
Submitted:
15 May 2023
Posted:
15 May 2023
You are already at the latest version
Abstract
Background: MOST-plus is a multicenter, randomized, open-label, adaptive Phase II trial evaluating the clinical benefit of targeted treatments matched to molecular alteration in advanced/metastatic solid tumors. Sorafenib was tested on patients with tumors harboring sorafenib-targeted genes Methods: The MOST-plus trial used a randomized discontinuation design. After 12 weeks of sorafenib (400 mg, po BID), patients with progressive disease dis-continued study, patients with objective response were proposed to continue sorafenib, whereas patients with stable disease (SD) were randomly assigned (1:1) to maintenance or interruption of treatment. Primary endpoint was RE-CIST version 1.1 progression-free rate at 16 weeks after randomization (PFR-16w). Secondary endpoints included progression-free survival (PFS), overall survival (OS), and toxicity. Statistical analyses used a sequential Bayes-ian approach with interim efficacy analyses. The enrolment could be stopped in the case of a 95% probability for the estimated PFR-16w to be higher in the maintenance than in the interruption arm (NCT02029001). Results: 151 patients were included, of whom 35 had SD at 12 weeks of Soraf-enib. For the 35 patients with SD on sorafenib, the PFR-16w was 65% [95% credibility interval 43.4–83.7] in the continuation arm and 25% [7.8–48.1] in the interruption arm. Median PFS and OS were improved in maintenance versus interruption arm (mPFS: 5.6 [95%CI 1.97–6.77] months versus 2.0 [95%CI 1.61–3.91] months (p =0.0231) and mOS : 14.3 [95%CI 8.9–23.8] versus 8.0 months [95%CI 3.5–15.2] (p =0.0857)). Conclusion: Sorafenib showed activity in progressive patients with solid tu-mors harboring somatic genomic alterations in sorafenib-targeted genes. Con-tinuing sorafenib when SD is achieved improves PFR compared to interruption
Keywords:
personalized medicine
; biologically-driven trial
; sorafenib
; randomised discontinuation design
1. Introduction
The emergence of Next Generation Sequencing [NGS] led to the identification of molecular alterations in genes involved in tumor progression in a variety of cancers [1]. Molecular profiling efforts from the International Cancer Genomics Consortium (ICGC), or The Cancer Genome Atlas (TCGA) have shown the multiplicity, complexity, diversity, and heterogeneity of genomic alterations within cancer types [2]. Several genomic-driven clinical trials have allowed to assess the therapeutic value of matching drugs with specific tumor characteristics [3-11]. It remains questionable whether disease stabilization results from drug efficacy or from intrinsic low proliferating tumors independently of targeted agent efficacy [12].
MOST-plus is a multicenter, randomized, open-label, adaptive platform phase II [13] aiming to assess the clinical benefit of continuing a biomarker-allocated treatment in advanced/metastatic solid tumors using a randomized discontinuation design (RDD [14]). This genomic-driven study evaluated seven targeted therapies: nilotinib, everolimus, sorafenib, lapatinib, pazopanib, olaparib, and durvalumab + tremelimumab. In this manuscript, we present the final analysis of the sorafenib cohort.
The oral multi-targeted tyrosine kinase inhibitor (TKI) sorafenib inhibits platelet-derived growth factor receptors (PDGFR), vascular endothelial growth factor (VEGF-R), Fms-like tyrosine kinase-3 (Flt-3), tyrosine-protein kinase KIT, RET [15] and fibroblast growth factor receptors FGFR1. Sorafenib inhibits tumor growth and angiogenesis through targeting both the RAF/MEK/ERK pathway and receptor tyrosine kinases [16]. Sorafenib treatment results in a cytostatic rather than a cytotoxic effect, thus the expected primary clinical benefit is disease stabilization rather than objective disease shrinkage. Sorafenib is approved for the treatment of unresectable hepatocellular carcinoma, advanced renal cell carcinoma, and locally advanced or metastatic, differentiated thyroid carcinoma [17-19].
The hypothesis of the MOST-plus trial is that continuing a targeted therapy such as sorafenib in patients with matched genomic alterations and stable disease after a 12-week induction period could improve clinical outcomes.
2. Materials and Methods
MOST-plus (My Own Specific Treatment, NCT02029001) is a multicenter, randomized, open-label, genomic-driven, adaptive phase II platform trial. The trial was conducted according to Good Clinical Practice guidelines, the Declaration of Helsinki, and relevant French and European laws and directives. All patients provided written informed consent.
Eligible patients were 18 years of age or older, with histologically confirmed advanced/metastatic solid tumors (any types) treated by at least one line of prior chemotherapy, and harboring at least one of the following molecular alterations according to local assessment: mutations or amplification/translocation in VEGFR1-3, PDGFRB, FLT3, BRAF (excluding V600E), CRAF, HRAS, KRAS, or RET, and/or cognate ligands. Patient genomic profiles were reviewed by a centralized virtual Molecular Tumor Board before patient enrolment. Other key eligibility criteria included adequate performance status according to Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2, presence of at least one measurable lesion as per the Response Evaluation Criteria In Solid Tumors, version 1.1 (RECIST 1.1 [20]), documented disease progression at inclusion, and normal organ and bone marrow functions confirmed within 7 days before sorafenib initiation.
Randomization was stratified according to Eastern Cooperative Oncology Group (ECOG) performance status (0-1 versus 2).
All eligible patients were initially treated with biomarker-allocated treatment for 12 weeks (sorafenib: 400mg, twice daily, per os). According to RDD, at the end of this induction period, patients with an objective response (OR) continue on therapy; patients with progressive disease (PD) permanently discontinue therapy, whereas patients with stable disease (SD) were randomly allocated (1:1) to either continue (maintenance arm) or discontinue (interruption arm) the allocated therapy. Patients in the interruption arm could reinitiate targeted therapy upon documented disease progression following treatment interruption.
Protocol-defined dose modifications, including interruptions and dose reductions, were used to manage adverse events (AEs) according to sorafenib summary of product characteristics. AE were graded according to the Common Terminology Criteria for Adverse Events (CTCAE -version 4.03). Disease assessments with computed tomography or magnetic resonance imaging were performed at baseline, at week 12 (W12) and then every 8 weeks until disease progression, death or withdrawal. Tumor responses were determined by the investigator according to RECIST V1.1.
