
Submitted:
17 May 2023
Posted:
17 May 2023
You are already at the latest version
Abstract
Excessive extracellular histones can damage cells despite their importance in DNA structure and gene regulation. We previously showed the harmful effect of histone 3.3 (H3.3) and its acetylated form (AcH3.3) in chronic obstructive pulmonary disease (COPD), but further research is needed to understand the in vivo underlying mechanisms. To investigate this, we administered a single dose of recombinant histones via intra-tracheal instillation in C57BL/6 mice and analyzed the outcomes after 48 hours. Both rH3.3 and rAcH3.3 effectively reached the bronchial and alveolar space. Lung histology revealed severe immune cell infiltration, rupture, damage, and thickening of many alveolar walls. Micro-CT scans showed macroscopic structure changes with a decreased tissue volume to total volume ratio. The administration of rH3.3 and rAcH3.3 induced lung apoptotic activity, as indicated by increased levels of cleavage caspase 3 and 9. Moreover, elevated plasma levels of pro-inflammatory cell mediators, including TNF-alpha, IL-6, MCP-3, and CXCL-1, were observed. The pathological changes and pro-inflammatory cytokine response were more pronounced in animals treated with rAcH3.3, which also exhibited leukocytosis and lymphocytosis. These findings emphasize the crucial role of extracellular H3.3 acetylation in inducing cytotoxicity and a robust, acute inflammatory response similar to what occurs in the lungs of COPD patients.
Keywords:
Histones
; COPD
; HDAC
; H3.3
; Inflammation
; Hyperacetylations
; Cytotoxicity
; Alveolar damage
; Cytokines.
1. Introduction
Chronic obstructive pulmonary disease (COPD) is a leading cause of disability and death worldwide [1,2,3,4,5]. COPD comprises two main conditions, namely emphysema and chronic bronchitis. Emphysema involves the destruction of the airways and alveoli, while chronic bronchitis is characterized by inflammation and narrowing of the airways. In recent decades, the cost of COPD-related healthcare has been exponentially increasing [6,7]. Disease management can relieve symptoms and prolong life; however, there are no treatments to stop disease progression. Cigarette smoke is the primary cause of COPD [8,9]. Other risk factors for developing COPD include exposure to occupational dust and chemicals, air pollution, genetic factors and respiratory infections [10].
The main mechanism involved in the pathogenesis of COPD is the persistent inflammation of the airways [10,11]. Factors such as ongoing exposure to cigarette smoke can exacerbate this inflammatory response. In addition to inflammation, other well-known mechanisms of COPD include the imbalance between proteases and anti-proteases, as well as oxidants and anti-oxidants [11,12]. These processes collectively lead to various pathogenic events, including inflammation, apoptosis, oxidative stress, damage of extracellular matrix structure, and abnormal tissue remodeling [10,11,12].
A common underlying factor in these pathologic processes is the presence of post-translational modifications (PTMs) in crucial lung proteins, which impair their normal function [13]. These proteins encompass acetyl- and methyltransferases, deacetyl- and demethylases, as well as histones [4,9,12,14,15]. Among the PTMs observed in COPD, acetylations and methylations are the most prevalent. We demonstrated that Histone 3 (H.3.3) and the acetylated form of H3.3 are significantly elevated in the lung of subjects with COPD [12]. Furthermore, investigations, using proteomics techniques have also revealed PTMs in histones H3 and H4 in lung cells [16,17]. Notably, histones, as well as non-histone proteins, can undergo PTMs that influence their function in COPD. These modifications include acetylation and deacetylation mediated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. The dysregulation of these PTMs and associated enzymes has been implicated in the pathogenesis of COPD [9,15,16,17].
Besides having nuclear functions, emerging studies suggest that histones can be released into extracellular spaces by activated immune, apoptotic, or necrotic cells. Histones could exhibit significant toxic or pro-inflammatory activity in vivo and in vitro [18,19,20]. Moreover, we found that extracellular H3.3 was cytotoxic to human primary lung structural cells in vitro through a mechanism driven by disruption of calcium homeostasis, endoplasmic reticulum (ER) stress, and mitochondrial membrane permeability that leads to the caspase activation and apoptosis[12]. These findings agreed with previous reports that showed calcium signaling mediated apoptosis in cells exposed to nucleosome histones [13,21].
Normally histones are the nuclei’s key components of chromatin. Histones build nucleosomes, octamers composed of two molecules or each core histones H2A, H2B, H3, and H4 that package the DNA and regulate the gene transcription by modifications that occur in its amino acid sequence[22]. COPD is associated with reduced HDAC activity and impaired proteasome degradation, resulting in increased histone acetylation, which can alter nucleosome histone composition[23]. Moreover, this hyperacetylation of histones has been related to inflammatory gene expression [9,15,24,25]. For those reasons, further studies of the pathologic effects and possible mechanisms of action of the aberrant extracellular histones in lung tissue are in need. In this study, we aim to analyze the direct lung toxicity of H3.3 and compare it with its acetylated form, AcH3.3, in mice in vivo. We hypothesize that aberrant extracellular H3.3 contributes to lung toxicity in COPD, and understanding its mechanisms of action could offer new insights into the disease's pathophysiology and potentially lead to the development of novel therapeutic interventions.
2. Results
2.1. Histone 3.3 Deposits at the Alveolar Epithelial Lining and the Surface of the Bronchial Lumen
To demonstrate the presence of the proteins at the alveolar tissue after the intratracheal instillation, a single dose of 25 uL of recombinant Histone 3.3 (rH3.3) (1ug/g of body weight in PBS) was administered to a mouse and euthanized 30 minutes after the lung instillation. Vehicle (25uL of PBS) instillation was used as a control. Both lungs were paraformaldehyde-fixed, paraffin-embedded, and sectioned. Slides were subjected to immunohistochemistry and tested for H3.3 using a polyclonal antibody. The presence of H3.3 after expossure of the biotinylated antibody at the bronchoalveolar surface confirmed the presence of the rH3.3 after the intratracheal administration when compared with control (Figure 1). Animals instilled with PBS showed minimal biotin-antibody-H3.3 staining at the nuclei of the alveolar cells with no presence in the extracellular space. In contrast, extracellular staining was increased in the mouse lungs instilled with the histones at the alveolar walls and the bronchial lumen.
2.2. Histone 3.3 and Hyperacetylated Histone 3.3 Severely Damage Lung Tissue after 48 Hours of Direct Instillation
We tested the in vivo toxicity of H3.3 in C57BL/6J mice. Thirty-two 10-week old, male mice obtained from Jackson laboratories were randomly separated into four groups of eight animals. Each animal received an intratracheal dose, in a volume that did not exceed 25 µL of the control vehicle, PBS, or 25uL of the recombinant proteins at 1µg/g body weight dose of rH2A, the non-cytotoxic histone; rH3.3 or rAcH3.3. Forty eight hours after the lung instillation, animals were euthanized and lung tissues were processed for a histochemistry analysis. Compared to controls (PBS- and rH2A-treated) the thickness of the epithelial airway was increased in the rH3.3- and rAcH3.3-treated lungs. Increase leukocyte infiltration at the bronchial and the alveolar interstitium was also observed in these same groups. Alveolar wall damage including enlargement of alveolar space and structural cell disruption was observed. These pathological changes were more pronounced in the rAcH3.3 group, compared to the other groups (Figure 2). Thus, rH3.3 and rAcH3.3 induce cytotoxicity and acute lung damage in mice similar to the one observed in human COPD lungs.
