
Submitted:
15 May 2023
Posted:
16 May 2023
You are already at the latest version
Abstract
The highly specialized structure and function of neurons depend on a sophisticated organization of the cytoskeleton, which supports a similarly sophisticated system to traffic organelles and cargo vesicles. Mitochondria sustain crucial functions by providing energy and buffering calcium where is needed. Accordingly, the distribution of mitochondria is not even in neurons and is regulated by a dynamic balance between active transport and stable docking events. This system is finely tuned to respond to changes in environmental conditions and neuronal activity. In this review, we summarize the mechanisms by which mitochondria are selectively transported in different compartments taking into account the structure of the cytoskeleton, the molecular motors and the metabolism of neurons. Remarkably, the motor proteins driving the mitochondrial transport in axons have been shown to mediate also their transfer between cells. This so-named intercellular transport of mitochondria is opening new exciting perspectives in the treatment of multiple diseases.
Keywords:
cytoskeleton
; microtubules
; mitochondria
; neuron
; transport
; TNTs
; mitochondrial transplantation
1. Introduction
Neurons are polarized structures divided in compartments, which are functionally distinct units. Organelles, proteins and RNA are transported along neuronal processes – named axons and dendrites – to distal areas as synapses, growth cones and branching points where mitochondria play different roles. Mitochondria are abundant in presynapses, while roughly 10% of dendritic spines contains mitochondria in basal conditions (1). Neuronal activity increases the transport of mitochondria in synapses (1). At a broader dimension, the somatodendritic and the axonal compartments have different distribution and trafficking properties of cargo vesicles and organelles, which do not diffuse but are actively selected at the pre-axonal exclusion zone (2). The majority of mitochondria are stationary in axons across different species (3). The other motile mitochondria travel long distances and the direction of the transport is named anterograde or retrograde if it is from the cell body to the distal area of axons or vice versa. Motile mitochondria show complex trajectories, including linear and oscillatory, with pauses and changes of direction (3). The fraction of mitochondria in a moving or a stationary state is associated to axonal growing: in the region of active growth cones there is a motile-to-stationary shift of mitochondria that is reversed when axonal growth is blocked. The consequence of this dynamic balance is that the net transport is anterograde in growing axons and retrograde in blocked axons (4). In this review, we focus on the mechanisms regulating the complex transport of mitochondria along the extraordinary distances over which neurons can extend.
Another type of long-distance transport contributing to the maintenance of neural homeostasis is represented by the transfer of mitochondria between adjacent cells. Different processes drive this transcellular communication, including the formation of tunneling nanotubes (TNTs). These structures, described for the first time ten years ago by Rustom and colleagues (5), seem to rely on the same molecular system used for mitochondrial transport and docking in axons. The intracellular transmission of mitochondria observed in astrocytes, microglia, and neurons, is important for the recovery of neural functions supporting both their viability and post-injury recovery (6-9). Similarly to spontaneous mitochondrial transfer between cells, stem cell-derived mitochondrial transplantation can provide an exogenous mitochondrial source thus restoring the mitochondrial functions in recipient cells (10, 11). As the primary hallmark in a wide range of brain states and pathologies is mitochondrial dysfunction, mitochondrial delivery into injured cells is opening a novel horizon for treating many diseases. Here we summarize the clinical applications highlighting the opportunities and challenges of mitochondrial transfer/transplantation, especially in brain disorders.
2. Mitochondria Move Along Microtubules and Actin Filaments
The cytoskeleton in neurons is composed of microtubules, actin filaments and neurofilaments. Neurofilaments are enriched in axons where they determine the diameter and the conductance. Microtubules and actin filaments regulate axonal maturation and growth and build the support for the transport of organelles, including mitochondria (12)(Figure 1) .
2.1. Microtubules
Microtubules are tubes assembled by dimers of α- and β-tubulin oriented head-to-tail in rapid phases of growth and collapse named “dynamic instability”(13). These structures confer the distinct polarity of microtubules, where α- and β-tubulins are exposed at the minus- and plus-end of microtubules, respectively. Microtubules have a peculiar organization and dynamics in neurons: they present a plus-end toward the distal part of axons and have a mixed polarity in dendrites (14). Microtubules polymerize at the plus-end via incorporation of fresh GTP to β-tubulin, which is hydrolysed to GDP in already incorporated tubulin dimers. However, GTP-bound tubulin dimers have been described also in the stable microtubule lattice (15-17) and are more enriched in axons than dendrites (17, 18). These so-named GTP islands protect microtubule depolymerisation and promote self-repair (19, 20) but also to regulate the local conformation of tubulin to modulate the transport of mitochondria(18). A recent study showed that anterograde mitochondria halt along GTP-bound elongated dimers within the microtubule bundle but they remain motile at the rim of the microtubule bundle(18). Furthermore, the affinity of the motor proteins kinesins linking organelles to microtubules depends on the different conformation of GDP- or GTP-bound dimers (17, 21, 22). For mitochondria, these elongated GTP islands increase the velocity of kinesin motor for mitochondria KIF5B (18).
Post-translational modifications of tubulin appear after polymerization of microtubules and include acetylation, detyrosination, glycylation and glutamylation. Developing axons grow in response to attractive and repulsive chemical guidance clues. This process is highly dynamic and involves cycles of de- and re-polymerization of actin and microtubules in the terminal part of axons named growth cone. In an initial phase, actin extends in protrusions, which are later invaded by microtubules and organelles as mitochondria and endoplasmic reticulum (engorgement). No acetylation is detected in growth cones, consistent with the presence of highly dynamic microtubules (23). Finally, the new formed structure is consolidated by depolymerization of actin and stabilization of microtubules (24). This process is regulated by kinases activated by growth factors as Slit, NGF or Wnt bound to the respective receptors Robo, TrakA and Frz/LRP. These GSK3β and Abl kinases regulate the localization of plus-end tracking proteins as CLASP2, APC and MAP1B which alter the stability of microtubules (25). Differentiated axons exhibit stable microtubules with high acetylation, glutamylation, and detyrosination (23). Over their role in regulating the stability of microtubules, post-translational modifications of tubulin regulate the affinity of motor adaptors to microtubules and alter the general transport of organelles rather than being specific to mitochondria (26, 27). Accessory proteins of microtubules regulate the transport of mitochondria in a similar way. For instance, MAP1B knock out neurons increase retrograde transport (28) and mitochondria of N2a cells or primary neurons overexpressing Tau do not travel in anterograde direction (29-31). This is probably due to a combination of Tau to generally destabilize microtubules (32) and to inhibit the kinesin-dependent transport of vesicles and organelles (31). Taken together, these studies seem to indicate that the transport of mitochondria is not specifically regulated at the level of the microtubule organization. However, it is possible that studies conducted so far analyzed general movement of mitochondria (stationary over motile mitochondria) and that broader methods to dissect the complex motility of mitochondria could show a subtle but specific role of accessory proteins and post-translational modifications of tubulin (33).
2.2. Actin
Actin filaments are formed by polarized globular monomers bound via weak interactions. Thus, actin polymers are intrinsically unstable and difficult to visualize in neurons. Electron microscopy allowed the visualization of patches of actin along axons, synapses and growth cones (12). Very little is known about the role of actin in the long range transport of mitochondria. It appears that microtubules do not have the exclusive role in this transport because the depolimerization of microtubules using nocodazole or vinblastine does not stop completely mitochondria in axons and dendrites (34, 35). In addition, disruption of the actin cytoskeleton using cytochalasin-D or lantruculin B had no effect (34). A more recent study shows that cytochalasin-D stabilizes axonal mitochondria and destabilizes dentritic mitochondria (36), suggesting that actin organization modulates the transport of mitochondria via different mechanisms in the two compartments. As we mentioned, actin is also present at synapses where it mediates the docking of mitochondria after administration of nerve growth factor (NGF) (37), regulating the short-range transport of mitochondria.