The primary endpoint was the proportion of randomized patients remaining progression-free at 16 weeks after randomization (PFR-16w) according to RECIST 1.1. Secondary endpoints include overall response rate, duration of response, progression-free survival (PFS), overall survival (OS), and safety.
The study used a Bayesian adaptive phase II design, allowing updating knowledge gradually rather than restricting revisions in a trial design with fixed sample sizes [21,22]. The analysis of the primary endpoint (PFR-16w) was carried out sequentially, with interim analyses planned after a 16-week follow-up for the first 20 randomized patients, then every 10 randomized patients. The probability of success (PFR-16w) was estimated from a beta-binomial model. In the absence of strong idea about the non-progression rate, a non-informative prior distribution beta (1,1) was considered. At each interim analysis, the trial could be stopped if there was a high posterior probability (≥95%) that the PFR-16w was higher in the maintenance than in the interruption arm. At the end of the trial, if no stopping rule occurred, the maintenance arm was considered superior if the posterior probability for the PFR-16w to be higher in the maintenance arm, was at least 90%. Mean PFR-16w estimated by the Bayesian method were described in each arm along with the associated 95% credibility intervals (CrI) (precision of the Bayesian estimation).
Maximum sample size was set at 50 randomized patients for each treatment cohort.
The median follow-up was calculated using the reverse Kaplan–Meier method. OS and PFS were estimated using the Kaplan–Meier method and described in terms of median along with associated 2-sided 95% confidence interval (CI) calculated by Brookmeyer and Crowley technique. Data for patients who were event-free at the time of analysis were censored at the date of the last follow-up for OS and at the time of last assessment for PFS. Patients randomized while they had disease progression were censored at the time of randomization for PFS analysis. For exploratory purposes, OS and PFS distributions were compared between the two randomization arms using a log-rank test stratified on ECOG Performance status at randomization.
Quantitative variables were described using the median and IQR. Qualitative variables were described using frequency and 95%CI. Efficacy data are presented according to disease status at the end of the induction period (i.e. at W12) for patients treated during at least one cycle and evaluable. All patients having received at least one cycle of sorafenib were assessed for safety. Evaluable patients for primary endpoint include all patients who received at least one cycle of sorafenib or discontinued treatment earlier for other reason than PD, death or related toxicity. Based on the ITT principle, PFS and OS analysis were performed including all randomly assigned patients. Data cutoff was February 27, 2020. Statistical analyses were performed using SAS software (version 9.4).
3. Results
3.1. Patient characteristics and trial profile
From April 2014 to May 2018, 151 patients with advanced/metastatic solid tumor were enrolled in the sorafenib cohort. Among them, 2 patients were not treated and 4 patients were treated but did not receive the full first cycle of sorafenib due to patient decision. These 6 patients were considered as non-evaluable for efficacy endpoints. All analyses were performed on the 145 patients having received at least complete cycle of sorafenib or discontinued earlier for other reason than PD, death or sorafenib-related toxicity (Figure 1).
Baseline characteristics are presented in Table 1. The most common (≥ 5%) primary tumor sites were lung (26.2%), colorectal (25.5%), gynecological (15.2%), and pancreas (12·4%). Almost all patients had metastatic disease at inclusion (94%) and were heavily pre-treated with a median number of prior treatment lines of 3 [1,2,3,4,5,6,7,8,9,10,11]. Chemotherapy was the main treatment type (62.1%) administered before inclusion. The molecular alterations that allowed patient inclusion were mainly KRAS hot-spot mutations (Figure 2). According to ESCAT classification [24], the majority of molecular alterations were ESCAT IV (71%) with only 1 patient with ESCAT I molecular alteration (a lung adenocarcinoma with RET translocation) and 22 patients with ESCAT II (14.5%) molecular alteration (mainly pancreatic cancer with KRAS host spot mutation).
3.2. Primary efficacy endpoint
At the end of the induction period: 5 (3.5%) patients with OR (5 PR) continued sorafenib, 105 (72.4%) discontinued sorafenib at/or before W12 mainly due to PD or death or unacceptable toxicity, whereas 35 (24.1%) patients with SD were randomized (1:1) to maintenance (N=18) or interruption (N=17) arms (Figure 2). In the interruption arm, sorafenib was re-introduced after treatment interruption and PD in 11 out of 17 patients (64.7%). One patient was randomized with a PR and discontinued the study one week after randomization. Among the 35 randomized patients, 3 patients with investigator-assessed SD at W12 were finally documented with PD at W12. They were randomly assigned to interruption arm, instead of discontinuing treatment and were therefore censored at the date of randomization for PFS analysis. At time of final analysis, all patients had discontinued sorafenib treatment, mainly due to PD (77.9%, Figure 1). At the second interim analysis, PFR-16w was 65% [95%CrI 43.4–83.7] for the maintenance arm and 25% [CrI 95%: 7.8–48·.] for the interruption arm (Figure 3A). With a probability that maintenance arm was superior to interruption arm of 99%, the stopping rule applied and the recruitment to sorafenib cohort prematurely stopped.
3.3. Secondary efficacy endpoints
Tumor response and median treatment duration post-randomization are presented in Figure 3B. In the maintenance arm, there was no objective response and the median duration of treatment post-randomization was 5.6 [1.9-31.4] months. Among the 11 patients of the interruption arm in whom sorafenib was re-started following PD, 2 (18.2%) patients reached PR after sorafenib reintroduction. Median time to treatment reinitiation was 2.2 [0.9–9.6] months and median duration of sorafenib treatment following restart was 2.6 [0.3–18.8] months. The median PFS was significantly higher in the maintenance arm than in the interruption arm (5.6 [95%CI 1.97–6.77] months and 2·0 [95%CI 1.61–3.91] months respectively [log-rank p=0.0231, Figure 4A]). Median OS from randomization was 14.3 [95%CI 8.9–23.8] months in the maintenance arm and 8.0 months [95%CI 3.5–15.2] in the interruption arm (log-rank p=0.0857, Figure 4B). Median PFS following sorafenib reinitiation was 2.8 [95%CI 1.2–4.1] months. Median OS was 11.8 [95%CI 6.4–19] months after sorafenib reinitiation, versus 3.8 [95%CI 0.6–19.6] months in patients who did not reinitiate sorafenib. Median PFS from inclusion (N=145) was 4.7 [3.7–6.3] months in the interruption arm and 8.3 [4.8–9.5] months in maintenance arm, 6.3 [3.1–8.2] months in patients with OR, and 2.2 [1.6–2.5] months in patients with PD at the end of the induction period (Figure 4C). Median OS from inclusion was 10.6 [6.3–18] months in the interruption arm, 17.1 months [11.6- 26.6] in the maintenance arm, 21.5 [14.7–NR] months in patients with OR, and 4.1 [2.9-4.9] months in patients with PD at the end of the induction period.
