fraefel, C.; Kubacki, J. Genome Sequence of Alongshan Virus from Ixodes ricinus Ticks Collected in Switzerland. Microbiol. Resour. Announc. 2023, 2022–2023. [Google Scholar]
- Jia, N.; Liu, H.B.; Ni, X.B.; Bell-Sakyi, L.; Zheng, Y.C.; Song, J.L.; Li, J.; Jiang, B.G.; Wang, Q.; Sun, Y.; Wei, R.; Yuan, T.T.; Xia, L.Y.; Chu, Y.L.; Wei, W.; Li, L.F.; Ye, J.L.; Lv, Q.Y.; Cui, X.M.; Guan, Y.; Tong, Y.G.; Jiang, J.F.; Lam, T.T.Y.; Cao, W.C. Emergence of human infection with Jingmen tick virus in China: A retrospective study. EBioMedicine 2019, 43, 317–324. [Google Scholar] [CrossRef]
- Colmant, A.M.G.; Charrel, R.N.; Coutard, B. Jingmenviruses : Ubiquitous, understudied, segmented flavi-like viruses. Front. Microbiol. 2022, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-D.; Wang, W.; Wang, N.N.; Qiu, K.; Zhang, X.; Tana, G.; Liu, Q. Prevalence of the emerging novel Alongshan virus infection in sheep and cattle in Inner Mongolia, northeastern China. Parasit. Vectors 2019, 12, 1–7. [Google Scholar] [CrossRef]
- Ebert, C.L.; Söder, L.; Kubinski, M.; Glanz, J.; Gregersen, E.; Dümmer, K.; Grund, D.; Wöhler, A.; Könenkamp, L.; Liebig, K.; Knoll, S.; Hellhammer, F.; Topp, A.; Becher, P.; Springer, A.; Strube, C.; Nagel-kohl, U.; Nordhoff, M.; Steffen, I.; Bauer, B.U.; Ganter, M.; Feige, K.; Becker, S.C.; Boelke, M. Detection and Characterization of Alongshan Virus in Ticks and Tick Saliva from Lower Saxony, Germany with Serological Evidence for Viral Transmission to Game and Domestic Animals. Microorganisms 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Temmam, S.; Bigot, T.; Chrétien, D.; Gondard, M.; Pérot, P.; Pommelet, V.; Dufour, E.; Petres, S.; Devillers, E.; Hoem, T.; Pinarello, V.; Hul, V.; Vongphayloth, K.; Hertz, J.C.; Loiseau, I.; Dumarest, M.; Duong, V.; Vayssier-Taussat, M.; Grandadam, M.; Albina, E.; Dussart, P.; Moutailler, S.; Cappelle, J.; Brey, P.T.; Eloit, M. Insights into the Host Range, Genetic Diversity, and Geographical Distribution of Jingmenviruses. mSphere 2019, 4, e00645–19. [Google Scholar] [CrossRef] [PubMed]
- Kholodilov, I.S.; Belova, O.A.; Ivannikova, A.Y.; Gadzhikurbanov, M.N.; Makenov, M.T.; Yakovlev, A.S.; Polienko, A.E.; Dereventsova, A.V.; Litov, A.G.; Gmyl, L.V.; Okhezin, E.V.; Luchinina, S.V.; Klimentov, A.S.; Karganova, G.G. Distribution and Characterisation of Tick-Borne Flavi-, Flavi-like, and Phenuiviruses in the Chelyabinsk Region of Russia. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Yu, Z.-M.; Chen, J.-T.; Qin, J.; Guo, J.-J.; Li, K.; Xu, Q.-Y.; Wang, W.; Lu, M.; Qin, X.-C.; Zhang, Y.-Z. Identification and characterization of Jingmen tick virus in rodents from Xinjiang, China. Infect. Genet. Evol. 2020, 84, 1–7. [Google Scholar] [CrossRef]
- Cai, X.; Cai, X.; Xu, Y.; Shao, Y.; Fu, L.; Men, X.; Zhu, Y. Virome analysis of ticks and tick-borne viruses in Heilongjiang and Jilin. Virus Res. 2023, 323, 199006. [Google Scholar] [CrossRef]
