
Submitted:
12 September 2023
Posted:
14 September 2023
You are already at the latest version
Abstract
Synthesizing evolution in the post-Darwinan era has been a very challenging endeavor, both because of the great number of perspectives regarding the same natural phenomena and because of the growing technology involving mapping the origin of novelties and selection of biological variation that surpass Darwin’s original proposal of evolution by means of natural selection. Since 1859 a lot has changed, and many have presented new perspectives on evolution; either dismissing, correcting or incorporating Darwin’s ideas. This review aims to approach these different theories focusing on virus host interactions and descriptions of host-switching events in 3 different viral lineages, as well as their implications on the understanding of host-switching phenomena itself.
Keywords:
evolution
; pos-Darwin
; virology
1. Introduction
(Co)evolutionary thinking through history
The understanding of coevolution, or the evolution of biological interactions, has changed dramatically over the last two centuries since Darwin’s work in the Origin of Species (Darwin, 1859) in the research field of Evolutionary Biology. However, these different perspectives have not been discussed in other areas of research-such as Medicine-keeping the most recent and complex understanding of symbiotic evolution unreached for those who directly address disease emergence, for example. When discussing the coevolutionary processes in human-pathogen interactions the medical understanding of such interactions has been simplistic when compared to the current host-parasite interaction frameworks, and require new perspectives involving coevolutionary concepts.
For this purpose we organize this review so as to introduce the history of coevolution focused on host-parasite interactions, and how these different understandings have been, even if unintentionally, applied into human-viral interactions. Moreover, we summarize the recent evolutionary frameworks both in the Evolutionary Biology and Medical fields, contrasting their focal point when addressing human-viral interactions.
The main goal of this work is to insert the human-viral interactions into the same conceptual context of host-parasite interactions, because, even though viruses are a special kind of parasites, and are still not unanimously seen as living organisms, they are still subjected to the same evolutionary limitations and processes as any other parasite, a fact routinely evidenced by the daily phylogeographic analysis we see of different viral strains-such as SARS-CoV-1-in the current epidemiological surveillance protocols.
By incorporating the recent coevolutionary frameworks that are being constructed in the Evolutionary Biology research into Medicine, we understand that it will enable future researchers of that area to understand host-viral interactions in a more organized, logical and robust manner, allowing them to explore these interactions with more complexity than it is now being handled.
1. Orthogeneticism-Eimer
More specifically, host-parasite interactions have been discussed under orthogeneticist ideas right after Darwin at the end of the XIX century. They described lineage evolution as a continuously specializing process, in a maximum efficiency scenario where the parasite symbiont should speciate towards a perfect specialist.
Parasite lineages should, therefore, be restricted to its host, inherited from its ancestor, until they reach an optimal host utilization capacity driven by a positive internal drive, usually accompanied by loss of structures and increased interaction complexity. The growing complexity of the interaction or morphological structures and consequential specialization would be followed by an acute dependence of the parasite to the host, with subsequent loss of morphological structures and rapid population decline to extinction (Figure 2; Eimer, 1898).
Orthogeneticists considered parasites the ultimate example of evolution, where the simplification or loss of structures-such as the loss of digestive tract frequently observed in intestinal tapeworms-must reflect the lineage’s imminent and self-imposed extinction due to the negative effect it caused to its resource. Self-imposed extinction being the consequence of maximum efficiency in resource utilization (Figure 2; Brooks, 2019; Agosta, 2020).
2. Neodarwinism
After orthogeneticism, the scientific community entered the modern era of biology, with the emergence of Mendelian’s ideas to the center of evolutionary thinking, giving rise to the neodarwinian era (Figure 1 and Figure 2; Agosta, 2020). Besides Mendel, the proposal of neodarwinism was significantly influenced by Herbert Spencer’s ideas, a sociologist with strong Lamarkian tendencies (later discussed in Paul, 1988) that opposed Darwin’s original “survival of the adequate” (Figure 2), replacing it with the famous “survival of the fittest” evolutionary perspective. Spencer’s apparently subtle change in the natural selection’s jargon stiffens the evolutionary process, previously being characterized as a sloppy restriction of diversity to what would later be defined as the Hardened Synthesis of Neodarwinism (Agosta, 2020).
Darwin’s original evolutionary theory describes a dynamic where the nature of the organism is the driving force behind diversity, one which balanced with and was limited by the conditions of the environment, or conditions of existence (Figure 1 and Figure 2). Neodarwinian thought, on the other hand, limited diversity to a reality of maximum efficiency, turning diversity of character states into a transitory condition to be replaced by the most efficient trait. Also, observable phenotypes should always be associated with a positive or negative fitness contribution, being neutrality or simple adequacy of character not an option [Spencer, 1852].
Figure 1.
General representation of the main developments in symbiont evolutionary theory.
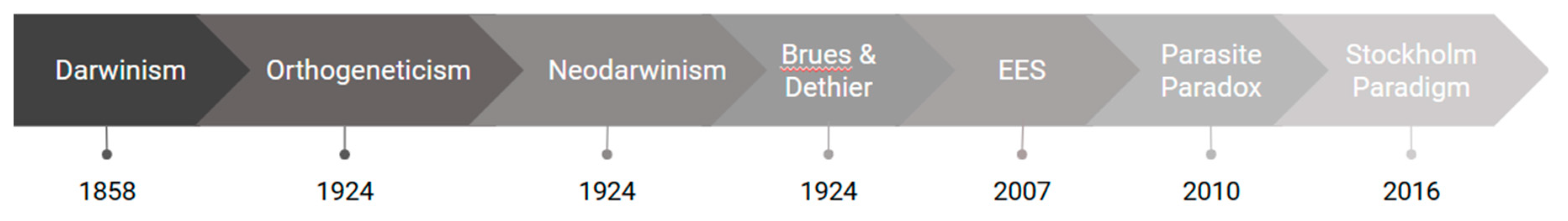
Figure 2.
Graphic representation of pre-21st century theory where 1 represent general distributions of an ancestral population’s phenotypes, where the y-axis depicts the frequency of a phenotype whereas the x-axis describes the phenotypic value of an organism; 2 schematic representation of a host’s lineage, before and after a speciation event; 3 Schematic representation of the parasite phenotypic distributions after the speciation event of the host. For Darwinism, the speciation could lead to a differentiation on the phenotypic distribution of the parasite population, that adjusts towards the optimum phenotype imposed by the host; for the Orthogeneticist, the speciation of the host should result in a rapid differentiation of the parasite population, with a acute change in phenotype distribution, as well as for Neodarwinism. 4 The phenotypic differentiation associated with host change-for the Darwinian view-could lead to speciation of the parasite, but not inevitably so; for orthogeneticist, the acute change in parasite phenotypic distribution should lead to a rapid specialization of parasites, usually accompanied by loss of structures or complexity, and with that, speciation. The same pattern is expected by the Neodarwinian perspective, where the phenotypic specialization would lead to loss of phenotypes with sub-optimal fitness to host’s selection. 5 simple representation of the self-imposed extinction described by the Orthogeneticism. In this case, the change in host’s natural selection pattern would lead to a fluctuation in the population size, with an initial oscillation with positive growth tendencies (selection of specialist parasite), followed by a period of maximum population size (optimum exploitation of resource), and a final decrease in population size, where the parasites become so specialized to that specific selection pressure that any change would lead to inaptitude to access the resource presented by the host, leading to parasite extinction.
Figure 2.
Graphic representation of pre-21st century theory where 1 represent general distributions of an ancestral population’s phenotypes, where the y-axis depicts the frequency of a phenotype whereas the x-axis describes the phenotypic value of an organism; 2 schematic representation of a host’s lineage, before and after a speciation event; 3 Schematic representation of the parasite phenotypic distributions after the speciation event of the host. For Darwinism, the speciation could lead to a differentiation on the phenotypic distribution of the parasite population, that adjusts towards the optimum phenotype imposed by the host; for the Orthogeneticist, the speciation of the host should result in a rapid differentiation of the parasite population, with a acute change in phenotype distribution, as well as for Neodarwinism. 4 The phenotypic differentiation associated with host change-for the Darwinian view-could lead to speciation of the parasite, but not inevitably so; for orthogeneticist, the acute change in parasite phenotypic distribution should lead to a rapid specialization of parasites, usually accompanied by loss of structures or complexity, and with that, speciation. The same pattern is expected by the Neodarwinian perspective, where the phenotypic specialization would lead to loss of phenotypes with sub-optimal fitness to host’s selection. 5 simple representation of the self-imposed extinction described by the Orthogeneticism. In this case, the change in host’s natural selection pattern would lead to a fluctuation in the population size, with an initial oscillation with positive growth tendencies (selection of specialist parasite), followed by a period of maximum population size (optimum exploitation of resource), and a final decrease in population size, where the parasites become so specialized to that specific selection pressure that any change would lead to inaptitude to access the resource presented by the host, leading to parasite extinction.
Regarding coevolution, neodarwinism describes symbiont evolution as a 1:1 relation, in a continuous process of innovation, selection and speciation. It presents a plot where parasites should become increasingly dependent on their host, while it adapts to present defenses against such parasites. Orthogeneticism and Neodarwinism seem to draw similar pictures of coevolution.
Neodarwinism was built under the pretense of uniting Mendelian and modern biology, and received contributions from authors such as Kellog, Fahrenholz, and Eichler, whose ideas on coevolution will now be succinctly introduced.
