Submitted:
12 May 2023
Posted:
16 May 2023
You are already at the latest version
Abstract
Oncogenic fusion proteins, arising from chromosomal rearrangements, have emerged as prominent drivers of tumorigenesis and crucial therapeutic targets in cancer research. In recent years, the potential of small molecular inhibitors in selectively targeting fusion proteins has exhibited significant prospects, offering a novel approach to combat malignancies harboring these aberrant molecular entities. This review provides a comprehensive overview of the current state of small molecular inhibitors as therapeutic agents for oncogenic fusion proteins. We discuss the rationale for targeting fusion proteins, elucidate the mechanism of action of inhibitors, assess the challenges associated with their utilization, and provide a summary of the clinical progress achieved thus far. The objective is to provide the medicinal community with current and pertinent information and to expedite the drug discovery programs in this area.
Keywords:
fusion proteins
; inhibitor
; therapeutic
; cancer
1. Introduction
Genomic instability is a prominent factor in driving tumorigenesis[1]. Specifically, gene fusions represent significant genomic events in the progression of cancer[2]. Gene fusions result from the physical juxtaposition of two or more previously separate genes, generating a novel chimeric gene that comprises the head of one gene and the tail of the other. These chimeric genes can be generated through several possible processes, including chromosomal inversions, tandem duplications, interstitial deletions or translocation events[3]. Gene fusions often encode fusion proteins that possess unique functional properties distinct from the original proteins, thereby leading to the alterations in cellular functions and promoting tumorigenesis. Gene fusions are frequently identified as primary tumor predisposing events in leukemias, lymphomas, solid malignancies, and benign tumors[4]. Various fusion proteins have emerged as critical biomarkers for clinical diagnosis and therapeutic targets[5]. Therefore, gene fusions have garnered substantial attention within the realm of cancer research.
The discovery of the BCR-ABL fusion protein in patients with chronic myeloid leukemia (CML) marked the inception of a new era in cancer research[6]. Subsequently, numerous gene fusions have been identified in various cancer types[7], with advancements in detection methods. The application of RNA sequencing technology has revealed a multitude of unique fusion events, surpassing a count of 2200, with 120 of them exhibiting high frequency. Among these, a substantial proportion encodes protein kinases, which are known to play crucial roles in multiple signaling pathways, with relatively high expression levels and kinase activity[8]. Fusion proteins have been recognized as pivotal drivers of cancer initiation and progression in variety of malignancies. For instance, the BCR-ABL fusion protein has been shown to induce the transformation of benign cells into malignant ones[9]. In fibrolamellar hepatocellular carcinoma (FL-HCC), the DNAJB1-PRKACA fusion drives tumorigenesis by hyperactivation the Wnt signaling pathway[10]. RET/PTC have been observed in papillary carcinomas and shown to activate the RAS-MAPK pathway in a ligand-independent way[11]. Furthermore, MAN2A1-FER has been identified in various tumor types and has been demonstrated to enhance the FER kinase activation[12]. An increasing number of studies have illustrated the pivotal role of kinase fusion proteins in the pathogenesis of cancer by aberrantly activating downstream signaling pathways. Notably, a mounting body of research has also highlighted the significance of non-kinase fusion proteins, particularly transcription factor-related non-kinase fusion proteins, in cancer development. For instance, PML-RARα and FUS-DDIT3 have been considered as the key drivers of acute myeloid leukemia (AML) and mucinous liposarcoma (MLS)[13,14]. Therefore, comprehending the mechanistic and evolutionary processes underlying fusion proteins is crucial for understanding their role in driving carcinogenesis.
The remarkable specificity exhibited by fusion genes towards cancer cells has propelled their emergence as diagnostic markers and promising targets for cancer therapies. By targeting fusion proteins, the potential arises to mitigate the off-target effects commonly associated with traditional cancer treatments, such as chemotherapy and radiation therapy, which frequently lead to many side effects and adversely impact the patients’ quality of life. Consequently, the development of selective inhibitors for fusion proteins has assumed critical goal in the realm of related-cancers therapies. Targeted therapies, particularly tyrosine-kinase inhibitors (TKIs), have demonstrated remarkably efficacy in treating specific cancer types, including the first-, second- and third-generation TKIs. For example, Imatinib, a drug designed to target BCR-ABL, has exhibited effectiveness in the treatment of CML[15]. Recently approved drugs, such as Larotrectinib and Entrectinib are capable of targeting fusion proteins harboring neurotrophic RTK (NRTK) genes[16]. Crizotinib has demonstrated efficient inhibition of ALK fusions[17]. Additionally, all-trans-retinoic acid (ATRA) and arsenic trioxide (ATO) can be employed to treat or alleviate the blockage of granulocyte differentiation caused by the RARA fusion protein[18]. Despite the success achieved by inhibitors in the treatment of fusion-driven tumors, resistance often emerges as a result of secondary mutations[19]. Remarkably, the therapeutic strategy for cancers has undergone a paradigm shift with the advent of molecular-targeted therapy for oncogenes-driven cancers.
As mentioned above, fusion proteins arising from fusion genes are important targets for cancer therapy. Therefore, in this article, we present a succinct categorization of fusions detected in diverse cancers, along with a summary of techniques employed for fusion gene detection. Furthermore, a comprehensive classification of kinase fusion and non-kinase fusion events is provided, primarily focused on their fusion forms, fusion inhibitors, mechanism of action, and drug resistance.
2. Fusion genes in diseases and the detection of fusion genes
At the genomic level, the process of chromosome breakage and subsequent re-splicing gives rise to gene fusions, which are novel genes formed through this mechanism[20]. Unlike other recurrent oncogenic mutations, gene fusions are often regarded as pathognomonic factors for malignancy, and many of them are strongly associated with specific cancer types, such as prostate cancer[21,22], lung cancer[23,24], breast cancer and so on (Figure 1).
Thus, the detection of gene fusions is essential for the identification of cancer and the development of targeted therapeutic approaches. These gene fusions generate novel chimeric proteins that can drive oncogenic transformation. The identification of gene fusions in cancer patients can provide valuable insight for personalized treatment strategies that target the aberrant fusion proteins or downstream signaling pathways, ultimately leading to improved therapeutic outcomes. Currently, a variety of techniques are wildly employed in clinical practice, include cell morphology, exon array analysis (EAA), immunohistochemistry (IHC), fluorescence in situ hybridization (FISH), reverse transcription polymerase chain reaction (RT-PCR), droplet digital PCR (DD PCR), and next-generation sequencing (NGS) (Figure 2).
NTRK, ALK, NTRK1, BRAF, RET fusions and many other gene fusions have previously been successfully detected by cytomorphological methods[26,27]. However, it may not always be sensitive enough to detect low-level fusion transcriptions or may not be able to differentiate between different fusion variants. The convenience and speed offered by IHC, the specificity provided by FISH, and the high detection sensitivity of RT-PCR have positioned them as widely utilized detection methods. These techniques have made great contribution to the identification of gene fusions, including ALK, NTRK, and RET. While IHC stands out for its cost-effectiveness and rapidity compared to other available assays, caution is warranted due to its tendency to yield false-positive results. Consequently, validation in combination with complementary techniques is often necessary to ensure reliable outcomes[28]. The emergence of NGS technology has facilitated targeted therapy for tumors by enabling the detection not only of fusions but also polygenes and fusion mutants[29]. In addition to the classical detection techniques, a large number of new assays are emerging, including targeted RNA sequencing (RNAseq) and full-length single-cell sequencing (scRNA-seq). Compared to FISH and RT-PCR methods, targeted RNAseq improves diagnostic yield by evaluating multiple genes simultaneously in a single assay, thereby increasing sequencing coverage and reducing diagnostic time. Additionally, it can also identify new gene fusions or complex structural rearrangements. However, the broader scope of detection in targeted RNAseq may lead to an increased likelihood of false-positive results[30]. The scRNA-seq[31], employing the detection algorithm scFusion, presents the opportunity to detect fusion genes at the single-cell level, thereby enhancing the detection power through co-analysis of relevant cells. However, the ability to detect rare fusions in highly heterogeneous tumor samples is limit. Furthermore, scRNAseq data often exhibit high levels of noise land contain various technical artifacts, which can also contribute to false-positive results[31].
3. The development of inhibitors targeted kinase fusion genes
3.1. ABL-fusion inhibitors
Abelson Murine Leukemia Viral Oncogene Homolo (ABL), a non-receptor tyrosine kinase, participated in many signaling pathways in both the cytoplasm and the nucleus, regulating critical cellular processes such as cell growth, survival, invasion, adhesion, and migration[32]. The most common fusion partner with ABL is the BCR gene, which gives rise to various forms of leukaemia in human (Figure 3A). The coiled-coil domain of the BCR facilitates dimerization and constitutive autophosphorylation of the ABL tyrosine kinase domain, resulting in uncontrolled tyrosine kinase activity. BCR-ABL has been identified as an anti-apoptotic gene, leading to abnormal cell proliferation and disruption of normal cell regulation. This fusion has been validated as a carcinogenic alteration[33,34]. BCR-ABL fusions are predominantly detected in CML, accounting for approximately 95% of cases. It has also been detected in patients with adult B-cell acute lymphoblastic leukemia (B-ALL), and with mixed phenotype acute leukemia (MPAA), with frequencies of approximately 25% and 30%, respectively. In contrast, it is rare in patients with AML, with less than1% of cases being observed. Additionally, occasional cases of this oncogenic fusion have been reported in patients with lymphoma and myeloma[35]. The primary treatment for BCR-ABL positive leukemias is the use of TKIs, such as Imatinib, Nilotinib, Dasatinib and Bosutinib (Figure 3B and 3C)[34].
3.1.1. Imatinib
Imatinib is a 2-phenylaminopyrimidine derivative neoplastic agent that has been approved by the FDA for the treatment of CML by inhibiting BCR-ABL tyrosine kinase[36]. Its mechanism of action involves competing the ATP binding sites of BCR-ABL, thus preventing downstream phosphorylation of target protein. Additionally, Imatinib is an inhibitor of platelet-derived growth factor (PDGF) and stem cell factor (SCF). Despite its effectiveness in treating CML, Imatinib has been reported to cause acquired drug resistance. The development of resistance to Imatinib in CML is a multifactorial process, including point mutations in the structural domain of BCR-ABL kinase, amplification of the BCR-ABL gene, and the presence of leukemic stem cells that can produce Imatinib-insensitive active mutants[36,37]. It is noteworthy that mutations in the structural domain of the BCR-ABL kinase, such as T315I, E255K, Y253F and M351T, represent a primary cause of drug resistance[36].
3.1.2. Dasatinib
Dasatinib is a new and potent multitargeted kinase inhibitor that has been approved as a second-generation TKI for the treatment of CML and ALL[38], which have not respond to Imatinib therapy. In contrast to Imatinib, Dasatinib represses both the active and inactive conformations of the ABL kinase domain. As a result of this unique characteristic, Dasatinib has emerged as a viable therapeutic alternative for patients who exhibit resistance to Imatinib. Currently, Dasatinib is being used as a treatment option for such patients[39]. Similarly, the therapeutic effect of Dasatinib is achieved through the ATP-competitive pathway[40]. And, the capability to cross the blood-brain barrier makes it a promising treatment for central nervous system (CNS) leukemia. Frequently occurrence of T315I and F317L mutations has been observed in patients who exhibit resistance to Dasatinib[38,41]. In recent years, Dasatinib has been found to induce autophagy in BCR-ABL positive leukemia cells, indicating that autophagy may be a potential direction for the development of novel drugs to treat BCR-ABL positive leukemia[42].
3.1.3. Nilotinib
Nilotinib is a transduction inhibitor that targets several proteins including BCR-ABL, c-kit and PDG, for the treatment of chronic phase CML. Nilotinib exhibits structural similarities to Imatinib, but is 20- to 50-fold more potent against BCR-ABL in phase I clinical trials. Consequently, Nilotinib has been regarded as a relatively safe treatment option that offers significant therapeutic advantages for patients with CML who exhibit resistance to Imatinib[43]. In contrast to Dasatinib, Nilotinib is a non-competitive inhibitor of ATP that effectively blocks the autophosphorylation of BCR-ABL at Tyr177, a GRB2 binding site involved in the regulation of multiple signaling pathways implicated in BCR-ABL pathogenesis[44]. Although Nilotinib is effective against most BCR-ABL mutations, it exhibits negligible efficacy against T315I and G250E mutations.
3.1.4. Asciminib
Asciminib is a novel BCR-ABL inhibitor that has been developed to overcome drug resistance and off-target toxicity associated with current inhibitors[45]. Unlike other inhibitors such as Imatinib that binds to the ATP binding site, Asciminib binds to the myristoyl pocket of BCR-ABL[45]. This unique mechanism of action makes Asciminib effective against many Imatinib-resistant BCR-ABL mutants, including the notoriously T315I mutation[46]. While some emerging myristate-site mutations remain sensitive to ATP-competitive inhibitors[47], a combination of Asciminib with an ATP-competitive TKI may prove to be a promising strategy to prevent the development of resistance[48].
3.1.5. Prospect
As the originally identified oncogenic fusion, research on ABL inhibitors is ongoing, with the development of new drugs to effectively treat CML. Recently developed ATP-competitive TKIs, such as Ponatinib and Vamotinib, have demonstrated the ability to target both the natural and mutant variants of BCR-ABL, including the T315I gatekeeper mutation. Furthermore, Bosutinib has exhibited high activity against BCR-ABL in Phase I and II clinical trials, reflected by a >100-fold increase in potency compared with Imatinib[49]. In addition to the continued progress in developing small molecule inhibitors, therapies targeting the BCR-ABL protein degradation pathway have shown efficacy in targeting not only naturally occurring kinase-active BCR-ABL protein, but also the mutant forms of BCR-ABL protein[35]. As a novel therapeutic approach, protein degradation strategies show the potential to outperform traditional protein activity inhibition methods.
3.2. ALK-fusion inhibitors
The identification of anaplastic lymphoma kinase (ALK) was originally accomplished in anaplastic large cell lymphoma (ALCL), which explains its name. The ALK fusions production is associated with the oncogenicity of ALK, which could be related to the altered conformation and phosphorylation of ALK protein[50]. Nevertheless, it is widely acknowledged that the identity of the fusion partner is the key determinant of oncogenic activity of ALK[51]. The retained kinase domain of ALK, along with the coiled-coil domain present in most fusion partners, enables the dimerization of ALK and its constitutive activation[52]. In addition to the frequently observed fusion types of ALK, such as EML4-ALK, NPM1-ALK, KIF5B-ALK, TFG-ALK, TPM3-ALK and TPM4-ALK[24,53,54,55], an increasing number of ALK fusion variants have been discovered (Figure 4). ALK fusion proteins are involved in several cellular signaling pathways, such as Ras/extracellular signal-regulated kinase (ERK), phosphatidylinositol 3 kinase (PI3K)/Akt, and Janus protein tyrosine kinase (JAK)/STAT [56] (Figure 5A). Thus compromised regulation may lead to the development of several diseases. And ALK fusion proteins have been reported in nonelastic ALCL, inflammatory myofibrosarcoma (IMT), diffuse large B-cell lymphoma (DLBCL) and non-small cell lung cancer (NSCLC)[57]. Furthermore, while EML4-ALK fusion is commonly observed in the majority of the 2-7% of NSCLC cases[58], ALK fusions are infrequent in SCLC. Nonetheless, there have been four rare instances of SCLS with ALK fusions reported recently, including ELM4-ALK, PLEKHM2-ALK and ALK-IR[59,60] (Figure 4). Fortunately, the first-generation drugs, including Crizotinib, a small molecule tyrosine kinase inhibitor initially designed to target cMET, along with the second-generation drugs, such as Ceritinib and Alectinib, and the third-generation agent Lorlatinib, were repurposed effectively for the treatment of cancer with ALK-fusion.
3.2.1. Crizotinib
Crizotinib, the first generation ALK-tyrosine kinase inhibitor, was initially designed as a cMET kinase inhibitor. It was subsequently discovered to inhibit ALK-rearranged and ROS-rearranged in NSCLC[61]. The use of Crizotinib leads to decreased phosphorylation of ALK fusion, resulting in an inactive protein conformation[62]. In various xenograft models using human-derived tumors or tumor cell lines expressing EML4- or NPM-ALK fusion proteins, Crizotinib inhibited tumor growth, decreased proliferation, increased apoptosis, and dose-dependent decreased phosphorylation. Based on its proven efficacy and safety in clinical trials, Crizotinib was granted approval by the U.S. FDA approval on 26 August 2011, and received full approval in 20 November 2013[63]. Despite the significant and long-lasting benefits of Crizotinib, the majority of patients eventually develop resistance, usually after a few years[64]. The main mechanisms of resistance to Crizotinib include secondary resistance mutations in ALK, such as L1196M, C1156Y[65], G1269A[66], L1152R[67], and I1171[68] (Figure 5B), as well as ALK copy number alterations. Interestingly, Crizotinib has demonstrated sensitivity to a large number of new ALK fusions, including HIP1-ALK[69], BIRC6-ALK[70], DYSF-ALK.
3.2.2. Ceritinib
Ceritinib is a kinase inhibitor for the treatment of patients with ALK-positive metastatic NSCLC who have failed prior Crizotinib therapy due to resistance or intolerance. It was approved by the FDA in April 2014, as a surprisingly high response rate (56%) towards Crizotinib-resistant tumors[71]. Mechanistically, Ceritinib suppress ALK-mediated phosphorylation of the downstream signaling proteins in an ATP-competitive manner. It has been demonstrated to be effective in treating NSCLC patients who have developed resistance to Crizotinib, specifically, mutations in key "gatekeeper" residues of the enzyme, L1196M and G1269M and G1269A[72]. These findings suggest that Ceritinib holds promise as a therapeutic option for NSCLC patients with ALK fusion and resistance to Crizotinib.
3.2.3. Alectinib
Alectinib, a highly selective inhibitor of ALK, is used specifically in the treatment of NSCLC expressing the EML4-ALK fusion protein[73]. It has been approved in Japan for the treatment of advanced, unresectable, or recurrent NSCLC with ALK rearrangement[74]. Alectinib is frequently used as a second-line treatment after Crizotinib therapy due to drug intolerance or resistance. Furthermore, it has exhibited remarkable effectiveness against several novel ALK fusion proteins, including VKORC1L1-ALK[75], SQSTM1-ALK[76], HIP1-ALK[77] and STRN-ALK[78]. For patients whose disease progression persists despite initial treatment with Crizotinib, converting to Alectinib may be a viable option that could enhance treatment compliance[79]. Notably, the presence of T1151K has been found to increase sensitivity to Alectinib[75]. However, secondary resistance mutations in ALK, including I1171T/N/S[68], G1202R[80], V1180L and F1174C, may lead to resistance to Alectinib (Figure 5B).
3.2.4. Brigatinib
Brigatinib is a tyrosine kinase inhibitor that selectively targets ALK and EGFR and has demonstrated efficacy against 9 different Crizotinib-resistant mutants of the EML4-ALK fusion[81]. It was developed by Ariad Pharmaceuticals, and received FDA-approved in 2017. In BaF3 cells expressing EML4-ALK, and these with gatekeeper mutations (G1269S and L1196M), Brigatinib has been shown to induce tumor regression. Furthermore, in EML4-ALK xenograft models in mice, Brigitanib exhibits dose-dependent inhibition of tumor growth, tumor burden and prolonged survival[81]. Currently, Brigatinib is primarily used as a treatment option for ALK-positive NSCLC who are intolerant to Crizotinib.
3.2.5. Prospect
Despite the availability of the first-, second-, and third-generation ALK-fusion inhibitors and a profound understanding of the resistance mechanisms, drug resistance to ALK-fusion inhibitors remains a major challenge[82]. As illustrated in Figure 4, drug design can be improved by considering the characteristics of the fusion type, particularly in the presence of the coiled-coil domain[83]. Above all, to effectively prevent or overcome drug resistance, a comprehensive understanding of the underlying mechanism is necessary. The development of small molecule drugs, such as Brigatinib, a potent and selective ALK inhibitor, has shown promise in treating metastatic NSCLC patients who have progressed but are intolerant to Crizotinib[84]. Similarly, NVP-TAE684 is also a potent and selective ALK inhibitor that effectively induces apoptosis and cell cycle arrest through rapid and prolonged inhibition of phosphorylation of NVM-ALK and its downstream effectors. Meanwhile, it is sensitive to the gatekeeper mutation L1196M[85].
3.3. ROS1 fusion inhibitors
ROS1 stands for c-ROS sarcoma carcinogen receptor tyrosine kinase (ROS proto-oncogene 1, receptor tyrosine kinase), which encodes a transmembrane tyrosine kinase receptor called ROS1 protein. However, the functional capabilities of ROS1 remain largely unknown[86]. Fusion events involving ROS1 have been detected in various cancers in adult and pediatric patients. The identified fusion partners include CD74, CCDC6, EZR, FIG, KDELR2 and others[87,88,89]. While fusion events may retain the kinase domains of ROS1, it has been reported that only certain fusion partners such as FIG, CCDC6, TMP, EZR, and GOPC contain coiled-coil domains[88]. In addition, it is not clear how several of the fusion partners of ROS1, such as CD74, induce oncogenicity as they lack the ability to induce dimerization in the N-terminal domain[88] (Figure 6). ROS1 fusion induces activation of several typical signaling pathways involved in cell survival and growth, for instance RAS-MAPK, JAK-STAT3, PI3K-AKT-mTOR[90] (Figure 7A). ROS1 fusion proteins account for approximately 1% of NSCLC[91], while in lung adenocarcinoma was about 3.3%. Currently, ROS1 inhibitors are classified into two classes, based on their preference for binding to different conformations of the ROS1 kinase domain. Type I inhibitors, such as Crizotinib and Entrectinib, preferentially bind to the “DFG-in” conformations of their targets. In contrast, type II ROS1 inhibitors, such as Cabozantinib and Foretinib, have a higher binding affinity for the “DFG-out” conformation [92].
3.3.1. Crizotinib
Crizotinib is a tyrosine kinase receptor inhibitor that has been used for the treatment of ROS1-positive NSCLC tumors. At present, it is the only clinical phase IV inhibitor available for this type of cancer[88]. By competing with ATP, it effectively inhibits the activity of ROS1 fusion with sub-nanomolar cellular potency[93]. Compared to other inhibitors, like Ceritinib and Alectinib, Crizotinib displayed significantly improved activity against ROS1 kinase[94]. However, it was invalid for G1202R, G2032R[95,96] (Figure 7B). Resistance to Crizotinib in cancers may develop due to various mechanism, including secondary mutations in the structural region of the ROS1 kinase or amplification of the ROS1 gene. Additionally, drug resistance may develop due to the activation of other signaling pathways that bypass the ROS1 pathway. To overcome Crizotinib resistance, several second-generation ROS1 inhibitors, including Ceritinib, Roritinib and Enretinide, are currently undergoning clinical trials[64].
3.3.2. Entrectinib
Entrectinib is a multi-kinase inhibitor that functions as an ATP competitor and has been demonstrated to be effective against ROS1, ALK, and pan-TRK[97]. In 2019, the FDA approved Entrectinib for the treatment of ROS1-positive metastatic NSCLC and NTRK gene fusion positive solid tumors. An integrated analysis of three-phase 1-2 trials indicated that Entrectinib shown promising results in the treatment of ROS1 fusion-containing cancers, and was found to be 40 times more potent than Crizotinib[98]. One of the advantages of Entrectinib is the ability to effectively cross the blood-brain barrier, making it a potentially effective treatment option for tumor that have spread to the brain[98]. However, it also encounters the obstacle of drug resistance mutations, such as G2032R, F2004C/I/V, L2086F[99], L2026M[100] (Figure 7B).
