Submitted:
12 May 2023
Posted:
15 May 2023
Read the latest preprint version here
Abstract
The kallikrein-kinin system consists of the two kininogen substrates, present in blood plasma, and of two serine proteases, the plasma and tissue kallikreins. The action of the latter on kininogens produce small peptides, short lived but endowed by powerful pharmacologic actions on blood vessels and other tissues. Many recent and exciting therapeutic developments in the field are briefly summarized. Notably, various novel strategies are being clinically developed to inhibit the formation of bradykinin or block its receptors in the management of hereditary angioedema. The interventions include orally bioavailable drugs, biotechnological proteins, and gene therapy. Several other medical indications are currently investigated. Harnessing controlled kinin formation is also of potential therapeutic interest as shown by the clinical development of recombinant tissue kallikrein for ischemic stroke and renal disease. Biomarkers of a kinin-mediated disorders, frequently implicating edemas, include the consumption of kininogen(s), plasma kallikrein activity, and the detection of circulating kinin metabolites such as fragments BK1-5 and BK2-9. Based on this, some opportunities to clinically apply the underexploited drugs of the kallikrein-kinin system are briefly reviewed. This personal perspective is offered by an observer of, and a participant in drug characterization during the last 4 decades.
Keywords:
kallikrein-kinin system
; kininogens
; bradykinin
; B1 receptor
; B2 receptor
; hereditary angioedema
1. The kallikrein kinin systems: formation and clearance of kinins
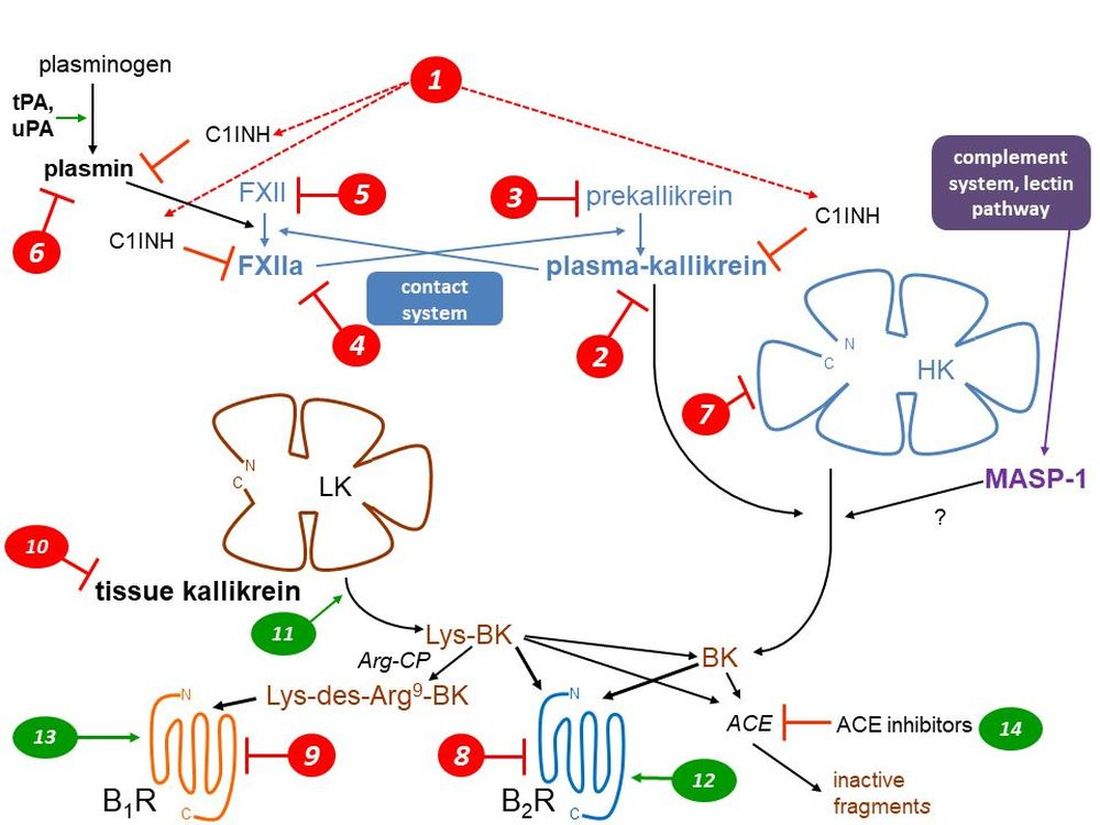
Both protective and pathogenic effects are mediated by two largely separate kallikrein-kinin systems (KKS; abbreviations are defined in Table 1) via the formation of small and unstable peptides, the kinins (Figure 1, schematic representation). Thus, vascular effects (vasodilation, increased microvascular permeability), inflammatory manifestations (edema, pain, increased local blood flow), smooth muscle contraction, and epithelial cell stimulation are potentially initiated by kinins [1]. The nonapeptide bradykinin (BK; H-Arg1-Pro2-Pro3-Gly4-Phe5-Ser6-Pro7-Phe8-Arg9-OH) is the reference kinin sequence found in domain 4 of two circulating proteins, the high molecular weight and low molecular weight kininogens (HK, LK; about 20 and 80% molar proportions, respectively). The hepatic synthesis of both kininogen forms is based on the alternative splicing of a single gene product, KNG1.
HK (110 kDa), circulating in a complexed form with prekallikrein and factor XI, is part of the contact system (Figure 1) along with coagulation factor XII (FXII, Hageman factor). When exposed to negatively charge surfaces, such as the basal membrane of denuded vascular endothelial cells, all these components assemble into a tetramolecular complex with ensuing proteolytic reactions: the mutual activation of FXII and prekallirein into their proteolytically active forms factor XIIa (FXIIa) and plasma kallikrein, respectively, the cleavage of HK releasing BK and the cleavage of factor XI that initiates the intrinsic coagulation pathway [2]. The contact system is tightly controlled by a circulating serpin inhibitor, C1-esterase inhibitor (C1INH) that is also part of the complement cascade. FXIIa and plasma kallikrein are irreversibly inhibited by C1INH [3]. Blood clots are cleared by the fibrinolytic system which is connected to the contact system (Figure 1): plasmin, the fibrinolytic enzyme, activates FXII into FXIIa to a certain extent, indirectly promoting BK production via secondarily activated plasma kallikrein. C1INH is a secondary inhibitor of plasmin [3]. Whether HK is directly cleaved by additional proteases has been suggested, but not well established in whole blood where endogenous inhibitors are present: plasmin and the complement associated protease MASP-1 may directly release BK from HK [4,5]. There is no evidence of BK release when platelets or neutrophils are activated in human whole blood [6], casting a doubt about previously suggested activation pathways demonstrated using purified components of the contact system.

Table 1.
List of abbreviations.
| Abbreviation | Standing for | Corresponding gene |
|---|---|---|
| ACE | angiotensin-I converting enzyme | ACE |
| Arg-CP | arginine carboxypeptidase | |
| B1R | bradykinin B1 receptor | BDKRB1 |
| B2R | bradykinin B2 receptor | BDKRB2 |
| BK | bradykinin | |
| C1INH | C1-esterase inhibitor | SERPING1 |
| FXII | coagulation factor XII | F12 |
| FXIIa | activated factor XII | |
| HAE | hereditary angioedema | |
| HAE-C1INH | HAE caused by C1INH haplodeficiency | |
| HK | high molecular weight kininogen | KNG1 |
| KKS | kallikrein-kinin system | |
| KLK-1 | tissue kallikrein | KLK1 |
| LK | low molecular weight kininogen | KNG1 |
| Lys-BK | kallidin | |
| mAb | therapeutic monoclonal antibody | |
| MASP-1 | mannan-binding lectin associated serine protease 1 | MASP1 |
| NPA | non-peptide antagonist | |
| tPA | tissue plasminogen activator | PLAT |
Tissue kallikrein (KLK-1; kallidinogenase) is a member of a family of 15 secreted proteases encoded on human chromosome locus 19q13.4 [7]. These serine proteases assume different, often uncertain physiological functions. Only KLK-1 is a relevant kininogenase in this family. This was verified with two KLKs normally found in the prostate: they release no kinins from purified HK (KLK-3), or very little (KLK-2 is 1000-fold less active than KLK-1 in this respect) [8]. KLK-1 releases the biologically active decapeptide Lys-BK (= kallidin) from both forms of kininogen, but mostly from the more abundant LK. KLK-1 is widely expressed and abundant in the kidney, pancreas, salivary glands, lungs, blood vessels and other tissues; its secretion and activation, via the removal of a N-terminal sequence, are not well understood. KLK-1 is regulated by its own irreversible inhibitor, kallistatin (SERPINA4 gene product). The previously claimed direct agonist effect of KLK-1 on human BK B2 receptor (B2R) has been disproved using the pure recombinant enzyme in its active form [9].
