
Submitted:
12 May 2023
Posted:
15 May 2023
You are already at the latest version
Abstract
After eight decades, the largest outbreak of sylvatic yellow fever virus (YFV) was recorded in Brazil between 2016-2018. Besides human and NHP surveillance, the entomo-virological approach is considered as a complementary tool. For this study, a total of 2904 mosquitoes of Aedes, Haemagogus and Sabethes genera from six Brazilian states (Bahia, Goiás, Mato Grosso, Minas Gerais, Pará, and Tocantins), were collected and grouped in 246 pools, which were tested for YFV using RT-qPCR. We detected 20 positive pools from Minas Gerais, 5 from Goiás, and 1 from Bahia, including 12 of Hg. janthinomys and 5 of Ae. albopictus. This is the first description of natural YFV infection in this species and warns of the likelihood of urban YFV re-emergence with the Ae. albopictus as a potential bridge vector. Three YFV sequences from Hg. janthinomys from Goiás and one from Minas Gerais, as well as one from Ae. albopictus from Minas Gerais clustered within the 2016-2018 outbreak clade, indicating YFV spread from Midwest and its infection in a main and in a likely-novel bridging vector species. Entomo-virological surveillance is critical in the YFV monitoring in Brazil, which could highlight the need to strengthen YFV surveillance, vaccination coverage, and vector control measures.
Keywords:
yellow fever virus
; vectors
; Aedes albopictus
; outbreak
; entomo-virological surveillance
1. Introduction
The yellow fever virus (YFV), included in the Orthoflavivirus flavi species, the prototype virus of the Orthoflavivirus genus and Flaviviridae family [1], is an endemic arbovirus in tropical and subtropical countries. In Brazil, YFV is historically maintained through urban and sylvatic transmission cycles. The urban cycle, which had not been recorded in Brazil since 1942 and in the Americas (Trinidad and Tobago) since 1954, involved transmission between Aedes aegypti mosquitoes, the main vector, and humans. In the sylvatic cycle, Haemagogus and Sabethes genera mosquitoes act as vectors and non-human primates (NHP) as amplifier hosts, with humans occasionally serving as unintentional hosts [2].
Despite the absence of urban cases due to the brief eradication of Ae. aegypti in Brazil until the 1960s, the virus remained endemic in forest areas in Northern and Midwestern Brazil (Amazon Forest and Cerrado, respectively) and is occasionally responsible for epizootics and human cases of yellow fever (YF) [2,3].
The overgrowing human action in natural landscapes provokes an imbalance in ecosystem dynamics, exposing hosts, vectors and known and unknown viruses to humans. In late 2016, the largest YF outbreak in Brazil in eight decades began in the countryside of Minas Gerais state and swiftly spread to other states of the Southeast region, which is the most populated in the country and is within the Atlantic Forest biome. Until then, this region, which registered 1865 human cases and 744 total deaths between 2016 and 2018, was not included under the Brazilian YFV vaccination program [4,5].
Since NHPs are used as sentinels in the sylvatic YFV surveillance due to their high susceptibility to the virus, the occurrence of epizootics implies viral circulation. It warns of the urgency to strengthen prevention and control measures, such as improving vaccination coverage in the area. The late detection and response could end in a severe outbreak, as the recently registered in Brazilian Atlantic Forest [4,6-8].
Entomo-virological surveillance can also be used as an important tool for the early detection of viral circulation and to set an epidemiological link to epizootics and human cases in investigation. Whereas entomological collection is focused on main and potential vectors of YFV in a specific region, viral detection in mosquitoes helps to foresee the escalation of an outbreak and could define the possible vector implicated in YFV transmission [7-10].
The possibility of urban YFV re-emergence in Brazil is feasible due to the YFV’s current dispersion to almost the entire country. In this scenario, sylvatic vectors are closer to urban areas and opportunistic mosquitoes with vectorial potential could act as bridge vectors between the two transmission cycles [11,12], reinforcing the importance of implementing entomo-virological surveillance. Here we present a retrospective study of YFV genomic investigation in 2904 Aedes, Haemagogus and Sabethes genera mosquitoes, grouped in 246 pools and collected by the entomo-virological surveillance of the Brazilian Ministry of Health in six Brazilian states between 2016 and 2017.
2. Material and Methods
2.1. Mosquitoes Collection and Taxonomic Identification
The mosquitos’ samples used in this study came from the YFV entomo-virological surveillance of the Brazilian Ministry of Health. They were collected in six states (Bahia, Goiás, Mato Grosso, Minas Gerais, Pará, and Tocantins) from four of the five Brazilian regions between January 2016 and April 2017.
Aedes, Haemagogus and Sabethes genera mosquitoes were collected by human attraction using hand nets, a polyester net bag of 30 cm in diameter with a 30 cm aluminum handle commonly used by our entomological surveillance team. Collected mosquitoes were transferred by oral suction to identified cryotubes which were stored in liquid nitrogen at -196 °C and transported to the Department of Arbovirology and Hemorrhagic Fevers of the Evandro Chagas Institute prior to taxonomic identification and further analysis.
In a -20 °C refrigerated table and using a stereo microscope Stemi 2000-C (Zeiss, Oberkochen, Germany), mosquitoes were morphologically identified by dichotomous keys [13-18] and organized in pools with 1 to 30 specimens, based on species, date and site of collection.
2.2. Mosquitoes maceration
Each pool was eluted in 1 mL of a solution composed of 77 mL of 1X Dulbecco’s Phosphate Buffered Salino (Thermo Fisher Scientific, Waltham, MA, USA), 20 mL of Fetal Bovine Serum (Thermo Fisher Scientific), and 3 mL of an antibiotic solution of penicillin (100 U/mL), streptomycin (10 mg/mL) and fungizone (2.5 mg/mL). Then, a 3 mm tungsten bead was added to each pool, which was macerated using the TissueLyser II system (Qiagen, Hilden, Germany) for 2 min at 25 Hz.
2.3. RNA extraction
Pools were centrifuged at 13,000 x g for 10 min and 200 µL of supernatants were used for RNA extraction, performed with the Maxwell® 16 Viral Total Nucleic Acid Purification Kit (Promega, Madison, WI, USA) in the Maxwell® 16 System (Promega) instrument. Alternatively, the QIAamp viral RNA Kit (Qiagen) was used. Since these are mosquito’s samples, we used the Escherichia coli bacteriophage MS2 as a noncompetitive internal control RNA, which was added in a 2 µl volume in each sample.