2.3. Micro-CT Scanning and 2D/3D Reconstruction of Murine Lungs Showed Decrease in Lung Tissue Density
To further evaluate the extent of the lung damage, 3D and 2D models of the architecture of the lungs were perfomed by micro-CT scanning. Micro-CT quantification algorithms allowed us to accurately measure the extent of the lung destruction and the distribution of air trapping. To visualize soft tissue in the micro-CT, lung tissues were immersed for three days in PTAH, a hematoxillin-phosphotungstic solution, which makes soft tissues radiopaque. Multiple sections were acquired (at 5.89 mm2 voxel resolution) using the Skyscan®1172, ex-vivo Micro-CT instrument. (a) A sagital section of the 3D reconstruction of the lung micro-CT images are shown (Figure 3A). There were no significant macroscopical changes between the lungs analyzed. However, when the small alveolar areas were analyzed in the 2D micro-CT sections, a decrease in the radiopacity of the PTAH-stained tissue with sporadic punctual condensations in the parenchymal tissue was observed in the rH3.3 and rAcH3.3-treated mice (Figure 3B). Significant increased alveolar air space with a reduction of the lung tissue volume was also detected in the lungs of rAcH3.3-treated mice when compared to rH2A controls (Figure 3C,D).
2.4. Leucocytosis and Lymphocytosis in Plasma after Instillation of rH3.3 and rAcH3.3
To evaluate systemic changes after the tracheal instillation of the histones, automated cell blood counts from blood samples were measured. rAcH3.3 instillation increased significantly the leucocyte count when compared with PBS control, **p < 0.01. The leucocytosis observed in the rAcH3.3 was at the expenses of lymphocytes, *p < 0.05. There were no changes in the white blood cells with rH3.3. However, an increase in neutrophil count was observed after the tracheal instillation of all of the histones, including the histone control H2A (Figure 4). There was a tendency toward an increase in platelet count after rAcH3.3 administration but did not reach statistical significance. Leucocyte, neutrophil, and platelet cell counts in the BALF were increased with all histones instilled, but did not reach statistical significance. No statistical changes in the lymphocyte cell count of the BALF were observed.
2.5. Extracellular rH3.3 and rAcH3.3 Induced Apoptosis at the Damaged Alveolar Tissue
There is an increase in the apoptotic activity and lung damage at the alveoli wall in COPD. Therefore, to evaluate the apoptotic activity in the alveolar wall, we investigate the caspase cleavage hallmarks expression of apoptosis, cleaved caspase-3 (cCaspase-3) and cleaved caspase-9 (cCaspase-9). cCaspase-3 is considered the “executioner of apoptosis” while cCaspase-9 is an activator of the cascade of caspases involved in the execution of apoptosis. The rH3.3- and rAcH3.3-treated animals showed an increase in pulmonary cells that were positive for cleaved caspase-3 and -9 located at the damaged alveolar wall (Figure 5).
2.6. High Plasma Levels of Pro-Inflammatory Cytokines Were Found after Instillation of rH3.3 and Even Higher with rAcH3.3
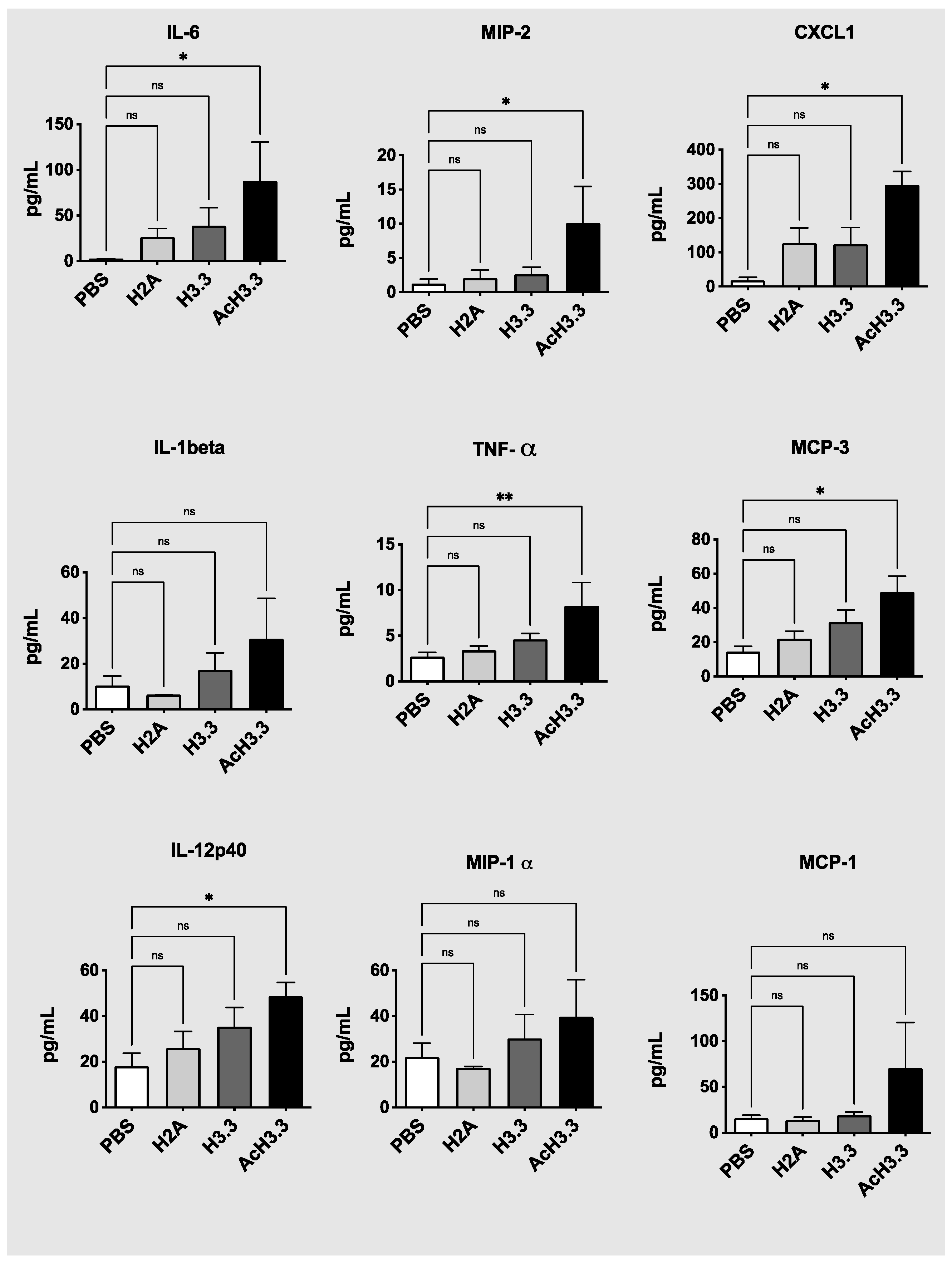
We then measured cytokine plasma levels from the animal groups to determine the systemic effect of the lung extracelullar histone administration. Many cytokines were increased after the instillation of rH3.3 and especially with rAcH3.3. Proinflamatory interleukine-6 (IL-6), and tumor necrosis factor (TNFα) were significantly elevated in the plasma of the animals intilled with rAcH3.3 * p < 0.05 vs control. Even though interleukine-1 beta (IL-1β) and macrophage inflammatory protein 2 (MIP-2) plasma levels didn’t reach statistical significant differences, there was a trend in increased levels after the administration of rH3.3 and rAcH3.3 (Figure 6). Chemokines including MCP3 and CXCL1 were also increased with histone administration and significantly in response to AcH3.3. Other cytokines were increased but didn’t reach statistical significance after AcH3.3 instillation include: macrophage inflammatory protein-1 alpha (MIP-1α), The cytokines that remained unchanged among groups include: human interferon-inducible protein 10 (IP-10), interleukine-18 (IL-18), macrophage inflammatory protein-1 beta (MIP-1β), Regulated upon Activation Normal T cell Expressed and Secreted (RANTES), and prostaglandin E2 (PGE2).