Kinins are inherently unstable, with a half-life well under 1 min [10], and metabolized by several metallopeptidases. A careful in vivo study showed that angiotensin converting enzyme (ACE, kininase II) is the dominant BK-inactivation pathway in rats, followed by aminopeptidase P [12]. Both these peptidases inactivate BK, initially producing the fragments BK1-7 and BK2-9, respectively. The fragment BK1-5 is the relatively stable product of a second cycle of BK1-7 cleavage by ACE. Arginine carboxypeptidases (Arg-CPs) (plasma carboxypeptidase N, cell membrane carboxypeptidase M; producing BK1-8 = des-Arg9-BK from BK) represent only a minor metabolic pathway in vivo [13] but connects the KKS with the pharmacological profile of the kinin B1 receptor (B1R; see below). Biomarkers of kinin-mediated disorders include the consumption of kininogen(s) and the detection of circulating kinin metabolites such as fragments BK1-5 and BK2-9, and the detection of plasma kallikrein activity, for instance using the synthetic substrate based on the C-terminal BK sequence HD-Pro-Phe-Arg-pNA. These assays are technically challenging, but one or more of them have been applied to hereditary angioedema (HAE), either during attacks or in remission [14,15], and to other edematous conditions such as ascites secondary to liver cirrhosis [16] and chronic urticaria [17,18] and to animal models of sepsis and sickle cell disease [19,20].
Figure 1.
Schematic representation of the KKS, featuring the 2 validated pathways of kinin generation, that of plasma kallikrein (part of the contact system) releasing bradykinin (BK) from high molecular weight kininogen (HK) and that mediated by secreted tissue kallikrein (KLK-1), generating Lys-BK mainly from low molecular weight kininogen (LK). A possible alternate BK generating pathway is indicated (purple). Two G protein coupled receptors (B1R, B2R) mediate the cellular effects of kinins. Two types of metallopeptidases that hydrolyze kinins are indicated (Arg-CP, ACE). Numerical markers indicate the mode of action of inhibitory (red) or stimulatory drugs (green) of the KKS and are referred to in Table 2, Table 3 and Table 4 and main text. See Table 1 for abbreviations. Modified from [10,11].
Figure 1.
Schematic representation of the KKS, featuring the 2 validated pathways of kinin generation, that of plasma kallikrein (part of the contact system) releasing bradykinin (BK) from high molecular weight kininogen (HK) and that mediated by secreted tissue kallikrein (KLK-1), generating Lys-BK mainly from low molecular weight kininogen (LK). A possible alternate BK generating pathway is indicated (purple). Two G protein coupled receptors (B1R, B2R) mediate the cellular effects of kinins. Two types of metallopeptidases that hydrolyze kinins are indicated (Arg-CP, ACE). Numerical markers indicate the mode of action of inhibitory (red) or stimulatory drugs (green) of the KKS and are referred to in Table 2, Table 3 and Table 4 and main text. See Table 1 for abbreviations. Modified from [10,11].
2. Kinin receptors
Before the era of molecular biology, the number and identity of kinin receptor subtypes in each mammalian species was uncertain. Historically, the first proposed kinin receptor subtype, the B1R, was discovered as the one mediating contraction in the isolated rabbit aorta and it possessed a typical order of potency for agonists and a first class of peptide antagonists [21]. Native kinins (BK and Lys-BK) from which the Arg9 residue has been removed by Arg-CPs (des-Arg9-BK, Lys-des-Arg9-BK, respectively) are the optimal agonists of the B1R, even if this kinin metabolic pathway is not prominent. Only Lys-des-Arg9-BK, also called des-Arg10-kallidin, has a subnanomolar affinity for the human (and rabbit) B1R [1]; this optimal agonist is presumably generated from Lys-BK (kallidin), itself derived from the cleavage of kininogens by tissue kallikrein (Figure 1). These reactions occur outside of the contact system. Early peptide antagonists, such as [Leu8]des-Arg9-BK, consolidated the pharmacological identity of the B1R; the other pharmacologic systems, directly responsive to BK and Lys-BK but insensitive to the des-Arg9 metabolites, were said to possess the B2R. The first B2R antagonists were discovered in the early 1980’s; they featured a constrained peptide backbone and were more or less protected from peptidases. Icatibant (Hoe 140; D-Arg-[Hyp3, Thi5, D-Tic7, Oic8]BK) is the success story among these early drugs [1,22] (Table 2). Selected modern, nonpeptide antagonists (NPAs) of both kinin receptor subtypes are presented in Figure 2. It is very typical that BK receptor antagonists exhibit species-dependent alterations of affinity and competitive behavior for their pharmacological targets [1]; thus, clinically developed antagonists have gone through a structural optimization process for the human forms of the B1R or B2R [1,23].
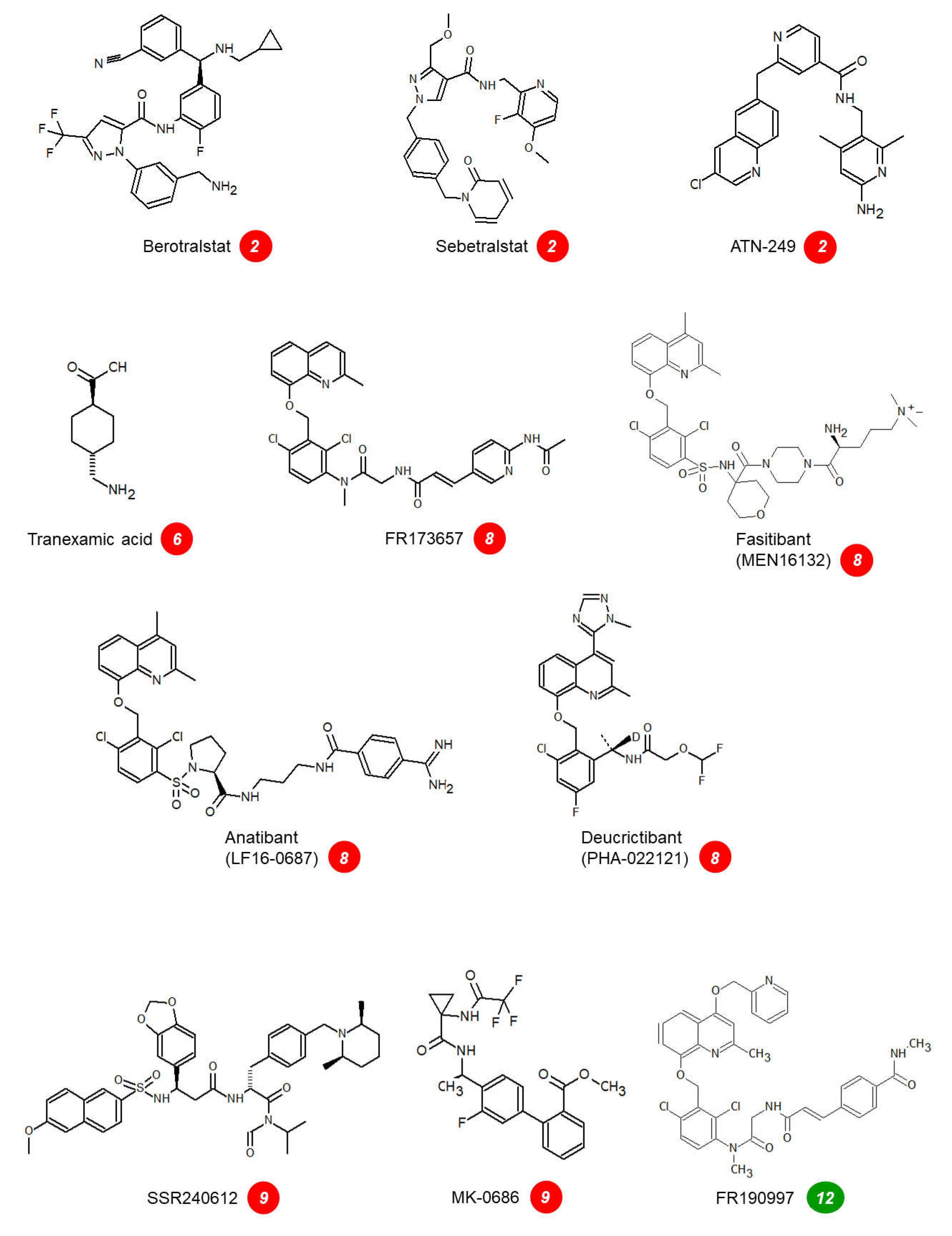
Figure 2.
Structure of the small molecule drugs cited in Table 2, Table 3 and Table 4 (except for KV998086 and BI1026706, currently undisclosed). The numerical markers indicate the mode of action as in Figure 1. The structure of the antagonists of kinin receptors is optimized for the human forms of these receptors. Note the structural similarities of the B2R antagonists (marker 8); only deucrictibant is developed as an orally administered drug in this class.