2.4. Real-Time Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
The assay was performed using the QuantiTect® Probe RT-PCR (Qiagen) (Thermo Fisher Scientific) and specific primers and probe for the YFV 5’ untranslated region [19]. The 25 µL reaction was composed of 12.5 µL of a 2X QuantiTect Probe RT-PCR Master Mix, 5.75 µL of nuclease-free water, 0.5 µL of forward primer (YFallF, 5’-GCTAATTGAGGTGYATTGGTCTGC-3’), 0.5 µL of reverse primer (YFallR, 5’-CTGCTAATCGCTCAAMGAACG-3’), 0.5 µL of probe (YFallP, 5’-FAM-ATCGAGTTGCTAGGCAATAAACAC-TMR-3’), 0.25 µL of the QuantiTect RT Mix enzyme and 5 µL of extracted RNA.
For the noncompetitive internal control RNA detection, the 25 µl reaction had the same composition as the YFV assay, but with the followings set of primers and probe: MS2 forward (5’-CATAAGTTAGATGGCCGTCTGT-3’), MS2 reverse (5’-TAGAGACGACAACCATGCCAAAC-3’) and MS2 probe (5’-VIC- TCCAGACAACGTGCAACATATCGCGACGTATCGTGATATGG -BHQ1-3’) [20].
In a 7500 Fast Real-Time PCR system (Thermo Fisher Scientific), the RT-qPCR assays were performed under the following cycling conditions: an initial RT step at 50 °C for 30 min, a denaturation step at 95 °C for 2 min, 45 cycles of 15 s at 95 °C and a final extension step of 1 min at 60 °C. Each sample was analyzed in duplicate and considered as positive when the average cycle threshold (Ct) value was less than 37 for both assays. The assay was validated by positive (YFV infected mice brain tissue) and negative (nuclease-free water) controls.
2.5. Nucleotide Sequencing
In a joint initiative, the AR843690 sample, a pool of Aedes albopictus mosquitoes, was sequenced using the MinION® sequencing device (Oxford Nanopore Technologies, Oxford, Oxfordshire, UK) by the Oswaldo Cruz Foundation (FIOCRUZ). The genome assembly was also performed by FIOCRUZ following the methodology described in Giovanetti et al. [21].
Other samples were prepared for sequencing by synthesizing first and second strands of complementary DNA, which were obtained with the cDNA Synthesis System Kit (Roche Diagnostics, Basel, Switzerland) and 400 µM Roche random primer. Agencourt AMPure XP Reagent Kit (Beckman Coulter, Brea, CA, USA) magnetic beads were used for cDNA purification and Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) for cDNA library preparation. Quantification of cDNA was assessed using Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and fragments size range was evaluated using 2100 Bioanalyzer Instrument (Agilent Technologies, Santa Clara, CA, USA). Sequencing was performed on the MiniSeq platform (Illumina) using the MiniSeq High Output Kit (300 cycles).
2.6. Bioinformatic Analysis
Genome assembly was carried out through de novo methodology by IDBA-UD (k-mers 20, 40, 60, 80, and 100) [22] and SPAdes (k-mers 21, 33, 55, and 77) [23]. Contigs were merged at Lasergene SeqMan Pro software [24], and then aligned against National Center for Biotechnology Information (NCBI) RefSeq database by DIAMOND [25] with a 10-3 e-value threshold. The contigs were inspected with MEGAN6 [26] to identify those corresponding to YFV. Using Geneious v.9.1.8 software [27], contigs were inspected and mapped to reference (NC_002031), and then to raw data to increase coverage, both by Geneious Mapper. A multiple sequence alignment (MSA) of YFV complete genome was performed using Mafft v.7 [28]. The five YFV genomes obtained were compared to 82 YFV sequences from arthropods, humans, and NHP.
The phylogenetic inference by maximum likelihood (ML) analysis with 1000 bootstrap iterations [29] was performed using GTR+F+R2 as a substitution model defined by IQ-TREE v.2 [30]. The resulting tree was rooted at midpoint. Visualization was performed using FigTree v.1.4.4 [31] and Inkscape v.1.1 [32].
3. Results
3.1. Collection and Taxonomic Identification
A total of 2904 mosquitoes of Aedes, Haemagogus, and Sabethes genera were collected between January 2016 and April 2017 (Figure 1A) from six Brazilian states: Goiás and Mato Grosso (Midwest), Pará and Tocantins (North), Bahia (Northeast), and Minas Gerais (Southeast) (Figures 1B-1C). They were grouped in 246 pools.
Mosquitoes of Aedes genus were the most collected in 2016 and 2017, representing 2088 specimens distributed in 155 pools, mostly from Goiás (38 pools/684 specimens) and Pará (47 pools/954 specimens). Although Minas Gerais was the state with more grouped pools (63 pools), Pará had most specimens collected during this study, with samples dating only from 2017. On the other hand, Mato Grosso was the state with fewer mosquitoes sampled, which were only from Aedes genus. Goiás was the only state with mosquitoes in both years.
Mosquitoes from Haemagogus genus were organized in 50 pools with 620 specimens. Goiás was the only state from which Haemagogus mosquitoes were collected in 2016 (7 pools/128 specimens), and most of those collected in 2017 were from Minas Gerais (32 pools/272 specimens). From the Sabethes genus, 196 specimens were collected and grouped in 41 pools. Following a similar pattern as the Haemagogus genus, in 2016, Sabethes mosquitoes were collected only in Goiás (13 pools/88 specimens) and mostly in Minas Gerais in 2017 (16 pools/57 specimens). Goiás was the only state with no Sabethes mosquitoes collected in 2017. A description of each pool from 2016 and 2017 is available in Tables S1 and S2, respectively.
3.2. RT-qPCR Detection
The 246 samples were tested for YFV and noncompetitive internal control RNA by RT-qPCR, from which the YFV amplicon were detected (positive) in 26 (Table 1). The MS2 were detected in all samples, validating our assays.
Fom the 26 positive pools, five had mosquitoes collected in 2016, all from Jandaia (Goiás – Cerrado), and sampled over a two-day interval. The other 21 samples were from 2017 and only one was sampled in Bahia state (Cocos – Cerrado), while all the others were captured in 10 different municipalities from Minas Gerais state (Atlantic Forest), sampled over a 10-day interval (Figure 2).
The five Jandaia positive samples consist of four pools of Hg. janthinomys and one Sa. glaucodaemon, and their Ct value ranged from 18.27 to 31.27. The only positive pool from Cocos, a Hg. janthinomys pool, had specimens collected during three days in March 2017 and a Ct value of 28.63.
Minas Gerais was the state with most positive samples, with 20 pools collected in 2017, mainly from Alvarenga, with five positive pools, and Caratinga, and Ituêta, with three positive pools each. Their Ct value ranged from 23.88 (pool of Ae. albopictus from Ituêta) to 35.61 (pool of Ae. albopictus from São Domingos das Dores). The 20 samples consist of seven pools of Hg. janthinomys, five of Ae. albopictus (female), two of Ae. argyrothorax, two of Ae. scapularis, two of Ae. serratus, one of Hg. leucocelaenus, and one of Aedes sp. Between those samples, there were nine, with only one to two specimens pooled, including two pools of Ae. albopictus, two of Ae. argyrothorax, and two of Ae. serratus.
