
Submitted:
12 May 2023
Posted:
15 May 2023
You are already at the latest version
Abstract
We hypothesize the possible route of introduction of the cosmopolitan DENV-2 genotype into Brazil through the border with Peru. The five sequences presented in this study create a geographical link with the three others sequences of this genotype already recorded on the continent.
Keywords:
arboviruses
; dengue virus serotype 2
; Cosmopolitan genotype
1. Introduction
Dengue virus (DENV) is a major cause of morbidity and mortality worldwide, causing recurrent epidemics in urban and rural areas. DENV includes four antigenically distinct and genetically similar serotypes, which contain about 65% identity of their genome [1].
The most recent epidemiological reports on epidemics in South America have described a wide circulation of serotypes DENV-1 and DENV-2, with DENV-2 seriously contributing to dengue-related mortality [2]. In Brazil, the dengue viruses circulate in all regions, causing outbreaks and epidemics alternately among Brazilian states [3]. According to Brazilian Ministry of Health reports, 2,956,149 suspected cases of dengue fever were registered from 2020 to 2022, with 1798 deaths. The co-circulation of serotypes DENV-1 and DENV-2 was also observed with a high frequency, such as in other South American countries [2-7]. In this period, the outbreaks occurred simultaneously with the SARS-CoV-2 pandemic, which contribute to a less detailed investigation and a consequent underreport of epidemiological surveillance data in several Brazilian states.
DENV-2 includes five non-sylvatic genotypes: American – Genotype I (restricted to Central and South America and already extinct); Asian I and Asian II – Genotypes IV and V (Asian continent countries); Asian/American – Genotype III (Central and South America, and Southeast Asia), and; Cosmopolitan – Genotype II (Asia-Pacific, Middle East, and Africa), this being the most widespread, with a significant contribution in global parameter to the total number of DENV-2 cases [8,9].
Much has been discussed about genomic diversity, evolution, and transmission dynamics in regions favorable for virus dissemination. The high geographic circulation creates potential conditions for the proliferation of viral agents and their genotypes and lineages. This determinant added to the high growth rate of spatial distribution set the favorable conditions for the dissemination of vectors, modifications of evolutionary dynamics, and remodeling of the enzootic amplification of DENV [10].
From the introduction of DENV-2 in Brazil in the 1990s to 2020, only the genotype III was detected in the country [3,11]. A new genotype, the Cosmopolitan (genotype II) was detected for the first time in Brazil from a human case reported in the state of Goiás (Midwest region) in November 2021 [12], being the second official record of this genotype in the Americas, following an outbreak in Peru in 2019 [9]. The increased dengue severity on the continent may be related to the shift from single serotype endemicity to hyperendemicity from the co-circulation of multiple serotypes and the introduction of new genotypes [9,13].
Based on the understanding of the evolutionary dynamics and global dispersion of DENV-2 in the Americas, we can presume that there has been a more expressive heterogeneity of the Cosmopolitan genotype when compared to the other DENV-2 genotypes. Despite the phylogenetical relationship between the single case of the cosmopolitan genotype in the Midwest region of Brazil and the cases described in Madre de Dios, Peru [9,12], no geographic link between these regions, which do not share borders, has been established [12,14,15].
Here we describe cases of the Cosmopolitan genotype in Brazil reported during a DENV-2 outbreak in the state of Acre (Northern region) in early 2021. These cases create a closer geographic link between the findings from Peru and the Midwest region of Brazil, clarifying the most likely route of introducing this genotype in Brazil from the border with Peru at the Madre de Dios region.
2. Materials and Methods
2.1. Study samples
The study samples were selected within an epidemiological context, in the period 2020-2021, during an outbreak in the state of Acre, northern Brazil, and totalize 163 sera samples from humans clinically suspected of dengue fever.
2.2. RNA extraction
For RNA extraction, 140 µL of cell culture supernatant was used using the QIAamp® Viral RNA Mini Kit (Qiagen, Hilden Germany), following the manufacturer's recommendations. After purification, the RNA was quantified using Qubit RNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) together with the Qubit 3.0 equipment (Thermo Fisher Scientific), according to the manufacturer's recommendations. They were subsequently stored in a -70 ºC freezer (under a range of -60 ºC to -80 ºC) until use.
