
Preprint
Review
Synthesis of Nitrogen Containing Heterocyclic Scaffolds through Sequential Reactions of Aminoalkynes with Carbonyls
Altmetrics
Downloads
112
Views
27
Comments
0
A peer-reviewed article of this preprint also exists.



This version is not peer-reviewed
Submitted:
16 May 2023
Posted:
17 May 2023
You are already at the latest version
Alerts
Abstract
Sequential reactions of aminoalkynes represent a powerful tool to easily assembly biologically important polyfunctionalized nitrogen heterocyclic scaffolds. Metal catalysis often plays a key role in terms of selectivity, efficiency, atom economy and green chemistry of these sequential approaches. This review examines the existing literature on the applications of reactions of aminoalkynes with carbonyls which are emerging for their synthetic potential. Aspect concerning the features of the starting reagents, the catalytic systems, alternative reaction conditions and the pathways as well as the possible intermediates are provided.
Keywords:
Subject: Chemistry and Materials Science - Organic Chemistry
1. Introduction
Aminoalkynes are bifunctional derivatives which can undergo a diverse array of transformations. They offer sequential reactions with an electrophile and a nucleophile and are ideal for cascade reactions. Sequential reactions represent a powerful tool to build up simple or more complex polyfunctionalized organic scaffolds from readily available reagents with high efficiency, selectivity, and atom economy [1-3]. Recently, applications of sequential reactions of aminoalkynes represent a very active research field in organic synthesis and medicinal chemistry. In particular, sequential reactions of β-, γ-, and δ-aminoalkynes to afford a variety heterocyclic scaffolds were explored. Inactivated alkynes moieties are not very reactive toward nucleophiles. Their behaviour changes by activation of the C-C triple bond by a metal catalyst. Various biologically important nitrogen heterocycles were directly synthesized in an easy way by means of intramolecular hydroamination of aminoalkynes in the presence several transition metal as well as lanthanide catalysts. [4-5] The aptitude to form π- and σ-complexes can help for the choice of catalysts for the desired transformations when bi- or polyfunctional substrates are involved [6]. The reaction of γ- and δ-aminoalkynes with sulfonyl azides in the presence of Ru3(CO)12 catalyst efficiently afforded led cyclic amidines of relevance in medicinal and coordination chemistry as well as materials science. [7] The gold(I)-catalyzed tandem cyclization of γ-aminoalkynes with alkynes in water afforded diversely substituted pyrrolo[1,2-a]quinolines. [8] Zhou et al. extended this reaction using less active terminal amidoalkynes in similar conditions. [9] The CuCl-catalyzed cascade transformation of internal β-aminoalkynes with alkynes under microwave irradiation gave diversely substituted tetrahydropyrrolo[1,2-a]quinolines. [10] An intramolecular gold-catalyzed hydroamination/aza-Diels-Alder tandem process of β-/γ-aminoalkynes with high regio and diastereoselectivity and up to almost complete chemoselectivity showed great efficiency in a one-pot approach to the complex nitrogen heterocyclic derivatives of medicinal importance such as the one-step synthesis of incargranine B aglycone and (±)-seneciobipyrrolidine (I). [11] Faňanás, Rodríguez, and co-workers described the preparation of complex pyrrolidines from readily available N-Boc-derived β-aminoalkynes and alkenes or alkynes through relay actions of PtII or Brønsted acids. [12-13] The reaction of α-aminoalkynes with carbon monoxide and selenium yielded 5-alkylideneselenazolin-2-ones stereoselectively via cycloaddition of in situ generated carbamoselenoates to the carbon-carbon triple bond. β-Aminoalkyne also afforded the corresponding six-membered selenium-containing heterocycle with the aid of CuI. [14] An operationally simple palladium-catalyzed intramolecular hydroaminocarbonylation of a variety of aminoalkynes directly provided a viable approach to a variety of valuable seven- and eight-membered lactams with high chemoselectivity and regioselectivity. [15] A sequential heterogeneous PtI2-catalyzed hydration of δ-aminoalkynes followed by intramolecular cyclization and intermolecular addition as well as ring-expansion cascade reaction with another electron-deficient alkynes was developed for the synthesis of various eight-membered nitrogen heterocycles in excellent yields under mild reaction conditions. The simple PtI2 could be easily recycled. [16] Moreover, a PtI2-catalyzed formal three-component cascade cycloaddition reactions between γ-aminoalkynes and electron-deficient alkynes gave highly functionalized cyclohexadiene-b-pyrrolidines in good yields.[17] Finally, among the domino and multicomponent processes that involve aminoalkynes, their cascade reactions with carbonyl derivatives stand out as a highly versatile tool to build up libraries of nitrogen-containing heterocyclic scaffolds with diversity and molecular complexity. This review will examine the literature on this last topic and is organized according to the structure of the aminoalkyne substrate. Aspects concerning the features of the catalytic systems, the substrate scope, insight into the reactions pathways, possible intermediates as well as alternative conditions are discussed.
2. Sequential Reactions of α-Aminoalkynes (Propargylamines) with Carbonyls
Propargylic amine derivatives represent useful α-aminoalkynes building blocks for the construction of nitrogen containing heterocyclic scaffolds through their sequential reactions with carbonyls. The gold-catalyzed reaction of propargylamine 1 with dialkyl acyclic/cyclic ketones, methyl, aryl/heteroaryl ketones and aldehydes bearing α-hydrogens 2 allowed a simple approach to pyridines 3 through a sequential amination / cyclization / aromatization cascade (Scheme 1). [18]
A variety of catalysts were tested in the reaction of 1 with 2. In particular, NaAuCl4·2H2O resulted a highly efficient catalyst. Moreover, Au 8 were synthesized and applied as efficient bi-functional catalysts. It was found that imidazolyl group acted as a Lewis base to catalyze the condensation of carbonyl compounds with propargylamine to form the imino intermediate, and the involved Au+-complex species activated the alkynyl moiety to give the dehydropyridine derivative which underwent auto-oxidation reaction to afford the target pyridines (Scheme 3). [19]
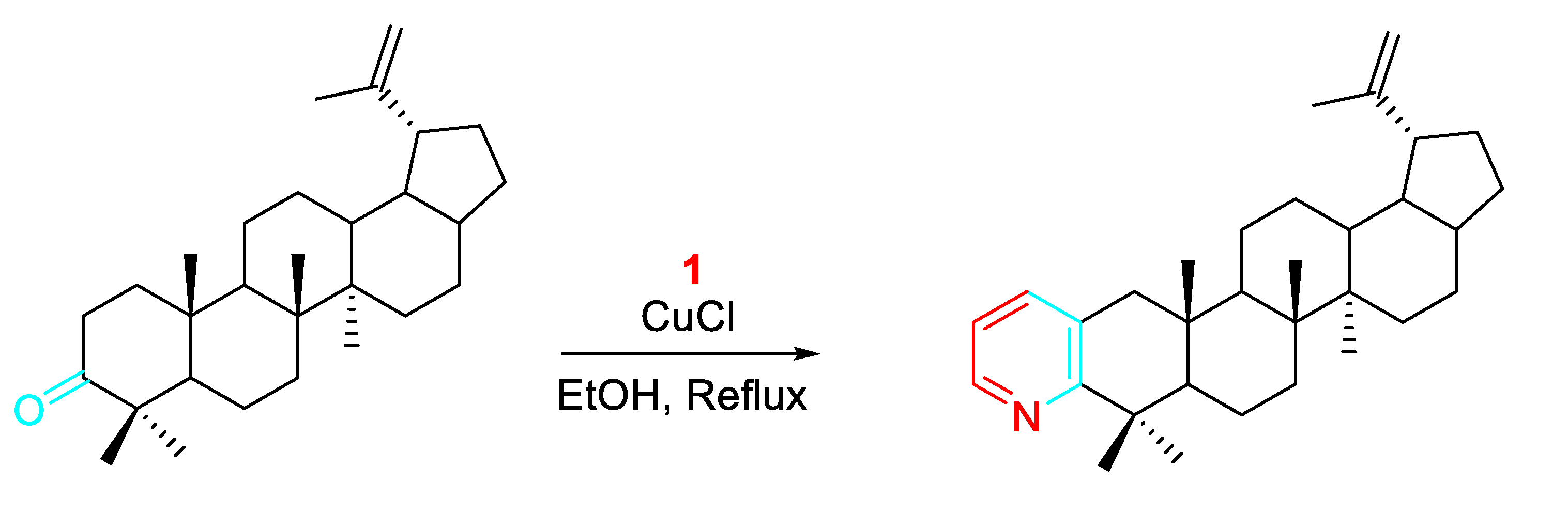