3.3. Phylogenetic Analysis
Five of the 26 positive samples met the quality control standards and were submitted to whole genome sequencing. The quality control standards are influenced by viral load, which is not usually high in mosquito’s samples. Three sequences were from Hg. janthinomys from Jandaia (Goiás), one of Hg. janthinomys from Alvarenga (Minas Gerais), and one of Ae. albopictus from Ituêta (Minas Gerais). Genomes were deposited in GenBank under the following accession numbers: OQ932914 (AR831907), OQ932915 (AR831908), OQ932916 (AR831909), MH329655 (AR843690), and MF370530 (AR843721).
The five new genome sequences belonged to the South America I clade. The two sequences from Minas Gerais were close to other sequences from the recent Brazilian outbreak and the three sequences from Goiás grouped together with another viral strain from the state of Goiás, collected in 2015 (Figure 3). Information about sequences included in this study are available in Table S3.
4. Discussion
In the last eight decades, YFV has been only maintained into sylvatic cycle in Brazil and occasionally re-emerged in North, Midwest and Southern regions, indicating its constant tendency to spread to Southeast, which was observed in the recent 2016-2018 outbreak [2-5,33-36].
In the Americas, forest-living mosquitoes of Haemagogus and Sabethes genera are considered primary and secondary vectors of sylvatic YFV, respectively, and have a wide geographical distribution in Brazil [2,8,10]. In our study, Haemagogus mosquitoes represent 21.35% of the 2904 collected mosquitoes and 20.3% of the 246 pools. On the other hand, mosquitoes of Sabethes genus represent 6.75% of the 2904 collected mosquitoes and 16.6% of the 246 pools. Most mosquitoes of these genera are from Goiás and Minas Gerais, collected in 2016 and 2017, respectively. In addition, half of the 26 positive samples are from mosquitoes of Haemagogus genus – and from these, only one is of Hg. leucocelaenus species – and another one is from mosquitoes of Sabethes genus, a pool of Sa. glaucodaemon from Jandaia.
In Brazil, the Hg. janthinomys species has been identified as the primary vector of sylvatic YFV [2,8,10]. There are descriptions of natural infection of this species by YFV during late-twentieth-century Brazilian outbreaks [37,38], and Hg. leucocelaenus, a commonly secondary vector defined as a primary vector in forests in Southern Brazil in the absence of Hg. janthinomys [33,34] were implicated as the main vectors in the recent outbreak in the Southeast region [10]. Our findings in Midwest Brazil (Cerrado biome) stress the potential for active transmission in the region, which is already considered an endemic area for YFV and has the presence of main vectors of the virus. On the other hand, the Haemagogus positive pools from Atlantic Forest support the vector presence and its participation in the maintenance of the 2016-2018 outbreak [10].
The positive Hg. janthinomys pool from Cocos, in the Bahia Cerrado, on the border with Northwestern Minas Gerais, corroborates the YFV spread from Minas Gerais to Bahia and the feasibility of YFV cases occurring in the region, which has a low vaccination coverage in humans, and the presence of main vectors and susceptible NHP [21].
Mosquitoes of Sabethes genus in general, are considered secondary vectors of YFV and could present a potential role in the transmission of YFV in the absence or in low density of primary vectors. Although, studies about the detection of YFV in Sa. glaucodaemon species and its role in the sylvatic YFV transmission cycle are scarce [2,8]. Due to the detection of YFV in Hg. janthinomys from the same municipality, it is unlikely the participation of Sa. glaucodaemon in the active transmission of the virus there.
Aedes mosquitoes represent the majority of samples analyzed in this study: they were 71.9% of the 2904 collected mosquitoes and 63% of the 246 pools. This genus comprises species of great importance in public health as Ae. aegypti and Ae. albopictus. The Ae. aegypti was the first vector related to a virus (YFV) [39,40], representing a revolution in the understanding of some viruses, afterwards named arboviruses, and was related to the urban transmission cycle of YFV in Brazil until 1942, when the last urban YFV outbreak occurred in the country [2]. Since the late 1980s, Ae. albopictus has been detected and spread in Brazil, and today more than 1000 municipalities have reported its presence in the peridomicile and adjacent natural or modified environments. Coincidentally, the highest infestation indexes for Ae. albopictus in Brazil is reported mainly in the Southeast region, where the outbreak occurred [41]. These species are very opportunistic and strongly anthropophilic species, able to colonize a wide range of habitats and adapted to tropical and temperate regions of the world [11,42].
The YFV was previously described in two of the Aedes species positive from the Atlantic Forest: Ae. scapularis [2,10,43], and Ae. serratus [34]. Indeed, this is the first YFV detection in Ae. argyrothorax, however, the real importance of YFV spillover in these three species in maintaining YFV in nature remains to be determined. The entomo-virological surveillance and vectorial competence experimental studies certainly could contribute to enlighten their role in sylvatic YFV maintenance.
Based on experimental studies, Ae. albopictus has been incriminated as a potential vector for YFV transmission [11,42,44]. Still, until 2018, when the Evandro Chagas Institute publicly announced these first detections [45], there was no scientific evidence of natural infection by YFV in this mosquito species.
For a mosquito species to be considered as a potential vector for arbovirus transmission, it is necessary to combine criteria of all of the physiological and ecological factors of vector, host, pathogen, and environment that determine the vector status of a given arthropod population. The criteria are: (a) isolation of a specific virus from specimens collected in nature; (b) demonstration of infection in the mosquito following experimental feeding on a viremic host or virus suspension; (c) demonstration of transmission of virus by bite to a vertebrate host or demonstration of transmission through excretion of virus in salivary fluids; and (d) field evidence confirming association of the mosquito species with the vertebrate population in which the virus infection is occurring [46,47]. Even with a considerable number of Ae. albopictus positive samples among the Minas Gerais pools, no subsequent detection of YFV naturally infected Ae. albopictus in the region was made, thus the species was not implicated as a vector involved in the recent Brazilian YFV outbreak, which reported cases were associated with the sylvatic transmission cycle [10].
Although it was not related to the outbreak, the Ae. albopictus confirmed experimental vector competence, and its biological characteristics, including the easy movement between both sylvatic and peri-urban environments associated with cases in humans and NHP, and its geographic distribution in Brazil reinforce the concern on the risk of YFV re-urbanization in the country [11,42,44].