2.3. Molecular detection
The assay was performed using the SuperScript III Platinum One-Step Quantitative RT-PCR Kit (Thermo Fisher Scientific), and included a set of oligonucleotide primers and dual-labeled fluorescent (Taqman) probes for in vitro, qualitative detection of DENV-1–4. Target amplification was recorded as increase and accumulation of fluorescence over time in contrast to background signal. The 25 µL reaction was composed of 12.5 µL of a 2X SuperScript III Platinum One-Step RT-PCR Master Mix, 2,2 µL of nuclease-free water, 1,0 µL of forward and reverse primers for DENV-1 (F – 5’, CAAAAGGAAGTCGYGCAATA, 3’; R – 5’, CTGAGTGAATTCTCTCTGCTRAAC, 3’), 0.5 µL for DENV-2 (F – 5’, CAGGCTATGGCACYGTCACGAT, 3’; R - 5’, CCATYTGCAGCARCACCATCTC, 3’), 1,0 for DENV-3 (F – 5’, GGACTRGACACACGCACCCA, 3’; R - 5’, CATGTCTCTACCTTCTCGACTTGYCT, 3’), 0,5 for DENV-4 (F – 5’, TTGTCCTAATGATGCTRGTCG, 3’; R - 5’, TCCACCYGAGACTCCTTCCA, 3’), 0.25 µL of DENV-1-DENV-4 probes, and 5 µL of extracted RNA [16].
In a 7500 Fast Real-Time PCR system (Thermo Fisher Scientific), the RT-qPCR assays were performed under the following cycling conditions: an initial RT step at 50 °C for 30 min, a denaturation step at 95 °C for 2 min, 45 cycles of 15 s at 95 °C and a final extension step of 1 min at 60 °C. Each sample was analyzed in duplicate and considered as positive when the average cycle threshold (Ct) value was less than 37. In total, 163 samples were tested. Positive samples for DENV-2 with Ct values less than 20 were selected for viral isolation and subsequent genomic analysis of the supernatants. The assay was validated by positive (DENV-1-4 lyophilized antigens) and negative (nuclease-free water) controls.
2.4. Viral isolation
To obtain the isolates, we used an Aedes albopictus derived cell line, clone C6/36 (ATCC: CRL1 660), which were seeded in 10 mL culture tubes containing 1.5 mL of glutamine-modified Leibowitz medium (L-15), plus 2.95% tryptose phosphate, non-essential amino acids, antibiotics (penicillin and streptomycin), and 2% fetal bovine serum. They were inoculated in a 1:10 ratio of serum in glutamine-modified Leibowitz medium (L-15) and were observed daily for a period of 7 days or until cytopathogenic effect (CPE) was verified. Upon completion they were forwarded for genomic sequencing.
2.5. Sequencing
Genomic analysis using Next-Generation Sequencing (NGS) was performed on five DENV-2 isolates of human sera samples screened by RT-qPCR. They were prepared for sequencing by synthesizing first and second strands of complementary DNA, which were obtained with the cDNA Synthesis System Kit (Roche Diagnostics, Basel, Switzerland) and 400 µM Roche random primer. Agencourt AMPure XP Reagent Kit (Beckman Coulter, Brea, CA, USA) magnetic beads were used for cDNA purification and library preparation was performed by amplicon using the DNAprep kit (Illumina, San Diego, CA, USA) adapted to the DENV-2 oligonucleotide set. Quantification of cDNA was assessed using Qubit 3.0 Fluorometer (Thermo Fisher Scientific) and fragments size range was evaluated using 2100 Bioanalyzer Instrument (Agilent Technologies, Santa Clara, CA, USA). Sequencing was performed using the MiniSeq sequencing system (Illumina) [17].