Copper salts were also effective catalysts in the reaction of cyclic ketones with propargylamine and the highest product yields were observed in i-PrOH in the presence of 5.0 mol % CuCl2 in air. Decreased yields among cyclic ketones were observed in the following order: six-membered ≫ eight-membered > five-membered ∼ seven-membered. However, the inexpensiveness of the catalyst and the tolerance to a wide number of protective functional groups in the ketone make the procedure very suitable for large scale preparation of fused pyridines. (Scheme 4). [20]
Selective aspects of the reaction of steroidal carbonyls with propargylamine were investigated. According to the results, the regioselective pyridine fusion to the cyclic skeleton was addressed by suitable choice between the substrate bearing a saturated or conjugated carbonyl group (Scheme 5). [18].
Analogously, new A-ring pyridine fused androstanes in 17a-homo-17-oxa (D-homo lactone), 17α-picolyl or 17(E)-picolinylidene series were obtained by reacting 4-en-3-one or 4-ene-3,6-dione D-modified androstane derivatives with propargylamine under the presence of a Cu(II) catalyst, and evaluated for potential anticancer activity in vitro (Scheme 6). [21]
Similarly, the efficient synthesis of pyridine rings fused to the 3,4-positions of the steroid nucleus was described via the Cu(II)-catalyzed reaction of propargylamine with 17β-hydroxyandrost-4-en-3-one, 17α-methyl-17β-hydroxyandrost-4-en-3-one, 17β-hydroxyestr-4-en-3-one. [22] The procedure was also applied to the synthesis of heterocyclic betulin derivatives (Scheme 7). [23-24]
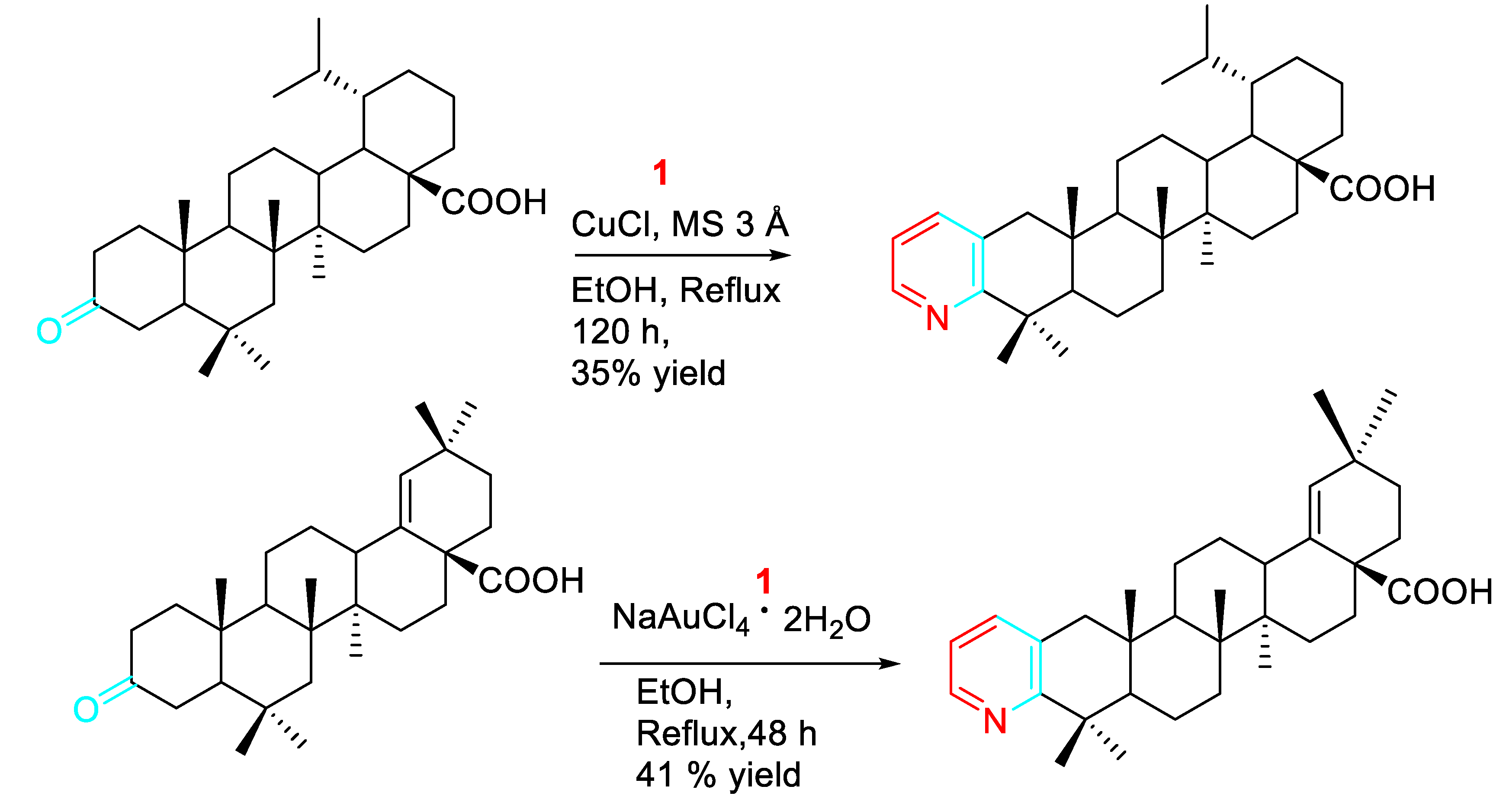
Optimization of the synthesis of steroidal pyridines was tried by prolonging the reaction time and varying the catalyst loading. In some cases the use of NaAuCl4⋅2H2O instead of CuCl and the addition of activated molecular sieves to the reaction mixture led to significant improvement (Scheme 8). [25]
Several other applications of the methodology accomplished the preparation of significant scaffolds. Indeed, the chemical synthesis of highly potent and acid-stable inhibitors of hedgehog signaling carbacyclopamine analog 11 was reported. The gold-catalyzed amination/annulation/aromatization sequence applied to the inseparable mixture of the mixture of the isomers 9 and 10 regioselectively furnished, after removal of the tert-butyldimethylsilyl ether (tetrabutylammonium fluoride, THF, 25 °C), carbacyclopamine analog 11 in 36% overall yield for the two steps. (Scheme 9). [26]
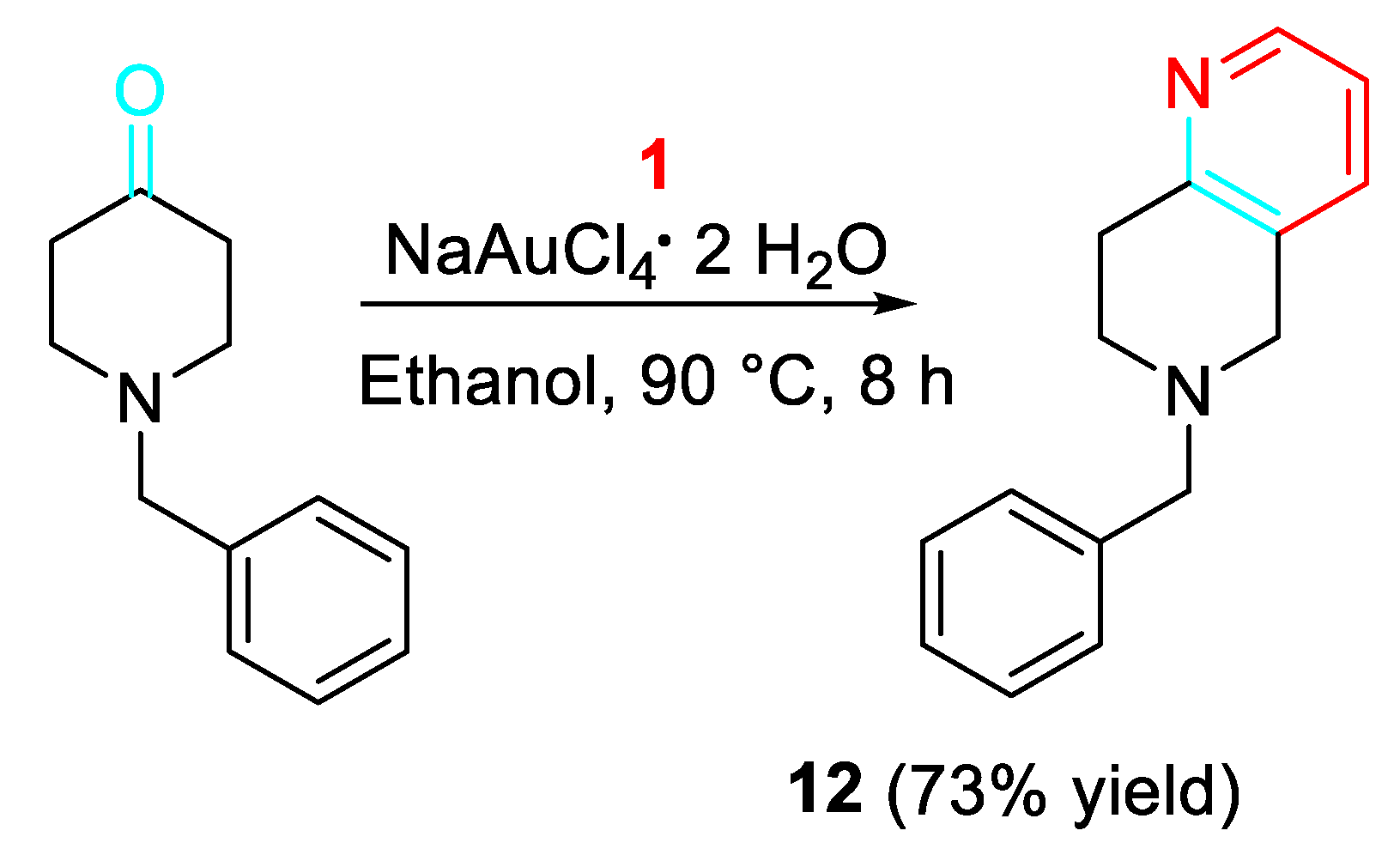
Furthermore, the gold-catalyzed reaction of 1-benzylpiperidin-4-one with propargylamine efficiently afforded the potassium channel modulator 6-benzyl-5,6,7,8-tetrahydro-1,6-naphthirine 12 (Scheme 10). [27]
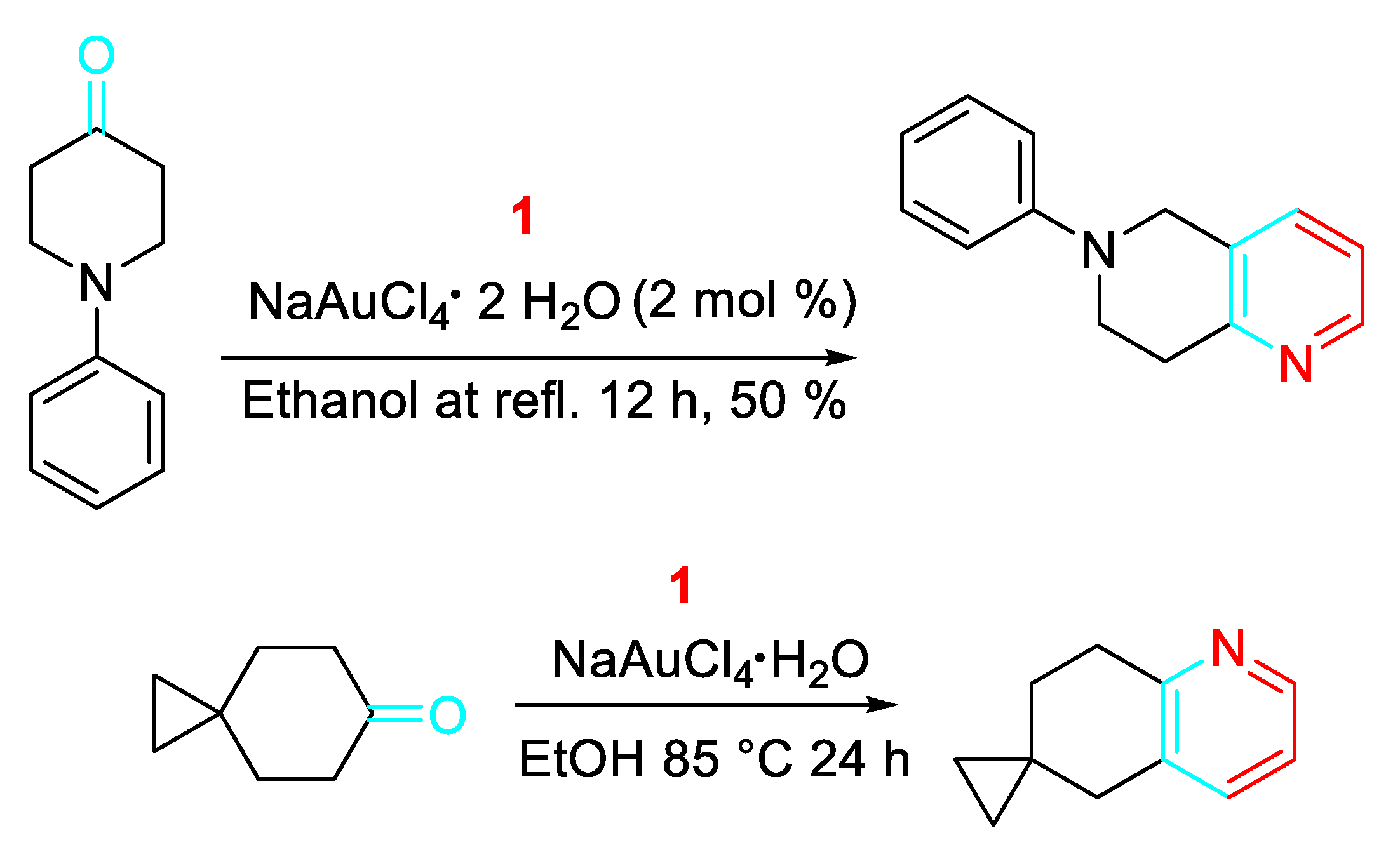
The methodology was extended to the synthesis of Wnt signal path inhibitors. (Scheme 11). [28-29]
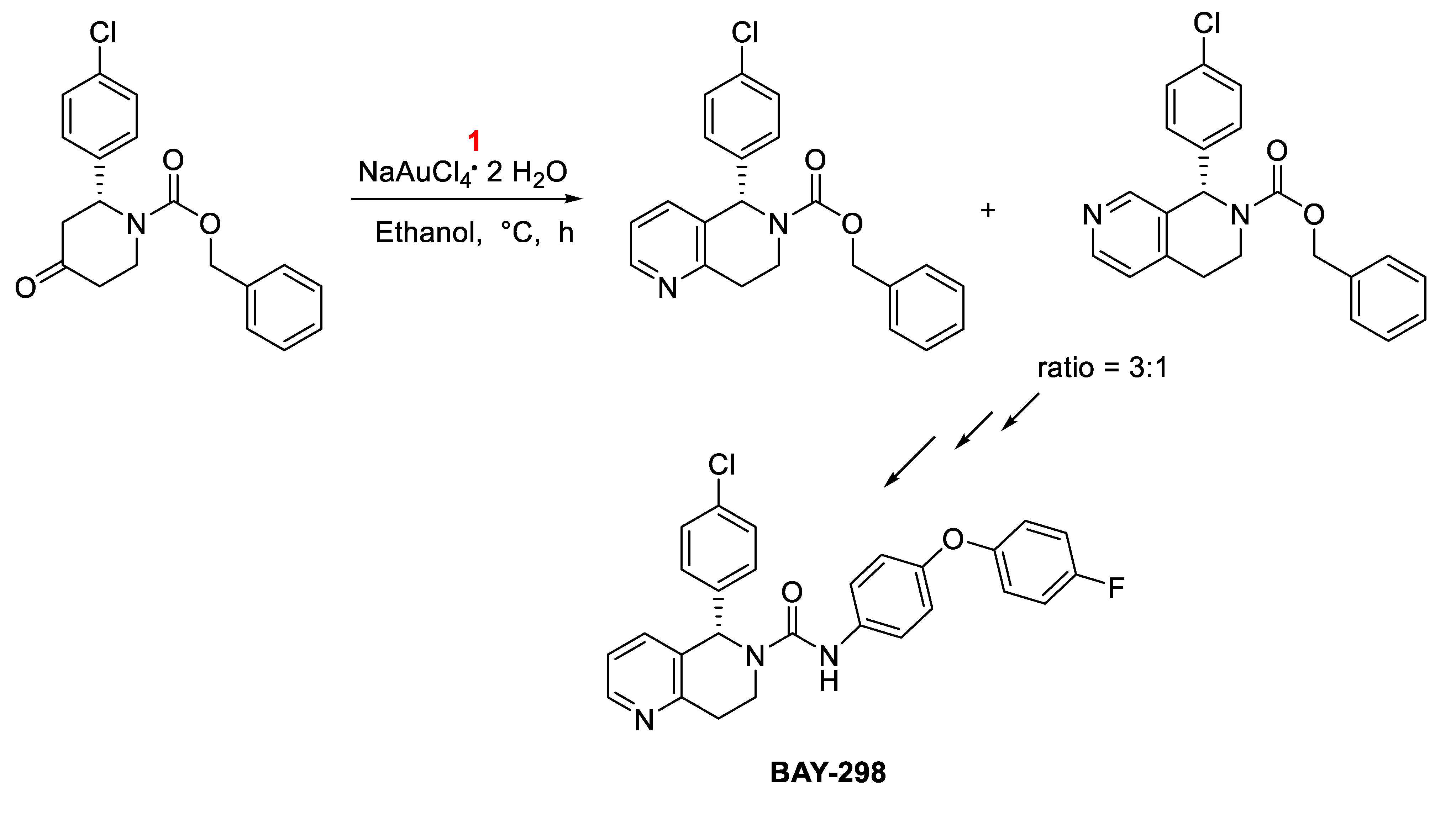
The gold-catalyzed synthesis of BAY-298, a nanomolar small molecule (SMOL) hLH-R antagonist reducing sex hormone levels in vivo was described (Scheme 12). [30]
Moreover, the gold-catalyzed sequential process of condensation/cyclization/aromatization procedure was extended as the key step for the preparation of BMS-846372, a potent and orally active human CGRP receptor antagonist employed for the migraine therapy (Scheme 13). [31]
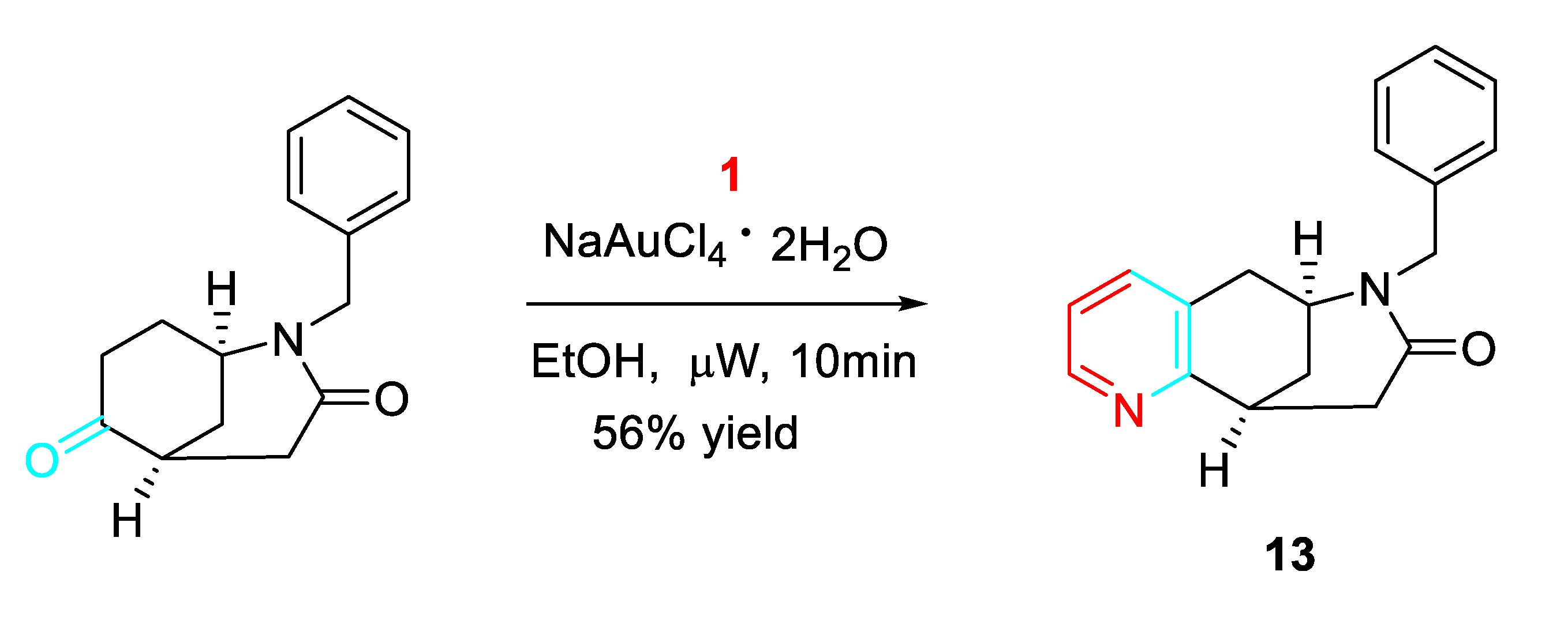
The methodology resulted, also, a viable tool obtain aryl and heteroaryl derivatives of benzomorphanes 13 pharmacologically active as inhibitors of 11 ß-hydroxysteroid dehydrogenase (HSD) 1 (Scheme 14). [32]
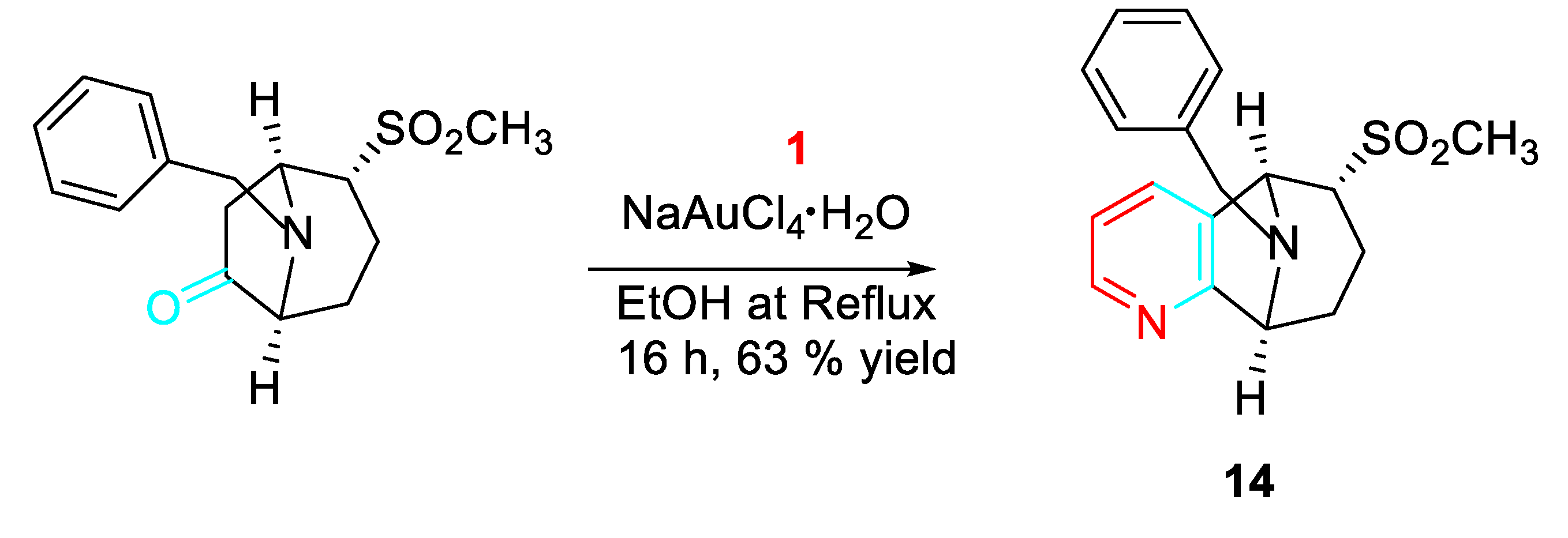
The synthetic approach had yielded a number of scaffolds suitable for the design of performance-diverse screening libraries (Scheme 15). [33]
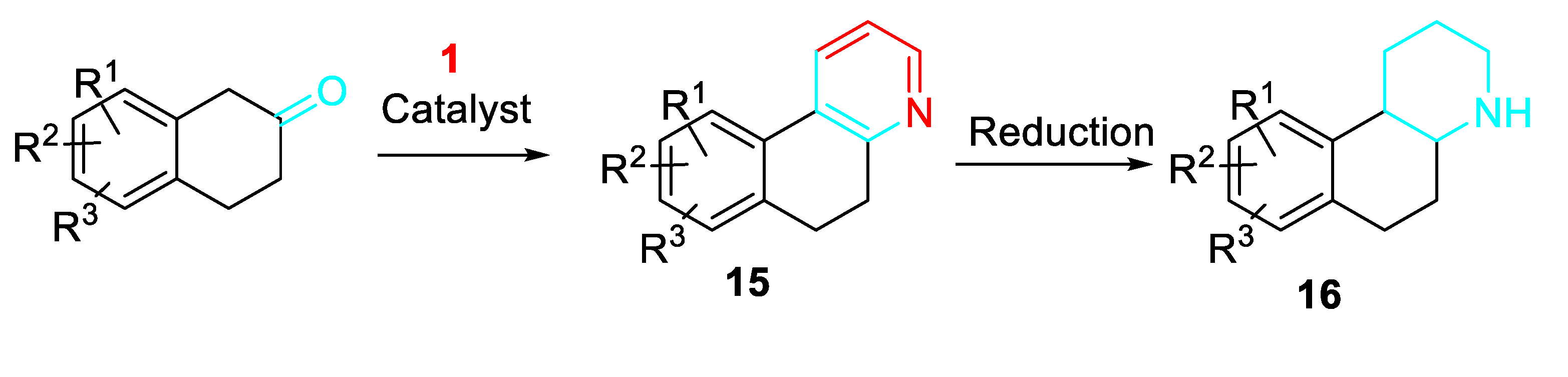
The reaction of 2-tetralones and propargylamine in the presence of complexes of gold or copper, preferably NaAuCl4 and CuC1 was employed to synthesize octahydrobenzoquinoline derivatives 16 as inhibitors of 11β-hydroxysteroid dehydrogenase for the treatment of metabolic disorders like metabolic syndrome, diabetes, obesity, and dyslipidemia. The reaction is usually run in alcohols at temperatures of 20 to 120° C through conventional heating or microwave irradiation. The resulting pyridine was reduced transformed to the corresponding piperidine (Scheme 16). [34]
The sequential gold-catalyzed condensation/annulation reaction of the 1,3-dihyrdo-2H-inden-2-one with the propargylamine provided the corresponding 9H-indeno[2,1-b]pyridine 17 as the ligand for the synthesis of an olefin polymerization catalyst (Scheme 17). [35]
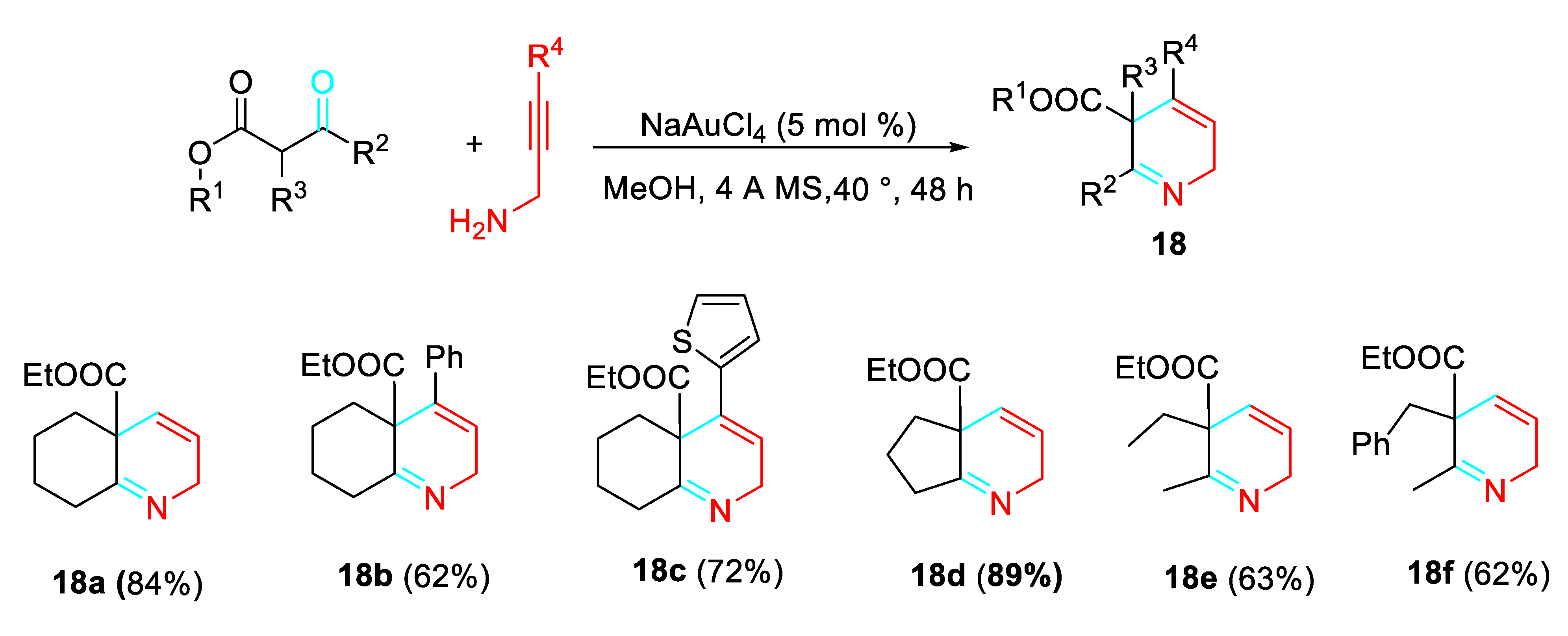
The gold(III)-catalyzed reaction of simple β-ketoesters with propargylamines achieved the synthesis of potentially bioactive 2,5-dihydropyridines 18 in satisfactory yields (Scheme 18). [36]
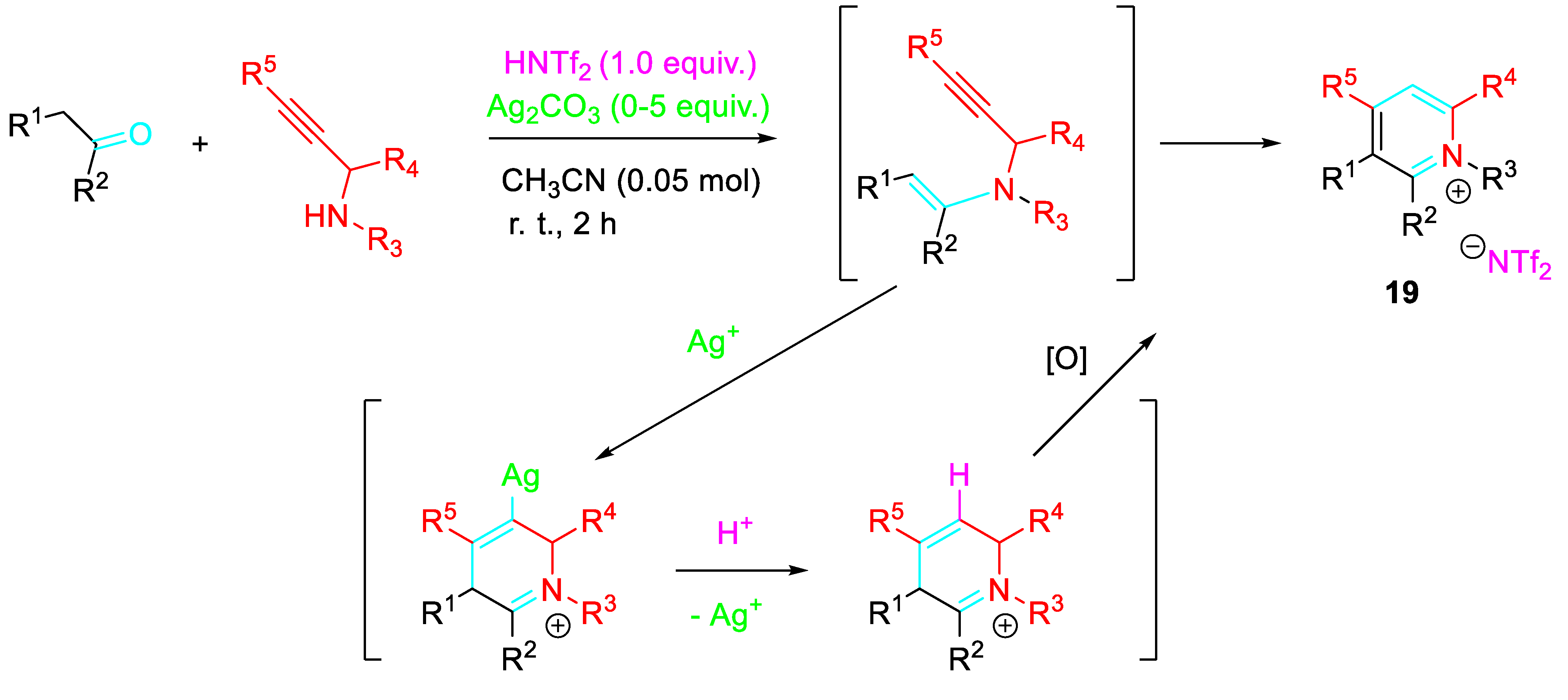
Substituted pyridinium salts 19 were obtained under mild conditions by a condensation reaction between carbonyls and propargylamine under the presence Ag2CO3/HNTf2 synergistically acting catalyst system. The one-pot transformation should proceed via sequential 6-endo-dig cyclization of the in situ generated propargylenamine / protonolysis of the resulting vinyl-silver intermediate (Scheme 19). [37]
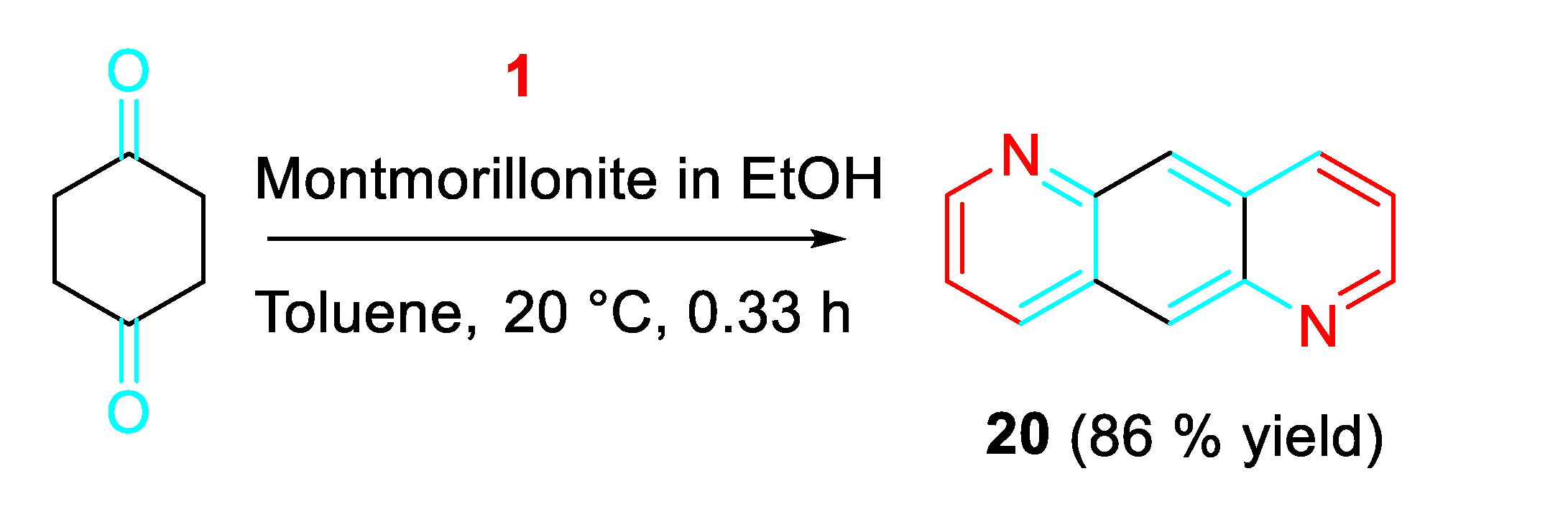
Interestingly, hetero-anthracene derivatives as 20, used in the preparation of organic light emitting devices, were practically obtained under metal-free conditions (Scheme 20). [38]
Moreover, substituted dihydrophenanthrolines 21 were easily obtained from 2-substituted 6,7-dihydroquinoline-8(5H)-ketones and propargylamine in alcohol at 70-130 °C. This metal-free method has the advantages of safety, cleanness and wide substrate applicability. The product can be efficiently isolated by adjusting the temperature or prolonging the reaction time (Scheme 21). [39]
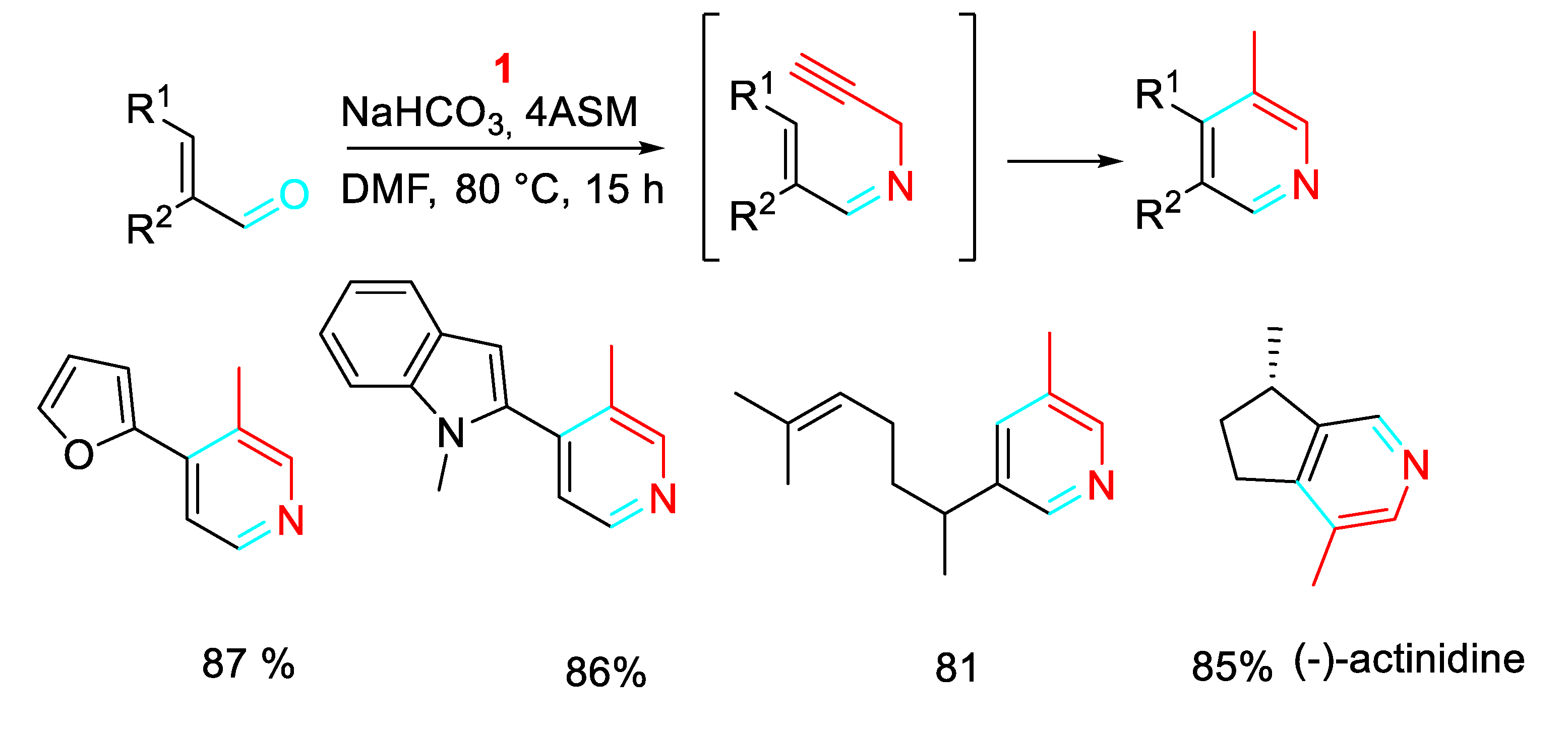
The reaction of readily available α,ß-unsaturated carbonyl compounds with propargylamine provided a high atom- and pot-economy strategy for the synthesis of polyfunctionalised pyridines under metal-free conditions with relevant functional group tolerance. (Scheme 22). [40]
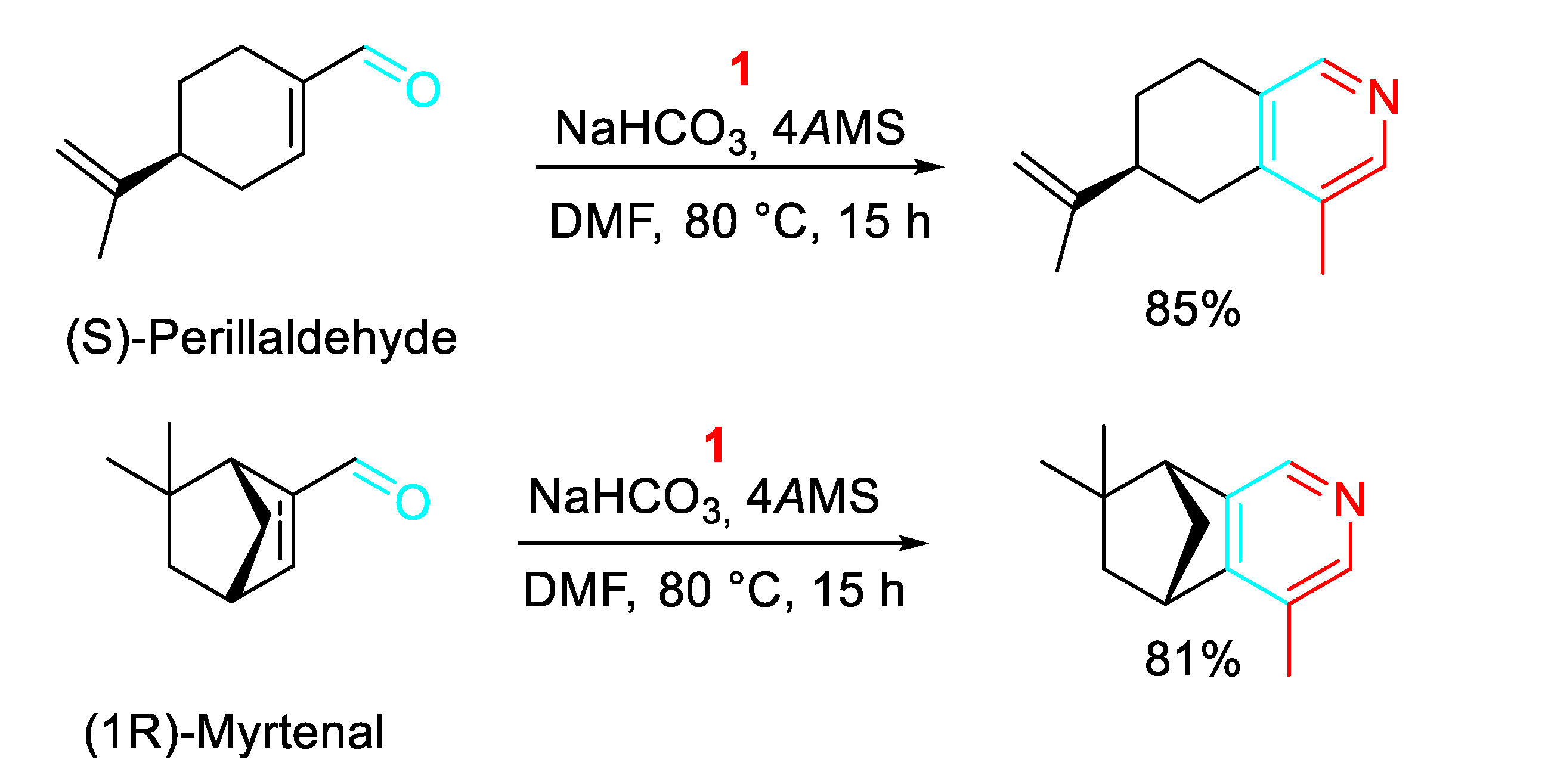
The method was applied to the synthesis of pyridines from the cosmetic, flavor and fragrance agent (S)-(-)-perillaldehyde and the flavoring agent found in cardamom, (1R)-myrtenal (Scheme 24).