3.3.3. Prospect
Regrettably, the number of developed inhibitors for the ROS1 fusion is relatively limited, with only two drugs, Crizotinib and Entrectinib, approved by the FDA. Based on the phase I/II clinical studies, Crizotinib has shown significant therapeutic potential against ROS1-fusion driven cancers[94]. Nevertheless, drug resistance remains an unavoidable challenge in cancer treatment, highlighting the need to overcome resistance[101]. In addition to the classical inhibitors, numerous drugs undergoing clinical trials have exhibited favorable inhibitory effects. Repotrectinib is a low-molecular-weight macrocyclic TKI with highly selective effects on ROS1 and ALK[102]. Brigatinib, initially used to treat ALK positive metastatic NSCLC intolerant to Crisotinib[103], and was later found to have inhibitory activity against ROS1 fusion positive tumors, making it a next-generation tyrosine kinase[104]. Additionally, besides targeted treatment, programmed death-ligand 1 (PD-L1), an immune-suppressive molecule, needs to be considered for therapeutic decision-making[105]. However, whether the administration of immune checkpoint inhibitor (ICI) therapy is suitable for ROS1 fusion after the development of Crizotinib resistance is uncertain.
3.4. NTRK fusion inhibitors
NTRK refers to a class of neurotrophic receptor tyrosine kinases that are involved in the regulation of various cellular processes through the activation of intracellular signaling pathways. The NTRK gene family is comprised of three distinct members, namely NTRK1, NTRK2 and NTRK3, which encode for the TRK family of receptor proteins, including TRKA, TRKB and TRKC[106]. The fusion types mainly include EML4-NTRK3, TPM3-NTRK1[107], QKI-NTRK2[108], etc. NTRK gene fusions results in the production of chimeric oncoproteins, such as ETV6-NTRK3[109] and MPRIP-NTRK1[110] (Figure 8). These oncoproteins function as oncogenic drivers that promote tumor cell proliferation and survival in vitro. Furthermore, immunohistochemical analysis of TRK expression in NTRK fusion-positive tumors indicates that the subcellular localization of the fusion protein may be determined by the partner[16]. NTRK fusions are frequently observed in rare cancer types including secretory breast cancer and congenital mesodermal nephroma, while they occur less frequently in more common tumors like breast, lung and colorectal cancers[111] (Figure 9A). Larotrectinib and Entrectinib have been approved by the FDA and EMA for as monotherapy for the treatment of patients with NTRK gene fusion positive solid tumors.
3.4.1. Entrectinib
Entrectinib is a multi-kinase inhibitor with ATP-competitive properties that targets TRKA, TRKB, TRKC, ROS1, and ALK. TRK receptors play a role in cell proliferation through downstream signaling of MAPK, PI3K and PLCγ. By inhibiting these pathways, Entrectinib can suppress cancer cell proliferation, leading to tumor size reduction[112]. The FDA has granted approval for the use of Entrectinib in the treatment of NTRK gene fusion solid tumors in both adult and pediatric patients. The systemic antitumor activity of this compound has also been observed[113]. Clinical studies has shown that Entrectinib is safe and well-tolerated in the patient with various tumor types, including NSCLC harboring SQSTM1-NTRK1[114], metastatic colorectal cancer harboring LMNA-NTRK1[115] , lung cancer harboring SQSTM1-NTRK1[116], mammary analogue secretory carcinoma harboring ETV6-NTRK3[117] and glioneuronal tumors harboring BCAN-NTRK1[118]. However, cases of resistance to Entrectinib have been reported, such as G595R,G623R[119], G667C[119], G623E, V573M, and G595L[119] (Figure 9B).
3.4.2. Larotrectinib
Larotrectinib is a highly selective TRK inhibitor that has been approved for the treatment of NTRK fusion-positive solid tumors in both adults and children[120]. By binding to TRK, Larotrectinib effectively obstructs neurotrophin-TRK interactions and TRK activation, resulting in the induction of cellular apoptosis and the inhibition of tumor cell growth in cases where TRK is overexpressed. Clinical studies have demonstrated that it exhibits potent antitumor activity in patients with NTRK fusion positive cancer, such as those with TPM3-NTRK1 in lung cancer[121] and CTRC-NTRK1 in pancreatic ductal adenocarcinoma[122]. Larotrectinib has demonstrated the ability to inhibit the growth of cell lines or xenografts containing TPM3-NTRK1, MPRIP-NTRK1 and ETV6-NTRK3[121]. Moreover, Larotrectinib exhibited favorable tolerability and demonstrated efficacy across all patients with tumors harboring NTRK gene fusions, thereby representing a promising therapeutic option for such individuals. However, Larotrectinib displayed limited activity in cell lines with point mutations in the TRK kinase domain, such as G667S, G696A, G595R, G623R, F617L[123], V573M[119] (Figure 9B).
3.4.3. Prospect
Mutations in NTRK genes are responsible for drug resistance, including solvent front mutations in TRKA G595R and TRKC G623 in the ATP-binding pocket of the kinase structural domain. To overcome acquired resistance to first-generation TKIs, next-generation TRK inhibitors such as Selitectinib is being developing. In preclinical studies, Merestinib has shown efficacy in tumor models with NTRK fusion and TRKA G667C variants[124]. Additionally, Cabozantinib can target NTRK fusions, but may also exhibit resistance against TRKA L564H, D679G, G595R, and G595L mutation[125]. It is crucial to note that treatment with TRK inhibitors can lead to overactivation of cancer-related pathways (MAPK pathway), including hotspot mutations, MET amplification, KRAS mutations and BRAF V600E mutations. This effect may result in treatment failure, and thus, combination therapy with drugs that regulate these pathways may represent a new paradigm in the fighting NTRK fusion diseases[125]. Moreover, combination therapy involving NTRK inhibitors and immune checkpoint inhibitors may also offer a promising treatment option.
3.5. RET fusion inhibitors
Rearranged during transfection (RET), a member of receptor tyrosine kinase family, has been demonstrated to have a crucial role in signal transduction. Perturbations in the expression of RET can lead to tumorigenesis. The most prevalent oncogenic variants include gene fusions and mutations. Notably, studies have highlighted that RET fusions occur with high frequently in populations exposed to ionizing radiation[126]. Various types of RET fusion have been identified, including CDCC6-RET, NCOA4-RET, KIF5B-RET[127], LSM14A-RET[128], TRIM33-RET, ZNF477P-RET[127], etc. (Figure 10). Typically, the gene fusion encodes a fusion protein that retains the kinase domain, which can induce the overactivation of downstream signaling pathways via partners dimerization or oligomerization, culminating in the development of multiple malignancies[129] (Figure 11A). RET fusion proteins have been detected in more than 20 cancer type, with thyroid and lung adenocarcinomas being the most prevalent[127] (Figure 10A). Cabozantinib, Vandetanib, Lenvatinib, Ponatinib, Sunitinib, and Sorafenib have been approved as therapeutic agents for treating RET fusions[130].
3.5.1. Lenvatinib
Lenvatinib, an oral tyrosine kinase inhibitor, has been demonstrated to target various receptors, including vascular endothelial growth factor receptor (VEGFR) 1-3, fibroblast growth factor receptor (FGFR) 1-3 and RET[131]. Current preclinical and clinical studies indicate that it can hinder the autophosphorylation of KIF5B-RET, CCDC6-RET and NCOA4-RET, which can ultimately prevent the development of tumors harboring RET fusion oncogenes. Its efficacy has been demonstrated in various cancers, including CCDC6-RET-positive human thyroid cancer[132] and lung cancers[131]. Despite these promising findings, targeted therapy for RET fusions has not produced favorable outcomes. Resistance to Lenvatinib has gradually developed after treatment. Specifically, mutations such as V738A, A807V and F998V, L730V and E732K, G810A have been identified as potential mechanisms underlying resistance to it[133] (Figure 11B).
3.5.2. Cabozantinib
Cabozantinib, a multi-kinase inhibitor, has been reported to bind to both the ATP binding site and adjacent hydrophobic/variable sites[129]. Phase II clinical trials in patients with RET-fusion-positive NSCLC demonstrated its efficacy[131]. However, a comparative analysis has revealed that Cabozantinib exhibits off-target effects and is not specific for RET gene fusions. The development of resistance to Cabozantinib during its use is a potential problem, and specific mutations such as L730I, E732K and V871I[133] (Figure 11B).
3.5.3. Vandetanib
Vandetanib is a multi-target inhibitor of several proteins, including VEGFR2-3, EGFR and RET[129]. It has been demonstrated to inhibit the autophosphorylation of these proteins, and then blocking the downstream signaling[131]. Inhibition of RET-fused cancers is achieved by competing with ATP[129], leading to reduce the colony-forming ability in RET-fused cell lines. Vandetanib has also been found to inhibit the growth of both KIF5B-RET-mediated tumor and multiple RET fusion positive NSCLC[131,134]. Several drug-resistant mutations have been identified as frequently occurring during use, including G810A, L881V[135], C634W, M918T[129], V804L/M/E[129,136], G810S[133], S904F[137] (Figure 11B).
3.5.4. Selpercatinib
Selpercatinib is an oral administered, small-molecule ATP-competitive agent known for its high selectivity[138]. While it is currently being investigation in clinical trials, Selpercatinib obtained accelerated approval from the FDA in 2020 for the treatment of specific RET-driven cancers. It exhibits greater sensitivity in patients with KIF5B-RET type, CCDC6-RET type and RET V804L/M and M918T resistant NSCLC[139]. Its antitumor activity in the CNS is notably prolonged as it is capable of crossing the blood-brain barrier[140]. Notably, the use of Selpercatinib in patients with RET fusion-positive NSCLC who have undergone platinum-based chemotherapy or remained untreated resulted in a significant reduction in tumor growth with minimal toxicity[141]. Selpercatinib has exhibited robust and durable anti-tumor activity in medullary thyroid carcinoma (MTC) cases harboring RET mutations[140]. Importantly, it has been associated with a favorable safety profile[142]. However, Recent reports have identified mutations in RET G810R/S/C/V that have the potential to result in treatment failures, despite not affecting ATP binding [95,96,137] (Figure 11B).
3.5.5. Pralsetinib
Pralsetinib is a highly selective, orally administered, small molecule drug that has demonstrated notable efficacy and tolerability in the treatment of various RET abnormalities, including wild-type and mutations such as V804L, V804M and M918T, as well as fusions like KIF5B-RET, CCDC6-RET[143,144]. Similar to Selpercatinib, the phase I/II trial of Pralsetinib is currently ongoing. Nevertheless, Pralsetinib received accelerated approval from the FDA approval in 2020, specifically for the treatment of metastatic NSCLC with RET fusion. It has exhibited considerable therapeutic efficacy in patients with RET fusion-positive metastatic NSCLC, RET mutant MTC and RET fusion-positive thyroid, with no significant off-target toxicity[131,143,145]. Moreover, Pralsetinib has been shown to have no genotoxicity both in vitro and in vivo[146] , making it a promising RET fusion inhibitor. However, caution must be taken when administering it with P-gp (P-glycoprotein) and CYP3A inhibitors, as they may decrease its effectiveness and increase the incidence and severity of adverse reactions[134,143]. Regrettably, it has been found to be insensitive to G810R/S mutation (Figure 11B), which may limit its effectiveness in certain patient populations[147].
3.5.6. Prospect
Multi-kinase inhibitors have demonstrated efficacy in the treatment of RET fusion-positive cancers, but their lack of specificity and susceptibility to drug resistance have been noted. The advent of highly selective targeted agents for RET has improved the treatment of RET fusion-positive patients. The selective inhibitors Pralsetinib and Selpercatinib have been approved by the FDA for the treatment of RET fusion-positive NSCLC[131]. Additionally, several RET fusions-targeting inhibitors are being developed and are currently undergoing clinical trials. These include TAS0953 (NCT04683250) for RET fusion positive cells[147], BOS172738 for RET fusion positive NSCLC[148], RXDX-105 for CCDC6-RET, NCOA4-RET and PRKAR1A-RET-containing cells[127], TPX-0046 for advanced solid tumors with RET fusions or mutations[129,148]. In addition, exploring combinations therapies may lead to new developments in the treatment of RET fusion mutations.
3.6. Others
3.6.1. EGFR fusion inhibitors
Epidermal Growth Factor Receptor (EGFR) is a transmembrane glycoprotein and a type I receptor tyrosine kinase that plays a key role in epithelial cell proliferation and signaling. Somatic mutations of EGFR have been found to be associated with various oncogenic processes, including tumor cell proliferation, angiogenesis, tumor invasion and metastasis[149]. Notably, EGFR fusions have also been observed among these mutations. The most common one is EGFR-RAD51[149], while other fusions include HMGA2-EGFR[150], KIF5B-EGFR[151], EGFR-SEPT14[152], EGFR-PURB[153], etc. (Figure 12A). The fusion of EGFR with RAD51/PURB may have the potential to enhance EGFR activity through two mechanisms, namely, asymmetric dimerization promotion and inhibition of receptor switching. The C-terminal tail of EGFR contains multiple autophosphorylation sites that participate in downstream signaling pathways, such as MAPK and PI3K/Akt (Figure 12A). However, in EGFR-RAD51 and EGFR-PURB fusion, the C-terminal domain (CTD) is absent, resulting in a disruption of signal regulation that can lead to pathological consequences[149,153]. For the treatment of EGFR aberrant disease, including EGFR fusions, EGFR TKIs of the first, second and third generation, such as Erlotinib, Afatinib and Osimertinib, have been developed[154] and reported to be effective. Erlotinib, a first-generation EGFR TKI, has been shown to inhibit the formation of glioma expressing HMGA2-EGFR and prolong survival in animal models. It has also demonstrated efficacy against the rare EGFR-SEPT14 fusion protein in patients with colorectal adenocarcinoma[150,155]. The second-generation Afatinib has exhibited a favorable responded in patients with KIF5B-EGFR fusion in NSCLC[156]. However, the T790M mutation remains the most common form of resistance to first- and second-generation inhibitor [157]. In contrast, the third-generation Osimertinib has demonstrated sustained CNS activity and the ability to inhibit tumor growth selectively, making it an attractive option for the treatment of EGFR-Fused NSCLC[158]. Lazertinib has been found to be therapeutically similar to Osimertinib[159]. In addition, the use of Cetuximab, an EGFR antibody, has shown promise in inhibiting the proliferation of Ba/F3 cells expressing EGFR RAD51[149,153]. Although EGFR fusions are not well understood due to their low incidence and lack of detection technology[158], it is critical to develop more accurate and effective detection methods to improve our understanding of EGFR fusion and its potential patient treatment.
3.6.2. RAF/BRAF-fusion inhibitors
B-Rapidly Accelerated Fibrosarcoma (BRAF) is a member of the serine/threonine protein kinase family that regulates the MAP kinase/ ERK signaling pathway, which is involved in various cellular processes, including cell division, differentiation and secretion. BRAF aberrations, including gene fusions, such as KIAA1549-BRAF[160,161], FAM131-BRAF[162], GIT2-BRAF[163], GTF2I-BRAF[164], LMO7-BRAF[165] have been identified, and the list of novel BRAF fusions continues to grow(Figure 12B). Among these, KIAA1549-BRAF is the most prevalent type of BRAF fusion. RRAF fusions are often found in low-grade gliomas such as dermatocytic astrocytoma(PA), dermatomyosarcoma-like astrocytoma (PMA) and diffuse meningeal glial neuron tumor (DLGNT)[166]. Sorafenib is a potent inhibitor of RAF1 and exhibits effects on BRAF, RET, PDGFRβ and VEGFR-3. In a phase I clinical trial, a combination of Sorafenib and Bevacizumab showed promising results in patients with a KIAA1549-BRAF fusion[167] (Figure 12B). Furthermore, in vitro and in vivo studies have shown that the combination of Trametinib with the PI3K inhibitor BKM120 or the CDK4/6 inhibitor LEE011 produces synergistic antitumor effects in melanoma cells harboring BRAF fusions[168]. While Vemurafenib, Dabrafenib, and Encorafenib are selective BRAF inhibitors used in BRAF V600E mutated melanomas[169], limited data exist on their effectiveness against BRAF fusion kinase due to the low occurrence of these alterations in cell lines[168]. Initial research on cell lines suggests that drug combinations may be more successful. Efforts have been made to utilize inhibitors of both BRAF and MEK to overcome drug resistance[170].
4. The development of inhibitors targeted non-kinase fusion genes
4.1. RARα-fusion inhibitors
Retinic acid receptors α (RARα) is a nuclear hormone receptor that binds to the retinoic acid response element (RARE) and forms a dimer with the retinoid X receptor protein (RXRA), collectively known as the RARα-RXRA complex. The RARα-RXRA complex is essential for the promyelocyte differentiation. Acute promyelocytic leukemia (APL) is a subtype of AML. Although, the molecular mechanisms of development and progression remain to be fully understood, the PML-RARα fusion, resulting from the chromosomal translocation t(15;17) (q24;q21), is considered as the hallmark of APL[171] (Figure 14). PML-RARα fusion have been identified in more than 98% of APL patients, while rare RARα fusion, including NPM-RARα and others, have also been identified. The production of PML-RARα fusion proteins results in the formation of stable homodimers. And then, these fusion proteins can disrupt normal RARα signaling, increase the recruitment of co-blockers, and suppress the transcription of target genes, ultimately preventing differentiation and leading to the development of APL[172,173]. As the potent transcriptional repressors of retinoic acid signaling, RARα fusion proteins can be overcome with therapeutic doses of ATO[174]. ATO is a common chemotherapeutic agent used primarily for the treatment of refractory or relapsed APL (Figure 13). Although the mechanism of action of ATO is not fully understood, it has been shown to damage or degrade PML-RARα fusion protein and induce apoptosis in MCF-7 cancer cells[175]. Studies have revealed that combining ATRA with chemotherapy has led to APL becoming a highly curable disease with high response rates (>90%)[176]. Furthermore, ATO has been shown to be effective in patients who have relapsed after clinical remission with ATRA[177,178,179] (Figure 13). However, patients carrying the ZBTB16-RARα or STAT5B-RARα fusion gene were naturally resistant to ATO[180,181]. Currently, there are fewer drugs targeting RARA fusion protein, and drug resistance remains a significant challenge in treating APL. Developing new therapeutic tools or drugs is necessary to overcome this challenge.
4.2. DDIT3-fusion inhibitors
DNA damage inducible transcript 3 (DDIT3), a cellular stress sensor, is expressed at low levels under normal physiological conditions. However, it can be rapidly induced in response to endoplasmic various stress, including reticulum stress, nutrient deprivation, DNA damage, cell growth arrest, or hypoxia. Additionally, DDIT3 is believed to be involved in the negative regulation of cell differentiation[182]. MLS is a prevalent of liposarcomas distinguished by a characteristic cytogenetic feature of chromosomal translocation. The most frequent gene fusion observed in MLS is FUS-DDIT3, found in over 90% of cases, while less commonly EWSR1-DDIT3 is identified[183,184,185] (Figure 14). The transcriptional regulation of FUS-DDIT3 is controlled by the FUS promoter, whereas the mRNA stability relies on the DDIT3 protein. The expression of FUS-DDIT3 protein is critical for MLS tumor development, and its expression level correlates with the expression of cell proliferation-related genes, although the underlying mechanism remains unknown[183,186]. It was suggested that FUS-DDIT3 could be a potential target for MLS therapy [281-283]. Furthermore, FUS-DDIT3 activates JAK-STAT signaling, leading to the development of targeted therapies for MLS based on this discovery[184]. Most JAK and GSK-3 inhibitors, such as Fedratinib, TG101209 and AT9283 have been found to be effective[186]. In addition, FUS-DDIT3 induces activation of IGF-IR and PI3K/AKT signaling and therapies targeting IGF-IR have shown efficacy in treating MLS both in vitro and in vivo. NVP-AEW541, BMS-754807 and Picropodophyllin have demonstrated effective inhibition of IGF-IR and PI3K/AKT signaling activity. These findings suggest that targeting this pathway could be a promising and specific therapeutic approach for the treatment of MLS[187]. Currently, the treatment landscape for MLS has expanded to include the utilization of new agents, such as Trabectedin or Eribulin in combination with traditional therapeutic approaches[185]. However, a notable deficiency persists in terms of pharmacological interventions that specifically target the FUS-DDIT3 fusion protein, which represents a characteristic molecular alteration in MLS. There is an urgent requirement to develop novel therapeutic strategies that directly address the oncogenic potential conferred by the FUS-DDIT3 fusion protein.
4.3. ETS-fusion inhibitors
ETS protein family encompasses 28 transcription factors that possess highly conserved DNA-binding ETS domains. These factors, including FLI1, ERG, FEV, ETV1, ETV4 and others, play a crucial role in regulating a range of physiological processes, including cell proliferation, differentiation and apoptosis[188]. The role of ETS gene fusions has been widely investigated in prostate cancer, breast cancer, lung cancer and colorectal cancer[188]. The most common form of ETS gene fusion observed in prostate cancer is TMPRSS2-ERG, accounting for approximately 50% of cases[189]. Also, other fusion such as EWS-FLI1, TMPRSG-ERG[190], HNRNPH1-ERG[191], and EWSR1-ETV1[192] have been observed in different types of cancers (Figure 14). The transcription of ETS is dependent on the expression and activation of poly ADP-ribose polymerase (PARP) and DNA protein kinase (DNAPK). This indicates that utilization of pharmacological agents that PARP and DNAPK shows promising potential in the inhibition of ETS fusion activity[189]. For instance, the PARP inhibitors, such as Olaparib[189], Veliparib and Rucaparib, have shown therapeutic efficacy against ETS fusions. Rucaparib has been found to be effective in cells with defective in the PTEN gene and expressing ETS gene fusion[193]. YK-4-279 inhibits the activity of EWS-FLI1 fusion protein by blocking RNA Helicase A (RHA) binding with EWS-FLI1[194]. TK216, a derivative of Yk-4-279, also inhibits the activation of EWS-FLI1[195]. Dutasteride inhibits the growth of TMPRSS2-ERG fusion-positive cells and significantly slows tumor growth when combined with Bicalutamide[196]. Additionally, the BET protein inhibitor, ABBV- 075, is a potent inhibitor of TMPRSS2-ETS fusion[197]. Studies have shown that the glucocorticoid receptor GR interacts with transcription factors of the ETS family. And the GR antagonist Mifepristone inhibits EWS-FLI1 and EWS-ERG[198]. The diversity of therapeutic approaches for ETS fusions provides valuable insights for follow-up studies.
4.4. MYB-fusion inhibitors
Avian myeloblastosis viral oncogene homolog (MYB) is a transcriptional regulator that plays a critical role in maintaining normal tissue homeostasis and development[199]. The presence of rearrangements and mutations in the MYB gene serves as compelling evidence for the involvement of MYB family members in human cancers. The most frequently occurring MYB fusion is MYB-NFIB[199,200] (Figure 14). In particular, the AKT-dependent IGF-IR signaling pathway regulates MYB-NFIB, making the IGF-1R-MYB-NFIB axis is a promising target for the treatment of ACC[201]. The administration of IGF-1R inhibitor, such as Linsitinib or BMS-754807, has been shown to downregulation of the expression of the MYB-NFIB fusion protein[200]. MYB-TYK2 fusion proteins are known to activate JAK/STAT signaling pathway. In cells expressing MYB-TYK2, a combination therapy involving Vorinostat, Tanespimycin and Cerdulatinib has exhibited notable efficacy[202]. And, transcriptional targets of MYB-QKI, such as KIT or CDK6, can be targeted for therapeutic intervention. ATR, a DNA damage-sensing kinase, has also been reported as a downstream therapeutic target for MYB and MYB-NFIB[200,203]. Monensin can induce MYB degradation and inhibition of MYB-NFIB at both the mRNA and protein levels[200].