2.6. Bioinformatic Analysis
The generated reads were initially mapped against the DENV-2 reference genome (NC_001474) available in the National Center for Biotechnology Information (NCBI) database to obtain the consensus sequences using the Geneious v.9.1.8 [18] software. The coding region of five obtained sequences (two complete and three partial) were aligned to other 124 sequences of different genotypes of DENV-2 available in GenBank, using Mafft v.7 [19] software. Then, a Maximum Likelihood (ML) phylogenetic inference was built [20] with 1000 bootstrap iterations using the IQ-TREE v.1.6.12 program [21]. The GTR+F+I+G4 was selected as the best nucleotide substitution model, according to ModelFinder embedded in IQ-TREE [22]. The ML tree was rooted on the midpoint with FigTree v.1.4.4 software (http://tree.bio.ed.ac.uk/software/figtree/) and edited with Inkscape v.1.1 (https://inkscape.org/release/inkscape-1.1/).
3. Results
We analyzed 163 sera samples from individuals clinically suspected to all four den-gue serotypes using a multiplex RT-qPCR and obtained DENV-2 detection in 144 (88%) sera, of which 139 were positive for DENV-2 and 5 for DENV-1. The results showed an in-crease in the number of cases for DENV-2 between January and March 2021. From the 139 positive samples to DENV-2, five presented Ct value less to 20 and were inoculated in C6/36 cells, from which we obtained five isolates.
The five isolates were sequenced and clustered in a monophyletic clade, including the sequence from the state of Goiás, Brazil, and two Peruvian sequences. Furthermore, these genomes belong to lineage 5 of the DENV-2 Cosmopolitan genotype (Figure 1). The sequences generated in this study were deposited in GenBank under the following accession numbers: OQ511271 to OQ511275. All sequences included in the phylogenetic inference are listed in the Supplementary Materials (Table S1).
4. Discussion
The first report of a DENV-2 cosmopolitan strain described in the Americas occurred in the late 1990’s in the Yucatan Peninsula of Mexico, but only one viral isolate was studied [9]. This genotype is circulating in other regions of the world and has been replacing the genotypes predominantly circulating in several regions of Asia [23]. We can infer from recent reports that there is probably a large-scale introduction of the cosmopolitan genotype of DENV-2 from Asian regions to Madre de Dios, Peru, which is derived from the strains reported in Dhaka, Bangladesh. The Cosmopolitan genotype has recently been reported in Madre de Dios and is circulating worldwide, presenting a wide dispersal pattern of its lineages (Figure 2) [9,12].
Until 2020, the Asian-American genotype was the only DENV-2 genotype circulating in Brazilian territory [3]. The single evidence of circulation of Cosmopolitan genotype was obtained from a patient from Goiás in November 2021 [12]. The five sequences obtained in this study clustered into a South American clade, well supported by high bootstrap values, with the two Peruvian sequences and the only previously sequence described from Brazil. The data presented here represents the first evidence of the detection of the DENV-2 Cosmopolitan genotype in the Northern region of Brazil and, more specifically, in the Acre state, months before the detection from Goiás.
The human sera samples were collected in February and March 2021, implying the circulation of this genotype in Brazil before the case reported in Goiás [12] and two years after the outbreak in Madre de Dios, Peru [9]. The phylogenetic inference also revealed that the South American clade was positioned among the sequences belonging to lineage 5 of the genotype [8], also described in the literature as Lineage C [24] or clade A [25]. These findings indicate a possible dispersal route of the DENV-2 cosmopolitan genotype in South America from Peru, with a feasible introduction into Brazil through the border of Acre and Madre de Dios, from where it may have spread to the Brazilian Midwest.
5. Conclusions
This study may contribute to filling gaps about the dispersion of the Cosmopolitan genotype from Peru [9] into Brazilian territory, highlighting the importance of strengthening the genomic surveillance of DENV-2 and conducting retrospective studies in Brazil and South America to monitoring the genotype dispersion and the precise time of its introduction into the continent and in Brazil.
Author Contributions
Conceptualization, M.T.A. and A.C.R.C.; methodology, L.H.A. H. and S. P.S.; validation, B.T.D.N. and S.P.S.; investigation, M.T.A. and L.H.A.H.; resources, I.T.S.P.; data curation, F.S.S. and E.V.P.S.; writing—original draft preparation, M.T.A and L.H.A.H.; writing—review and editing, A.C.R.C and F.G.N.; visualization, A.L.M.W. and F.G.N.; supervision, L.M.N.C. and A.C.R.C.; project administration, A.C.R.C.; funding acquisition, A.C.R.C.. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Coordination for the Improvement of Higher Education Personnel (CAPES) and the Evandro Chagas Institute, Health and Environment Surveillance Secretariat, Ministry of Health, Brazil. Grant number “88887.612806/2021-00”.