The natural YFV infection detected in Ae. albopictus pools in this study suggest that this mosquito species could play the role of bridge vector linking the sylvatic YFV to the urban cycle and establishing an intermediary transmission cycle, as documented in Africa with Ae. simpsoni and other Aedes mosquito species [3,12], once the low vector capacity could be overcome by other factors such as high vector density, high human-biting rate, and high daily survival rates [48].
In the phylogenetic analysis, the five sequences obtained in this study are clade-related to others from the 2016-2018 outbreak within South America I genotype of YFV [5,49-51]. The three Midwest sequences, previous to the outbreak, are related to a 2015 sequence obtained from an NHP sample collected in Novo Brasil, a municipality from Goiás, located 175 km away from Jandaia. These sequences from Goiás clustered in a sister clade to the outbreak clade, confirming the already descripted topology of previous phylogeny positioning the NHP as related to the recent Southeast sequences [50] and could reinforce the possibility of spread from the Midwest to the Southeast [21].
The other two sequences are from geographically close Minas Gerais municipalities and clustered within the outbreak clade, close to a sequence which were obtained from a Hg. janthinomys and two other sequences obtained from NHP, all in 2017 from the Espírito Santo state. The proximity of Minas Gerais municipalities to Espírito Santo could explain their closeness due to virus spread, even with the multiple viral exchanges observed through the deposited sequences during the outbreak, which actually justifies the existence of several sub-clades with the 2017-2018 sequences in the phylogenetic analysis [51].
5. Conclusions
Entomo-virological surveillance proves to be a crucial strategy in the YFV surveillance in Brazil, linking and confirming data of virus active transmission to NHP and humans, besides shedding light on the involvement of unusual or potential vectors in the maintenance of YFV, which may pave the way for new studies.
Analysis of main and potential vectors helps to understand their participation in the spread and maintenance of sylvatic YFV and the possibility of an oncoming re-emergence of urban YFV in Brazil. Furthermore, it could highlight the urgency to strengthen the monitoring of syndromic surveillance and NHP deaths, vaccination coverage, and vector control measures, including Ae. aegypti and Ae. albopictus.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Description of pools of mosquitoes from 2016; Table S2: Description of pools of mosquitoes from 2017; Table S3: Sequences used in the phylogenetic analysis.
Author Contributions
Conceptualization, A.C.R.C., L.H.A.H. and J.P.N.N; methodology, L.H.A.H., T.Y.B.P., C.F.A., S.P.S., F.S.S., A.A.A., G.J.G.P.C., B.L.S.N., J.W.R.J., C.N.E., C.G.N., V.F., M.G., L.C.J.A. and B.T.D.N.; software, L.H.A.H. and S.P.S.; validation, A.C.R.C., S.P.S., B.T.D.N., P.F.C.V., L.C.M. and J.P.N.N.; formal analysis, A.C.R.C.; investigation, A.C.R.C., L.H.A.H., T.Y.B.P and S.P.S.; data curation, S.P.S. and B.T.D.N.; writing—original draft preparation, A.C.R.C, L.H.A.H., C.F.A. and A.A.A.; writing—review and editing, A.C.R.C., L.H.A.H., T.Y.B.P., V.F., M.G., L.C.J.A. and J.P.N.N.; visualization, A.C.R.C., S.P.S., V.F., M.G., L.C.J.A., B.T.D.N., P.F.C.V., L.C.M. and J.P.N.N.; supervision, P.F.C.V., L.C.M. and J.P.N.N.; project administration, A.C.R.C.
Funding
This work was partially supported by Brazilian National Council for Scientific and Technological Development (CNPq), grant numbers 166720/2017-8 and 106256/2018-1, to L.H.A.H.
Data Availability Statement
The five sequences were deposited in GenBank under the following accession numbers: OQ932914 (AR831907), OQ932915 (AR831908), OQ932916 (AR831909), MH329655 (AR843690), and MF370530 (AR843721).
Acknowledgments
We thank the Health and Environment Surveillance Secretariat from Brazilian Ministry of Health, and the Health State Secretariats of Bahia, Goiás, Mato Grosso, Minas Gerais, Pará, and Tocantins.
Conflicts of Interest
The authors declare no conflict of interest.
References
- International Committee on Taxonomy of Viruses. Available online: https://ictv.global/taxonomy (accessed on 09 April 2023).
- Vasconcelos, P.F.C. Febre amarela. Rev. Soc. Bras. Med. Trop. 2003, 36, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Vasconcelos, P.F.C. Yellow fever. J. Clin. Virol. 2015, 64, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Possas, C.; Lourenço-de-Oliveira, R.; Tauil, P.L.; Pinheiro, F.P.; Pissinatti, A.; Cunha, R.V.; Freire, M.; Martins, R.M.; Homma, A. Yellow fever outbreak in Brazil: the puzzle of rapid viral spread and challenges for immunization. Mem. Inst. Oswaldo Cruz 2018, 113, e180278. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Mendonça, M.C.L.; Fonseca, V.; Mares-Guia, M. A.; Fabri, A.; Xavier, J.; Jesus, J. G; Gräf, T; Rodrigues, C.D.S.; Santos, C.C.; et al. Yellow Fever Virus Reemergence and Spread in Southeast Brazil, 2016-2019. J. Virol. 2019, 94, e01623–19. [Google Scholar] [CrossRef]
- Silva, N.I.O.; Sacchetto, L.; Rezende, I.M.; Trindade, G.S.; LaBeaud, A.D.; de Thoisy, B.; Drumond, B.P. Recent sylvatic yellow fever virus transmission in Brazil: the news from an old disease. Virol. J. 2020, 17, 9. [Google Scholar] [CrossRef]
- Brazil. Ministry of Health. Guia de vigilância em saúde, 5th ed.; Ministry of Health: Brasilia, Brazil, 2022; pp. 623–649. ISBN 978-65-5993-102-6. [Google Scholar]
- Brazil. Ministry of Health. Guia de vigilância de epizootias em primatas não humanos e entomologia aplicada à vigilância da febre amarela., 2nd ed.; Ministry of Health: Brasilia, Brazil, 2017; ISBN 978-85-334-2102-8. [Google Scholar]
- Câmara, D.C.P.; Codeço, C.T.; Ayllón, T.; Nobre, A.A.; Azevedo, R.C.; Ferreira, D.F.; Pinel, C.S.; Rocha, G.P.; Honório, N.A. Entomological Surveillance of Aedes Mosquitoes: Comparison of Different Collection Methods in an Endemic Area in RIO de Janeiro, Brazil. Trop. Med. Infect. Dis. 2022, 7, 114. [Google Scholar] [CrossRef]
- Abreu, F.V.S.; Ribeiro, I.P.; Ferreira-de-Brito, A.; Santos, A.A.C.; Miranda, R.M.; Bonelly, I.S.; Neves, M.S.A.S.; Bersot, M.I.; Santos, T.P.; Gomes, M.Q.; et al. Haemagogus leucocelaenus and Haemagogus janthinomys are the primary vectors in the major yellow fever outbreak in Brazil, 2016-2018. Emerg. Microbes Infect. 2019, 8, 218–231. [Google Scholar] [CrossRef]