4.5. MLL-fusion inhibitors
Myeloid/lymphoid leukemia (MLL), is a kind of histone methyltransferases in the maintenance of hematopoietic stem cells. MLL gene fusions can be generated due to chromosomal translocation or epigenetic changes, which promote the occurrence and development of leukemia. MLL rearrangement is one of the most prevalent chromosomal abnormalities in AML[204]. Multiple types of MLL fusions were identified, such as MLL-AF4[205], MLL-AF9[206,207], MLL-AF6[208] and others (Figure 14). The oncogenic activity of MLL fusion proteins depends on the binding of the CXXC domain to the promoter and/or enhancer of the target gene. Disulfiram has been shown to disrupt the CXXC domain and inhibit the binding of MLL fusion protein to DNA, thereby attenuating their activity[209]. In addition, most MLL fusion proteins rely on transcriptional regulatory mechanisms mediated by asparagine endopeptidase (AEP) and histone methyltransferase (DOT1L)[207]. A selective DOT1L inhibitor, EPZ004777, has demonstrated efficacy against various MLL fusion proteins, including MLL-AEP [210], MLL-AF9, MLL-AF4 and MLL- ENL fusions[211,212]. Rabeprazole, a protonpump inhibitors, selectively inhibits the growth of MLL-AF4 and MLL-AF9-positive cells[213]. KO-382, a small molecule inhibitor of Menin-MLL interaction, binds to Menin and inhibits the proliferation of leukemic cells transformed by MLL-AF9[214]. Mocetinostat, the inhibitor of the histone deacetylase complex (HDAC), is used to inhibit MLL-AF9[212]. The immunoproteasome inhibitor ONX-0914 exhibits therapeutic activity against ALL expressing MLL-AF4 fusion protein[215]. The HSP90 inhibitors Radicol and Ganetespib induce degradation of MLL fusion protein[216]. In conclusion, the treatment of MLL fusions holds great promise.
4.6. FOXO1-fusion inhibitors
The Forkhead Box O (FOXO) family, comprising FOXO1, FOXO3, FOXO4 and FOXO6, represents a group of transcription factors that play critical roles in various physiological processes and responses to environmental stimuli[217]. Among the FOXO family members, FOXO1 fusions are the most prevalent, with notable example including PAX3-FOXO1[218] and PAX7-FOXO1[218] (Figure 14). Inhibition of key target genes regulated by PAX3-FOXO1 using the small molecule inhibitor JQ1, which blocks the BRD4 protein, have been shown to induce cell death and effectively suppresses tumor growth in vivo[217]. The potent and selective histone deacetylase inhibitor, Entinostat, reduces the mRNA and protein expression levels of PAX3-FOXO1 in tumor cells. JARID2, a direct transcriptional target of PAX3-FOXO1 fusion protein and its downstream effector, shows reduced cell proliferation upon inhibition[219]. Chromodomain decapping enzyme DNA-binding protein 4 (CHD4) is a key co-regulator of PAX3-FOXO1, meaning it a potential therapeutic target for rhabdomyosarcoma (RMS)[220]. ATR inhibitors show greater activity in PAX7-FOXO1 fusions. Furthermore, PAX3-FOXO1 protein levels can be regulated by proteasome inhibitors such as MG-132. Small molecule inhibitors against fusion transcription factors remain a challenging area of research. Therefore, indirect inhibition strategies against such carcinogens hold promising as a more pragmatic approach.
4.7. YAP1-fusion inhibitors
Yes-associated protein 1 (YAP1) is an essential transcriptional co-activator that acts as a downstream effector of the HIPPO pathway primarily through the TEAD family of transcription factors[221,222]. Its crucial roles in normal tissue homeostasis, development, stem cell maintenance, making it an attractive target for cancer therapy[222]. YAP1 fusion types, include YAP1-TFE3[222], YAP1-MAML2[222], YAP1-NUTM1[221] have been identified (Figure 14). The activity of the YAP1 fusion protein is reliant on its interaction with TEAD, mediated through TEAD binding sites, which has been shown to hinder the development of associated tumors[223]. Therefore, disrupting the interaction between YAP1 and TEAD may hold promise as an effective strategy for cancer therapy. In addition, Verteporfin, a small molecule inhibitor, disrupts the interaction between YAP1 and TEAD, leading to the inhibition of YAP1 function[222]. Pharmacological and genetic disruption of YAP-TEAD interactions reduced the oncogenic potential of the fusion, suggesting the potential of targeting this interaction as a future approach for cancer treatment[224,225].
5. Conclusions
In recent years, advancements in detection methods have yielded an expanding repertoire of identified gene fusion events, encompassing both known and novel types. These discoveries have provided valuable insights into potential therapeutic targets, fueling further investigations in this field. Although the efficiency of the assay technology has improved, the challenge of false-positive results persists[30,31]. Hence, continuous refinement of detection methods is critical to deepen our understanding of gene fusions and to foster the development of effective strategies for tumor treatment. Tumors harboring gene fusions usually exhibit heightened malignancy, and although targeted drugs have been proven effective against certain fusions, many tumors remain without effective treatment options. We have compiled a comprehensive list of drugs currently undergoing clinical trials with the specific objective of targeting fusion proteins. The data comes from the DrugBank database[226](Table 1). Despite the availability of several small molecule inhibitors approved by the FDA, drug resistance remains a pervasive clinical issue[227]. While small molecule inhibitors have undeniably advanced cancer treatment, the exploration and development of alternative therapeutic options are imperative, given the eventual emergence of tumor cells resistance over time[228,229,230].
The Protein Targeting Chimeras (PROTACs) is a novel technique that effectively induces protein degradation through the ubiquitin-proteasome degradation system (UPS). PROTACs offer potential advantages in addressing target protein mutations, overexpression, and resistance to small molecule inhibitors or monoclonal antibodies[231,232,233]. Moreover, PROTACs hold promise in targeting non-drug or drug-resistant targets, including nuclear receptors, transcription factors and folded proteins[231,233]. These attributes make PROTACs an attractive approach for treating fusion proteins associated with diseases such as NPM/EML4-ALK, BCR-ABL[233,234], presenting a viable strategy for combating drug resistance. Furthermore, considering that fusion partners often possess coiled-coil domains, crucial for the function of resulting fusion proteins (Figure 4), these domains could serve as potential targets for developing inhibitors in future designs. Moreover, gene fusions have demonstrated remarkable potential in generating tumor-specific neoantigens, making immunotherapy centered around these neoantigens a promising approach for cancer treatment[235]. In particular, certain fusions can modulate the expression of PD-L1 through downstream pathways, as observed with ALK fusions. Given the correlation between PD-L1 expression on tumor cell surfaces and the response to PD-1/PD-L1 antibody, it may be a prudent choice for tumors harboring such gene fusions[130,236]. Among these therapeutic strategies, immune checkpoint blockade (ICB) therapy utilizing PD-1/PD-L1 blockade is currently one of the most prevalent immunotherapeutic approaches. Overall, there is growing optimism that cancers driven by gene fusion can be effectively addressed through continued research and the development of new treatments in the future[237].
Author Contributions
Conceptualization, T.Q., Y.K., G.W., K.S., R.W. and C.J.; writing—original draft preparation, T.Q., Y.K. and C.J.; writing—review and editing, T.Q., Y.K., G.W., C.J., K.S., R.W., Y.W. and Y.C; funding acquisition, T.Q.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (No. 32000363) and The National Science Foundation of Zhejiang Province (No. LQ21C060006).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; Imielinski, M.; Weischenfeldt, J.; Beroukhim, R.; Campbell, P.J. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; Boyault, S.; Burkhardt, B.; Butler, A.P.; Caldas, C.; Davies, H.R.; Desmedt, C.; Eils, R.; Eyfjörd, J.E.; Foekens, J.A.; Greaves, M.; Hosoda, F.; Hutter, B.; Ilicic, T.; Imbeaud, S.; Imielinski, M.; Jäger, N.; Jones, D.T.; Jones, D.; Knappskog, S.; Kool, M.; Lakhani, S.R.; López-Otín, C.; Martin, S.; Munshi, N.C.; Nakamura, H.; Northcott, P.A.; Pajic, M.; Papaemmanuil, E.; Paradiso, A.; Pearson, J.V.; Puente, X.S.; Raine, K.; Ramakrishna, M.; Richardson, A.L.; Richter, J.; Rosenstiel, P.; Schlesner, M.; Schumacher, T.N.; Span, P.N.; Teague, J.W.; Totoki, Y.; Tutt, A.N.; Valdés-Mas, R.; van Buuren, M.M.; van 't Veer, L.; Vincent-Salomon, A.; Waddell, N.; Yates, L.R.; Zucman-Rossi, J.; Futreal, P.A.; McDermott, U.; Lichter, P.; Meyerson, M.; Grimmond, S.M.; Siebert, R.; Campo, E.; Shibata, T.; Pfister, S.M.; Campbell, P.J.; Stratton, M.R. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Heim, S. Interstitial deletions generating fusion genes. Cancer Genomics Proteomics 2021, 18, 167–196. [Google Scholar] [CrossRef]
- Pederzoli, F.; Bandini, M.; Marandino, L.; Ali, S.M.; Madison, R.; Chung, J.; Ross, J.S.; Necchi, A. Targetable gene fusions and aberrations in genitourinary oncology. Nat. Rev. Urol. 2020, 17, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Heydt, C.; Wölwer, C.B.; Velazquez Camacho, O.; Wagener-Ryczek, S.; Pappesch, R.; Siemanowski, J.; Rehker, J.; Haller, F.; Agaimy, A.; Worm, K.; Herold, T.; Pfarr, N.; Weichert, W.; Kirchner, T.; Jung, A.; Kumbrink, J.; Goering, W.; Esposito, I.; Buettner, R.; Hillmer, A.M.; Merkelbach-Bruse, S. Detection of gene fusions using targeted next-generation sequencing: a comparative evaluation. BMC Med. Genomics 2021, 14, 62. [Google Scholar] [CrossRef] [PubMed]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; Pau, G.; Reeder, J.; Cao, Y.; Mukhyala, K.; Selvaraj, S.K.; Yu, M.; Zynda, G.J.; Brauer, M.J.; Wu, T.D.; Gentleman, R.C.; Manning, G.; Yauch, R.L.; Bourgon, R.; Stokoe, D.; Modrusan, Z.; Neve, R.M.; de Sauvage, F.J.; Settleman, J.; Seshagiri, S.; Zhang, Z. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef]
- Cilloni, D.; Saglio, G. Molecular pathways: BCR-ABL. Clin. Cancer Res. 2012, 18, 930–937. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lalazar, G.; Houlihan, S.L.; Tschaharganeh, D.F.; Baslan, T.; Chen, C.C.; Requena, D.; Tian, S.; Bosbach, B.; Wilkinson, J.E.; Simon, S.M.; Lowe, S.W. DNAJB1-PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 13076–13084. [Google Scholar] [CrossRef]
- Asai, N.; Murakami, H.; Iwashita, T.; Takahashi, M. A mutation at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret impairs their transforming activity and association with shc adaptor proteins. J. Biol. Chem. 1996, 271, 17644–17649. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Yu, Y.P.; Tao, J.; Liu, S.; Tseng, G.; Nalesnik, M.; Hamilton, R.; Bhargava, R.; Nelson, J.B.; Pennathur, A.; Monga, S.P.; Luketich, J.D.; Michalopoulos, G.K.; Luo, J.H. MAN2A1-FER fusion gene is expressed by human liver and other tumor types and has oncogenic activity in mice. Gastroenterology 2017, 153, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wang, X.; Song, H.; Zhang, Y.; Zhang, R.; Li, S.; Jin, W.; Chen, S.; Fang, H.; Chen, Z.; Wang, K. A PML/RARα direct target atlas redefines transcriptional deregulation in acute promyelocytic leukemia. Blood 2021, 137, 1503–1516. [Google Scholar] [CrossRef] [PubMed]
- Zullow, H.J.; Sankar, A.; Ingram, D.R.; Samé Guerra, D.D.; D'Avino, A.R.; Collings, C.K.; Lazcano, R.; Wang, W.L.; Liang, Y.; Qi, J.; Lazar, A.J.; Kadoch, C. The FUS::DDIT3 fusion oncoprotein inhibits BAF complex targeting and activity in myxoid liposarcoma. Mol. Cell 2022, 82, 1737–1750. [Google Scholar] [CrossRef] [PubMed]
- Capdeville, R.; Silberman, S. Imatinib: a targeted clinical drug development. Semin. Hematol. 2003, 40, 15–20. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; McTigue, M.; Grodsky, N.; Ryan, K.; Padrique, E.; Alton, G.; Timofeevski, S.; Yamazaki, S.; Li, Q.; Zou, H.; Christensen, J.; Mroczkowski, B.; Bender, S.; Kania, R.S.; Edwards, M.P. Structure based drug design of Crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Zhu, J.; Chen, Z.; de Thé, H. Curing APL through PML/RARA degradation by As2O3. Trends Mol. Med. 2012, 18, 36–42. [Google Scholar] [CrossRef]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef]
- Hahn, Y.; Bera, T.K.; Gehlhaus, K.; Kirsch, I.R.; Pastan, I.H.; Lee, B. Finding fusion genes resulting from chromosome rearrangement by analyzing the expressed sequence databases. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 13257–13261. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; Lee, C.; Montie, J.E.; Shah, R.B.; Pienta, K.J.; Rubin, M.A.; Chinnaiyan, A.M. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; Yu, J.; Wang, L.; Montie, J.E.; Rubin, M.A.; Pienta, K.J.; Roulston, D.; Shah, R.B.; Varambally, S.; Mehra, R.; Chinnaiyan, A.M. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; Bando, M.; Ohno, S.; Ishikawa, Y.; Aburatani, H.; Niki, T.; Sohara, Y.; Sugiyama, Y.; Mano, H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Koivunen, J.P.; Mermel, C.; Zejnullahu, K.; Murphy, C.; Lifshits, E.; Holmes, A.J.; Choi, H.G.; Kim, J.; Chiang, D.; Thomas, R.; Lee, J.; Richards, W.G.; Sugarbaker, D.J.; Ducko, C.; Lindeman, N.; Marcoux, J.P.; Engelman, J.A.; Gray, N.S.; Lee, C.; Meyerson, M.; Jänne, P.A. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin. Cancer Res. 2008, 14, 4275–4283. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, Q.; Tang, M.; Barthel, F.; Amin, S.; Yoshihara, K.; Lang, F.M.; Martinez-Ledesma, E.; Lee, S.H.; Zheng, S.; Verhaak, R.G.W. TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 2018, 46, D1144–D1149. [Google Scholar] [CrossRef]
- Amin, S.M.; Haugh, A.M.; Lee, C.Y.; Zhang, B.; Bubley, J.A.; Merkel, E.A.; Verzi, A.E.; Gerami, P. A comparison of morphologic and molecular features of BRAF, ALK, and NTRK1 fusion Spitzoid Neoplasms. Am. J. Pathol. 2017, 41, 491–498. [Google Scholar] [CrossRef]
- De la Fouchardière, A.; Tee, M.K.; Peternel, S.; Valdebran, M.; Pissaloux, D.; Tirode, F.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C.; Yeh, I. Fusion partners of NTRK3 affect subcellular localization of the fusion kinase and cytomorphology of melanocytes. Mod. Pathol. 2021, 34, 735–747. [Google Scholar] [CrossRef]
- Batra, U.; Nathany, S.; Sharma, M.; Pasricha, S.; Bansal, A.; Jain, P.; Mehta, A. IHC versus FISH versus NGS to detect ALK gene rearrangement in NSCLC: all questions answered? J. Clin. Pathol. 2022, 75, 405–409. [Google Scholar] [CrossRef]
- Mangolini, A.; Rocca, C.; Bassi, C.; Ippolito, C.; Negrini, M.; Dell'Atti, L.; Lanza, G.; Gafà, R.; Bianchi, N.; Pinton, P.; Aguiari, G. Detection of disease-causing mutations in prostate cancer by NGS sequencing. Cell Biol. Int. 2022, 46, 1047–1061. [Google Scholar] [CrossRef]
- Wagener-Ryczek, S.; Pappesch, R. Targeted RNA-sequencing for the evaluation of gene fusions in lung tumors: current status and future prospects. Expert Rev. Mol. Diagn. 2021, 21, 531–534. [Google Scholar] [CrossRef]
- Jin, Z.; Huang, W.; Shen, N.; Li, J.; Wang, X.; Dong, J.; Park, P.J.; Xi, R. Single-cell gene fusion detection by scFusion. Nat. Commun. 2022, 13, 1084. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O.; Superti-Furga, G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004, 5, 33–44. [Google Scholar] [CrossRef]
- Carofiglio, F.; Trisciuzzi, D.; Gambacorta, N.; Leonetti, F.; Stefanachi, A.; Nicolotti, O. Bcr-Abl allosteric inhibitors: Where we are and Where we are going to. Molecules 2020, 25, 4210. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. Breakthrough in degradation of BCR-ABL fusion protein for the treatment of cancer. ACS Medicinal Chem. Lett. 2020, 11, 2359–2360. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.Q.; Gao, F.H. Regulatory molecules and corresponding processes of BCR-ABL protein degradation. J. Cancer 2019, 10, 2488–2500. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Yang, Y.; Gupta, P.; Wang, A.; Zhao, M.; Zhao, Y.; Qu, M.; Ke, Y.; Liu, Y.; Liu, H.M.; Xu, X.; Sun, Y.; Chen, Z.S.; Hu, Z. A small molecule inhibitor, OGP46, is effective against Imatinib-resistant BCR-ABL mutations via the BCR-ABL/JAK-STAT pathway. Mol. Ther. Oncolytics. 2020, 18, 137–148. [Google Scholar] [CrossRef]
- Chandrasekhar, C.; Kumar, P.S.; Sarma, P. Novel mutations in the kinase domain of BCR-ABL gene causing Imatinib resistance in chronic myeloid leukemia patients. Sci. Rep. 2019, 9, 2412. [Google Scholar] [CrossRef]
- Hochhaus, A. Management of Bcr-Abl-positive leukemias with Dasatinib. Expert Rev. Anticancer Ther. 2007, 7, 1529–1536. [Google Scholar] [CrossRef]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O'Brien, S.; Nicaise, C.; Bleickardt, E.; Blackwood-Chirchir, M.A.; Iyer, V.; Chen, T.T.; Huang, F.; Decillis, A.P.; Sawyers, C.L. Dasatinib in Imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar] [CrossRef]
- Müller, M.C.; Erben, P.; Schenk, T.; Lauber, S.; Kruth, J.; Hoffmann, J.; Ernst, T.; Lahaye, T.; Metzgeroth, G.; Acevedo, M.; Nicaise, C.; Hehlmann, R.; Hochhaus, A. Response to Dasatinib after Imatinib Failure According to Type of Preexisting BCR-ABL Mutations. Blood 2006, 108, 748. [Google Scholar] [CrossRef]
- Krijanovski, Y.; Donato, N.; Cardama, A.Q.; Cortes, J.; Talpaz, M. Dasatinib resistance in CML patients. Identification of novel BCR-ABL kinase domain mutation. Blood 2007, 110, 1957. [Google Scholar] [CrossRef]
- Makiko, M.; Yoko, N.; Itaru, K.; Satoshi, S.; Hidefumi, H.; Yasuhiko, K.; Toshio, H.; Tatsutoshi, N.; Souichi, A. Dasatinib induces autophagy in mice with Bcr-Abl-positive leukemia. Int. J. Hematol. 2017, 105, 335–340. [Google Scholar]