3.4. Safety endpoints
Almost all treated patients (87%) experienced at least one sorafenib-related AE including 46% with at least one grade ≥3 sorafenib-related AE (Table 2). Consistently with the known safety profile of sorafenib, the most common (⩾5% in overall population) grade ≥3 sorafenib-related AE were vomiting (6.2%), fatigue (7·6%), hand and skin syndrome (6.2%), and hypertension (12.4%). A total of 3 unexpected deaths were reported by investigators: a fatal dyspnea with various hypothesis of death pulmonary embolism, arrhythmia or stroke all possibly related to sorafenib, (N=1), a cardio-respiratory arrest of unknown etiology (N=1), and an acute coronary syndrome in a patient with typical ventriculography of Tako-Tsubo (N=1).

Table 1.
Patient demographics and baseline characteristics.
| Disease status at end of induction period | TOTAL | ||||||||||
| OR | PD | SD | |||||||||
| Maintenance | Interruption | ||||||||||
| N=5 | N=105 | N=18 | N=17 | N=145 | |||||||
| Age (years) | |||||||||||
| Median (IQR) | 66.0 (61.0-70.0) |
63.0 (56.0-68.0) |
60.0 (55.0-65.0) |
65.0 (57.0-68.0) |
63.0 (56.0-68.0) |
||||||
| Sex | |||||||||||
| M | 3 | (60.0) | 49 | (46.7%) | 8 | (44.4%) | 6 | (35.3%) | 66 | (45.5%) | |
| F | 2 | (40.0) | 56 | (53.3%) | 10 | (55.6%) | 11 | (64.7%) | 79 | (54.5%) | |
| PS ECOG, n (%) | |||||||||||
| 0 | 3 | (60.0) | 28 | (26.7%) | 10 | (55.6%) | 6 | (35.3%) | 47 | (32.4%) | |
| 1 | 2 | (40.0) | 63 | (60.0%) | 7 | (38.9%) | 9 | (52.9%) | 81 | (55.9%) | |
| 2 | 0 | (0.0%) | 14 | (13.3%) | 1 | (5.6%) | 2 | (11.8%) | 17 | (11.7%) | |
| Main primary tumor site (≥10% in at least one subgroup), n (%) | |||||||||||
| CRC | 0 | (0.0%) | 31 | (29.5%) | 4 | (22.2%) | 2 | (11.8%) | 37 | (25.5%) | |
| H & N | 1 | (20.0%) | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (0.7%) | |
| Retroperitoneal | 0 | (0.0%) | 0 | (0.0%) | 2 | (11.1%) | 0 | (0.0%) | 2 | (1.4%) | |
| Gynecological. | 0 | (0.0%) | 18 | (17.1%) | 6 | (33.3%) | 5 | (29.4%) | 29 | (20.0%) | |
| Lung | 3 | (60.0%) | 22 | (21.0%) | 5 | (27.8%) | 8 | (47.1%) | 38 | (26.2%) | |
| Prostate | 1 | (20.0%) | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (0.7%) | |
| Pancreas | 0 | (0.0%) | 16 | (15.2%) | 0 | (0.0%) | 2 | (11.8%) | 18 | (12.4%) | |
| Prior number of lines in advanced/metastatic stage | |||||||||||
| Median (IQR) | 5.0 (3.0-6.0) | 3.0 (2.0-4.0) | 2.5 (2.0-4.0) | 3.0 (2.0-3.0) | 3.0 (2.0-4.0) | ||||||
| 1L or 2L | 1 (20%) | 50 (47.6%) | 9 (50.0%) | 8 (47.1%) | 68 (46.9%) | ||||||
| 3L to 5L | 2 (40%) | 43 (41.0%) | 8 (44.4%) | 8 (47.1%) | 61 (42.1%) | ||||||
| ≥ 6L | 2 (40%) | 12 (11.4%) | 1 (5.6%) | 1 (5.8%) | 16 (11.0%) | ||||||
| Type of prior line before inclusion, n (%) | |||||||||||
| CT | 3 | (60.0) | 68 | (64.8%) | 7 | (38.9%) | 12 | (70.6%) | 90 | (62.1%) | |
| CT + Anti-angiogenic | 0 | (0.0%) | 27 | (25.7%) | 5 | (27.8%) | 2 | (11.8%) | 34 | (23.4%) | |
| Targeted therapy | 1 | (20%) | 7 | (6.6%) | 5 | (27.8%) | 2 | (11.8%) | 15 | (10.3%) | |
| Immunotherapy | 0 | (0.0%) | 2 | (1.9%) | 1 | (5.6%) | 0 | (0.0%) | 3 | (2.1%) | |
| Hormonotherapy | 0 | (0.0%) | 1 | (1.0%) | 0 | (0.0%) | 1 | (5.9%) | 2 | (1.4%) | |
| Best response to prior line according to RECIST V1.1, n (%) | |||||||||||
| CR | 0 | (0.0%) | 2 | (1.9%) | 0 | (0.0%) | 0 | (0.0%) | 2 | (1.4%) | |
| PR | 1 | (20.0) | 8 | (7.6%) | 0 | (0.0%) | 2 | (11.8%) | 11 | (7.6%) | |
| SD | 0 | (0.0%) | 36 | (34.3%) | 7 | (38.9%) | 12 | (70.6%) | 55 | (37.9%) | |
| PD | 2 | (40.0) | 56 | (53.3%) | 10 | (55.6%) | 2 | (11.8%) | 70 | (48.3%) | |
| NE | 1 | (20.0) | 3 | (2.9%) | 1 | (5.6%) | 1 | (5.9%) | 6 | (4.1%) | |
In grey: patients randomized at the end of induction period. OR: objective response, PD: progressive disease; SD: Stable disease, CR: complete response, PR: partial response, NE: non-evaluable, CRC: colorectal, H&N: Head and neck squamous cell carcinoma, L: lines of treatment. Data are n (%), and median.