2.1. Kellog
Kellog was one of the most influential presenters of the vicariant mode of speciation, proposing that lineage divergence should always be preceded by geographical isolation, with subsequent differential selection of the isolated populations. This classical vicariant model relies on interruption of gene flow between subpopulations and selection promoting character differentiation to the point of speciation (Kellog, 1907).
Regarding host-parasite interactions, Kellog presented two main assumptions: (1) parasites are highly attuned to their host species; (2) the parasite’s environment is so uniform (host) that it becomes isolated from external selection forces, that is, geographical isolation of hosts does not affect their parasites’ populations.
Parasite evolution should be buffered by their host’s uniformity.
With these two assumptions Kellog then recognizes three types of host-parasite interactions: 1) Sister parasite species occurring in sister host species that are in disjunct regions – commonness of genealogy scenario. 2) Parasite species occurring in sister host species that are in disjunct regions. In this case the isolation of both symbionts led to speciation of the host but not of the parasite, due to a buffer-like effect of the host on the evolution of parasites – the presumed uniformity of environment provided by the host would block parasite selection.
The third type of host-parasite interaction describes that parasite species associated with two or more phylogenetically distant host species that occur in the same geographical region. Such a scenario is not clarified by the orthogeneticists, being an important case in geographical isolation studies.
In order to address the third scenario that bothered the general view of neodarwinian parasite evolution, Kellogg addresses three possible factors that could enable or disable the generalist behavior observed in those cases: (1) co-occurrence of hosts; (2) differential speciation of hosts and parasites and (3) inherent incapacity to expand in host use. These hypotheses were explored no further, but Kellogg’s idea of right adaptation for each environment remained in the neodarwinian school of thought.
2.2. Fahrenholz
Fahrenholz also made important contributions to the neodarwinian perspective of host-parasite interactions, proposing the utilization of parasite occurrence as a tool to infer host phylogenetic relationship. The presence of the same or closely related parasite species in different host groups should allow researchers to infer the relationship between host groups, regardless of their geographical context. Parasites are assumed to be as dependent on their hosts as free-living organisms are to their environment. Therefore, members of the same host species should be utilized by individuals of the same parasite species, while members of different species should be utilized by parasites whose phylogenetic relationship reflected the hosts’, including the degree of divergence between them [Hopkins, 1942].
In Fahrenholz’s rule [Brooks, 2019; Eichler, 1948, 1966], if parasites produce a new species in response to its environment – as is with every biological system – and the host is its environment, they must follow the new host species after a speciation event, and the parasite phylogeny must reflect the host’s. The author does not address Kellogg’s third scenario of multiple hosts for a parasite species, highlighting cospeciation as the most adequate perspective of parasite evolution.
2.3. Eichler
Eichler agrees with the cospeciation view shown in Fahrenholz’s rule (Eichler, 1948) presenting the so-called Divergence rule, where one should expect – based on parasite occurrence diversity in one host – that host species that are more isolated phylogenetically should carry less parasites, while when finding a great number of parasite groups in one host species, one can expect that the host group it belongs to is algo great in diversity (Eichler, 1948). The evidence of asymmetry between host and parasite lineage is simply overlooked, being the use of parasites to work as a tool to study host evolution, the main goal of Eichler in this work.
The author also discourses on the continuous specialization of parasites to hosts, going as far as discussing Szidat’s rule – basal parasite lineages utilize basal host groups, while diversified parasites should inhabit diversified hosts (Eichler, 1948). Neodarwinian thought kept the cospeciation perspective of symbiont evolution, incorporating all the previously mentioned ideas.
3. Brues’s turning point
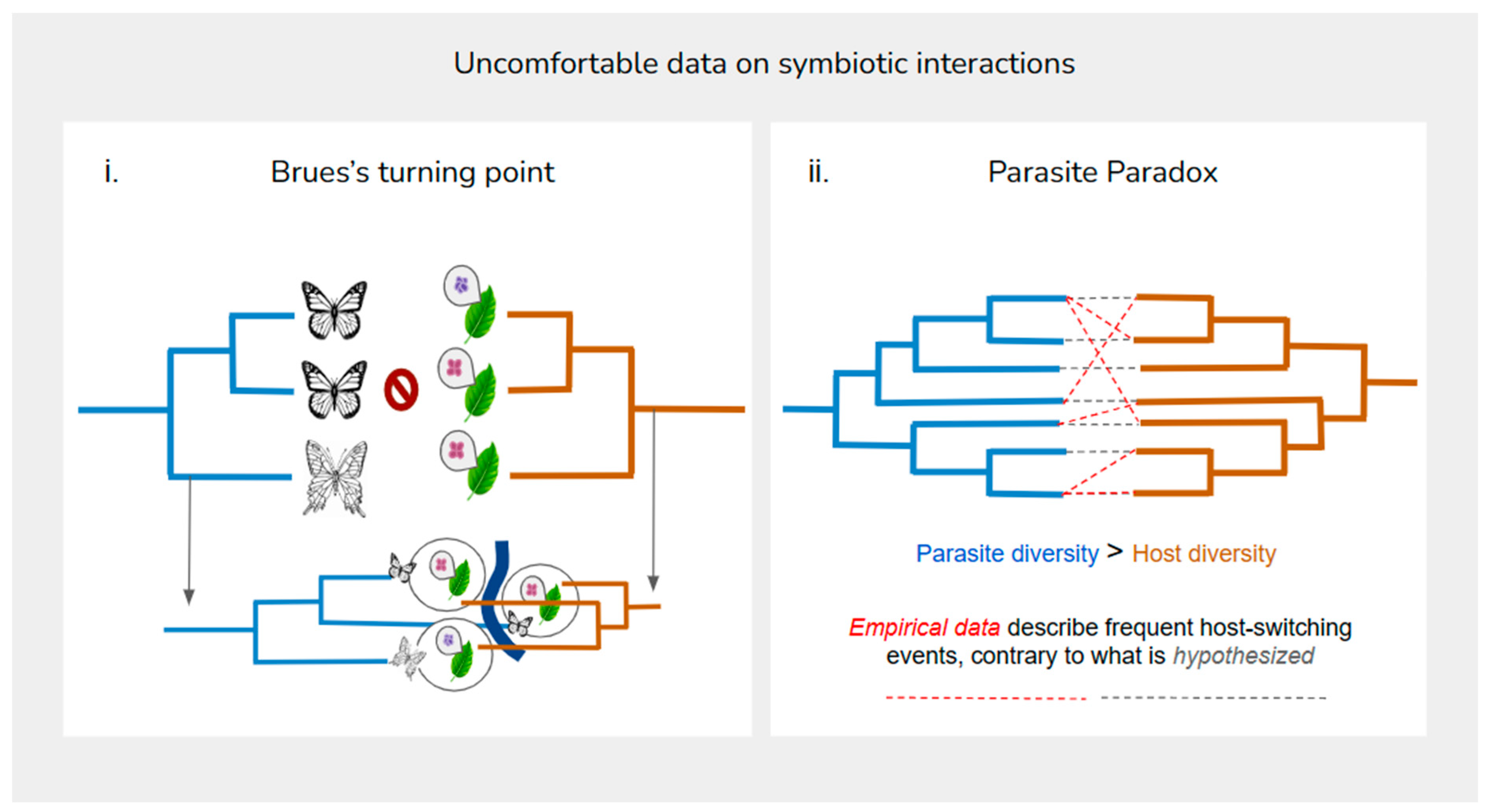
With time, the importance of parasites to the understanding of host-parasite systems were taken into consideration, especially considering Brues’s work on the evolution of phytophagous insects (Brues, 1921; Brues, 1924; Figure 3). The author discusses at length how the observed associations seemed to be ruled by resource specificity with no clear phylogenetic or geographic dependency. This evidence pointed to the fact that the host was not the focus of the association, but the character it carried – resource over taxonomy (Brooks, 2019).
This particular puzzle was later addresses by Dethier in his book-Chemical Insect Attractants and Repellents – being the first exploration of pheromones in insect-plant interaction, its functions as an attractor and a repeller, being conserved but widespread phylogenetically (Dethier, 1948). Plant choice by insects, therefore, was no longer based on taxonomy, but a complex synergy of resources that enabled host use by a symbiont, not necessarily leading to identical phylogenies.
4. 21st century evolutionary theory
The modern evolutionary biologists are now required to understand evolution considering all these different perspectives, and moreover, approach the biological uproar climate change is promoting across the globe. Evolutionary thinking has now been required as an applied science, more than ever before. Community evolution has, in this scenario, been required to solve immediate problems in society, instead of its past, and more philosophical, utility. In order to understand and manage the consequences of climate change, two recent perspectives have been presented, the Extended Evolutionary Synthesis and the Stockholm Paradigm.
4.1. Extended Evolutionary Synthesis
The extended synthesis (Figure 1), originally proposed by Pigliucci and Muller, is majorly based on an earlier hypothesis, the Modern Synthesis, that was presented along the years of 1920 to 1942, with the official proposition by Huxley (Huxley, 1942). The Modern Synthesis was built under great influence of the concepts of population genetics, centered on the near dogma of “variation-natural selection-fixation", with strong tendencies towards gradualism (evolution should by slow and gradual), externalism, where species form should be defined by the external forces, as well as gene centrism, that is, character evolution could be reduced to gene evolution (Pigliucci, 2007; Huxley, 1942; Figure 1).