- Kantarjian, H.; Giles, F.; Wunderle, L.; Bhalla, K.; O'Brien, S.; Wassmann, B.; Tanaka, C.; Manley, P.; Rae, P.; Mietlowski, W.; Bochinski, K.; Hochhaus, A.; Griffin, J.D.; Hoelzer, D.; Albitar, M.; Dugan, M.; Cortes, J.; Alland, L.; Ottmann, O.G. Nilotinib in Imatinib-resistant CML and Philadelphia chromosome-positive ALL. N. Engl. J. Med. 2006, 354, 2542–2551. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Mohi, M.G.; Pride, Y.B.; Quinnan, L.R.; Malouf, N.A.; Podar, K.; Gesbert, F.; Iwasaki, H.; Li, S.; Van Etten, R.A.; Gu, H.; Griffin, J.D.; Neel, B.G. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell 2002, 1, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Breccia, M.; Colafigli, G.; Scalzulli, E.; Martelli, M. Asciminib: an investigational agent for the treatment of chronic myeloid leukemia. Expert Opin. Investig. Drugs 2021, 30, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Eide, C.A.; Zabriskie, M.S.; Savage Stevens, S.L.; Antelope, O.; Vellore, N.A.; Than, H.; Schultz, A.R.; Clair, P.; Bowler, A.D.; Pomicter, A.D.; Yan, D.; Senina, A.V.; Qiang, W.; Kelley, T.W.; Szankasi, P.; Heinrich, M.C.; Tyner, J.W.; Rea, D.; Cayuela, J.M.; Kim, D.W.; Tognon, C.E.; O'Hare, T.; Druker, B.J.; Deininger, M.W. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell 2019, 36, 431–443.e435. [Google Scholar] [CrossRef] [PubMed]
- A. , E.C.; S., Z.M.; L., S.S.; Orlando, A.; A., V.N.; Hein, T.; Anna, R.S.; M., C.P.; D., B.A.; D., P.A.; Dongqing, Y.; V., S.A.; Qiang, W.; W., K.T.; Philippe, S.; C., H.M.; W., T.J.; Delphine, R.; Michel, C.J.; Wook, K.D.; E., T.C.; Thomas, O.H.; J., D.B.; W., D.M. Combining the allosteric ABL1 inhibitor Asciminib (ABL001) with Ponatinib suppresses emergence of and restores efficacy against highly resistant BCR-ABL1 compound mutants. Blood 2019, 134, 431–443.e435. [Google Scholar]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; Groell, J.M.; Grotzfeld, R.M.; Hassan, A.Q.; Henry, C.; Iyer, V.; Jones, D.; Lombardo, F.; Loo, A.; Manley, P.W.; Pelle, X.; Rummel, G.; Salem, B.; Warmuth, M.; Wylie, A.A.; Zoller, T.; Marzinzik, A.L.; Furet, P. Discovery of Asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. [Google Scholar] [CrossRef]
- Keller, V.A.G.; Brümmendorf, T.H. Novel aspects of therapy with the dual Src and Abl kinase inhibitor Bosutinib in chronic myeloid leukemia. Expert Rev. Anticancer Ther. 2012, 12, 1121–1127. [Google Scholar] [CrossRef]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The transcriptional roles of ALK fusion proteins in tumorigenesis. Cancers (Basel) 2019, 11, 1074. [Google Scholar] [CrossRef]
- Wang, D.; Li, D.; Qin, G.; Zhang, W.; Ouyang, J.; Zhang, M.; Xie, L. The structural characterization of tumor fusion genes and proteins. Comput. Math. Methods Med. 2015, 2015, 912742. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; Hu, Y.; Tan, Z.; Stokes, M.; Sullivan, L.; Mitchell, J.; Wetzel, R.; Macneill, J.; Ren, J.M.; Yuan, J.; Bakalarski, C.E.; Villen, J.; Kornhauser, J.M.; Smith, B.; Li, D.; Zhou, X.; Gygi, S.P.; Gu, T.L.; Polakiewicz, R.D.; Rush, J.; Comb, M.J. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef]
- Takeuchi, K.; Choi, Y.L.; Togashi, Y.; Soda, M.; Hatano, S.; Inamura, K.; Takada, S.; Ueno, T.; Yamashita, Y.; Satoh, Y.; Okumura, S.; Nakagawa, K.; Ishikawa, Y.; Mano, H. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin. Cancer Res. 2009, 15, 3143–3149. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.; Shapiro, D.N.; Look, A.T.; Saltman, D.L. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1995, 267, 316–317. [Google Scholar] [CrossRef]
- Lamant, L.; Dastugue, N.; Pulford, K.; Delsol, G.; Mariamé, B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 1999, 93, 3088–3095. [Google Scholar] [CrossRef]
- Qin, Z.; Sun, H.; Yue, M.; Pan, X.; Chen, L.; Feng, X.; Yan, X.; Zhu, X.; Ji, H. Phase separation of EML4-ALK in firing downstream signaling and promoting lung tumorigenesis. Cell Discov. 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Toyokawa, G.; Takenoyama, M.; Taguchi, K.; Toyozawa, R.; Inamasu, E.; Kojo, M.; Shiraishi, Y.; Morodomi, Y.; Takenaka, T.; Hirai, F.; Yamaguchi, M.; Seto, T.; Shimokawa, M.; Ichinose, Y. An extremely rare case of small-cell lung cancer harboring variant 2 of the EML4-ALK fusion gene. Lung Cancer 2013, 81, 487–490. [Google Scholar] [CrossRef]
- Li, T.; Zhang, F.; Wu, Z.; Cui, L.; Zhao, X.; Wang, J.; Hu, Y. PLEKHM2-ALK: A novel fusion in small-cell lung cancer and durable response to ALK inhibitors. Lung Cancer 2020, 139, 146–150. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Crizotinib resistance: implications for therapeutic strategies. Ann. Oncol. 2016, 27 Suppl 3, iii42–iii50. [Google Scholar] [CrossRef]
- Timm, A.; Kolesar, J.M. Crizotinib for the treatment of non-small-cell lung cancer. Am. J. Health-Syst. Pharm. 2013, 70, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.M.; Maher, V.E.; Bijwaard, K.E.; Becker, R.L.; Zhang, L.; Tang, S.W.; Song, P.; Liu, Q.; Marathe, A.; Gehrke, B.; Helms, W.; Hanner, D.; Justice, R.; Pazdur, R.U.S. Food and Drug Administration approval: Crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin. Cancer Res. 2014, 20, 2029–2034. [Google Scholar] [CrossRef]
- Katayama, R.; Shaw, A.; Khan, T.; Mino-Kenudson, M.; Solomon, B.; Halmos, B.; Jessop, N.; Wain, J.; Yeo, A.; Benes, C.; Drew, L.; Saeh, J.; Crosby, K.; Sequist, L.; Iafrate, A.; Engelman, J. Mechanisms of acquired Crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra117. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; Ishikawa, Y.; Kimura, H.; Mitsudomi, T.; Tanio, Y.; Mano, H. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [PubMed]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; Varella-Garcia, M.; Camidge, D.R. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; Capelletti, M.; Shimamura, T.; Ercan, D.; Stumpfova, M.; Xiao, Y.; Weremowicz, S.; Butaney, M.; Heon, S.; Wilner, K.; Christensen, J.G.; Eck, M.J.; Wong, K.K.; Lindeman, N.; Gray, N.S.; Rodig, S.J.; Jänne, P.A. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar] [CrossRef]
- Toyokawa, G.; Hirai, F.; Inamasu, E.; Yoshida, T.; Nosaki, K.; Takenaka, T.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Secondary mutations at I1171 in the ALK gene confer resistance to both Crizotinib and Alectinib. J. Thorac. Oncol. 2014, 9, e86–e87. [Google Scholar] [CrossRef]
- Fang, D.D.; Zhang, B.; Gu, Q.; Lira, M.; Xu, Q.; Sun, H.; Qian, M.; Sheng, W.; Ozeck, M.; Wang, Z.; Zhang, C.; Chen, X.; Chen, K.X.; Li, J.; Chen, S.H.; Christensen, J.; Mao, M.; Chan, C.C. HIP1-ALK, a novel ALK fusion variant that responds to Crizotinib. J. Thorac. Oncol. 2014, 9, 285–294. [Google Scholar] [CrossRef]
- Shan, L.; Jiang, P.; Xu, F.; Zhang, W.; Guo, L.; Wu, J.; Zeng, Y.; Jiao, Y.; Ying, J. BIRC6-ALK, a novel fusion gene in ALK break-apart FISH-negative lung adenocarcinoma, responds to Crizotinib. J. Thorac. Oncol. 2015, 10, e37–e39. [Google Scholar] [CrossRef]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; Sarkisova, Y.; Sun, F.; Steffy, A.; Pferdekamper, A.C.; Li, A.G.; Joseph, S.B.; Kim, Y.; Liu, B.; Tuntland, T.; Cui, X.; Gray, N.S.; Steensma, R.; Wan, Y.; Jiang, J.; Chopiuk, G.; Li, J.; Gordon, W.P.; Richmond, W.; Johnson, K.; Chang, J.; Groessl, T.; He, Y.-Q.; Phimister, A.; Aycinena, A.; Lee, C.C.; Bursulaya, B.; Karanewsky, D.S.; Seidel, H.M.; Harris, J.L.; Michellys, P.-Y. Synthesis, structure–activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-Chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in Phase 1 and Phase 2 Clinical Trials. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; Riely, G.J.; Solomon, B.J.; Wolf, J.; Thomas, M.; Schuler, M.; Liu, G.; Santoro, A.; Lau, Y.Y.; Goldwasser, M.; Boral, A.L.; Engelman, J.A. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef]
- Latif, M.; Saeed, A.; Kim, S.H. Journey of the ALK-inhibitor CH5424802 to phase II clinical trial. Arch. Pharm. Res. 2013, 36, 1051–1054. [Google Scholar] [CrossRef]
- McKeage, K. Alectinib: A Review of Its Use in Advanced ALK-Rearranged Non-Small Cell Lung Cancer. Drugs 2015, 75, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Zhu, V.W.; Schrock, A.B.; Bosemani, T.; Benn, B.S.; Ali, S.M.; Ou, S.I. Dramatic response to alectinib in a lung cancer patient with a novel VKORC1L1-ALK fusion and an acquired ALK T1151K mutation. Lung Cancer (Auckl) 2018, 9, 111–116. [Google Scholar] [CrossRef]
- Honda, K.; Kadowaki, S.; Kato, K.; Hanai, N.; Hasegawa, Y.; Yatabe, Y.; Muro, K. Durable response to the ALK inhibitor alectinib in inflammatory myofibroblastic tumor of the head and neck with a novel SQSTM1-ALK fusion: A case report. Invest. New Drugs 2019, 37, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Klempner, S.J.; Greenbowe, J.R.; Azada, M.; Schrock, A.B.; Ali, S.M.; Ross, J.S.; Stephens, P.J.; Miller, V.A. Identification of a novel HIP1-ALK fusion variant in Non-Small-Cell Lung Cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to Alectinib. J. Thorac. Oncol. 2014, 9, 1821–1825. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Jiang, Y.; Jiang, W.; Wang, H.; Liu, S.; Shao, Y.; Zhao, W.; Ning, R.; Yu, Q. STRN-ALK fusion in lung adenocarcinoma with excellent response upon Alectinib treatment: A case report and literature review. Onco. Targets Ther. 2020, 13, 12515–12519. [Google Scholar] [CrossRef]
- Pan, Y.; Xiao, W.; Ye, F.; Wang, H.; Shen, Y.; Yu, X.; Han, X.; Chu, Q.; Zhou, C.; Zhang, Z.; Ren, S. Outcomes of switching from Crizotinib to Alectinib in patients with advanced non-small cell lung cancer with anaplastic lymphoma kinase fusion. Ann. Transl. Med. 2021, 9, 1014. [Google Scholar] [CrossRef]
- Ignatius Ou, S.H.; Azada, M.; Hsiang, D.J.; Herman, J.M.; Kain, T.S.; Siwak-Tapp, C.; Casey, C.; He, J.; Ali, S.M.; Klempner, S.J.; Miller, V.A. Next-generation sequencing reveals a novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to Alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on Crizotinib. J. Thorac. Oncol. 2014, 9, 549–553. [Google Scholar]
- Gettinger, S.N.; Bazhenova, L.; Salgia, R.; Langer, C.J.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Narasimhan, N.I.; Dorer, D.J.; Rivera, V.M.; Clackson, T.; Haluska, F.G.; Camidge, D.R. Updated efficacy and safety of the ALK inhibitor AP26113 in patients (pts) with advanced malignancies, including ALK+ non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2014, 32, 8047. [Google Scholar] [CrossRef]
- Li, Z.; Liu, F.; Wu, S.; Ding, S.; Chen, Y.; Liu, J. Research progress on the drug resistance of ALK kinase inhibitors. Curr. Med. Chem. 2021, 29, 2456–2475. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; Piazza, R.; Gambacorti-Passerini, C.; Mologni, L. Lorlatinib treatment elicits multiple on- and off-Target mechanisms of resistance in ALK-driven cancer. Cancer Res. 2018, 78, 6866–6880. [Google Scholar] [CrossRef] [PubMed]
- Siaw, J.T.; Wan, H.; Pfeifer, K.; Rivera, V.M.; Guan, J.; Palmer, R.H.; Hallberg, B. Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK-positive neuroblastoma cells, Drosophila and mice. Oncotarget 2016, 7, 29011–29022. [Google Scholar] [CrossRef]
- Galkin, A.V.; Melnick, J.S.; Kim, S.; Hood, T.L.; Li, N.; Li, L.; Xia, G.; Steensma, R.; Chopiuk, G.; Jiang, J.; Wan, Y.; Ding, P.; Liu, Y.; Sun, F.; Schultz, P.G.; Gray, N.S.; Warmuth, M. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 270–275. [Google Scholar] [CrossRef]
- El-Deeb, I.M.; Yoo, K.H.; Lee, S.H. ROS receptor tyrosine kinase: a new potential target for anticancer drugs. Med. Res. Rev. 2011, 31, 794–818. [Google Scholar] [CrossRef]
- Park, S.; Ahn, B.C.; Lim, S.W.; Sun, J.M.; Kim, H.R.; Hong, M.H.; Lee, S.H.; Ahn, J.S.; Park, K.; Choi, Y.; Cho, B.C.; Ahn, M.J. Characteristics and outcome of ROS1-positive Non-Small Cell Lung Cancer patients in routine clinical practice. J. Thorac. Oncol. 2018, 13, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. ROS1 protein-tyrosine kinase inhibitors in the treatment of ROS1 fusion protein-driven non-small cell lung cancers. Pharmacol. Res. 2017, 121, 202–212. [Google Scholar] [CrossRef]
- Zeng, L.; Li, Y.; Xiao, L.; Xiong, Y.; Liu, L.; Jiang, W.; Heng, J.; Qu, J.; Yang, N.; Zhang, Y. Crizotinib presented with promising efficacy but for concomitant mutation in next-generation sequencing-identified ROS1-rearranged non-small-cell lung cancer. Onco. Targets Ther. 2018, 11, 6937–6945. [Google Scholar] [CrossRef]
- Davies, K.D.; Doebele, R.C. Molecular pathways: ROS1 fusion proteins in cancer. Clin. Cancer Res. 2013, 19, 4040–4045. [Google Scholar] [CrossRef]
- Rimkunas, V.M.; Crosby, K.E.; Li, D.; Hu, Y.; Kelly, M.E.; Gu, T.L.; Mack, J.S.; Silver, M.R.; Zhou, X.; Haack, H. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin. Cancer Res. 2012, 18, 4449–4457. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Jenkins, C.; Iyer, S.; Schoenfeld, A.; Keddy, C.; Davare, M.A. ROS1-dependent cancers - biology, diagnostics and therapeutics. Nat. Rev. Clin. Oncol. 2021, 18, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; de Figueiredo-Pontes, L.L.; Kobayashi, S.; Costa, D.B. Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor Crizotinib in ROS1-translocated lung cancer. J. Thorac. Oncol. 2012, 7, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.Y.; Li, Q.; Engstrom, L.D.; West, M.; Appleman, V.; Wong, K.A.; McTigue, M.; Deng, Y.L.; Liu, W.; Brooun, A.; Timofeevski, S.; McDonnell, S.R.; Jiang, P.; Falk, M.D.; Lappin, P.B.; Affolter, T.; Nichols, T.; Hu, W.; Lam, J.; Johnson, T.W.; Smeal, T.; Charest, A.; Fantin, V.R. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking Crizotinib-resistant ROS1 mutations. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 3493–3498. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Liu, S.V.; McCoach, C.E.; Zhu, V.W.; Tan, A.C.; Yoda, S.; Peterson, J.; Do, A.; Prutisto-Chang, K.; Dagogo-Jack, I.; Sequist, L.V.; Wirth, L.J.; Lennerz, J.K.; Hata, A.N.; Mino-Kenudson, M.; Nardi, V.; Ou, S.I.; Tan, D.S.; Gainor, J.F. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann. Oncol. 2020, 31, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhou, Q. Diagnostics, therapeutics and RET inhibitor resistance for RET fusion-positive non-small cell lung cancers and future perspectives. Cancer Treat. Rev. 2021, 96, 102153. [Google Scholar] [CrossRef]
- Rolfo, C.; Ruiz, R.; Giovannetti, E.; Gil-Bazo, I.; Russo, A.; Passiglia, F.; Giallombardo, M.; Peeters, M.; Raez, L. Entrectinib: a potent new TRK, ROS1, and ALK inhibitor. Expert Opin. Investig. Drugs 2015, 24, 1493–1500. [Google Scholar] [CrossRef]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; De Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; Avanzi, N.; Amboldi, N.; Saccardo, M.B.; Casero, D.; Orsini, P.; Bandiera, T.; Mologni, L.; Anderson, D.; Wei, G.; Harris, J.; Vernier, J.M.; Li, G.; Felder, E.; Donati, D.; Isacchi, A.; Pesenti, E.; Magnaghi, P.; Galvani, A. Entrectinib, a Pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol. Cancer Ther. 2016, 15, 628–639. [Google Scholar] [CrossRef]
- Keddy, C.; Shinde, P.; Jones, K.; Kaech, S.; Somwar, R.; Shinde, U.; Davare, M.A. Resistance profile and structural modeling of next-generation ROS1 tyrosine kinase inhibitors. Mol. Cancer Ther. 2022, 21, 336–346. [Google Scholar] [CrossRef]
- Facchinetti, F.; Loriot, Y.; Kuo, M.S.; Mahjoubi, L.; Lacroix, L.; Planchard, D.; Besse, B.; Farace, F.; Auger, N.; Remon, J.; Scoazec, J.Y.; Andre, F.; Soria, J.C.; Friboulet, L. Crizotinib-resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1- and ALK-rearranged lung cancers. Clin. Cancer Res. 2016, 22, 5983–5991. [Google Scholar] [CrossRef]
- Gandara, D.R.; Li, T.; Lara, P.N.; Kelly, K.; Riess, J.W.; Redman, M.W.; Mack, P.C. Acquired resistance to targeted therapies against oncogene-driven non-small-cell lung cancer: approach to subtyping progressive disease and clinical implications. Clin. Lung Cancer 2014, 15, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Lin, J.J.; Nagasaka, M.; Zhu, V.W.; Gabrail, N.Y.; Bazhenova, L.; Anderson, P.M.; Solomon, B.J.; Dudek, A.Z.; Pippas, A.W.; Shirinian, M.; Baik, C.S.; Stopatschinskaja, S.; Camidge, D.R.; Cho, B.C.; Drilon, A.E. TRIDENT-1: A global, multicenter, open-label Phase II study investigating the activity of repotrectinib in advanced solid tumors harboring ROS1 or NTRK1-3 rearrangements. J. Clin. Oncol. 2020, 38, TPS9637–TPS9637. [Google Scholar] [CrossRef]
- Markham, A. Brigatinib: first global approval. Drugs 2017, 77, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Haruko, D.; Seiji, N.; Jun, S.K.; Hiroshi, T.; Yasushi, G.; Kadoaki, O.; Ryo, T.; Masahiro, K.; Toshiaki, T.; Yoshihiro, H.; Masahiro, M.; Takaya, I.; Shingo, M.; Kiyotaka, Y.; Shogo, N.; Koichi, G. Phase II study of Brigatinib in ROS1 positive non-small cell lung cancer (NSCLC) patients previously treated with Crizotinib: Barossa cohort 2. J. Clin. Oncol. 2021, 39, 9040. [Google Scholar]
- Cai, L.; Duan, J.; Qian, L.; Wang, Z.; Wang, S.; Li, S.; Wang, C.; Zhao, J.; Zhang, X.; Bai, H.; Wang, J. ROS1 fusion mediates immunogenicity by upregulation of PD-L1 after the activation of ROS1-SHP2 signaling pathway in Non-Small Cell Lung Cancer. Front. Immunol. 2020, 11, 527750. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023–e000023. [Google Scholar]
- Pekova, B.; Sykorova, V.; Mastnikova, K.; Vaclavikova, E.; Moravcova, J.; Vlcek, P.; Lastuvka, P.; Taudy, M.; Katra, R.; Bavor, P.; Kodetova, D.; Chovanec, M.; Drozenova, J.; Astl, J.; Hrabal, P.; Vcelak, J.; Bendlova, B. NTRK fusion genes in thyroid carcinomas: clinicopathological characteristics and their impacts on prognosis. Cancers (Basel) 2021, 13, 1932. [Google Scholar]
- Ni, J.; Xie, S.; Ramkissoon, S.H.; Luu, V.; Sun, Y.; Bandopadhayay, P.; Beroukhim, R.; Roberts, T.M.; Stiles, C.D.; Segal, R.A.; Ligon, K.L.; Hahn, W.C.; Zhao, J.J. Tyrosine receptor kinase B is a drug target in astrocytomas. Neuro. Oncol. 2017, 19, 22–30. [Google Scholar] [CrossRef]
- Osako, T.; Takeuchi, K.; Horii, R.; Iwase, T.; Akiyama, F. Secretory carcinoma of the breast and its histopathological mimics: value of markers for differential diagnosis. Histopathology 2013, 63, 509–519. [Google Scholar] [CrossRef]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; Berge, E.M.; Kim, J.; Sasaki, H.; Park, S.; Kryukov, G.; Garraway, L.A.; Hammerman, P.S.; Haas, J.; Andrews, S.W.; Lipson, D.; Stephens, P.J.; Miller, V.A.; Varella-Garcia, M.; Jänne, P.A.; Doebele, R.C. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef]
- Marcus, L.; Donoghue, M.; Aungst, S.; Myers, C.E.; Helms, W.S.; Shen, G.; Zhao, H.; Stephens, O.; Keegan, P.; Pazdur, R. FDA approval summary: Entrectinib for the treatment of NTRK gene fusion solid tumors. Clin. Cancer Res. 2021, 27, 928–932. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; Besse, B.; Chawla, S.P.; Bazhenova, L.; Krauss, J.C.; Chae, Y.K.; Barve, M.; Garrido-Laguna, I.; Liu, S.V.; Conkling, P.; John, T.; Fakih, M.; Sigal, D.; Loong, H.H.; Buchschacher, G.L., Jr.; Garrido, P.; Nieva, J.; Steuer, C.; Overbeck, T.R.; Bowles, D.W.; Fox, E.; Riehl, T.; Chow-Maneval, E.; Simmons, B.; Cui, N.; Johnson, A.; Eng, S.; Wilson, T.R.; Demetri, G.D. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet. Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Emiliano, C.; Maurizio, S.; Alexander, D. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar]