3. Molecular Motors Transport Mitochondria via Microtubules
Mitochondria associate with the microtubule network through kinesin and dynein, the molecular motors which drive mitochondria respectively in anterograde and retrograde direction (Figure 1). Therefore, kinesins move mitochondria towards the plus-ends of microtubules and dynein mediates minus-end directed mitochondrial transport. Among the kinesin superfamily proteins (also known as KIFs) encoded in humans and mice by 45 different genes (38), KIF5/kinesin-1, KIF1B/kinesin-3 and Kinesin-Like Protein 6 (KLP6) are the main kinesins that mediate mitochondrial transport in neurons (39-41). Two kinesin heavy chains (KHCs) and two kinesin light chains (KLCs) form a 380 kDal heterotetramer complex. It is composed of a conserved globular motor domain (or head) consisting of an ATP-binding motif and a microtubule-binding domain, attached to a stalk domain for dimerization and to a tail domain with binding and regulatory functions. While the motor domains are highly conserved, the remaining sequences are unique for each kinesin determining the cargo specificity and the direction of the transport. In contrast to the large specialized kinesin superfamily, only the cytoplasmatic dynein 1 drives the minus-end-directed microtubule transport. Then, different mechanisms must explain how dynein specifically transport the numerous different cargos. Dynein is a very large protein complex (1.2MD) composed of distinct polypeptides all of which are present in two copies. The dynein heavy chain (DHC) contains the motor domain with six distinct AAA domains folded into a ring-shaped structure, together with a microtubule binding domain and a tail for assembling of the other components: the intermediate chains (DIC), the light intermediate chains (DLIC), and three different light chains (DLC) (42, 43). To be fully active, dynein requires the interaction with dynactin complex and a coiled-coil cargo adaptor (44). Dynactin anchors dynein to microtubules through its larger subunit p150Glued. In addition, dynactin contains two actin-related proteins, Arp1 and Actr10/Arp11 and the subunits p62, p25, and p27 (45). Loss of Actr10 reduces selectively the mitochondrial retrograde transport leading to accumulation of mitochondria in axon terminals (45). Specific adaptor proteins link kinesin and dynein-dynactin to each cargo for its transport along microtubules. On the outer mitochondrial membrane, the Mitochondrial Rho GTPase protein (Miro 1 and Miro 2, in humans RHOT1 and RHOT2) recruits motor proteins by Milton/TRAK adaptors and metaxins (MTX) proteins. Miro contains a transmembrane domain in its C-terminus and two GTPase domains, one at the N-terminal and one near the C-terminal, flanking two calcium binding EF hand motifs which regulate the motility of mitochondria depending on calcium. The GTPase domains are crucial for regulating mitochondrial distribution: when GDP is bound Miro does not recruit adaptor and motor proteins(46). In mammals the different binding specificity of the two TRAK proteins targets mitochondria to dendrites and axons: while TRAK1 binds to both kinesin and dynein components and is responsible for axonal localization of mitochondria, TRAK2 primarily interacts with dynein and plays a critical role in targeting mitochondria to dendrites (47).
Interestingly, as in Miro1/2 double-knockout cells TRAKs are still recruited to the outer mitochondrial membrane to drive mitochondrial trafficking, Miro cannot be the only outer membrane protein that links mitochondria to motor proteins (48). In fact, loss of Drosophila Miro (dMiro) cannot fully block mitochondrial movement (49). Thus, several other kinesin-containing complexes have been discovered in neurons, including syntabulin (SYBU), fasciculation and elongation protein zeta 1 (FEZ1), or Ran-binding protein 2 (RanBP2) (50-52). Interestingly all these adaptors contribute to the maintenance and remodeling of synapses (53, 54). Another interactor of the Miro/Milton complex is Mitofusin 2 (Mfn2) which activity is disrupted in presence of pathogenic mutants, which arrest mitochondria independently from Mfn2 profusion activity (55). In contrast with the anterograde transport machineries, the players that link dynein to mitochondria are not well characterized. One suggested system is represented by the direct interaction of the OMM protein voltage-dependent anion-selective channel (VDAC) with dynein motor protein (56). In conclusion, the main molecular actors involved in anterograde mitochondrial transport form a complex with MIRO (receptor), KIF5 (motor), and Milton/TRAKs (adaptors). Oppositely, dynein in complex with dynactin mediate retrograde mitochondrial transport by interacting with Milton/TRAK2 and MIRO.
4. Mitochondrial Docking and Anchoring Machineries in Neurons
Mitochondria localize in specific regions of the neuron that need most energy and high ions flux, such as the synapses or the distal regions of actively growing axons (57). While a large proportion of mitochondria is moving during neuronal development, in mature neurons the stationary pool of mitochondria represent more than two-thirds. Remarkably, more than 30% of synapse is occupied by anchored mitochondria serving as ‘power stations’ (58). In addition, as mitochondria can temporarily stop and start moving again at various subcellular localizations, specific docking mechanisms are needed. The mitochondria-associated protein syntaphilin (SNPH) is a central microtubules calcium-dependent docking system (Figure 1). Syntaphilin directly interacts with microtubules through its N-terminal microtubule-binding domain while its C-terminal tail inserts into the OMM (59). The dynein light chain (DLC) LC8 binds to SNPH, thus enhancing the SNPH-based docking of mitochondria to microtubules (60). SNPH can also interact with the myosin VI (MYO6, Jaguar in Drosophila) and anchor mitochondria on presynaptic filamentous (F)-actin. The AMPK-dependent phosphorylation of MYO6 drives the capture of mobile axonal mitochondria at presynaptic terminals switching them from microtubule-dependent transport to actin-mediated tethering (61). Other myosin motor proteins in association with the actin cytoskeleton have been shown to interrupt mitochondrial transport on the microtubule tracks facilitating their positioning. For example, Myo19, stops mitochondria to actin filaments through Miro (62) (Figure 1). In general, where mitochondria are more needed, actin is enriched and docks mitochondria away from microtubules.
Neurons are polarized cells divided in soma, dendrites and axons. Microtubules are plus-ending oriented toward axonal terminals and are mixed oriented in dendrites. The orientation of microtubules in axons determines the transport of mitochondria towards the distal part (retrograde) or the soma (anterograde). GTP, post-translational modifications (PTMs) and microtubule associated proteins (MAPs) regulate the movement of mitochondria acting on the stability of microtubules and the affinity of motor proteins that bind mitochondria. Kynesin and dynein mediate the anterograde and retrograde transport, respectively, of organelles and vesicle cargoes. Specific receptors link these motor proteins to mitochondria. Syntabulin and the Miro/Trak complex bind kinesin and Acrt10 and VDAC1 bind the dynein/dynactin complex. Mitochondria travel also along actin filaments but the mechanisms regulating long-range transport are largely unknown. Finally, actin filaments dock mitochondria at synapses using the adaptors Myo19 and Snph.
5. Metabolic Control of Mitochondrial Transport
Mitochondria produce ATP and buffer calcium for the functioning and survival of neurons. Thus, it is not surprising that mitochondria are more abundant in neurons than in other cell types and that calcium and ADP/ATP are a prime signaling molecules to regulate the distribution of mitochondria. Glutamate increases calcium and immobilizes mitochondria at synapses. A similar effect is obtained injecting ADP or in hypoxia (63). It seems that the localization of mitochondria is largely regulated by stationarity rather than transport as the signaling molecules studies pointed out so far.