The Modern Synthesis also embraces ideas like allopatric speciation and biological species concept (both by Mayr), and other names of population genetics such as Simpson (1994), Fisher and Dobzhansky, presenting itself as a fusion of Darwinian, Neodarwinian and Mendelian genetics. The Extended Synthesis does not diverge, in its core, from the Modern Synthesis view of evolution, adding to it in terms of molecular and technological advancements of the last century.
Pugliucci & Muller’s proposal comes as a long process of dissatisfaction with the original synthesis of evolution, since modern issues in evolutionary biology aim to address topics in evo-devo, phenotypic plasticity, epigenetics and character evolution, subjects that are not bound by gene evolution. Questions regarding the mechanisms of evolution could neither be discussed nor tested by using the general concepts presented by the Modern Synthesis (Pugliucci & Muller, 2010). The group of authors who now contribute to the development of the Extended Evolutionary Synthesis justify their endeavor by describing it as a mechanistic-causal basis of the general evolutionary concepts presented by the Modern Synthesis, without causing great changes to the evolutionary framework that was previously built (Brooks, 2011).
4.2. Parasite Paradox
The Parasite Paradox, discussed at length by Agosta, 2020 and Brooks, 2019 raises the debate on the divergence between observed and expected parasite diversity according to the Neodarwinian framework (Figure 3). Both views on parasite evolution state that parasites are ultimate specialists, bound by the evolutionary trajectory of their hosts and soon to be extinct through self imposed selection. Beyond that evidence, the parasite paradox also discusses the empirical evidence that parasite diversity is exceedingly higher than host diversity, something which does not agree with the prevalent understanding of symbiont – and parasite – evolution.
By presuming the extreme specialist view on parasites, host-switching events are seen either as an error in identification by the observer, or an early and rare event in parasite diversification (Kellog’s third scenario). The Paradox discourses on that second divergence between evidence and Neodarwinian evolution, highlighting the absence of symmetry between parasite and host diversity and phylogenetic relatedness.
Such evidence was then partly addressed by the Neodarwinians, who argued that parasite diversity was explained by a decreasing pathogenicity promoted by selection. By continuously reducing pathogenicity, parasites should be able to become more generalist, expand and locally adapt (Read, 1994). Host-switching, however, was still bound to the emergence of a novelty that enabled parasite lineages to jump from a host species to another.
This particular view was later incorporated into virology as the avirulent hypothesis of evolution-a traditional perspective on viral evolution that assumes the loss of pathogenicity associated with high viral diversity. Nowadays, the trade-off hypothesis has replaced the virulence hypothesis when discussing virulence evolution (Alizon, 2009; Marquez, 2012).
Figure 3.
Defining evidence in coevolution. i) Brues’s work on phytophagous species evolution set the stage for an important paradigm shift, where resource should not converge with, necessarily, taxon. Phytophagous insects would interact with plants that carried the necessary resource, not the species itself. ii) The Parasite Paradox, on the other hand, describes the incongruence between the expected host-parasite interaction described by the time’s prevailing evolutionary framework (Neodarwinism) and the empirical data.
Figure 3.
Defining evidence in coevolution. i) Brues’s work on phytophagous species evolution set the stage for an important paradigm shift, where resource should not converge with, necessarily, taxon. Phytophagous insects would interact with plants that carried the necessary resource, not the species itself. ii) The Parasite Paradox, on the other hand, describes the incongruence between the expected host-parasite interaction described by the time’s prevailing evolutionary framework (Neodarwinism) and the empirical data.
5. Modern Evolutionary Theory – Post Neodarwinism – later 20th century evolutionary theory
5.1. Charles Mode
The emergence of coevolution in the sense that both host and parasite influence each other’s evolutionary path was first introduced by Charles Mode (Mode, 1958), being the first proposal of symbiont evolution that diverges from Neodarwinian cospeciation. Although it still discusses a dual evolution, disregarding the complexity of interactions, Mode’s work is one of the first ones that actually address parasite’s influence on host selection, instead of it playing a passive role in symbiont evolution.
5.2. Ehrich and Raven
Ehrich and Raven then argued that plant-insect diversification must be driven by a complex interaction between both lineages, where both parts of the association should be influenced by the symbiosis that in time could leave phylogenetic marks, but not in a mirrored manner, necessarily. The authors do not explore in detail the evolutionary implications of coevolution in the final topology of the lineages, focusing, instead, in the flexibility of coevolution, and diverging from the general Neodarwinian view (Ehrich & Raven, 1964).
By exploring the diversification of butterflies and their plant groups, Ehrich and Raven structured a series of questions regarding coevolutionary thinking, being the first authors in community evolution who express a discomfort in their current perspective of coevolution-to assume a state of neutrality for one of the symbionts in diversification (Ehrich & Raven, 1964). In their perspective, coevolution should involve the study of groups of organisms with a close ecological relationship instead of the traditional 1:1 exploration of lineages. This perspective, although initially simple, enables the understanding of a complexity in coevolution, that instead of a strict symbiosis, embraces events such as host-switching, generalization (i.e. host-repertoire expansion (Braga, 2020) and an escape from ideas like co-extinction in community evolution.
By 1960s, the coevolutionary perspective described by Ehrich and Raven gained supporters as they evidenced more “mistakes” in symbiont utilization, both in host-parasite interactions and in plant-insect investigations (Ehrich, 1948; Ehrich & Raven,1964, Verschaffelt, 1910). The new perspective was accompanied by the essential works of Bigelow and Hennig (Bigelow 1956) who actually enabled community evolution to investigate the phylogenetic history of symbionts, something which before them lacked method and robustness (Hennig, 1965).
By the end of the twentieth century, evolutionary biology stopped searching for deterministic rules in diversification and coevolution and began to look for general guidelines on evolution. Mode’s and Ehrich and Raven’s ideas, therefore, took a more central stage in coevolutionary thinking, due to their less strict views on diversification and selection. This scenario was later met with the discussions on climate change, general biological reassembly, and the emergent diseases crisis, reported by authors such as Elton (1958) and Carson (1962).
6.2. Stockholm Paradigm
As opposed to the Extended Synthesis, the Stockholm Paradigm aims to recover Darwinian ideas in order to discuss evolution, claiming that the historical construction of Neodarwinism lost essential information on the study of diversification of life, originally proposed by Darwin (Brooks, 2019). The authors aim to discuss coevolution as a process that entails the synergy of capacity and opportunity in the determination of biological interactions. Capacity is by them defined as the general ability of an organism to utilize resources presented to them (being a free-living or symbiont organism), given by the Information Space accumulated through inheritance, being genetic, epigenetic and developmental information but also information acquired by other means, such as education. Opportunity, however, is defined as the coincidence of resource availability (either environmental or host-wise) in time and space, or in other words, the realization of capacity is conditioned by the opportunity.
The authors discuss at length on the framework that entails this Paradigm, all of which being built on past Darwinian proposals as a conglomerate of thought that opposes the general Neodarwinian view previously described. It relies on three general pillars: (1) Ecological Fitting; (2) Oscillation Hypothesis; (3) Taxon Pulse, where Ecological Fitting addresses the initial stages of an interaction, describing the interaction in terms of realization of capacity limited by opportunity, selection of phenotypic variations, and establishment of an interaction.
Oscillation Hypothesis and Taxon pulse, on the other hand, discuss the phylogenetic and phylogeographic aspects of evolution, the first exploring the alternating aspect of specialism and generalism, while the second describes the spatio-temporal reality of evolution, and how the exploration of hosts by parasites, for example, relies on cycles of stability and perturbation, or better yet, on the scenario of opportunity. This paradigm aims, therefore, to present evolution from the populational to the phylogeographic perspectives.
7. Applied evolution in host-virus interactions
Medicine has attempted to absorb evolutionary ideas in two different moments: (1) during the 1881-1940s period, the so-called Medical Darwinism, and (2) in 1991 with the proposal of Darwinian Medicine by Willians & Nesse, which will now be succinctly described.
7.1. Medical Darwinism (1881 – 1940s)
Medicine and evolution have been previously described as interdisciplinary, but profoundly different from each other (Zampieri, 2009), where the first does not aim to deeply understand the theories behind the scientific data, but instead takes from it what is applicable to solve society’s issues involving biology of pathogens and human diseases. Therefore, it has not accompanied the changes in evolutionary theory previously described, although it has previously been invested in the specific subjects of evolution of infectious diseases and immunology (Zampieri 2009) during the so-called Medical Darwinism (Zampieri 2009), from 1881 to the 1940s. For the duration of World War 2, Medical Darwinism was skewed towards eugenistic ideals and was thus forgotten by many research groups.
After that period there was a steep decrease in medical works addressing the evolutionary perspective of diseases (Zampieri, 2009), until the official proposal of Darwinian Medicine in 1991 (Willians & Nesse, 1991). Medical Darwinism (1881 – 1940s) was mainly focused of on typology of human types – based on internal characteristics, humans could be defined along the lines of dominance of diathesis, that is, a general temperament such as lymphatic, choleric and sanguine, that should lead to the unbalance of different organs and systems, consequently defining groups of people with tendencies to specific diseases and capabilities (Waller, 2002; Zampieri, 2009).