Figure 2.
Structure of the small molecule drugs cited in Table 2, Table 3 and Table 4 (except for KV998086 and BI1026706, currently undisclosed). The numerical markers indicate the mode of action as in Figure 1. The structure of the antagonists of kinin receptors is optimized for the human forms of these receptors. Note the structural similarities of the B2R antagonists (marker 8); only deucrictibant is developed as an orally administered drug in this class.
The receptor classification was confirmed by the discovery of a kinin receptor locus: in human chromosome region 14q32, genes encoding the G protein coupled receptors B2R and B1R, respectively termed BDKRB2 and BDKRB1, are in found tandem [24]; a similar organization is found in the genome of other mammals. While the expression of both genes is regulated, the B2R generally accounts for the in vivo effects of kinins in healthy laboratory animals: this receptor subtype is constitutively expressed in many cell types, including vascular endothelial cells, smooth muscle cells, some epithelia, sensory neurons, and other cell types [1]. The B1R, initially discovered in rabbit isolated blood vessels maintained in vitro for several hours, is not generally detectable in healthy animals. The paradox was resolved when the B1R was found to be expressed following tissue trauma, inflammation (such as the injection of bacterial lipopolysaccharide to animals) under the control of inflammatory cytokines (e.g., interleukin-1, tumor necrosis factor-α, interferon-γ) and signaling (e.g., mitogen activated protein kinases, NF-κB, Jak/Stat) [24,25,26,27]. While B1R and B2R are structurally related, only the latter is phosphorylated, internalized, and recycled following agonist stimulation [1,24]. Thus, the B1R is potentially important in sustained inflammatory states. It does not follow that B1R should be systematically blocked in tissue injury situations: for instance, the development of an adaptative collateral circulation is mediated by this receptor subtype following an arterial occlusion in a rodent model [28].
Both kinin receptor subtypes are coupled mainly to protein Gq and calcium signaling, leading typically to smooth muscle contraction, but also prominently to vascular endothelial cell stimulation, including calcium-dependent prostanoid and nitric oxide release and fluid leakage secondary to gap formation between endothelial cells [1,24].
Table 2.
Inhibiting the kallikrein-kinin system for treating or preventing attacks of hereditary angioedema.
Table 2.
Inhibiting the kallikrein-kinin system for treating or preventing attacks of hereditary angioedema.
| Type of agentmode of action marker in Figure 1 | Drug or intervention | Development status | Ref. |
|---|---|---|---|
| Parenteral replacement of C1INH 1 | various C1INH concentrates, natural or recombinant | approved, widely used | 29 |
| Gene therapy to increase the endogenous synthesis of C1INH 1 | BMN 311 HAE | clinical trials | 30 |
| OTL-105 HAE | preclinical | 31 | |
| Kunitz-domain based peptide inhibitor of plasma kallikrein 2 | ecallantide | approved | 32 |
| Small molecule inhibitors of plasma kallikrein 2 | berotralstat (BCX7353) | approved | 33 |
| sebetralstat (KVD-900) | clinical trials | 34 | |
| ATN-249, ATN-111 | clinical trials | 35 | |
| Anti-plasma kallikrein mAb 2 | lanadelumab | approved | 36 |
| STAR-0215 | clinical trials | 37 | |
| Transfer of a gene encoding an anti-plasma kallikrein mAb 2 | RegenxBio undisclosed | preclinical | 38 |
| Antisense suppressor of hepatic plasma prekallikrein production 3 | donidalorsen (PKK-L Rx) | clinical trials | 39 |
| Gene therapy to disrupt hepatic plasma prekallikrein production 3 | NTLA-2002 | clinical trials | 40 |
| Small molecule inhibitor of factor XIIa 4 | KV998086 | preclinical | 41 |
| Anti-factor XII mAb 4 | Garadacimab (CSL312) | clinical trials | 42 |
| Small interfering RNA targeting factor XII mRNA 5 | ALN-F12 | preclinical, halted? | 43 |
| ARC-F12 | preclinical, halted? | 44 | |
| Plasmin/tPA inhibitor 6 | tranexamic acid | approved, 2nd line prophylactic agent | 45 |
| Bradykinin B2R antagonists 8 | peptide icatibant | approved | 46 |
| NPA deucrictibant (PHA-022121, PHA-121) | clinical trials | 23, 47 |
3. Pharmacological inhibition of the KKS: hereditary angioedema (HAE)
Figure 1 is a schematic representation of the kinin-generating pathways and their receptors; numerical markers indicate the mode of action of the numerous drugs of the KKS. Earlier achievements, such as the early peptide receptor antagonists, are reviewed elsewhere [48]. The present emphasis is on drugs that are currently in use, have reached clinical development (successfully or not), or are in preclinical development.
The therapeutic showcase of the KKS is presently hereditary angioedema (HAE), a rare disease most often caused by the haplodeficiency of C1INH (numerous mutations transmitted in an autosomal dominant manner are known in the corresponding gene SERPING1) [49]. HAE is characterized by recurrent episodes (attacks) of swelling due to fluid extravasation; limbs, the orofacial and genital areas, and the intestine can be affected. Attacks may be life-threatening (suffocation), painful and incapacitating. The physiopathology of HAE and its management have been recently reviewed [49,50,51]. While C1INH inhibits multiple proteases in the contact, fibrinolytic and complement systems, bradykinin is believed to be the ultimate mediator of HAE-C1INH attacks. Variants of six additional causal genes very rarely cause HAE independently from SERPING1: PLG, F12, KNG1, HS3ST6, ANGPT1, MYOF. The 3 first ones belong to the connected contact or fibrinolytic systems, also plausibly leading to the unregulated production of kinins, while the physiopathology of HAE caused by the exceedingly rare variants of the last 3 genes is essentially unknown. There are certainly other causal genes not currently identified in HAE patients with normal C1INH levels [52]. Age, sex, and hormonal status modulate the frequency and intensity of HAE attacks, the facilitatory effect of estrogens being particularly well documented.
Let us limit ourselves here to a few remarks concerning the therapy of HAE, an admittedly crowded market for such a rare disease. Drugs and biotechnological treatments are used or proposed for attack prevention (prophylaxis), to abort attacks (“on demand” treatments), or both. While numerous HAE therapies that affect the KKS are approved or under development (Table 2), the number of validated molecular target is limited. The parenteral administration of C1INH, or gene therapy to increase the hepatic biosynthesis of normal C1INH, is physiologically sound for HAE-C1INH (and proven by multiple clinical trials for C1INH concentrates). Of note, this intervention also works in many HAE patients with normal C1INH levels. The heart of the contact system is also targeted in HAE (Figure 1, Table 2): plasma kallikrein or its proenzyme prekallikrein, FXIIa or its proenzyme FXII can be suppressed or pharmacologically inhibited by several pharmacological or biotechnological interventions. The proof of concept for a further level of intervention on the contact system has been recently reported in a preclinical study: the mAb 3E8 targets domain 6 of HK, thus inhibiting the assembly of the trimolecular complex HK-prekallikrein-factor XI (mode of action 7 in Figure 1). In transgenic mice that express human HK, mAb 3E8 inhibits dextran sulfate-induced BK formation and FXII activation [53].
On the effector side, the BK B2R antagonists inhibit the vascular manifestations of HAE (Table 2, Figure 1). The injectable and rapidly cleared peptide antagonist icatibant is widely used to abort HAE attacks. The nonpeptide B2R antagonist deucrictibant [23] (Figure 2) is orally bioavailable, more potent, and longer lived than icatibant in vivo; it is currently developed for on demand treatment of HAE attacks (a potentially convenient substitute to subcutaneous icatibant, Table 2). Chronically administered deucrictibant will also be tested for prophylaxis. Both icatibant and deucrictibant are competitive and reversible antagonists at the human B2R [23]; the affinity and/or competitive behavior of these drugs varies for B2Rs from other mammalian species. Kinin B1R antagonists have not been tested for HAE, and the rationale for them may be weak, considering that the human form of the B1R is not effectively coupled to the contact system (see above).
There is clear evidence of fibrinolytic system activation during HAE attacks: the D-dimers that are fibrin degradation products are elevated in blood during attacks, but without thrombotic risks [54]. Whether fibrinolysis initiates the attacks is a complex question that has been reviewed [55]. Let us add two remarks to the debate. Indicating a particular vulnerability, adding recombinant tPA (10 nM) to human whole blood or citrated plasma from HAE-C1INH patients selectively increased the early BK production in vitro (measured by enzyme immunoassay and corroborated with bioassay) [6,56]. In addition, in a survey of patients concerning the circumstances preceding HAE attacks, most identified triggering factors could be related to limited coagulation followed by fibrinolysis: physical exertion, mechanical trauma, infection, menstruation, dental procedures, etc. [57]. Mental stress is one of the top patient-identified triggering factor of HAE attacks [57]. In well controlled clinical psychology studies in healthy volunteers, mental stress activates tPA secretion and the turnover of fibrin (detected by D-dimer formation) [58], further supporting the case for fibrinolysis-induced attacks.