- Couto-Lima, D.; Madec, Y.; Bersot, M.I.; Campos, S.S.; Motta, M.A.; Santos, F.B.; Vazeille, M.; Vasconcelos, P.F.C.; Lourenço-de-Oliveira, R.; Failloux, A.B. Potential risk of re-emergence of urban transmission of Yellow Fever virus in Brazil facilitated by competent Aedes populations. Sci. Rep. 2017, 7, 4848. [Google Scholar] [CrossRef]
- Santos, T.P.; Roiz, D.; Abreu, F.V.S.; Luz, S.L.B.; Santalucia, M.; Jiolle, D.; Neves, M.S.A.S.; Simard, F.; Lourenço-de-Oliveira, R.; Paupy, C. Potential of Aedes albopictus as a bridge vector for enzootic pathogens at the urban-forest interface in Brazil. Emerg. Microbes Infect. 2018, 7, 191. [Google Scholar] [CrossRef]
- Lane, J. Neotropical Culicidae, Volume 1; Edusp: São Paulo, Brazil, 1953. [Google Scholar]
- Lane, J. Neotropical Culicidae, Volume 2; Edusp: São Paulo, Brazil, 1953. [Google Scholar]
- Forratini, O.P. Entomologia médica. Culicini: Culex, Aedes e Psorophora. Volume 2; Edusp: São Paulo, Brazil, 1965. [Google Scholar]
- Forratini, O.P. Entomologia médica. Culicini: Haemagogus, Mansonia, Culiseta, Sabethini, Toxorhynchitini, Arboviroses, Filariose bancroftiana, Genética. Volume 3; Edusp: São Paulo, Brazil, 1965. [Google Scholar]
- Forratini, O.P. Culicidologia médica: identificação, biologia, epidemiologia; Edusp: São Paulo, Brazil, 2002; ISBN 85-314-0699-4. [Google Scholar]
- Consoli, R.A.G.B.; Lourenço-de-Oliveira, R. Principais mosquitos de importâcia sanitaria no Brasil, 1st ed.; Editora Fiocruz: Rio de Janeiro, Brazil, 1994; ISBN 85-85676-03-5. [Google Scholar]
- Domingo, C.; Patel, P.; Yillah, J.; Weidmann, M.; Méndez, J.A.; Nakouné, E.R.; Niedrig, M. Advanced yellow fever virus genome detection in point-of-care facilities and reference laboratories. J. Clin. Microbiol. 2012, 50, 4054–4060. [Google Scholar] [CrossRef]
- Menting, S.; Thai, K.T.D.; Nga, T.T.T.; Phuong, H.L.; Klatser, P.; Wolthers, K.C.; Binh, T.Q.; de Vries, P.J.; Beld, M. Internally Controlled, Generic Real-Time PCR for Quantification and Multiplex Real-Time PCR with Serotype-Specific Probes for Serotyping of Dengue Virus Infections. Adv. Virol. 2011, 2011, 514681. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Pinotti, F.; Zanluca, C.; Fonseca, V.; Nakase, T.; Koishi, A.C.; Tscha, M.; Soares, G.; Dorl, G.G.; Marques, A.E.M.L.; et al. Genomic epidemiology sheds light on the recent spatio-temporal dynamics of yellow fever virus and the spatial corridor that fueled its ongoing emergence in southern Brazil. medRxiv 2023, 23284525. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pharm, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- DNASTAR. Available online: https://www.dnastar.com (accessed on 05 March 2023).
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Figtree. Available online: https://tree.bio.ed.ac.uk/software/figtree/ (accessed on 14 April 2023).
- Inkscape. Available online: https://inkscape.org/release/inkscape-1.1/ (accessed on 14 April 2023).
- Vasconcelos, P.F.; Sperb, A.F.; Monteiro, H.A.; Torres, M.A.; Sousa, M.R.; Vasconcelos, H.B.; Mardini, L.B.; Rodrigues, S.G. Isolations of yellow fever virus from Haemagogus leucocelaenus in Rio Grande do Sul State, Brazil. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, J.C.; Almeida, M.A.B.; Santos, E.; Fonseca, D.F.; Sallum, M.M.; Noll, C.A.; Monteiro, H.A.O.; Cruz, A.C.R.; Carvalho, V.L.; Pinto, E.V.; et al. Yellow fever virus in Haemagogus leucocelaenus and Aedes serratus mosquitoes, Southern Brazil, 2008. Emerg. Infect. Dis. 2010, 16, 1918–1924. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, M.A.; dos Santos, E.; da Cruz Cardoso, J.; da Fonseca, D.F.; Noll, C.A.; Silveira, V.R.; Maeda, A.Y.; de Souza, R.P.; Kanamura, C.; Brasil, R.A. Yellow fever outbreak affecting Allouata populations in southern Brazil (Rio Grande do Sul State), 2008-2009. Am. J. Primatol. 2012, 74, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kumm, H.W.; Cerqueira, N.L. The role of Aëdes leucocelaenus in the epidemiology of jungle yellow fever in Brazil. Bull. Ent. Res. 1952, 42, 195–199. [Google Scholar] [CrossRef]
- Vasconcelos, P.F.; Rodrigues, S.G.; Degallier, N.; Moraes, M.A.; da Rosa, J.F.; da Rosa, E.S.; Mondet, B.; Barros, V.L.; da Rosa, A.P. An epidemic of sylvatic yellow fever in the Southeast region of Maranhao State, Brazil, 1993-1994: epidemiologic and entomologic findings. Am. J. Trop. Med. Hyg. 1997, 57, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, P.F.; Costa, Z.G.; Travassos da Rosa, E.S.; Luna, E.; Rodrigues, S.G.; Barros, V.L.; Dias, J.P.; Monteiro, H.A.; Oliva, O.F.; Vasconcelos, H.B.; et al. Epidemic of jungle yellow fever in Brazil, 2000: implications of climatic alterations in disease spread. J. Med. Virol. 2001, 65, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Finlay, C. Yellow fever: its transmission by means of the Culex mosquito. Am. J. Med. Sci. 1986, 92, 395–409. [Google Scholar] [CrossRef]
- Reed, W.; Carrol, J.; Agramonte, A. The etiology of yellow fever. J. Am. Med. Assoc. 1901, 36, 415–440. [Google Scholar] [CrossRef]
- Goldani, L.Z. Yellow fever outbreak in Brazil. Braz. J. Infect. Dis. 2017, 21, 123–124. [Google Scholar] [CrossRef]
- Amraoui, F.; Vazeille, M.; Failloux, A.B. French Aedes albopictus are able to transmit yellow fever virus. Euro Surveill. 2016, 21, 30361. [Google Scholar] [CrossRef]
- Cunha, M.S.; Faria, N.R.; Caleiro, G.S.; Candido, D.S.; Hill, S.C.; Claro, I.M.; da Costa, A.C.; Nogueira, J.S.; Maeda, A.Y.; da silva, F.G.; et al. Genomic evidence of yellow fever virus in Aedes scapularis, southeastern Brazil, 2016. ACTA Trop 2020, 205, 105390. [Google Scholar] [CrossRef] [PubMed]
- Damasceno-Caldeira, R.; Nunes-Neto, J.P.; Aragão, C.F.; Freitas, M.N.O.; Ferreira, M.S.; Castro, P.H.G.d.; Dias, D.D.; Araújo, P.A.d.S.; Brandão, R.C.F.; Nunes, B.T.D.; et al. Vector competence of Aedes albopictus for yellow fever virus: risk of reemergence of urban yellow fever in Brazil. Viruses 2023, 15, 1019. [Google Scholar] [CrossRef] [PubMed]
- Agência Brasil. Available online: https://agenciabrasil.ebc.com.br/geral/noticia/2018-02/pesquisa-detecta-virus-da-febre-amarela-em-novo-tipo-de-mosquito (accessed on 18 April 2023).