Data Availability Statement
The sequences generated in this study were deposited in GenBank under the following accession numbers: OQ511271 to OQ511275.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Weaver, S.C. , Barret A.D.T. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2004, 2, 789–801. [Google Scholar] [CrossRef]
- WHO (World Health Organization). 2022. Epidemiological Update for Dengue, Chikungunya and Zika in 2022 - Epidemiological Bulletin. Washington: Pan American Health Organization.
- Fritsch, H.; Moreno, K.; Lima, I.A.B.; Santos, C.S.; Costa, B.G.G.; Almeida, B.L.; Santos, R.A.; Francisco, M.V.L.O.; Sampaio, M.P.S.; Lima, M.M.; et al. Phylogenetic Reconstructions Reveal the Circulation of a Novel Dengue Virus-1V Clade and the Persistence of a Dengue Virus-2 III Genotype in Northeast Brazil. Viruses, 2023, 15, 1073. [Google Scholar] [CrossRef]
- Brazilian Government. 2020. Monitoramento dos casos de arboviruses urbanas transmitidas pelo Aedes (dengue, chikungunya e Zika) – vol. 51, n. 2. Brasília: Ministry of Health.
- Brazilian Government. 2020. Monitoramento dos casos de arboviruses urbanas transmitidas pelo Aedes (dengue, chikungunya e Zika) – vol. 51, n. 18. Brasília: Ministry of Health.
- Brazilian Government. 2022. Monitoramento dos casos de arboviruses até a semana epidemiológica 45 de 2022 – vol. 53, n. 43. Brasília: Ministry of Health.
- Brazilian Government. 2023. Monitoramento dos casos de arboviruses até a semana epidemiológica 52 de 2022 – vol. 54, n. 1. Brasília: Ministry of Health.
- Yenamandra, S.P.; Koo, C.; Chiang, S.; Lim, H.S.J.; Yeo, Z.Y.; Ng, L.C.; Hapuarachchi, H.C. Evolution, heterogeneity and global spread of the cosmopolitan genotype of Dengue virus type 2. Sci. Rep. 2021, 11, 13496. [Google Scholar] [CrossRef] [PubMed]
- García, M.P.; Padilla, C.; Figueroa, D.; Manrique, C.; Cabezas, C. Emergence of the Cosmopolitan genotype of dengue virus serotype 2 (DENV2) in Madre de Dios, Peru, 2019. Rev Peru Med Exp Salud Publica 2022, 39, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Donalisio, M.R.; Freitas, A.R.R.; Zuben, A.P.B.V. Arboviruses Emerging in Brazil: challenges for the clinic and implications for public health. Revista de Saúde Pública 2017, 51, 30–40. [Google Scholar] [CrossRef]
- Nogueira, R.M.R.; Miagostovich, M.P.; Lampe, E.; Souza, R.W.; Zagne, S.M.O.; Schatzmayr, H.G. Dengue epidemic in the state of Rio de Janeiro, Brazil, 1990–1: Co-circulation of dengue 1 and dengue 2 serotypes. Epidemiol. Infec. 1993, 111, 163–170. [Google Scholar] [CrossRef]
- Giovanetti, M.; Pereira, L.A.; Santiago, G.A.; Fonseca, V.; Mendoza, M.P.G.; Oliveira, C.; Moraes, L.; Xavier, J.; Tosta, S.; Fristch, H.; et al. . Emergence of Dengue Virus Serotype 2 Cosmopolitan Genotype, Brazil. Emerg. Infect. Dis. 2022, 28, 1725–1727. [Google Scholar] [CrossRef] [PubMed]
- Lopes, N.; Nozawa, C.; Linhares, R.E.C. Características gerais e epidemiologia dos arbovírus emergentes no Brasil. ver. Pan-Amaz. Saude. 2015, 5, 55–64. [Google Scholar]
- Williams, M.; Mayer, S.V.; Johnson, W.L.; Chen, R.; Volkova, E.; Vilcarromero, S.; Widen, S.G.; Wood, T.G.; Ognio-Suarez, L.; Long, K.C.; et al. Lineage II of Southeast Asian/American DENV-2 Is Associated with a Severe Dengue Outbreak in the Peruvian Amazon, Am. J. Trop. Med. Hyg. 2014, 91, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Falconi-Agapito, F.; Selhorst, P.; Merino, X.; Torres, F.; Michiels, J.; Fernandez, C.; Talledo, M.; Ariën, K.K. A new genetic variant of dengue serotype 2 virus circulating in the Peruvian Amazon. Int. J. Infect. Dis. 2020, 96, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Santiago, G.A; Vergne, E.; Quiles, Y.; Cosme, J.; Vazquez, J.; Medina, J.F.; Medina, F.; Colón, C.; Margolis, H.; Muñoz-Jordán, J. Analytical and Clinical Performance of the CDC Real Time RT-PCR Assay for Detection and Typing of Dengue Virus. PLoS Negl. Trop. Dis. 2013, 7, 36–38. [Google Scholar] [CrossRef]
- Illumina Inc. MiniSeq System User Guide 2016. Available online: support.illumina.com/content/dam/illuminesupport/ documents/documentation/system_documentation/miniseq/miniseq-system-guide- 1000000002695-04.pdf (accessed on 27 April 2023).