Oral tranexamic acid, an inhibitor of plasmin and tissue plasminogen activator, is approved as a second line prophylactic treatment of HAE. However, it lacks validation through extensive clinical trials for this indication, does not normalize the patient lives as well as more recent therapies and does not work well in some patients [45].
Table 3.
Selected additional indications for the inhibition of the KKS.
| Indication | Drug mode of action marker in Figure 1 | Status | Ref. |
|---|---|---|---|
| ACE-inhibitor induced acquired angioedema | C1INH concentrate 1 | ineffective in a small clinical trial | 59 |
| Idiopathic pulmonary fibrosis, interstitial lung disease | mAb garadacimab (CSL312) 4 | clinical trials | 60 |
| Thrombosis prevention | ALN-F12 5 | preclinical | 61 |
| COVID-19 pneumonia | peptide B2R antagonist icatibant 8 | ineffective in a clinical trial | 62 |
| Cerebral edema/head trauma | B2R NPA anatibant (LF16-0687) 8 | ineffective in an interrupted clinical trial | 63 |
| Osteoarthritis pain | B2R NPA fasitibant (MEN16132) 8 | largely ineffective in a clinical trial | 64 |
| Diabetic macula edema | B1R NPA BI1026706 8 | ineffective in a clinical trial | 65 |
| Glioma | intracerebroventricular icatibant 8 | preclinical | 66 |
| ACE-inhibitor induced acquired angioedema | icatibant 8 | ineffective in a clinical trial | 67 |
| Diarrheal states induced by dextran sulfate | oral icatibant, oral FR173657 8 | preclinical | 68, 69 |
| Pancreatitis | icatibant, FR173657 8 | preclinical | 70, 71 |
| Circulatory complications of burns | icatibant 8 | preclinical | 72, 73 |
| Inflammatory edema of various causes | FR173657, icatibant 8 | preclinical | 74, 75 |
| Breast cancer | B1R NPA SSR240612, etc. 8 | preclinical, in vitro | 76 |
| Inflammatory pain | B1R NPA MK-0686, SS240612 9 | ineffective in clinical trials | 77 |
| Inflammatory bowel disease | B1R NPA SSR240612 9 | preclinical | 78 |
| Airway disease | mAb DX-2300 10 | preclinical | 79 |
| Aortic aneurysm expansion | mAb DX-2300, etc. 10 | preclinical | 80 |
4. Other application of KKS inhibitors
Table 3 presents a selection of therapeutic ongoing or terminated therapeutic projects that exploited inhibitors of the KKS. Some comments are offered here concerning specific indications. Pain is one of the cardinal signs of inflammation; despite good preclinical evidence, the clinical development of sophisticated and orally bioavailable B1R antagonists has failed due to their lack of efficacy in phase 2 trials (Table 3, Figure 2) [77]. Fasitibant, a B2R antagonist injected in an intraarticular manner, as also failed to relieve pain associated with knee osteoarthritis (Figure 2) [64]. These trials were a major disappointment, but preclinical research continues for a specific form of pain associated with breast cancer and its chemotherapy; the blockade of either or both kinin receptor subtypes exert favorable effects on the associated mechanical and cold allodynia [81]. The badly underdeveloped clinical research concerning the B1R as a druggable target could benefit from the repurposing of exquisitely specific drugs that have passed successfully clinical phase 1 development (Figure 2), for instance for the prevention of COVID-19 complications [82].
Gliomas, including the highly malignant glioblastoma multiforme, overexpress the B2R in a manner correlated to clinical aggressivity; it is coupled to several pro-tumoral signaling systems [83]. Inhibiting B2R may be of therapeutic value in this condition, based on a preclinical study that also shows the difficulty of reaching the tumor through the blood-brain barrier with the presently available B2R antagonists (intracerebroventricular icatibant was used in animals) [66]. Interestingly, the B1R is also often present in glioma, but rather supports tumor growth, based on pharmacological blockade or results obtained BDKRB1 knock out mice [66].
ACE inhibitors (enalapril, ramipril, many others) constitute an important class of antihypertensive drugs with benefits on several organs. ACE is a carboxydipeptidase that activates the formation of the pressor hormone angiotensin II from its precursor angiotensin I, but also mediates the most important inactivation pathway for kinins such as BK and Lys-des-Arg9-BK. A certain fraction of the beneficial effects of ACE inhibitors in human patients seems to derive from the potentiation of the vasodilator effect of kinins (mode of action 14 in Figure 1) [84]. Many drugs are associated with acquired angioedema, an unpredictable and potentially life-threatening side effect. The angioedema induced by ACE inhibitors is hypothesized by several investigators to be BK-dependent [85]; for instance, increased blood concentration of the BK metabolite BK1-5 was measured during such episodes [86]. However, such attacks did not respond to administration of icatibant [67] or of a C1INH concentrate [59] in clinical settings that are comparable to HAE attacks for which these interventions are therapeutic. Possible explanations include the lack of involvement of the contact system in kinin generation, a prominent role for the kinin B1R or, simply, the lack of developing fluid extravasation when the therapies were initiated relatively late during such unexpected episodes. Of note, thrombolytic therapy for ischemic stroke based on a plasminogen activator such as tPA is sometimes associated with brain edema, angioedema, and hypotension: these are serious side effects presumably mediated by excessive BK formation, due to the connection between fibrinolysis and the contact system [4]. A beneficial effect of icatibant has been reported in such episodes [87,88].
An efficient monoclonal antibody that blocks the enzymatic action of tissue kallikrein, DX-2300, has been developed and shown of potential interest in preclinical research (Table 3). Whether this physiological regulator of circulation and renal function could be inhibited in vivo without major side effects remain to be seen; however, DX-2300 is a powerful tool to selectively block the quantitatively important KLK-1-induced kinin formation that is independent from the contact system [6].
5. Therapeutic value of KKS stimulation?
Kinin generation via the contact system can be chronically blocked without apparent harm, as implied above when discussing the therapy of hereditary angioedema. The alternate kallikrein-kinin system, mediated by tissue kallikrein, appears to be adaptative in various ways. Tissue kallikrein promotes reparative neovascularization following experimental ischemia and protects the heart in animal models of pathologies [89,90]. This enzyme, produced in a regulated manner in the kidney, is released in urine and protects from sodium overload and salt-sensitive hypertension [91]. Tissue kallikrein also participates to flow-dependent vasodilation, a local circulatory adaptative mechanism [92]. So, why not consider the parenteral administration of tissue kallikrein in therapeutics? In China, active KLK-1 purified from human urine has reached clinical use for acute ischemic stroke (Table 4). When added to standard thrombolytic therapy, parenteral tissue kallikrein improved the neurological recovery in a significant manner [93]. A pharmaceutically refined recombinant tissue kallikrein, DM199, is being clinically developed for cerebrovascular and renal indications [94,95].
Is intravenous infusion of kinins feasible in intensive care situations where the stimulation of vascular B2Rs has been proposed to have therapeutic value, such as myocardial infarction and ischemic stroke? A preclinical project addressed this possibility by characterizing peptides that are both prodrugs and “soft drugs” [96,97,98] (Table 4). For instance, BK-Arg (BK sequence prolonged with an Arg residue) has virtually no affinity for the B2R, but releases BK in vivo following the action of Arg-CPs in the circulation, with typical vasodilator responses [97]; thus BK-Arg is a prodrug. This peptide is also a soft drug, because its active form, BK, is rapidly cleared with minimal extra-vascular effects. Other activating peptidases and agonist designs have been explored.
Table 4.