3. Discussion
Histones are indispensable protein complexes that play a critical role in packaging and organizing DNA within the nuclei of cells [12]. These histones undergo various post-translational modifications (PTMs), including phosphorylation, methylation, ubiquitination, and acetylation [14]. Under normal circumstances, histones primarily reside within the nucleus and are tightly regulated. Although, some histones such as H1 can remain in the cytoplasm and play different functions [26]; most of these chromatin unbound histones are rapidly degraded. The excess of histones is efficiently degraded by the proteasome through detection of site-specific modifications, predominantly phosphorylation, and ubiquitination [27,28]. However, this degradation process can be disrupted by an excess of PTMs at different sites of the histones. In senescent cells, the autophagy/lysosomal pathway assumes a major role in chromatin and histone degradation [29]. It is worth noting that alterations in the structure of histone proteins can compromise their functionality[14,30]. Notably, cigarette smoke has been identified as a significant contributor to the induction of PTMs, thereby promoting the transcription of inflammatory factors in the context of COPD[16,17,31]. Histones with excessive PTMs can impede their degradation and accumulate in the cytoplasm [12,32,33]. In apoptosis and cell death, the accumulated histones are released and reach the extracellular space, causing tissue damage [12,18,34]. In this study, we tested in vivo the effect of PTMs in histones in the pathogenesis of lung damage. We investigated if the presence of extracellular H3.3, and PTMs of H3.3, particularly hyperacetylation of H3.3, are responsible for lung damage.
The results of this study demonstrated that the acetylated form of H3.3 induces greater lung structural damage compared to unmodified H3.3. Histological studies revealed increased leukocyte infiltration, alveolar thickening, and inflammation in lungs treated with AcH3.3. Additionally, Micro-CT studies demonstrated a reduction in lung tissue volume. While all evaluated histones significantly increased neutrophil count, AcH3.3 also elevated total leukocytes and lymphocytes. The acute systemic cellular response triggered by AcH3.3 was characterized by a pro-inflammatory and chemotactic systemic cytokine profile. Notably, elevated levels of the proinflammatory mediators IL-6, TNF-alpha and the chemokines CXCL1 and MCP3 were detected in the plasma of the AcH3.3-treated mouse. While previous studies have reported lung structural damage associated with nucleosome proteins[18,35], our findings highlight the specific detrimental effect of a modified histone, implicating it as a causative factor in lung damage.
Moreover, the change in the alveolar architecture induced by AcH3.3 was associated to cell death, mediated by active caspase activation when evaluated at the molecular level. Cleaved caspases 3 and 9 were detected in the disrupted alveolar cells treated with H3.3 and AcH3.3. These findings suggest that alveolar epithelial cells could be more sensitive to the toxicity mediated by extracellular histones, especially H3.3 and its acetylated isoform. These findings agree with our previous in vitro work [12] and by others[33,36] that extracellular histones and specifically H3.3 induce a repetitive calcium flux that altered the mitochondrial membrane potential and promote the initiation of the caspase cascade[37]
Therefore, our investigation has conclusively demonstrated that both rH3.3 and rAcH3.3 induce cytotoxicity, inflammation and acute lung damage in C57BL/6J mice. Notably, the detrimental effects were more pronounced with rAcH3.3, which is consistent with observations made in human COPD lungs. These findings are of great significance for the advancement of pharmacological interventions aimed at treating COPD. Strategies involving the activation of HDACs or the inhibition of histone acetyltransferases (HATs) hold promise for reducing the accumulation of acetylated H3.3. Furthermore, monoclonal antibodies targeting AcH3.3 could potentially mitigate the lung damage caused by extracellular acetylated H3.3. In summary, this study provides valuable insights into the role of extracellular histones in COPD and establishes a foundation for further research investigating their potential as therapeutic targets.
4. Materials and Methods
4.1. Reagents
rH2A and rH3.3 proteins were obtained from New England Bio Lab, Ipswich, MA (Cat. #s M2502S, M2507S; respectively).. The rAcH3.3 from Active Motif, Carlsbad, CA(Cat. #31289) is a recombinant H3 pan-acetyl synthetic modified histone, containing acetylation at K4, K9, K14, K18 and K23 aa. Cleaved caspase (cCas)-3 and cCas-9 antibodies from Cell signaling, Danver, MA. Biotin antibody, streptavidin and DAB Substrate Kit (Cat#550880) were obtained from BD Bioscience, San Jose, CA.
4.2. Mouse Lung Instillation
The Institutional Animal Care and Use Committee of Temple University approved this animal protocol. C57BL/6 mice of 8-weeks of age obtained from Jackson’s laboratories were anesthetized using intraperitoneal injection of Ketamine/Xylazine (100/40 mg/g of body weight) and positioned in a rodent-tilted work stand, Hallowell EMC. Each animal was suspended from the upper teeth and the tongue placed aside for accessibility of the airway. A fiber optic, light source-equipped arm was used to visualize the airway. Proteins were administered with a MicroSprayer® model I-1C (PennCentury™), which was placed into the tracheal opening between the vocal cords. Animals were closely monitored and, under anesthesia, euthanized by exsanguination 48 hours after protein instillation.