- Mitchell, C.J. Mosquito vector competence and arboviruses. In Current topics in vector research; Harris, K.F., Ed.; Praeger: New York, NY, USA, 1983; Volume 1, pp. 63–92. ISBN 0030586372. [Google Scholar]
- Mitchell, C.J. The role of Aedes albopictus as an arbovirus vector. Parassitologia 1995, 37, 109–113. [Google Scholar] [PubMed]
- Kramer, L.D.; Ebel, G.D. Dynamics of flavivirus infection in mosquitoes. Adv. Virus Res. 2003, 60, 187–232. [Google Scholar] [PubMed]
- Bonaldo, M.C.; Gómez, M.M.; Santos, dos Santos, A.C.C.; Abreu, F.V.S.; Ferreira-de-Brito, A.; Miranda, R.M.; Castro, M.G.; Lourenço-de-Oliveira, R. Genome analysis of yellow fever virus of the ongoing outbreak in Brazil revealspolymorphisms. Mem. Inst. Oswaldo Cruz, 2017, 112, 447–451. [Google Scholar] [CrossRef]
- Delatorre, E.; Abreu, F.V.S.; Ribeiro, I.P.; Gómez, M.M.; dos Santos, A.A.C.; Ferreira-de-Brito, A.; Neves, M.S.A.S.; Bonelly, I.; de Miranda, R.M.; Furtado, N.D.; et al. Distinct YFV lineages co-circulated in the Central-Western and Southeastern Brazilian Regions from 2015 to 2018. Front. Microbiol. 2019, 10, 1079. [Google Scholar] [CrossRef]
- Gómez, M.M.; Abreu, F.V.S.; Santos, A.A.C.D.; Mello, I.S.; Santos, M.P.; Ribeiro, I.P.; Ferreira-de-Brito, A.; Miranda, R.M.; Castro, M.G.; Ribeiro, M.S.; et al. Genomic and structural features of the yellow fever virus from the 2016-2017 Brazilian outbreak. J. Gen. Virol. 2018, 99, 536–548. [Google Scholar] [CrossRef]
Figure 1.
Total of collected mosquitoes per year and genera (A), distribution of total pools of mosquitoes (above) and total of specimens (below) per genera and Brazilian state of collection (B), and map of location of the respective Brazilian states of collection (C). BA: Bahia; GO: Goiás; MT: Mato Grosso; MG: Minas Gerais; PA: Pará; and TO: Tocantins.
Figure 1.
Total of collected mosquitoes per year and genera (A), distribution of total pools of mosquitoes (above) and total of specimens (below) per genera and Brazilian state of collection (B), and map of location of the respective Brazilian states of collection (C). BA: Bahia; GO: Goiás; MT: Mato Grosso; MG: Minas Gerais; PA: Pará; and TO: Tocantins.
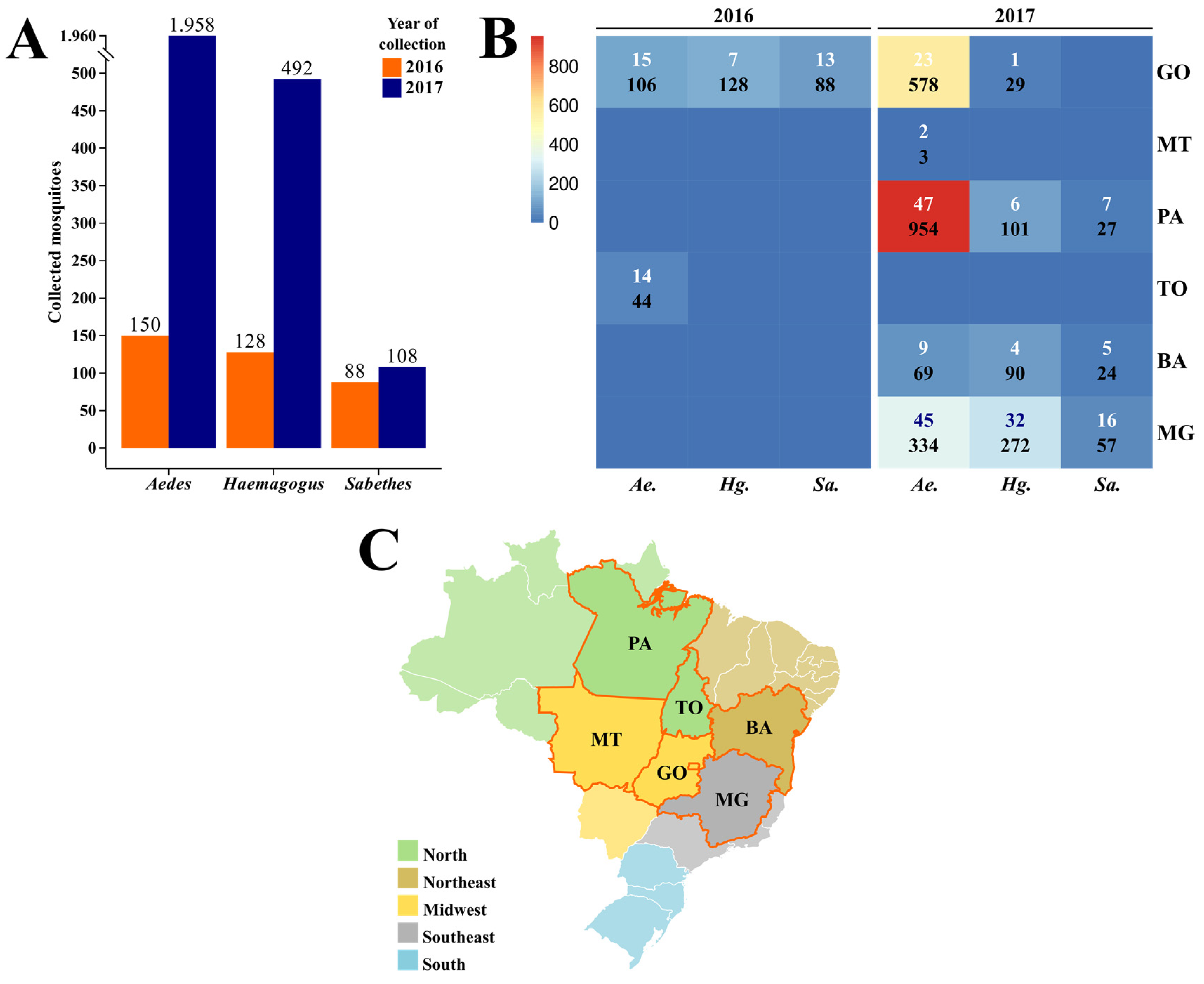
Figure 2.