Therapeutic stimulation of kinin formation or of kinin receptor signaling.
| Type of agentmode of action marker in Figure 1 | Drug | status | Ref. |
|---|---|---|---|
| Activated tissue kallikrein injection for cerebrovascular or renal indications 11 | purified from human urine: Kailikang | approved in China, stroke | 93 |
| recombinant: DM199 | clinical trials | 94,95 | |
| B2R agonist to open the blood-brain barrier: adjuvant to brain tumor chemotherapy 12 | labradimil, others | clinical trials of labradimil failed | 99, 100 |
| Combined B1R and B2R agonists to open the blood-brain barrier: adjuvant to brain tumor chemotherapy 12, 13 | co-administered peptide agonists or kinin heterodimer | preclinical | 101 |
| Peptide pro-drugs that release bradykinin via the action of peptidases 12 | bradykinin-Arg, D-Arg-bradykinin-Arg-Arg, others | preclinical | 96-98 |
| B2R NPA for breast cancer 12 | FR-190997 and analogs | preclinical, in vitro | 102 |
| Attenuation of Alzheimer’s disease development, B2R agonist 12 | [Hyp3,Thi5,NChg7,Thi8]-BK | preclinical, animal model | 103 |
| ACE inhibition 14 | enalapril, ramipril, many others | a fraction of therapeutic effects mediated by kinins | 84 |
6. Conclusions
In a certain sense, the medicinal chemistry related to the KKS has reached maturity, with the development of modern drugs, injectable biotechnological proteins, and advanced gene therapy projects (Table 2, Table 3 and Table 4). In addition to C1INH replacement therapy, HAE has been the focus of intense drug development efforts based on a limited number of validated targets (plasma kallikrein, FXIIa and their respective zymogens, the B2R). A recent effort to transition to oral therapies is also noted. Concerning other indications, efficacy in various animal models has led to disappointing clinical outcomes, as in many other therapeutic areas. The existence of orally bioavailable drugs that have at least passed clinical phase 1 development (B1R and B2R antagonists, plasma kallikrein inhibitors; Figure 2) could facilitate their repurposing for additional therapeutic indications. For instance, there is evidence for both neuroprotective and detrimental effects of kinins, mediated by either B1R or B2R, in various neurological disorders [103,104]. Let us hope that the underexploited pharmacopeia overviewed in the present paper finds novel clinical applications.
Funding
This research received no external funding.
Conflicts of Interest
The author served as a consultant for Pharvaris B.V. and received research funds from Shire/Takeda, DiaMedica and Pharvaris B.V. The funders had no role in the writing of the manuscript, or in the decision to publish the results.
References
- Leeb-Lundberg, L.M.; Marceau, F.; Müller-Esterl, W.; Pettibone, D.J.; Zuraw, B.L. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol, Rev. 2005, 57, 27–77. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.P.; Joseph, K.; Ghebrehiwet, B. The complex role of kininogens in hereditary angioedema. Front. Allergy 2022, 3, 952753. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.P. Enzymatic pathways in the pathogenesis of hereditary angioedema: the role of C1 inhibitor therapy. J. Allergy Clin. Immunol. 2010, 126, 918–925. [Google Scholar] [CrossRef]
- Gauberti, M.; Potzeha, F.; Vivien, D.; Martinez de Lizarrondo, S. Impact of Bradykinin Generation During Thrombolysis in Ischemic Stroke. Front. Med. (Lausanne) 2018, 5, 195. [Google Scholar] [CrossRef] [PubMed]
- Dobó, J.; Major, B.; Kékesi, K.A.; Szabó, I.; Megyeri, M.; Hajela, K.; Juhász, G.; Závodszky, P.; Gál, P. Cleavage of kininogen and subsequent bradykinin release by the complement component: mannose-binding lectin-associated serine protease (MASP)-1. PLoS One 2011, 6, e20036. [Google Scholar] [CrossRef] [PubMed]
- Charest-Morin, X.; Hébert, J.; Rivard, G.É.; Bonnefoy, A.; Wagner, E.; Marceau, F. Comparing Pathways of Bradykinin Formation in Whole Blood From Healthy Volunteers and Patients With Hereditary Angioedema Due to C1 Inhibitor Deficiency. Front. Immunol. 2018, 9, 2183. [Google Scholar] [CrossRef] [PubMed]
- Yousef, G.M.; Chang, A.; Scorilas, A.; Diamandis, E.P. Genomic organization of the human kallikrein gene family on chromosome 19q13.3-q13.4. Biochem. Biophys. Res. Commun. 2000, 276, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Deperthes, D.; Marceau, F.; Frenette, G.; Lazure, C.; Tremblay, R.R.; Dubé, J.Y. Human kallikrein hK2 has low kininogenase activity while prostate-specific antigen (hK3) has none. Biochim. Biophys. Acta 1997, 1343, 102–106. [Google Scholar] [CrossRef]
- Charest-Morin, X.; Raghavan, A.; Charles, M.L.; Kolodka, T.; Bouthillier, J.; Jean, M.; Robbins, M.S.; Marceau, F. Pharmacological effects of recombinant human tissue kallikrein on bradykinin B2 receptors. Pharmacol. Res. Perspect. 2015, 3, e00119. [Google Scholar] [CrossRef]
- Marceau, F.; Rivard, G.E.; Gauthier, J.M.; Binkley, K.E.; Bonnefoy, A.; Boccon-Gibod, I.; Bouillet, L.; Picard, M.; Levesque, G.; Elfassy, H.L.; et al. Measurement of Bradykinin Formation and Degradation in Blood Plasma: Relevance for Acquired Angioedema Associated With Angiotensin Converting Enzyme Inhibition and for Hereditary Angioedema Due to Factor XII or Plasminogen Gene Variants. Front Med (Lausanne) 2020, 7, 358. [Google Scholar] [CrossRef]
- Marceau, F.; Bachelard, H.; Charest-Morin, X.; Hébert, J.; Rivard, G.E. In vitro modeling of bradykinin-mediated angioedema states. Pharmaceuticals (Basel) 2020, 13, 201. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.M.; Segreti, J.; Banfor, P.N.; Widomski, D.L.; Backes, B.J.; Lin, C.W.; Ballaron, S.J.; Cox, B.F.; Trevillyan, J.M.; Reinhart, G.A.; et al. Effect of bradykinin metabolism inhibitors on evoked hypotension in rats: rank efficacy of enzymes associated with bradykinin-mediated angioedema. Br. J. Pharmacol. 2008, 153, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Scicli, A.G.; Carretero, O.A. Contributions of various rat plasma peptidases to kinin hydrolysis. J. Pharmacol. Exp. Ther., 1989, 251, 817–820. [Google Scholar] [PubMed]
- Defendi, F.; Charignon, D.; Ghannam, A.; Baroso, R.; Csopaki, F.; Allegret-Cadet, M.; Ponard, D.; Favier, B.; Cichon, S.; Nicolie, B.; et al National Reference Centre for Angioedema, C.R.E.A.K.; et al. Enzymatic assays for the diagnosis of bradykinin-dependent angioedema. PLoS One 2013, 8, e70140. [Google Scholar] [CrossRef] [PubMed]
- Marceau, F.; Rivard, G.E.; Hébert, J.; Gauthier, J.; Bachelard, H.; Gangnus, T.; Burckhardt, B.B. Picomolar Sensitivity Analysis of Multiple Bradykinin-Related Peptides in the Blood Plasma of Patients With Hereditary Angioedema in Remission: A Pilot Study. Front. Allergy 2022, 3, 837463. [Google Scholar] [CrossRef]
- Cugno, M.; Salerno, F.; Nussberger, J.; Bottasso, B.; Lorenzano, E.; Agostoni, A. Bradykinin in the ascitic fluid of patients with liver cirrhosis. Clin Sci (Lond) 2001, 101, 651–657. [Google Scholar] [CrossRef]
- Hofman, Z.L.M.; van den Elzen, M.T.; Kuijpers, J.; de Maat, S.; Hack, C.E.; Knulst, A.C.; Röckmann, H.; Maas, C. Evidence for bradykinin release in chronic spontaneous urticaria. Clin. Exp. Allergy. 2020, 50, 343–351. [Google Scholar] [CrossRef]
- Mostmans, Y.; De Smedt, K.; Richert, B.; Elieh Ali Komi, D.; Maurer, M.; Michel, O. Markers for the involvement of endothelial cells and the coagulation system in chronic urticaria: A systematic review. Allergy 2021, 76, 2998–3016. [Google Scholar] [CrossRef]
- Barratt-Due, A.; Johansen, H.T.; Sokolov, A.; Thorgersen, E.B.; Hellerud, B.C.; Reubsaet, J.L.; Seip, K.F.; Tønnessen, T.I.; Lindstad, J.K.; Pharo, A.; et al. The role of bradykinin and the effect of the bradykinin receptor antagonist icatibant in porcine sepsis. Shock 2011, 36, 517–523. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.M.; Kasztan, M.; Henderson, M.W.; Ellsworth, P.; Davis, P.R.; Wilson, K.J.; Reeves, B.; Key, N.S.; Strickland, S.; McCrae, K.; et al. High molecular weight kininogen contributes to early mortality and kidney dysfunction in a mouse model of sickle cell disease. J. Thromb. Haemost. 2020, 18, 2329–2340. [Google Scholar] [CrossRef]
- Regoli, D.; Barabé, J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980, 32, 1–46. [Google Scholar]
- Hock, F.J.; Wirth, K.; Albus, U.; Linz, W.; Gerhards, H.J.; Wiemer, G.; Henke, S.; Breipohl, G.; König, W.; Knolle, J.; et al. Hoe 140 a new potent and long acting bradykinin-antagonist: in vitro studies. Br. J. Pharmacol. 1991, 102, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Lesage, A.; Marceau, F.; Gibson, C.; Loenders, B.; Katzer, W.; Ambrosi, H.D.; Saupe, J.; Faussner, A.; Pardali, E.; Knolle, J. In vitro pharmacological profile of PHA-022121, a small molecule bradykinin B2 receptor antagonist in clinical development. Int. Immunopharmacol. 2022, 105, 108523. [Google Scholar] [CrossRef] [PubMed]
- Marceau, F.; Bachelard, H.; Bouthillier, J.; Fortin, J.P.; Morissette, G.; Bawolak, M.T.; Charest-Morin, X.; Gera, L. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int. Immunopharmacol. 2020, 82, 106305. [Google Scholar] [CrossRef]
- Larrivée, J.-F.; Bachvarov, D.R.; Houle, F.; Landry, J.; Huot, J.; Marceau, F. Role of the mitogen-activated protein kinases in the expression of the kinin B1 receptors induced by tissue injury. J. Immunol. 1998, 160, 1419–1426. [Google Scholar] [CrossRef]
- Moreau, M.E.; Bawolak, M.T.; Morissette, G.; Adam, A.; Marceau, F. Role of nuclear factor-kappaB and protein kinase C signaling in the expression of the kinin B1 receptor in human vascular smooth muscle cells. Mol. Pharmacol. 2007, 71, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Koumbadinga, G.A.; Désormeaux, A.; Adam, A.; Marceau, F. Effect of interferon-γ on inflammatory cytokine-induced bradykinin B1 receptor expression in human vascular cells. Eur. J. Pharmacol. 2010, 647, 117–125. [Google Scholar] [CrossRef]
- Emanueli, C.; Bonaria Salis, M.; Stacca, T.; Pintus, G.; Kirchmair, R.; Isner, J.M.; Pinna, A.; Gaspa, L.; Regoli, D.; Cayla, C.; et al. Targeting kinin B1 receptor for therapeutic neovascularization. Circulation 2002, 105, 360–366. [Google Scholar] [CrossRef]
- Longhurst, H.; Farkas, H. Biological therapy in hereditary angioedema: transformation of a rare disease. Expert Opin. Biol. Ther. 2020, 20, 493–501. [Google Scholar] [CrossRef]
- Biomarin. https://www.biomarin.com/our-treatments/pipeline/bmn-331-for-hae/ [last accessed 1 May 2023].