4.3. Tissue Sample Collection and Blood and BALF Cell Counts
Under general anesthesia, total blood was collected by cardiac puncture and mixed with 1/10 v/v of 3.8% sodium citrate as anticoagulant. Fifty microliters of anticoagulated blood were for hematology counts using Hemavet® automatic counter, as previously described[38,39]. Plasma was separated by centrifugation for cytokine measurements. Lungs in situ were flushed with 1mL PBS to collect the BALF. Collected liquid was centrifuged, cellular content was measured and separated from the cell-free fluid. BALF pellet was suspended in 50 microliters of PBS and cells were count using the Hemavet® automatic counter. The left lung was clamped and dissected for RNA and protein studies and the right lung was fixed by intratracheal infusion of 4% paraformaldehyde solution. After fixation the upper lobe of the right lung was transferred to a phosphotungstic acid hematoxylin solution (PTAH) for visualization of soft tissue in micro-CT, using similar methodology previously described by this group [40]. The lower lobe of the right lung was paraffin embedded for histology, immunofluorescence and immunohistochemistry (IHC) studies.
4.4. Immunohistochemistry of H3.3
IHC studies were performed to evaluate the effectiveness of the intra-tracheal instillation of the histones into the alveolar and bronchial spaces. To accomplish this, animals were anesthetized and histones were instilled as explained above. After 30 minutes, the animals were euthanized, blood was extracted by cardiac puncture and tissues were perfused with PBS. Lungs were extracted, fixed and paraffin-embedded for sectioning. Tissue slides were deparaffinized, rehydrated and epitope-retrieved as previously described ([39,41]). Samples were blocked with 10% goat serum in PBS and then incubated overnight with biotinylated-antiH3.3 antibody. Samples were washed and incubated with streptavidin. Signal was detected using the BD Pharmingen DAB substrate kit, following manufacturer’s recommendations.
4.5. Lung Tissue Damage Studies
After BALF was collected, the right lung was perfused and inflated with a fixative solution and the right main bronchus was collapsed using a suture to maintain the normal structure of the lung. Tissue was paraffinized and section. Stained H&E lung, 5 um, sections were used to determine accumulation of inflammatory cells, small airway wall thickening, mucus accumulation, alveolar cellular infiltration and wall destruction. Analysis of the cellularity and thickness of the alveolar wall was performed by taking measurements from multiple pictures of different regions of the lung. Areas with large bronchi or large blood vessels were excluded. To evaluate the ratio of the alveolar tissue over total lung area, several taken photographs were transferred into a black and white image 8-bit type. In image J, a threshold was selected where most of the tissue was acquired and measurement of the selected area was taken. The measurement was divided by the area (Integrated Density Value IDV/area of the lung tissue).
4.6. Micro-CT Analysis of Murine Lung Tissue
Paraformaldehyde (4% in PBS) fixed lung tissue was immersed in 2 % phosphotungstic acid, 0.02 % potassium permanganate, and 0.1 % hematoxylin solution (PTAH) for an average of 5 days in order to visualize soft tissue using micro-CT. PTAH-prepared tissues were scanned in a Skyscan 1172, 12 megapixel, high resolution image pixel resolution size of 5.89 µm, x-ray source spot size of 300 nm, Al 0.5mm filter, voltage of 59 kV, current of 167 µA, rotation step of 0.40o, frame averaging of 5. Lung tissue structure was 3D reconstructed using Skyscan N-recon (reconstruction), visualized using CTVox volume rendering software, and analyzed using Skyscan CT-An software. Micro-CT scanning was useful to demonstrate and measure the presence of tissue damage and air trapped inside of the lungs. Radiopaque portions of the images were considered pulmonary tissue, or tissue volume, while radiolucent sections were considered as alveolar space. Indirect measurement of the lung tissue volume/total tissue volume ratio was calculated by Image J.
4.7. Apoptosis Measurement
Structural lung cells were searched for apoptosis markers by IHC as described above. The number of apoptosis-positive alveolar cells/40x fields will be counted in a minimum of 20 fields for each lung section in order to have a representative sample for statistical analysis.
4.8. Inflammatory Markers
Levels of inflammatory cells including neutrophils, macrophages and lymphocytes and their cytokine mediators were measured in blood and BALF. Cytokines including TNF a, IL-1b, IL 6, IL-8, MIP-2, MCP-1, MIP-1a, MIP-1b, MCP-3, CXCL1, PGE2, IL-12, IL-18, RANTES, and IP-10 were measured using Multiplexed Protein Assay (AssayGate, Inc; Ijamsville, MD)
4.9. Statistical Analysis
Differences among groups were statistically analyzed using one-way ANOVA followed by Holm Sidak’s Multiple Comparisons Test. P < 0.05 was considered to be significant. Data are reported as mean ± standard error of the mean (S.E.M.) for each group.
Author Contributions
Conceptualization, M.C.R. and C.A.B.; methodology, M.C.R, O.P.L. and C.B.A.; software, M.C.R., M.L.F., M.F.B. and C.A.B.; validation, M.C.R., D.J.C., M.L.F., V.O. and C.A.B.; formal analysis, M.C.R., M.F.B., M.A., V.O., O.P.L. and C.A.B.; investigation, M.C.R., M.F.B., M.A., D.J.C., M.L.F., V.O., O.P.L. and C.A.B.; resources, M.C.R. and C.A.B.; data curation, M.C.R., M.L.F., M.A. and C.A.B.; writing—original draft preparation, M.C.R. and C.A.B.; writing—review and editing, M.R., O.P. and C.B.; visualization, M.R. and C.B.; supervision, M.R. and C.B.; project administration, M.C.R. and C.A.B.; funding acquisition, M.C.R. and C.A.B. All authors have read and agreed to the published version of the manuscript.”