The practicality of the protocol for the sustainable synthesis of these kinds of molecules through a tandem condensation/alkyne isomerization/6π-3-azatriene electrocyclization sequence was highlighted (scheme 26). [43]
The reaction of propargylamines with (hetero)aromatic aldehydes efficiently afforded β-carbolines, γ-carbolines and other fused azaheteroaromatics under metal-free conditions (Scheme 27). [44]
A one-pot, three-component method allowed the preparation of 3-substituted pyridines and carbolines 22 via copper-free, palladium-catalyzed Sonogashira cross-coupling with aryl iodides, followed by 6π-aza cyclization. This method provided selectively the fused pyridines in good yields (67−92%) (Scheme 28). [45]
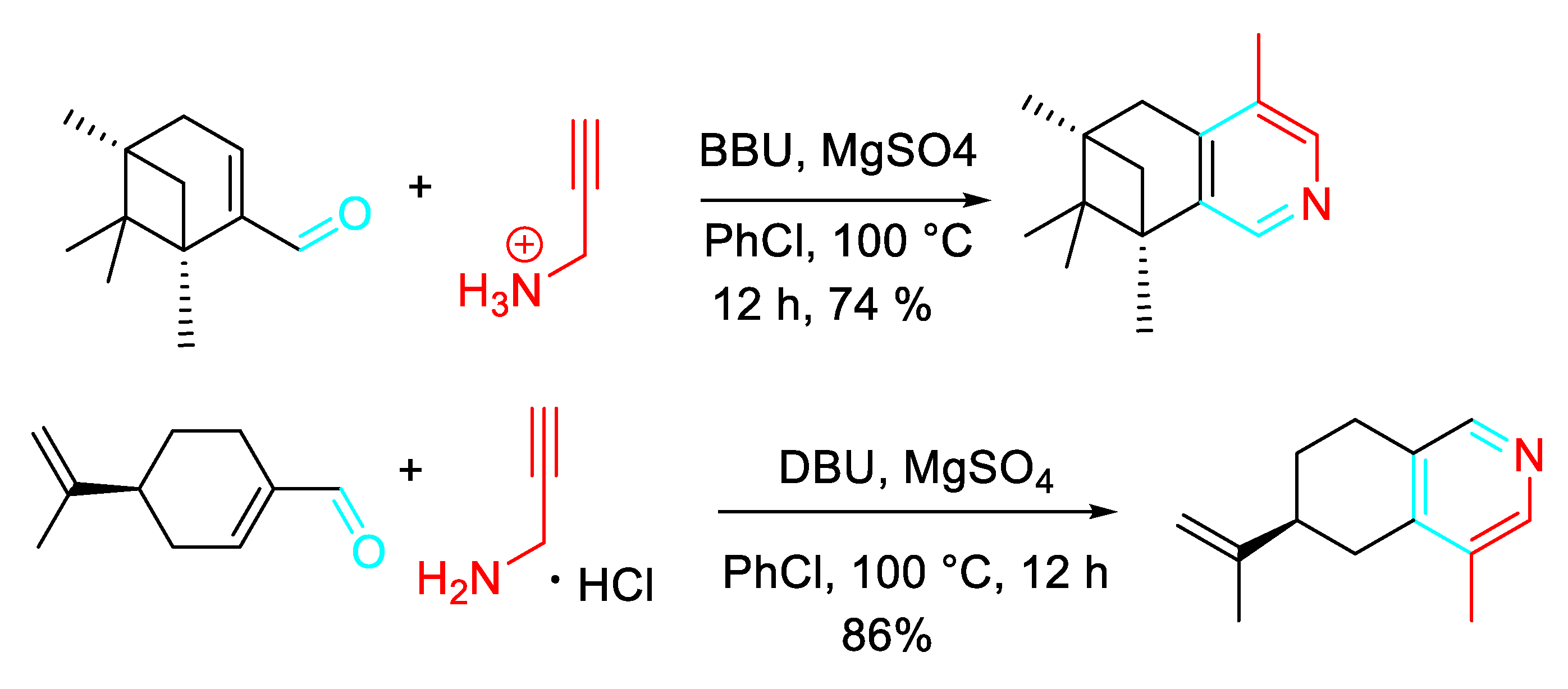
Alternatively, a further preparation method for the polysubstituted pyridine derivatives comprised the employment of an α,β-unsaturated carbonyl compound and propargylamine hydrochloride as raw materials in chlorobenzene (PhCl) with the sequential addition of 1,8-diaza-bicycloundecylenic-7-alkene (DBU) and magnesium sulfate (MgSO4) (Scheme 29). The advantage of this alternative procedure is a relatively strong industrial application prospect. [46]
Accordingly, an easy synthesis of Onychine 24, an azafluorenone alkaloid isolated from a plant of the Annonaceae family, was reported to occur through Aza-Claisen rearrangement, tautomerization, 1,5-sigmatropic hydrogen shift, 6π electron cyclization, and oxidation of the N-propargyl enamine, obtained in a yield of 61% by dehydration condensation of but-2-yn-1-amine with 1,3-indanedione (Scheme 30). [47]
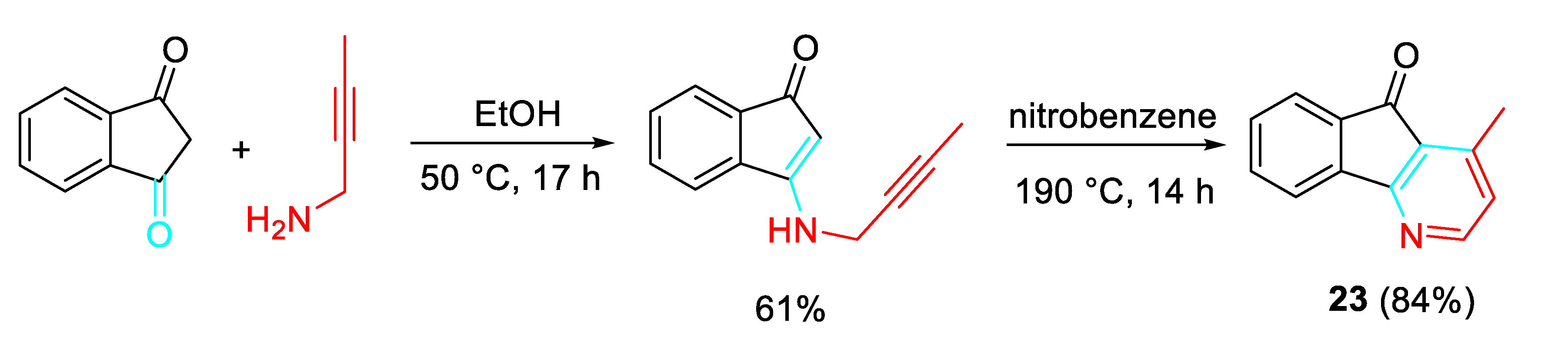
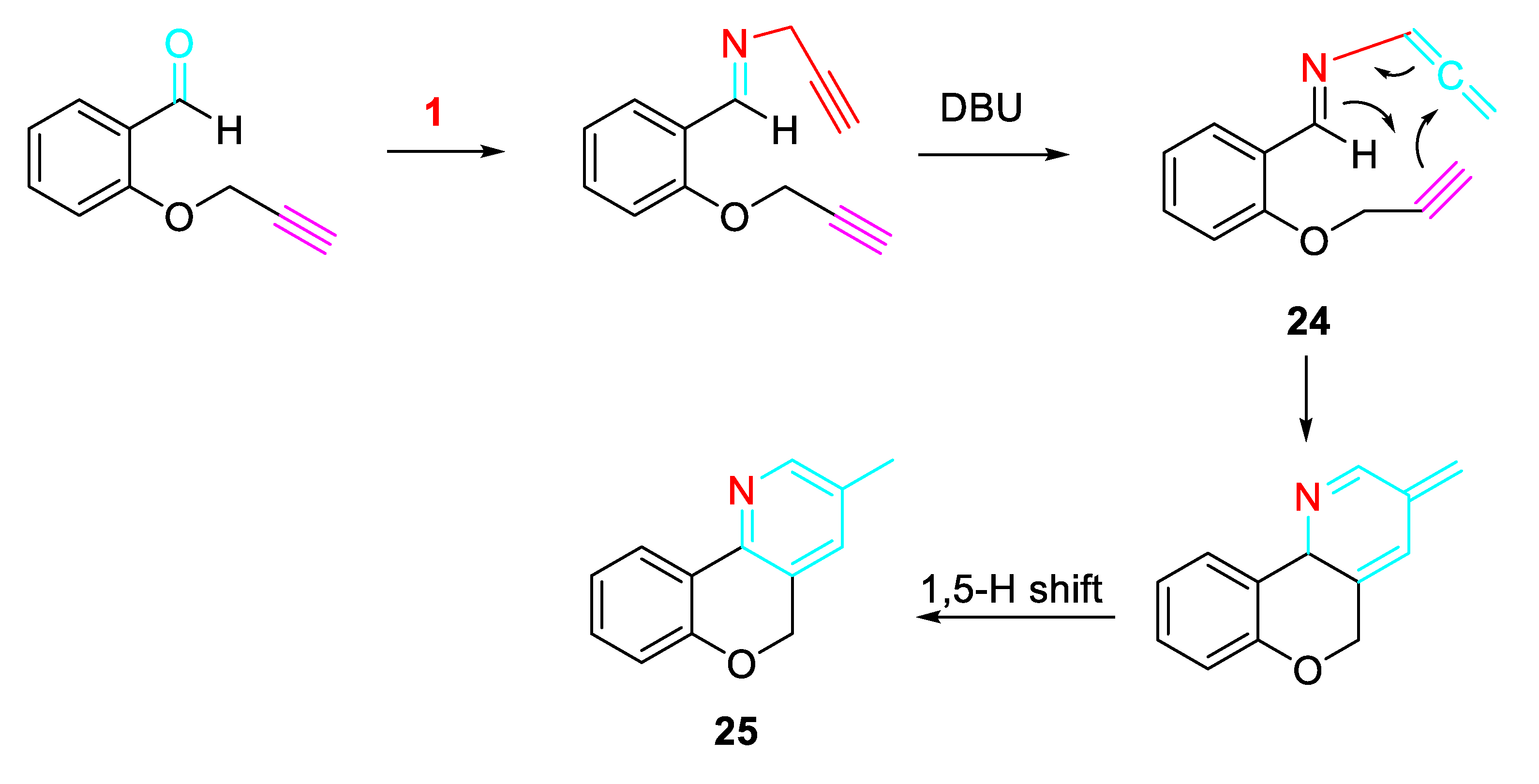
The sequential O-propargylation of aromatic hydroxyaldehydes/ condensation reaction with propargylamine allowed a simple approach the synthesis of chromenopyridine of and chromenopyridinone derivatives. The intramolecular cycloaddition reaction between the alkyne and azadiene of 24, which is formed as an intermediate, furnished the desired skeleton of chromenopyridine 25 (Scheme 31). [48]
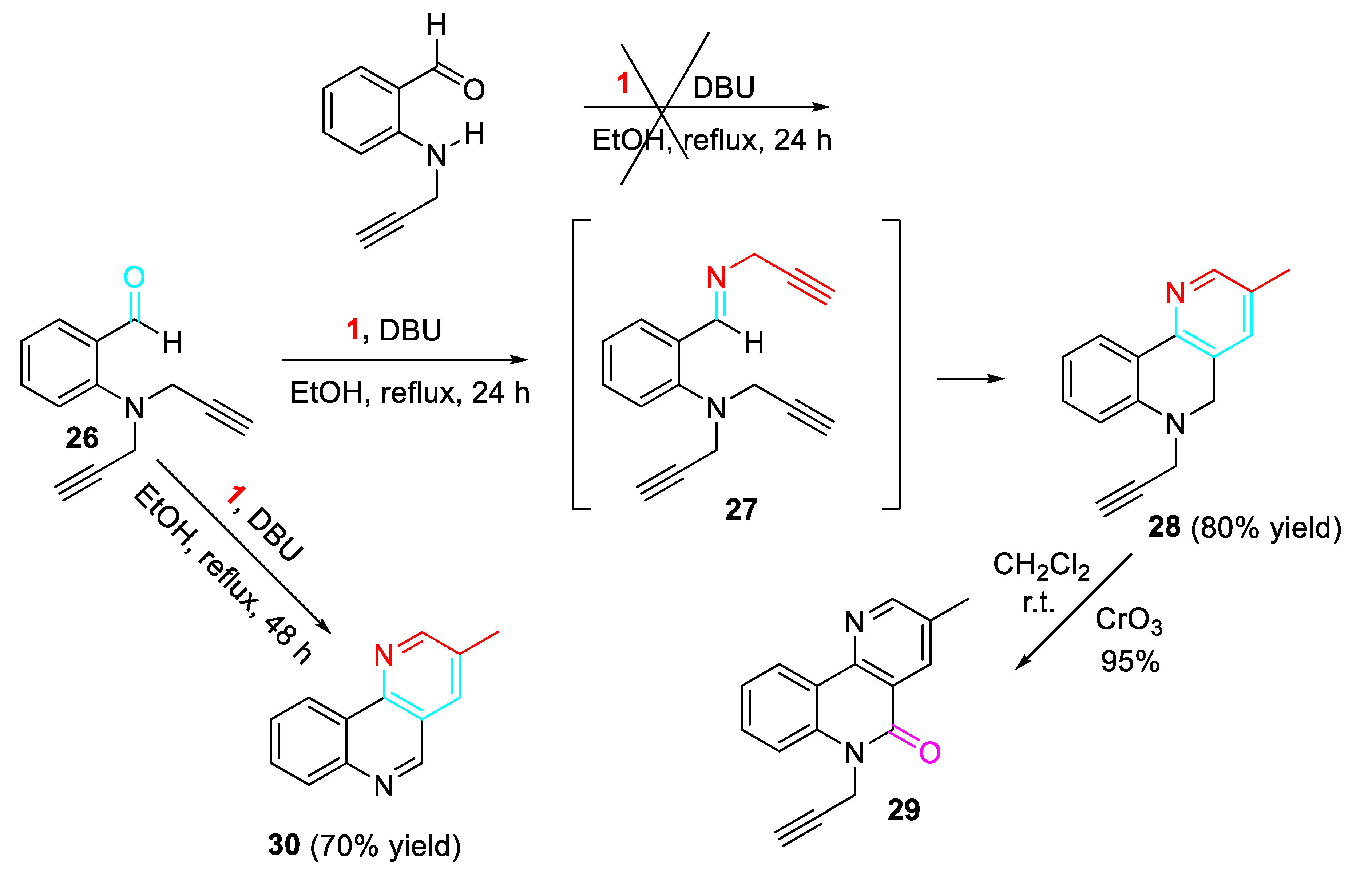
Moreover, the N-propargylation of aromatic aminobenzaldehydes, followed by reaction with propargylamine in the presence of DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) gave the corresponding benzo[h][1,6]-naphthyridines 30 (Scheme 32). [49] The lack of reactivity of the 2-(prop-2-yn-1-ylamino)benzaldehyde was surmounted by double propargylation of the aniline derivative leading to the intermediate 27 which cyclized in refluxing ethanol to afford the N-propargyl derivative 28 in 78 % yield. The 3-methylbenzo[h][1,6]-naphthyridine 30 was isolated by increasing the reaction time to 48 h. Oxidation of 28 with CrO3 in pyridine in dichloromethane at room temperature gave the desired product 29 in 95 % yield.
Moreover, a variety of starting materials synthesized by Sonogashira coupling reactions 31 afforded the corresponding naphthyridine derivatives 32 by reacting with propargylamine in refluxing EtOH in the presence of DBU (Scheme 33).
The approach was extended to the synthesis of the chromenopyrazinone derivatives 33 (Scheme 34).
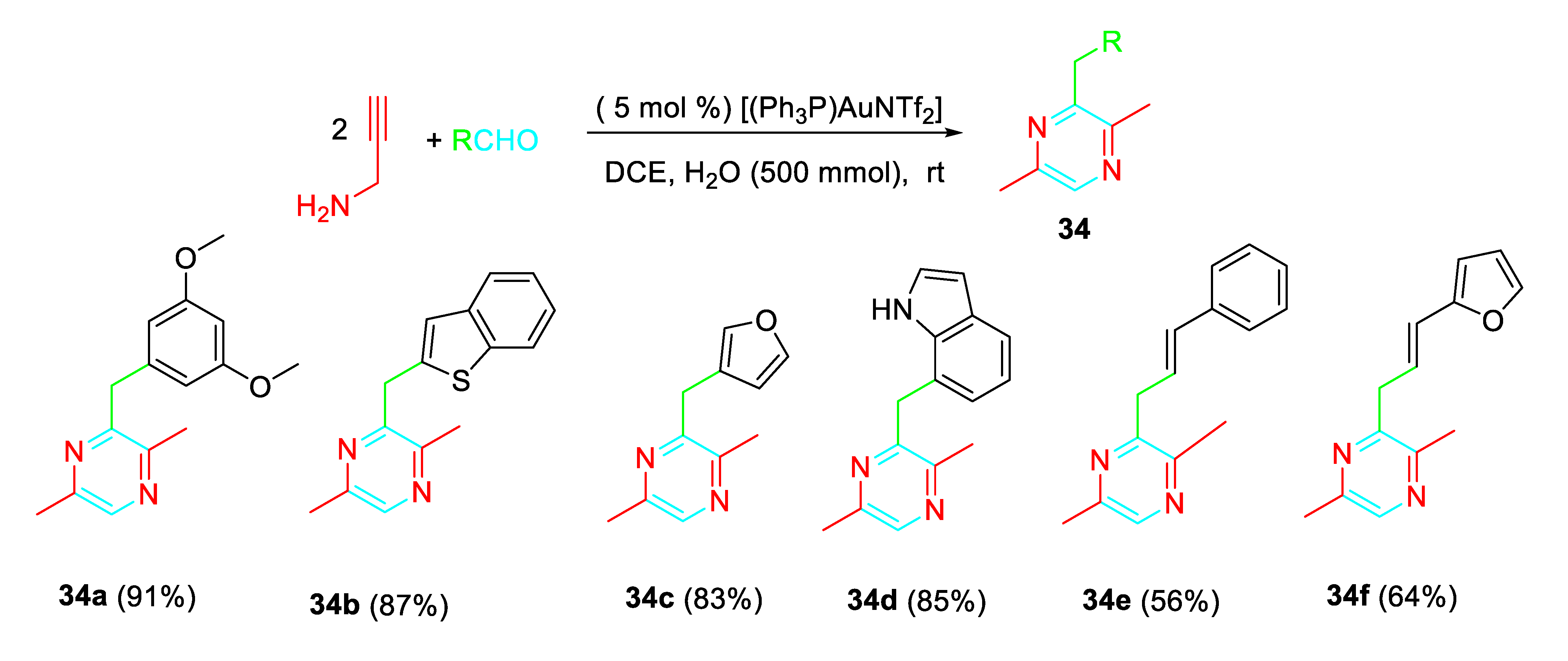
Pyrazines 34 were also synthesized through the gold-catalysed coupling reaction of aldehydes with propargylamine by means of a different sequential process (Scheme 35). [50]
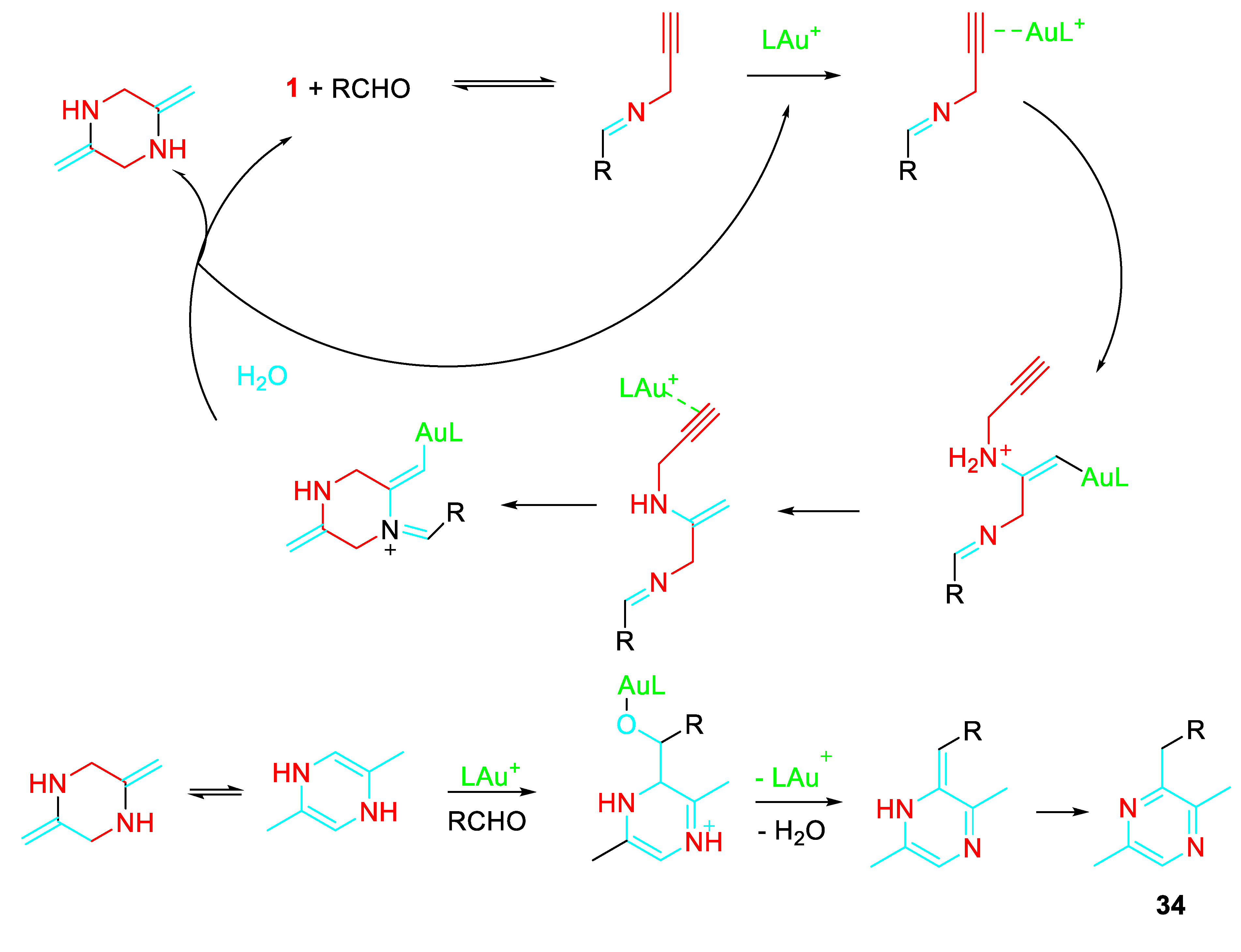
The following reaction mechanism was suggested. (Scheme 36).