5.1. Calcium
As mentioned above, mitochondria rely on the adaptor proteins Milton in Drosophila and TRAKs in mammals for anterograde movement along axonal microtubules. These adaptors bridge the receptor proteins Miro1 and 2 located in the outer mitochondrial membrane to kinesin-1 (62). Miro contains two EF-hands that bind calcium. In absence of these domains, mitochondria fail to stop in presence of calcium in axons and dendrites (62, 64), placing Miro as a regulator of mitochondrial arrest in sites where cytoplasmic calcium concentration is high. Two models explain a conformational change of Miro in presence of calcium but the mechanism is opposite. According to Wang and Schwarz, kinesin-1 binds microtubules with its N-terminal domain and the Miro/Milton complex via its C-terminal region (64). This complex transports mitochondria but calcium induces the sequestration of the whole complex from microtubules. In MacAskill et al., Miro directly binds kinesin-1 and calcium detaches kinesin-1 from mitochondria but not from microtubules (62). In contrast, another study shows that Miro1 binds the mitochondrial calcium uniporter (MCU) through its N-terminal domain on the outer mitochondrial membrane (65). This interaction is required for the transport of mitochondria and it depends on the MCU-dependent calcium influx in the mitochondrial matrix rather than cytoplasmic calcium (66). E208K/E328K mutations in the EF-hands of Miro abolished calcium entry in the matrix (66). Curiously, a different mutation (R272Q) in the EF-hands of Miro has no effect on mitochondrial movement although it disrupts calcium handling into mitochondria (67). The discrepancies on the role of Miro could be explained by the use of different mutants and raise the possibility that Miro is regulated by accessory proteins bound to the EF-hands or that the regulation of the transport complex is yet to be fully addressed (68). Indeed, the EF1 domain of Miro1 senses cytosolic calcium and changes the shape of mitochondria independently of MCU-dependent calcium uptake and fusion/fission proteins in fibroblasts (69). Furthermore, the analysis performed in neurites of iPSCs and in axons and dendrites of primary neurons could also indicate different functions of Miro depending on its location in neurons. For example, HDAC6 deacetylates Miro and stops mitochondria in axons rich in calcium (70) (Figure 2). Altough intriguing, given the broad functions of Mfn2 it is difficult to link directly mitochondrial transport to the levels of mitochondrial calcium (71). SNPH is another calcium sensor that arrests mitochondria in axons (60, 72). In presence of elevated calcium, SNPH dissociates kinesin-1 from mitochondria to enhance their docking at presynapses: high calcium levels disrupt the Miro-Trak-Kinesin complex favoring the anchoring of mitochondria by SNPH that inhibits the activity of kinesin (73). The anchoring of mitochondria to microtubules by SNPH is reversible since in absence of calcium Miro can rapidly resume its calcium-free conformation and form the Miro/Trak/Kinesin complex to drive mitochondrial motility (62).
5.2. Glucose
Elevated electric activity mobilizes the Glut3 and Glut4 transporters at presynapses to increase the availability of glucose to boost local metabolism (74-76). Thus it is not surprising that glucose additionally halts mitochondria via an active mechanism (77). O-GlcNAcylation is a post-translational modification important for proteins involved in neuronal signaling and synaptic plasticity (78, 79). Glucose activates O-GlcNAc transferase and increases the O-GlcNAcylation of Milton, which inhibits the transport of mitochondria (77). The protein four and a half LIM domains protein 2 (FHL2) associates to O-GlcNAcylated Milton and favors the docking of mitochondria to actin (80) (Figure 2).
5.3. ATP
The ratio of ADP and ATP regulates the positioning of mitochondria in primary neurons (63, 81), possibly involving mechanisms to sense and drive mitochondria to sites of high energy consumption. Since motor proteins require ATP to transport mitochondria, it is possible that in sites of high ATP consumption mitochondria halt because kinesin and dynein are not functional. The contribution of ATP and calcium signals to mitochondrial motility are difficult to disentangle because mitochondria are major regulators of the concentration of these two molecules. A study shows that neuronal depolarization decreases ATP levels and activate AMP-activated protein kinase (AMPK), increasing the anterograde transport of mitochondria into axons (81). Similar results were obtained when lactate uptake was inhibited locally (82). These studies suggest that the motility of mitochondria can be regulated by ATP independently of calcium. However, it is difficult to understand if this transport is directly regulated by AMPK or if it is the consequence of the broad mechanisms downstream of AMPK (Figure 2).
5.4. Hypoxia
Hypoxia is an important factor during brain ischemia and it has been shown to regulate mitochondrial motility in neurons (83, 84) (Figure 2). Decreased oxygen induces hypoxia-inducible factor 1α (HIF-1α) that, among its other targets, increases the expression of hypoxia up-regulated mitochondrial movement regulator (HUMMR). Reduced HUMMR in normoxia does not change the motility of mitochondria but diminishes the amount of motile mitochondria with anterograde movement in hypoxia, consistent with a defect in kinesin-1 transport (83). Indeed, HUMMR coimmunoprecipitates with Miro and Milton in normoxia (83). It would be interesting to understand if HUMMR is also a sensor of hypoxia and if it increases its binding the Miro/Milton complex. Interestingly, nitric oxide (NO) inhibitors recovered the motility of mitochondria during hypoxia and NO administration halted mitohondria (84, 85). It is not clear if this is a direct or indirect effect nor if HUMMR is involved.
5.6. Reactive Oxygen Species (ROS)
Sources of ROS in the CNS are mitochondria and reactive microglia during inflammation. Intracellular and extracellular ROS equally block mitochondria without affecting other organelles (86-88). In these experiments, axons are more vulnerable than dendrites (87). Two opposite studies show that ROS inhibit the Miro/Milton complex by activating p38α MAPK independently from calcium (88) and that ROS increase calcium and the activity of the c-Jun N-terminal Kinase (86).
5.7. Growth Factors and Neurotransmitters
In addition, molecules for neuronal communication and growth seem to modulate the transport of mitochondria (Figure 2). In developing neurons, administration of NGF accumulates mitochondria close to the site of treatment (37). Although the mechanism is not clear, it seems to involve the PI3 kinase pathway (89). Moreover, serotonin and dopamine act via their respective receptors on the AKT-GSK3β pathway with opposite effects on mitochondria. Dopamine halts and serotonin mobilizes mitochondria (90, 91). Interestingly, GSK3β was found to be localized to HDAC6 and HDAC6 activity and phosphorylation were regulated by GSK3β in primary neurons (92).
6. Mitophagy
Mitochondria can be also arrested when they are sequestered and degraded by autophagic engulfment, a process known as mitophagy. Damaged and depolarized mitochondria are cleared by the Pink1/Parkin pathway (Figure 2). In this conditions, Pink1 is recruited and stabilized by Parkin on the outer mitochondrial membrane. This complex interacts with the Miro/Milton complex and induces the proteasomal degradation of Miro, thus releasing kinesin and arresting mitochondria (93, 94). It is under debate where autophagy occurs, if it involves the transport of mitochondria to the soma or if it happens in distal axons (95, 96). Nevertheless, it is clear that transport and degradation of mitochondria are intertwined processes regulated by metabolism (97, 98).