Funding
“This research was funded by Flight Attendant Medical Research Institute, grant number Young Clinical Scientist Award (YCSA_142023) to C.A.B.; and the National Heart, Lung, and Blood Institute of the National Institutes of Health, K01 award HL103197 to M.C.R.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ntritsos, G.; Franek, J.; Belbasis, L.; Christou, M.A.; Markozannes, G.; Altman, P.; Fogel, R.; Sayre, T.; Ntzani, E.E.; Evangelou, E. Gender-specific estimates of COPD prevalence: a systematic review and meta-analysis. Int J Chron Obstruct Pulmon Dis 2018, 13, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Halpin, D.M.G.; Criner, G.J.; Papi, A.; Singh, D.; Anzueto, A.; Martinez, F.J.; Agusti, A.A.; Vogelmeier, C.F. Global Initiative for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease. The 2020 GOLD Science Committee Report on COVID-19 and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 2021, 203, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.; Criner, G.J. Chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013, 187, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Chronic obstructive pulmonary disease: a growing but neglected global epidemic. PLoS Med 2007, 4, e112. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Prevention of death in COPD. N Engl J Med 2007, 356, 2211. [Google Scholar] [CrossRef]
- Ford, E.S. Hospital discharges, readmissions, and ED visits for COPD or bronchiectasis among US adults: findings from the nationwide inpatient sample 2001-2012 and Nationwide Emergency Department Sample 2006-2011. Chest 2015, 147, 989–998. [Google Scholar] [CrossRef]
- Ford, E.S.; Murphy, L.B.; Khavjou, O.; Giles, W.H.; Holt, J.B.; Croft, J.B. Total and state-specific medical and absenteeism costs of COPD among adults aged ≥ 18 years in the United States for 2010 and projections through 2020. Chest 2015, 147, 31–45. [Google Scholar] [CrossRef]
- Duffy, S.P.; Criner, G.J. Chronic Obstructive Pulmonary Disease: Evaluation and Management. Med Clin North Am 2019, 103, 453–461. [Google Scholar] [CrossRef]
- Barnes, P.J. Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol 2009, 71, 451–464. [Google Scholar] [CrossRef]
- Bagdonas, E.; Raudoniute, J.; Bruzauskaite, I.; Aldonyte, R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int J Chron Obstruct Pulmon Dis 2015, 10, 995–1013. [Google Scholar] [CrossRef]
- Yoshida, T.; Tuder, R.M. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 2007, 87, 1047–1082. [Google Scholar] [CrossRef] [PubMed]
- Barrero, C.A.; Perez-Leal, O.; Aksoy, M.; Moncada, C.; Ji, R.; Lopez, Y.; Mallilankaraman, K.; Madesh, M.; Criner, G.J.; Kelsen, S.G.; et al. Histone 3.3 participates in a self-sustaining cascade of apoptosis that contributes to the progression of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013, 188, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Szatmary, P.; Huang, W.; Criddle, D.; Tepikin, A.; Sutton, R. Biology, role and therapeutic potential of circulating histones in acute inflammatory disorders. J Cell Mol Med 2018, 22, 4617–4629. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Qiu, T.; Wei, G.; Que, Y.; Wang, W.; Kong, Y.; Xie, T.; Chen, X. Role of Histone Post-Translational Modifications in Inflammatory Diseases. Front Immunol 2022, 13, 852272. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Histone deacetylase-2 and airway disease. Ther Adv Respir Dis 2009, 3, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Nevid, M.Z.; Friedman, A.E.; Rahman, I. Cigarette smoke induces distinct histone modifications in lung cells: implications for the pathogenesis of COPD and lung cancer. J Proteome Res 2014, 13, 982–996. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Rahman, I. Gene expression profiling of epigenetic chromatin modification enzymes and histone marks by cigarette smoke: implications for COPD and lung cancer. Am J Physiol Lung Cell Mol Physiol 2016, 311, L1245–L1258. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Fan, X.G.; Tang, D. Release and activity of histone in diseases. Cell Death Dis 2014, 5, e1370. [Google Scholar] [CrossRef]
- Allam, R.; Kumar, S.V.; Darisipudi, M.N.; Anders, H.J. Extracellular histones in tissue injury and inflammation. J Mol Med (Berl) 2014, 92, 465–472. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat Med 2009, 15, 1318–1321. [Google Scholar] [CrossRef]
- Wu, D.; Ingram, A.; Lahti, J.H.; Mazza, B.; Grenet, J.; Kapoor, A.; Liu, L.; Kidd, V.J.; Tang, D. Apoptotic release of histones from nucleosomes. J Biol Chem 2002, 277, 12001–12008. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Yang, Y.; Ma, D.; He, L.; Xiao, W. Expression level of histone deacetylase 2 correlates with occurring of chronic obstructive pulmonary diseases. Mol Biol Rep 2013, 40, 3995–4000. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc 2009, 6, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Thimmulappa, R.; Vij, N.; Navas-Acien, A.; Sussan, T.; Merali, S.; Zhang, L.; Kelsen, S.G.; Myers, A.; Wise, R.; et al. Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: the role of Nrf2-regulated proteasomal activity. Am J Respir Crit Care Med 2009, 180, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Zlatanova, J.S.; Srebreva, L.N.; Banchev, T.B.; Tasheva, B.T.; Tsanev, R.G. Cytoplasmic pool of histone H1 in mammalian cells. J Cell Sci 1990, 96 ( Pt 3), 461–468. [Google Scholar] [CrossRef]
- Singh, R.K.; Kabbaj, M.H.; Paik, J.; Gunjan, A. Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat Cell Biol 2009, 11, 925–933. [Google Scholar] [CrossRef]
- Shmueli, M.D.; Sheban, D.; Eisenberg-Lerner, A.; Merbl, Y. Histone degradation by the proteasome regulates chromatin and cellular plasticity. FEBS J 2022, 289, 3304–3316. [Google Scholar] [CrossRef]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J Cell Biol 2013, 202, 129–143. [Google Scholar] [CrossRef]
- Rajendrasozhan, S.; Yao, H.; Rahman, I. Current perspectives on role of chromatin modifications and deacetylases in lung inflammation in COPD. COPD 2009, 6, 291–297. [Google Scholar] [CrossRef]
- Seiler, C.L.; Song, J.U.M.; Kotandeniya, D.; Chen, J.; Kono, T.J.Y.; Han, Q.; Colwell, M.; Auch, B.; Sarver, A.L.; Upadhyaya, P.; et al. Inhalation exposure to cigarette smoke and inflammatory agents induces epigenetic changes in the lung. Sci Rep 2020, 10, 11290. [Google Scholar] [CrossRef] [PubMed]
- Hoeksema, M.; van Eijk, M.; Haagsman, H.P.; Hartshorn, K.L. Histones as mediators of host defense, inflammation and thrombosis. Future Microbiol 2016, 11, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Wen, T.; Song, J.; Xie, D.; Wu, L.; Jiang, X.; Jiang, P.; Wen, Z. Extracellular histones are clinically relevant mediators in the pathogenesis of acute respiratory distress syndrome. Respir Res 2017, 18, 165. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Hajji, N.; Joseph, B. Cracking the death code: apoptosis-related histone modifications. Cell Death Differ 2010, 17, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med 2013, 187, 160–169. [Google Scholar] [CrossRef]
- Karki, P.; Birukov, K.G.; Birukova, A.A. Extracellular histones in lung dysfunction: a new biomarker and therapeutic target? Pulm Circ 2020, 10, 2045894020965357. [Google Scholar] [CrossRef] [PubMed]
- Collier, D.M.; Villalba, N.; Sackheim, A.; Bonev, A.D.; Miller, Z.D.; Moore, J.S.; Shui, B.; Lee, J.C.; Lee, F.K.; Reining, S.; et al. Extracellular histones induce calcium signals in the endothelium of resistance-sized mesenteric arteries and cause loss of endothelium-dependent dilation. Am J Physiol Heart Circ Physiol 2019, 316, H1309–H1322. [Google Scholar] [CrossRef] [PubMed]
- Liverani, E.; Rico, M.C.; Garcia, A.E.; Kilpatrick, L.E.; Kunapuli, S.P. Prasugrel metabolites inhibit neutrophil functions. J Pharmacol Exp Ther 2013, 344, 231–243. [Google Scholar] [CrossRef]
- Garcia, A.E.; Rico, M.C.; Liverani, E.; DeLa Cadena, R.A.; Bray, P.F.; Kunapuli, S.P. Erosive arthritis and hepatic granuloma formation induced by peptidoglycan polysaccharide in rats is aggravated by prasugrel treatment. PLoS One 2013, 8, e69093. [Google Scholar] [CrossRef]
- Ali, S.; Cunningham, R.; Amin, M.; Popoff, S.N.; Mohamed, F.; Barbe, M.F. The extensor carpi ulnaris pseudolesion: evaluation with microCT, histology, and MRI. Skeletal Radiol 2015, 44, 1735–1743. [Google Scholar] [CrossRef]
- Rico, M.C.; Castaneda, J.L.; Manns, J.M.; Uknis, A.B.; Sainz, I.M.; Safadi, F.F.; Popoff, S.N.; Dela Cadena, R.A. Amelioration of inflammation, angiogenesis and CTGF expression in an arthritis model by a TSP1-derived peptide treatment. J Cell Physiol 2007, 211, 504–512. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Recombinant histone 3.3 reaches the alveolar tissue after tracheal instillation. Thirty minutes after tracheal aerosolization of 25 µL of PBS or 25 µL (30 µg) of rH3.3, lungs were flushed with PBS, fixed, embedded, and sectioned for immunohistochemistry for H3.3. Lung instillation of (a) PBS or (b) rH3.3.