The heterogeneous gold(I)-catalyzed version of the cascade reaction of aldehydes with propargylamine occurred in 1,2-dichloroethane at 40 °C under the presence of the readily available MCM-41-immobilized phosphine gold(I) complex [MCM-41-PPh3-AuNTf2]. The easily preparable heterogeneous gold(I) catalyst could be recovered by filtration and recycled (Scheme 37). [51]
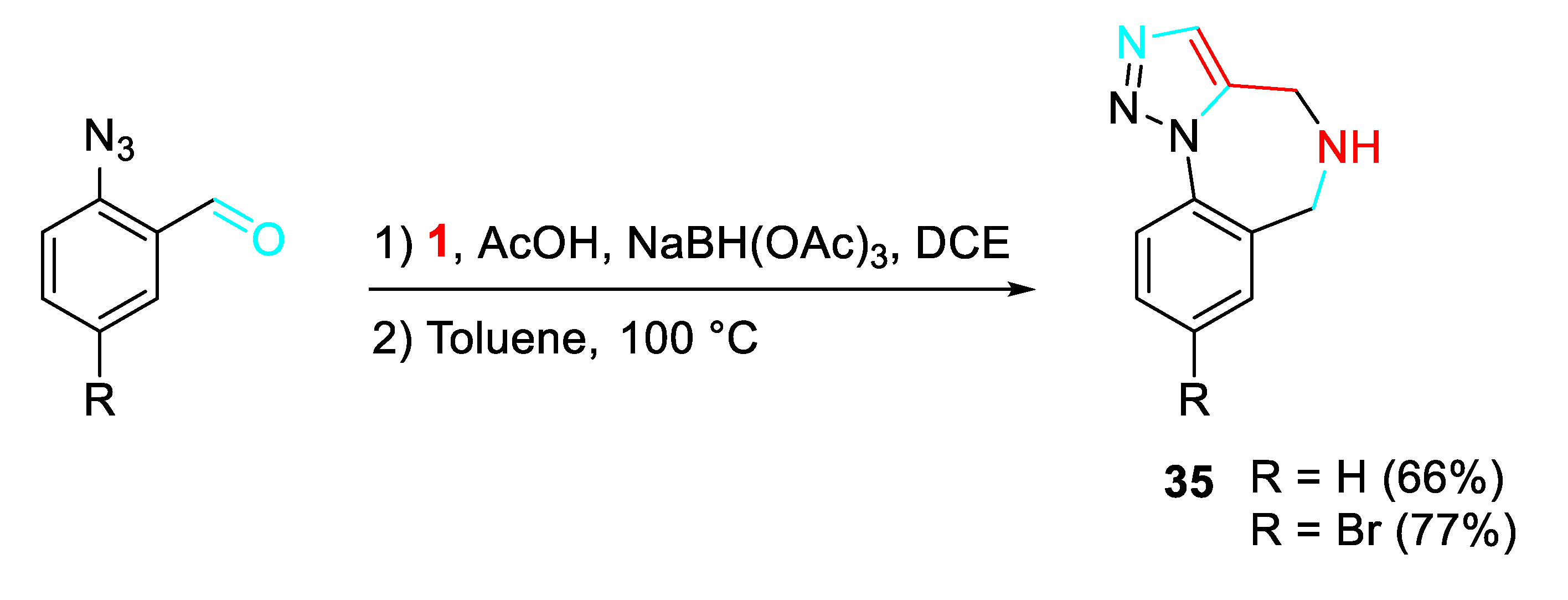
A variant of sequential multicomponent assembly processes (MCAPs)-cyclization approach in accord with the plan outlined in Scheme 38 was explored for preparing a variety of 1,2,3-triazolo-1,4-benzodiazepines 35 of possible medical relevance by a sequential reductive amination of 2-azidobenzaldeyde derivatives with propargylamine / intramolecular Huisgen cycloaddition. [52]
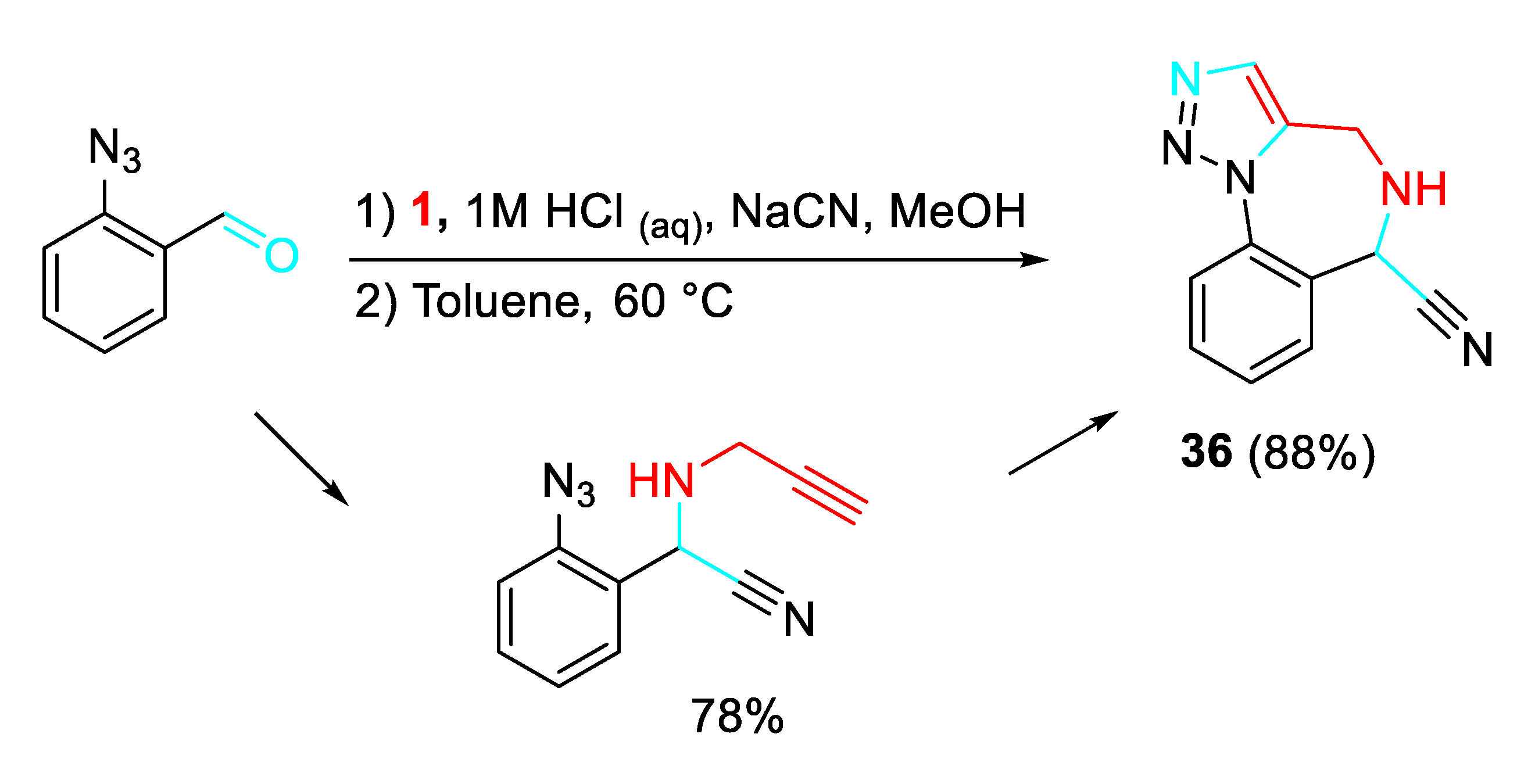
A wide library was obtained through N-functionalizations, palladium-catalyzed cross-coupling reactions, and applications of α-aminonitrile chemistry (Scheme 39). [53]
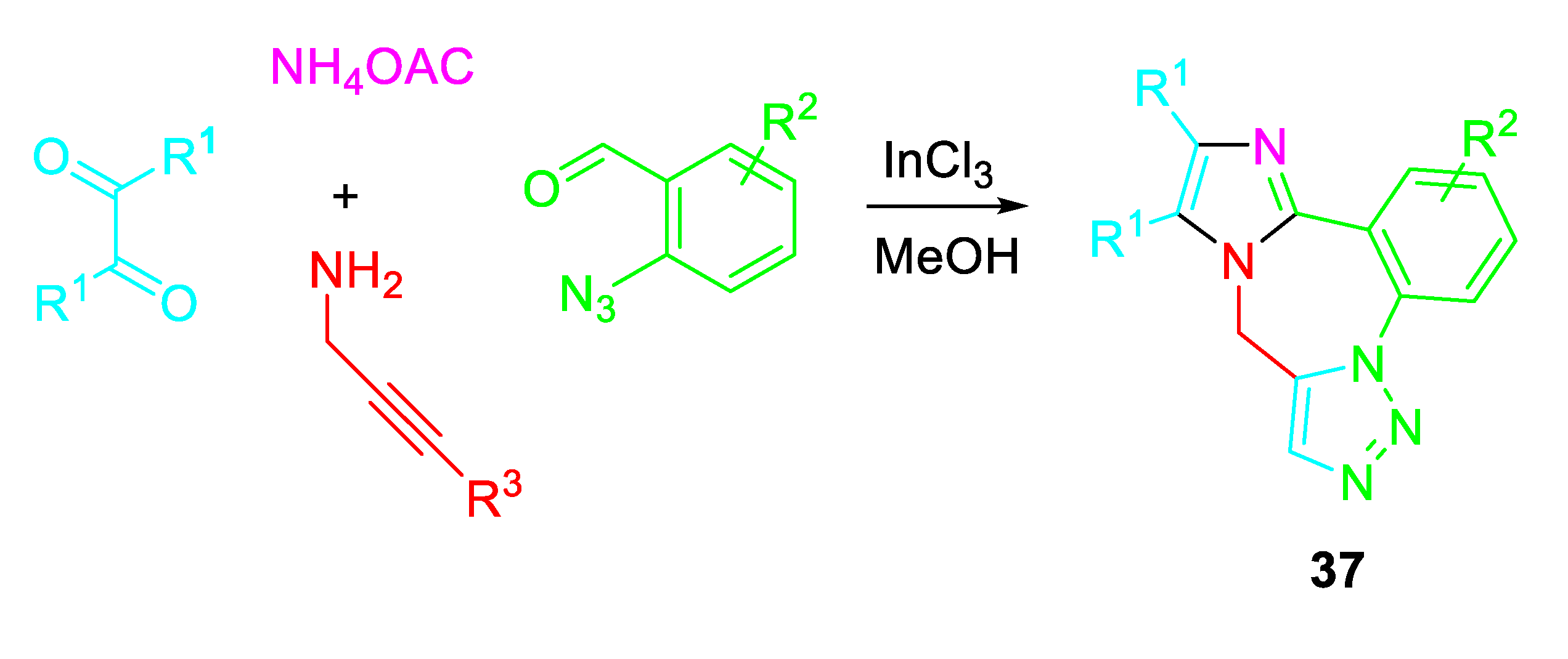
An atom-economical multicomponent sequential InCl3-catalyzed cyclocondensation / azide-alkyne 1,3-dipolar cycloaddition of 2-azidobenzaldehydes with propargylamines under the presence of α-diketone and ammonium acetate efficiently afforded the corresponding 9H-benzo[f]imidazo[1,2-d][1,2,3]triazolo[1,5-a][1,4]diazepines 37 (Scheme 40). [54].
3. Sequential Reactions of β-Aminoalkynes with Carbonyls
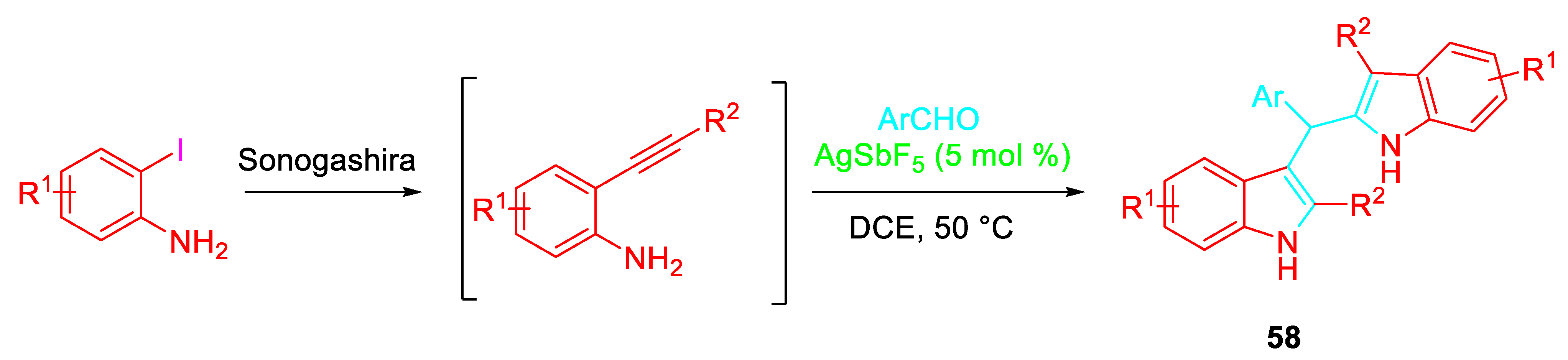
Versatile β-aminoalkynes building blocks for the synthesis of nitrogen containing heterocyclic compounds are represented by 2-alkynylanilines 38 [55-57]. Their sequential reaction with carbonyl derivatives was directed towards the formation of different scaffolds by changing the reaction conditions. The reaction of 38 with simple ketones or β-ketoesters selectively afforded the corresponding N-(Z)- alkenyl indoles 40 under the presence of InBr3 catalyst. The sequential reaction was considered to proceed through the activation of the β-keto esters / formation of β-enamino esters 39 / intramolecular 5-endo-dig cyclization promoted by activation of the acetylene (Scheme 41). [58]
Conversely, the divergent cyclization/alkenylation sequence to give the indole derivative 41 occurred by reacting the 2-alkynylanilines 38a with 1,3-dicarbonyls under presence of NaAuCl4·2H2O as the catalyst (Scheme 42). [59]
Moreover, reactions between readily available 2-alkynylanilines and activated ketones promoted by p-toluenesulfonic acid (p-TsOH) afforded 4-alkyl-2,3-disubstituted quinolines 42. The features of substituents at the other end of the triple bond of 2-alkynylanilines achieved to access to diversified 4-alkylquinolines, achievable with difficulty by classical Friedländer reaction (Scheme 43). [60]
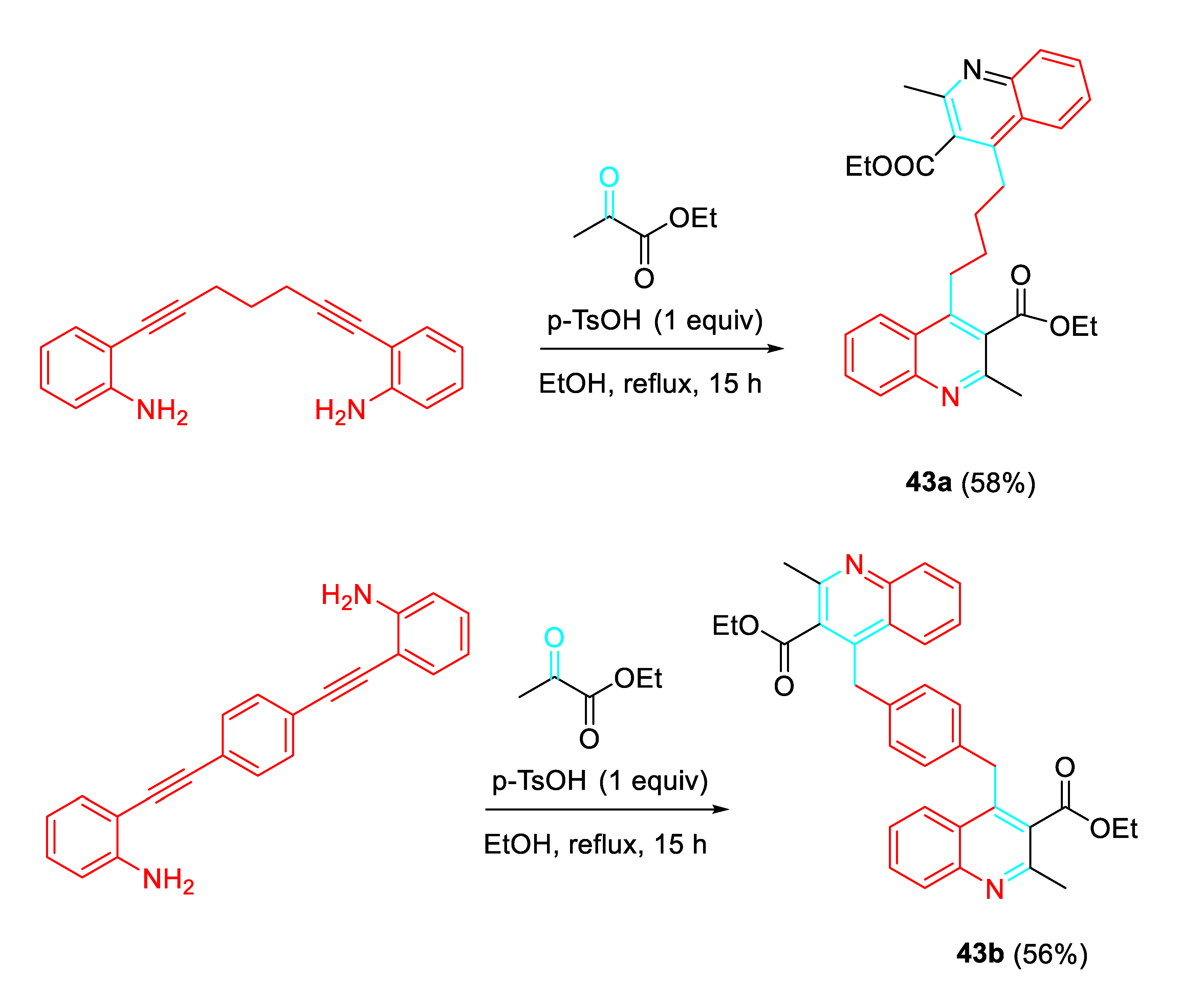
The procedure also achieved the preparation of quinoline dimers 43 with alkyl or aryl linkers at C-4 (Scheme 44).
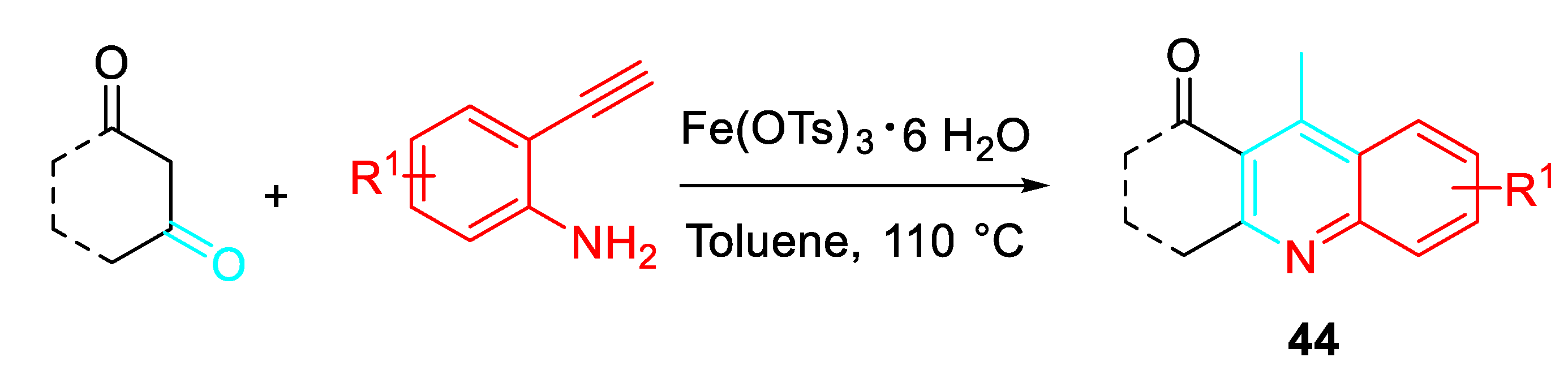
Alternatively, the one-pot synthesis of 4-methyl-2,3-disubstituted quinolines 44 was achieved by means of the inexpensive iron(III)catalyzed sequential condensation, cyclization and aromatization of 1,3-diketones with 2-ethynylaniline derivatives (Scheme 45). [61]
Furthermore, a cost-effective p-TsOH promoted synthetic strategy for the synthesis of substituted quinolines was achieved by the reaction between levulinic acid with different 2-alkynylanilines under mild metal-free solventless conditions (Scheme 46). [62]
The combination of CuBr2 and trifluoroacetic acid (TFA) directly afforded the corresponding quinolines/naphthyridines by reacting the 2-ethynylaniline with ethyl glyoxylate (Scheme 47). [63]
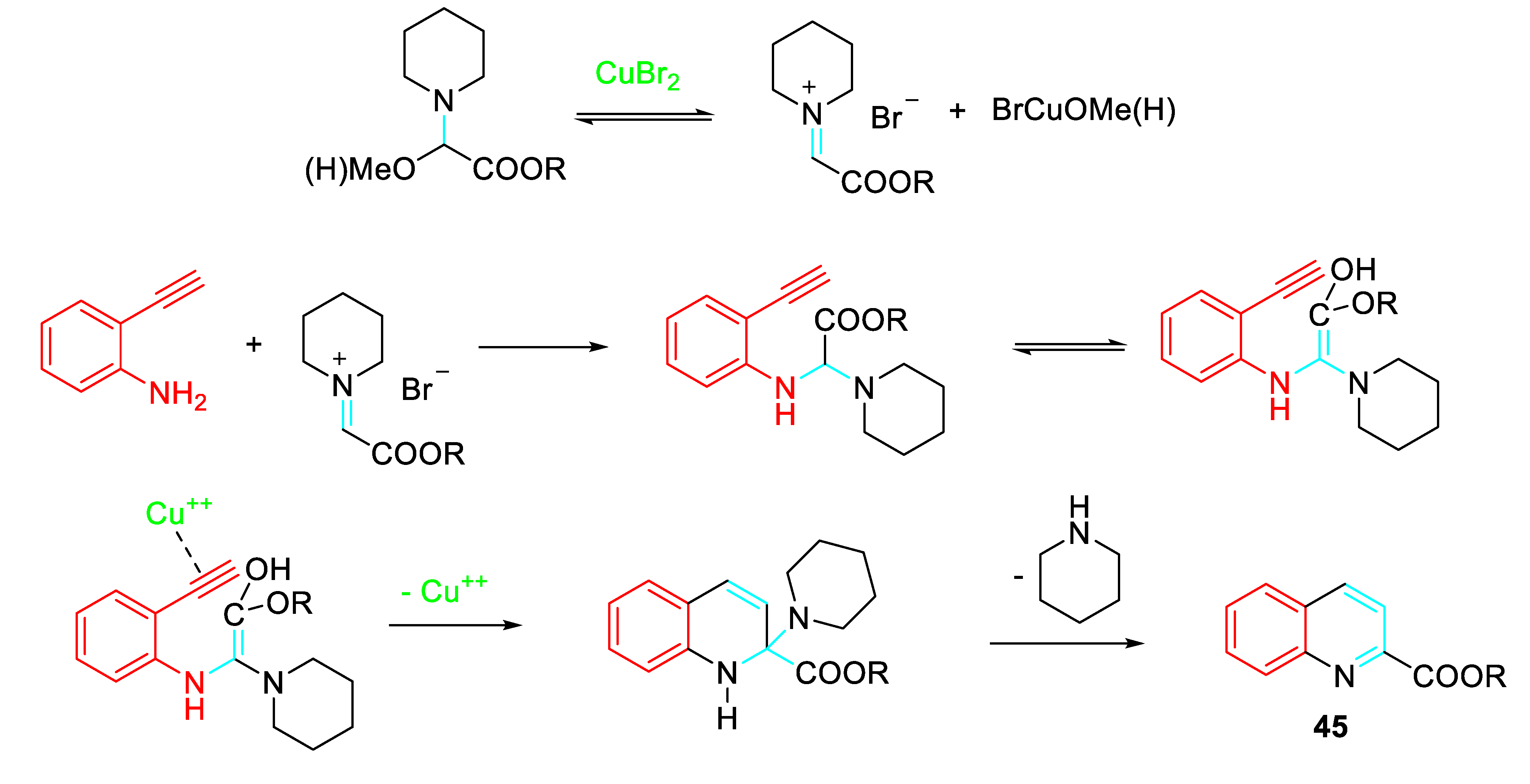
N, O- acetals also functioned as a C1 part leading to the preparation of quinoline derivatives 45 according to the following path (Scheme 48). [64]
A three-component, one-pot sulfuric acid mediated reaction of 2-(2-(trimethylsilyl) ethynyl)anilines with arylaldehydes in alcohol efficiently provided 4-alkoxy-2-arylquinolines 46 (Scheme 49). [65]
This strategy was extended to efficiently afford the unusual formation of 8-aryl-8,9-dihydro-3H-pyrano[3,2-f]quinoline-3,10(7H)-dione derivatives 48 by condensative cyclization of 6-amino-5-[(trimethylsilyl)ethynyl]-2H-chromen-2-one 47 with aromatic aldehydes (Scheme 50). [66]
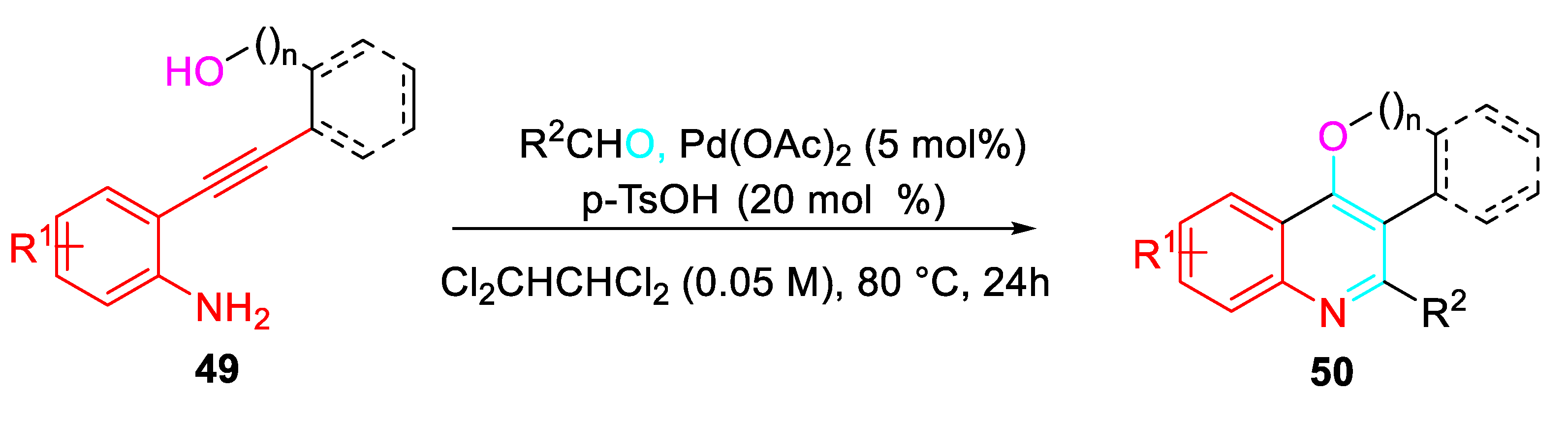
Synthesis It was envisioned that the in situ generated N-(2-alkynylphenyl)imine might be cyclized to give ring-fused quinoline derivatives. Indeed, a tandem reaction of 2-alkynylanilines 49 with aldehydes catalyzed by the combination of Pd(OAc)2 and p-TsOH allowed the regioselective synthesis of ring-fused quinolines 50 (Scheme 51). [67]
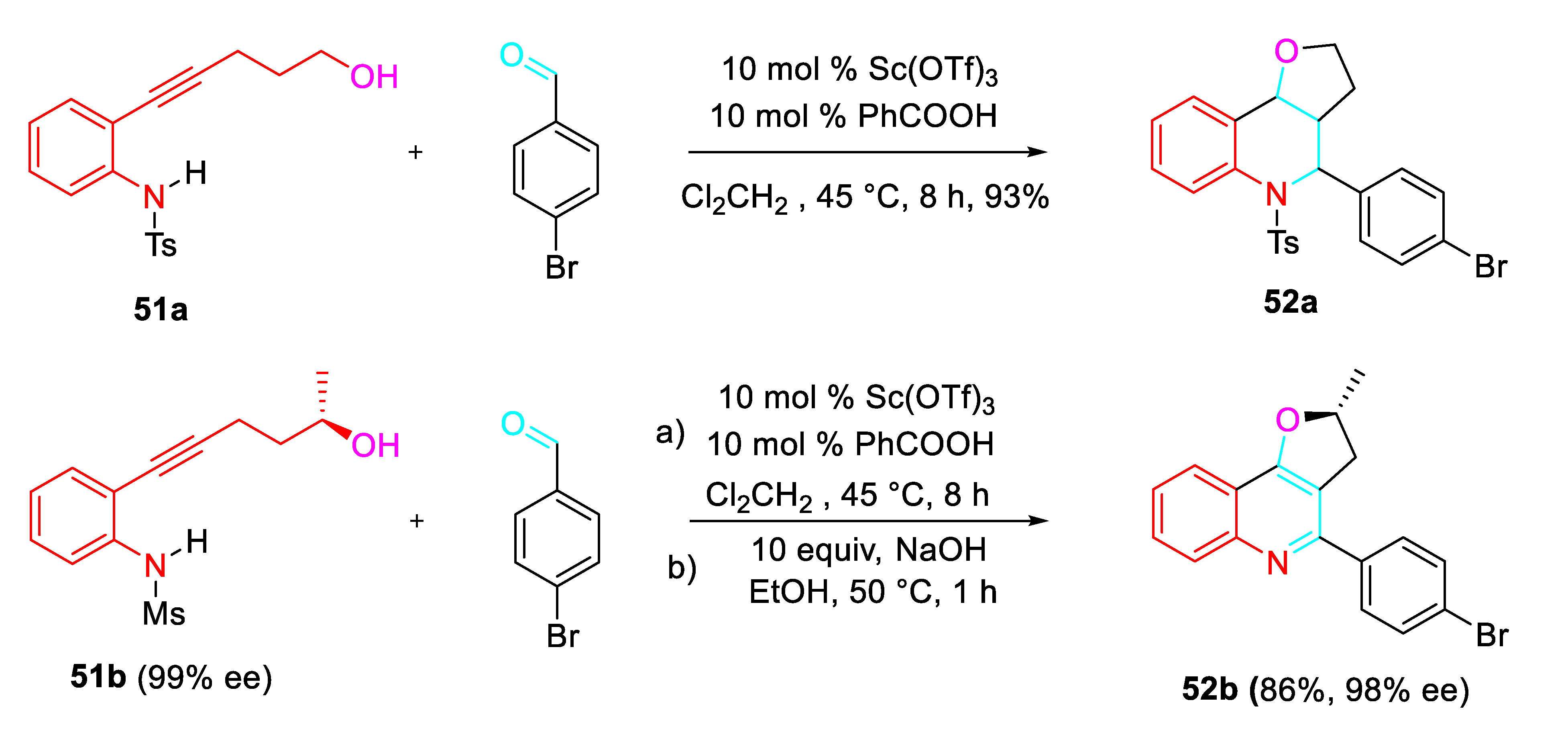
Moreover, a Sc(OTf)3-catalyzed tandem aza-Prins cyclization reaction of 2-alkynylaniline derivatives 51 with aldehydes afforded under mild reaction conditions fused tricyclic derivatives 52. Interestingly, when the enantiopure optically active 2-alkynylaniline (R)-51b, having a central chirality (99% ee), was subjected to the optimized reaction conditions followed by subsequent treatment with NaOH, the quinoline derivative (R)-52b was obtained directly (86% yield, 98% ee) (Scheme 52). Six- and seven-membered oxacyclo-fused products were also easily synthesized. [68]
It is worth noting that the N-(2-alkynylphenyl) imines 53 readily available by means of condensation of 2-iodoanilines with ketones or aldehydes followed by Sonogashira coupling with acetylenes were prone to undergo different sequential processes. Ring-fused indoles 54 were obtained from N-(2-alkynylphenyl) imines 53a in high yields under mild conditions under the presence of a gold (III) as a catalyst (Scheme 53). [69]
Furthermore, the N-(2-alkynylphenyl) imines intermediates 55 treated with NIS in DCM induced novel iodonium mediated domino reaction cascades, which provided ring-fused indole compounds 56 or simply by changing the reaction conditions ring fused quinoline compounds 57 (Scheme 54). [70]
The application of the methodology to the synthesis of iodoquinolones from suitable N-(2-alkynylphenyl)imine [71] as well as to the construction of polycyclic indole derivatives through the [3 +2] cycloaddition of metal-containing azomethine ylides generated from N-(o-alkynylphenyl)imine derivatives and W(CO)5(L) [72] were also reported. A Different sequential cycloisomerization/C3-functionalization of the in situ generated 2-alkynylanilines via Sonogashira coupling of 2-iodoanilines achieved a one-pot synthesis of 2,2’-disubstituted diindolylmethanes (Scheme 55). [73]
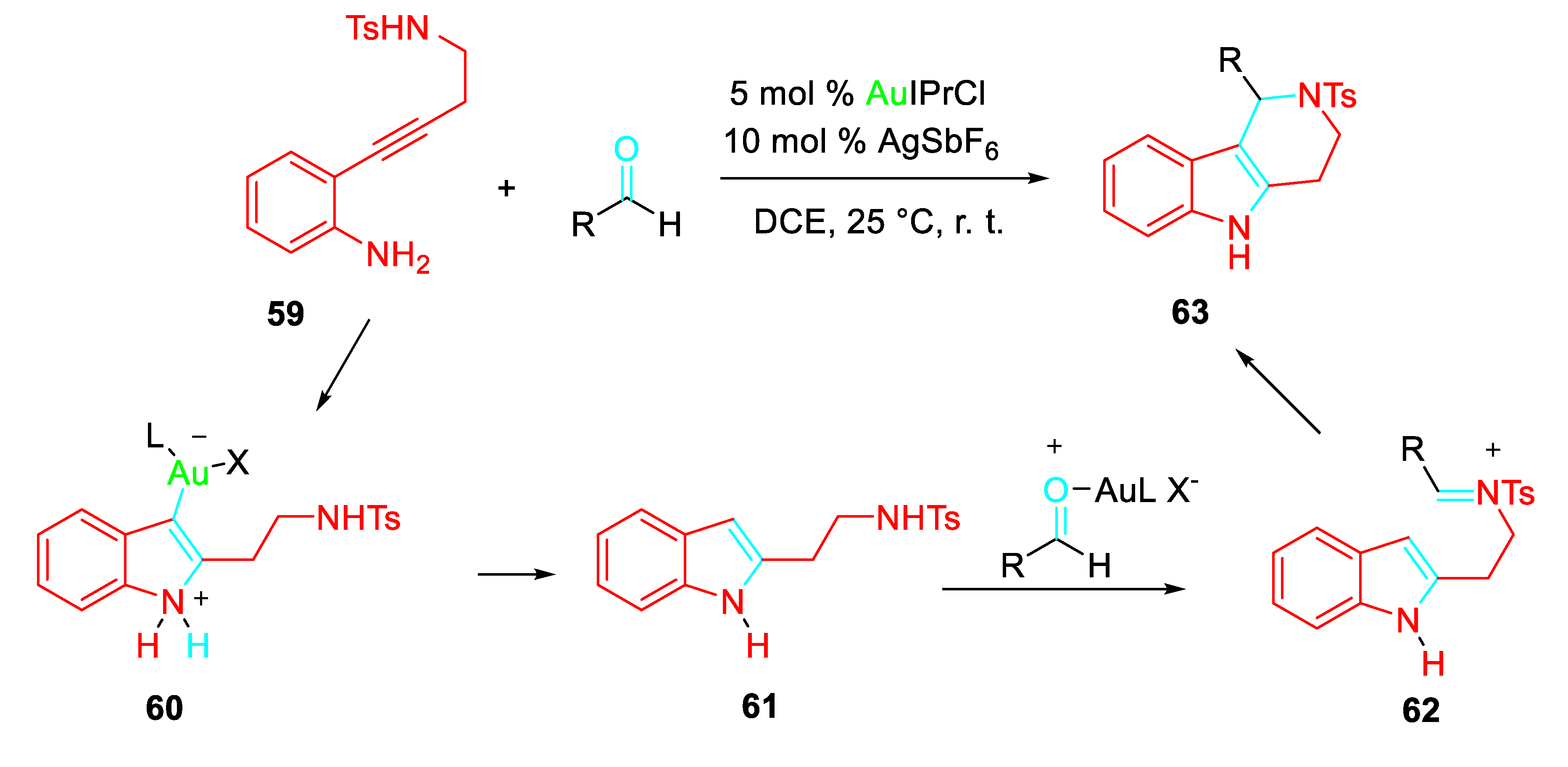
A one-pot strategy for the synthesis of 1-substituted 2-tosyl-2,3,4,5-tetrahydropyrido[4,3-b]indole scaffolds 63 through a sequential gold-catalyzed hydroamination/Pictet-Spengler cyclization of 2-(4-aminobut-1-yn-1-yl)aniline with aldehydes was demonstrated (Scheme 56). [74] The initial π-coordination of cationic Au(I) species with the alkyne moiety of 2-(4-aminobut-1-yn-1-yl)aniline 59 forms a π-complex which gave the cyclic intermediate 60. The following protodemetalation affords the isotryptamine 61. Subsequently, activation of aldehyde by Au(I) species followed by an intramolecular nucleophilic addition of indole moiety of 61 to a highly reactive N-sulfonyliminium intermediate 62 provides the tetrahydropyridoindole 63 with regeneration of the catalyst. Ag(I) also promotes the Pictet-Spengler reaction.