Table 2.
– Adverse Events summary.
| Disease status at end of induction period | TOTAL | ||||||||||||
| OR | PD | SD | |||||||||||
| Maintenance | Interruption | ||||||||||||
| N=5 | N=105 | N=18 | N=17 | N=145 | |||||||||
| Number of patients with at least, n (%) | |||||||||||||
| One AE (all grades) | 5 | (100.0%) | 105 | (100.0%) | 18 | (100.0%) | 17 | (100.0%) | 145 | (100.0%) | |||
| One sorafenib related AE (all grades) | 5 | (100.0%) | 86 | (81.9%) | 18 | (100.0%) | 17 | (100.0%) | 126 | (86.9%) | |||
| One Grade ≥3 AE | 1 | (20.0%) | 84 | (80.0%) | 13 | (72.2%) | 14 | (82.4%) | 112 | (77.2%) | |||
| One Grade ≥3 sorafenib-related AE | 1 | (20.0%) | 46 | (43.8%) | 12 | (66.7%) | 8 | (47.1%) | 67 | (46.2%) | |||
| One related SAE | 0 | (0.0%) | 33 | (31.4%) | 6 | (33.3%) | 4 | (23.5%) | 43 | (29.7%) | |||
| One SUSAR | 0 | (0.0%) | 10 | (9.5%) | 0 | (0.0%) | 1 | (5.9%) | 11 | (7.6%) | |||
| Main (≥ 5%)Grade ≥3 related AE, n (%) | |||||||||||||
| Abdominal pain | 0 | (0.0%) | 1 | (1.0%) | 1 | (5.6%) | 0 | (0.0%) | 2 | (1.4%) | |||
| Intestinal perforation | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.6%) | 0 | (0.0%) | 1 | (0.7%) | |||
| Vomiting | 0 | (0.0%) | 4 | (3.8%) | 5 | (27.8%) | 0 | (0.0%) | 9 | (6.2%) | |||
| Fatigue | 0 | (0.0%) | 9 | (8.6%) | 2 | (11.1%) | 0 | (0.0%) | 11 | (7.6%) | |||
| QT prolonged | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.9%) | 1 | (0.7%) | |||
| GGT increased | 0 | (0.0%) | 3 | (2.9%) | 1 | (5.6%) | 0 | (0.0%) | 4 | (2.8%) | |||
| Weight decreased | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.6%) | 0 | (0.0%) | 1 | (0.7%) | |||
| WBC decreased | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.9%) | 1 | (0.7%) | |||
| Hypocalcemia | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.6%) | 0 | (0.0%) | 1 | (0.7%) | |||
| Dyspnea | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.9%) | 1 | (0.7%) | |||
| Pulmonary embolism | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (5.9%) | 1 | (0.7%) | |||
| Hand Skin syndrome | 0 | (0.0%) | 4 | (3.8%) | 4 | (22.2%) | 1 | (5.9%) | 9 | (6.2%) | |||
| Rash | 0 | (0.0%) | 2 | (1.9%) | 0 | (0.0%) | 1 | (5.9%) | 3 | (2.1%) | |||
| Hypertension | 1 | (20.0%) | 10 | (9.5%) | 3 | (16.7%) | 4 | (23.5%) | 18 | (12.4%) | |||
AE: adverse event, Gr.: Grade according to NCI-CTCAE v4.03, SAE: Serious adverse event.
Figure 1.
Trial profile. Following an induction period of 12 weeks, 145 treated and evaluable patients were categorized according to disease status: patient with objective response (OR, n =5)) continued sorafenib, patients with stable disease (SD, n =35)) were randomized (R) with a 1:1 ratio to maintenance or interruption arm; while patients with progressive disease (PD, n =105) permanently discontinued sorafenib. Randomization was stratified according to ECOG PS: 0-1 versus 2 at randomization. Non-evaluable patients were defined as patients with less than 1 cycle of treatment due to other reason than disease progression, sorafenib-related toxicities, or death. 1All patients with OR at W12 have permanently discontinued sorafenib due to PD at time of database cutoff. 2Five patients in the interruption arm were randomized without documented SD: PD (N=3), PR (N=1), and NE (N=1), and 1 patient was randomized in maintenance arm despite >28 days sorafenib temporary discontinuation before randomization. 3This subgroup also includes patients with sorafenib permanent discontinuation before or at W12 due to toxicity (N=34), investigator or patient decision (N=14), death (N=10), other reason (N=1 with abnormal ECG at time of randomization).
Figure 1.
Trial profile. Following an induction period of 12 weeks, 145 treated and evaluable patients were categorized according to disease status: patient with objective response (OR, n =5)) continued sorafenib, patients with stable disease (SD, n =35)) were randomized (R) with a 1:1 ratio to maintenance or interruption arm; while patients with progressive disease (PD, n =105) permanently discontinued sorafenib. Randomization was stratified according to ECOG PS: 0-1 versus 2 at randomization. Non-evaluable patients were defined as patients with less than 1 cycle of treatment due to other reason than disease progression, sorafenib-related toxicities, or death. 1All patients with OR at W12 have permanently discontinued sorafenib due to PD at time of database cutoff. 2Five patients in the interruption arm were randomized without documented SD: PD (N=3), PR (N=1), and NE (N=1), and 1 patient was randomized in maintenance arm despite >28 days sorafenib temporary discontinuation before randomization. 3This subgroup also includes patients with sorafenib permanent discontinuation before or at W12 due to toxicity (N=34), investigator or patient decision (N=14), death (N=10), other reason (N=1 with abnormal ECG at time of randomization).