- Farago, A.F.; Le, L.P.; Zheng, Z.; Muzikansky, A.; Drilon, A.; Patel, M.; Bauer, T.M.; Liu, S.V.; Ou, S.-H.I.; Jackman, D.; Costa, D.B.; Multani, P.S.; Li, G.G.; Hornby, Z.; Chow-Maneval, E.; Luo, D.; Lim, J.E.; Iafrate, A.J.; Shaw, A.T. Durable clinical response to Entrectinib in NTRK1-rearranged Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2015, 10, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Ardini, E.; Bosotti, R.; Amatu, A.; Valtorta, E.; Somaschini, A.; Raddrizzani, L.; Palmeri, L.; Banfi, P.; Bonazzina, E.; Misale, S.; Marrapese, G.; Leone, A.; Alzani, R.; Luo, D.; Hornby, Z.; Lim, J.; Veronese, S.; Vanzulli, A.; Bardelli, A.; Martignoni, M.; Davite, C.; Galvani, A.; Isacchi, A.; Siena, S. Sensitivity to Entrectinib associated with a novel LMNA-NTRK1 gene fusion in metastatic colorectal cancer. J. Natl. Cancer Inst. 2015, 108, djv306. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Siena, S.; Ou, S.-H.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; Doebele, R.; Giannetta, L.; Cerea, G.; Marrapese, G.; Schirru, M.; Amatu, A.; Bencardino, K.; Palmeri, L.; Sartore-Bianchi, A.; Vanzulli, A.; Cresta, S.; Damian, S.; Duca, M.; Ardini, E.; Li, G.; Christiansen, J.; Kowalski, K.; Johnson, A.D.; Patel, R.; Luo, D.; Chow-Maneval, E.; Hornby, Z.; Multani, P.S.; Shaw, A.T.; De Braud, F.G. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor Entrectinib: combined results from two Phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef]
- Drilon, A.; Li, G.; Dogan, S.; Gounder, M.; Shen, R.; Arcila, M.; Wang, L.; Hyman, D.M.; Hechtman, J.; Wei, G.; Cam, N.R.; Christiansen, J.; Luo, D.; Maneval, E.C.; Bauer, T.; Patel, M.; Liu, S.V.; Ou, S.H.I.; Farago, A.; Shaw, A.; Shoemaker, R.F.; Lim, J.; Hornby, Z.; Multani, P.; Ladanyi, M.; Berger, M.; Katabi, N.; Ghossein, R.; Ho, A.L. What hides behind the MASC: clinical response and acquired resistance to Entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC). Ann. Oncol. 2016, 27, 920–926. [Google Scholar] [CrossRef]
- Alvarez-Breckenridge, C.; Miller, J.J.; Nayyar, N.; Gill, C.M.; Kaneb, A.; D'Andrea, M.; Le, L.P.; Lee, J.; Cheng, J.; Zheng, Z.; Butler, W.E.; Multani, P.; Chow Maneval, E.; Ha Paek, S.; Toyota, B.D.; Dias-Santagata, D.; Santagata, S.; Romero, J.; Shaw, A.T.; Farago, A.F.; Yip, S.; Cahill, D.P.; Batchelor, T.T.; Iafrate, A.J.; Brastianos, P.K. Clinical and radiographic response following targeting of BCAN-NTRK1 fusion in glioneuronal tumor. NPJ Precis. Oncol. 2017, 1, 5. [Google Scholar] [CrossRef]
- Rogers, C.; Morrissette, J.J.D.; Sussman, R.T. NTRK point mutations and their functional consequences. Cancer Genet. 2022, 262-263, 5–15. [Google Scholar] [CrossRef]
- Kummar, S.; Lassen, U.N. TRK inhibition:A new tumor-agnostic treatment strategy. Target Oncol. 2018, 13, 545–556. [Google Scholar] [CrossRef]
- Doebele, R.C.; Davis, L.E.; Vaishnavi, A.; Le, A.T.; Estrada-Bernal, A.; Keysar, S.; Jimeno, A.; Varella-Garcia, M.; Aisner, D.L.; Li, Y.; Stephens, P.J.; Morosini, D.; Tuch, B.B.; Fernandes, M.; Nanda, N.; Low, J.A. An oncogenic NTRK fusion in a patient with soft-tissue Sarcoma with response to the Tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. 2015, 5, 1049–1057. [Google Scholar] [CrossRef]
- O'Reilly, E.M.; Hechtman, J.F. Tumour response to TRK inhibition in a patient with pancreatic adenocarcinoma harbouring an NTRK gene fusion. Ann. Oncol. 2019, 30, viii36–viii40. [Google Scholar] [CrossRef]
- Liu, F.; Wei, Y.; Zhang, H.; Jiang, J.; Zhang, P.; Chu, Q. NTRK fusion in Non-Small Cell Lung Cancer: diagnosis, therapy, and TRK inhibitor resistance. Front. Oncol. 2022, 12, 864666. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; Larsen, P.D.; Bouhana, K.S.; Winski, S.L.; Hamor, R.; Wu, W.I.; Parker, A.; Morales, T.H.; Sullivan, F.X.; DeWolf, W.E.; Wollenberg, L.A.; Gordon, P.R.; Douglas-Lindsay, D.N.; Scaltriti, M.; Benayed, R.; Raj, S.; Hanusch, B.; Schram, A.M.; Jonsson, P.; Berger, M.F.; Hechtman, J.F.; Taylor, B.S.; Andrews, S.; Rothenberg, S.M.; Hyman, D.M. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef]
- Yuekun, W.; Piaopiao, L.; Yu, W.; Wenbin, M. NTRK fusions and TRK inhibitors: potential targeted therapies for adult glioblastoma. Front. Oncol. 2020, 10, 593578. [Google Scholar]
- Krishnan, A.; Berthelet, J.; Renaud, E.; Rosigkeit, S.; Distler, U.; Stawiski, E.; Wang, J.; Modrusan, Z.; Fiedler, M.; Bienz, M.; Tenzer, S.; Schad, A.; Roth, W.; Thiede, B.; Seshagiri, S.; Musholt, T.J.; Rajalingam, K. Proteogenomics analysis unveils a TFG-RET gene fusion and druggable targets in papillary thyroid carcinomas. Nat. Commun. 2020, 11, 2056. [Google Scholar] [CrossRef]
- Li, A.Y.; McCusker, M.G.; Russo, A.; Scilla, K.A.; Gittens, A.; Arensmeyer, K.; Mehra, R.; Adamo, V.; Rolfo, C. RET fusions in solid tumors. Cancer Treat. Rev. 2019, 81, 101911. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Ling, F.; Zhang, J.; Xiao, M.; Mao, W. A novel intergenic LSM14A-RET fusion variant in a patient with lung adenocarcinoma. J. Thorac. Oncol. 2020, 15, e52–e53. [Google Scholar] [CrossRef]
- Vodopivec, D.M.; Hu, M.I. RET kinase inhibitors for RET-altered thyroid cancers. Ther. Adv. Med. Oncol. 2022, 14, 17588359221101691. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, T.; Song, X.; Liu, B.; Wei, J. Gene fusion neoantigens: Emerging targets for cancer immunotherapy. Cancer Lett. 2021, 506, 45–54. [Google Scholar] [CrossRef]
- Takamori, S.; Matsubara, T.; Haratake, N.; Toyokawa, G.; Fujishita, T.; Toyozawa, R.; Ito, K.; Yamaguchi, M.; Taguchi, K.; Okamoto, T.; Seto, T. Targeted therapy for RET fusion lung cancer: breakthrough and unresolved issue. Front. Oncol. 2021, 11, 704084. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kodama, K.; Takase, K.; Sugi, N.H.; Yamamoto, Y.; Iwata, M.; Tsuruoka, A. Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion-driven tumor models. Cancer Lett. 2013, 340, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shen, T.; Mooers, B.H.M.; Hilberg, F.; Wu, J. Drug resistance profiles of mutations in the RET kinase domain. Br. J. Pharmacol. 2018, 175, 3504–3515. [Google Scholar] [CrossRef]
- M. , D.L.; Estelamari, R.; Richa, D.; V., I.C. Therapeutic strategies in RET gene rearranged non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Terzyan, S.S.; Shen, T.; Liu, X.; Huang, Q.; Teng, P.; Zhou, M.; Hilberg, F.; Cai, J.; Mooers, B.H.M.; Wu, J. Structural basis of resistance of mutant RET protein-tyrosine kinase to its inhibitors nintedanib and vandetanib. J. Biol. Chem. 2019, 294, 10428–10437. [Google Scholar] [CrossRef]
- Mologni, L.; Redaelli, S.; Morandi, A.; Plaza-Menacho, I.; Gambacorti-Passerini, C. Ponatinib is a potent inhibitor of wild-type and drug-resistant gatekeeper mutant RET kinase. Mol. Cell Endocrinol. 2013, 377, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Thein, K.Z.; Velcheti, V.; Mooers, B.H.M.; Wu, J.; Subbiah, V. Precision therapy for RET-altered cancers with RET inhibitors. Trends Cancer 2021, 7, 1074–1088. [Google Scholar] [CrossRef]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; Peled, N.; Weiss, J.; Kim, Y.J.; Ohe, Y.; Nishio, M.; Park, K.; Patel, J.; Seto, T.; Sakamoto, T.; Rosen, E.; Shah, M.H.; Barlesi, F.; Cassier, P.A.; Bazhenova, L.; De Braud, F.; Garralda, E.; Velcheti, V.; Satouchi, M.; Ohashi, K.; Pennell, N.A.; Reckamp, K.L.; Dy, G.K.; Wolf, J.; Solomon, B.; Falchook, G.; Ebata, K.; Nguyen, M.; Nair, B.; Zhu, E.Y.; Yang, L.; Huang, X.; Olek, E.; Rothenberg, S.M.; Goto, K.; Subbiah, V. Efficacy of Selpercatinib in RET fusion-positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef]
- Bradford, D.; Larkins, E.; Mushti, S.L.; Rodriguez, L.; Skinner, A.M.; Helms, W.S.; Price, L.S.L.; Zirkelbach, J.F.; Li, Y.; Liu, J.; Charlab, R.; Turcu, F.R.; Liang, D.; Ghosh, S.; Roscoe, D.; Philip, R.; Zack-Taylor, A.; Tang, S.; Kluetz, P.G.; Beaver, J.A.; Pazdur, R.; Theoret, M.R.; Singh, H. FDA approval summary: Selpercatinib for the treatment of lung and thyroid cancers with RET gene mutations or fusions. Clin. Cancer Res. 2021, 27, 2130–2135. [Google Scholar] [CrossRef]
- Anthony, M. Selpercatinib: First Approval. Drugs 2020, 80, 1119–1124. [Google Scholar]
- Loong, H.; Goto, K.; Park, K.; Ohe, Y.; Nishio, M.; Cho, B.C.; Kim, Y.J.; French, P.; Soldatenkova, V.; Tan, D. FP14.10 efficacy and safety of Selpercatinib (LOXO-292) in East Asian patients with RET fusion-positive NSCLC. J. Thorac. Oncol. 2021, 16, S231–S232. [Google Scholar] [CrossRef]
- Wirth, L.J.; Sherman, E.J.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.H.; Worden, F.; Brose, M.S.; Leboulleux, S.; Godbert, Y.; Meurer, M.; Morris, J.; Owonikoko, T.K.; Shao-Weng Tan, D.; Gautschi, O.; Patel, J.; Yang, L.; Kherani, J.; Cabanillas, M.E.; Shah, M.H. O10-3 Selpercatinib (LOXO-292) in patients (pts) with RET-altered thyroid cancer. Ann. Oncol. 2021, 32, S289. [Google Scholar] [CrossRef]
- Ali, F.; Neha, K.; Chauhan, G. Pralsetinib: chemical and therapeutic development with FDA authorization for the management of RET fusion-positive non-small-cell lung cancers. Arch. Pharm. Res. 2022, 45, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Pralsetinib: A Review in Advanced RET Fusion-Positive NSCLC. Drugs 2022, 82, 811–816. [Google Scholar] [CrossRef]
- Subbiah, V.; Cassier, P.A.; Siena, S.; Garralda, E.; Paz-Ares, L.; Garrido, P.; Nadal, E.; Vuky, J.; Lopes, G.; Kalemkerian, G.P.; Bowles, D.W.; Seetharam, M.; Chang, J.; Zhang, H.; Green, J.; Zalutskaya, A.; Schuler, M.; Fan, Y.; Curigliano, G. Pan-cancer efficacy of pralsetinib in patients with RET fusion-positive solid tumors from the phase 1/2 ARROW trial. Nat. Med. 2022, 28, 1640–1645. [Google Scholar] [CrossRef]
- Kim, J.; Bradford, D.; Larkins, E.; Pai-Scherf, L.H.; Chatterjee, S.; Mishra-Kalyani, P.S.; Wearne, E.; Helms, W.S.; Ayyoub, A.; Bi, Y.; Sun, J.; Charlab, R.; Liu, J.; Zhao, H.; Liang, D.; Ghosh, S.; Philip, R.; Pazdur, R.; Theoret, M.R.; Beaver, J.A.; Singh, H. FDA approval summary: Pralsetinib for the treatment of lung and thyroid cancers with RET gene mutations or fusions. Clin. Cancer Res. 2021, 27, 5452–5456. [Google Scholar] [CrossRef]
- Shabbir, A.; Kojadinovic, A.; Shafiq, T.; Mundi, P.S. Targeting RET alterations in cancer: recent progress and future directions. Crit. Rev. Oncol. Hematol. 2023, 181, 103882. [Google Scholar] [CrossRef]
- Drusbosky, L.M.; Rodriguez, E.; Dawar, R.; Ikpeazu, C.V. Therapeutic strategies in RET gene rearranged non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 50. [Google Scholar] [CrossRef]
- Di Federico, A.; Filetti, M.; Palladini, A.; Giusti, R.; Piras, M.; De Giglio, A.; Ardizzoni, A.; Gelsomino, F. EGFR-RAD51 gene fusion NSCLC responsiveness to different generation EGFR-TKIs: two cases and review of the literature. Transl. Lung Cancer Res. 2022, 11, 497–503. [Google Scholar] [CrossRef]
- Komuro, A.; Raja, E.; Iwata, C.; Soda, M.; Isogaya, K.; Yuki, K.; Ino, Y.; Morikawa, M.; Todo, T.; Aburatani, H.; Suzuki, H.; Ranjit, M.; Natsume, A.; Mukasa, A.; Saito, N.; Okada, H.; Mano, H.; Miyazono, K.; Koinuma, D. Identification of a novel fusion gene HMGA2-EGFR in glioblastoma. Int. J. Cancer 2018, 142, 1627–1639. [Google Scholar] [CrossRef]
- Wang, X.; Huang, L.; Cai, J.; Liu, A. A novel KIF5B-EGFR fusion variant in Non-Small-Cell Lung Cancer and response to Afatinib: A case report. Onco. Targets Ther. 2021, 14, 3739–3744. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, H.B.; Chen, X.; Yang, X.; Ye, Y.; Bekaii-Saab, T.; Zheng, Y.; Zhang, Y. A Rare EGFR-SEPT14 Fusion in a Patient with Colorectal Adenocarcinoma Responding to Erlotinib. Oncologist 2020, 25, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Kartik, K.; Jean-Nicolas, G.; Kwang, C.Y.; J, G.F.; J, G.B.; Kyle, G.; Eiki, I.; K, O.T.; Vijay, P.; S, R.S.; K, R.S.; Beth, E.-S.; Tiziana, V.; Andrew, W.; Heidi, C.; Yingjun, Y.; H, S.J.; Jens, M.; Deborah, M.; S, R.J.; J, S.P.; A, M.V.; M, A.S.; M, L.C. EGFR fusions as novel therapeutic targets in lung cancer. Cancer Discov. 2016, 6, 601–611. [Google Scholar]
- Paik, P.K. Something old, Something new, Something borrowed, Something fused: novel EGFR rearrangements in lung adenocarcinomas. Cancer Discov. 2016, 6, 574–575. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Lazertinib: First Approval. Drugs 2021, 81, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Shao, C. KIF5B-EGFR fusion: a novel EGFR mutation in lung adenocarcinoma. Onco. Targets Ther. 2020, 13, 8317–8321. [Google Scholar] [CrossRef] [PubMed]
- Zhu, V.W.; Klempner, S.J.; Ou, S.I. Receptor Tyrosine Kinase Fusions as an Actionable Resistance Mechanism to EGFR TKIs in EGFR-Mutant Non-Small-Cell Lung Cancer. Trends Cancer 2019, 5, 677–692. [Google Scholar] [CrossRef]
- Xia, W.; Weiwei, P.; Zhimin, Z.; Jing, C.; Anwen, L. Emerging a novel VOPP1-EGFR fusion coexistent with T790M as an acquired resistance mechanism to prior Icotinib and sensitive to osimertinib in a patient With EGFR L858R lung adenocarcinoma: a case report. Front. Oncol. 2021, 11, 720819. [Google Scholar]
- Heppner, D.E.; Wittlinger, F.; Beyett, T.S.; Shaurova, T.; Urul, D.A.; Buckley, B.; Pham, C.D.; Schaeffner, I.K.; Yang, B.; Ogboo, B.C.; May, E.W.; Schaefer, E.M.; Eck, M.J.; Laufer, S.A.; Hershberger, P.A. Structural basis for inhibition of mutant EGFR with Lazertinib (YH25448). ACS Med. Chem. Lett. 2022, 13, 1856–1863. [Google Scholar] [CrossRef]
- Jones, D.T.; Kocialkowski, S.; Liu, L.; Pearson, D.M.; Bäcklund, L.M.; Ichimura, K.; Collins, V.P. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008, 68, 8673–8677. [Google Scholar] [CrossRef]
- Dahiya, S.; Yu, J.; Kaul, A.; Leonard, J.R.; Gutmann, D.H. Novel BRAF alteration in a Sporadic Pilocytic Astrocytoma. Case Rep. Med. 2012, 2012, 418672. [Google Scholar] [CrossRef]
- Cin, H.; Meyer, C.; Herr, R.; Janzarik, W.G.; Lambert, S.; Jones, D.T.; Jacob, K.; Benner, A.; Witt, H.; Remke, M.; Bender, S.; Falkenstein, F.; Van Anh, T.N.; Olbrich, H.; von Deimling, A.; Pekrun, A.; Kulozik, A.E.; Gnekow, A.; Scheurlen, W.; Witt, O.; Omran, H.; Jabado, N.; Collins, V.P.; Brummer, T.; Marschalek, R.; Lichter, P.; Korshunov, A.; Pfister, S.M. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Helgager, J.; Lidov, H.G.; Mahadevan, N.R.; Kieran, M.W.; Ligon, K.L.; Alexandrescu, S. A novel GIT2-BRAF fusion in pilocytic astrocytoma. Diagn. Pathol. 2017, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Tomić, T.T.; Olausson, J.; Wilzén, A.; Sabel, M.; Truvé, K.; Sjögren, H.; Dósa, S.; Tisell, M.; Lannering, B.; Enlund, F.; Martinsson, T.; Åman, P.; Abel, F. A new GTF2I-BRAF fusion mediating MAPK pathway activation in pilocytic astrocytoma. PLoS One 2017, 12, e0175638. [Google Scholar] [CrossRef]
- He, H.; Li, W.; Yan, P.; Bundschuh, R.; Killian, J.A.; Labanowska, J.; Brock, P.; Shen, R.; Heerema, N.A.; de la Chapelle, A. Identification of a recurrent LMO7-BRAF fusion in Papillary Thyroid Carcinoma. Thyroid 2018, 28, 748–754. [Google Scholar] [CrossRef]
- Penman, C.L.; Faulkner, C.; Lowis, S.P.; Kurian, K.M. Current understanding of BRAF alterations in diagnosis, prognosis, and therapeutic targeting in pediatric low-grade gliomas. Front. Oncol. 2015, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Westin, S.N.; Wang, K.; Araujo, D.; Wang, W.L.; Miller, V.A.; Ross, J.S.; Stephens, P.J.; Palmer, G.A.; Ali, S.M. Targeted therapy by combined inhibition of the RAF and mTOR kinases in malignant spindle cell neoplasm harboring the KIAA1549-BRAF fusion protein. J. Hematol. Oncol. 2014, 7, 8. [Google Scholar] [CrossRef]
- Kim, H.S.; Jung, M.; Kang, H.N.; Kim, H.; Park, C.W.; Kim, S.M.; Shin, S.J.; Kim, S.H.; Kim, S.G.; Kim, E.K.; Yun, M.R.; Zheng, Z.; Chung, K.Y.; Greenbowe, J.; Ali, S.M.; Kim, T.M.; Cho, B.C. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 2017, 36, 3334–3345. [Google Scholar] [CrossRef]
- Kulkarni, A.; Al-Hraishawi, H.; Simhadri, S.; Hirshfield, K.M.; Chen, S.; Pine, S.; Jeyamohan, C.; Sokol, L.; Ali, S.; Teo, M.L.; White, E.; Rodriguez-Rodriguez, L.; Mehnert, J.M.; Ganesan, S. BRAF fusion as a novel mechanism of acquired resistance to Vemurafenib in BRAFV600E mutant melanoma. Clin. Cancer Res. 2017, 23, 5631–5638. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; Kudchadkar, R.; Burris, H.A., 3rd; Falchook, G.; Algazi, A.; Lewis, K.; Long, G.V.; Puzanov, I.; Lebowitz, P.; Singh, A.; Little, S.; Sun, P.; Allred, A.; Ouellet, D.; Kim, K.B.; Patel, K.; Weber, J. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Grimwade, D.; Lo Coco, F. Acute promyelocytic leukemia: a model for the role of molecular diagnosis and residual disease monitoring in directing treatment approach in acute myeloid leukemia. Leukemia 2002, 16, 1959–1973. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Stewart, D.; Koeffler, H.P. Differentiation therapy of leukemia: 3 decades of development. Blood 2009, 113, 3655–3665. [Google Scholar] [CrossRef]
- Sirulnik, A.; Melnick, A.; Zelent, A.; Licht, J.D. Molecular pathogenesis of acute promyelocytic leukaemia and APL variants. Best Pract. Res. Clin. Haematol. 2003, 16, 387–408. [Google Scholar] [CrossRef] [PubMed]
- De Braekeleer, E.; Douet-Guilbert, N.; De Braekeleer, M. RARA fusion genes in acute promyelocytic leukemia: a review. Expert Rev. Hematol. 2014, 7, 347–357. [Google Scholar] [CrossRef]
- Lu, J.; Chew, E.H.; Holmgren, A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 12288–12293. [Google Scholar] [CrossRef]
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhoa, L.; Gu, L.J.; Wang, Z.Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Chomienne, C.; Degos, L. Treatment of acute promyelocytic leukaemia. Best Pract. Res. Clin. Haematol. 2001, 14, 153–174. [Google Scholar] [CrossRef]
- Marasca, R.; Zucchini, P.; Galimberti, S.; Leonardi, G.; Vaccari, P.; Donelli, A.; Luppi, M.; Petrini, M.; Torelli, G. Missense mutations in the PML/RARalpha ligand binding domain in ATRA-resistant As(2)O(3) sensitive relapsed acute promyelocytic leukemia. Haematologica 1999, 84, 963–968. [Google Scholar]