The theory of diathesis, or human types, relied on the evolution of human variation. The internal drivers of human types – and their specific diseases-were then confronted with the rising Germs Theory, strongly evidenced by Pasteur’s experiments in microbiology (Pasteur, 1881). Instead of diathesis, human diseases should actually be caused by microorganisms that invaded the organism. The decline of Medical Darwinism was accelerated by the expansion of medical research based on experimentation, its manipulation by eugenistic propaganda, and religious opposition. It also diverged from original Darwinian ideas by replacing the fundamental idea of population – and individual variation – with the utility of types, simplifying evolution of diseases to a phenomenon with one or few causes (Zampieri, 2009). Diseases were then approached as norms, or universal types instead of individual pathogens, or an inconsequential variation of the pure form (Nesse & Williams, 1995).
From the constitutionalist perspective of medicine, that is, the definition of a type to be considered as the ideal form (the healthy), would then be used to identify the disease, or the variation from the norm. The definition of the ideal type was then explained by Natural Selection, without considering the reality of variation that is inherent to nature. The persistence of the definition of types in this scenario leads to the understanding of variation from the norm as lesser, or negative variation. Natural selection must, then, be the driving force enabling the conservation of higher types and elimination of lower forms. It is not difficult to see how this was readily received by eugenic groups in World War 2 (Tracy, 1992; Méthod, 2015).
Considering Medical Darwinism, the duality built by the rejection of the populational perspective of species and pathogens was in consonance with the inherent duality of healthy and sick as two isolated and incompatible entities. It extensively discussed the idea of hereditary diseases, and its incongruence with the view of Darwinism that was dominant – Neodarwinism-explaining that civilization and medicine blocked the elimination of such traits (Zampieri, 2009).
Regarding diseases caused by pathogens, which is our focus here, one can see how the definition of types, as well as the elimination of the less fit variants influences the current view of pathogen evolution. The severity of natural selection described by Medical Darwinism was described as the reason why it was then replaced by Darwinian Medicine, although it seems to still somehow influence medicine’s view on pathogens (Nesse & Williams, 1991).
7.2. Darwinian Medicine
Proposed in 1991 by Nesse and Willians, this approach aimed to replace the typological view with the populational, based especially on the advancements of evolutionary biology, with the discovery of genetic polymorphisms and the non-directionality of evolution, as well as on the advancements regarding genetic variation. The concept of trade-off became paramount to the understanding of pathogenesis and disease, whereas genes should become more prevalent if it leads to higher reproductive success, not necessarily to better health. Concepts like linkage of characters and trade-off were then used to explain the persistence of hereditary diseases, what is currently described in evolution as Genetic Load (Wallace, 1991).
Darwinian Medicine focuses on the origin of vulnerability to disease, rather than the evolution of disease, per se, since natural selection must shape vulnerability, or the malfunction of traits leading to disease vulnerability, and not the disease itself. This vulnerability should vary between host groups or populations, due to their phenotypic variation, something which not necessarily escaped selection, but could have been maintained by it due to trade-off phenomena (Nesse & Williams, 1991; Wallace, 1991).
It is interesting to note that disease vulnerability (external origin) is described by the authors as dependent on 6 principles: (1) the response to natural selection can be slow relative to the rate of environmental change, causing a mismatch between design and environment; 2) natural selection can be slower in the host than in the pathogen, this being crucial especially in competition with a pathogen that reproduces more quickly than humans; 3) natural selection cannot solve some problems no matter how much time it is given, for trade-offs force compromises or because there are constraints peculiar to living systems (see Gerhart and Kirschner 1997; Minelli 2003); 4) we misunderstand what selection shapes, not seeing traits that increase reproductive success at the cost of disease vulnerability; and finally 6) we may misunderstand what selection shapes, as defenses can be readily mistaken for diseases.
The description of Darwinian Medicine was further explored in 1991, where Williams & Nesse describe the evolutionary perspective of humans and their diseases in a less strict way, where human traits are not the evolutionary construction of a perfect design, but the best possible reproducer. Darwinian Medicine does not seem to see selection as a process that maximizes strength, health and longevity (Williams & Nesse, 1991). Regarding infectious diseases, the authors describe disease dynamics considering the different changes infection induce in the host’s health-(1) direct damage; (2) Impairment of function; (3) Repair by the host; (4) Compensatory adjustments; (5) hygienic measures; (6) host defenses that expel, destroy or sequester pathogen; (7) evasion of host defenses by parasites; (8) attack on host defenses by parasite; (9) trophic mechanisms by the parasite; (10) dispersal mechanisms – transmission dynamics and (11) manipulation of host’s behavior or metabolism by the parasite.
In terms of pathogen evolution, Darwinian Medicine seems to follow the coevolutionary arms race perspective, where pathogens evolve in the course of the infection, developing resistance to medication, for example. Coevolution is also discussed in terms of development of novelties through host and parasite generations. Virulence is another pathogen trait that is commonly discussed in this scenario, being the avirulent hypothesis (Dobsanksy, 1951) described as defective by the authors, since it considers pathogen evolution as a slow and gradual process, as well as the fact that some of parasite’s success must rely on some level of virulence (factors such as 6, 10 and 11).
This aforementioned perspective of the avirulent hypothesis seems to disagree with the current evolutionary perspective of parasite evolution because the authors suppose an evolutionary equilibrium in parasites, where virulence-as well as other parasite traits-should not vary unless ecological circumstances change rapidly and extremely. Ewald (1987 and 1991) lengthily describes and predicts virulence as a process that varies according to transmission characteristics, where in indirect transmission scenarios (human host and insect vector) virulence should be high in humans and low in vector; whereas in direct transmission virulence should be lower in order to enable contact between hosts.
Another prediction presented by Ewald and Schubert (1989) is that diseases transmitted by contact with inanimate vectors (water, i. e), should be more virulent, since the high density of pathogen population in a host must be released into the media and overcome dilution, a problem that does not emerge in diseases that involve only hosts. Another mode of transmission discussed by them that could lead to changes in virulence is alterations in mode of transmission, from person-person to transmission through water, is expected to lead to an increase in virulence.
It is important to note that the variation in virulence due to host-switching events or changes in transmission dynamics presume, necessarily, the evolution of such virulence, as a novel character that rose from these disruption events. The emergence of such novelties is then explained by the rapid evolutionary rate of pathogens, due to their short life cycle and high fertility rate, being mutation and multiple infections enabling recombination events, great sources of novelty. Ewald discusses many other scenarios of virulence evolution, which are not relevant here (Williams & Nesse, 1991).
The second event of merging evolutionary thinking into medicine has led to important changes in the practical aspect of evolution, and how to understand and handle emerging infectious diseases, but one can see how Darwinian Medicine has kept a more Neodarwinian view of evolution, where evolution of pathogens implies in the directional and opportunistic emergence of evolutionary novelties, instead of admitting evolutionary processes such as plasticity and conservatism to pathogen evolution. Increase of virulence, or host-switching events are, in this perspective, associated with rapid evolution of a novelty in order to realize host infection, instead of the possibility of a preexisting capacity. The six principles described by their perspective are interesting in order to comprehend the selective pressures in host-parasite interactions, but it does not discuss their implications for host and parasite diversification.
The understanding of evolution in these terms seems to agree with the general understanding of viral evolution, being host-switching events seen as rare and “unexpected”, as observed in many articles describing the evolution of emerging and reemerging pathogens (Lai, 2003; Benvenuto, 432 2020; Wu, 2020). The general strategy when handling the “new” viruses usually involves exploring the closely related species that are better understood by the scientific community, and through the concept of homology and phylogenetic conservatism, develop and test antiviral drugs and vaccines while knowledge of the actual emergent variant is not made available (Lai, 2003; Adler, 2022). Such an approach is evolutionarily sound and agrees with modern evolution.
Regarding the specific understanding of the Stockholm Paradigm, however, the traditional understanding of emerging infectious diseases seems to highlight the origin of novel capacity in the viral lineage, where mutations and recombination events that are evidenced in the new strain seem to be readily described as the causative agent of the species jump – as described in Darwinian Medicine-without discussing the possibility of preexisting capacity. The Stockholm Paradigm proposes that the emergence of an important capacity that conveniently enables the realization of a host-switching event should be less probable than the existence of capacity that was already part of the information space of the pathogen, that was expressed at the time due to opportunity (Brooks, 2019; Feronato, 2021; Boeger, 2022).
That congruence of capacity between emergent and non-emergent strains should be especially evidenced by the phylogenetic analysis of the group. When the phylogeny portrays the similarity between the “new” emergent disease and their sister taxa through small branch lengths or close monophyly, for example, these topological measures should highlight that their capacity space, or general information space, are more similar than different. When a host-switching event is readily associated with few mutations, they seem to end up receiving greater importance than the overall genetic similarity between sister viruses.
The discussion on the overestimation of mutations in emerging diseases is accompanied by the empirical evidence of the great frequency of host-switching events based on data from helminth host and insect-plant coevolutionary analyses, also presented by the authors of the Stockholm Paradigm (Brooks, 2019). Phylogenetic models that analyze coevolutionary processes seem to maximize cospeciation between symbiont species, which also reinstate the rarity of species jumps.
Viral evolution, therefore, could be more complex than is currently viewed. The current pandemic SARS-CoV-2, for example, has been extensively analyzed molecularly, phylogenetically and ecologically, and these incongruences between phylogenetic divergence and the general understanding of the viral group. We will now compare the phylogenetic data com four current host switching events: the new monkeypox virus (MPXV-2022), pandemic SARS-CoV-2, and pmd H1N1 (2009-2010), and raise the question of previous capacity space, in opposition with the emergence of a key novelty for species jump realization on differential host utilization.