- Pharming Group, N.V. https://www.pharming.com/pipeline [last accessed 1 May 2023].
- Duffey, H.; Firszt, R. Management of acute attacks of hereditary angioedema: role of ecallantide. J. Blood Med. 2015, 6, 115–123. [Google Scholar] [CrossRef]
- Ahuja, M.; Dorr, A.; Bode, E.; Boulton, A.P.R.; Buckland, M.; Chee, S.; Dalley, C.; Denman, S.; Ekbote, A.; Elkhalifa, S.; et al. Berotralstat for the prophylaxis of hereditary angioedema-Real-world evidence data from the United Kingdom. Allergy 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Aygören-Pürsün, E.; Zanichelli, A.; Cohn, D.M.; Cancian, M.; Hakl, R.; Kinaciyan, T.; Magerl, M.; Martinez-Saguer, I.; Stobiecki, M.; Farkas, H.; et al. An investigational oral plasma kallikrein inhibitor for on-demand treatment of hereditary angioedema: a two-part, randomised, double-blind, placebo-controlled, crossover phase 2 trial. Lancet 2023, 401, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Kalfus, I.; Offman, E.; McDonald, A. Pharmacokinetics, safety, and potency of ATN-249, a novel oral plasma kallikrein inhibitor for hereditary angioedema. Allergy Asthma Clin. Immunol. 2019; 15(suppl 4), 45. [Google Scholar]
- Riedl, M.A.; Maurer, M.; Bernstein, J.A.; Banerji, A.; Longhurst, H.J.; Li, H.H.; Lu, P.; Hao, J.; Juethner, S.; Lumry, W.R.; et al. Lanadelumab demonstrates rapid and sustained prevention of hereditary angioedema attacks. Allergy 2020, 75, 2879–2887. [Google Scholar] [CrossRef] [PubMed]
- Astria Therapeutics. https://astriatx.com/our-science/scientific-presentations-and-publications/ [last accessed 26 April 2023]/.
- REGENXBIO Inc. https://ir.regenxbio.com/news-releases/news-release-details/regenxbio-reports-continued-progress-across-programs-year-end-0/ [last accessed 1 May 2023].
- Ferrone, J.D.; Bhattacharjee, G.; Revenko, A.S.; Zanardi, T.A.; Warren, M.S.; Derosier, F.J.; Viney, N.J.; Pham, N.C.; Kaeser, G.E.; Baker, B.F.; et al. IONIS-PKKRx a Novel Antisense Inhibitor of Prekallikrein and Bradykinin Production. Nucleic Acid Ther. 2019, 29, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Intellia Therapeutics, Inc. https://www.intelliatx.com/our-science/publications-and-presentations/ [last accessed 26 April 2023].
- KalVista Pharmaceuticals. https://www.kalvista.com/products-pipeline/factor-xiia [last accessed 26 April 2023].
- Craig, T.J.; Reshef, A.; Li, H.H.; Jacobs, J.S.; Bernstein, J.A.; Farkas, H.; Yang, W.H.; Stroes, E.S.G.; Ohsawa, I.; Tachdjian, R.; et al. Efficacy and safety of garadacimab, a factor XIIa inhibitor for hereditary angioedema prevention (VANGUARD): a global, multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2023, 401, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qin, J.; Borodovsky, A.; Racie, T.; Castoreno, A.; Schlegel, M.; Maier, M.A.; Zimmerman, T.; Fitzgerald, K.; Butler, J.; Akinc, A. An investigational RNAi therapeutic targeting Factor XII (ALN-F12) for the treatment of hereditary angioedema. RNA 2019, 25, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Arrowhead Pharmaceuticals, Inc. http://ir.arrowheadpharma.com/news-releases/news-release-details/arrowhead-pharmaceuticals-presents-new-data-arc-f12-and-arc-lpa [Last accessed 1 May 2023].
- Wintenberger, C.; Boccon-Gibod, I.; Launay, D.; Fain, O.; Kanny, G.; Jeandel, P.Y.; Martin, L.; Gompel, A.; Bouillet, L. Tranexamic acid as maintenance treatment for non-histaminergic angioedema: analysis of efficacy and safety in 37 patients. Clin. Exp. Immunol. 2014, 178, 112–117. [Google Scholar] [CrossRef]
- Maurer, M.; Aberer, W.; Caballero, T.; Bouillet, L.; Grumach, A.S.; Botha, J.; Andresen, I.; Longhurst, H.J.; IOS Study Group. The Icatibant Outcome Survey: 10 years of experience with icatibant for patients with hereditary angioedema. Clin. Exp. Allergy 2022, 52, 1048–1058. [Google Scholar] [CrossRef]
- Maurer, M.; Anderson, J.; Aygören-Pürsün, E.; Bouillet, L.; Baeza, M.L.; Chapdelaine, H.; et al. Efficacy And Safety of Bradykinin B2 Receptor Inhibition With Oral PHVS416 In Treating Hereditary Angioedema Attacks: Results Of RAPIDe-1 Phase 2 Trial. J. Allergy Clin. Immunol. 2023, 151, Supplement–AB134. [Google Scholar] [CrossRef]
- Marceau, F.; Regoli, D. Bradykinin receptor ligands: therapeutic perspectives. Nat. Rev. Drug Discov. 2004, 3, 845–852. [Google Scholar] [CrossRef]
- Petersen, R.S.; Fijen, L.M.; Levi, M.; Cohn, D.M. Hereditary Angioedema: The Clinical Picture of Excessive Contact Activation. Semin. Thromb. Hemost. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Valerieva, A.; Longhurst, H.J. Treatment of hereditary angioedema-single or multiple pathways to the rescue. Front. Allergy 2022, 3, 952233. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Magerl, M.; Betschel, S.; Aberer, W.; Ansotegui, I.J.; Aygören-Pürsün, E.; Banerji, A.; Bara, N.A.; Boccon-Gibod, I.; Bork, K.; et al. The international WAO/EAACI guideline for the management of hereditary angioedema - The 2021 revision and update. World Allergy Organ J. 2022, 15, 100627. [Google Scholar] [CrossRef] [PubMed]
- Bork, K.; Wulff, K.; Witzke, G.; Staubach, P.; Hardt, J.; Meinke, P. Gene Mutations Linked to Hereditary Angioedema in Solitary Angioedema Patients With Normal C1 Inhibitor. J. Allergy Clin. Immunol. Pract. 2023, 12. in press. [Google Scholar] [CrossRef]
- Chen, Z.L.; Singh, P.K.; Horn, K.; Calvano, M.R.; Kaneki, S.; McCrae, K.R.; Strickland, S.; Norris, E.H. Anti-HK antibody inhibits the plasma contact system by blocking prekallikrein and factor XI activation in vivo. Blood Adv. 2023, 7, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Reshef, A.; Zanichelli, A.; Longhurst, H.; Relan, A.; Hack, C.E. Elevated D-dimers in attacks of hereditary angioedema are not associated with increased thrombotic risk. Allergy 2015, 70, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Maas, C. Plasminflammation-An Emerging Pathway to Bradykinin Production. Front. Immunol. 2019, 10, 2046. [Google Scholar] [CrossRef]
- Marceau, F.; Bachelard, H.; Rivard, G.É.; Hébert, J. Increased fibrinolysis-induced bradykinin formation in hereditary angioedema confirmed using stored plasma and biotechnological inhibitors. BMC Res. Notes 2019, 12, 291. [Google Scholar] [CrossRef]
- Zotter, Z.; Csuka, D.; Szabó, E.; Czaller, I.; Nébenführer, Z.; Temesszentandrási, G.; Fust, G.; Varga, L.; Farkas, H. The influence of trigger factors on hereditary angioedema due to C1-inhibitor deficiency. Orphanet J. Rare Dis. 2014; 9, 44. [Google Scholar] [CrossRef]
- von Känel, R. Acute mental stress and hemostasis: When physiology becomes vascular harm. Thromb Res. 2015, 135 Suppl 1, S52–S55. [Google Scholar] [CrossRef]
- Strassen, U.; Bas, M.; Wirth, M.; Wirth, M.; Gröger, M.; Stelter, K.; Volkenstein, S.; Kehl, V.; Kojda, G.; Hoffmann, T.K.; et al. Efficacy of human C1 esterase inhibitor concentrate for treatment of ACE-inhibitor induced angioedema. Am. J. Emerg. Med. 2023, 64, 121–128. [Google Scholar] [CrossRef]
- CSL, Limited. https://www.csl. 1 May.