Figure 1.
Recombinant histone 3.3 reaches the alveolar tissue after tracheal instillation. Thirty minutes after tracheal aerosolization of 25 µL of PBS or 25 µL (30 µg) of rH3.3, lungs were flushed with PBS, fixed, embedded, and sectioned for immunohistochemistry for H3.3. Lung instillation of (a) PBS or (b) rH3.3.
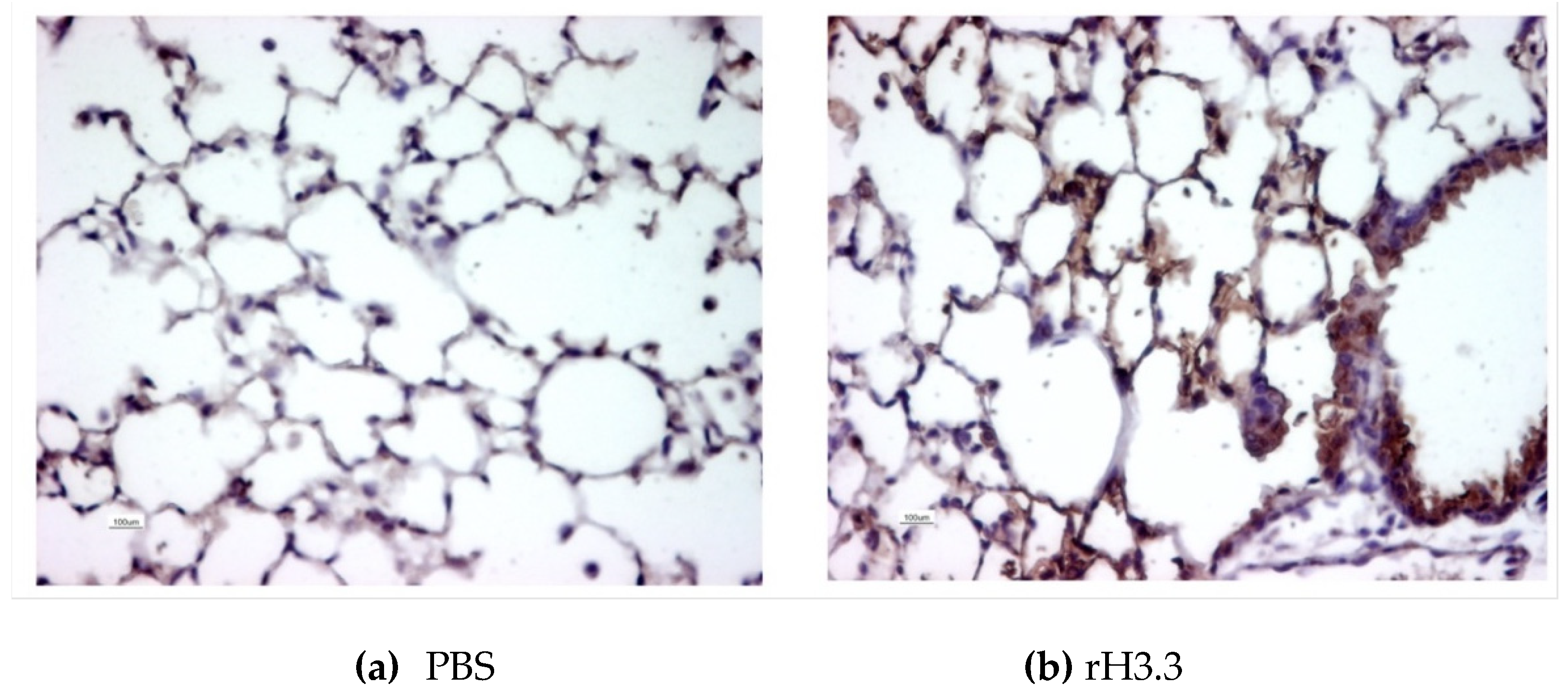
Figure 2.
Intratracheal-aerosolized rH3.3 and rAcH3.3 induce structural lung tissue damage. Representative photographs of lung tissue stained with hematoxylin & eosin (H&E) at magnifications of 4X (top panel, (a)), 10X (at bronchial area (b); the alveolar area (c)) and 40X at the small bronchial branches (d) Representative inverted photographs used to calculated lung tissue area analyzed by ImageJ) (e) 40X selected alveolar and brochila sections comparing specifically rAcH3.3 lung tissue areas respect PBS control. (f) quantitation of the integrated density value IDVarea of the lung tissue by ImageJ analysis *p < 0.05 vs. PBS control.
Figure 2.
Intratracheal-aerosolized rH3.3 and rAcH3.3 induce structural lung tissue damage. Representative photographs of lung tissue stained with hematoxylin & eosin (H&E) at magnifications of 4X (top panel, (a)), 10X (at bronchial area (b); the alveolar area (c)) and 40X at the small bronchial branches (d) Representative inverted photographs used to calculated lung tissue area analyzed by ImageJ) (e) 40X selected alveolar and brochila sections comparing specifically rAcH3.3 lung tissue areas respect PBS control. (f) quantitation of the integrated density value IDVarea of the lung tissue by ImageJ analysis *p < 0.05 vs. PBS control.