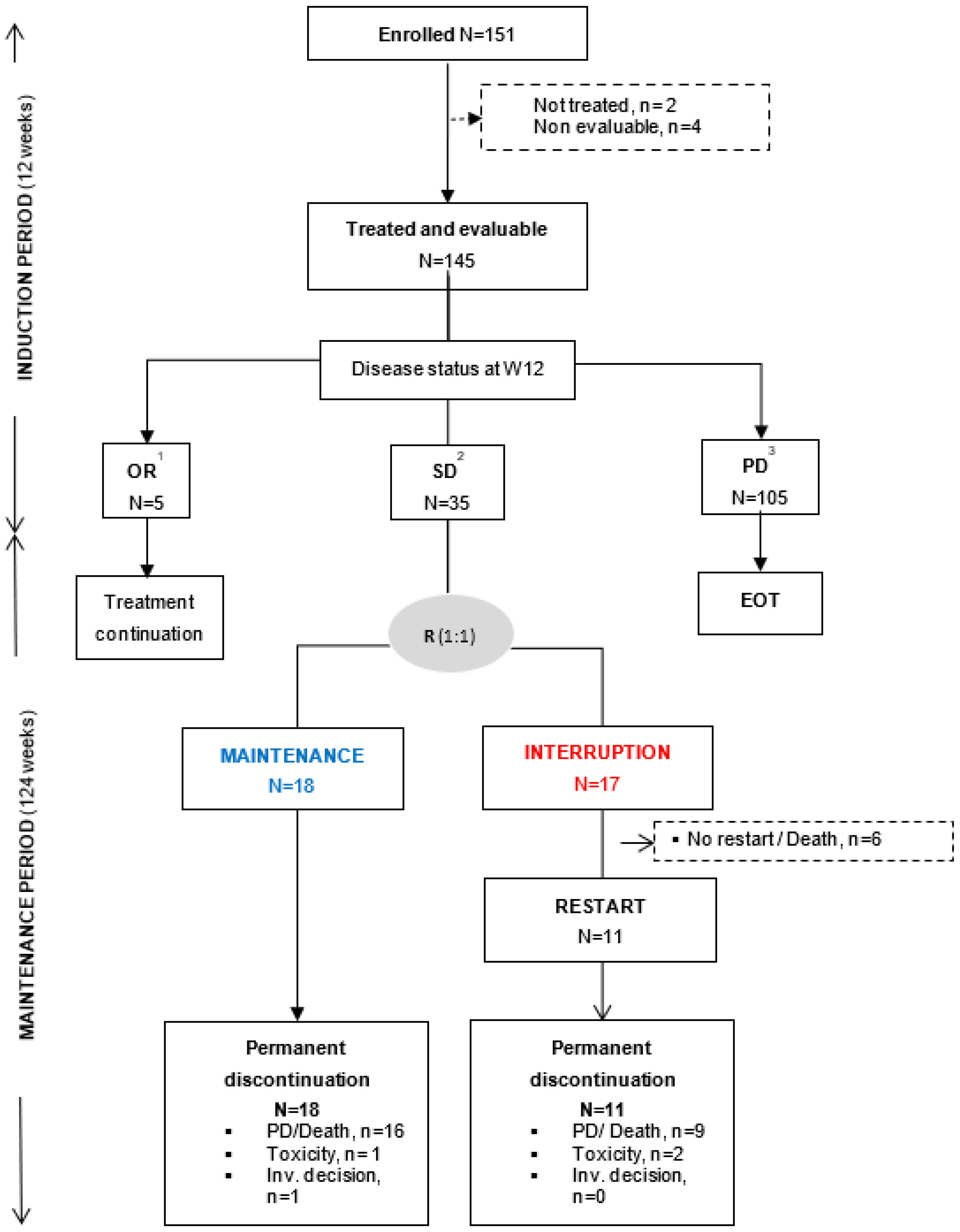
Figure 2.
Detailed of molecular alteration having led to sorafenib recommendation. Molecular alterations per gene name and type of alteration according to disease status at Week 12.
Figure 2.
Detailed of molecular alteration having led to sorafenib recommendation. Molecular alterations per gene name and type of alteration according to disease status at Week 12.
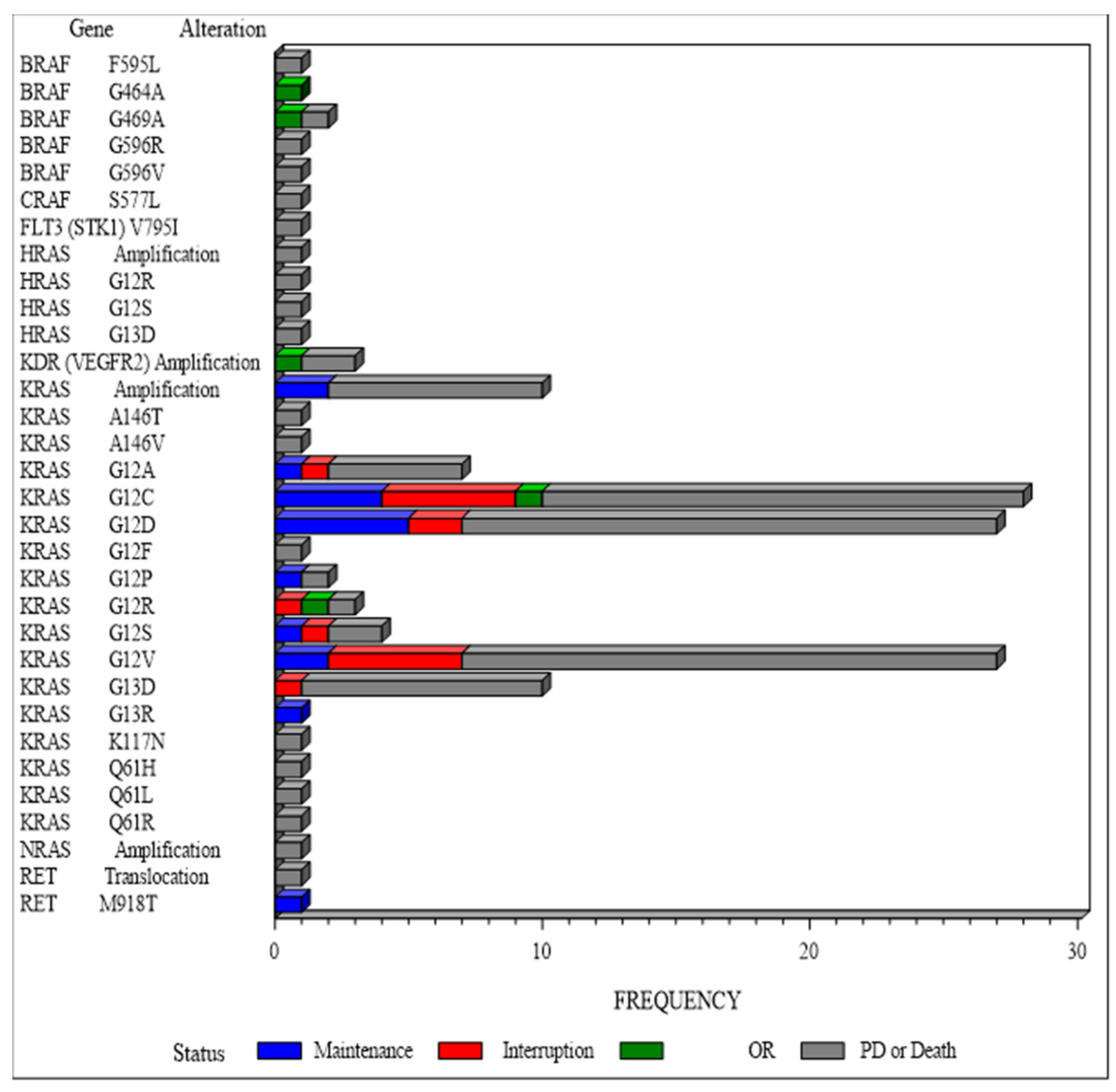
Figure 3.
A. Bayesian estimates of the probability distribution of being progression free (success) at 16 weeks after randomization. Prior and posterior density functions of the probability of success were updated after each successive interim analysis. Success was defined as being progression free at 16 weeks. B. Duration of treatment and best response from randomization. Primary tumor site and molecular alterations having led to inclusion in the sorafenib cohort are listed for each patient. In the