- Bell-Sakyi, L. Continuous cell lines from the tick Hyalomma anatolicum anatolicum. J. Parasitol. 1991, 77, 1006–1008. [Google Scholar] [CrossRef]
- Bell-Sakyi, L.; Zweygarth, E.; Blouin, E.F.; Gould, E.A.; Jongejan, F. Tick cell lines: tools for tick and tick-borne disease research. Trends Parasitol. 2007, 23, 450–457. [Google Scholar] [CrossRef]
- Maikova, G.B.; Chernokhaeva, L.L.; Rogova, Y.V.; Kozlovskaya, L.I.; Kholodilov, I.S.; Romanenko, V.V.; Esyunina, M.S.; Ankudinova, A.A.; Kilyachina, A.S.; Vorovitch, M.F.; Karganova, G.G. Ability of inactivated vaccines based on far-eastern tick-borne encephalitis virus strains to induce humoral immune response in originally seropositive and seronegative recipients. J. Med. Virol. 2019, 91, 190–200. [Google Scholar] [CrossRef]
- Bugmyrin, S.V.; Romanova, L.Y.; Belova, O.A.; Kholodilov, I.S.; Bespyatova, L.A.; Chernokhaeva, L.L.; Gmyl, L.V.; Klimentov, A.S.; Ivannikova, A.Y.; Polienko, A.E.; Yakovlev, A.S.; Ieshko, E.P.; Gmyl, A.P.; Karganova, G.G. Pathogens in Ixodes persulcatus and Ixodes ricinus ticks ( Acari, Ixodidae ) in Karelia ( Russia ). Ticks Tick. Borne. Dis. 2022, 13, 102045. [Google Scholar] [CrossRef] [PubMed]
- Kholodilov, I.; Belova, O.; Burenkova, L.; Korotkov, Y.; Romanova, L.; Morozova, L.; Kudriavtsev, V.; Gmyl, L.; Belyaletdinova, I.; Chumakov, A.; Chumakova, N.; Dargyn, O.; Galatsevich, N.; Gmyl, A.; Mikhailov, M.; Oorzhak, N.; Polienko, A.; Saryglar, A.; Volok, V.; Yakovlev, A.; Karganova, G. Ixodid ticks and tick-borne encephalitis virus prevalence in the South Asian part of Russia (Republic of Tuva). Ticks Tick. Borne. Dis. 2019, 10, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Scaramozzino, N.; Crance, J.; Jouan, A.; Briel, D.A.D.E. Comparison of Flavivirus Universal Primer Pairs and Development of a Rapid, Highly Sensitive Heminested Reverse Transcription-PCR Assay for Detection of Flaviviruses Targeted to a Conserved Region of the NS5 Gene Sequences. 2001, 39, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Saksida, A.; Jakopin, N.; Jelovšek, M.; Knap, N.; Fajs, L.; Lusa, L.; Lotri, S.; Bogovi, P.; Arnež, M.; Strle, F.; Avšič-županc, T. Virus RNA Load in Patients with Tick-borne Encephalitis, Slovenia. Emerg. Infect. Dis. 2018, 24, 1315–1323. [Google Scholar] [CrossRef]
- Zou, S.; Foster, G.A.; Dodd, R.Y.; Petersen, L.R.; Stramer, S.L. West Nile Fever Characteristics among Viremic Persons Identified through Blood Donor Screening. J. Infect. Dis. 2010, 20855, 1354–1361. [Google Scholar] [CrossRef]
- Brown, J.A.; Factor, D.L.; Tkachenko, N.; Templeton, S.M.; Crall, N.D.; Pape, W.J.; Bauer, M.J.; Ambruso, D.R.; Dickey, W.C.; Marfin, A.A. West Nile Viremic Blood Donors and Risk Factors for Subsequent West Nile Fever. VECTOR-BORNE ZOONOTIC Dis. 2007, 7, 479–488. [Google Scholar] [CrossRef]
Figure 1.
RNA molecule quantification formula.