- Lengfelder, E.; Hofmann, W.K.; Nowak, D. Impact of arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia 2012, 26, 433–442. [Google Scholar] [CrossRef]
- Strehl, S.; König, M.; Boztug, H.; Cooper, B.W.; Suzukawa, K.; Zhang, S.J.; Chen, H.Y.; Attarbaschi, A.; Dworzak, M.N. All-trans retinoic acid and arsenic trioxide resistance of acute promyelocytic leukemia with the variant STAT5B-RARA fusion gene. Leukemia 2013, 27, 1606–1610. [Google Scholar] [CrossRef]
- Mistry, A.R.; Pedersen, E.W.; Solomon, E.; Grimwade, D. The molecular pathogenesis of acute promyelocytic leukaemia: implications for the clinical management of the disease. Blood Rev. 2003, 17, 71–97. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription factor C/EBP homologous protein in health and diseases. Front. Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef] [PubMed]
- Zullow, H.J.; Sankar, A.; Ingram, D.R.; Same Guerra, D.D.; D'Avino, A.R.; Collings, C.K.; Lazcano, R.; Wang, W.L.; Liang, Y.; Qi, J.; Lazar, A.J.; Kadoch, C. The FUS::DDIT3 fusion oncoprotein inhibits BAF complex targeting and activity in myxoid liposarcoma. Mol. Cell 2022, 82, 1737–1750. [Google Scholar] [CrossRef] [PubMed]
- Dolatabadi, S.; Jonasson, E.; Andersson, L.; Luna Santamaria, M.; Linden, M.; Osterlund, T.; Aman, P.; Stahlberg, A. FUS-DDIT3 fusion oncoprotein expression affects JAK-STAT signaling in Myxoid Liposarcoma. Front. Oncol. 2022, 12, 816894. [Google Scholar] [CrossRef] [PubMed]
- Marcel, T.; Jasmin, M.; Christian, B.; Magdalene, C.; Ilka, I.; Konrad, S.; Sandra, E.; Inga, G.; Bianca, A.; Claudia, R.; Stefan, F.; Susanne, H.; Thomas, S.; Pierre, Å.; Eva, W.; Sebastian, H.; Wolfgang, H. FUS-DDIT3 fusion protein-driven IGF-IR signaling is a therapeutic target in myxoid liposarcoma. Clin. Cancer Res. 2017, 23, 6227–6238. [Google Scholar]
- Svec, D.; Dolatabadi, S.; Thomsen, C.; Cordes, N.; Shannon, M.; Fitzpatrick, P.; Landberg, G.; Aman, P.; Stahlberg, A. Identification of inhibitors regulating cell proliferation and FUS-DDIT3 expression in myxoid liposarcoma using combined DNA, mRNA, and protein analyses. Lab. Invest. 2018, 98, 957–967. [Google Scholar] [CrossRef]
- Schenkl, C.; Schrepper, A.; Heyne, E.; Doenst, T.; Schwarzer, M. The IGF-1R inhibitor NVP-AEW541 causes insulin-independent and reversible cardiac contractile dysfunction. Biomedicines 2022, 10, 2022. [Google Scholar] [CrossRef]
- Gasi Tandefelt, D.; Boormans, J.; Hermans, K.; Trapman, J. ETS fusion genes in prostate cancer. Endocr. Relat. Cancer 2014, 21, R143–R152. [Google Scholar] [CrossRef]
- Feng, F.Y.; Brenner, J.C.; Hussain, M.; Chinnaiyan, A.M. Molecular pathways: targeting ETS gene fusions in cancer. Clin. Cancer Res. 2014, 20, 4442–4448. [Google Scholar] [CrossRef]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef]
- L. , S.J.; E., R.R.; Yi-Cheng, W.; R., L.A.; A., A.T.; S., G.A.; Richard, A.; E., K.A.; J., H.B.; Xiaotu, M.; I., S.T.; Soheil, M. ETS family transcription factor fusions in childhood AML: distinct expression networks and clinical implications. Blood 2021, 138, 2356. [Google Scholar] [CrossRef]
- Matjasic, A.; Zupan, A.; Bostjancic, E.; Pizem, J.; Popovic, M.; Kolenc, D. A novel PTPRZ1-ETV1 fusion in gliomas. Brain Pathol. 2020, 30, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Choudhary, G.; Heston, W.D.; Klein, E.A.; Almasan, A. Abstract B27: The PARP inhibitor rucaparib radiosensitizes prostate cancer cells, most effectively those that are PTEN-deficient and are expressing ETS gene fusion proteins, which inhibit NHEJ DNA repair. Cancer Res. 2012, 72. [Google Scholar] [CrossRef]
- Qian, C.; Li, D.; Chen, Y. ETS factors in prostate cancer. Cancer Lett. 2022, 530, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Katayoun, J.; Eric, M.; Lin, C.E.Y.; Lauren, O.; Chiara, T.; Eugenio, G.; James, B.; James, F.; Jeffrey, T.; Francesco, B.; J, L.B. TK216, a Novel, small molecule inhibitor of the ETS-Family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood 2016, 128, 4035. [Google Scholar]
- Ateeq, B.; Vellaichamy, A.; Tomlins, S.A.; Wang, R.; Cao, Q.; Lonigro, R.J.; Pienta, K.J.; Varambally, S. Role of dutasteride in pre-clinical ETS fusion-positive prostate cancer models. Prostate 2012, 72, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.J.; Wilcox, D.; Lin, X.; Hessler, P.; Torrent, M.; He, W.; Uziel, T.; Albert, D.H.; McDaniel, K.; Kati, W.; Shen, Y. Exploitation of Castration-resistant Prostate Cancer transcription factor dependencies by the novel BET inhibitor ABBV-075. Mol. Cancer Res. 2017, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Nataraj, N.B.; Sekar, A.; Ghosh, S.; Bornstein, C.; Drago-Garcia, D.; Roth, L.; Romaniello, D.; Marrocco, I.; David, E.; Gilad, Y.; Lauriola, M.; Rotkopf, R.; Kimchi, A.; Haga, Y.; Tsutsumi, Y.; Mirabeau, O.; Surdez, D.; Zinovyev, A.; Delattre, O.; Kovar, H.; Amit, I.; Yarden, Y. ETS proteins bind with glucocorticoid receptors: relevance for treatment of ewing sarcoma. Cell Rep. 2019, 29, 104–117 e104. [Google Scholar] [CrossRef]
- Humtsoe, J.O.; Kim, H.S.; Jones, L.; Cevallos, J.; Boileau, P.; Kuo, F.; Morris, L.G.T.; Ha, P. Development and characterization of MYB-NFIB fusion expression in adenoid cystic carcinoma. Cancers (Basel) 2022, 14, 2263. [Google Scholar] [CrossRef]
- Wagner, V.P.; Bingle, C.D.; Bingle, L. MYB-NFIB fusion transcript in adenoid cystic carcinoma: Current state of knowledge and future directions. Crit. Rev. Oncol. Hematol. 2022, 176, 103745. [Google Scholar] [CrossRef]
- Andersson, M.K.; Aman, P.; Stenman, G. IGF2/IGF1R signaling as a therapeutic target in MYB-positive adenoid cystic carcinomas and other fusion gene-driven tumors. Cells 2019, 8, 913. [Google Scholar] [CrossRef]
- Shirazi, P.T.; Eadie, L.N.; Heatley, S.L.; Page, E.C.; Francois, M.; Hughes, T.P.; Yeung, D.; White, D.L. Exploring the oncogenic and therapeutic target potential of the MYB-TYK2 fusion gene in B-cell acute lymphoblastic leukemia. Cancer Gene Ther. 2022, 29, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.K.; Mangiapane, G.; Nevado, P.T.; Tsakaneli, A.; Carlsson, T.; Corda, G.; Nieddu, V.; Abrahamian, C.; Chayka, O.; Rai, L.; Wick, M.; Kedaigle, A.; Stenman, G.; Sala, A. ATR is a MYB regulated gene and potential therapeutic target in adenoid cystic carcinoma. Oncogenesis 2020, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Hidemasa, M.; Kenichi, Y.; Kana, N.; Yutarou, H.; Moe, H.; Yuri, I.; Yasuhiko, K.; Yusuke, S.; Yuichi, S.; Kenichi, C.; Hiroko, T.; Ai, O.; Yasuhito, N.; June, T.; Hiroo, U.; Nobutaka, K.; Daisuke, T.; Takashi, T.; Akio, T.; Satoru, M.; Manja, M.; Claudia, H.; Seishi, O.; Souichi, A. KRAS mutations frequently coexist with high-risk MLL fusions and are independent adverse prognostic factors in MLL-rearranged acute Myeloid Leukemia. Blood 2020, 136, 28–29. [Google Scholar]
- Huang, W.; Fang, K.; Chen, T.Q.; Zeng, Z.C.; Sun, Y.M.; Han, C.; Sun, L.Y.; Chen, Z.H.; Yang, Q.Q.; Pan, Q.; Luo, X.Q.; Wang, W.T.; Chen, Y.Q. CircRNA circAF4 functions as an oncogene to regulate MLL-AF4 fusion protein expression and inhibit MLL leukemia progression. J. Hematol. Oncol. 2019, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Slany, R.K. MLL fusion proteins and transcriptional control. Biochim. Biophys. Acta, Gene Regul. Mech. 2020, 1863, 194503. [Google Scholar] [CrossRef]
- Takahashi, S.; Yokoyama, A. The molecular functions of common and atypical MLL fusion protein complexes. Biochim. Biophys. Acta, Gene Regul. Mech. 2020, 1863, 194548. [Google Scholar] [CrossRef]
- Marschalek, R. The reciprocal world of MLL fusions: A personal view. Biochim. Biophys. Acta, Gene Regul. Mech. 2020, 1863, 194547. [Google Scholar] [CrossRef]
- Cantilena, S.; Gasparoli, L.; Pal, D.; Heidenreich, O.; Klusmann, J.H.; Martens, J.H.A.; Faille, A.; Warren, A.J.; Karsa, M.; Pandher, R.; Somers, K.; Williams, O.; de Boer, J. Direct targeted therapy for MLL-fusion-driven high-risk acute leukaemias. Clin. Transl. Med. 2022, 12, e933. [Google Scholar] [CrossRef]
- Yokoyama, A. Transcriptional activation by MLL fusion proteins in leukemogenesis. Exp. Hematol. 2017, 46, 21–30. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; Waters, N.J.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; Richon, V.M.; Pollock, R.M. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.M.; So, C.W.E. Novel therapeutic strategies for MLL-rearranged leukemias. Biochim. Biophys. Acta, Gene Regul. Mech. 2020, 1863, 194584. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Li, D.D.; Chen, X.; Wang, Y.Z.; Xu, J.J.; Jiang, Z.Y.; You, Q.D.; Guo, X.K. Proton pump inhibitors selectively suppress MLL rearranged leukemia cells via disrupting MLL1-WDR5 protein-protein interaction. Eur. J. Med. Chem. 2020, 188, 112027. [Google Scholar] [CrossRef] [PubMed]
- Kessler, L.; Wu, T.; Grembecka, J.; Cierpicki, T.; Purohit, T.; Miao, H.; Kempinska, K.; Ely, T.; Hensen, R.; Li, S.; Patricelli, M.; Li, S.; Kucharski, J.; Zhang, J.; Yao, Y.; Yu, K.; Wang, Y.; Li, L.; Ren, P.; Liu, Y. Discovery of novel menin-MLL small molecule inhibitors that display high potency and selectivity in vitro and in vivo. Eur. J. Cancer 2016, 69, S88. [Google Scholar] [CrossRef]
- Jenkins, T.W.; Downey-Kopyscinski, S.L.; Fields, J.L.; Rahme, G.J.; Colley, W.C.; Israel, M.A.; Maksimenko, A.V.; Fiering, S.N.; Kisselev, A.F. Activity of immunoproteasome inhibitor ONX-0914 in acute lymphoblastic leukemia expressing MLL-AF4 fusion protein. Sci. Rep. 2021, 11, 10883. [Google Scholar] [CrossRef]
- Sandra, C.; Nicholas, G.; Owen, W.; Jasper, d.B. Drug induced MLL fusion degradation via Hsp90 inhibition. Blood 2015, 126, 4850. [Google Scholar]
- Wachtel, M.; Schafer, B.W. PAX3-FOXO1: zooming in on an "undruggable" target. Semin. Cancer Biol. 2018, 50, 115–123. [Google Scholar] [CrossRef]
- Raze, T.; Lapouble, E.; Lacour, B.; Guissou, S.; Defachelles, A.S.; Gaspar, N.; Delattre, O.; Pierron, G.; Desandes, E. PAX-FOXO1 fusion status in children and adolescents with alveolar rhabdomyosarcoma: Impact on clinical, pathological, and survival features. Pediatr. Blood Cancer 2023, 70, e30228. [Google Scholar] [CrossRef]
- Walters, Z.S.; Villarejo-Balcells, B.; Olmos, D.; Buist, T.W.; Missiaglia, E.; Allen, R.; Al-Lazikani, B.; Garrett, M.D.; Blagg, J.; Shipley, J. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene 2014, 33, 1148–1157. [Google Scholar] [CrossRef]
- Bohm, M.; Wachtel, M.; Marques, J.G.; Streiff, N.; Laubscher, D.; Nanni, P.; Mamchaoui, K.; Santoro, R.; Schafer, B.W. Helicase CHD4 is an epigenetic coregulator of PAX3-FOXO1 in alveolar rhabdomyosarcoma. J. Clin. Invest. 2016, 126, 4237–4249. [Google Scholar] [CrossRef]
- Anderson, W.J.; Fletcher, C.D.M.; Hornick, J.L. Loss of expression of YAP1 C-terminus as an ancillary marker for epithelioid hemangioendothelioma variant with YAP1-TFE3 fusion and other YAP1-related vascular neoplasms. Mod. Pathol. 2021, 34, 2036–2042. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Holland, E.C.; Vasioukhin, V. YAP1 and its fusion proteins in cancer initiation, progression and therapeutic resistance. Dev. Biol. 2021, 475, 205–221. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Okonechnikov, K.; Vouri, M.; Brabetz, S.; Jones, D.T.W.; Korshunov, A.; Capper, D.; Chavez, L.; Pfister, S.M.; Kool, M.; Kawauchi, D. EPEN-24. YAP1 fusion proteins mediate oncogenic activity in ependymoma via interaction with tead transcription factors. Neuro-Oncology 2018, 20, i78. [Google Scholar] [CrossRef]
- Sekine, S.; Kiyono, T.; Ryo, E.; Ogawa, R.; Wakai, S.; Ichikawa, H.; Suzuki, K.; Arai, S.; Tsuta, K.; Ishida, M.; Sasajima, Y.; Goshima, N.; Yamazaki, N.; Mori, T. Recurrent YAP1-MAML2 and YAP1-NUTM1 fusions in poroma and porocarcinoma. J. Clin. Invest. 2019, 129, 3827–3832. [Google Scholar] [CrossRef]
- Frank, S.; Pia, H.; Hua-Jun, W.; J, C.P.; Franziska, M.; Patrick, P.; Valeri, V.; Eric, H. GENE-04. The oncogenic functions of yap1-gene fusions can be inhibited by disruption of yap1-tead interaction. Neuro-Oncology 2019, 21, vi98. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; Assempour, N.; Iynkkaran, I.; Liu, Y.; Maciejewski, A.; Gale, N.; Wilson, A.; Chin, L.; Cummings, R.; Le, D.; Pon, A.; Knox, C.; Wilson, M. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic. Acids. Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Wu, W.; Haderk, F.; Bivona, T.G. Non-canonical thinking for targeting ALK-Fusion onco-proteins in lung cancer. Cancers (Basel) 2017, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Cai, M.; Shao, L.; Zhang, J. Targeting protein kinases degradation by PROTACs. Front. Chem. 2021, 9, 679120. [Google Scholar] [CrossRef]
- Zhou, X.; Dong, R.; Zhang, J.Y.; Zheng, X.; Sun, L.P. PROTAC: A promising technology for cancer treatment. Eur. J. Med. Chem. 2020, 203, 112539. [Google Scholar] [CrossRef]
- Liu, J.P. Novel strategies for molecular targeting to cancer. Clin. Exp. Pharmacol. Physiol. 2016, 43, 287–289. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Q.; Xu, J.; Tang, L.; Zhang, Y.; Du, F.; Zhao, Y.; Wu, X.; Li, M.; Shen, J.; Ding, R.; Cao, H.; Li, W.; Li, X.; Chen, M.; Wu, Z.; Cho, C.H.; Du, Y.; Wen, Q.; Xiao, Z. PROTACs in gastrointestinal cancers. Mol. Ther. Oncolytics 2022, 27, 204–223. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Du, Y.; Huang, L.; Cui, J.; Niu, J.; Xu, Y.; Zhu, Q. Discovery of novel potent covalent inhibitor-based EGFR degrader with excellent in vivo efficacy. Bioorg. Chem. 2022, 120, 105605. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ma, J.; Liu, Y.; Xia, J.; Li, Y.; Wang, Z.P.; Wei, W. PROTACs: A novel strategy for cancer therapy. Semin. Cancer Biol. 2020, 67, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pu, W.; Zheng, Q.; Ai, M.; Chen, S.; Peng, Y. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol. Cancer 2022, 21, 99. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Xu, R.; Yan, B.; Li, F.; Liu, H. Elevation of tumor mutation burden in ROS1-fusion lung adenocarcinoma resistant to crizotinib: A case report. Medicine 2018, 97, e13797. [Google Scholar] [CrossRef]
- Hattori, T.; Maso, L.; Araki, K.Y.; Koide, A.; Hayman, J.; Akkapeddi, P.; Bang, I.; Neel, B.G.; Koide, S. Creating MHC-restricted neoantigens with covalent inhibitors that can be targeted by immune therapy. Cancer Discov. 2023, 13, 132–145. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, E.; Bai, L.; Li, Y. Autophagy in cancer immunotherapy. Cells 2022, 11, 2996. [Google Scholar] [CrossRef]
Figure 1.
The data in the bar chart show the percentage of gene fusions by cancer types (A) and gene fusion types (B). The data comes from the TCGA Fusion Gene Database[25]. BRCA (Breast invasive carcinoma); PRAD (Prostate adenocarcinoma); SARC (Sarcoma); BLCA (Bladder Urothelial Carcinoma); LUAD (Lung adenocarcinoma); LUSC (Lung squamous cell carcinoma); SKCM (Skin Cutaneous Melanoma); LIHC (Liver hepatocellular carcinoma); LGG (Brain Lower Grade Glioma); UCEC (Uterine Corpus Endometrial Carcinoma); STAD (Stomach adenocarcinoma); HNSC (Head and Neck squamous cell carcinoma); CESC (Cervical squamous cell carcinoma and endocervical adenocarcinoma); OV (Ovarian serous cystadenocarcinoma); UCS (Uterine Carcinosarcoma); ESCA (Esophageal carcinoma); COAD (Colon adenocarcinoma); KIRC (Kidney renal clear cell carcinoma); GBM (Glioblastoma multiforme); ACC (Adrenocortical carcinoma);THCA (Thyroid carcinoma); KIRP (Kidney renalpapillary cell carcinoma); PAAD (Pancreatic adenocarcinoma); MESO (Mesothelioma); READ (Rectum adenocarcinoma); PCPG (Pheochromocytoma and Paraganglioma); AML (Acute Myeloid Leukemia); TGCT (Testicular Germ Cell Tumors); CHOL (Cholangiocarcinoma); THYM (Thymoma); KICH (Kidney Chromophobe); DLBC (Lymphoid Neoplasm Diffuse Large B-cell Lymphoma); UVM (Uveal Melanoma).
Figure 1.
The data in the bar chart show the percentage of gene fusions by cancer types (A) and gene fusion types (B). The data comes from the TCGA Fusion Gene Database[25]. BRCA (Breast invasive carcinoma); PRAD (Prostate adenocarcinoma); SARC (Sarcoma); BLCA (Bladder Urothelial Carcinoma); LUAD (Lung adenocarcinoma); LUSC (Lung squamous cell carcinoma); SKCM (Skin Cutaneous Melanoma); LIHC (Liver hepatocellular carcinoma); LGG (Brain Lower Grade Glioma); UCEC (Uterine Corpus Endometrial Carcinoma); STAD (Stomach adenocarcinoma); HNSC (Head and Neck squamous cell carcinoma); CESC (Cervical squamous cell carcinoma and endocervical adenocarcinoma); OV (Ovarian serous cystadenocarcinoma); UCS (Uterine Carcinosarcoma); ESCA (Esophageal carcinoma); COAD (Colon adenocarcinoma); KIRC (Kidney renal clear cell carcinoma); GBM (Glioblastoma multiforme); ACC (Adrenocortical carcinoma);THCA (Thyroid carcinoma); KIRP (Kidney renalpapillary cell carcinoma); PAAD (Pancreatic adenocarcinoma); MESO (Mesothelioma); READ (Rectum adenocarcinoma); PCPG (Pheochromocytoma and Paraganglioma); AML (Acute Myeloid Leukemia); TGCT (Testicular Germ Cell Tumors); CHOL (Cholangiocarcinoma); THYM (Thymoma); KICH (Kidney Chromophobe); DLBC (Lymphoid Neoplasm Diffuse Large B-cell Lymphoma); UVM (Uveal Melanoma).

Figure 2.
Methods of detection of gene fusions.
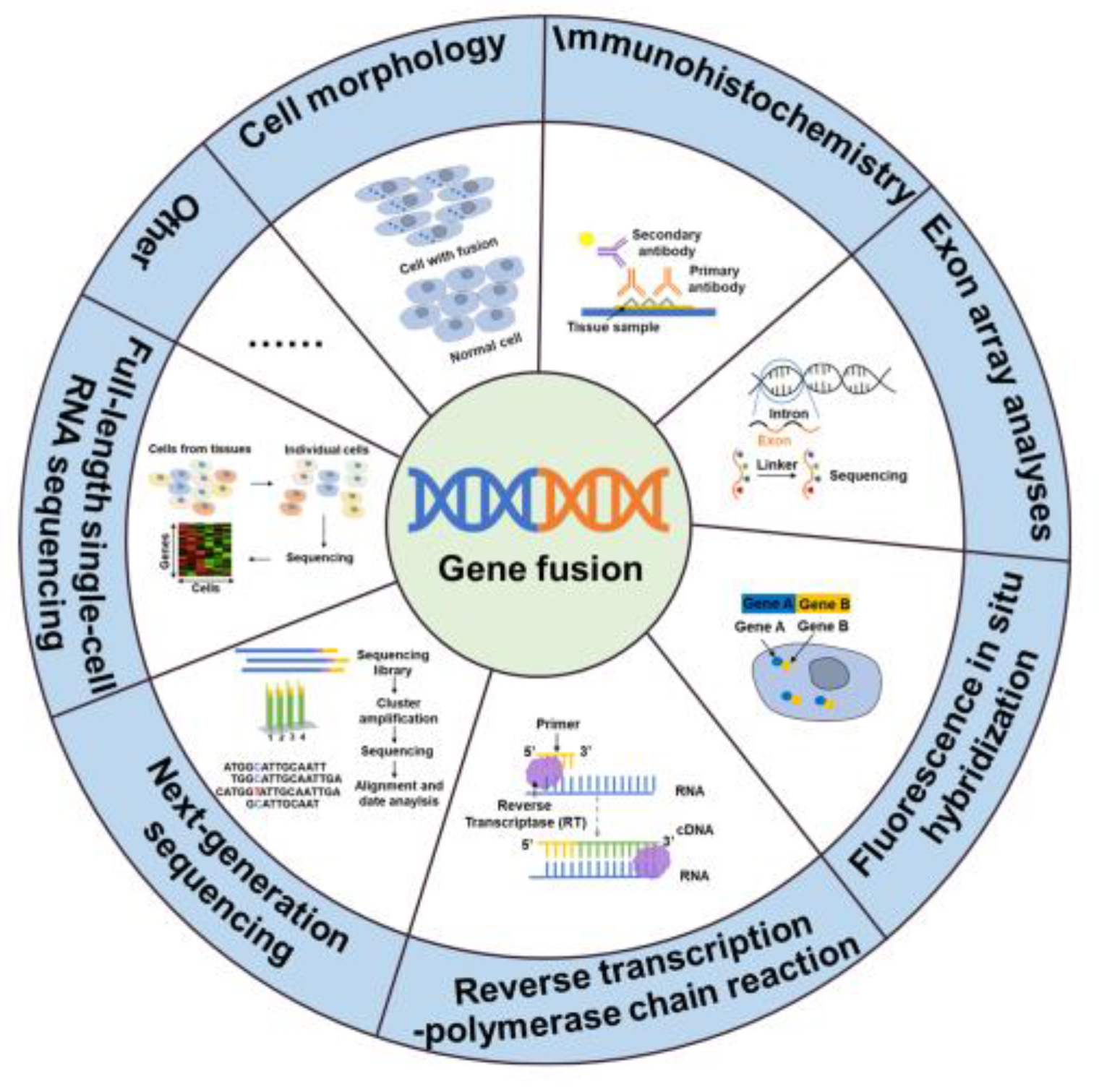
Figure 3.
The structure of BCR-ABL fusion protein, its impact on downstream signaling pathways, and the current inhibitors used to target BCR-ABL. (A) Schematic structural composition of BCR-ABL fusion proteins. (B) the activation of molecular pathways downstream of BCR-ABL. Upon dimerization, BCR-ABL fusion proteins undergo autophosphorylation at tyrosine 177 of BCR. The dimerization event facilitates the recruitment of various proteins, such as GRB2, to activate multiple downstream signaling pathways, including PI3K/AKT, MAPK and JAK/STAT pathways. (C) The current inhibitors of BCR-ABL and major drug resistance mutations associated with them.
Figure 3.
The structure of BCR-ABL fusion protein, its impact on downstream signaling pathways, and the current inhibitors used to target BCR-ABL. (A) Schematic structural composition of BCR-ABL fusion proteins. (B) the activation of molecular pathways downstream of BCR-ABL. Upon dimerization, BCR-ABL fusion proteins undergo autophosphorylation at tyrosine 177 of BCR. The dimerization event facilitates the recruitment of various proteins, such as GRB2, to activate multiple downstream signaling pathways, including PI3K/AKT, MAPK and JAK/STAT pathways. (C) The current inhibitors of BCR-ABL and major drug resistance mutations associated with them.
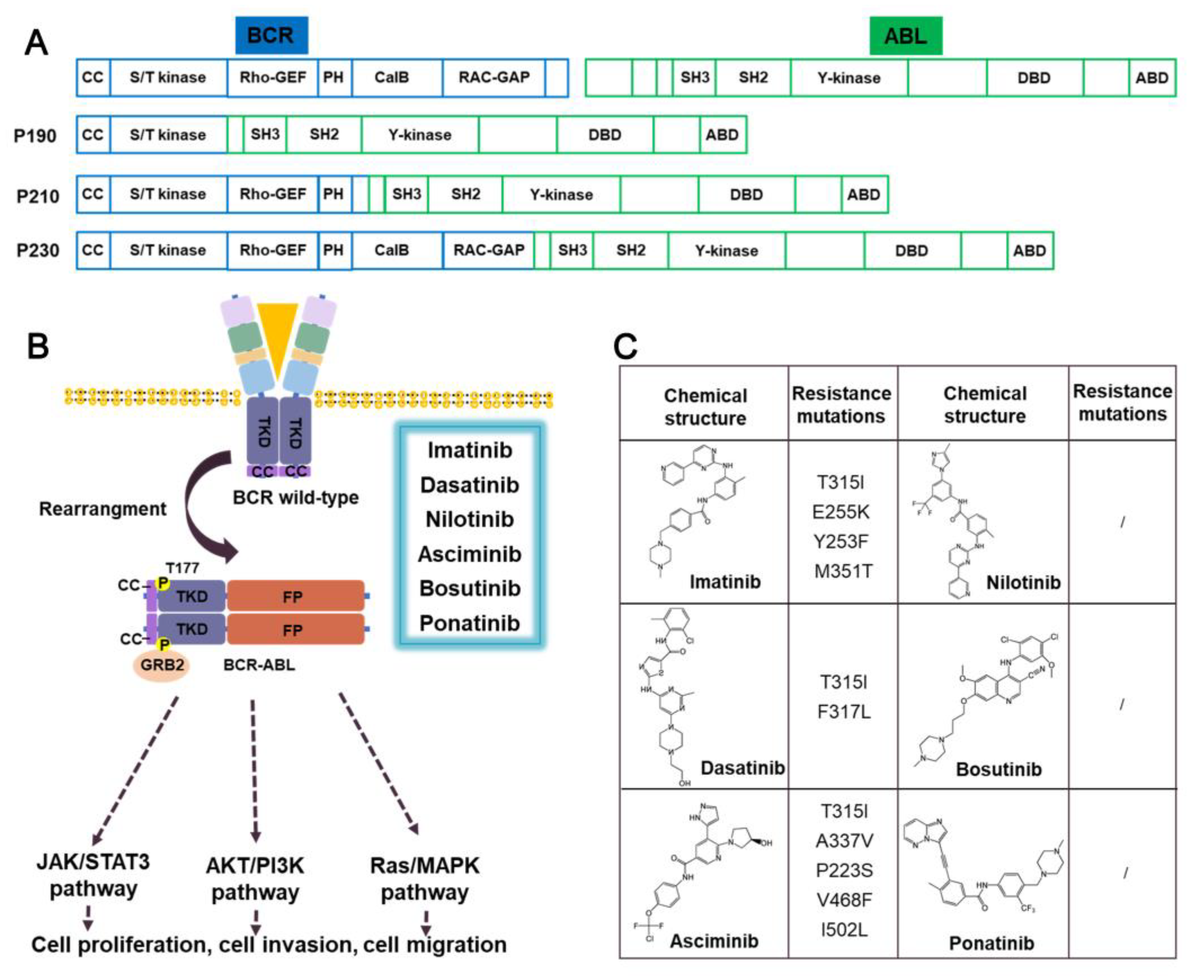
Figure 4.