8. Viruses and coevolutionary perspectives
Monkeypox
Monkeypox virus (MPXV) is a member of the Poxviridae family, and the Orthopoxvirus genus, along with smallpox, camelpox, cowpox, among other important pathogens for human and animal health. Like all orthopoxviruses, monkeypox is a double stranded DNA virus, being sister to smallpox virus, varying, however, in genome size, organization and ORFs (open reading frames) content. Identity with the 2 main Variola strains being 84.6% (with VARV-India) and 84.5% (VARV-Gar), with evidence of duplication of four ORFs in terminal regions of the genome, making MPXV genome slightly larger than that of VARV. The higher variation of monkeypox relative to smallpox is observed in the terminal regions of the genome, being the structural ORFs the most conserved portions of the DNA (Shchelkunov, 2005).
The terminal regions of the orthopoxvirus genome contains the majority of the virulence, immunomodulatory and host cell infection genes, previously investigated in VARV and camelpox (CPXV), where in MPXV two interferon resistance genes have been affected, resulting in lower efficiency of virus transmission via aerosol (Massung, 1995). Central African MPXV immunomodulatory response has also been previously described as less efficient than VARV, leading to stronger host inflammatory response (Shchelkunov et al., 1998). Lastly, MPXV genome, although not being the shortest, is one of the orthopoxviruses with fewer immune evasion genes-after two variola viruses-being camelpox the species with the greatest number of ORFs (18) (Shchelkunov, 2005; Kugelman, 2014). These and other investigations point to a lower transmission efficiency, and human-to-human transmission of monkeypox virus, supporting the 1980’s vaccination cessation (Shchelkunov, 2005; Fine, 1988).
Monkeypox is a disease first described in 1958 in a Danish laboratory, first studied in monkey hosts. In 1970, there was the first human outbreak of monkeypox, identified in a 9-month-old infant, with subsequent spread throughout the Democratic Republic of Congo in 1970. The cases have since been associated with close contact with wild hosts, being monkeys, or more recently, rodents (Giulio, 2004). Previous American introductions of the disease have been linked to close contact with infected prairie dogs, raising concern on monkeypox spread in the rodent population of the country.
As of the year 2000 there has been a clear increase in monkeypox reports, both confirmed and suspected, the second category bearing the majority of the cases. Between January and September of 2020, more than 4,000 suspected cases were reported in Nigeria (183 being confirmed cases, two of them diagnosed in Israel and Singapore) (Bunge, 2022).
The virus strains have been previously genetically characterized in two large clades, the Central and West African monkeypox, where the disease is endemic. The recent cases of disease in other regions are described as exportation of the disease. The African case fatality rate is considered low, being 3.6% in Western and 10.6% in Central African clades (Bunge, 2022). The largest epidemic spread of the disease before the year 2000 was documented in the Democratic Republic of Congo in 1996, with 520 confirmed cases of the Central African clade (Bunge, 2022).
Only in 2003 there was the identification of monkeypox cases outside of the African continent, being the US outbreak, which was later associated with infection by close contact with prairie dogs. The rodents seemed to be transported with infected hosts from Ghana, before being sold in Wisconsin (Reed, 2004; Bunge, 2022). All 47 cases were linked to exposure with the same shipment of prairie dogs, and after the event, the FDA and CDC banned any importation of rodents from Africa (Wisconsin Department of Health and Family Services).
The modern disease cases have been seen as a consequence of smallpox vaccination cessation after its eradication in 1980 in many countries, not being part of the routine vaccination program of many regions (Yong, 2020; Alankunle, 2020). It is also known that the traditional smallpox vaccine, designed with Vaccinia strains (Orthopox virus) also provided 85% protection against monkeypox (Fine, 1988). Unfortunately, Fine’s work, although reinstating the potential growth of monkeypox cases in the future as a consequence of the cessation of vaccination, agreed with the Global Commission for the Certification of Smallpox Eradication to discontinue vaccination based on the reduced transmission of monkeypox compared to smallpox viruses (Parker, 2007).
The reemergence of the disease identified in May 2022 (ECDC, 2022) was, from its beggining, characterized by two general scenarios: (1) infections associated with close contact with infected wild hosts; and (2) human-to-human infections through contact with large droplets of saliva, contaminated objects and with skin lesions (Velavan, 2022). The USA infection in 2003 as well as the UK epidemic of 2018-2019 have both been compared to the West and Central African clades, and traced back to West African MXPV based on genome identity.
Monkeypox outbreaks outside of its endemic region are also usually linked to travel-related events, which enables international spread of the disease, both among humans and its establishment in wild populations, especially rodents (Velavan, 2022). The most recent reports of the disease in several countries are yet to be linked by a source infection, suggesting human-human transmission. The emergence of monkeypox, in humans, however, needs to be better characterized according to the context that precluded the host-switch from the wild host to the human population.
Animal-human transmission is knowingly present in the endemic regions of the disease (Kugelman, 2014; Fuller, 2011), either by monkeys during the first identification of the virus (Giulio, 539 2004) or more recently, and especially in the American emergence of the disease, by rodents such as squirrels and prairie dogs (Guarner, 2004; Reed, 2004). In these cases, infection has been traced back to content with a sick wild host. The human-to-human cases, however, have been understood as phenomena where the specific MPXV genotype has suffered gene losses or frame-shifting mutations leading to its establishment in the human population. Gene losses and other mutations have been documented in Central African viruses (Kugelman, 2014; Vaughan, 2020), where the authors associate the genetic changes to higher transmission capacity in human hosts, but the USA and UK cases do not seem to present such evidence.
Several phylogenetic analyses have been performed in order to better understand general MPXV evolution, distinguishing two large clades of the viruses that are endemic to the Central and Western African countries. Regarding the recent outbreaks outside of Africa, however, the most recent publications that also include the May 2022 case from Portugal (Isidro, 2022) trace all the international strains as part of the Western MPX clade. It is important to note that the data shows a distinct separation of the USA strain of 2003 from the cases of Singapore 2020, Israel 2018 and strains from Europe of 2022, where almost all cases of the USA 2003 epidemic were associated with a shipment of infected prairie dogs to be kept as pets (Reed, 2004), whereas the other cases of 2018-2022 were not traced back to contact with a wild host, but associated with human-human transmission through sexual activity (Thornhill, 2022).
Finally, it seems that the emergence of monkeypox disease in non-endemic regions since 2018 are associated with a genomic change in the transmission dynamics of a subclade within the West African monkeypox. This well-determined phylogenetic difference (Isidro, 2022) points to a general change in capacity involving more than one monkeypox gene, instead to a punctual change in surface proteins that enable host-cell entrance, as has been discussed in SARS-CoV-2 reemergence of 2020 (Jain, 2020).
Influenza A H1N1
Influenza viruses are generally distinguished in 3 groups-A, B and C – which comprise half of the genera of Orthomyxoviridae family. As described by the International Committee on Viral Taxonomy (Shaw, 2013), viruses that belong to this family are segmented, negative-sense RNA viruses. A putative Influenza D virus (Collin, 2015), as well as the Wellfleet Bay virus, recently found infecting cattle in North America (Allison, 2015), are being included in the family (Suarez, 2016). Influenza A viruses (IAVs) are the most dominant taxon of the family, with the greatest host range, being wild birds considered the primordial reservoir of Influenza A – bears 16 HA subtypes of the 18 known – specially the Anseriformes and Charadriiformes orders. The centrality of wild birds as reservoirs for the viruses make these pathogens a constant and potential threat to human and animal populations.
Influenza A can be transmitted through host-switching events that either do not persist in the new species or become endemic in the new host population. In the latter case, the colonization of the new host species seems to reduce the rate of spillback, as is seen in equine, human and swine Influenza A (Suarez, 2016). Influenza A infections that are not established in the new host population are considered sporadic, although reinfection usually occurs.
Its morphology is well known, although not well understood, since its general form varies with the medium/ tissue. It can be visualized in two forms: spherical, around 80-120 nm when in cellular culture; and filamentous, with several micrometers size when observed in clinical isolates. It is composed of 8 RNA, packaged segments, which translate to 10 basic proteins, many of which present accessory proteins produced by alternative splicing and reading frame shift of themselves. The accessory proteins have knowingly acted as virulence factors (Chen, 2001) or immune response modulators (Jagger, 2012), however, many of them have no known function (Suarez, 2016).
The complexity of IAVs replication strategy has been extensively explored, and mechanisms involving host-switching have also been elegantly described. The accessory binding of the HA (hemagglutinin) protein to the surface glycoproteins containing either 2-3 or 2-6 sialic acids has been robustly described as determinants to viral entry in the host cell (Suzuki, 2005). The abundance and diversity of sialic acid type by tissue and host species have so far explained the colonization of swine (both 2-3 and 2-6), human (2-6) and avian (2-3) hosts, as well as the mixing vessel role swine species have presented in the ecology and evolution of IAVs. The pmdH1N1 fits in that epidemiological scenario, where its emergence in human, swine, turkeys, ferrets, and other species in a sporadic manner (Pantin Jackwood, 2010; Hinshaw, 1983; Vincent, 2014).
At the same time, studies involving wild avian Influenza viruses have revealed the extensive amino-acid diversity of genotypes in this group of hosts, which withhold almost the entirety of IAVs subtypes, except the recently described bat IAVs. For these viruses, previous studies have revealed that, among all 16 HA subtypes, around 25% of the amino acid sequence is conserved, whereas the subtype divergence, when in a pairwise comparison, can vary between 20 – 63% (Nobusawa, 1991). Both the amino acidic and the nucleotide (Stallknecht, 1998; Suarez, 2000) sequences discuss a geographical clusterization of IAVs lineages among American and Eurasian groups. Meanwhile, the American/Eurasian hypothesis is met with exceptions such as the shorebird and gull H2 lineages, although they have been explained as unique subpopulations of the hemagglutinin gene, associated with the migratory nature of the hosts (Makarova 1999, Suarez, 2016).