interruption arm (red lines), patients were proposed to reinitiate sorafenib in case of PD (black star). One patient was enrolled without molecular alteration and randomized in the interruption arm.
Figure 3.
A. Bayesian estimates of the probability distribution of being progression free (success) at 16 weeks after randomization. Prior and posterior density functions of the probability of success were updated after each successive interim analysis. Success was defined as being progression free at 16 weeks. B. Duration of treatment and best response from randomization. Primary tumor site and molecular alterations having led to inclusion in the sorafenib cohort are listed for each patient. In the interruption arm (red lines), patients were proposed to reinitiate sorafenib in case of PD (black star). One patient was enrolled without molecular alteration and randomized in the interruption arm.
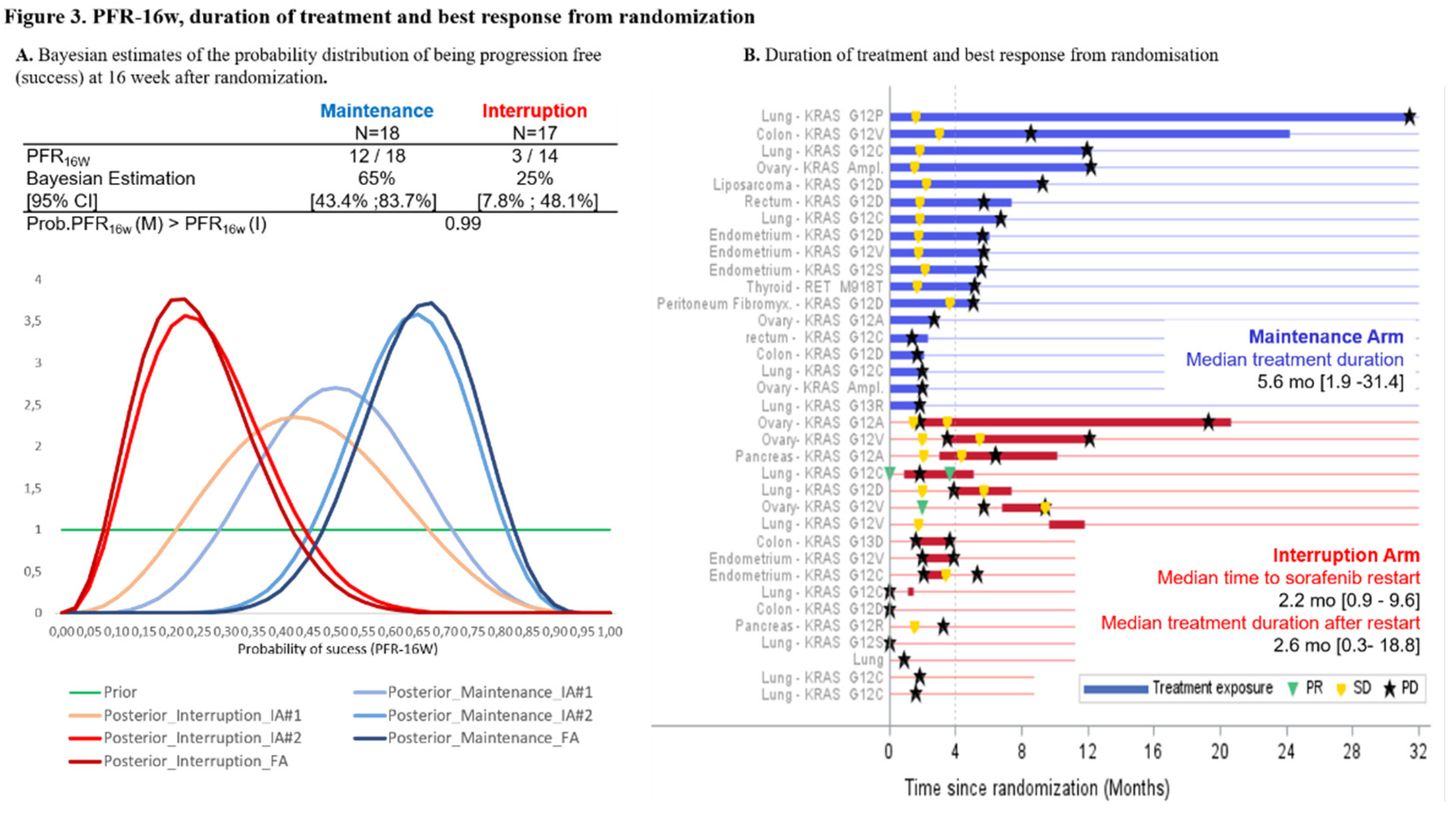
Figure 4.