A Cu(OTf)2-catalyzed intramolecular radical cascade reactions efficiently enabled the synthesis of quinoline-annulated compounds 65. [75] The method represents an effective route to natural products and a variety drug like libraries (Scheme 57).
The proposed mechanism for the synthesis of the polyheterocyclic scaffolds is shown in the following Scheme 58. The intermediate 66 undergoes a copper salt promoted one-electron oxidation to generate the radical cation 67. Subsequent radical addition into the C-C bond of 68 affords the radical 69, which cyclizes to give the radical 70. Finally, trapping of the nitrogen radical by iodine radical generated from the oxidation of iodide affords the complex 71 which after iodide elimination furnishes the product 65.
A regio- and stereoselective three-component, one-pot cascade reaction involving an imination/annulation/cyanation sequence was achieved by combining palladium(II) trifluoroacetate and copper(II) acetate with the readily available 2-alkynylanilines, cyclic ketones and trimethylsilyl cyanide in dimethyl sulfoxide to efficiently afford the corresponding 1-benzoazepine carbonitrile derivatives 72 (Scheme 59). [76]
The construction of spirocyclic quinolones 73 which are difficult to synthesize through traditional methodologies was explored by selectively directing the reaction of 2-alkynylanilines with ketones under suitable reaction conditions. Interestingly, the same starting reagents selectively afforded the quinolines 74 or the N-alkenyl indoles 75 under different reaction conditions (Scheme 60). [77]
Very likely, the condensation reaction under the Brønsted acid mediated conditions in EtOH led to the iminium ion intermediate [A] which undergoes aza-Prins to afford the intermediate which after quenching by ethanol, hydrolysis and tautomerization reactions gave the quinolinone 73 (Scheme 61).
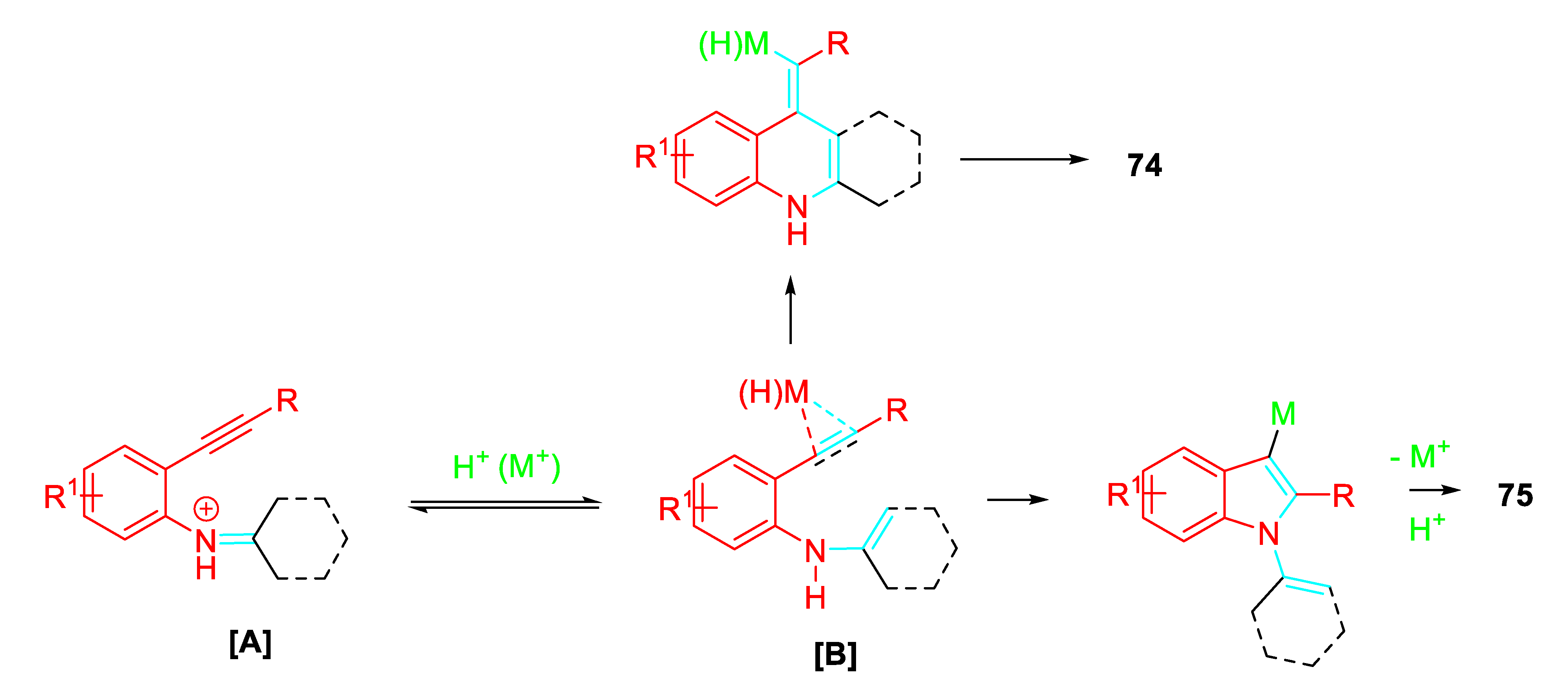
Conversely, isomerization of the iminium ion intermediate [A] to the intermediate [B] should led selectively to the indoles 75 via a 5-endo-dig cyclization or to the quinoline 74 via a regio-divergent 6-exo-dig cyclization (Scheme 62).
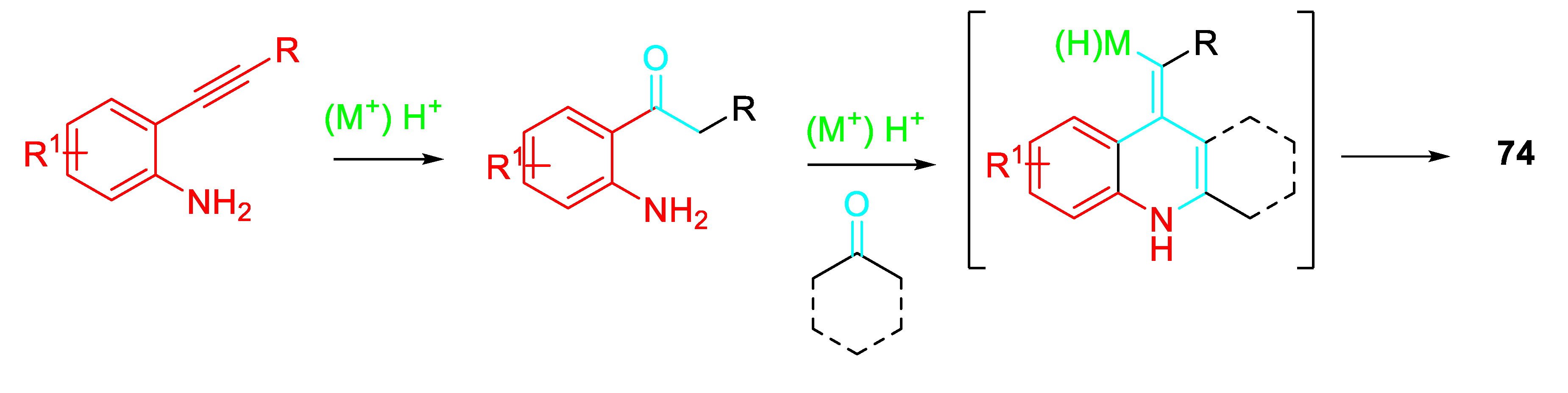
Alternatively, the quinolines 74 could be generated from the 2-aminoaryl ketone obtained by the fast hydration reaction of the 2-alkynylaniline both in the presence of a stoichiometric amount of p-TsOH·H2O or 0.2 equiv. FeCl3 in Toluene at 110 °C (Scheme 63).
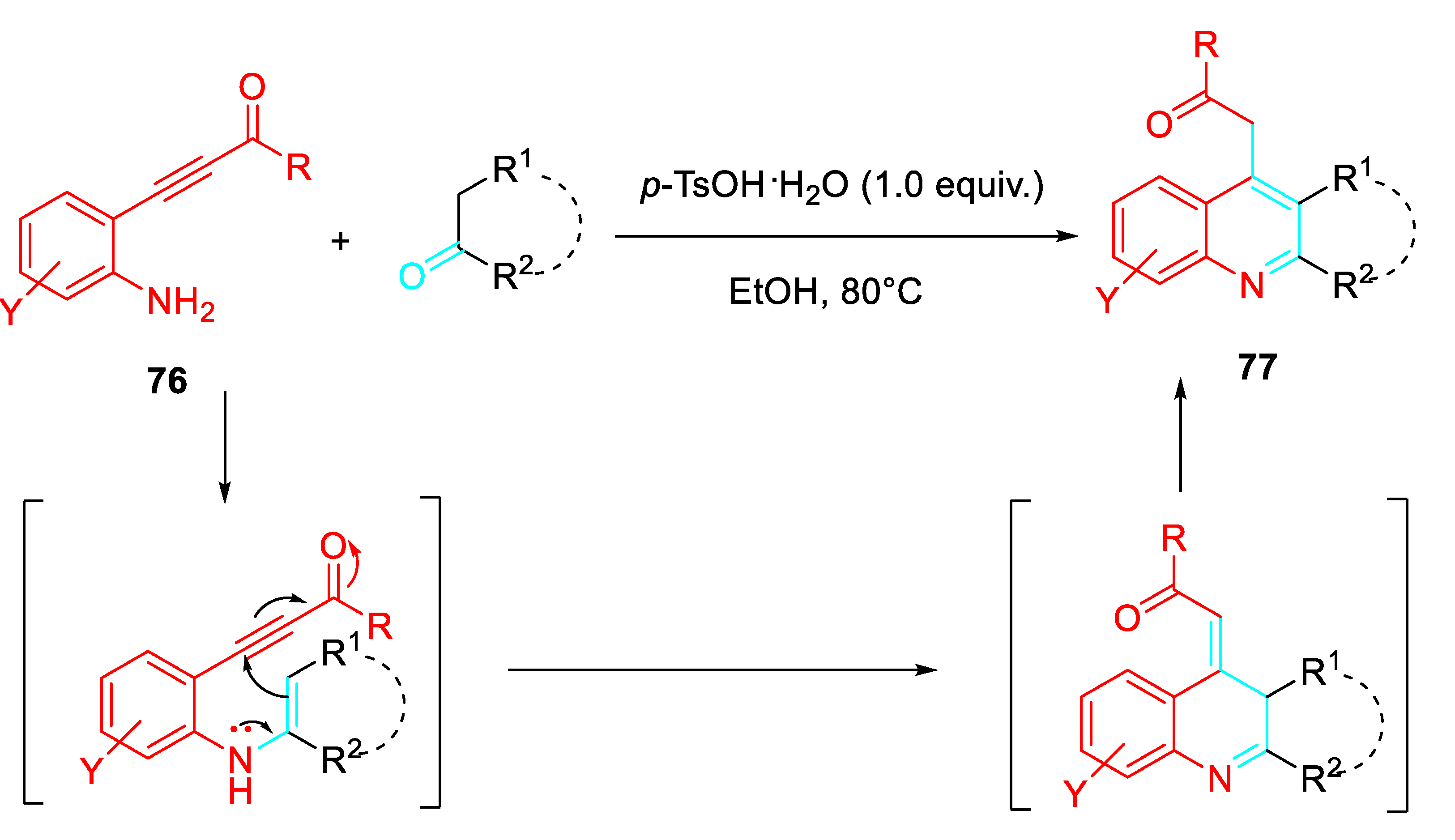
Interestingly, the features of the substituent bonded at the terminal position of the triple bond of the 2-alkynylaniline and of the reaction medium determined the reaction path. Internal alkynes allowed the p-TsOH·H2O mediated preparation of quinolones 73 in EtOH at reflux or the formation of the quinolines 74 in Toluene at 110°C both in the presence of a stoichiometric amount of p-TsOH·H2O or FeCl3 as the catalyst. Conversely, the ZnBr2 -catalyzed reaction in Toluene at 110 °C of the same internal alkyne derivatives gave only the N-alkenylindoles 75. The presence of a trimethylsilyl group or no substituent at the terminal position of the starting aminoalkyne resulted in the formation of the corresponding quinolines. Moreover, the sequential Brønsted acid mediated reaction with enolisable ketones in EtOH of the starting aminoalkynes β-(2-aminophenyl)-α,β-ynones 76 resulted in an efficient approach to only polycyclic quinolines 77 (Scheme 64). [78]
Steroids bearing a simple ketone group at position 3, such as 5α-cholestan-3-one, produced only the corresponding linear cholestanoquinoline derivative in moderate yield. On the contrary, the optimised methodology allowed the divergent generation of the angular quinoline derivative, whose synthesis is generally considered more challenging and demanding, from 3-keto-Δ4-polycyclic steroidal derivatives. Interestingly, with steroidal dicarbonyl derivatives, the condensation reaction took place selectively only on the conjugated carbonyl group at position 3, leaving the ketone group at position 17 unreacted (Scheme 65). A- and D-ring fused steroidal quinoline analogues represent potential antibacterial agents. [79]
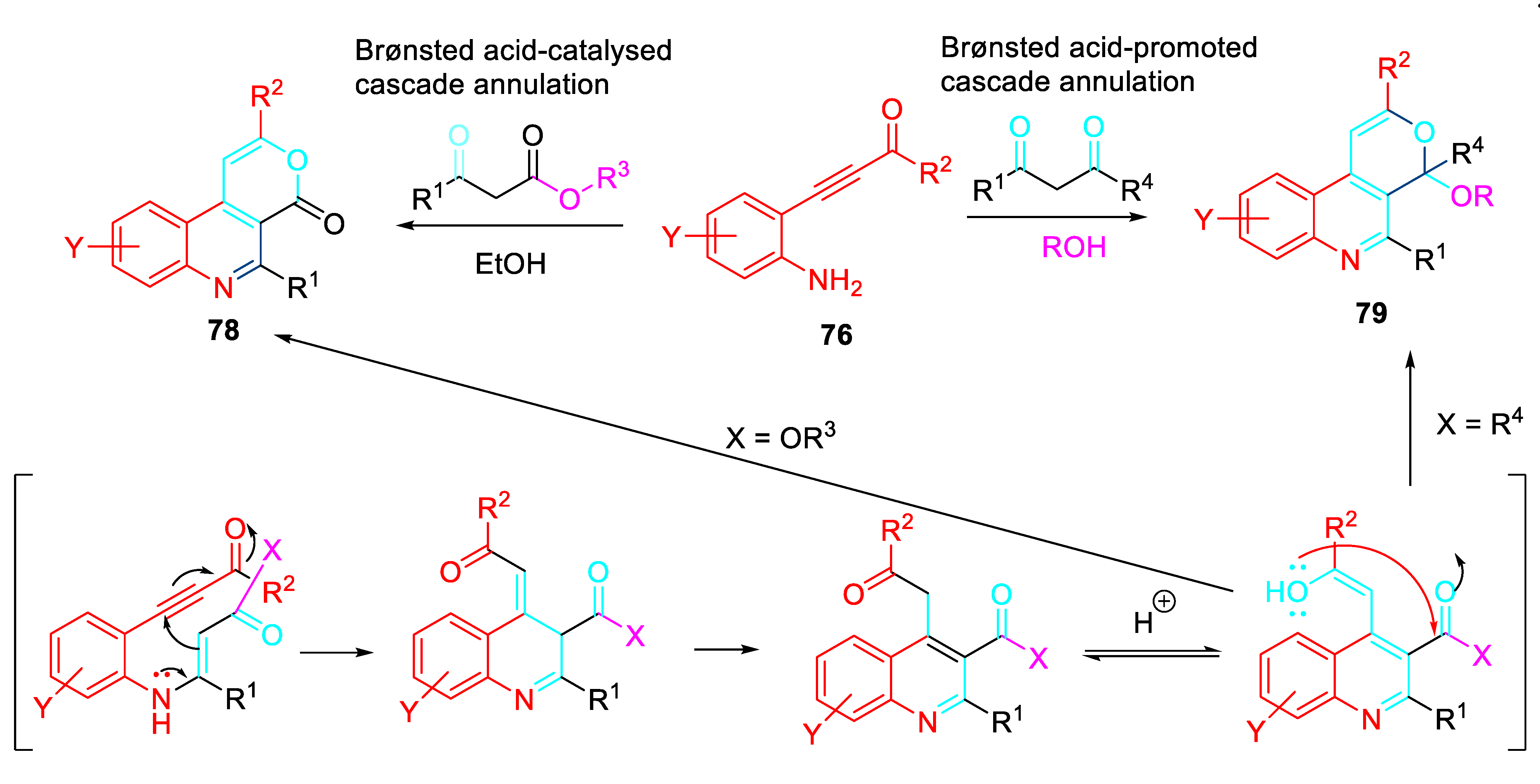
The Brønsted acid-promoted reaction of β-(2-aminophenyl)-α,β-ynones with ketones was expanded to activated carbonyl compounds such as β-ketoesters and β-diketones. The carbonyl group at position 3 of the quinoline nucleus could further react with the other keto functionality in the alkyl-substituent at position 4, generating an additional [3,4]-fused 6-membered ring whose structure depends on the type of β-dicarbonyl compound used. Indeed, for β-ketoesters, a thorough screening of reaction conditions revealed that catalytic amounts of p-TsOH.H2O were sufficient to efficiently promote a cascade double-cyclisation leading to 4H-pyrano[3,4-c]quinoline-4-one derivatives 78. On the contrary, with β-diketones, a stoichiometric amount of p-TsOH.H2O triggered a three-component reaction, involving a molecule of the alcoholic solvent to afford 79. Both procedures appear to be simple and versatile and are expected to be of great impact since the multiple potential applications of the obtained organic compounds (Scheme 66). [80]
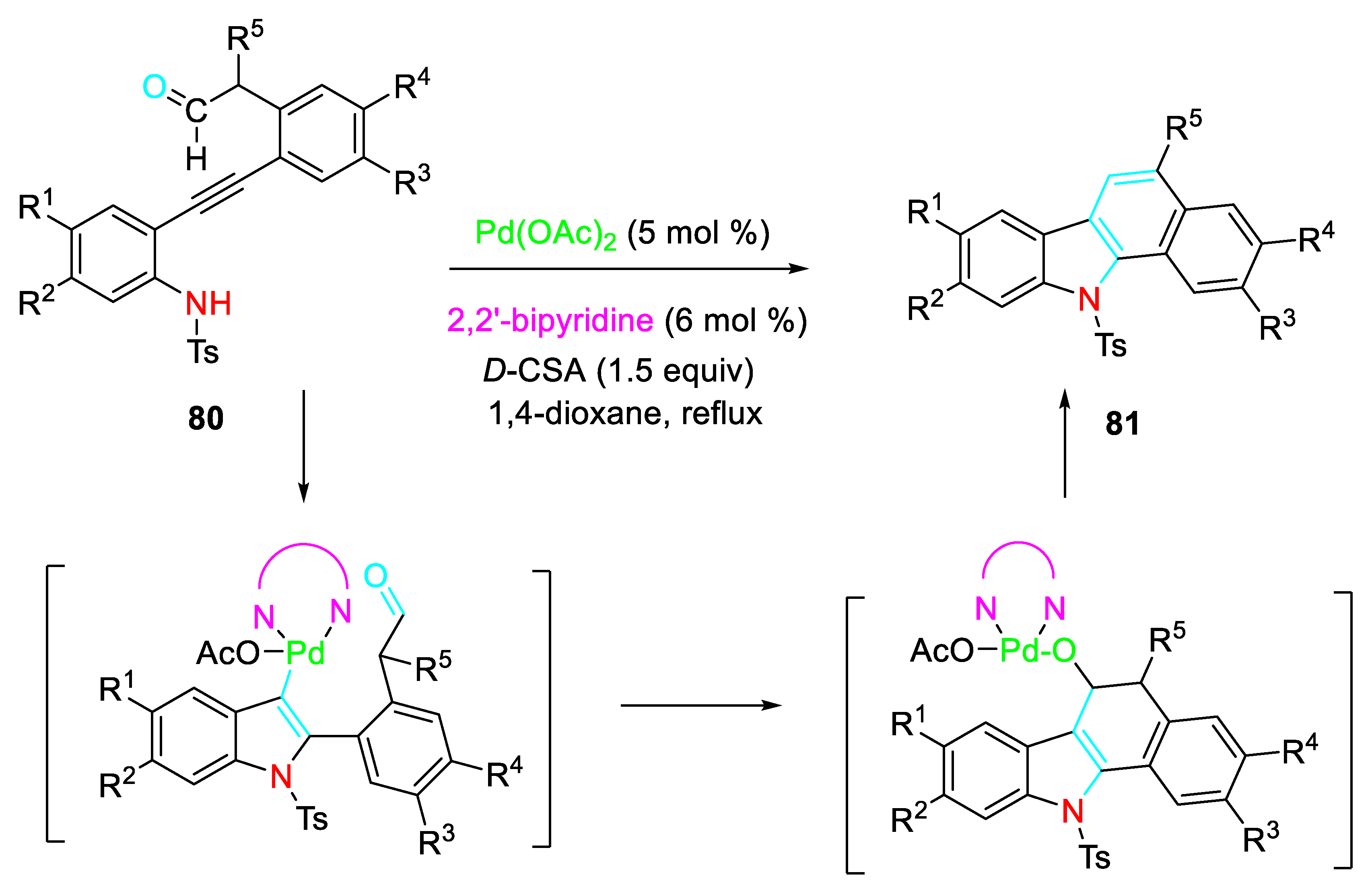
A sequential aminopalladation of β-amino alkyne derivatives 80, followed by intramolecular nucleophilic addition of the generated carbon-palladium bond to a tethered aldehyde group achieved the synthesis of a variety of benzo[a]carbazoles 81 with remarkable diversification (Scheme 67). [81]
The ongoing research activity devoted to the synthesis of indole derivatives encouraged to explore a high flexible approach to 11H-indolo[3,2-c]quinolines 83 according to the retrosynthetic Scheme 68.