Mitochondria are transported along microtubules with the Kynesin/Milton/Miro complex. Cytoplasmatic calcium can bind directly to Miro EF-hands or increase calcium in the matrix of mitochondria through MCU. It is not clear if mitochondria halt following the dissociation of the whole complex or if kinesin remains attached to microtubules. This process involves a change of the shape of mitochondria (mitochondrial shape transition, MiST) independent of fusion and fission proteins. Also, mitochondria can stop following deacetylation of Miro by histone deacetylates 6 (HDAC6) in presence of calcium or probably by active GSK3β. Mitochondria can be transferred to syntaphilin (SNPH) to enhance their docking. Glucose induces the O-GlcNAcylation of Miro and the recruitment of half LIM domains protein 2 (FHL2) to dock mitochondria. Hypoxia induces the expression of hypoxia up-regulated mitochondrial movement regulator (HUMMR) and nitric oxide (NO) levels. Reactive oxygen species (ROS) activate p38α MAPK which inhibits Miro and activate c-Jun N-terminal Kinase (JNK) which stops mitochondria. Depolarized and damage mitochondria can recruit and stabilize the Pink1/Parkin complex, which targets Miro to proteasomal degradation and induces mitophagy. Finally, PI3K and AMPK regulate the movement of mitochondria by unknown mechanisms.
7. Specialized Cytoskeleton Structures Allow Mitochondria to Cross Cell Boundaries
Proper neuronal homeostasis is maintained not only by intracellular trafficking of mitochondria but also by intercellular exchange of mitochondria. For example, astrocytes can transfer healthy mitochondria to damaged neurons and provide neuroprotection and neurorepair (7-9, 99). Vice versa, astrocytes may internalize damaged mitochondria. Davis and colleagues described this process, named transmitophagy, at the optic nerve head where retinal ganglion cell axons shed the damaged mitochondria to be degraded by astrocytes (100). Interestingly, in vivo transplanted stem cells have been shown to perform a similar bidirectional mitochondrial exchange (101-103). Therefore, stem cells can both donate their healthy mitochondria and take up damaged mitochondria from somatic stressed cells for degradation (104, 105). Today, this intercellular mitochondrial transfer represents probably the most exciting therapeutic option for multiple diseases. Different mechanisms for transcellular transfer of mitochondria have been identified, including the formation of tunneling nanotubes (TNTs) (106). These structures form transient cytoplasmic bridges containing a skeleton mainly composed of F-actin, microtubules or both. Thinner TNTs (<100 nm in diameter) only contain F-actin, whereas thicker TNTs (>100 nm in diameter) are composed of both F-actin and microtubules. TNTs may polymerize and depolymerize rapidly in 30-60 seconds, spanning distances of up to 300 µm. The movement of mitochondria along these structures is mediated by transport complexes (107). As for intracellular trafficking, the key protein that modulates intercellular mitochondria transfer is Miro. It interacts directly with the motor protein KIF5, or through adaptor proteins, including TRAK1 and TRAK2 and MYO10 and MYO19. The knocking down or overexpression of Miro affect the efficiency of the transfer of mitochondria from mesenchymal stem cells (MSCs) to nerve cells in order to repair injury (103, 108). The horizontal mitochondrial transfer has been nominated by Liu et al. as the “find me” and “save me” intercellular comunication, since damaged cells take up functional mitochondria from healthy donor cells (109). However, the signaling mechanisms that initiate it remain unknown. A calcium-dependent mechanisms involving CD38 has been proposed by Hayakawa who studied the role of transcellular mitochondrial trafficking in post-stroke recovery (7).
Many studies show that it is feasible to treat many diseases in brain associated with mitochondrial dysfunctions by the described mitochondrial transfer process between cells or even by direct transplantation of isolated mitochondria (Table 1). It will be important to decipher in details the mechanisms regulating mitochondria entering in damaged cells including TNTs formation, to understand the contributing molecular elements and make mitochondrial transport-based therapy successful.
8. Conclusions
In this review, we summarized the complexity of the transport of mitochondria in neurons and the similarity of this system to the intracellular transport of mitochondria. Although the first has been broadly characterized, it could give the basis to understand how the latter happens and ameliorate its potential benefits in therapeutic approaches. The comprehension we have of this transport is quite extensive but it is clear that some of the conclusions established in the past were oversimplified. Recent advances in technology and analysis will help to investigate the fine regulation of mitochondrial motility, especially regarding poorly studied movements as duration of pauses and oscillatory movements. These mechanisms will possibility link microtubule dynamics to motor proteins at a more refined resolution, scaling down to micron-size compartments as spines. It will be intriguing to understand how metabolism regulates such movement at a single-mitochondrion scale.
Author Contributions
Conceptualization, CB and MZ; writing—original draft preparation, CB and MZ.; writing—review and editing, CB and MZ; supervision, CB and MZ. All authors have read and agreed to the published version of the manuscript.
Funding
MZ was supported by the Deutsche Forschungsgemeinschaft (Project number 269925409).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We gratefully acknowledge the substantial contribution of all the investigators in the field, that we could not include in this review caused space limitations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Farias, G.G.; Guardia, C.M.; Britt, D.J.; Guo, X.; Bonifacino, J.S. Sorting of Dendritic and Axonal Vesicles at the Pre-axonal Exclusion Zone. Cell Rep. 2015, 13, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron. 2017, 96, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.L.; Hollenbeck, P.J. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993, 104 Pt 3, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bukoreshtliev, N.V.; Gerdes, H.-H. Developing neurons form transient nanotubes facilitating electrical coupling and calcium signaling with distant astrocytes. PLoS ONE 2012, 7, e47429. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Huang, L.; Nakamura, Y.; Lo, E.H.; Hayakawa, K. Astrocyte signaling in the neurovascular unit after central nervous system injury. Int. J. Mol. Sci. 2019, 20, 282. [Google Scholar] [CrossRef]
- Lippert, T.; Borlongan, C.V. Prophylactic treatment of hyperbaric oxygen treatment mitigates inflammatory response via mitochondria transfer. CNS Neurosci. Ther. 2019, 25, 815–823. [Google Scholar] [CrossRef]