- Kearse, M.; Moir, R.; Wilson, A.; Havas-Stones, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; Thierer, T.; Ashton, B.; Meintjes, P.; Drummond, A. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Myung, J. Tutorial on maximum likelihood estimation. Journal of Mathematical Psychology 2003, 47, 90–100. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Haeseler, A.V.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.; Wong, T.; Jermiin, L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Phadungsombat, J.; Nakayama, E.E.; Saito, A.; Egawa, A.; Sato, T.; Rahim, R.; Hasan, A.; Lin, M.Y.; Takasaki, T.; et al. Genotype replacement of dengue virus type 3 and clade replacement of dengue virus type 2 genotype Cosmopolitan in Dhaka, Bangladesh in 2017. Infect. Genet. Evol. 2019, 75, 103977. [Google Scholar] [CrossRef]
- Phadungsombat, J.; Lin, M.Y.C.; Srimark, N.; Yamanaka, A.; Nakayama, E.E.; Moolasart, V.; Suttha, P.; Shioda, T.; Uttayamakul, S. Emergence of genotype Cosmopolitan of dengue virus type 2 and genotype III of dengue virus type 3 in Thailand. PLoS One 2018, 13, e0207220. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kong, Q.; Wang, J.; Qiu, X.; Wen, Y.; Yu, X.; Liu, M.; Pan, H.W.J.; Sun, Z. Multiple Lineages of Dengue Virus Serotype 2 Cosmopolitan Genotype Caused a Local Dengue Outbreak in Hangzhou, Zhejiang Province, China, in 2017. Sci. Rep. 2019, 9, 7345. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Maximum Likelihood phylogenetic tree based on the alignment of the coding region of a DENV-2 dataset. The five sequences presented in the study (highlighted in red) and 124 others belonging to the five non-sylvatic genotypes of the virus. The nucleotide substitution model selected was GTR+F+I+G4. The genotypes are identified on the right side of the tree, and a different color represents each lineage of the Cosmopolitan genotype. Each branch of the highlighted lineage 5 clade contains the accession number, country, and collection date of the sequence.
Figure 1.
Maximum Likelihood phylogenetic tree based on the alignment of the coding region of a DENV-2 dataset. The five sequences presented in the study (highlighted in red) and 124 others belonging to the five non-sylvatic genotypes of the virus. The nucleotide substitution model selected was GTR+F+I+G4. The genotypes are identified on the right side of the tree, and a different color represents each lineage of the Cosmopolitan genotype. Each branch of the highlighted lineage 5 clade contains the accession number, country, and collection date of the sequence.