Subsequently, the selective build-up of the 11H-indolo[3,2-c]quinoline 83 was carried out through a two-step one-pot gold-catalyzed reaction in CH3CN at room temperature of aldehydes with the 2,2’-(ethyne-1,2-diyl)dianiline derivative 82a as the starting β-aminoalkynes which was cyclised under the presence of Au catalyst (5 mol %). Then, the aldehyde (2 equiv.) was added and the reaction mixture was stirred till completion. This alternative high regioselective protocol is of wide applicability in mild neutral reaction conditions (Scheme 69). [82]
Surprisingly, the reaction of inexpensive aryl(heteroaryl)aldehydes with the same starting 2,2’-(ethyne-1,2-diyl)dianiline derivatives in the presence of a catalytic amount of HCl achieved the synthesis of 2,2'-disubstituted-1H,1'H-3,3'-biindoles 84 (Scheme 70). [83]
Accordingly, a class of double D–p–A branched organic dyes based on 2,2’-disubstituted-1H,10H-3,3’-biindole moiety have been synthesised as photosensitizers for dye-sensitized solar cells. [84] Alternatively, the 2,2′-disubstituted 3,3′-biindoleswere were obtained by using an acidic DES able to exploit a double activity, i.e. solvent and BA catalyst under microwave heating at 70 °C. [85] Very likely, the Brønsted acid promotes the formation of the iminium ion I by reaction of 83 with two equiv. of the aldehyde. The iminium ion I undergo an aza-Prins type 5-exo-dig cyclization to the intermediate (II) quenched by the nucleophilic addition of water to give intermediate III. The subsequent cyclization generates the stabilized benzylic carbocation intermediate IV which provides the desired biindoles 85 by the loss of a molecule of water and the proton regeneration (Scheme 71).
Analogously, the tandem gold-catalyzed 5-endo-dig / spirocyclization of 2-[(2-aminophenyl)ethynyl]phenylamines with isatins regioselectively afforded the corresponding 5’,11’-dihydrospiro-[indoline-3,6’-indolo[3,2-c]quinolin]-2-one derivatives 85 in good yields at room temperature. The reaction with ketones gave the 6,6-disubstituted-6,11-dihydro-5H-indolo[3,2-c]-quinolones 86 (Scheme 72). [86]
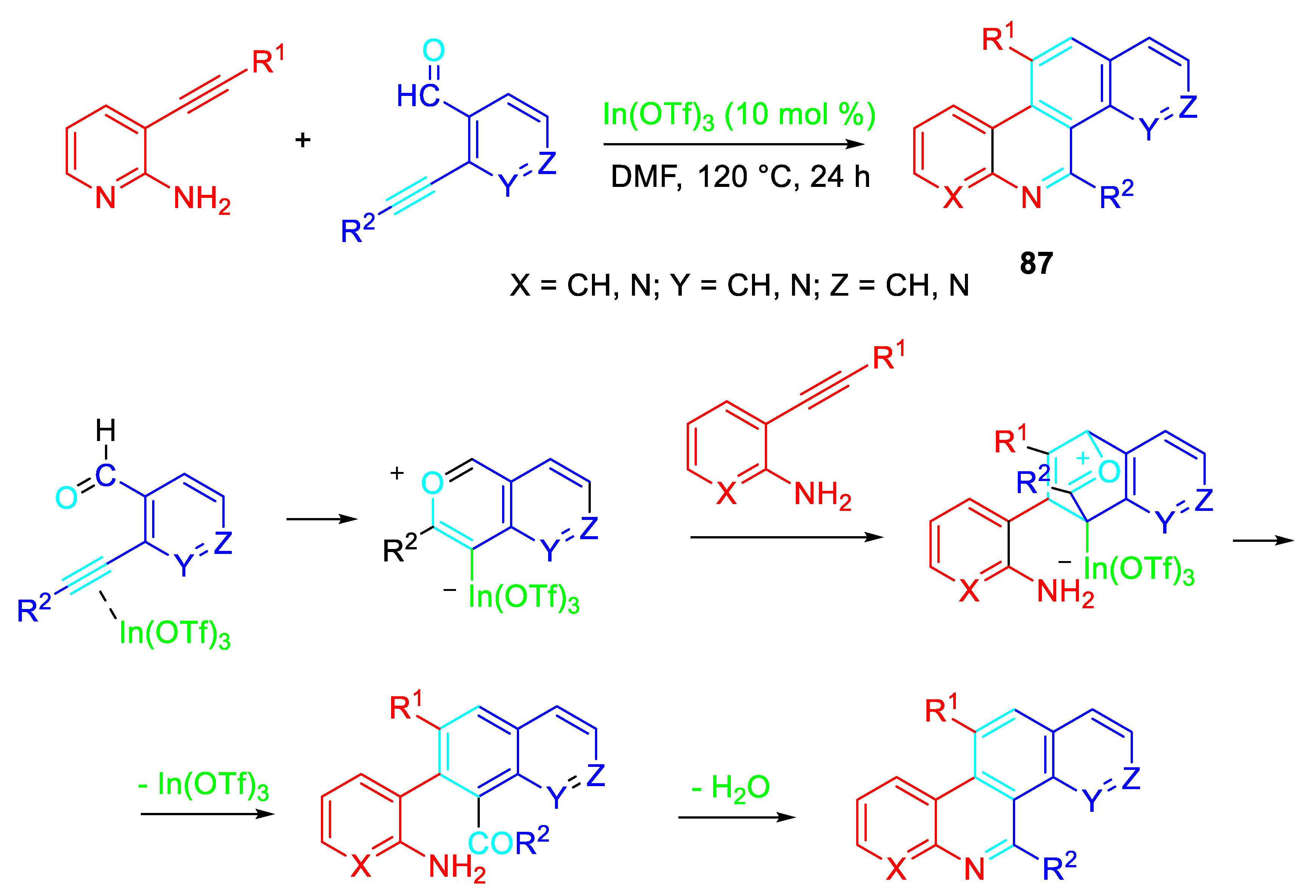
A sequential intramolecular nucleophilic attack/intermolecular cycloaddition/ dehydration reaction accomplished the synthesis of ring-condensed heteroaromatic compounds 87 starting from 2-alkynylbenzaldehydes and 2-alkynylanilines under the presence In(OTf)3 catalyst (Scheme73). [87]
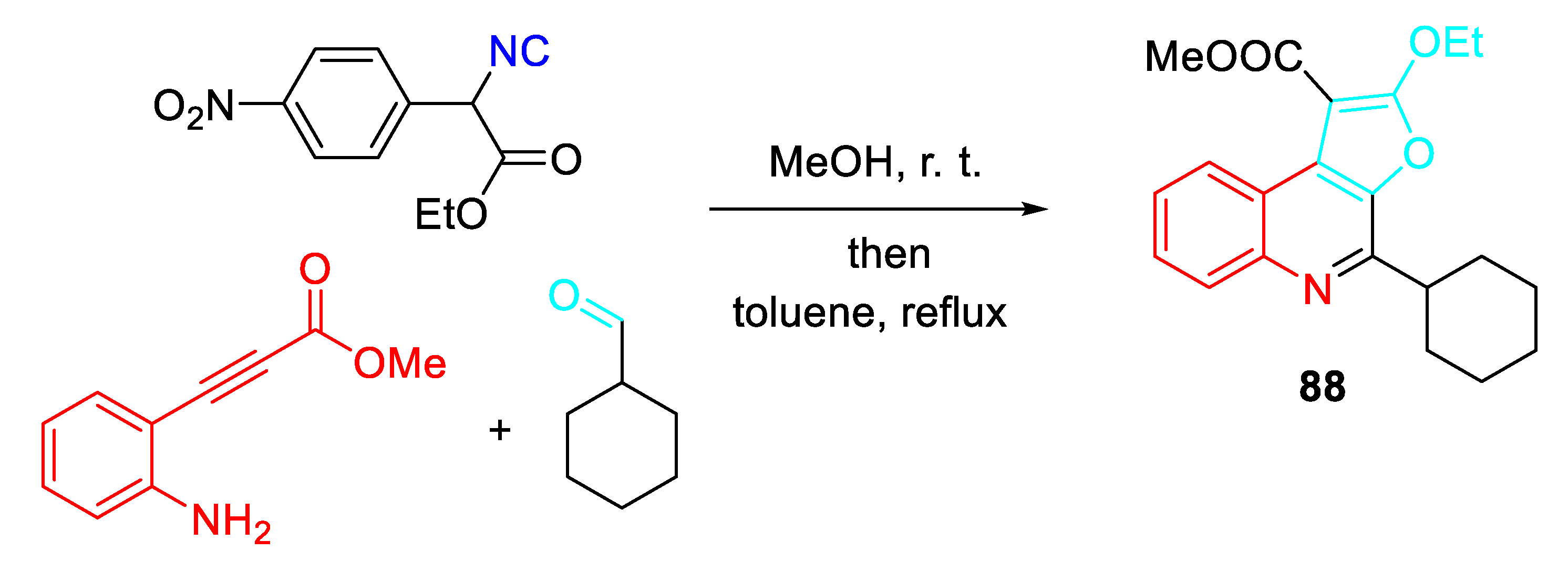
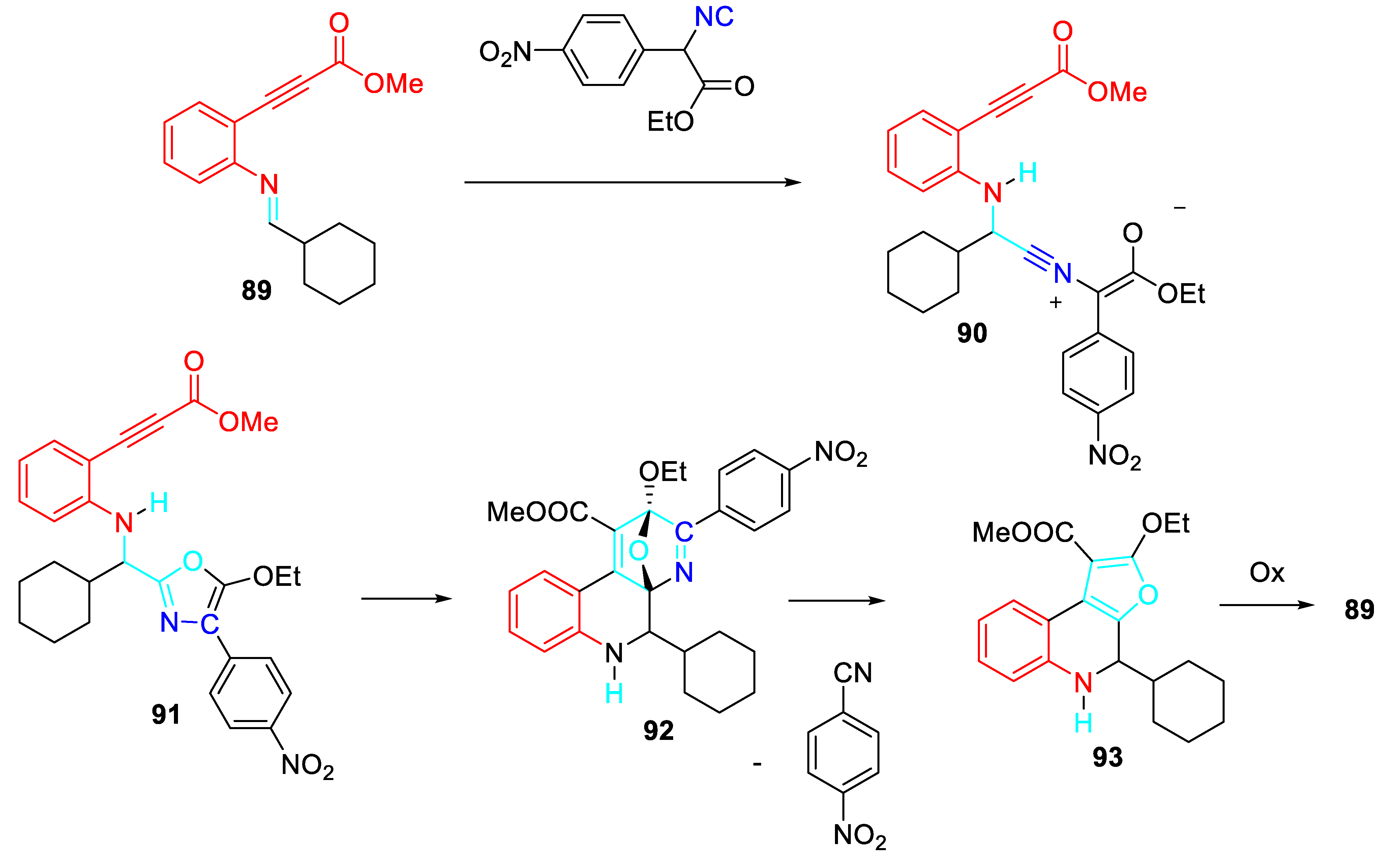
An intriguing three-component reaction of 2-alkynylanilines, aldehydes and α-(4-nitrophenyl)-α-isocyanoacetates in methanol at room temperature, followed by addition of toluene and heating to reflux, provided the polysubstituted furo[2,3-c]quinolines 88 in satisfactory yields (Scheme 74). [88]
The reaction of in the situ formed imine intermediate 89 with α-isocyanoacetates produces the 5-alkoxyoxazoles 91 which through an intramolecular Diels-Alder cycloaddition between the oxazole and the tethered triple bond would furnish oxa-bridged heterocycles 92. Their subsequent fragmentation by a retro Diels-Alder process furnishes derivatives 93 and benzonitrile. Finally, oxidation mediated by atmospheric oxygen could generates the target aromatic 2-alkoxyfuro[2,3-c]- quinolones 88 (Scheme 75).
4. Sequential Reactions of γ- and δ-Aminoalkynes with Carbonyls
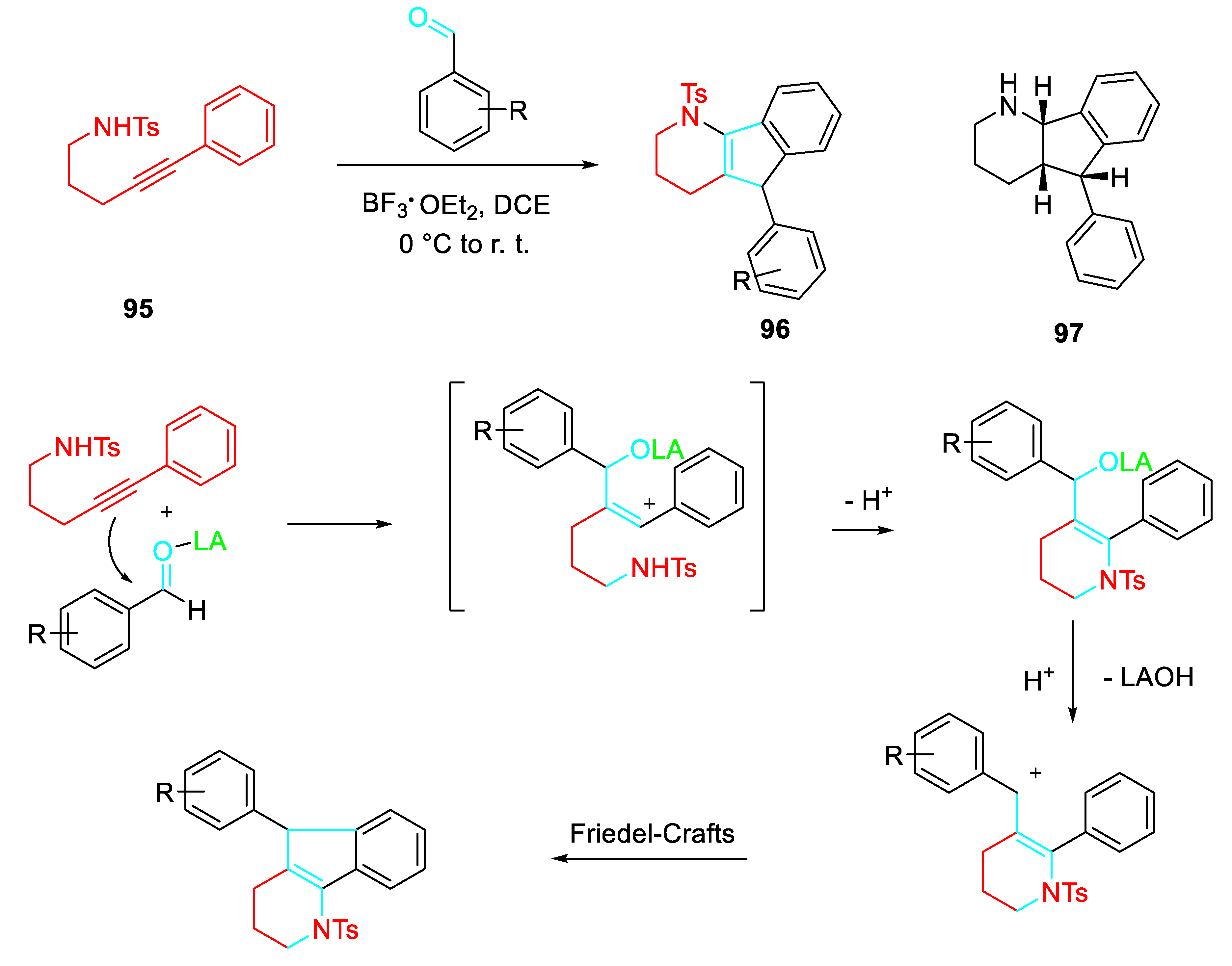
Sequential reactions of of γ- and δ-aminoalkynes with carbonyls were less investigated. A library of 1-tosyl-2,3,4,5-tetrahydro-1H-indeno[1,2-b]-pyridines 96 has been established by cascade cyclization / Friedel-Crafts reaction of 4-methyl-N-(pent-4-yn-1-yl)benzenesulfonamides 95 and aldehydes in good yields. The methodology was applied to the total synthesis of the antidepressant agent (±)-5-phenyl-2,3,4,4a,5,9b-hexahydro-1H-indeno[1,2-b]pyridine 97 (Scheme 77). [90]
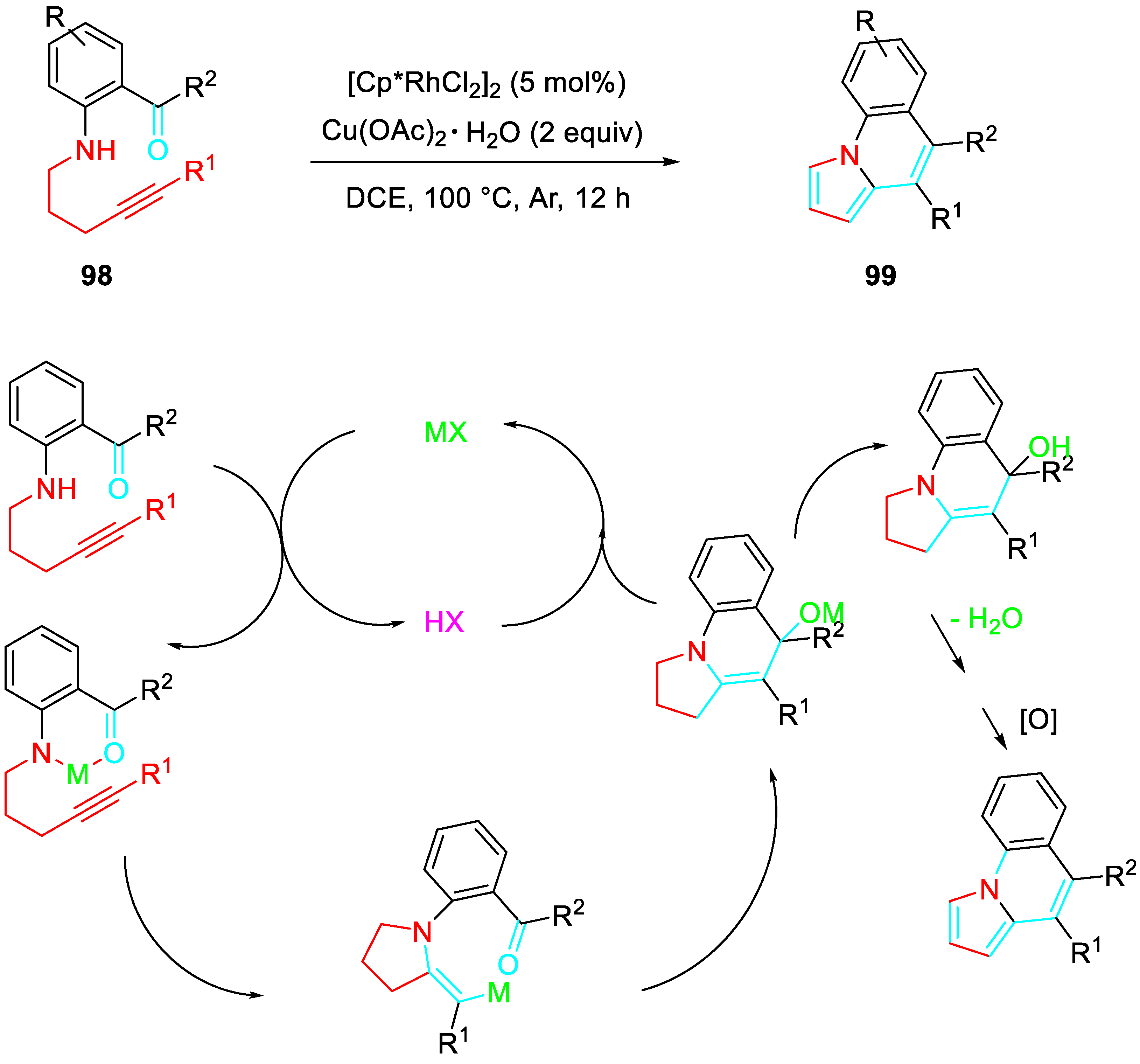
The sequential rhodium(III)-catalyzed intramolecular annulation of o-alkynyl amino aromatic ketones 98 / aromatization achieved the one pot building-up the pyrrolo[1,2-a]quinolines 99. The protocol could be scaled-up and allowed the synthesis of challenging products suitable for further elaboration (Scheme 78). [91]
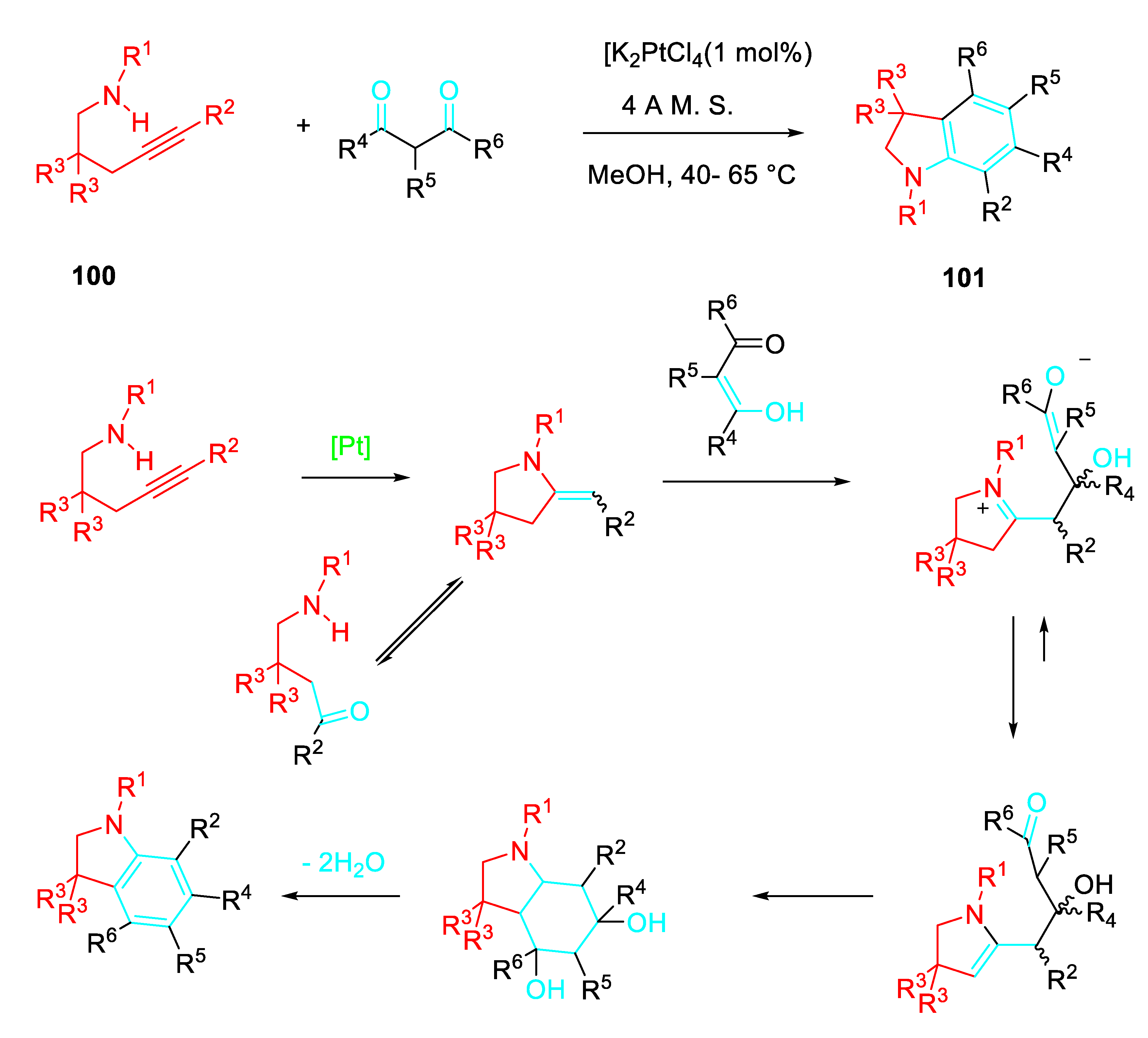
Although the metal-catalyzed reaction of γ-aminoalkynes 100 with 1,3-diketones is expected to afford a wide variety of products, unexpectedly the reaction accomplished the isolation of only indolines 101 in up to 99% yield (Scheme 79). [92]
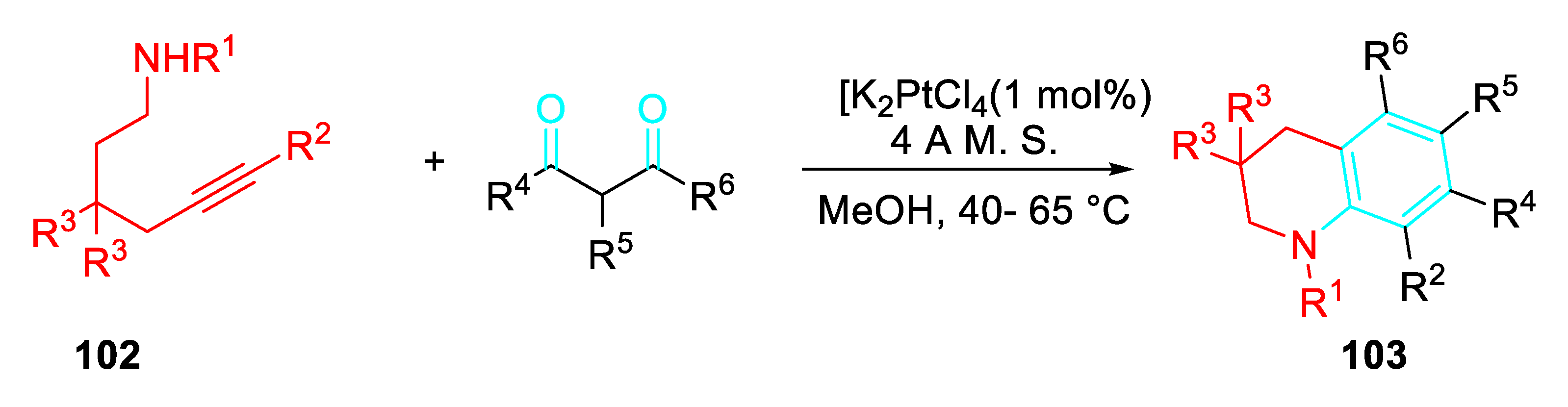
The extension of the reaction to δ-aminoalkynes 102 gave in high yields the 1,2,3,4-tetrahydroquinolines 103 of importance in medicinal chemistry.