- Rocca, C.J.; Goodman, S.M.; Dulin, J.N.; Haquang, J.H.; Gertsman, I.; Blondelle, J.; et al. Transplantation of wild-type mouse hematopoietic stem and progenitor cells ameliorates deficits in a mouse model of Friedreich’s ataxia. Sci. Transl. Med. 2017, 9, eaaj2347. [Google Scholar] [CrossRef]
- Liu, K.; Guo, L.; Zhou, Z.; Pan, M.; Yan, C. Mesenchymal stem cells transfer mitochondria into cerebral microvasculature and promote recovery from ischemic stroke. Microvasc. Res. 2019, 123, 74–80. [Google Scholar] [CrossRef]
- Kevenaar, J.T.; Hoogenraad, C.C. The axonal cytoskeleton: From organization to function. Front Mol Neurosci. 2015, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Mitchison, T.J. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997, 13, 83–117. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Black, M.M.; Banker, G.A. Changes in microtubule polarity orientation during the development of hippocampal neurons in culture. J. Cell Biol. 1989, 109, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, A.; Quesnoit, M.; Moutel, S.; Cantaloube, I.; Pous, C.; Perez, F. Detection of GTP-tubulin conformation in vivo reveals a role for GTP remnants in microtubule rescues. Science 2008, 322, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Aumeier, C.; Schaedel, L.; Gaillard, J.; John, K.; Blanchoin, L.; Thery, M. Self-repair promotes microtubule rescue. Nat Cell Biol. 2016, 18, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Nakata, T.; Niwa, S.; Okada, Y.; Perez, F.; Hirokawa, N. Preferential binding of a kinesin-1 motor to GTP-tubulin-rich microtubules underlies polarized vesicle transport. J Cell Biol. 2011, 194, 245–255. [Google Scholar] [CrossRef]
- Van Steenbergen, V.; Lavoie-Cardinal, F.; Kazwiny, Y.; Decet, M.; Martens, T.; Verstreken, P.; et al. Nano-positioning and tubulin conformation contribute to axonal transport regulation of mitochondria along microtubules. Proc. Natl. Acad. Sci. USA 2022, 119, e2203499119. [Google Scholar] [CrossRef]
- Tropini, C.; Roth, E.A.; Zanic, M.; Gardner, M.K.; Howard, J. Islands containing slowly hydrolyzable GTP analogs promote microtubule rescues. PLoS ONE 2012, 7, e30103. [Google Scholar] [CrossRef]
- Vemu, A.; Szczesna, E.; Zehr, E.A.; Spector, J.O.; Grigorieff, N.; Deaconescu, A.M.; et al. Severing enzymes amplify microtubule arrays through lattice GTP-tubulin incorporation. Science 2018, 361, eaau1504. [Google Scholar] [CrossRef]
- Guedes-Dias, P.; Nirschl, J.J.; Abreu, N.; Tokito, M.K.; Janke, C.; Magiera, M.M.; et al. Kinesin-3 Responds to Local Microtubule Dynamics to Target Synaptic Cargo Delivery to the Presynapse. Curr Biol. 2019, 29, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Shima, T.; Morikawa, M.; Kaneshiro, J.; Kambara, T.; Kamimura, S.; Yagi, T.; et al. Kinesin-binding-triggered conformation switching of microtubules contributes to polarized transport. J Cell Biol. 2018, 217, 4164–4183. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Brady, S.T. Post-translational modifications of tubulin: Pathways to functional diversity of microtubules. Trends Cell Biol. 2015, 25, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Dent, E.W.; Gertler, F.B. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003, 40, 209–227. [Google Scholar] [CrossRef]
- Geraldo, S.; Gordon-Weeks, P.R. Cytoskeletal dynamics in growth-cone steering. J Cell Sci. 2009, 122 Pt 20, 3595–3604. [Google Scholar] [CrossRef]
- Hammond, J.W.; Huang, C.F.; Kaech, S.; Jacobson, C.; Banker, G.; Verhey, K.J. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol Biol Cell. 2010, 21, 572–583. [Google Scholar] [CrossRef]
- Nirschl, J.J.; Magiera, M.M.; Lazarus, J.E.; Janke, C.; Holzbaur, E.L. alpha-Tubulin Tyrosination and CLIP-170 Phosphorylation Regulate the Initiation of Dynein-Driven Transport in Neurons. Cell Rep. 2016, 14, 2637–2652. [Google Scholar] [CrossRef]
- Jimenez-Mateos, E.M.; Gonzalez-Billault, C.; Dawson, H.N.; Vitek, M.P.; Avila, J. Role of MAP1B in axonal retrograde transport of mitochondria. Biochem J. 2006, 397, 53–59. [Google Scholar] [CrossRef]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer's disease. J Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef]
- Shahpasand, K.; Uemura, I.; Saito, T.; Asano, T.; Hata, K.; Shibata, K.; et al. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J Neurosci. 2012, 32, 2430–2441. [Google Scholar] [CrossRef]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef] [PubMed]
- Bodakuntla, S.; Magiera, M.M.; Janke, C. Measuring the Impact of Tubulin Posttranslational Modifications on Axonal Transport. Methods Mol Biol. 2020, 2101, 353–370. [Google Scholar] [PubMed]
- Ligon, L.A.; Steward, O. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J Comp Neurol. 2000, 427, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.L.; Hollenbeck, P.J. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995, 131, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, V.; Lauterbach, M.; Schuman, E.M. Spatially Stable Mitochondrial Compartments Fuel Local Translation during Plasticity. Cell. 2019, 176, 73–84. [Google Scholar] [CrossRef]
- Chada, S.R.; Hollenbeck, P.J. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr Biol. 2004, 14, 1272–1276. [Google Scholar] [CrossRef]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef]
- Lin, M.-Y.; Sheng, Z.-H. Regulation of mitochondrial transport in neurons. Exp. Cell Res. 2015, 334, 35–44. [Google Scholar] [CrossRef]
- Nangaku, M.; Sato-Yoshitake, R.; Okada, Y.; Noda, Y.; Takemura, R.; Yamazaki, H.; et al. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 1994, 79, 1209–1220. [Google Scholar] [CrossRef]
- Wozniak, M.J.; Melzer, M.; Dorner, C.; Haring, H.-U.; Lammers, R. The novel protein KBP regulates mitochondria localization by interaction with a kinesin-like protein. BMC Cell Biol. 2005, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Canty, J.T.; Tan, R.; Kusakci, E.; Fernandes, J.; Yildiz, A. Structure and mechanics of dynein motors. Annu. Rev. Biophys. 2021, 50, 549–574. [Google Scholar] [CrossRef] [PubMed]
- McKenney, R.J.; Huynh, W.; Tanenbaum, M.E.; Bhabha, G.; Vale, R.D. Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 2014, 345, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Drerup, C.M.; Herbert, A.L.; Monk, K.R.; Nechiporuk, A.V. Regulation of mitochondria-dynactin interaction and mitochondrial retrograde transport in axons. Elife, 2223. [Google Scholar]
- Davis, K.; Basu, H.; Izquierdo-Villalba, I.; Shurberg, E.; Schwarz, T.L. Miro GTPase domains regulate the assembly of the mitochondrial motor–adaptor complex. Life Sci. Alliance 2023, 6, e202201406. [Google Scholar] [CrossRef]
- Van Spronsen, M.; Mikhaylova, M.; Lipka, J.; Schlager, M.A.; van den Heuvel, D.J.; Kuijpers, M.; et al. TRAK/Milton Motor-Adaptor Proteins Steer Mitochondrial Trafficking to Axons and Dendrites. Neuron. 2013, 77, 485–502. [Google Scholar] [CrossRef]