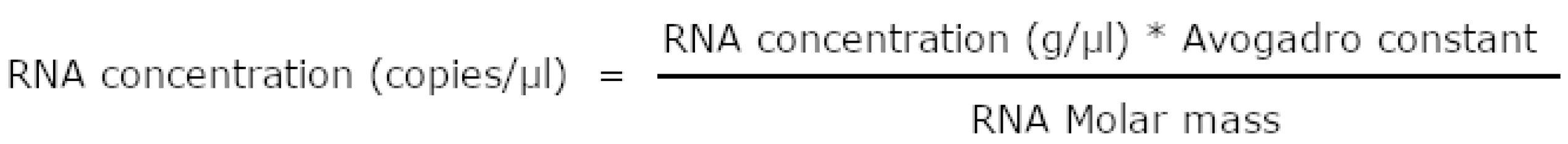
Figure 2.
Amplification curve of the standard RNA of known amount.
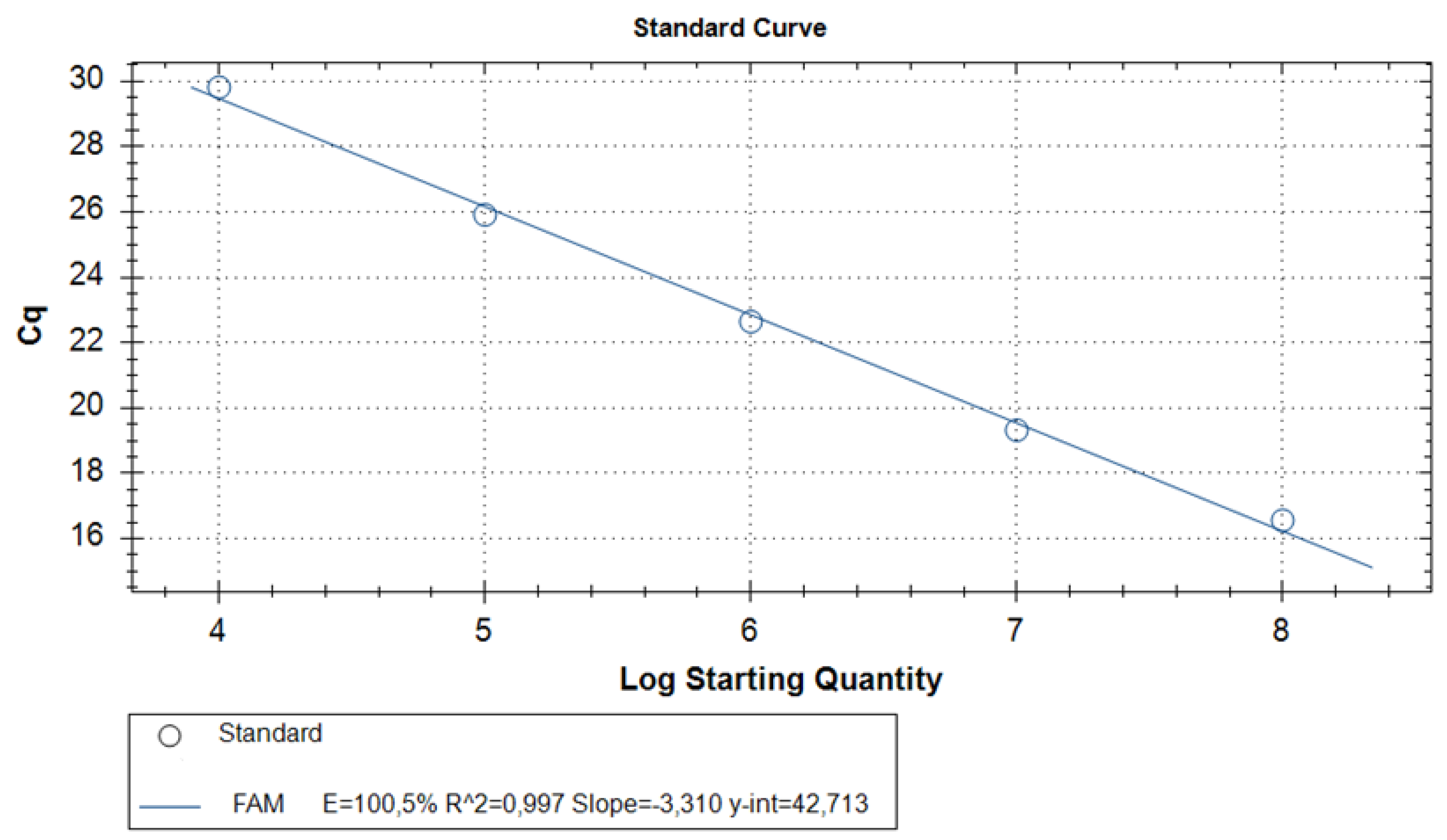

Table 1.