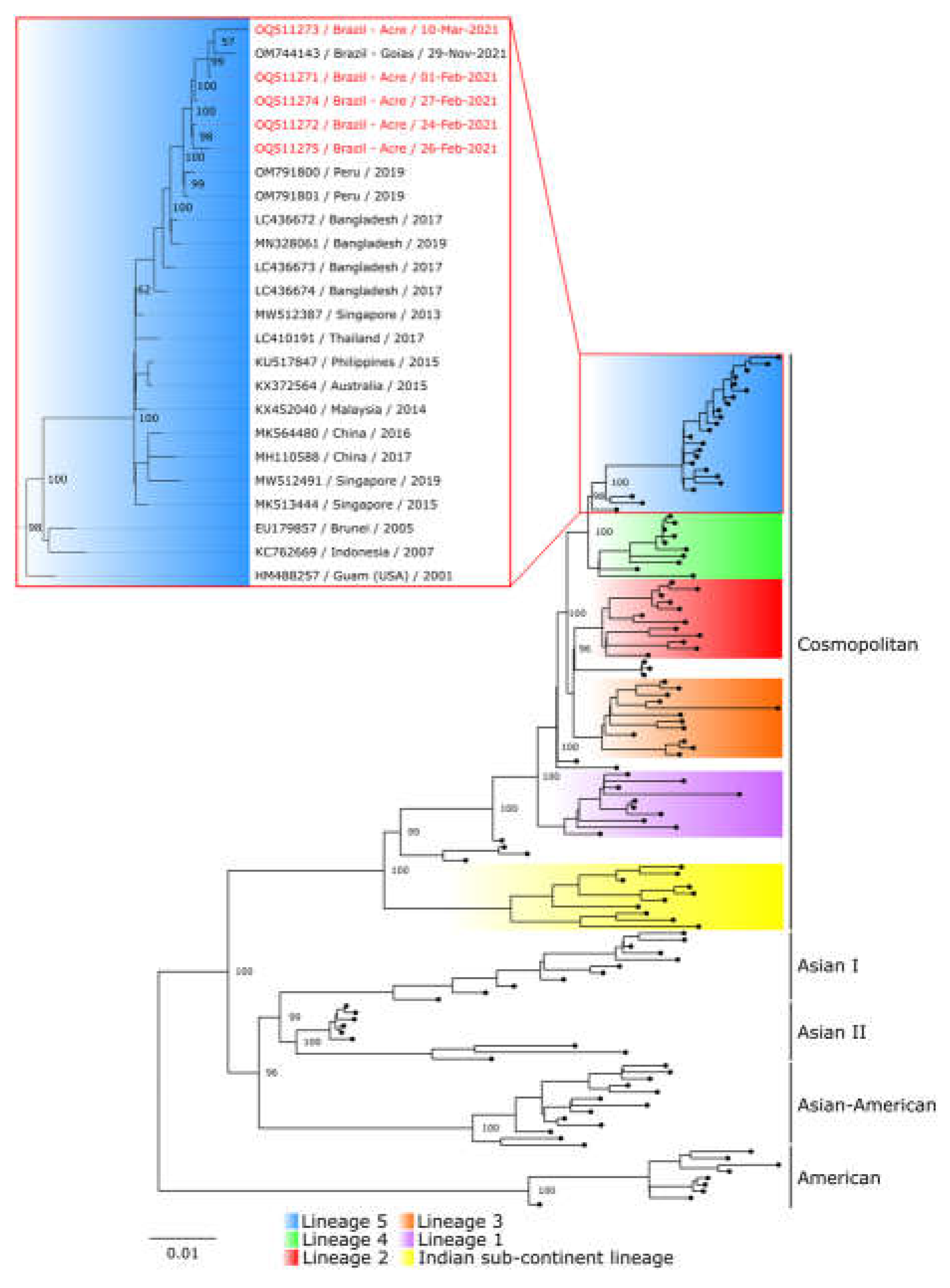
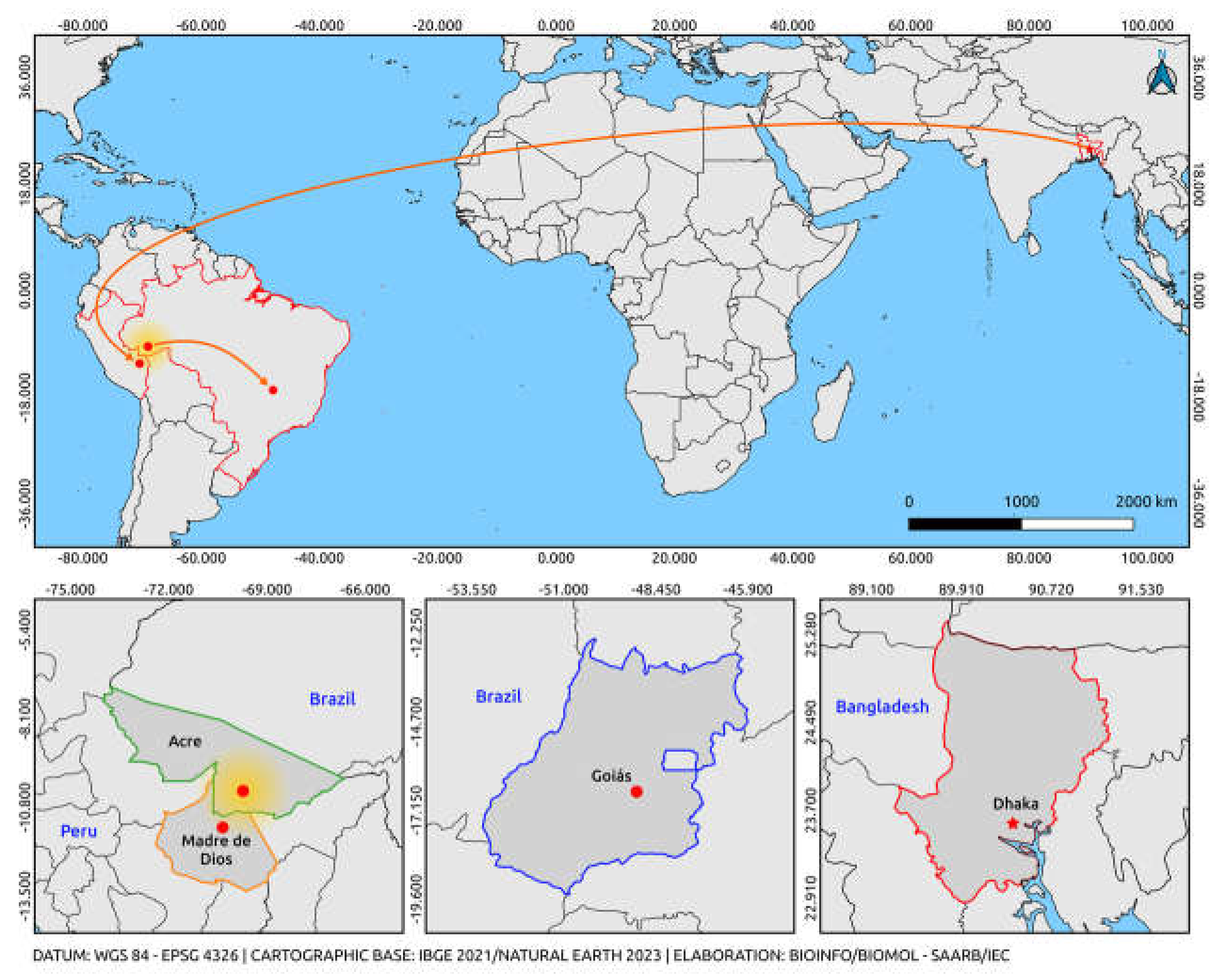
Figure 2.
Map illustrating the introduction route of DENV-2 suggested in this study. The orange arrows indicate the routes of entry of DENV-2 based on reports from the last five years (Dakha/Bangladesh - Acre/Brazil - Goiás/Brazil). The colors delineate the states and the intersections between the country regions. The circle in yellow gradient indicates the occurrence of a possible dispersion of the lineage to other regions of Brazil and border countries. The map was built using the Qgis v.3.28 software, available at https://qgis.org/pt_BR/site/.
Figure 2.
Map illustrating the introduction route of DENV-2 suggested in this study. The orange arrows indicate the routes of entry of DENV-2 based on reports from the last five years (Dakha/Bangladesh - Acre/Brazil - Goiás/Brazil). The colors delineate the states and the intersections between the country regions. The circle in yellow gradient indicates the occurrence of a possible dispersion of the lineage to other regions of Brazil and border countries. The map was built using the Qgis v.3.28 software, available at https://qgis.org/pt_BR/site/.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