- López-Doménech G, Covill-Cooke C, Ivankovic D; et al. Miro proteins coordinate microtubule-and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef]
- Guo, X.; Macleod, G.T.; Wellington, A.; Hu, F.; Panchumarthi, S.; Schoenfield, M.; et al. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005, 47, 379–393. [Google Scholar] [CrossRef]
- Cai, Q.; Gerwin, C.; Sheng, Z.-H. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol. 2005, 170, 959–969. [Google Scholar] [CrossRef]
- Cho Ki Cai, Y.; Yi, H.; Yeh, A.; Aslanukov, A.; Ferreira, P.A. Association of the kinesin-binding domain of RanBP2 to KIF5B and KIF5C determines mitochondria localization and function. Traffic. 2007, 8, 1722–1735. [Google Scholar]
- Ikuta, J.; Maturana, A.; Fujita, T.; Okajima, T.; Tatematsu, K.; Tanizawa, K.; et al. Fasciculation and elongation protein zeta-1 (FEZ1) participates in the polarization of hippocampal neuron by controlling the mitochondrial motility. Biochem. Biophys. Res. Commun. 2007, 353, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.-J.; Cheng, X.-T.; Sun, T.; Xie, Y.; Huang, N.; Li, S.; et al. Defects in syntabulin-mediated synaptic cargo transport associate with autism-like synaptic dysfunction and social behavioral traits. Mol. Psychiatry 2021, 26, 1472–1490. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fok, A.H.K.; Fan, R.; Kwan, P.-Y.; Chan, H.-L.; Lo, L.H.-Y.; et al. Specific depletion of the motor protein KIF5B leads to deficits in dendritic transport, synaptic plasticity and memory. eLife 2020, 9, e53456. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070. [Google Scholar] [CrossRef]
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304. [Google Scholar] [CrossRef]
- Wilhelm, B.G.; Mandad, S.; Truckenbrodt, S.; Kröhnert, K.; Schäfer, C.; Rammner, B.; et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 2014, 344, 1023–1028. [Google Scholar] [CrossRef]
- Kang, J.-S.; Tian, J.-H.; Pan, P.-Y.; Zald, P.; Li, C.; Deng, C.; et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef]
- Chen, Y.-M.; Gerwin, C.; Sheng, Z.-H. Dynein light chain LC8 regulates syntaphilin-mediated mitochondrial docking in axons. J. Neurosci 2009, 29, 9429–9438. [Google Scholar] [CrossRef]
- Li, S.; Xiong, G.-J.; Huang, N.; Sheng, Z.-H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020, 2, 1077–1095. [Google Scholar] [CrossRef]
- MacAskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; et al. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009, 61, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Mironov, S.L. ADP regulates movements of mitochondria in neurons. Biophys. J. 2007, 92, 2944–2952. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Schwarz, T.L. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009, 136, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Niescier, R.F.; Hong, K.; Park, D.; Min, K.T. MCU Interacts with Miro1 to Modulate Mitochondrial Functions in Neurons. J Neurosci. 2018, 38, 4666–4677. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.T.; Niescier, R.F.; Min, K.T. Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc Natl Acad Sci U S A 2011, 108, 15456–15461. [Google Scholar] [CrossRef]
- Schwarz, L.; Sharma, K.; Dodi, L.D.; Rieder, L.S.; Fallier-Becker, P.; Casadei, N.; et al. Miro1 R272Q disrupts mitochondrial calcium handling and neurotransmitter uptake in dopaminergic neurons. Front Mol Neurosci. 2022, 15, 966209. [Google Scholar] [CrossRef]
- Zinsmaier, K.E. Mitochondrial Miro GTPases coordinate mitochondrial and peroxisomal dynamics. Small GTPases. 2021, 12, 372–398. [Google Scholar] [CrossRef]
- Nemani, N.; Carvalho, E.; Tomar, D.; Dong, Z.; Ketschek, A.; Breves, S.L.; et al. MIRO-1 Determines Mitochondrial Shape Transition upon GPCR Activation and Ca(2+) Stress. Cell Rep. 2018, 23, 1005–1019. [Google Scholar] [CrossRef]
- Kalinski, A.L.; Kar, A.N.; Craver, J.; Tosolini, A.P.; Sleigh, J.N.; Lee, S.J.; et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J Cell Biol. 2019, 218, 1871–1890. [Google Scholar] [CrossRef]
- Zaman, M.; Shutt, T.E. The Role of Impaired Mitochondrial Dynamics in MFN2-Mediated Pathology. Front Cell Dev Biol. 2022, 10, 858286. [Google Scholar] [CrossRef]
- Kang, J.S.; Tian, J.H.; Pan, P.Y.; Zald, P.; Li, C.; Deng, C.; et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008, 132, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sheng, Z.H. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol. 2013, 202, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.M.; Burnett, A.L.; Rameau, G.A. Activity-dependent regulation of surface glucose transporter-3. J Neurosci. 2011, 31, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Weisova, P.; Concannon, C.G.; Devocelle, M.; Prehn, J.H.; Ward, M.W. Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J Neurosci. 2009, 29, 2997–3008. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Wu, Z.; Farrell, R.J.; Ryan, T.A. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron. 2017, 93, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Pekkurnaz, G.; Trinidad, J.C.; Wang, X.; Kong, D.; Schwarz, T.L. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell. 2014, 158, 54–68. [Google Scholar] [CrossRef]
- Lagerlof, O.; Hart, G.W.; Huganir, R.L. O-GlcNAc transferase regulates excitatory synapse maturity. Proc Natl Acad Sci U S A. 2017, 114, 1684–1689. [Google Scholar] [CrossRef]
- Yang, Y.R.; Song, S.; Hwang, H.; Jung, J.H.; Kim, S.J.; Yoon, S.; et al. Memory and synaptic plasticity are impaired by dysregulated hippocampal O-GlcNAcylation. Sci Rep. 2017, 7, 44921. [Google Scholar] [CrossRef]
- Basu, H.; Pekkurnaz, G.; Falk, J.; Wei, W.; Chin, M.; Steen, J.; et al. FHL2 anchors mitochondria to actin and adapts mitochondrial dynamics to glucose supply. J Cell Biol. 2021, 220, e201912077. [Google Scholar] [CrossRef]
- Tao, K.; Matsuki, N.; Koyama, R. AMP-activated protein kinase mediates activity-dependent axon branching by recruiting mitochondria to axon. Dev Neurobiol. 2014, 74, 557–573. [Google Scholar] [CrossRef]
- Watters, O.; Connolly, N.M.C.; Konig, H.G.; Dussmann, H.; Prehn, J.H.M. AMPK Preferentially Depresses Retrograde Transport of Axonal Mitochondria during Localized Nutrient Deprivation. J Neurosci. 2020, 40, 4798–4812. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lim, S.; Hoffman, D.; Aspenstrom, P.; Federoff, H.J.; Rempe, D.A. HUMMR, a hypoxia- and HIF-1alpha-inducible protein, alters mitochondrial distribution and transport. J Cell Biol. 2009, 185, 1065–1081. [Google Scholar] [CrossRef] [PubMed]
- Zanelli, S.A.; Trimmer, P.A.; Solenski, N.J. Nitric oxide impairs mitochondrial movement in cortical neurons during hypoxia. J Neurochem. 2006, 97, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Rintoul, G.L.; Bennett, V.J.; Papaconstandinou, N.A.; Reynolds, I.J. Nitric oxide inhibits mitochondrial movement in forebrain neurons associated with disruption of mitochondrial membrane potential. J Neurochem. 2006, 97, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.C.; Tandarich, L.C.; Hollenbeck, P.J. ROS regulation of axonal mitochondrial transport is mediated by Ca2+ and JNK in Drosophila. PLoS ONE 2017, 12, e0178105. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Bourdette, D.; Banker, G. Oxidative stress inhibits axonal transport: Implications for neurodegenerative diseases. Mol Neurodegener. 2012, 7, 29. [Google Scholar] [CrossRef]