When addressing, more specifically, the host-switching event seen in the pmdH1N1, one can realize that the swine-like H1N1 IAV is actually a result of serial processes of reassortment between swine, avian and human viruses that occurred in swine populations, was undetected for decades in the population, and emerged in humans in the host-switching event that occurred in Mexico in 2009 (CDC, 2009).
The complex segment interchange is accompanied by one simple conclusion: the pandemic strain must have been maintained in low frequencies in the original host population, and the substitutions described in the strain’s genome, for the 5 sublineages, have no known, or important, functional benefit that enabled human colonization (Tumpey, 2004; Garten, 2009). At the same time, previous studies have found not one marker associated with adaptation to human hosts (mapped using 1918 H1N1 and HPIAV H5N1) (Zamarin, 2006; Jackson, 2008; Garten, 2009). The authors of these important works conclude by highlighting that the pmdH1N1 has no classical “human adaptation” substitutions, and yet successfully infected and established itself in the human population, also being antigenically (in terms of H1 segment) similar to other Eurasian swine Influenza viruses, with no notable difference in their antigenic properties (Russell, 2008).
Experimental HIA have shown little to no cross-reactivity, however, when comparing ferret HI response to pmdH1N1 pos-infection with a closely related swine H1N1 (Garten, 2009). Dunham et al. Also analyze and conclude the low genetic novelty of pmdH1N1, compared to other swine-origin H1N1, and describe the same lack of “human adaptation” substitutions (Dunham, 2009), although they did not analyze the antigenic properties of the pmdH1N1 as Garten has.
Finally, one can see how the host-switching event of pmdH1N1/2009 paints a more complicated scenario than that of the previous cases discussed here. The colonization of humans has not been linked to substitutions associated with human utilization, whereas its long-term – and undetected -survival in swine hosts, along with multiple reassortments between Eurasian swine and avian Influenza has given ample opportunity for virus transmission in Mexico. It is also important to note that the homogeneous nature of the pmdH1N1 suggests a single or few events of colonization, both suggested by Garten et al (2009) and as predicted by the classical understanding of Founder’s effect (Mayr, 1963).
9. Host information space
Although the main focus of this work is to insert viral lineages within the parasite symbiont context discussed by the different coevolutionary frameworks, it is important to also discuss the diversity in the host lineage, and how it can influence the emergence of a new symbiosis. The host is usually represented in the Stockholm Paradigm as the selective pressure being imposed on the phenotypic variety of the parasite population or lineage. This vision, however, is used in order to enable a better understanding of the Ecological Fitting dynamics, when it can also be understood as a complex system both considering the parasite’s phenotypic diversity and the host’s phenotypic (and resource availability) variation (Brooks, 2019; Sax, 2007).
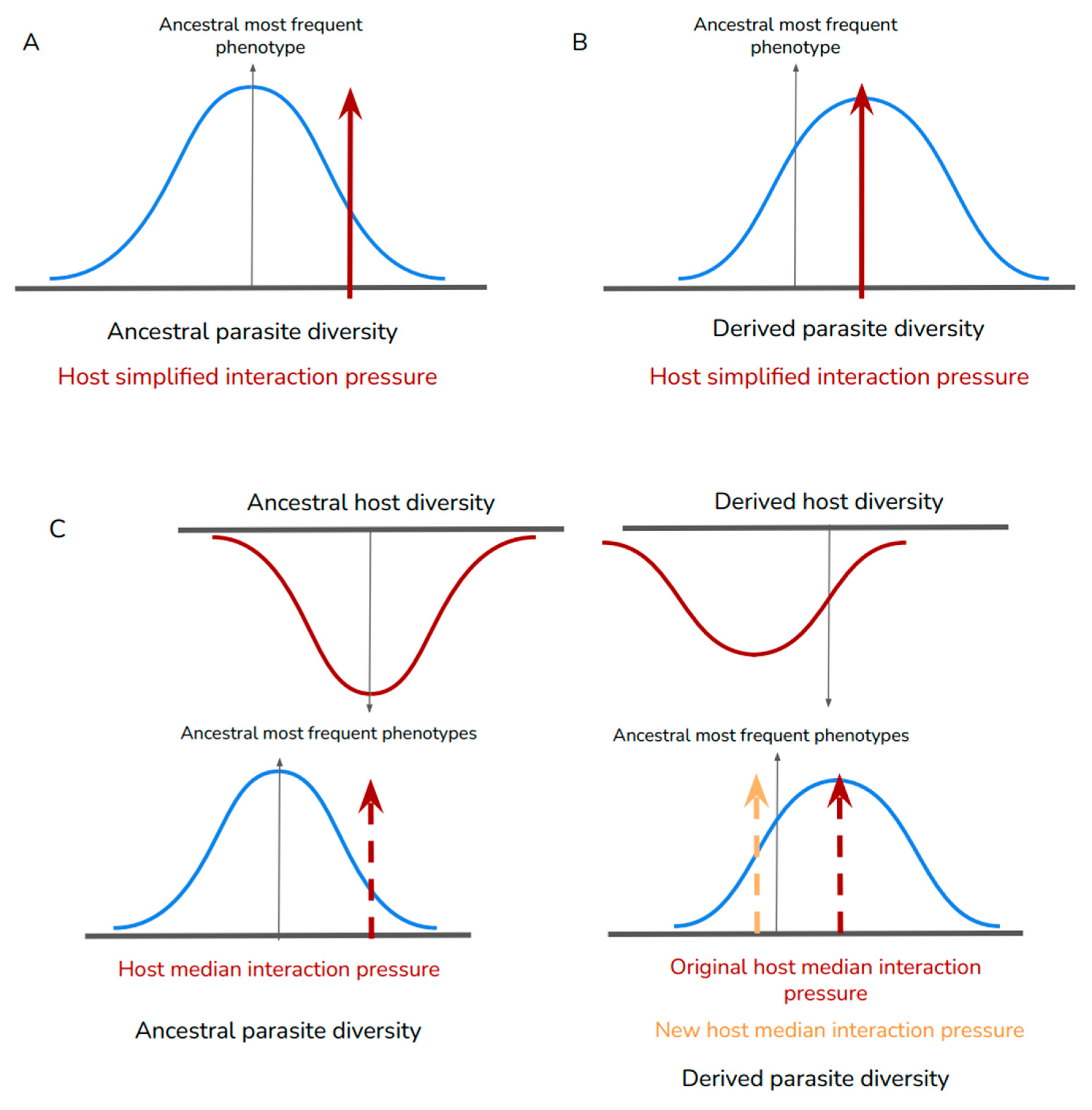
We can describe the host’s complexity in the same way we describe parasite’s capacity diversity, where the host capacity space is not defined by a single phenotype (Fig. 4A and B), but varies in the population (Fig. 4C). In this way, the interaction pressure during the establishment of a new biological interaction is actually a gradient, considering resource availability by the host and the viral exploitation capacity (Brooks, 2019). While the most frequent selection pressure will induce an alteration in the parasite lineage, the host’s lineage can also be influenced by the interaction, resulting in differential selection pressure on both sides of the symbiosis (Agosta, 2006).
Figure 4.
Adding complexity to the host information space change in a new symbiosis. In A and B we represent a hypothetical alteration in the variety (and capacity space) of a pathogen population, where the host interaction pressure is represented by an optimal phenotype application, and the optimal pathogen phenotype is the one which exploits the host’s resources more efficiently. A represents the initial stage of the new interaction, and B represents the later phenotypic distribution, once the pathogen population has evolved under the host interaction pressure. In C we represent the host’s phenotype diversity, showing that it also varies in terms of compatibility with the pathogen. The median host interaction pressure is represented by the red line, whereas the orange line represents the new median interaction pressure, since both populations (and capacity spaces) are influenced by the interaction.
Figure 4.
Adding complexity to the host information space change in a new symbiosis. In A and B we represent a hypothetical alteration in the variety (and capacity space) of a pathogen population, where the host interaction pressure is represented by an optimal phenotype application, and the optimal pathogen phenotype is the one which exploits the host’s resources more efficiently. A represents the initial stage of the new interaction, and B represents the later phenotypic distribution, once the pathogen population has evolved under the host interaction pressure. In C we represent the host’s phenotype diversity, showing that it also varies in terms of compatibility with the pathogen. The median host interaction pressure is represented by the red line, whereas the orange line represents the new median interaction pressure, since both populations (and capacity spaces) are influenced by the interaction.
Because of the accumulation of complexity in the symbiotic interaction, described in terms of Ecological Fitting, one can see how both lineages influence each other, however it is important to note that this process is not necessarily protein-related, or unidirectional. The mutual adjustment involves a complexity of phenotype alteration both on the pathogen and in the host population phenotypes, where in the pathogens these changes occur more rapidly due to its shorter generation time and simpler structure which result in a more flexible information space (Feronato, 2021, Brooks, 2019) and higher adaptability.