Schematic diagram of ALK fusion proteins in different cancers and their structural composition. (A) Illustration of ALK fusion events frequently observed in various cancers. Each colored dot represents a distinct ROS1 fusion event, as depicted in (B). (B) Schematic structural composition of ALK fusion proteins. CC (Coiled coil); KD (Kinase domain).
Figure 4.
Schematic diagram of ALK fusion proteins in different cancers and their structural composition. (A) Illustration of ALK fusion events frequently observed in various cancers. Each colored dot represents a distinct ROS1 fusion event, as depicted in (B). (B) Schematic structural composition of ALK fusion proteins. CC (Coiled coil); KD (Kinase domain).
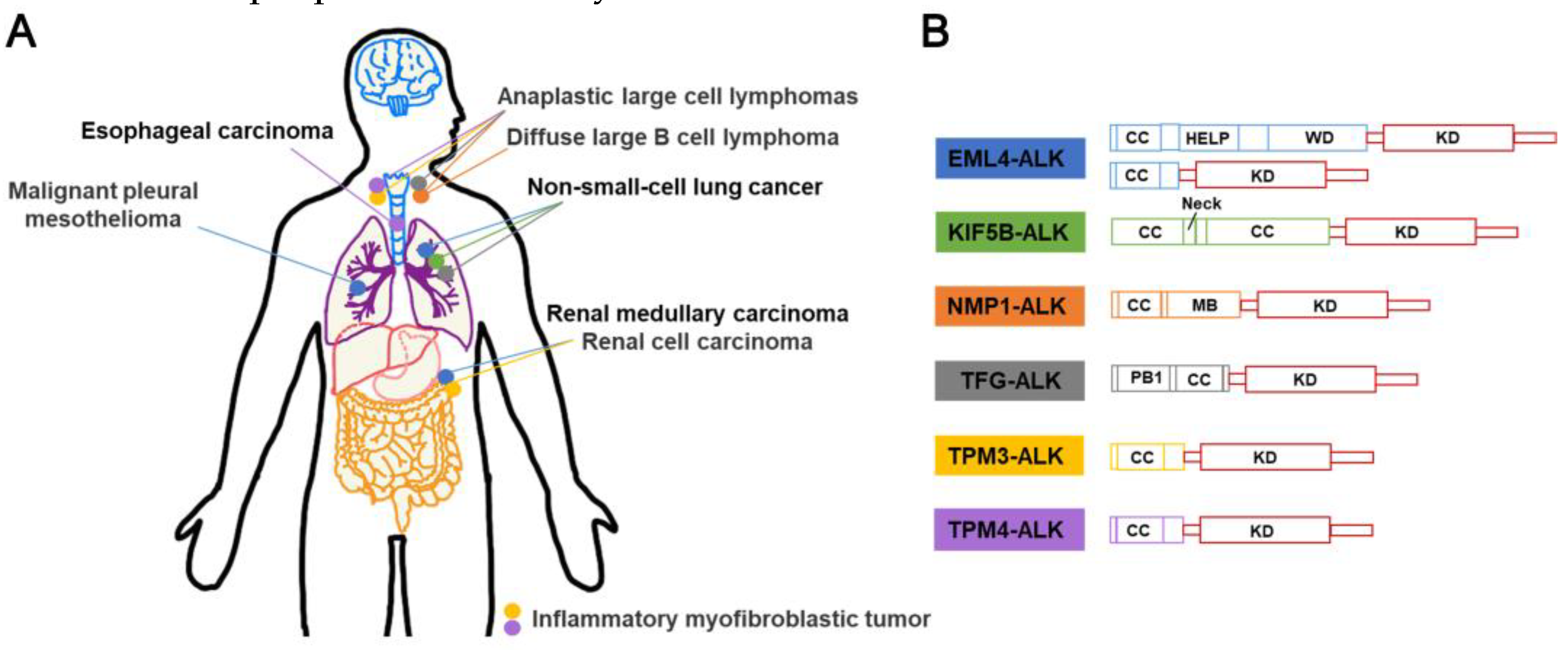
Figure 5.
The downstream signaling pathways of ALK fusion proteins and current inhibitors. (A) Activation of molecular pathway downstream of ALK fusion proteins. Unlike wild-type ALK, ALK fusion facilitate the ligand-independent dimerization of ALK, resulting in its constitutive activation and the initiation of multiple downstream signaling pathways. (B) Inhibitors targeting ALK fusions and major drug resistance mutations. MAM1/2 (Multiple receptor protein-tyrosine phosphatase mu 1/2); LDL (Low-density lipoprotein receptor domain); TM (Transmembrane domain); FP (Fusion partner); TRD (Intracellular kinase domain).
Figure 5.
The downstream signaling pathways of ALK fusion proteins and current inhibitors. (A) Activation of molecular pathway downstream of ALK fusion proteins. Unlike wild-type ALK, ALK fusion facilitate the ligand-independent dimerization of ALK, resulting in its constitutive activation and the initiation of multiple downstream signaling pathways. (B) Inhibitors targeting ALK fusions and major drug resistance mutations. MAM1/2 (Multiple receptor protein-tyrosine phosphatase mu 1/2); LDL (Low-density lipoprotein receptor domain); TM (Transmembrane domain); FP (Fusion partner); TRD (Intracellular kinase domain).
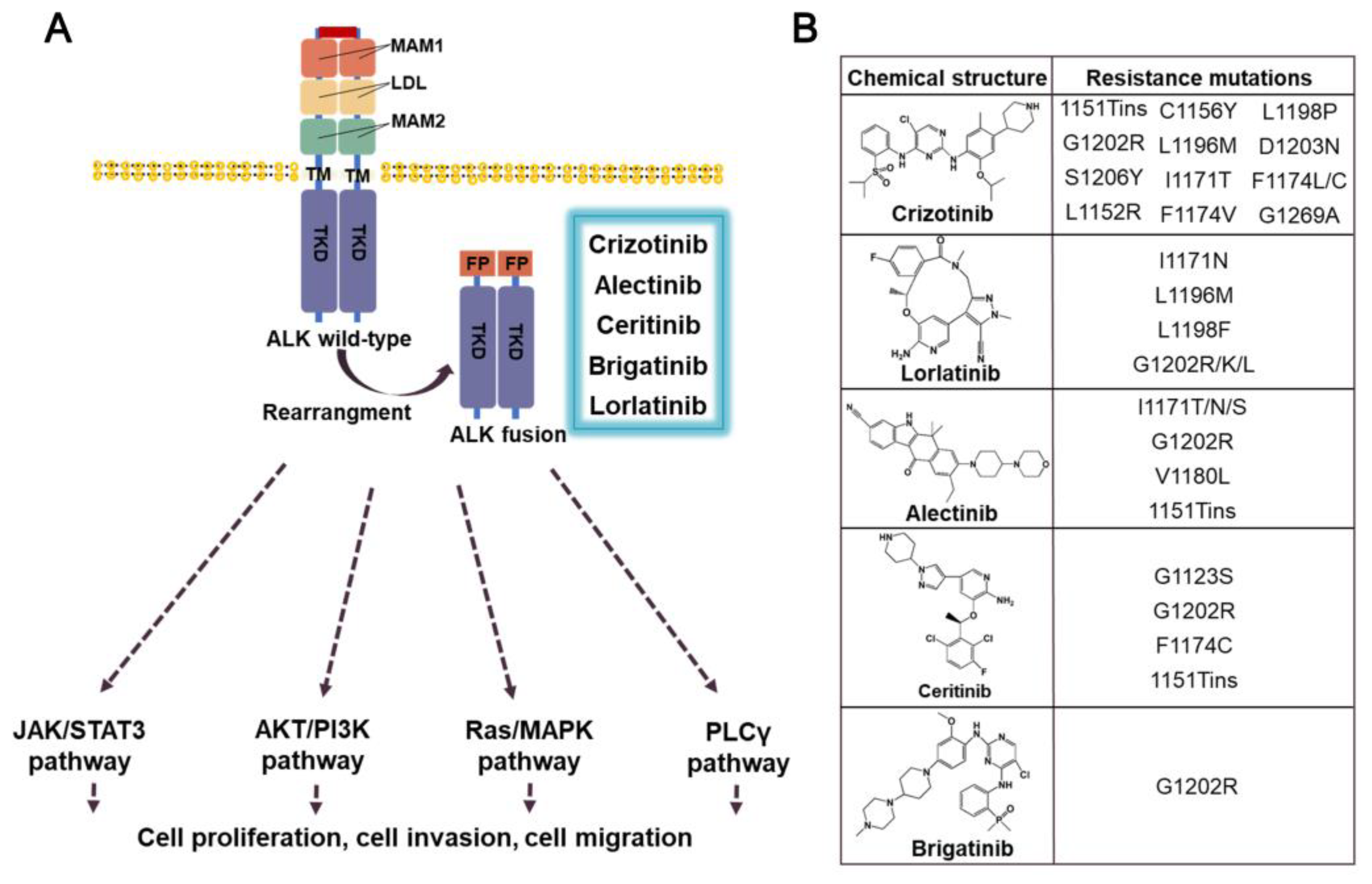
Figure 6.
Schematic diagram of ROS1 fusion proteins in cancers and their structural composition. (A) Illustration of the frequent occurrence of ROS1 fusion in different cancers. Each colored dot represents a distinct ROS1 fusion event, as depicted in (B). (B) Schematic structural composition of ROS1 fusion proteins. CC (Coiled coil); KD (Kinase domain).
Figure 6.
Schematic diagram of ROS1 fusion proteins in cancers and their structural composition. (A) Illustration of the frequent occurrence of ROS1 fusion in different cancers. Each colored dot represents a distinct ROS1 fusion event, as depicted in (B). (B) Schematic structural composition of ROS1 fusion proteins. CC (Coiled coil); KD (Kinase domain).
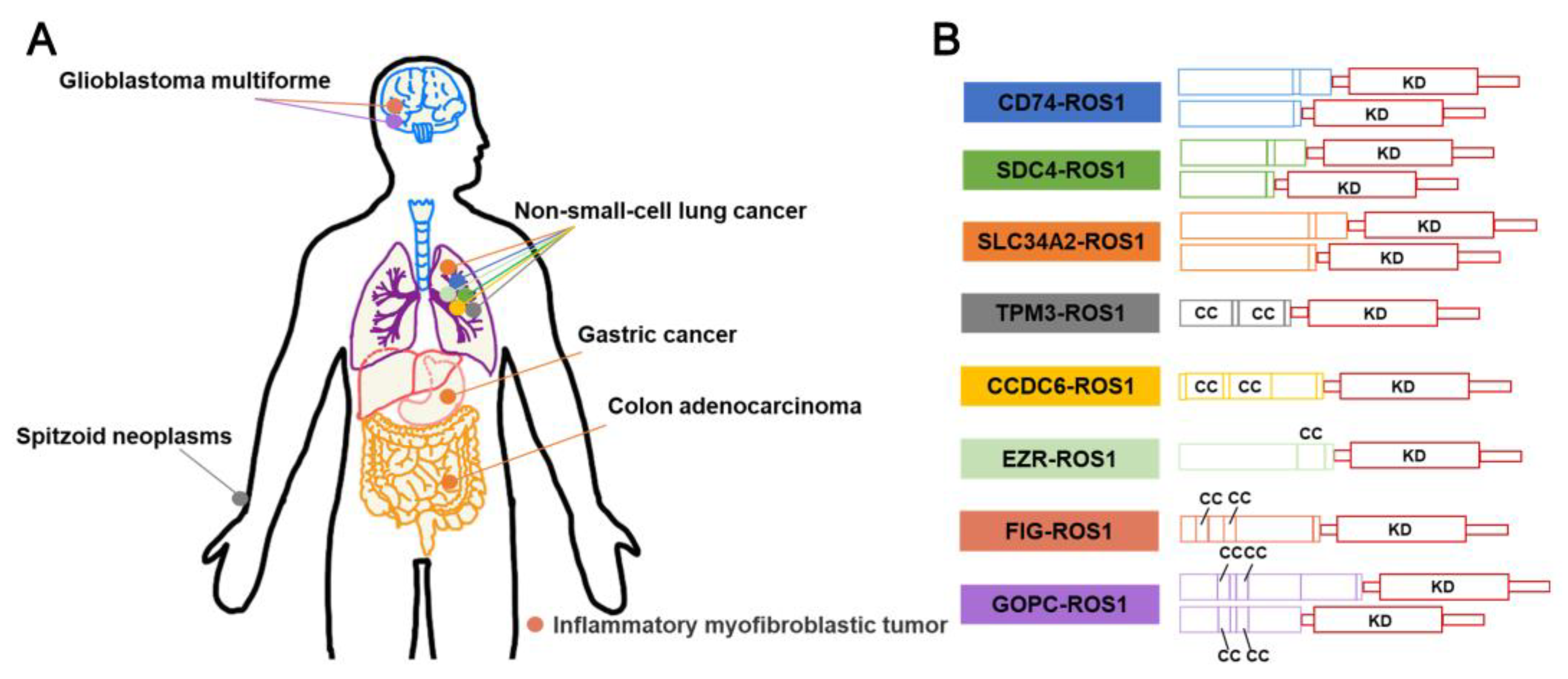
Figure 7.
The downstream signaling pathways of ROS1 fusion proteins and current inhibitors. (A) Activation of molecular pathways downstream of ROS1 fusion proteins. (B) Inhibitors targeting ROS1 fusions and major drug resistance mutations. FN (Fibronectin type-III domain); TM (Transmembrane domain); FP (Fusion partner); TRD (Intracellular kinase domain).
Figure 7.
The downstream signaling pathways of ROS1 fusion proteins and current inhibitors. (A) Activation of molecular pathways downstream of ROS1 fusion proteins. (B) Inhibitors targeting ROS1 fusions and major drug resistance mutations. FN (Fibronectin type-III domain); TM (Transmembrane domain); FP (Fusion partner); TRD (Intracellular kinase domain).
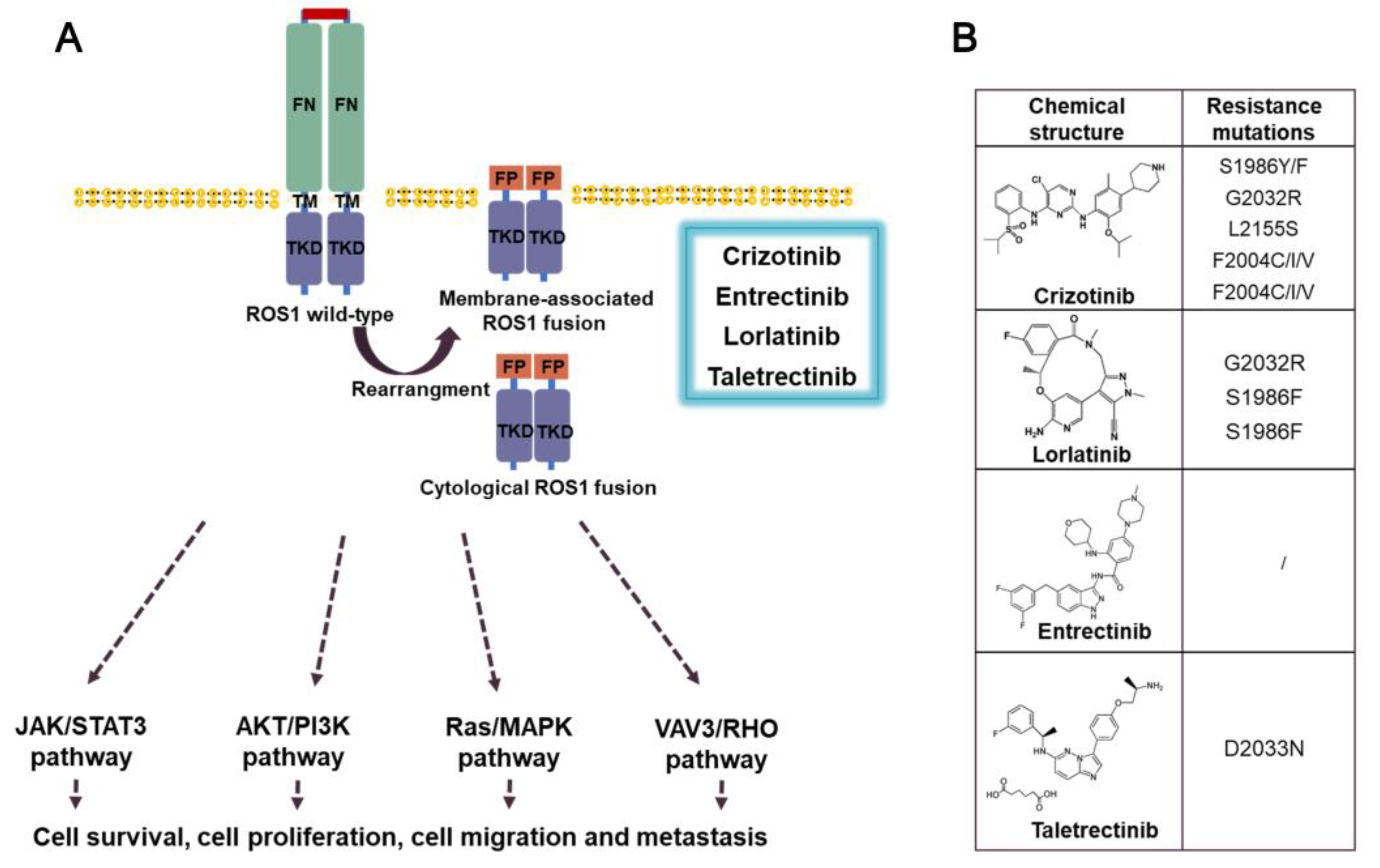
Figure 8.
Schematic diagram of NTRK fusions in cancers and their structural composition. (A) Illustration of the prevalent NTRK fusion events observed in different cancers. Each distinct colored dot correspond to a specific NTRK fusion, as depicted in (B). (B) Schematic structural composition of NTRK fusion. CC (Coiled coil); KD (Kinase domain); TM (Transmembrane domain).
Figure 8.
Schematic diagram of NTRK fusions in cancers and their structural composition. (A) Illustration of the prevalent NTRK fusion events observed in different cancers. Each distinct colored dot correspond to a specific NTRK fusion, as depicted in (B). (B) Schematic structural composition of NTRK fusion. CC (Coiled coil); KD (Kinase domain); TM (Transmembrane domain).
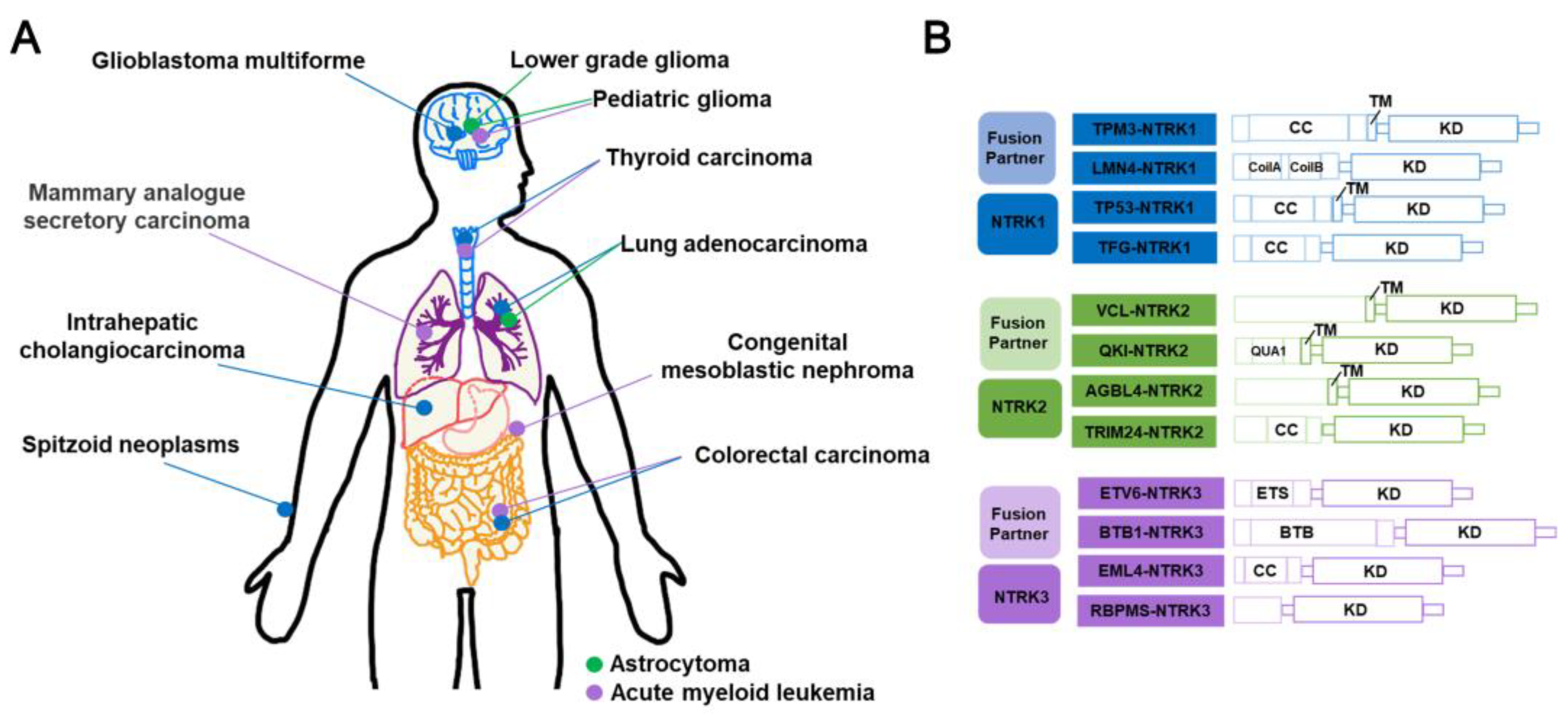
Figure 9.
The downstream signaling pathways of NTRK fusions and current inhibitors. (A) Activation of molecular pathways downstream of NTRK fusion. (B) Inhibitors targeting NTRK fusions and major drug resistance mutations. TM (Transmembrane domain); FP (Fusion partner); TRD (Intracellular kinase domain).
Figure 9.
The downstream signaling pathways of NTRK fusions and current inhibitors. (A) Activation of molecular pathways downstream of NTRK fusion. (B) Inhibitors targeting NTRK fusions and major drug resistance mutations. TM (Transmembrane domain); FP (Fusion partner); TRD (Intracellular kinase domain).