Municipalities of origin of the positive samples for YFV at RT-qPCR.
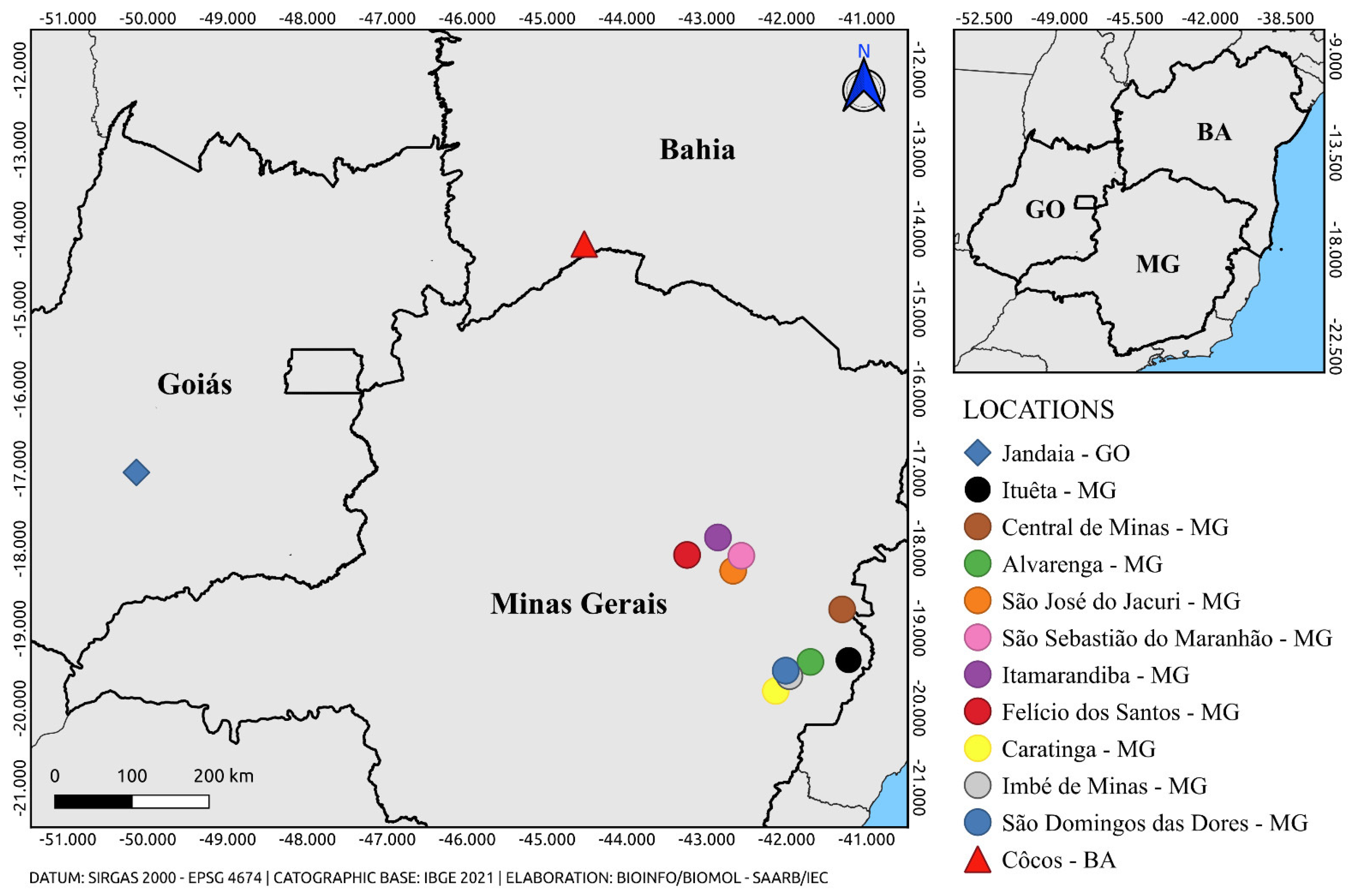
Figure 3.
Maximum likelihood phylogenetic inference using the GTR+F+R2 nucleotide substitution model based on the YFV complete genome of 87 sequences, including five obtained from mosquitoes in our study (in red). African genotypes clades are collapsed. Each record includes the sequence accession number, host species, country (and state for Brazilian sequences), and collection year. GO: Goiás; ES: Espírito Santo; MG: Minas Gerais; MS: Mato Grosso do Sul; PA: Pará; RJ: Rio de Janeiro; RO: Rondônia; RR: Roraima; RS: Rio Grande do Sul.
Figure 3.
Maximum likelihood phylogenetic inference using the GTR+F+R2 nucleotide substitution model based on the YFV complete genome of 87 sequences, including five obtained from mosquitoes in our study (in red). African genotypes clades are collapsed. Each record includes the sequence accession number, host species, country (and state for Brazilian sequences), and collection year. GO: Goiás; ES: Espírito Santo; MG: Minas Gerais; MS: Mato Grosso do Sul; PA: Pará; RJ: Rio de Janeiro; RO: Rondônia; RR: Roraima; RS: Rio Grande do Sul.