Final Remarks
This review aims to discuss the main frameworks on coevolution since Darwin, as well as discuss some of the various recent host-switching events involving virus-host interactions. With this we intended to highlight the complexity of these symbioses, which outreach the simple processes of selection described by the early coevolutionary frameworks such as Orthogeneticism and Neodarwinism, symbioses that are today being addressed by modern theories of evolution.
Of the three host-pathogen interactions succinctly explored here, one can see how the evolution of the pathogen does not follow a simple process of selection of the best trait, nor can it be fully explained by the emergence of a mutation that enabled the species jump. The colonization of a new host seems to rely on both the compatibility of symbionts and their ecological congruence, as described by the Stockholm Paradigm.
The Extended Synthesis, while modern, does not address the host-pathogen evolution beyond its gradual, gene centered and externalist perspective. Because of these general views of the theory, Influenza A H1N1 virus evolution, for example, cannot be fully understood, since its host switching events are not associated with any specific gene evolution, but involves a complex net of capacity/opportunity interaction.
Finally, highlighting the importance of the coevolutionary understanding of emerging diseases by the medical field of research was also one of the objectives of this work, since human-virus interactions seem to be increasingly more complex than described by traditional 1 pathogen: 1 host evolutionary understanding. We find it of extreme importance to further incorporate coevolution into the problem-solving framework of health, and hope that this work attests to it.
Acknowledgements
The authors would like to express their appreciation for all the researchers who critically reviewed this work, including: Juliano Bordignon, Tiago Graf, Patricia Shigunov, Wander R. Pavanelli and Walter A. P. Boeger. Your input greatly enriched this work. We also thank Instituto Carlos Chagas for the continuous support of this research.
Conflict of Interest
the authors would like to state that there is no conflict of interest involving this work and agree that the submitted work is the final version of this article.
References
- Darwin, Charles. On the origin of species, 1859.
- Eimer, G. H. T. 1898. On orthogenesis and the impotence of natural selection in species formation. The Open Court Publishing Company, Chicago, 56 p. [Translated by J. M. McCormack.].
- Brooks, Daniel R., Eric P. Hoberg, and Walter A. Boeger. "The Stockholm Paradigm." The Stockholm Paradigm. University of Chicago Press, 2019.
- Agosta, Salvatore J., and Daniel R. Brooks. The Major Metaphors of Evolution. Series in Evolutionary Biology: New Perspectives in Its Development. Cham: Springer Nature Switzerland, 2020.
- Eichler, Wolfdietrich (1948) XLI.— Some rules in Ectoparasitism. Annals and Magazine of Natural History, 1:8, 588-598. [CrossRef]
- Kellog, 1907. Darwinism to-day: A discussion of present-day scientific criticism of the Darwinian selection theories, together with a brief account of the principal other proposed auxiliary and alternative theories of species-forming. Henry Holt and Company, New York, 403 p.
- Brues, C. T. 1924. The specificity of food-plants in the evolution of phy-tophagous insects. American Naturalist 58:127–144.
- Brues, C. T. 1921. Correlation of taxonomic affinities with food habits in Hymenoptera, with special reference to parasitism. American Naturalist 55:134–164. [CrossRef]
- Herbert Spencer, “The Development Hypothesis” (Leader, March 20, 1852; repr. in Essays, I [New York: Appleton, 19071, l-7).
- Dethier, Vincent J. Chemical insect attractants and repellents. Vol. 65. No. 2. LWW, 1948. [CrossRef]
- Braga, Mariana P., et al. "Bayesian inference of ancestral host–parasite interactions under a phylogenetic model of host repertoire evolution." Systematic biology 69.6 (2020): 1149-1162. [CrossRef]
- Pigliucci, Massimo, and Gerd B. Müller. "Elements of an extended evolutionary synthesis." Evolution: The extended synthesis (2010): 3-17.
- Brooks, Daniel R. "The major metaphors of evolution: Visualizing the extended synthesis." Evolution: Education and Outreach 4.3 (2011): 446-452. [CrossRef]
- Verschaeffelt, E. 1910. The cause determining the selection of food in some herbivorous in-sects. Proc. Acad. Sci., Amsterdam, 13: 536-542.
- R. S. Bigelow, Monophyletic Classification and Evolution, Systematic Biology, Volume 5, Issue 4, December 1956, Pages 145–146. [CrossRef]
- Nesse R. M., Williams G. C. 1999. Research designsthat address evolutionary questions about medicaldisorders. Pages 16–22 inEvolution in Health &Disease, edited by S. Stearns. New York: OxfordUniversity Press.
- Ewald, Paul W., et al. "Vertical and vector-borne transmission of insect endocytobionts and the evolution of benignity." Insect Endocytobiosis: Morphology, Physiology, Genetics, and Evolution. CRC, Boca Raton, FL (1989): 21-35.
- Nesse R. M., Williams G. C. 1995.Evolution and Heal-ing: New Science of Darwinian Medicine. London(UK): Weidenfeld and Nicholson.
- Tracy, Sarah W. "George Draper and American constitutional medicine, 1916-1946: reinventing the sick man." Bulletin of the History of Medicine 66.1 (1992): 53-89.
- Adams, Joseph. A Treatise on the Supposed Hereditary Properties of Diseases: Containing Remarks on the Unfounded Terrors. J. Callow, 1814.
- Wallace, Bruce. "Fifty years of genetic load." Fifty Years of Genetic Load. Cornell University Press, 2019.
- Elton, Charles S., and Charles S. Elton. The reasons for conservation. Springer US, 1958.
- Carson, Rachel. "Silent spring. 1962." (2009).
- Zampieri, Fabio. "Medicine, evolution, and natural selection: an historical overview." The Quarterly Review of Biology 84.4 (2009): 333-355. [CrossRef]
- Williams, George C., and Randolph M. Nesse. "The dawn of Darwinian medicine." The Quarterly review of biology 66.1 (1991): 1-22. [CrossRef]
- Gerhart, John, and Marc Kirschner. "The theory of facilitated variation." Proceedings of the National Academy of Sciences 104.suppl_1 (2007): 8582-8589. [CrossRef]
- Hennig, Willi. "Phylogenetic systematics." Annual review of entomology 10.1 (1965): 97-116.
- Waller, John. "'The illusion of an explanation': The concept of hereditary disease, 1770-1870." Journal of the History of medicine and Allied Sciences 57.4 (2002): 410-448. [CrossRef]
- Feronato, Sofia G., Sabrina Araujo, and Walter A. Boeger. "‘Accidents waiting to happen’—Insights from a simple model on the emergence of infectious agents in new hosts." Transboundary and Emerging Diseases 69.4 (2022): 1727-1738.. [CrossRef]
- Shchelkunov, Sergei Nikolaevich, Svetlana S. Marennikova, and Richard W. Moyer. Orthopoxviruses pathogenic for humans. Springer Science & Business Media, 2006.
- Pasteur, Louis. "On the germ theory." Science 62 (1881): 420-422. [CrossRef]
- European Centre for Disease Prevention and Control. Monkeypox multi-country outbreak – 23 May 2022. ECDC: Stockholm; 2022.
- Isidro, J., Borges, V., Pinto, M. et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat Med 28, 1569–1572 (2022). [CrossRef]
- Méthot, Pierre-Olivier. "Darwin, evolution, and medicine: Historical and contemporary perspectives." Handbook of evolutionary thinking in the sciences (2015): 587-617.
- Thornhill, John P., et al. "Monkeypox virus infection in humans across 16 countries—April–June 2022." New England Journal of Medicine 387.8 (2022): 679-691. [CrossRef]
- Minelli A. 2003. The Development of Animal Form: Ontogeny, Morphology, and Evolution. Cambridge (UK): Cambridge University Press .