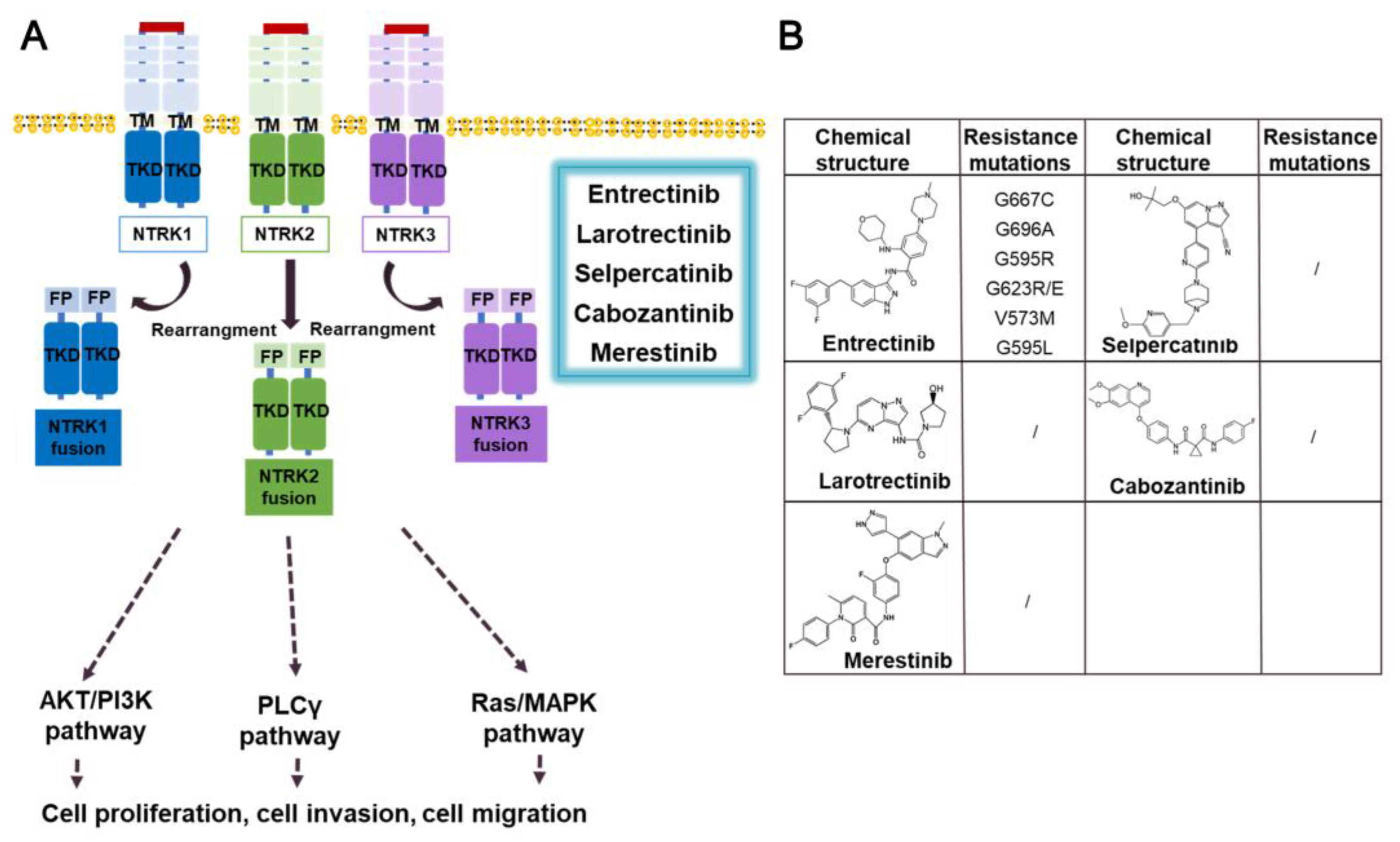
Figure 10.
Schematic diagram of RET fusion proteins in different cancers and their structural composition. (A) illustration of the RET fusion events identified in different cancers. Each distinct colored dot corresponds to a specificRET fusion in (B). (B) Schematic structural composition of RET fusion proteins. CC (Coiled coil); KD (Kinase domain); TM (Transmembrane domain).
Figure 10.
Schematic diagram of RET fusion proteins in different cancers and their structural composition. (A) illustration of the RET fusion events identified in different cancers. Each distinct colored dot corresponds to a specificRET fusion in (B). (B) Schematic structural composition of RET fusion proteins. CC (Coiled coil); KD (Kinase domain); TM (Transmembrane domain).
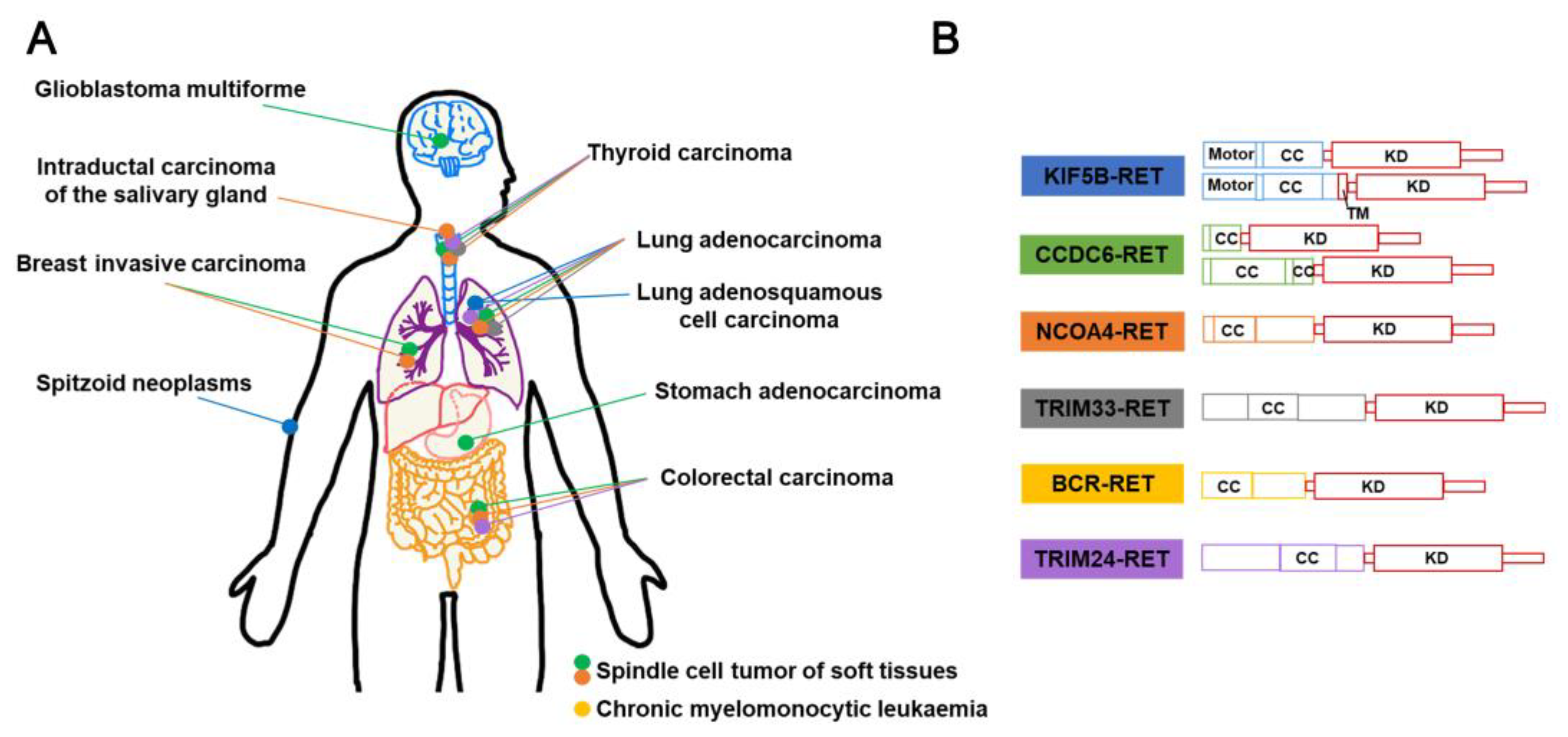
Figure 11.
The downstream signaling pathways of RET fusion proteins and current inhibitors. (A) Activation of molecular pathway downstream of RET fusion proteins. (B) Inhibitors targeting RET fusion proteins and major drug resistance mutations. CLD (Cadherin-like domains); CRD (Cysteine-rich domain); TM (Transmembrane domain); FP (Fusion partner); TRD (Intracellular kinase domain).
Figure 11.
The downstream signaling pathways of RET fusion proteins and current inhibitors. (A) Activation of molecular pathway downstream of RET fusion proteins. (B) Inhibitors targeting RET fusion proteins and major drug resistance mutations. CLD (Cadherin-like domains); CRD (Cysteine-rich domain); TM (Transmembrane domain); FP (Fusion partner); TRD (Intracellular kinase domain).
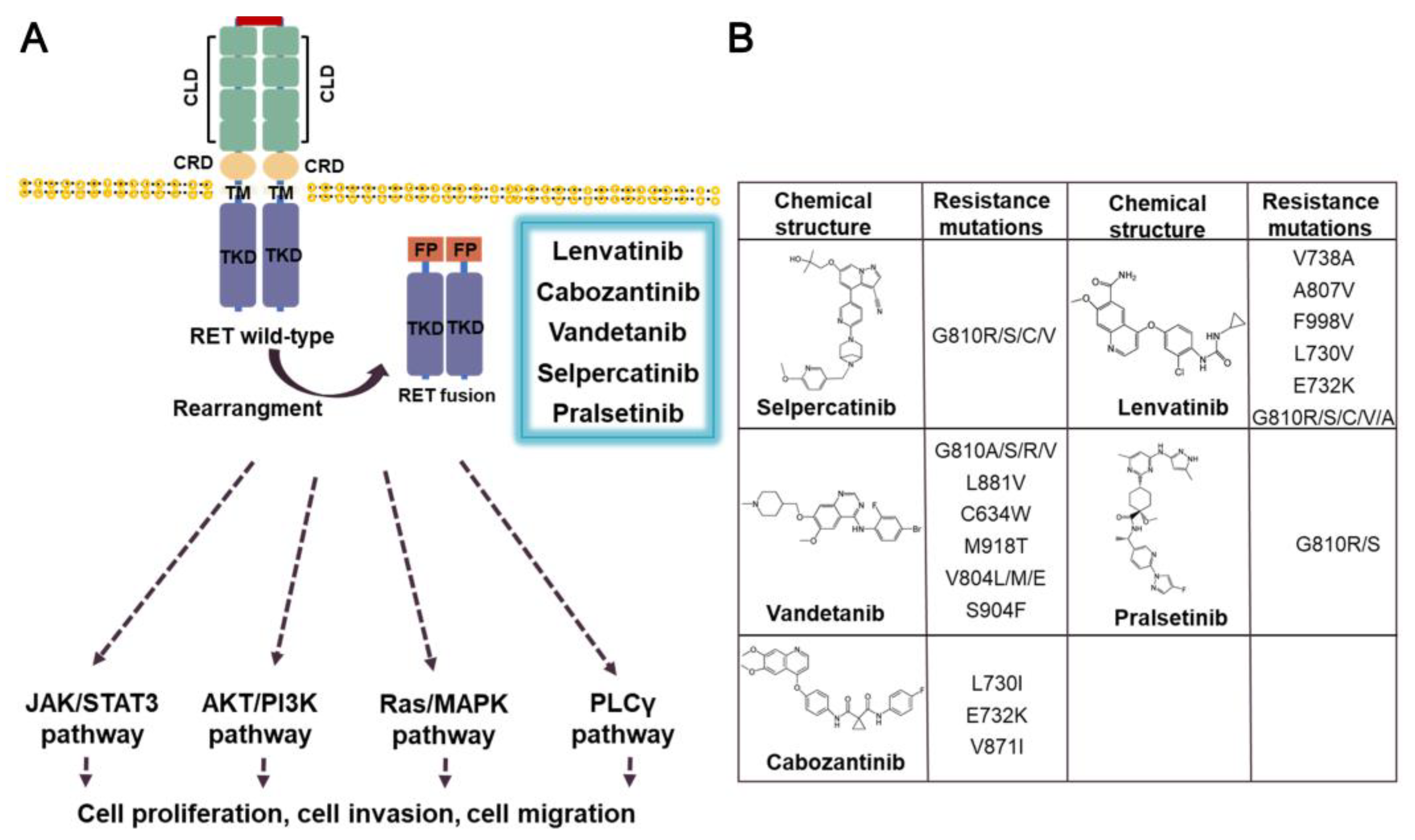
Figure 12.
The downstream signaling pathways of EGFR and BRAF fusion proteins, current inhibitors and major drug resistance mutations. (A) EGFR fusion proteins, (B) BRAF fusion proteins. RTK (Receptor tyrosine kinase); FP (Fusion partner). The upper right corner of Osimertinib refers to its main drug resistant mutations.
Figure 12.
The downstream signaling pathways of EGFR and BRAF fusion proteins, current inhibitors and major drug resistance mutations. (A) EGFR fusion proteins, (B) BRAF fusion proteins. RTK (Receptor tyrosine kinase); FP (Fusion partner). The upper right corner of Osimertinib refers to its main drug resistant mutations.
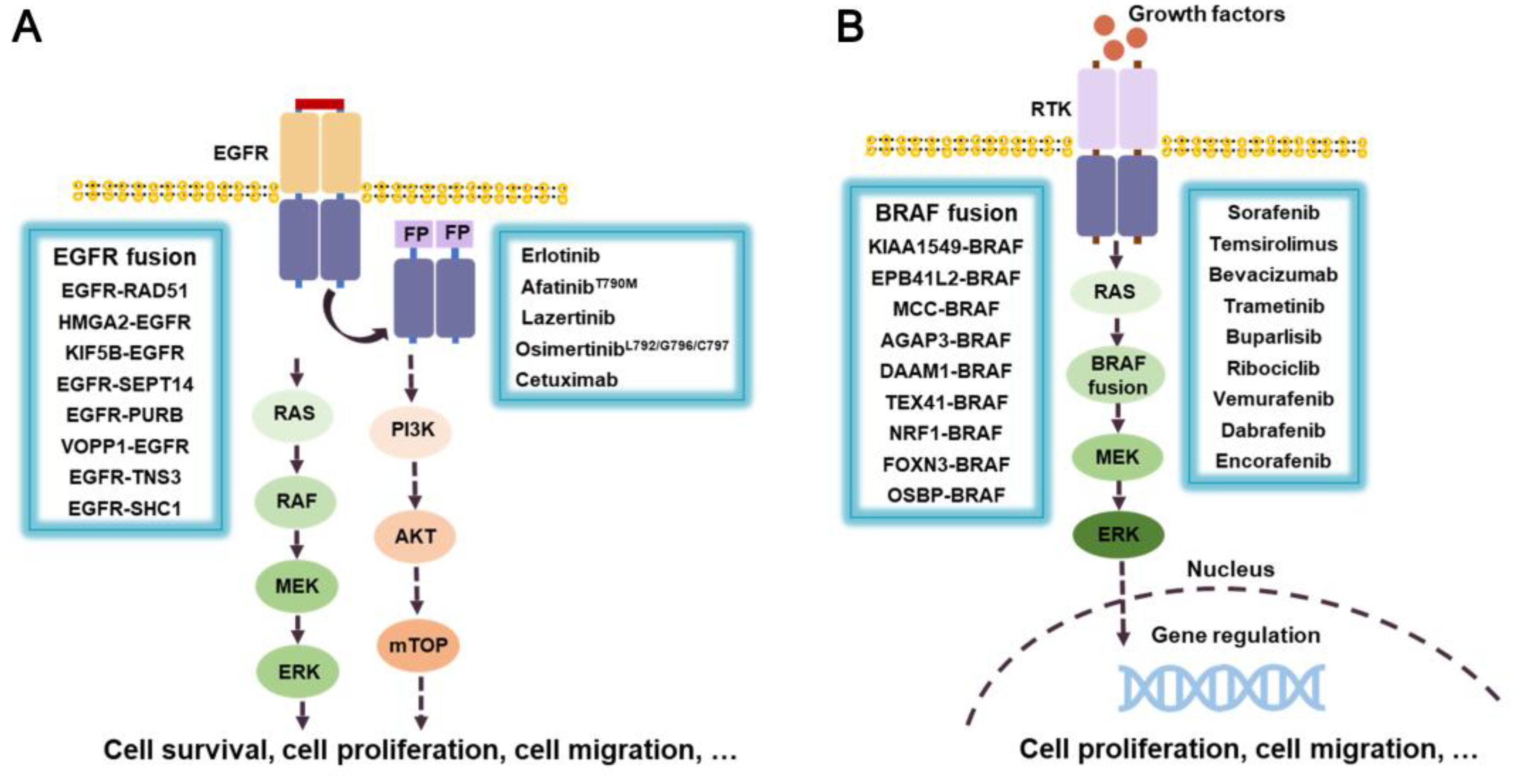
Figure 13.
Distinct pathways regulating PML/ RARα degradation. RA: Retinoic acid.
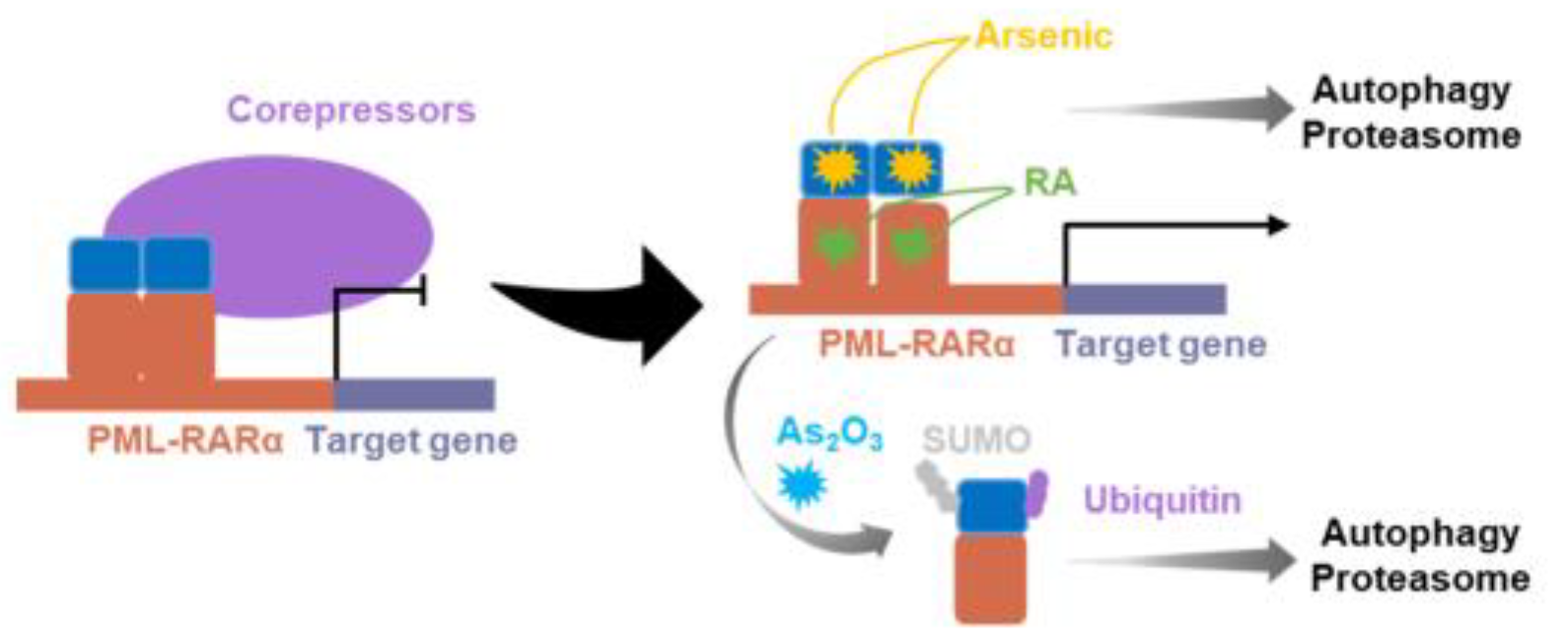
Figure 14.
Schematic structural composition of common non-kinase fusion genes.
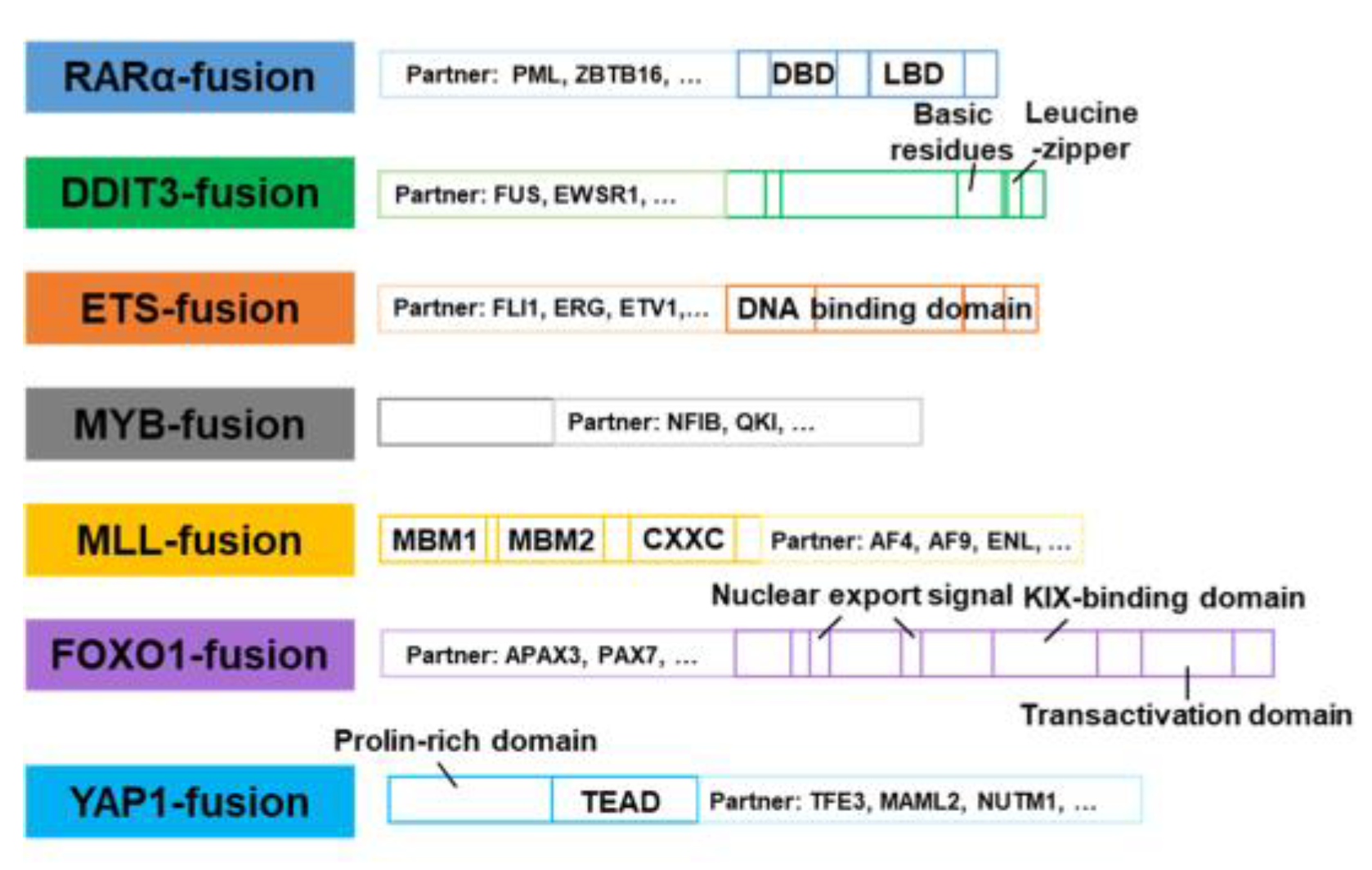

Table 1.
Disease types and inhibitors of fusion proteins.
| Fusion proteins | Cancers | Inhibitors |
|---|---|---|
| ABL fusion (Mainly BCR-ABL) |
CML, ALL | Imatinib***, Dasatinib***, Nilotinib***, Bosutinib***, Ponatinib**, Radotinib**, Asciminib**, Vamotinib*/** |
| ALK-fusion | NSCLC, IMT | Crizotinib***, Ceritinib***, Alectinib***, Lorlatinib***, Brigatinib** |
| ROS1 fusion | NSCLC and Other solid tumors | Crizotinib***, Entrectinib** |
| NTRK1/2/3 fusion | GIST, NSCLC, IFS | Entrectinib***, Larotrectinib**, Taletrectinib* |
| RET fusion | NSCLC, THCA, HCC, MTC | Lenvatinib**, Vandetanib**, Cabozantinib**, Sorafenib**, Ponatinib*, Regorafenib*, Selpe Vamotinib rcatinib**, Pralsetinib** |
| EGFR fusion | NSCLC, PAAD | Gefitinib**, Erlotinib**, Afatinib**, Osimertinib** |
| BRAF fusion | NSCLC, CRC, HCC, GIST | Sorafenib**, Vemurafenib**, Dabrafenib**, Encorafenib** |
| RARα fusion | APL | Arsenic trioxide*** |
*: Phase I Clinical; **: Phase II/III Clinical; ***: Phase IV Clinical. GIST: Gastrointestinal stromal tumor; IFS: Infantile Fibrosarcoma.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