5. Conclusions and Outlook
A variety of aminoalkynes can trigger sequential reactions with carbonyls to generate valuable heterocyclic scaffolds. The increasing number of aminoalkynes as starting building blocks have greatly widened the scope of sequential approaches to large libraries of valuable nitrogen containing heterocyclic compounds which can be obtained from easily available reagents. Coinage metals dominated the field and in particular gold complexes demonstrated superior performance as catalysts for these transformations. Inexpensive and less toxic iron and zinc salts are growing in importance as efficient catalysts. As for reaction media, greener alternatives such water, ionic liquids and solventless reactions have been reported. Advantages of microwave irradiation over the conventional heating have also been highlighted. Extensive mechanistic studies allowed the identification of several intermediates and aid to explain the key role of the catalyst and the additives employed. Often, he activation of the alkyne moiety by metal catalysis is essential to boost the sequential process. We foresee that further advancements will achieve straightforward alternative easy access to a wide array of polyheterocyclic scaffolds with potential remarkable biological activity.
Author Contributions
V.M. contributed to searching and collating of the relevant literature. Co-author L.P. and corresponding author A.A. wrote the body of the article. All authors read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We gratefully acknowledge the University of L’Aquila for financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hwang, E. T.; Lee, S. Multienzymatic Cascade Reactions via Enzyme Complex by Immobilization. ACS Catal. 2019, 9, 4402–4425. [Google Scholar] [CrossRef]
- Nicolaou, K. C.; Edmonds, D. J.; Bulger, P. G. Cascade Reactions in Total Synthesis. Angew. Chem. Int. Ed. 2006, 45, 7134–7186. [Google Scholar] [CrossRef]
- Nicolaou, K. C.; Chen, J. S. The art of total synthesis through cascade reactions. Chem. Soc. Rev. 2009, 38, 2993–3009. [Google Scholar] [CrossRef]
- Oishi, M.; Nakanishi, Y.; Suzuki, H. Selective Incorporation of Primary Amines into a Trizirconium Imido System and Catalytic Cyclization of Aminoalkynes. Inorg. Chem. 2017, 56, 9802–9813. [Google Scholar] [CrossRef]
- Arcadi, A. Gold-Catalyzed Synthesis of Nitrogen Heterocyclic Compounds via Hydroamination Reactions. In: Bandini, M. (eds) Au-Catalyzed Synthesis and Functionalization of Heterocycles. Top Heterocycl Chem. 2016, 46, 53–86. [Google Scholar] [CrossRef]
- Arcadi, A.; Abbiati, G.; Rossi, E. Tandem imination/annulation of γ- and δ-ketoalkynes in the presence of ammonia/amines. J. Organomet. Chem. 2011, 696, 87e98. [Google Scholar] [CrossRef]
- Chang, S.; Lee, M.; Jung, D. Y.; Yoo, E. J.; Cho, S. H.; Han, S. K. Catalytic One-Pot Synthesis of Cyclic Amidines by Virtue of Tandem Reactions Involving Intramolecular Hydroamination under Mild Conditions. J. Am. Chem. Soc. 2006, 128, 12366–12367. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Che, C.-M. A Highly Efficient and Selective AuI-Catalyzed Tandem Synthesis of Diversely Substituted Pyrrolo[1,2-a]quinolines in Aqueous Media. Angew. Chem. Int. Ed. 2008, 47, 3805–3810. [Google Scholar] [CrossRef]
- Zhou, Y.; Feng, E.; Liu, G.; Ye, D.; Li, J.; Jiang, H.; Liu, H. Gold-Catalyzed One-Pot Cascade Construction of Highly Functionalized Pyrrolo[1,2-a]quinolin-1(2H)-ones. J. Org. Chem. 2009, 74, 7344–7348. [Google Scholar] [CrossRef]
- Ma, C.-L.; Zhao, J.-H.; Yang, Y.; Zhang, M.-K.; Shen, C.; Sheng, R.; Dong, X.-W.; Hu, Y.-Z. A Copper-Catalyzed Tandem Cyclization Reaction of Aminoalkynes with Alkynes for the Construction of Tetrahydropyrrolo[1,2-a]quinolines Scaffold. Scientific Reports 2017, 7, 16640. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.-L.; Li, X.-H.; Yu, X.-L.; Zhu, X.-L.; Hu, Y.-Z.; Dong, X.-W.; Tan, B.; Liu, X.-Y. Gold-catalyzed tandem synthesis of bioactive spiro-dipyrroloquinolines and its application in the one-step synthesis of incargranine B aglycone and seneciobipyrrolidine (I). Org. Chem. Front. 2016, 3, 324–329. [Google Scholar] [CrossRef]
- Galvàn, A.; Calleja, J.; Faňanás, F. J.; Rodríguez, F. Synthesis of Pyrrolidine Derivatives by a Platinum/Brønsted Acid Relay Catalytic Cascade Reaction Chem. Eur. J. 2015, 21, 3409–3414. [Google Scholar] [CrossRef]
- Huple, D. B.; Liu, R.-S. One-Pot Stereocontrolled Synthesis of Bicyclic Pyrrolidine Derivatives by a Platinum-Brønsted Acid Relay Cascade Reaction. ChemCatChem 2015, 7, 2824–2825. [Google Scholar] [CrossRef]
- Fujiwara, S.-i.; Shikano, Y.; Shin-ike, T.; Kambe, N.; Sonoda, N. Stereoselective Synthesis of New Selenium-Containing Heterocycles by Cyclocarbonylation of Aminoalkynes with Carbon Monoxide and Selenium. J. Org. Chem. 2002, 67, 6275–6278. [Google Scholar] [CrossRef]
- Hu, Y.; Huang, H. Highly Selective Construction of Medium-Sized Lactams by Palladium-Catalyzed Intramolecular Hydroaminocarbonylation of Aminoalkynes. Org. Lett. 2017, 19, 5070–5073. [Google Scholar] [CrossRef]
- Li, X.; Wang, S.; Wang, H.; Wang, W.; Liu, L.; Chang, W.; Lia, J. Synthesis of Eight-Membered Nitrogen Heterocycles via a Heterogeneous PtI2-Catalyzed Cascade Cycloaddition Reaction of δ-Aminoalkynes with Electron-Deficient Alkynes. Adv. Synth. Catal. 2020, 362, 1525–1531. [Google Scholar] [CrossRef]
- Li, X.; Jiang, C.; Wang, X.; Ren, J.; Zeng, T.; Xu, X.; Li, J.; Liu, L. Platinum Iodide-Catalyzed Formal Three-Component Cascade Cycloaddition Reactions between γ-Aminoalkynes and Electron-Deficient Alkynes. J. Org. Chem. 2021, 86, 16614–16624. [Google Scholar] [CrossRef]
- Abbiati, G.; Arcadi, A.; Bianchi, G.; Di Giuseppe, S.; Marinelli, F.; Rossi, E. Sequential Amination/Annulation/Aromatization Reaction of Carbonyl Compounds and Propargylamine: A New One-Pot Approach to Functionalized Pyridines. J. Org. Chem. 2003, 68, 6959–6966. [Google Scholar] [CrossRef]
- Yang, D.; Liu, H.; Wang, D.-L.; Lu, Y.; Zhao, X.-L.; Liu, Y. Au complex containing phosphino and imidazolyl moieties as a bifunctional catalyst for one-pot synthesis of pyridine derivatives. J. Mol. Catal. A: Chem. 2016, 424, 323–330. [Google Scholar] [CrossRef]
- Sotnik, S. O.; Subota, A.I; Kliuchynskyi, A. Y.; Yehorov, D. V.; Lytvynenko, A. S.; Rozhenko, A. B.; Kolotilov, S. V.; Ryabukhin, S. V.; Volochnyuk, D. M. Cu-Catalyzed Pyridine Synthesis via Oxidative Annulation of Cyclic Ketones with Propargylamine. J. Org. Chem. 2021, 86, 7315–7325. [Google Scholar] [CrossRef] [PubMed]
- Savić, M. P.; Ajduković, J. J.; Plavša, J.J.; Bekić, S. S.; Ćelić, A. S.; Klisurić, O. R.; Jakimov, D. S.; Edward, T. Petri, E. T.; Djurendić, E. A. Evaluation of A-ring fused pyridine D-modified androstane derivatives for antiproliferative and aldo-keto reductase 1C3 inhibitory activity. Med. Chem. Commun. 2018, 9, 969–981. [Google Scholar] [CrossRef]
- Yan, J.-Z.; Li, J.; Rao, G.-W. One-pot synthesis of new A-ring fused steroidal pyridines. Steroids 2007, 7 2, 736–739. [Google Scholar] [CrossRef]
- Laavola, M.; Haavikko, R.; Hämäläinen, M.; Leppänen, T.; Nieminen, R.; Sami Alakurtti, S.; Moreira, V. M.; Yli-Kauhaluoma, J.; Eeva Moilanen, E. Betulin Derivatives Effectively Suppress Inflammation in Vitro and in Vivo. J. Nat. Prod. 2016, 79, 274–280. [Google Scholar] [CrossRef]
- Haavikko, R.; Nasereddin, A.; Sacerdoti-Sierra, N.; Kopelyanskiy, D.; Sami Alakurtti, S.; Mari Tikka, M.; Charles, L. Jaffe, C. L.; Yli-Kauhaluoma, J. Heterocycle-fused lupane triterpenoids inhibit Leishmania donovani amastigotes. Med. Chem. Commun. 2014, 5, 445–451. [Google Scholar] [CrossRef]
- Hodoň, J.; Frydrych, I.; Trhlíková, Z.; Pokorný, J.; Borková, L.; Benická, S.; Vlk, M.; Lišková, B.; Kubíčková, A.; Medvedíková, M.; Pisár, M.; Šarek, J.; Das, V.; Ligasová, A.; Koberna, K.; Džubák, P.; Hajdúch, M.; Urban, M. Triterpenoid pyrazines and pyridines – Synthesis, cytotoxicity, mechanism of action, preparation of prodrugs. Eur. J. Med. Chem. 2022, 243, 114777. [Google Scholar] [CrossRef]
- Rabe, S.; Moschner, J.; Bantzi, M.; Heretsch, P.; Giannis, A. C-H-Functionalization logic guides the synthesis of a carbacyclopamine analog. Beilstein J. Org. Chem. 2014, 10, 1564–1569. [Google Scholar] [CrossRef]
- Simcere Pharmaceutical Group, Compound as potassium channel modulating agents. CN10825 0128, 2018, A.
- Suzhou Yunxuan Pharmaceutical Co Ltd, Heterocyclic compound with Wnt signal path inhibitory activity and application thereof. CN10525 4613, 2016, A.
- Zhang, X.; Zheng, J.; Ma, H. Heteroaryl compounds as cxcr4 inhibitors, composition and method using the same. Patent WO2019/6 0860, 2019, A1. [Google Scholar]
- Wortmann, L.; Lindenthal, B.; Muhn, P.; Walter, A.; Nubbemeyer, R.; Heldmann, D.; Sobek, L.; Morandi, F.; Schrey, A. K.; Moosmayer, D.; Günther, J.; Kuhnke, J.; Koppitz, M.; Lücking, U.; Röhn, U.; Schäfer, M.; Nowak-Reppel, K.; Kühne, R.; Weinmann, H. , Langer, G. Discovery of BAY-298 and BAY-899: Tetrahydro-1,6-naphthyridine-Based, Potent, and Selective Antagonists of the Luteinizing Hormone Receptor Which Reduce Sex Hormone Levels in Vivo. J. Med. Chem. 2019, 62, 10321–10341. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Chen, L.; Conway, C. M.; Denton, R.; Keavy, D.; Gulianello, M.; Huang, Y.; Kostich, W.; Lentz, K. A.; Mercer, S. E.; Schartman, R.; Signor, L.; Browning, M.; Macor, J. E.; Dubowchik, G. M. Discovery of BMS-846372, a Potent and Orally Active Human CGRP Receptor Antagonist for the Treatment of Migraine. ACS Med. Chem. Lett. 2012, 3, 337–341. [Google Scholar] [CrossRef]
- C. H. Boehringer Sohn AG & Co. KG. Aryl- and heroarylcarbonyl derivatives of benzomorphanes and related scaffolds, medicaments containing such compounds and their use. EP222 0048, 2017, B1.
- Lowe, R. A.; Taylor, D.; Chibale, K.; Nelson, A.; Marsden, S. P. Synthesis and evaluation of the performance of a small molecule library based on diverse tropane-related scaffolds. Biorg. & Med. Chem. 2020, 28, 115442. [Google Scholar] [CrossRef]
- Eckhardt, M.; Peters, S.; Nar, H.; Himmelsbach, F.; Zhuang, L. Boehringer Ingheleim Int, Aryl-and heteroarylcarbonyl derivatives of hexahydroindenopyridine and octahydrobenzoquinoline. US 2011/0136800 A1, 2011.
- Park, N. Y.; Lee, H. S.; Piao, L. H.; Park, S. Y.; Jeong, W. Transition Metal Compound for Olefin Polymerization Catalyst, and Olefin Polymerization Catalyst Including Same, Hanwha Solutions Corporation (Seoul), United. States Patent US 11,254,759 B2, 2022. [Google Scholar]
- Faňanás, F. J.; Arto, T.; Mendoza, A.; Rodríguez, F. Synthesis of 2,5-Dihydropyridine Derivatives by Gold-Catalyzed Reactions of β-Ketoesters and Propargylamines. Org. Lett. 2011, 13, 4184–4187. [Google Scholar] [CrossRef]
- Lee, S. Yoo, H.; Park, S.; Yoon, R.; Kim, S. Facile One-Pot Synthesis of Polysubstituted Pyridinium Salts by Annulation of Enamines with Alkynes. Chem. Eur. J. 2023, e202300059. [Google Scholar] [CrossRef]
- Changchun Haipurunsi Tech co ltd, A kind of miscellaneous anthracene derivant and preparation method thereof and organic luminescent device. CN10821 8860A, 2018.
- Univ Zhejlang Technology, Method for synthesizing substituted dihydrophenanthroline compound. CN11248 0112 A, 2021.
- Watkins, E. B.; Uredi, D.; Motati, D. R. Novel Methods for Preparation of Substituted Pyridines and Related Novel Compounds. US Patent 2020/0095245 A1, 2020. [Google Scholar]
- Uredi, D.; Motati, D. R.; Watkins, E. B. A simple, tandem approach to the construction of pyridine derivatives under metal-free conditions: a one-step synthesis of the monoterpene natural product, (-)-actinidine. Chem. Comm. 2019, 55, 3270–3273. [Google Scholar] [CrossRef]
- Zhao, Z.; Wei, H.; Xiao, K.; Cheng, B.; Zhai, H.; Li, Y. Facile Synthesis of Pyridines from Propargyl Amines: Concise Total Synthesis of Suaveoline Alkaloids. Angew. Chem. Int. Ed. 2019, 58, 1148–1152. [Google Scholar] [CrossRef]
- Wei, H.; Li, Y. Quick Access to Pyridines through 6p-3-Azatriene Electrocyclization: Concise Total Synthesis of Suaveoline Alkaloids. Synlett 2019, 30, 1615–1620. [Google Scholar] [CrossRef]
- Uredi, D.; Motati, D. R.; Watkins, E. B. A Unified Strategy for the Synthesis of β-Carbolines, γ-Carbolines, and Other Fused Azaheteroaromatics under Mild, Metal-Free Conditions. Org. Lett. 2018, 20, 6336–6339. [Google Scholar] [CrossRef]
- Uredi, D.; Burra, A. G.; Watkins, E. B. Rapid Access to 3-Substituted Pyridines and Carbolines via a Domino, Copper-free, Palladium-Catalyzed Sonogashira Cross-Coupling/6π-Aza Cyclization Sequence. J. Org. Chem. 2021, 86, 17748–17761. [Google Scholar] [CrossRef]
- Lanzhou University, A kind of polysubstituted pyridine derivative and preparation method thereof. CN10835 8834, 2018, A.