A - PFS from randomization. B - OS from randomization. C- PFS from inclusion. D- OS from inclusion.
Figure 4.
A - PFS from randomization. B - OS from randomization. C- PFS from inclusion. D- OS from inclusion.

4. Discussion
This multicenter, randomized, open-label, phase II demonstrates that maintaining sorafenib treatment for molecularly selected patients, when the disease is not progressing during an induction period, improves PFS. Based on the previous MD Anderson Cancer Center experience, it was initially expected that approximately 65% of enrolled patients would experience a SD after 12 weeks of treatment [23]. In our sorafenib cohort, the randomization rate at 12 weeks was lower than expected (i.e. 24%). However, this was consistent with the data obtained by Ratain et al [12] in a RDD phase II trial reporting a non-progression rate of 32% following an induction period of 12 weeks with sorafenib in metastatic renal cell carcinoma. In agreement with our results, the authors showed that maintaining sorafenib significantly improved the rate of non-progression compared to the placebo group (PFR-24w: 50% versus 18% [p=0.0077]). Nevertheless, the efficacy of the sorafenib maintenance treatment may depend on the histology of the primary tumor. Indeed, during the course of our trial, the low percentage of colorectal cancer patients achieving SD after the induction period, led to the interruption of the sorafenib recommendation for patients with colorectal cancer, as supported by Samalin et al [25].
Molecular screening program in the MOST-plus trial was not centralized. Each institution performed its own genomic analysis; however, a centralized molecular tumor board reviewed the sorafenib treatment indication before enrollment. The vast majority of our patients (87%) had hot-spot KRAS-mutated tumors, including 28% KRASG12C (OR group: N=1/5, SD group: N=9/35, PD group: N=18/105 [Figure 2]). Analysis of this cohort did not allow to identify association between a given molecular alteration and outcomes. Results from precision medicine clinical trials are often biased by the fact that tumor profiling before inclusion frequently used heterogeneous tumor samples and different sequencing technologies. Furthermore, the predictive value of known mutations for targeted treatment depends on the tumor type and the presence of other relevant, potentially resistant, alterations. In line with this, several studies have shown that the activation of the MAP kinase pathway downstream of a RAS mutation is mutation- and tissue-specific [26]. Several mechanisms were described as being involved in the acquired resistance to sorafenib, such as reactivation of wild-type KRAS, crosstalk between PI3K/Akt and JAK-STAT pathways, or the activation of hypoxia-inducible pathways and epithelial-mesenchymal transition [27]. This was not analyzed in our study.
Sorafenib is a multi-target kinase inhibitor, not selective for KRAS mutation [15]. Several drugs that more specifically target components of the MAP kinase pathway have been developed, including allele-specified. Sotorasib is a specific and irreversible inhibitor of KRASG12C that covalently traps KRASG12C in the inactive state, thus inhibiting KRAS oncogenic signaling [28]. A recent phase II has demonstrated that sotorasib as single agent had significant clinical activity in KRASG12C non–small-cell lung cancer, which has led to its recent approval by Food and Drug administration (N=126 patients, with objective response rate of 37.1% [95%CI 28.6–46.2] and disease control rate of 80·6% [95%CI 72.6–87.2] [29]. The NEXIRI trial suggests that the efficacy of sorafenib may also depend on histological tumor type [25]. This is also supported by other studies on MAP kinase pathway inhibitors in CRC, though differences in KRAS alleles may also be at play [26,30].
As an adaptive approach, the Bayesian method adopted in the MOST-plus platform trial was designed to quickly modify the course of each ongoing cohort through regular updating in information during the study, and more specifically to allow early termination of uninteresting cohorts. To be effective and deliver the required results, such approach requires a predefined number of steps with regards to the planning of the trial. First, a reasonable hypothesis about the non-progression rate for each cohort needs to be determined with clinicians to reflect a clinically relevant desirable outcome. A non-informative prior distribution should only be considered in the absence of strong idea about the probability of success, which was the case in the sorafenib cohort of MOST-plus, but should remain as much as possible an exception. Second, the adaptive algorithms must be pre-specified with precise stopping rules in order to minimize operational biases. Finally, the requirement for computer-based simulations requires more planning time than experimental designs based on a frequentist methodology. The second methodological choice implemented in the MOST-plus trial was a randomization discontinuation design. RDD which allows the enrichment of study population with selected patients following an initial induction treatment period and the conduct of a controlled trial with reduced sample size and limited use of placebo (compared to upfront randomized trials). In addition, all patients in RDD initially receive the targeted agent (during the induction period), which partially explain the few partial responses observed and prolonged survival in this sub-population.
5. Conclusions
In conclusion, the MOST-plus sorafenib cohort validates that agents targeting Ras/Raf/MEK/ERK signaling pathway may achieve prolonged tumor control. In such situation, the continuation of targeted therapy should be the rule. Novel KRAS-specific inhibitors should be further explored in non-histology-specific tumors bearing specific genomic alterations.