- Liu, J.; Cooley, B.C.; Akinc, A.; Butler, J.; Borodovsky, A. Knockdown of liver-derived factor XII by GalNAc-siRNA ALN-F12 prevents thrombosis in mice without impacting hemostatic function. Thromb. Res. 2020, 196, 200–205. [Google Scholar] [CrossRef] [PubMed]
- ISPY COVID Consortium. Report of the first seven agents in the I-SPY COVID trial: a phase 2, open label, adaptive platform randomised controlled trial. EClinicalMedicine 2023, 58, 101889. [Google Scholar] [CrossRef] [PubMed]
- Shakur, H.; Andrews, P.; Asser, T.; Balica, L.; Boeriu, C.; Quintero, J.D.; Dewan, Y.; Druwé, P.; Fletcher, O.; Frost, C.; et al. The BRAIN TRIAL: a randomised, placebo controlled trial of a Bradykinin B2 receptor antagonist (Anatibant) in patients with traumatic brain injury. Trials 2009, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.G.; Pavelka, K.; Nizzardo, A.; Rossi, C.; Scartoni, S.; Contini, M.P.; di Molfetta, S.; Bertolotti, M.; Capriati, A.; Maggi, C.A. A Double-Blind, Randomized, Controlled, Four parallel Arm, Dose-Finding Study to Evaluate the Efficacy, Safety, Tolerability, and Pharmacokinetics of Single Intra-Articular (IA) Injections of Fasitibant in Patients with Symptomatic OA of the Knee [abstract]. Arthritis Rheumatol. 2015, 67 (suppl 10). https://acrabstracts.org/abstract/a-double-blind-randomized-controlled-four-parallel-arm-dose-finding-study-to-evaluate-the-efficacy-safety-tolerability-and-pharmacokinetics-of-single-intra-articular-ia-injections-of-fas/. [Last Accessed 1 May 2023].
- Lang, G.E.; Tadayoni, R.; Tang, W.; Barth, C.; Weiss-Haljiti, C.; Chong, V. BI 1026706 Study Group. Bradykinin 1 Receptor Antagonist BI1026706 Does Not Reduce Central Retinal Thickness in Center-Involved Diabetic Macular Edema. Transl. Vis. Sci. Technol. 2020, 9, 25. [Google Scholar] [CrossRef]
- Nicoletti, N.F.; Sénécal, J.; da Silva, V.D.; Roxo, M.R.; Ferreira, N.P.; de Morais, R.L.T.; Pesquero, J.B.; Campos, M.M.; Couture, R.; Morrone, F.B. Primary Role for Kinin B1 and B2 Receptors in Glioma Proliferation. Mol. Neurobiol. 2017, 54, 7869–7882.e2. [Google Scholar] [CrossRef] [PubMed]
- Straka, B.T.; Ramirez, C.E.; Byrd, J.B.; Stone, E.; Woodard-Grice, A.; Nian, H.; Yu, C.; Banerji, A.; Brown, N.J. Effect of bradykinin receptor antagonism on ACE inhibitor-associated angioedema. J. Allergy Clin. Immunol. 2017, 140, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Takanashi, H.; Kitagawa, H.; Wirth, K.J.; Okayasu, I. Effect of icatibant, a bradykinin B2 receptor antagonist, on the development of experimental ulcerative colitis in mice. Dig. Dis. Sci. 1999, 44, 845–851. [Google Scholar] [CrossRef]
- Kamata, K.; Hayashi, I.; Mizuguchi, Y.; Arai, K.; Saeki, T.; Ohno, T.; Saigenji, K.; Majima, M. Suppression of dextran sulfate sodium-induced colitis in kininogen-deficient rats and non-peptide B2 receptor antagonist-treated rats. Jpn. J. Pharmacol. 2002, 90, 59–66. [Google Scholar] [CrossRef]
- Hirata, M.; Hayashi, I.; Yoshimura, K.; Ishii, K.; Soma, K.; Ohwada, T.; Kakita, A.; Majima, M. Blockade of bradykinin B2 receptor suppresses acute pancreatitis induced by obstruction of the pancreaticobiliary duct in rats. Br. J. Pharmacol. 2002, 135, 29–36. [Google Scholar] [CrossRef]
- Kanbe, T.; Naruse, S.; Kitagawa, M.; Nakae, Y.; Hayakawa, T. Effects of a bradykinin receptor antagonist (HOE140) on taurocholate-induced acute pancreatitis in rats. Pancreas 1996, 13, 283–288. [Google Scholar] [CrossRef]
- Jonkam, C.C.; Enkhbaatar, P.; Nakano, Y.; Boehm, T.; Wang, J.; Nussberger, J.; Esechie, A.; Traber, L.D.; Herndon, D.; Traber, D.L. Effects of the bradykinin B2 receptor antagonist icatibant on microvascular permeability after thermal injury in sheep. Shock 2007, 28, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; O’Halloran, E.B.; Ippolito, J.A.; Choudhry, M.A.; Kovacs, J. Alcohol potentiates postburn remote organ damage through shifts in fluid compartments mediated by bradykinin. Shock 2015, 43, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Griesbacher, T.; Legat, F.J. Effects of the non-peptide B2 receptor antagonist FR173657 in models of visceral and cutaneous inflammation. Inflamm. Res. 2000, 49, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.J.; Alpermann, H.G.; Satoh, R.; Inazu, M. The bradykinin antagonist Hoe 140 inhibits carrageenan- and thermically induced paw oedema in rats. Agents Actions Suppl. 1992, 38, 428–431. [Google Scholar]
- Dubuc, C.; Savard, M.; Bovenzi, V.; Lessard, A.; Côté, J.; Neugebauer, W.; Geha, S.; Chemtob, S.; Gobeil, F. Jr. Antitumor activity of cell-penetrant kinin B1 receptor antagonists in human triple-negative breast cancer cells. J. Cell. Physiol. 2019, 234, 2851–2865. [Google Scholar] [CrossRef] [PubMed]
- Fincham, C.I.; Bressan, A.; Paris, M.; Rossi, C.; Fattori, D. Bradykinin receptor antagonists - a review of the patent literature 2005-2008. Expert Opin. Ther. Pat. 2009, 19, 919–941. [Google Scholar] [CrossRef]
- Hara, D.B.; Leite, D.F.; Fernandes, E.S.; Passos, G.F.; Guimarães, A.O.; Pesquero, J.B.; Campos, M.M.; Calixto, J.B. The relevance of kinin B1 receptor upregulation in a mouse model of colitis. Br. J. Pharmacol. 2008, 154, 1276–1286. [Google Scholar] [CrossRef]
- Sexton, D.J.; Chen, T.; Martik, D.; Kuzmic, P.; Kuang, G.; Chen, J.; Nixon, A.E.; Zuraw, B.L.; Forteza, R.M.; Abraham, W.M.; et al. Specific inhibition of tissue kallikrein 1 with a human monoclonal antibody reveals a potential role in airway diseases. Biochem J. 2009, 422, 383–392. [Google Scholar] [CrossRef]
- Moran, C.S.; Biros, E.; Krishna, S.M.; Morton, S.K.; Sexton, D.J.; Golledge, J. Kallikrein-1 Blockade Inhibits Aortic Expansion in a Mouse Model and Reduces Prostaglandin E2 Secretion From Human Aortic Aneurysm Explants. J. Am. Heart Assoc. 2021, 10, e019372. [Google Scholar] [CrossRef]
- Brusco, I.; Becker, G.; Palma, T.V.; Pillat, M.M.; Scussel, R.; Steiner, B.T.; Sampaio, T.B.; Ardisson-Araújo, D.M.P.; de Andrade, C.M.; Oliveira, M.S.; et al. Kinin B1 and B2 receptors mediate cancer pain associated with both the tumor and oncology therapy using aromatase inhibitors. Sci. Rep. 2023, 13, 4418. [Google Scholar] [CrossRef]