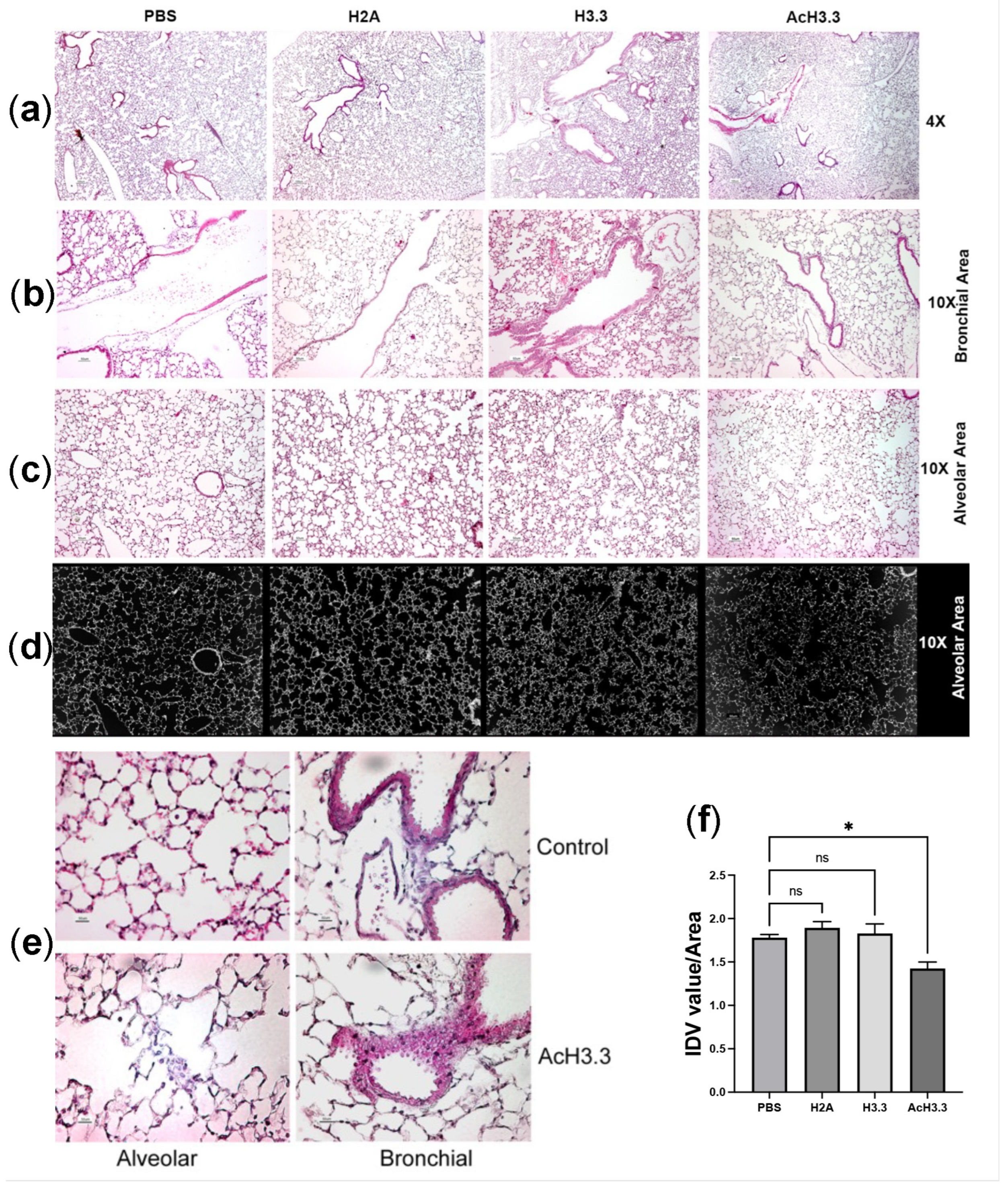
Figure 3.
MicroCT scan reconstruction of the anatomical structure of the rAcH3.3-aerosolized lungs demonstrated alteration of the parenchyma/air space ratio when compared to H2A control. (a) 3D reconstruction of the micro-CT images of the lungs aerosolized with histones (CT Vox). (b) 2D sections (i.e., micro-CT slides) of the upper lobe where the main bronchi are located. Inside each panel is a magnification of the lung parenchyma. White arrows are pointing radiopaque areas observed only in the rH3.3- and rAcH3.3-aerosolized lungs. (c) Representative further magnification of the lung parenchyma areas. (d) Quantification of the lung tissue/total lung volume. ****p < 0.0001 vs. rH2A control.
Figure 3.
MicroCT scan reconstruction of the anatomical structure of the rAcH3.3-aerosolized lungs demonstrated alteration of the parenchyma/air space ratio when compared to H2A control. (a) 3D reconstruction of the micro-CT images of the lungs aerosolized with histones (CT Vox). (b) 2D sections (i.e., micro-CT slides) of the upper lobe where the main bronchi are located. Inside each panel is a magnification of the lung parenchyma. White arrows are pointing radiopaque areas observed only in the rH3.3- and rAcH3.3-aerosolized lungs. (c) Representative further magnification of the lung parenchyma areas. (d) Quantification of the lung tissue/total lung volume. ****p < 0.0001 vs. rH2A control.

Figure 4.
rAcH.3.3 increases white blood cell counts at the expenses of lymphocytes. (a) Automated blood counts from anti-coagulated blood collected from cardiac puncture demonstrated significant leukocytosis and lymphocytosis in mice treated with rAcH3.3 when compared to control (*p < 0.05, **p < 0.001). (b) Automated BALF cell counts showed a tendency of elevation in white blood counts but did not reach statical significance.
Figure 4.
rAcH.3.3 increases white blood cell counts at the expenses of lymphocytes. (a) Automated blood counts from anti-coagulated blood collected from cardiac puncture demonstrated significant leukocytosis and lymphocytosis in mice treated with rAcH3.3 when compared to control (*p < 0.05, **p < 0.001). (b) Automated BALF cell counts showed a tendency of elevation in white blood counts but did not reach statical significance.
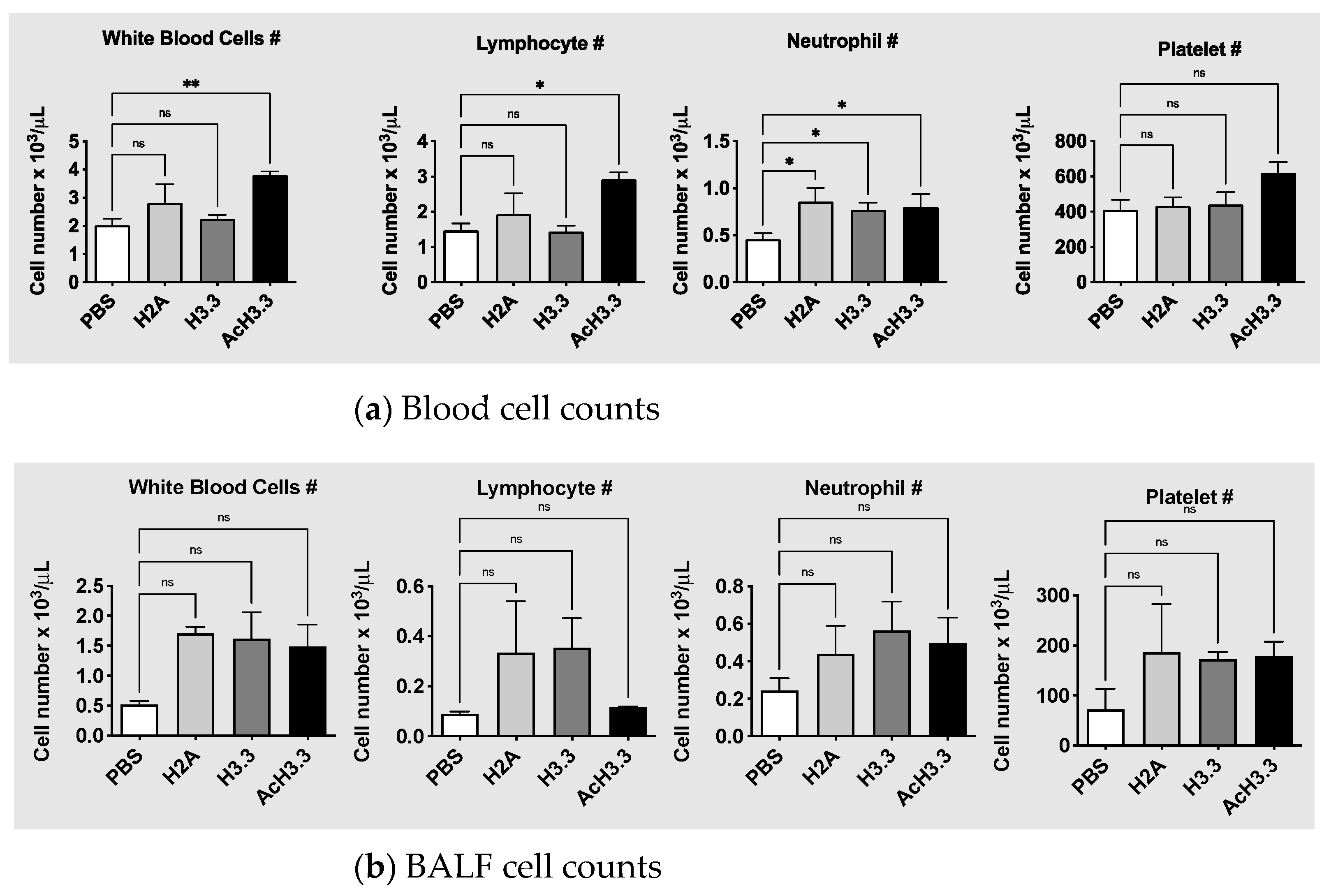
Figure 5.