Author Contributions
Conceptualization, O.T, JYB, DP, LM, and SC; methodology, DP,LM, SC;; validation, JYB and OT.; formal analysis, LM and DP.; investigation, OT, MT, CLT, AI, IRC, CDF, FB, AG, CGR, BY, PC, AD and JYB; resources, OT, MT, CLT, AI, IRC, CDF, FB, AG, CGR, BY, PC, AD and JYB.; data curation, LM, GG, DP; writing—original draft preparation, OT, GG, LM, DP, JYB; writing—review and editing, OT, MT, CLT, AI, IRC, CDF, FB, AG, CGR, BY, PC, AD and JYB; project administration, GG and DP.; funding acquisition, GG, OT, JYB, and DP. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Fondation ARC contre le Cancer (Grant PGA120140200809). LYRICAN (INCA-DGOS-INSERM 12563), NetSARC (INCA & DGOS), InterSARC (INCA), LabEx DEvweCAN (ANR-10-LABX 0061), PIA Institut Convergence Francois Rabelais PLAsCAN (PLASCAN, 17-CONV-0002), La Ligue contre le Cancer (Canopée) and EURACAN (EC 739521).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee CPP Sud-EST IV (protocol code ET12-081 and 17/01/2023).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to legal, ethical and contractual reasons.
Acknowledgments
We thank the patients and their families for participating in this trial, all participating center staff as well as iDMC members Pr Dietrich, Pr Penel, and Pr Franck Bonnetain. We thank S Darnis for editing of our manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dancey J.E., Bedard P.L., Onetto N., et al.: The genetic basis for cancer treatment decisions. Cell. 2012; 148: 409-20. [CrossRef]
- Vogelstein B, Kinzler K.W. The Path to Cancer --Three Strikes and You're Out. New England Journal of Medicine. 2015; 373:1895-98, 20. [CrossRef]
- Knepper T.C., Bell G.C., Hicks J.K., et al. Key Lessons Learned from Moffitt’s Molecular Tumor Board: The Clinical Genomics Action Committee Experience. Oncologist. 2017; 22:144-51. [CrossRef]
- Massard C, Michiels S, Ferté C, et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discovery. 2017; 7:586-95. [CrossRef]
- Tsimberidou A.M., Iskander N.G., Hong D.S., et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clinical Cancer Research. 2012; 18:6373-83. [CrossRef]
- Ross J.S., Wang K, Gay L, et al. Comprehensive Genomic Profiling of Carcinoma of Unknown Primary Site: New Routes to Targeted Therapies. JAMA Oncol. 2015; 1:40-9. [CrossRef]
- Tsimberidou A.M., Wen S, Hong D.S., et al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clinical Cancer Research. 2014; 20:4827-36. [CrossRef]
- Le Tourneau C, Delord J.P., Gonçalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. The Lancet Oncology. 2015 ; 16:1324-34. [CrossRef]
- Jiang X, Pissaloux D, De La Fouchardiere C, et al. The sum of gains and losses of genes encoding the protein tyrosine kinase targets predicts response to multi-kinase inhibitor treatment: Characterization, validation, and prognostic value. Oncotarget. 2015 ; 6:26388-99. [CrossRef]
- André F, Bachelot T, Commo F, et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncology. 2014 ;15:267-274. [CrossRef]
- Bernichon E, Vallard A, Wang Q, et al. Genomic alterations and radioresistance in breast cancer: an analysis of the ProfiLER protocol. Annals of Oncology. 2017; 28:2773-79. [CrossRef]
- Ratain M.J., Eisen T, Walter M. et al. Phase II Placebo-Controlled Randomized Discontinuation Trial of Sorafenib in Patients With Metastatic Renal Cell Carcinoma. Journal of Clinical Oncology. 2006; 24:2505-12. [CrossRef]
- Saville BR, Berry SM. Efficiencies of platform clinical trials: A vision of the future. Clinical Trials. 2016; 13:358-66. [CrossRef]
- Stadler W. Other paradigms: randomized discontinuation trial design. Cancer Journal. 2009; 15 :431-34. [CrossRef]
- Carlomagno F, Anaganti S, Guida T, et al. BAY 43-9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst. 2006; 98:326-34. [CrossRef]
- Wilhelm SM, Carter C, Tang L, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Research. 2004; 64:7099-09. [CrossRef]
- National Cancer Institute: FDA Approval for Sorafenib Tosylate. 2015. http://www.cancer.gov/about-cancer/treatment/drugs/fda-sorafenib-tosylate (accessed June 8, 2015).
- Llovet, J.M. Sorafenib in Advanced Hepatocellular Carcinoma. New England Journal of Medicine; 2008; 359:378-90. [CrossRef]
- Kane R.C., Farrell A.T., Saber H, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clinical Cancer Research. 2006; 12:7271-78. [CrossRef]
- Eisenhauer E.A., Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). European Journal of Cancer. 2009; 45:228-47. [CrossRef]
- Berry D.A. Bayesian clinical trials. Nature Review. Drug Discovery. 2006; 5:27-36. [CrossRef]
- Zohar S, Teramukai S, Zhou Y. Bayesian design and conduct of phase II single-arm clinical trials with binary outcomes: a tutorial. Contemp Clinical Trials. 2008; 29:608-16. [CrossRef]
- Mateo J, Chakravarty D, Dienstmann R, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Annals of Oncology. 2018; 29:1895-02. [CrossRef]
- Tsimberidou A.M., Hong S.D., Ye Y et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): An MD Anderson Precision Medicine Study. JCO Precis Oncol. 2017;2017:PO.17.00002. [CrossRef]
- Samalin E, Fouchardière C, Thézenas S, et al. Sorafenib Plus Irinotecan Combination in Patients With RAS-mutated Metastatic Colorectal Cancer Refractory To Standard Combined Chemotherapies: A Multicenter, Randomized Phase 2 Trial (NEXIRI-2/PRODIGE 27). Clinical Colorectal Cancer. 2020; 19:301-10. [CrossRef]
- Cook J.H., Melloni G.E.M, Gulhan D.C, et al. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nature Communications. 2021; 12:1808. [CrossRef]
- Zhu YJ, Zheng B, Wang HY, et al. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017; 38:614-22. [CrossRef]
- Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019; 575:217-23. [CrossRef]
- Skoulidis F, Li B., Dy GK, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. New England Journal of Medicine. 2021; 384:2371-81. [CrossRef]
- Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012; 483:100-03. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