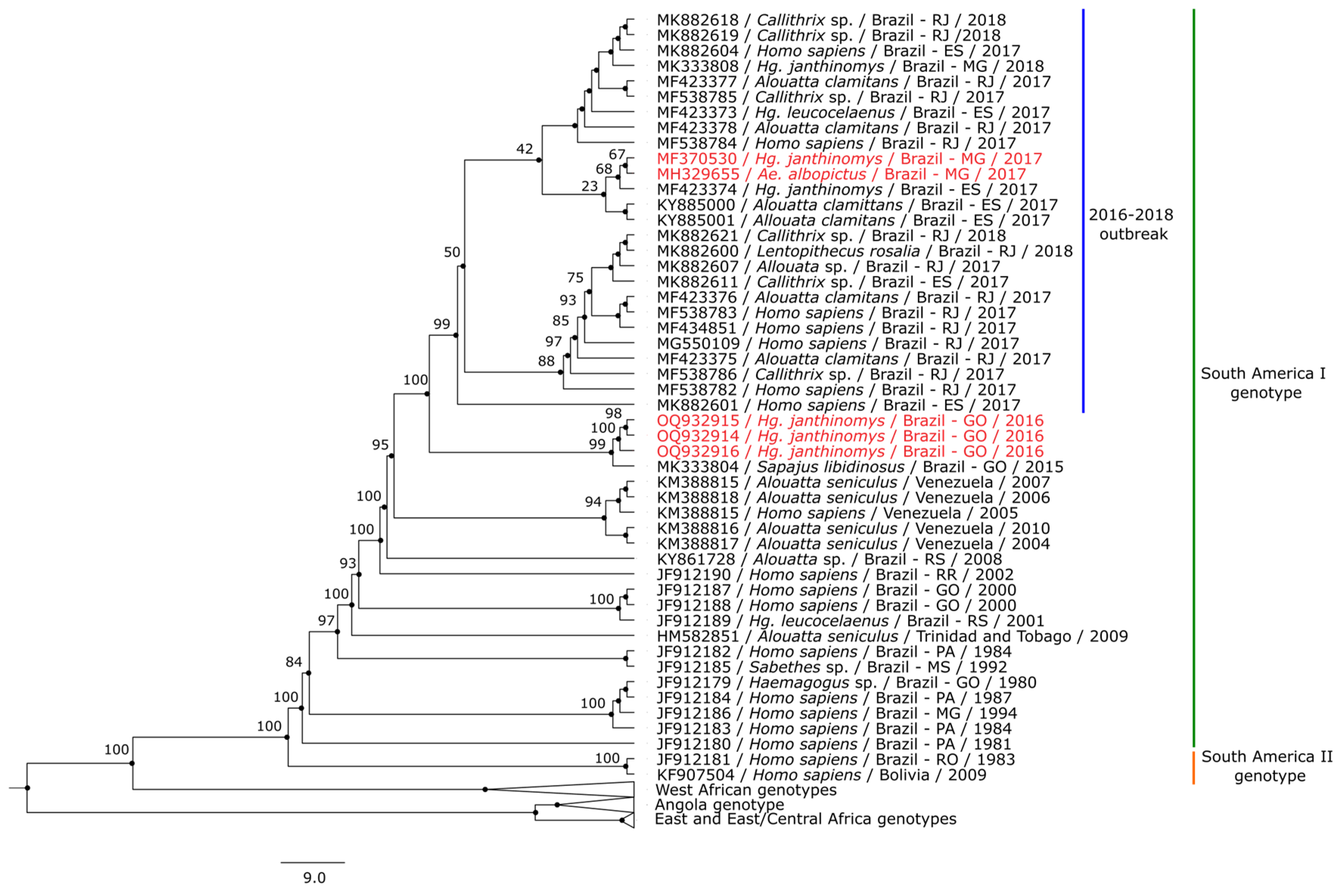

Table 1.
Samples with YFV amplicon detection at RT-qPCR.
| Sample ID |
Species | Specimens per pool | Location | Collection Date | RT-qPCR Ct value |
|---|---|---|---|---|---|
| AR831906 | Hg. janthinomys | 30 | Jandaia – GO | 24-Sep-2016 | 26.17 |
| AR831907 | Hg. janthinomys | 30 | 24-Sep-2016 | 23.52 | |
| AR831908 | Hg. janthinomys | 28 | 24-Sep-2016 | 21.26 | |
| AR831909 | Hg. janthinomys | 7 | 26-Sep-2016 | 18.27 | |
| AR831914 | Sa. glaucodaemon | 30 | 24-Sep-2016 | 31.27 | |
| AR843690 | Ae. albopictus ♀ | 25 | Ituêta – MG | 14-Jan-2017 | 23.88 |
| AR843692 | Ae. scapularis | 25 | 31.08 | ||
| AR843693 | Ae. argyrothorax | 2 | 33.79 | ||
| AR843713 | Hg. janthinomys | 15 | Central de Minas – MG | 13-Jan-2017 | 32.23 |
| AR843715 | Ae. albopictus ♀ | 4 | Alvarenga – MG | 15-Jan-2017 | 34.28 |
| AR843716 | Ae. argyrothorax | 2 | 32.88 | ||
| AR843717 | Aedes sp. | 1 | 35.55 | ||
| AR843720 | Hg. janthinomys | 25 | 29.08 | ||
| AR843721 | Hg. janthinomys | 25 | 28.44 | ||
| AR843728 | Hg. janthinomys | 19 | São José do Jacuri – MG | 16-Jan-2017 | 32.09 |
| AR843738 | Ae. scapularis | 3 | São Sebastião do Maranhão – MG |
17-Jan-2017 | 24.08 |
| AR843741 | Hg. leucocelaenus | 2 | 24.09 | ||
| AR843745 | Ae. serratus | 1 | Itamarandiba – MG | 18-Jan-2017 | 35.46 |
| AR843765 | Hg. janthinomys | 6 | Felício dos Santos – MG | 20-Jan-2017 | 32.52 |
| AR843771 | Ae. albopictus ♀ | 2 | Caratinga – MG | 10-Jan-2017 | 35.12 |
| AR843772 | Ae. serratus | 2 | 32.16 | ||
| AR843777 | Hg. janthinomys | 1 | 27.60 | ||
| AR843807 | Hg. janthinomys | 4 | Imbé de Minas – MG | 16-Jan-2017 | 34.81 |
| AR843821 | Ae. albopictus ♀ | 1 | São Domingos das Dores – MG |
19-Jan-2017 | 35.61 |
| AR843829 | Ae. albopictus ♀ | 4 | 35.18 | ||
| AR845803 | Hg. janthinomys | 18 | Cocos – BA | 17,18,20-Mar-2017 | 28.63 |
♀: female; Ae.: Aedes; Hg.: Haemagogus; Sa.: Sabethes; sp.: species not defined; BA: Bahia; GO: Goiás; MG: Minas Gerais; Ct: cycle threshold value.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