Positive cells of cCaspase-3 and -9 by immunohistochemistry were increased after 48 h of rH3.3- and rAcH3.3-lung aerosolization. A-D: PBS, E-H: rH2A, I-L: rH3.3 and M-P: rAcH3.3.
Figure 5.
Positive cells of cCaspase-3 and -9 by immunohistochemistry were increased after 48 h of rH3.3- and rAcH3.3-lung aerosolization. A-D: PBS, E-H: rH2A, I-L: rH3.3 and M-P: rAcH3.3.
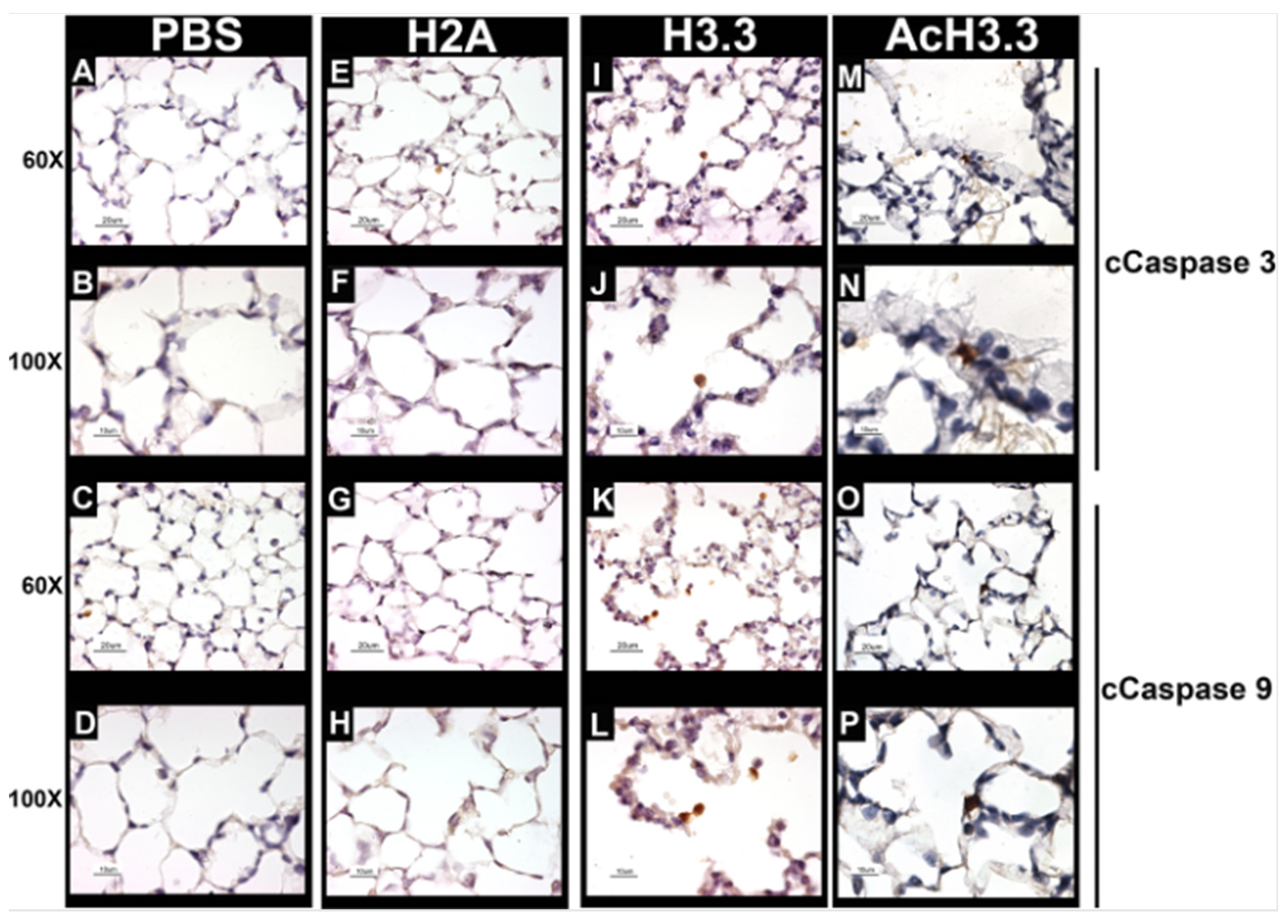
Figure 6.
Pro-inflammatory cytokines were increased after rAcH3.3 treatment. Graphic representation of the plasma levels of CXCL1, IL-1beta, MIP-2 TNF-alpha, IL-6, MCP-3, IL-12p40, MIP-1α, and MCP-1 cytokines from animals instilled with PBS (white bars), rH2A (light grey bars), rH3.3 (dark grey bars) and rAcH3.3 (black bars). *p < 0.05, **p < 0.01 (rHistone vs PBS).
Figure 6.
Pro-inflammatory cytokines were increased after rAcH3.3 treatment. Graphic representation of the plasma levels of CXCL1, IL-1beta, MIP-2 TNF-alpha, IL-6, MCP-3, IL-12p40, MIP-1α, and MCP-1 cytokines from animals instilled with PBS (white bars), rH2A (light grey bars), rH3.3 (dark grey bars) and rAcH3.3 (black bars). *p < 0.05, **p < 0.01 (rHistone vs PBS).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