- Ewald, Paul W. "Transmission modes and the evolution of virulence: with special reference to cholera, influenza, and AIDS." Human Nature 2.1 (1991): 1-30. [CrossRef]
- Woo PC, Lau SK, Wernery U, Wong EY, Tsang AK, Johnson B, Yip CC, Lau CC, Sivakumar S, Cai JP, Fan RY, Chan KH, Mareena R, Yuen KY. Novel betacoronavirus in dromedaries of the Middle East, 2013. Emerg Infect Dis. 2014 Apr;20(4):560-72. PMID: 24655427; PMCID: PMC3966378. [CrossRef]
- Woo PC, Wang M, Lau SK, Xu H, Poon RW, Guo R, Wong BH, Gao K, Tsoi HW, Huang Y, Li KS, Lam CS, Chan KH, Zheng BJ, Yuen KY. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J Virol. 2007 Feb;81(4):1574-85. Epub 2006 Nov 22. PMID: 17121802; PMCID: PMC1797546. [CrossRef]
- Ji, W., Wang, W., Zhao, X., Zai, J., & Li, X. (2020). Homologous recombination within the spike glycoprotein of the newly identified coronavirus may boost cross-species transmission from snake to human. Journal of Medical Virology. [CrossRef]
- Benvenuto D, Giovanetti M, Ciccozzi A, Spoto S, Angeletti S, Ciccozzi M. The 2019-new coronavirus epidemic: Evidence for virus evolution. J Med Virol. 2020 Apr;92(4):455-459. Epub 2020 Feb 7. PMID: 31994738; PMCID: PMC7166400. [CrossRef]
- Naqvi AAT, Fatima K, Mohammad T, Fatima U, Singh IK, Singh A, Atif SM, Hariprasad G, Hasan GM, Hassan MI. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim Biophys Acta Mol Basis Dis. 2020 Oct 1;1(10):165. Epub 2020 Jun 13. PMID: 32544429; PMCID: PMC7293463. [CrossRef]
- Gonzalez, J. M., et al. "A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae." Archives of virology 148.11 (2003): 2207-2235. [CrossRef]
- Helmy, Y.A.; Fawzy, M.; Elaswad, A.; Sobieh, A.; Kenney, S.P.; Shehata, A.A. The COVID-19 Pandemic: A Comprehensive Review of Taxonomy, Genetics, Epidemiology, Diagnosis, Treatment, and Control. J. Clin. Med. 2020, 9, 1225. [CrossRef]
- Fagbami AH, Monath TP, Fabiyi A. Dengue virus infections in Nigeria: a survey for antibodies in monkeys and humans. Trans R Soc Trop Med Hyg. 1977;71(1):60-5. PMID: 404737. [CrossRef]
- Vasilakis N, Tesh RB, Weaver SC. Sylvatic dengue virus type 2 activity in humans, Nigeria, 1966. Emerg Infect Dis. 2008 Mar;14(3):502-4. PMID: 18325274; PMCID: PMC2570833. [CrossRef]
- Weaver SC, Tesh RB, Vasilakis N. Sylvatic Dengue Virus Type 2 Activity in Humans, Nigeria, 1966. Emerging Infectious Diseases. 2008;14(3):502-504. [CrossRef]
- Wang E, Ni H, Xu R, Barrett AD, Watowich SJ, Gubler DJ, Weaver SC. Evolutionary relationships of endemic/epidemic and sylvatic dengue viruses. J Virol. 2000 Apr;74(7):3227-34. PMID: 10708439; PMCID: PMC111823. [CrossRef]
- Vasilakis, N., Cardosa, J., Hanley, K. et al. Fever from the forest: prospects for the continued emergence of sylvatic dengue virus and its impact on public health. Nat Rev Microbiol 9, 532–541 (2011). [CrossRef]
- Vasilakis N, Shell EJ, Fokam EB, Mason PW, Hanley KA, Estes DM, Weaver SC. Potential of ancestral sylvatic dengue-2 viruses to re-emerge. Virology. 2007 Feb 20;358(2):402-12. Epub 2006 Oct 2. PMID: 17014880; PMCID: PMC3608925. [CrossRef]
- Forattini, O.P. (2003). Epidemiology and Phylogenetic Relationships of Dengue Viruses. Dengue Bulletin – Vol 27, 2003 91.
- Weaver SC, Barrett AD. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat Rev Microbiol. 2004 Oct;2(10):789-801. PMID: 15378043; PMCID: PMC7097645. [CrossRef]
- Valentine MJ, Murdock CC, Kelly PJ. Sylvatic cycles of arboviruses in non-human primates. Parasit Vectors. 2019 Oct 2;12(1):463. PMID: 31578140; PMCID: PMC6775655. [CrossRef]
- Weaver SC, Vasilakis N. Molecular evolution of dengue viruses: contributions of phylogenetics to understanding the history and epidemiology of the preeminent arboviral disease. Infect Genet Evol. 2009 Jul;9(4):523-40. Epub 2009 Feb 13. PMID: 19460319; PMCID: PMC3609037. [CrossRef]
- Halstead SB. Dengue. Lancet. 2007 Nov 10;370(9599):1644-52. PMID: 17993365. [CrossRef]
- Shaw, M. L. and P. Palese. 2013. Orthomyxoviridae. In: Fields Virology, 6th edition, D. M. Knipe and P. M. Howley, eds. Wolter Luwer: Baltimore, MD. 1151–1185.
- Ewald, Paul W. "Transmission modes and evolution of the parasitism-mutualism continuum." Annals of the New York Academy of Sciences 503 (1987): 295-306.. [CrossRef]
- Collin, E. A., Z. Sheng, Y. Lang, W. Ma, B. M. Hause, and F. Li. 2015. Co-circulation of two distinct genetic and antigenic lineages of proposed influenza D virus in cattle. Journal of Virology 89:1036–1042. 922. [CrossRef]
- Allison, A. B., J. R. Ballard, R. B. Tesh, J. D. Brown, M. G. Ruder, M. K. Keel, B. A. Munk, R. M. Mickley, S. E. Gibbs, A. P. Travassos da Rosa, J. C. Ellis, H. S. Ip, V. I. Shern-Bochsler, M. B. Rogers, E. Ghedin, E. C. Holmes, C. R. Parrish, and C. Dwyer. 2015. Cyclic avian mass mortality in the northeastern United States is associated with a novel orthomyxovirus. Journal of Virology 89:1389–1403. [CrossRef]
- Suarez, D.L. (2016). Influenza A virus. In Animal Influenza, D.E. Swayne (Ed.). [CrossRef]
- Chen, W., P. A. Calvo, D. Malide, J. Gibbs, U. Schubert, I. Bacik, S. Basta, R. O’Neill, J. Schickli, P. Palese, P. Henklein, J. R. Bennink, and J. W. Yewdell. 2001. A novel influenza A virus mitochondrial protein that induces cell death. Nature Medicine 7:1306–1312. [CrossRef]
- CHEUNG, T.K.W. and POON, L.L.M. (2007), Biology of Influenza A Virus. Annals of the New York Academy of Sciences, 1102: 1-25. [CrossRef]
- Jagger, B. W., H. M. Wise, J. C. Kash, K. A. Walters, N. M. Wills, Y. L. Xiao, R. L. Dunfee, L. M. Schwartzman, A. Ozinsky, G. L. Bell, R. M. Dalton, A. Lo, S. Efstathiou, J. F. Atkins, A. E. Firth, J. K. Taubenberger, and P. Digard. 2012. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204. [CrossRef]
- Pantin-Jackwood, M., J. L. Wasilenko, E. Spackman, D. L. Suarez, and D. E. Swayne. 2010. Susceptibility of turkeys to pandemic-H1N1 virus by reproductive tract insemination. Virology Journal 7:27. [CrossRef]
- Hinshaw, V. S., R. G. Webster, W. J. Bean, J. Downie, and D. A. Senne. 1983. Swine influenza-like viruses in turkeys: potential source of virus for humans? Science 220:206–208. [CrossRef]
- Vincent, A., L. Awada, I. Brown, H. Chen, F. Claes, G. Dauphin, R. Donis, M. Culhane, K. Hamilton, N. Lewis, E. Mumford, T. Nguyen, S. Parchariyanon, J. Pasick, G. Pavade, A. Pereda, M. Peiris, T. Saito, S. Swenson, K. Van Reeth, R. Webby, F. Wong, and J. Ciacci-Zanella. 2014. Review of influenza A virus in swine worldwide: a call for increased surveillance and research. Zoonoses and Public Health 61:4–17. [CrossRef]
- Nobusawa, E., T. Aoyama, H. Kato, Y. Suzuki, Y. Tateno, and K. Nakajima. 1991. Comparison of complete amino acid sequences and receptor-binding properties among 13 serotypes of hemagglutinins of influenza A viruses. Virology 182:475–485. [CrossRef]
- Stallknecht, D. E. 1998. Ecology and epidemiology of avian influenza viruses in wild bird populations: waterfowl, shorebirds, pelicans, cormorants, etc. In: Proceedings of the Fourth International Symposium on Avian Influenza, D. E. Swayne and R. D. Slemons, eds. United States Animal Health Association: Athens, GA. 61–69.
- Suzuki, Y. 2005. Sialobiology of influenza: molecular mechanism of host range variation of influenza viruses. Biological and Pharmaceutical Bulletin 28:399–408. [CrossRef]
- Suarez, D. L. 2000. Evolution of avian influenza viruses. Veterinary Microbiology 74:15–27. [CrossRef]
- Makarova, N. V., N. V. Kaverin, S. Krauss, D. Senne, and R. G. Webster. 1999. Transmission of Eurasian avian H2 influenza virus to shorebirds in North America. Journal of General Virology 80:3167–3171. [CrossRef]
- Garten, Rebecca J., et al. "Antigenic and genetic characteristics of swine-origin 2009 A (H1N1) influenza viruses circulating in humans." science 325.5937 (2009): 197-201. [CrossRef]
- Zamarin, Dmitriy, Mila B. Ortigoza, and Peter Palese. "Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice." Journal of virology 80.16 (2006): 7976-7983. [CrossRef]
- Jackson, David, et al. "A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity." Proceedings of the National Academy of Sciences 105.11 (2008): 4381-4386. [CrossRef]
- Russell, Colin A., et al. "The global circulation of seasonal influenza A (H3N2) viruses." Science 320.5874 (2008): 340-346. [CrossRef]
- Dunham, Eleca J., et al. "Different evolutionary trajectories of European avian-like and classical swine H1N1 influenza A viruses." Journal of virology 83.11 (2009): 5485-5494. [CrossRef]
- Mayr, Ernst. Animal Species and Evolution, Cambridge, MA and London, England: Harvard University Press, 1963. [CrossRef]
- Centers for Disease Control and Prevention (CDC). Outbreak of swine-origin influenza A (H1N1) virus infection-Mexico, March-April 2009. MMWR Morb Mortal Wkly Rep. 2009 May 8;58(17):467-70. PMID: 19444150.
- Sax, Dov F., et al. "Ecological and evolutionary insights from species invasions." Trends in ecology & evolution 22.9 (2007): 465-471. [CrossRef]
- Agosta, S.J. (2006) On ecological fitting, plant-insect associations, herbivore host shifts, and host plant selection. Oikos 114, 556–565.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