- Debattisti, V.; Gerencser, A.A.; Saotome, M.; Das, S.; Hajnoczky, G. ROS Control Mitochondrial Motility through p38 and the Motor Adaptor Miro/Trak. Cell Rep. 2017, 21, 1667–1680. [Google Scholar] [CrossRef]
- Chada, S.R.; Hollenbeck, P.J. Mitochondrial movement and positioning in axons: The role of growth factor signaling. J Exp Biol. 2003, 206 Pt 12, 1985–1992. [Google Scholar] [CrossRef]
- Chen, S.; Owens, G.C.; Crossin, K.L.; Edelman, D.B. Serotonin stimulates mitochondrial transport in hippocampal neurons. Mol Cell Neurosci. 2007, 36, 472–483. [Google Scholar] [CrossRef]
- Chen, S.; Owens, G.C.; Edelman, D.B. Dopamine inhibits mitochondrial motility in hippocampal neurons. PLoS ONE 2008, 3, e2804. [Google Scholar] [CrossRef]
- Chen, S.; Owens, G.C.; Makarenkova, H.; Edelman, D.B. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS ONE 2010, 5, e10848. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Weihofen, A.; Thomas, K.J.; Ostaszewski, B.L.; Cookson, M.R.; Selkoe, D.J. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 2009, 48, 2045–2052. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.H. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012, 22, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, X.; Wu, X.; Jiang, L.; Ahsan, A.; Ma, S.; et al. Somatic autophagy of axonal mitochondria in ischemic neurons. J Cell Biol. 2019, 218, 1891–1907. [Google Scholar] [CrossRef] [PubMed]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sa, J.; da Cruz, E.S.O. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011, 18, 732–742. [Google Scholar] [CrossRef]
- Davis, C.-h.O.; Kim, K.-Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Scis. SA 2014, 111, 9633–9638. [Google Scholar] [CrossRef]
- Boukelmoune, N.; Chiu, G.S.; Kavelaars, A.; Heijnen, C.J. Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol. Commun. 2018, 6, 1–13. [Google Scholar] [CrossRef]
- Caicedo, A.; Fritz, V.; Brondello, J.-M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, 9073. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; et al. Miro1 enhances mitochondria transfer from multipotent mesenchymal stem cells (MMSC) to neural cells and improves the efficacy of cell recovery. Molecules. 2018, 23, 687. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Kami, D.; Maeda, R.; Murata, Y.; Jo Ji Kitani, T.; et al. TAT-dextran–mediated mitochondrial transfer enhances recovery from models of reperfusion injury in cultured cardiomyocytes. J. Cell. Mol. Med. 2020, 24, 5007–5020. [Google Scholar] [CrossRef]
- Mahrouf-Yorgov, M.; Augeul, L.; Da Silva, C.C.; Jourdan, M.; Rigolet, M.; Manin, S.; et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. 2017, 24, 1224–1238. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Gao, Y.; Liu, J.; Huang, Y.; Yin, J.; Feng, Y.; et al. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct. Target. Therapy 2021, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [PubMed]
- Gao, L.; Zhang, Z.; Lu, J.; Pei, G. Mitochondria are dynamically transferring between human neural cells and alexander disease-associated GFAP mutations impair the astrocytic transfer. Front. Cell. Neurosci. 2019, 13, 316. [Google Scholar] [CrossRef]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; et al. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res. 2014, 92, 10–18. [Google Scholar] [CrossRef]
- Chang, J.C.; Wu, S.L.; Liu, K.H.; Chen, Y.H.; Chuang, C.S.; Cheng, F.C.; et al. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson's disease: Restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl Res. 2016, 170, 40–56. [Google Scholar] [CrossRef]
- Shi, X.; Zhao, M.; Fu, C.; Fu, A. Intravenous administration of mitochondria for treating experimental Parkinson's disease. Mitochondrion 2017, 34, 91–100. [Google Scholar] [CrossRef]
- Robicsek, O.; Ene, H.M.; Karry, R.; Ytzhaki, O.; Asor, E.; McPhie, D.; et al. Isolated Mitochondria Transfer Improves Neuronal Differentiation of Schizophrenia-Derived Induced Pluripotent Stem Cells and Rescues Deficits in a Rat Model of the Disorder. Schizophr Bull. 2018, 44, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, Z.; Yan, C.; Pu, K.; Wu, M.; Bai, J.; et al. Muscle-derived autologous mitochondrial transplantation: A novel strategy for treating cerebral ischemic injury. Behav Brain Res. 2019, 356, 322–331. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Zorova, L.D.; Pevzner, I.B.; Khutornenko, A.A.; Plotnikov, E.Y.; et al. Improving the Post-Stroke Therapeutic Potency of Mesenchymal Multipotent Stromal Cells by Cocultivation With Cortical Neurons: The Role of Crosstalk Between Cells. Stem Cells Transl Med. 2015, 4, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.-J.; Kuo, C.-C.; Lee, H.-C.; Shen, C.-I.; Cheng, F.-C.; Wu, S.-F.; et al. Transferring Xenogenic Mitochondria Provides Neural Protection against Ischemic Stress in Ischemic Rat Brains. Cell Transp. 2016, 25, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Pourmohammadi-Bejarpasi, Z.; Roushandeh, A.M.; Saberi, A.; Rostami, M.K.; Toosi, S.M.R.; Jahanian-Najafabadi, A.; et al. Mesenchymal stem cells-derived mitochondria transplantation mitigates I/R-induced injury, abolishes I/R-induced apoptosis, and restores motor function in acute ischemia stroke rat model. Brain Res. Bull. 2020, 165, 70–80. [Google Scholar] [CrossRef]
- Gollihue, J.L.; Patel, S.P.; Eldahan, K.C.; Cox, D.H.; Donahue, R.R.; Taylor, B.K.; et al. Effects of mitochondrial transplantation on bioenergetics, cellular incorporation, and functional recovery after spinal cord injury. J. Neurotraum. 2018, 35, 1800–1818. [Google Scholar] [CrossRef]
- Bamshad, C.; Habibi Roudkenar, M.; Abedinzade, M.; Yousefzadeh Chabok, S.; Pourmohammadi-Bejarpasi, Z.; Najafi-Ghalehlou, N.; et al. Human umbilical cord-derived mesenchymal stem cells-harvested mitochondrial transplantation improved motor function in TBI models through rescuing neuronal cells from apoptosis and alleviating astrogliosis and microglia activation. Int Immunopharmacol. 2023, 118, 110106. [Google Scholar] [CrossRef]
- Kuo, C.C.; Su, H.L.; Chang, T.L.; Chiang, C.Y.; Sheu, M.L.; Cheng, F.C.; et al. Prevention of Axonal Degeneration by Perineurium Injection of Mitochondria in a Sciatic Nerve Crush Injury Model. Neurosurgery 2017, 80, 475–488. [Google Scholar] [CrossRef]
Figure 1.
Organization of cytoskeleton and molecular components for the transport of mitochondria in neurons.
Figure 1.
Organization of cytoskeleton and molecular components for the transport of mitochondria in neurons.
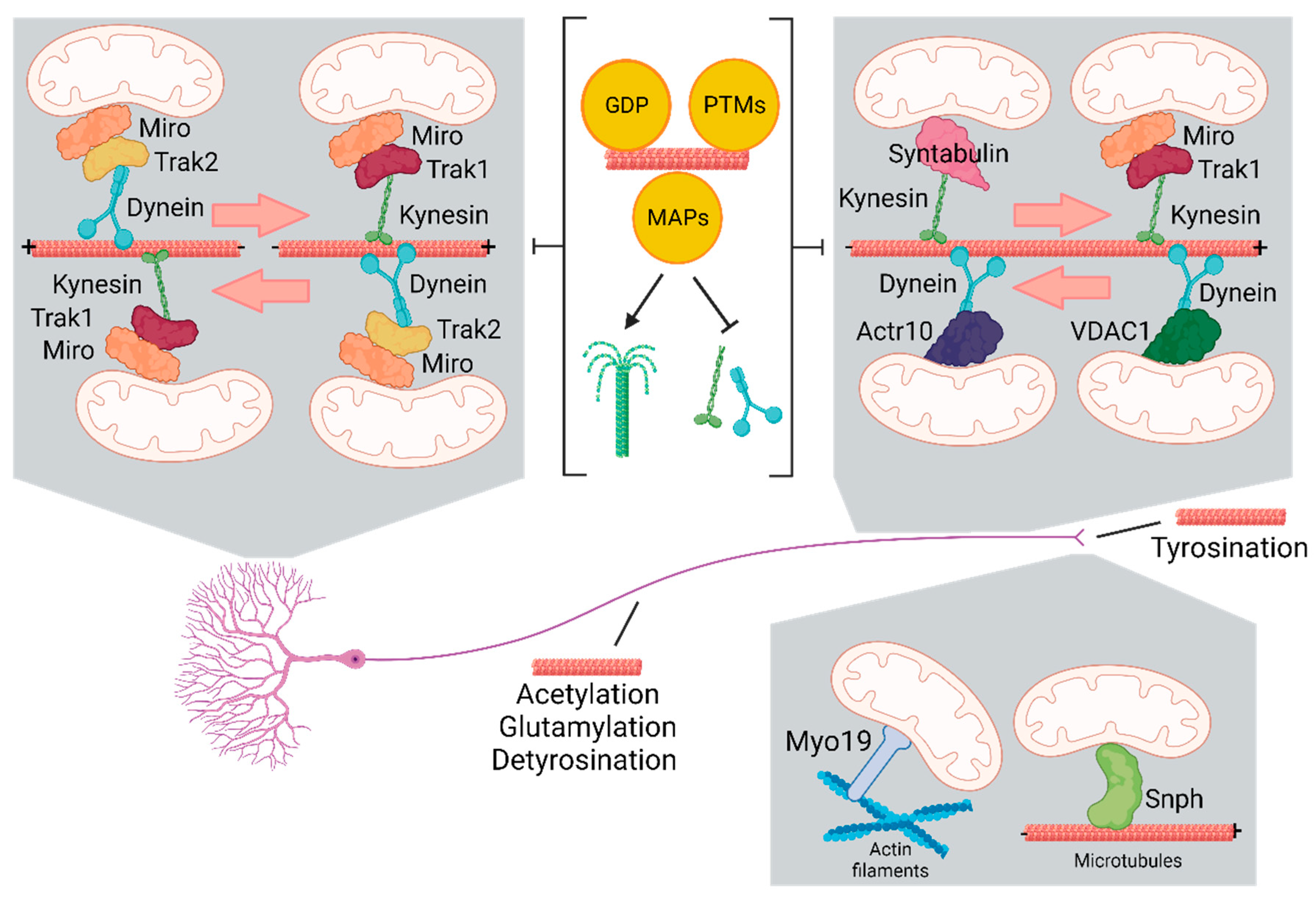
Figure 2.