- Mendes, G.M.M.; Do Nascimento, I.J.B.; Marazzi-Diniz, P.H.; Da Silveira, I. .B.; Itaborahy, M.F.; Viana, L.E.; Silva, F.A.; Santana, M.F.; Pinto, R.A.; Dutra, B.G.; et al. The des-Arg9-bradykinin/B1R axis: Hepatic damage in COVID-19. Front, Physiol. 2022, 13, 1080837. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, J.; Shi, F.; Shan, A.; Xu, S.; Lv, W. BDKRB2 is a novel EMT-related biomarker and predicts poor survival in glioma. Aging (Albany NY), 2021, 13, 7499–7516. [Google Scholar] [CrossRef] [PubMed]
- Gainer, J.V.; Morrow, J.D.; Loveland, A.; King, D.J.; Brown, N.J. Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N. Engl. J. Med. 1998, 339, 1285–1292. [Google Scholar] [CrossRef]
- Hoover, T.; Lippmann, M.; Grouzmann, E.; Marceau, F.; Herscu, P. Angiotensin converting enzyme inhibitor induced angio-oedema: a review of the pathophysiology and risk factors. Clin. Exp. Allergy 2010, 40, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Hubers, S.A.; Kohm, K.; Wei, S.; Yu, C.; Nian, H.; Grabert, R.; Sexton, D.J.; Brown, N.J. Endogenous bradykinin and B1-B5 during angiotensin-converting enzyme inhibitor-associated angioedema. J. Allergy Clin. Immunol. 2018, 142, 1636–1639.e5. [Google Scholar] [CrossRef]
- Cheong, E.; Dodd, L.; Smith, W.; Kleinig, T. Icatibant as a Potential Treatment of Life-Threatening Alteplase-Induced Angioedema. J. Stroke Cerebrovasc. Dis. 2018, 27, e36–e37. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.; Campana, C.; Zimmerman, J.; Brooks, S. Icatibant for the treatment of orolingual angioedema following the administration of tissue plasminogen activator. Am. J. Emerg. Med. 2018, 36, 1125.e1–1125.e2. [Google Scholar] [CrossRef]
- Stone, O.A.; Richer, C.; Emanueli, C.; van Weel, V.; Quax, P.H.; Katare, R.; Kraenkel, N.; Campagnolo, P.; Barcelos, L.S.; Siragusa, M.; et al. Critical role of tissue kallikrein in vessel formation and maturation: implications for therapeutic revascularization. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 657–664. [Google Scholar] [CrossRef]
- Koch, M.; Spillmann, F.; Dendorfer, A.; Westermann, D.; Altmann, C.; Sahabi, M.; Linthout, S.V.; Bader, M.; Walther, T.; Schultheiss, H.P.; et al. Cardiac function and remodeling is attenuated in transgenic rats expressing the human kallikrein-1 gene after myocardial infarction. Eur. J. Pharmacol. 2006, 550, 143–148. [Google Scholar] [CrossRef]
- Katori, M.; Majima, M. Renal (tissue) kallikrein-kinin system in the kidney and novel potential drugs for salt-sensitive hypertension. Prog. Drug Res. 2014, 69, 59–109. [Google Scholar] [CrossRef]
- Bergaya, S.; Meneton, P.; Bloch-Faure, M.; Mathieu, E.; Alhenc-Gelas, F.; Lévy, B.I.; Boulanger, C.M. Decreased flow-dependent dilation in carotid arteries of tissue kallikrein-knockout mice. Circ. Res. 2001, 88, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, J.; Wang, L.; Liu, J. Urinary Kallidinogenase plus rt-PA Intravenous Thrombolysis for Acute Ischemic Stroke: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Comput. Math. Methods Med. 2022, 2022, 1500669. [Google Scholar] [CrossRef]
- Alexander-Curtis, M.; Pauls, R.; Chao, J.; Volpi, J.J.; Bath, P.M.; Verdoorn, T.A. Human tissue kallikrein in the treatment of acute ischemic stroke. Ther. Adv. Neurol. Disord. 2019, 12, 1756286418821918. [Google Scholar] [CrossRef] [PubMed]
- Shehjar, F.; Maktabi, B.; Rahman, Z.A.; Bahader, G.A.; James, A.W.; Naqvi, A.; Mahajan, R.; Shah, Z.A. Stroke: Molecular mechanisms and therapies: Update on recent developments. Neurochem. Int. 2023, 162, 105458. [Google Scholar] [CrossRef]
- Charest-Morin, X.; Roy, C.; Fortin, E.J.; Bouthillier, J.; Marceau, F. Pharmacological evidence of bradykinin regeneration from extended sequences that behave as peptidase-activated B2 receptor agonists. Front. Pharmacol. 2014, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Jean, M.; Gera, L.; Charest-Morin, X.; Marceau, F.; Bachelard, H. In Vivo Effects of Bradykinin B2 Receptor Agonists with Varying Susceptibility to Peptidases. Front. Pharmacol. 2016, 6, 306. [Google Scholar] [CrossRef] [PubMed]
- Bachelard, H.; Charest-Morin, X.; Marceau, F. D-Arg0-Bradykinin-Arg-Arg, a Latent Vasoactive Bradykinin B2 Receptor Agonist Metabolically Activated by Carboxypeptidases. Front. Pharmacol. 2018, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Prados, M.D.; Schold, S.C.; Fine, H.A.; Jaeckle, K.; Hochberg, F.; Mechtler, L.; Fetell, M.R.; Phuphanich, S.; Feun, L.; Janus, T.J.; et al. A randomized, double-blind, placebo-controlled, phase 2 study of RMP-7 in combination with carboplatin administered intravenously for the treatment of recurrent malignant glioma. Neuro. Oncol. 2003, 5, 96–103. [Google Scholar] [CrossRef]
- Warren, K.; Jakacki, R.; Widemann, B.; Aikin, A.; Libucha, M.; Packer, R.; Vezina, G.; Reaman, G.; Shaw, D.; Krailo, M.; et al. Phase II trial of intravenous lobradimil and carboplatin in childhood brain tumors: A report from the Children’s Oncology Group. Cancer Chemother. Pharmacol. 2006, 58, 343–347. [Google Scholar] [CrossRef]
- Côté, J.; Savard, M.; Neugebauer, W.; Fortin, D.; Lepage, M.; Gobeil, F. Dual kinin B1 and B2 receptor activation provides enhanced blood-brain barrier permeability and anticancer drug delivery into brain tumors. Cancer Biol. Ther. 2013, 14, 806–811. [Google Scholar] [CrossRef]
- Rassias, G.; Leonardi, S.; Rigopoulou, D.; Vachlioti, E.; Afratis, K.; Piperigkou, Z.; Koutsakis, C.; Karamanos, N.K.; Gavras, H.; Papaioannou, D. Potent antiproliferative activity of bradykinin B2 receptor selective agonist FR-190997 and analogue structures thereof: A paradox resolved? Eur. J. Med. Chem. 2021, 210, 112948. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.A.; Toricelli, M.; Schöwe, N.M.; Malerba, H.N.; Dong-Creste, K.E.; Farah, D.M.A.T.; De Angelis, K.; Irigoyen, M.C.; Gobeil, F.; Araujo Viel, T.; et al. Kinin B2 Receptor Activation Prevents the Evolution of Alzheimer's Disease Pathological Characteristics in a Transgenic Mouse Model. Pharmaceuticals (Basel) 2020, 13, 288. [Google Scholar] [CrossRef] [PubMed]
- Nokkari, A.; Abou-El-Hassan, H.; Mechref, Y.; Mondello, S.; Kindy, M.S.; Jaffa, A.A.; Kobeissy, F. Implication of the Kallikrein-Kinin system in neurological disorders: Quest for potential biomarkers and mechanisms. Prog. Neurobiol. 2018, 165–167, 26–50. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