Viruses used for the testing of the qPCR system.
| Virus | Strain | Virus amount, PFU/ml | Virus Origin |
|---|---|---|---|
| ALSV | Miass519 | + 1 | HAE/CTVM8 2 cells |
| ALSV | Miass527 | + | IRE/CTVM19 3 cells |
| YGTV | Plast-T22438 | + | HAE/CTVM8 cells |
| YGTV | Bredy-T22181 | + | HAE/CTVM8 cells |
| TBEV | Sofjin | 6.7 | Mouse brain |
| TBEV | EK-328 | 7.1 | PEK 4 cells |
| TBEV | Absettarov | 9.5 | Mouse brain |
| OHFV | Nikitina | 7.6 | PEK cells |
| WNV | SHUA-3 | 7.7 | Vero cells |
| LIV | S1 | 6.2 | Mouse brain |
| POWV | Pow-24 | 7.6 | PEK cells |
| JEV | Gagar | 8.1 | PEK cells |
| Dengue-4 | Cambodja | + | Mouse brain |
| KEMV | KEM-21/10 | 7.1 | PEK cells |
Table 2.
Ticks used in the work.
| Tick species | Number of Ticks in a Pool |
Collection site | Collection Date |
|---|---|---|---|
| Ixodes ricinus | 6 ♂ | Russia, Kaliningrad Region | 2017 |
| Ixodes ricinus | 5 ♀ | Russia, Kaliningrad Region | 2017 |
| Ixodes persulatus | 5 ♀ | Russia, Primorsky Territory | 2021 |
| Dermacentor retuculatus | 3 ♀ | Russia, Chelyabinsk Region | 2015 |
| Dermacentor marginatus | 4 ♀ | Russia, Chelyabinsk Region | 2015 |
| Haemaphysalis conccina | 2 ♂ | Russia, Primorsky Territory | 2021 |
| Haemaphysalis japonica | 2 ♀ | Russia, Primorsky Territory | 2021 |
Table 3.
Oligonucleotides used for the standard RNA samples preparation.
| Oligonucleotide | Sequence |
|---|---|
| BHT7_Miass_VP1a_F3 | 5′-ATGACTGGATCCTAATACGACTCACTATAGGCTTGTAAAGCTAGCGACTGGA-3′ |
| Miass_gly_2R | 5′-AAAGCCTCATGGACGGTCTG-3′ |
Table 4.
Oligonucleotides used for ALSV-specific qPCR.
| Oligonucleotide | Sequence | Location |
|---|---|---|
| Miass_gly_3F | 5`-TGGATCAGCTCACACCACAC-3` | VP1a |
| Miass_gly_3R | 5`-TCACCGTCACAGTGGAATGG-3` | VP1a |
| Miass_gly3_PROBE | (FAM)-TTGCGACCCCGTTGTCGTCG-(BHQ-1) | VP1a |
Table 5.
Samples where amplification was detected during qPCR.
| Probe | Cq | Quantity | Detection result |
|---|---|---|---|
| 108 Standard RNA | 16.4±0.9 * | 108 | - |
| 107 Standard RNA | 19.0±0.6 * | 107 | - |
| 106 Standard RNA | 22.1±1.2 * | 106 | - |
| 105 Standard RNA | 25.5±1* | 105 | - |
| 104 Standard RNA | 29.7±0.9* | 104 | - |
| ALSV strain Miass519 | 15.8 | 9.7*107 | positive |
| ALSV strain Miass527 | 7.4 | 3.6*1010 | positive |
| Human serum, spiked with ALSV strain Miass519 | 18.5 | 1.7*107 | positive |
| Sheep serum, spiked with ALSV strain Miass519 | 19 | 1.2*107 | positive |
* Average Cq among several qPCR runs.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