Regulation of mitochondrial transport.
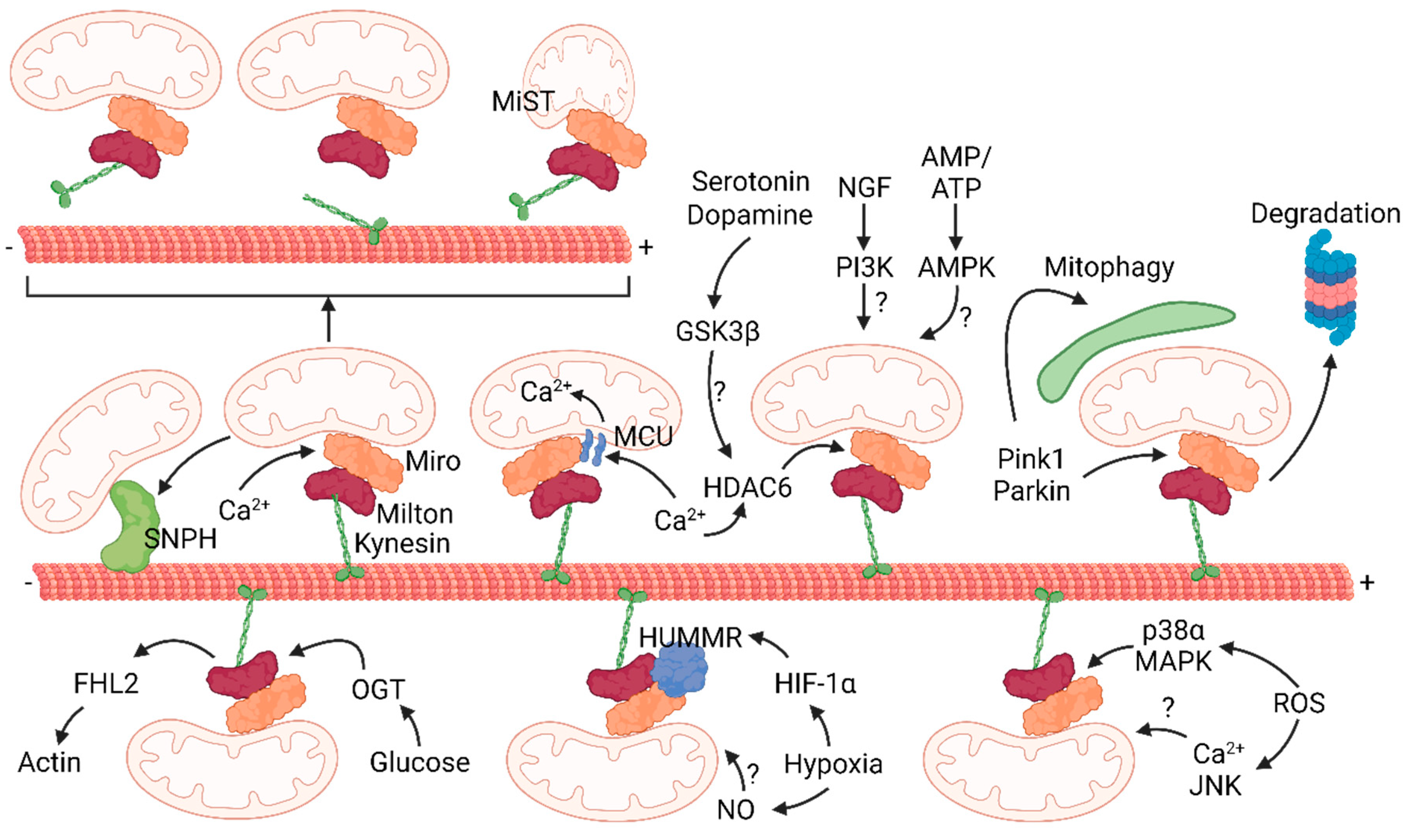

Table 1.
Mitochondrial transfer therapy for brain diseases.
| Disease | Treatment | Clinical Outcome | Ref. |
|---|---|---|---|
| Rat model of Parkinson Disease (PD) | Mitochondria | Restored mitochondrial functions and reduced oxitative damage in dopaminergic neurons | (110) |
| Mouse model of PD | Mitochondria | Increased electron transport chain activity, reduced ROS level and prevented apoptosis and necrosis. | (111) |
| Rat model of schizophrenia (SZ) | Mitochondria | Prevented mitochondrial dysfunction in intra-prefrontal cortex neurons and emergence of attention deficit | (112) |
| Middle cerebral artery occlusion (MCAO) in rats | Mitochondria | Decreased brain infarct volume and reversed neurological deficits | (113) |
| MCAO in rats | Mesenchymal multipotent stromal cells | Reduced infarct volume in the brain and partial restoration of neurological status*. | (114) |
| Ischemic Stress in rats | Mitochondria | Restored motor performance; attenuated brain infarct area and neuronal cell death | (115) |
| MCAO in rats | MSCs-derived mitochondria | Declined blood creatine phosphokinase level, abolished apoptosis, decreased astroglyosis and microglia activation, reduced infarct size, and improved motor function | (116) |
| Ischemia-reperfusion stroke injury | MSCs | Rescued damaged cerebrovascular system in stroke | (109) |
| Spinal cord injury (SCI) in rats | Mitochondria | Maintenance of normal bioenergetics without recovery of motor and sensory functions | (117) |
| Traumatic brain injury (TBI) in rats | MSCs-derived mitochondria | Improved sensorimotor functions | (118) |
| Nerve crush injury in rats | Mitochondria | Improved neurobehaviors, electrophysiology of nerve conduction, and muscle activities | (119)Kuo |
* More profound neuroprotective effects have been obtained when MSCs were injected after cocultivation with neurons.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