- Chikayuki, Y.; Miyashige, T.; Yonekawa, S.; Kirita, A.; Matsuo, N.; Teramoto, H.; Sasaki, S.; Higashiyama, K.; Yamauchi, T. Transition-Metal-Free Synthesis of Pyridine Derivatives by Thermal Cyclization of N-Propargyl Enamines. Synthesis 2020, 52, 1113–1121. [Google Scholar] [CrossRef]
- Keskin, S.; Balci, M. Intramolecular Heterocyclization of O-Propargylated Aromatic Hydroxyaldehydes as an Expedient Route to Substituted Chromenopyridines under Metal-Free Conditions. Org. Lett. 2015, 17, 964–967. [Google Scholar] [CrossRef]
- Hoplamaz, E.; Keskin, S.; Balci, M. Regioselective Synthesis of Benzo[h][1,6]-naphthyridines and Chromenopyrazinones through Alkyne Cyclization. Eur. J. Org. Chem. 2017, 1489–1497. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Alonso, J. M.; Israel Fernández, I.; Gómez-Campillosa, G.; Torres, M. R. A gold-catalysed imine-propargylamine cascade sequence: synthesis of 3-substituted-2,5-dimethylpyrazines and the reaction mechanism. Chem. Commun. 2014, 50, 4567–4570. [Google Scholar] [CrossRef]
- Nie, Q.; Yao, F.; Yi, F.; Cai; M. A heterogeneous gold(I)-catalyzed cascade annulation of aldehydes with propargylamine leading to 3-substituted 2,5-dimethylpyrazines. J. Organomet. Chem. 2017, 846, 343–350. [Google Scholar] [CrossRef]
- Donald, J. R.; Martin, S. F. Synthesis and Diversification of 1,2,3- Triazole-Fused 1,4-Benzodiazepine Scaffolds. Org. Lett. 2011, 13, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Donald, J. R.; Wood, R. R.; Martin, S. F. Application of a Sequential Multicomponent Assembly Process/ Huisgen Cycloaddition Strategy to the Preparation of Libraries of 1,2,3-Triazole-Fused 1,4-Benzodiazepines. ACS Comb. Sci. 2012, 14, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H. H.; Palazzo, T. A.; Kurth, M- J. Facile One-Pot Assembly of Imidazotriazolobenzodiazepines via Indium(III)-Catalyzed Multicomponent Reactions. Org. Lett. 2013, 15, 4492–4495. [Google Scholar] [CrossRef]
- Festa, A. A.; Raspertov, P. V.; Voskressensky, L. G. 2-(Alkynyl)anilines and Derivatives-Versatile Reagents for Heterocyclic Synthesis. Adv. Synth. Catal. 2022, 364, 466–486. [Google Scholar] [CrossRef]
- Vavsari, V. F.; Nikbakht, A.; Balalaie, S. Annulation of 2-Alkynylanilines: The Versatile Chemical Compounds. Asian J. Org. Chem. 2022; e202100772. [Google Scholar] [CrossRef]
- Kamble, O. S.; Khatravath, M.; Dandela, R. Applications of Ethynylanilines as Substrates for Construction of Indoles and Indole-Substituted Deivatives. ChemistrySelect 2021, 6, 7408–7427. [Google Scholar] [CrossRef]
- Murai, K.; Hayashi, S.; Takaichi, N.; Kita, Y.; Fujioka, H. Tandem β-Enamino Ester Formation and Cyclization with o-Alkynyl Anilines Catalyzed by InBr3: Efficient Synthesis of-(N-Indolyl)-α,β-unsaturated Esters. J. Org. Chem. 2009, 74, 1418–1421. [Google Scholar] [CrossRef]
- Arcadi, A.; Alfonsi, M.; Bianchi, G.; D’Anniballe, G.; Marinelli, F. Gold-Catalysed Direct Couplings of Indoles and Pyrroles with 1,3-Dicarbonyl Compounds. Adv. Synth. Catal. 2006, 348, 331–338. [Google Scholar] [CrossRef]
- Peng, C.; Wang, Y.; Liu, I.; Wang, H.; Zhao, J.; Q. Zhu, Q. p-Toluenesulfonic Acid Promoted Annulation of 2-Alkynylanilines with Activated Ketones: Efficient Synthesis of 4-Alkyl-2,3-Disubstituted Quinolines. Eur. J. Org. Chem. 2010, 818–822. [Google Scholar] [CrossRef]
- Sivaraman, M.; T. Perumal, P. T. Synthesis of 4-methyl-2,3-disubstituted quinoline scaffolds via environmentally benign Fe(III) catalysed sequential condensation, cyclization and aromatization of 1,3-diketone and 2-ethynylaniline. RSC Adv. 2014, 4, 52060–55266. [CrossRef]
- Ortiz-Cervantes, C.; Flores-Alamo, M.; García, J. J. Synthesis of pyrrolidones and quinolines from the known biomass feedstock levulinic acid and amines. Tetrahedron Lett. 2016, 57, 766–771. [Google Scholar] [CrossRef]
- Wang, G.; Jia, J.; Liu, G.; Yu, M.; Chu, X.; Liu, X.; Zhao, X. Copper(I)-catalyzed tandem synthesis of 2-acylquinolines from 2-ethynylanilines and glyoxals. Chem. Commun. 2021, 57, 11811–11814. [Google Scholar] [CrossRef]
- Sakai, N.; Tamura, K.; Shimamura, K.; Ikeda, R.; Konakahara, T. Copper-Catalyzed [5+1] Annulation of 2-Ethynylanilines with an N,O-Acetal Leading to Construction of Quinoline Derivatives. Org. Lett. 2012, 14, 836–839. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Peng, C.; Liu, L.; Zhao, J.; Su, L.; Zhu, Q. Sulfuric acid promoted condensation cyclization of 2-(2-(trimethylsilyl)ethynyl)anilines with arylaldehydes in alcoholic solvents: an efficient one-pot synthesis of 4-alkoxy-2-arylquinolines. Tetrahedron Lett. 2009, 50, 2261–2265. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Taher, A.; Ponra, S. Unusual Product from Condensative Cyclization: Pyrano[3,2-f]quinolin-3,10-diones from 6-Amino-5-[(trimethylsilyl)ethynyl]-2H-chromen-2-one and Aryl Aldehydes. Synlett 2010, 735–740. [Google Scholar] [CrossRef]
- Ock, S. K.; Youn, S. W. Synergistic Effect of Pd(II) and Acid Catalysts on Tandem Annulation Reaction for the Regioselective Synthesis of Ring-Fused Quinolines. Bull. Korean Chem. Soc. 2010, 31, 704–707. [Google Scholar] [CrossRef]
- Zhu, C.; Ma, S. Sc(OTf)3-Catalyzed Bicyclization of o-Alkynylanilines with Aldehydes:Ring-Fused 1,2-Dihydroquinolines. Angew. Chem. Int. Ed. 2014, 53, 13532–13535. [Google Scholar] [CrossRef]
- Fu, W.; Xu, C.; Zou, G.; Hong, D.; Deng, D.; Wang, Z.; Ji, B. AuCl3-Catalyzed Tandem Reaction of N-(o-Alkynylphenyl)imines: A Modular Entry to Polycyclic Frameworks Containing an Indole Unit. Synlett 2009, 5, 763–766. [Google Scholar] [CrossRef]
- Halim, R.; Scammells, P. J.; Flynn, B. L. Alternating Iodonium-Mediated Reaction Cascades Giving Indole- And Quinoline-Containing Polycycles. Org. Lett. 2008, 10, 1967–1970. [Google Scholar] [CrossRef]
- Halim, R.; Aurelio, L.; Scammells, P. J.; and Bernard, L. Flynn, B. L. Scaffold-Divergent Synthesis of Ring-Fused Indoles, Quinolines, and Quinolones via Iodonium-Induced Reaction Cascades. J. Org. Chem. 2013, 78, 4708–4718. [Google Scholar] [CrossRef]
- Kusama, H.; Takaya, J.; Iwasawa, N. A Facile Method for the Synthesis of Polycyclic Indole Derivatives: The Generation and Reaction of Tungsten-Containing Azomethine Ylides. J. Am. Chem Soc. 2002, 124, 11592–11593. [Google Scholar] [CrossRef]
- Kayeta, A.; Singh, V. K. A one-pot synthesis of 2,2’-disubstituted diindolylmethanes (DIMs) via a sequential Sonogashira coupling and cycloisomerization/C3-functionalization of 2-iodoanilines. Org. Biomol. Chem. 2017, 15, 6997–7007. [Google Scholar] [CrossRef] [PubMed]
- Subba Reddy, B. V.; Swain, M.; Madhusudana Reddy, S.; Yadav, J. S.; Sridhar, B. Gold-Catalyzed Domino Cycloisomerization/Pictet-Spengler Reaction of 2-(4-Aminobut-1-yn-1-yl)anilines with Aldehydes: Synthesis of Tetrahydropyrido[4,3-b]indole Scaffolds. J. Org. Chem. 2012, 77, 11355–11361. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Zhang, J.; Zhang, D.; Wei, D.; Zuo, J.; Song, J.; Yu, B.; Liu, H.-M. Cu(OTf)2-Catalyzed Intramolecular Radical Cascade Reactions for the Diversity-Oriented Synthesis of Quinoline-Annulated Polyheterocyclic Frameworks Org. Lett. 2021, 23, 1445–1450. [Google Scholar] [CrossRef]
- G. K. Dhandabani, M. R. Mutra, J.-J. Wang, Palladium-Catalyzed Regioselective Synthesis of 1-Benzoazepine Carbonitriles from o-Alkynylanilines via 7-endo-dig Annulation and Cyanation Adv. Synth. Catal. 2018, 360, 4754–4763. [Google Scholar] [CrossRef]
- Marsicano, V.; Arcadi, A.; Chiarini, M.; Fabrizi, G.; Goggiamani, A.; Iazzetti, A. Synthesis of 2,2,3-substituted-2,3-Dihydroquinolin-4(1H)-ones vs. Functionalised Quinoline or N-Alkenylindole Derivatives through Sequential Reactions of 2-Alkynylanilines with Ketones. Org. Biomol. Chem. 2021, 19, 421–438. [Google Scholar] [CrossRef]
- Marsicano, V.; Chiarini, M.; Marinelli, F.; Arcadi, A. Synthesis of Polycyclic Quinolines by Means of Brønsted Acid Mediated Reaction of β-(2-Aminophenyl)-α,β-Ynones with Ketones. Adv. Synth. Catal. 2019, 361, 2365–2370. [Google Scholar] [CrossRef]
- Gogoi, S.; Shekarrao, K.; Duarah, A.; Bora, T.C.; Gogoi, S.; Boruah, R.C. A microwave promoted solvent-free approach to steroidal quinolines and their in vitro evaluation for antimicrobial activities. Steroids 2012, 77, 1438–1435. [Google Scholar] [CrossRef]
- Marsicano, V.; Arcadi, A.; Chiarini, M.; Fabrizi, G.; Goggiamani, A.; Iazzetti, A. Sequential condensation/biannulation reactions of β-(2-aminophenyl)-α,β-ynones with1,3-dicarbonyls. Org. Biomol. Chem. 2021, 19, 5177–5190. [Google Scholar] [CrossRef] [PubMed]
- Jash, M.; Das, B.; Chowdhury, C. One-Pot Access to Benzo[a]carbazoles via Palladium(II)-Catalyzed Hetero- and Carboannulations. J. Org. Chem. 2016, 81, 10987–10999. [Google Scholar] [CrossRef] [PubMed]
- Abbiati, G.; Arcadi, A.; Chiarini, M.; Marinelli, F.; Pietropaolo, E.; Rossi, E. An alternative one-pot gold-catalyzed approach to the assembly of 11H-indolo-[3,2-c]quinolines. Org. Biomol. Chem. 2012, 10, 7801–7808. [Google Scholar] [CrossRef]
- Arcadi, A.; Chiarini, M.; D’Anniballe, G.; Marinelli, F.; Pietropaolo, E. Brønsted Acid Catalyzed Cascade Reactions of 2-[(2-Aminophenyl)ethynyl]phenylamine Derivatives with Aldehydes: A New Approach to the Synthesis of 2,2′-Disubstituted 1H,1′H-3,3′-Biindoles. Org. Lett. 2014, 16, 1736–1739. [Google Scholar] [CrossRef]
- Qian, X.; Gao, H.-H.; Zhu, Y.-Z.; Lu, L.; Zheng, J.-Y. Biindole-based double D–p–A branched organic dyes for efficient dye-sensitized solar cells RSC Adv. 2015, 5, 4368–4375. [CrossRef]
- Brambilla, E.; Gritti, A.; Pirovano, V.; Arcadi, A.; Germani, R.; Tiecco, M.; Abbiati, G. Acidic DESs as Active Media for a Sustainable Synthesis of Biindoles Starting from 2,2’-Diaminotolanes and Aldehydes. Eur. J. Org. Chem. 2023, e202300204. [Google Scholar] [CrossRef]
- Subba Reddy, B.V.; Swain, M.; Madhusudana Reddy, S.; Yadav, J. S.; Sridhar, B. Gold-Catalyzed 5-endo-dig Cyclization of 2-[(2-Aminophenyl)-ethynyl]phenylamine with Ketones for the Synthesis of Spiroindolone and Indolo[3,2-c]quinolone Scaffolds. Eur. J. Org. Chem. 2014, 3313–3318. [Google Scholar] [CrossRef]
- Yanada, R.; Hashimoto, K.; Tokizane, R.; Miwa, Y.; Minami, H.; Yanada, K.; Ishikura,M. ; Takemoto, Y. Indium(III)-Catalyzed Tandem Reaction with Alkynylbenzaldehydes and Alkynylanilines to Heteroaromatic Compounds. J. Org. Chem. 2008, 73, 5135–5138. [Google Scholar] [CrossRef] [PubMed]
- Bouma, M. J.; Masson, G.; Zhu, J. Exploiting the Divergent Reactivity of Isocyanoacetates: One-Pot Three-Component Synthesis of Functionalized Angular Furoquinolines. Eur. J. Org. Chem. 2012, 475–479. [Google Scholar] [CrossRef]
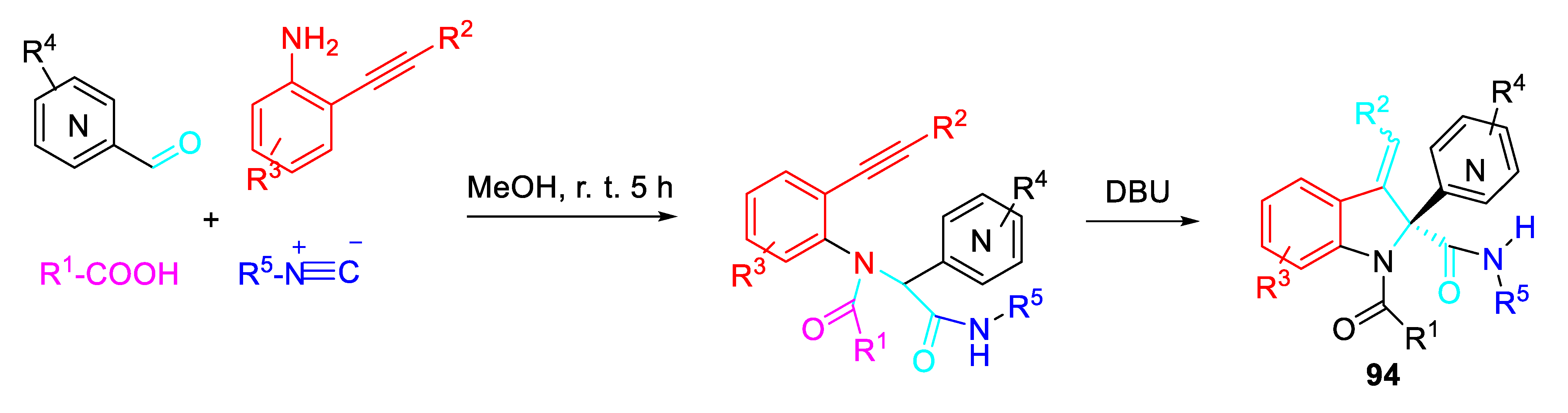
- Zhao, S.; He, Y.; Gao, F.; Wei, Y.; Zhang, J.; Chen, M.; Gao, Y.; Zhang, Y.; Liu, J.-Y.; Guo, Z.; Li, Z.; Nie, S. Rapid access to C2-quaternary 3-methyleneindolines via base-mediated post-Ugi Conia-ene cyclization. Chem. Commun. 2023, 59, 3099–3102. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, U.; Borah, M.; Deka, M. J.; Saikia, A. K. Synthesis of Tetrahydro-1H-indeno[1,2-b]pyridine via Cascade Cyclization and Friedel-Crafts Reaction. J. Org. Chem. 2016, 81, 8736–8743. [Google Scholar] [CrossRef]
- Hu, Y.; Jia, Y.; Tuo, Z.; Zhou, W. Rhodium(III)-Catalyzed Intramolecular Annulation and Aromatization for the Synthesis of Pyrrolo[1,2-a]quinolines. Org. Lett. 2023, 25, 1845–1849. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Che, C.-M. Highly Efficient and Regioselective Platinum(II)-Catalyzed Tandem Synthesis of Multiply Substituted Indolines and Tetrahydroquinolines. Angew. Chem. Int. Ed. 2009, 48, 2367–2371. [Google Scholar] [CrossRef]
Scheme 1.
The catalyst was envisaged to promote both the amination of carbonyl compounds 2 and the regioselective 6-endo-dig cyclization of the N-propargylenamine (N-propargyldienamine) intermediate 5. (Scheme 2).
Scheme 1.
The catalyst was envisaged to promote both the amination of carbonyl compounds 2 and the regioselective 6-endo-dig cyclization of the N-propargylenamine (N-propargyldienamine) intermediate 5. (Scheme 2).
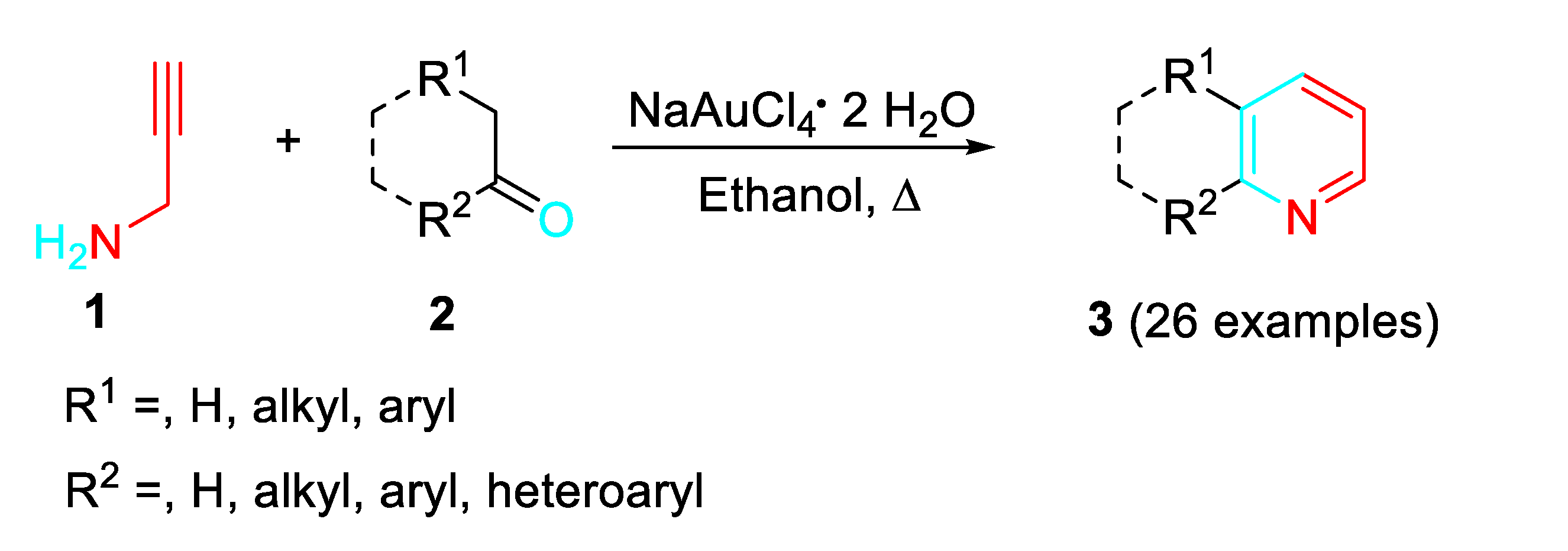
Scheme 2.
Transition metal-catalyzed sequential amination/cyclization/aromatization reaction.
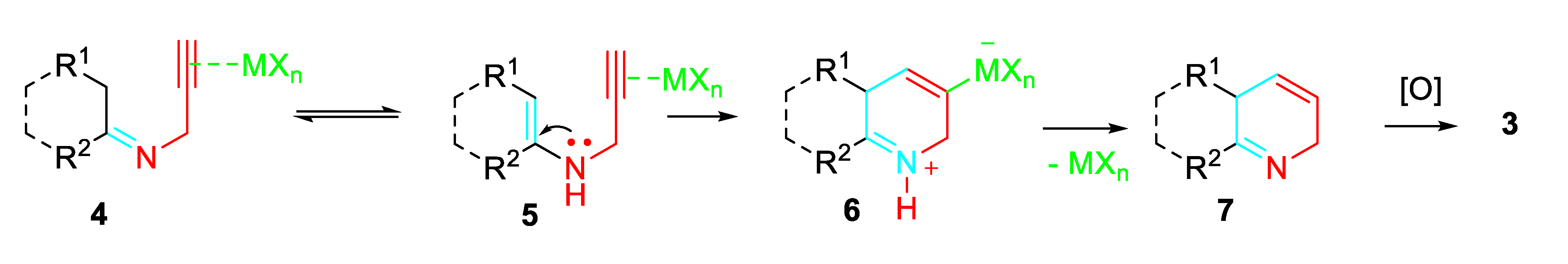
Scheme 3.
Sequential reactions of carbonyl compounds and propargylamine catalyzed by Au-complex 8.
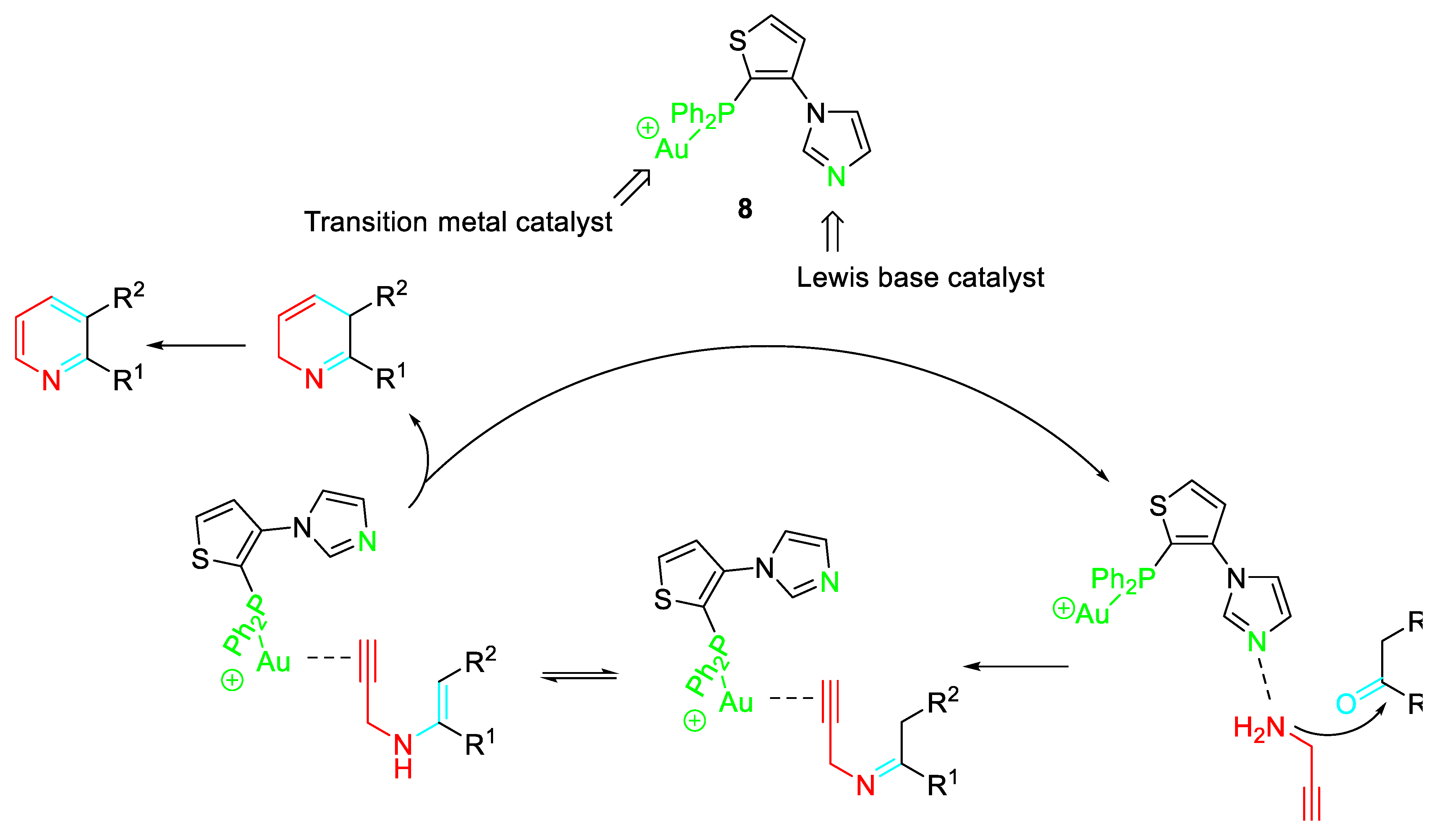
Scheme 4.
Cu-catalyzed pyridine synthesis from cyclic ketones and propargylamine.
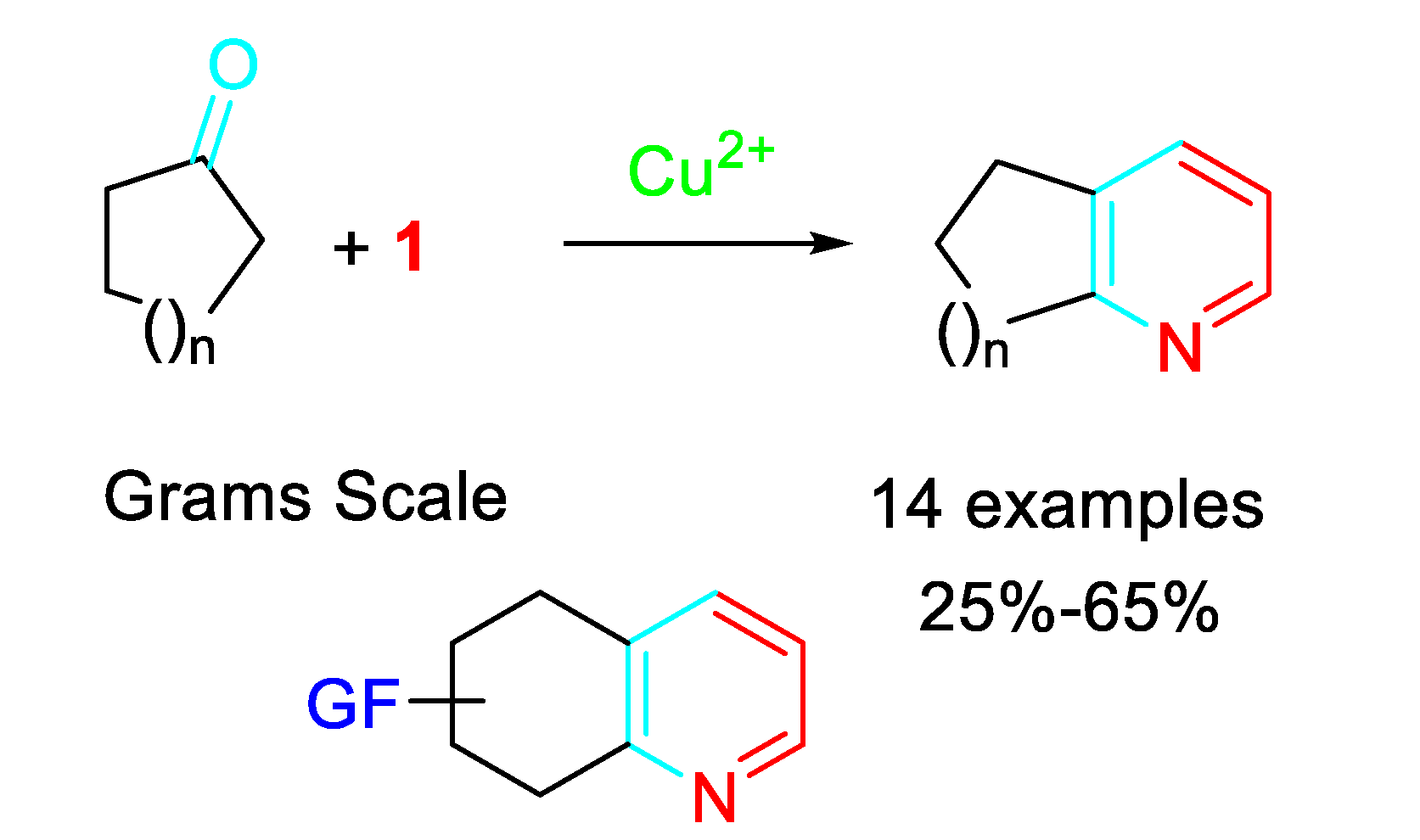
Scheme 5.
Linear vs angular steroidal pyridines.
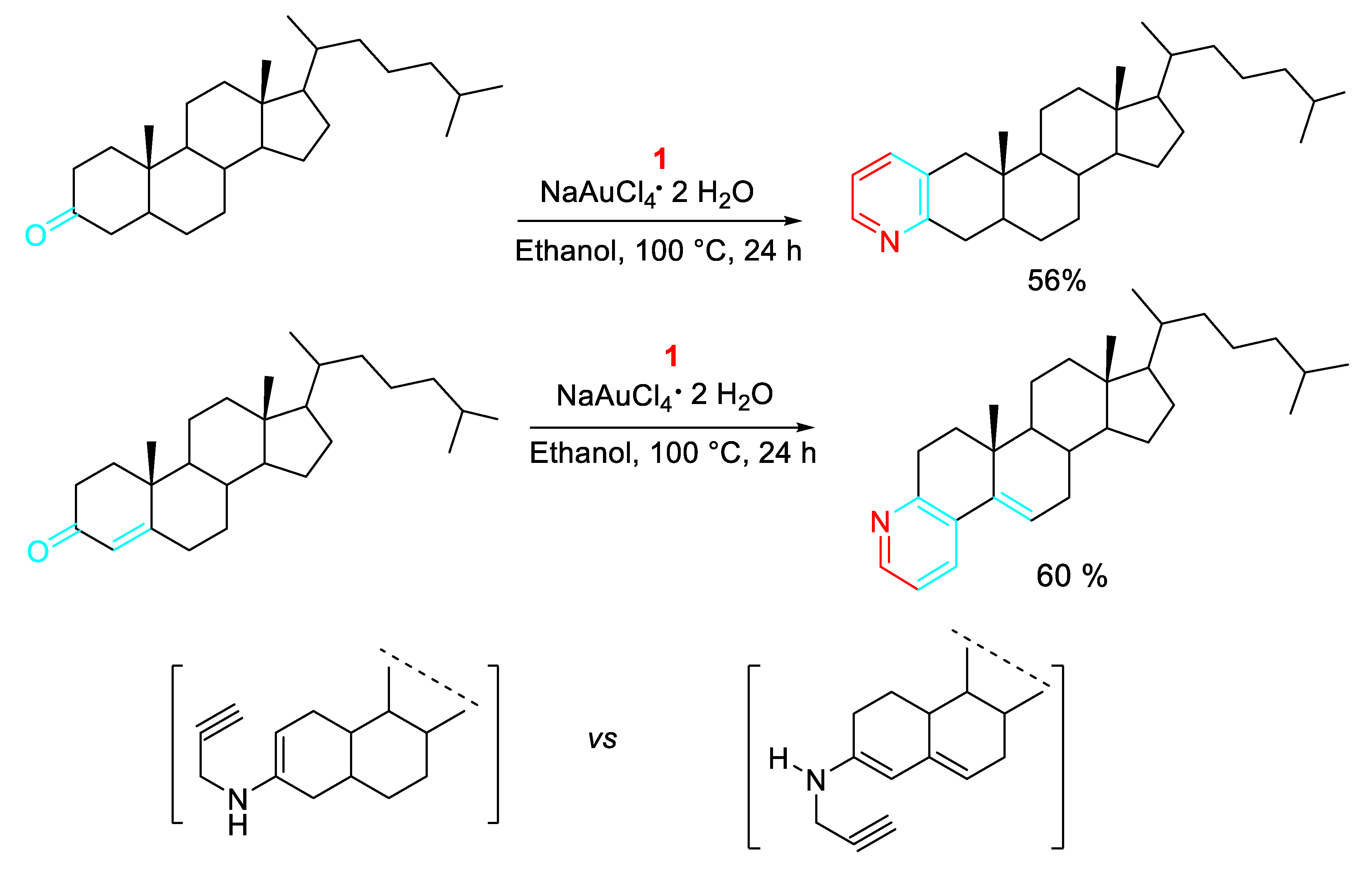
Scheme 6.
Copper-catalyzed synthesis of A-ring fused pyridine D-modified androstane derivatives.
Scheme 7.
Copper-catalyzed synthesis of betulin derivatives.
Scheme 8.
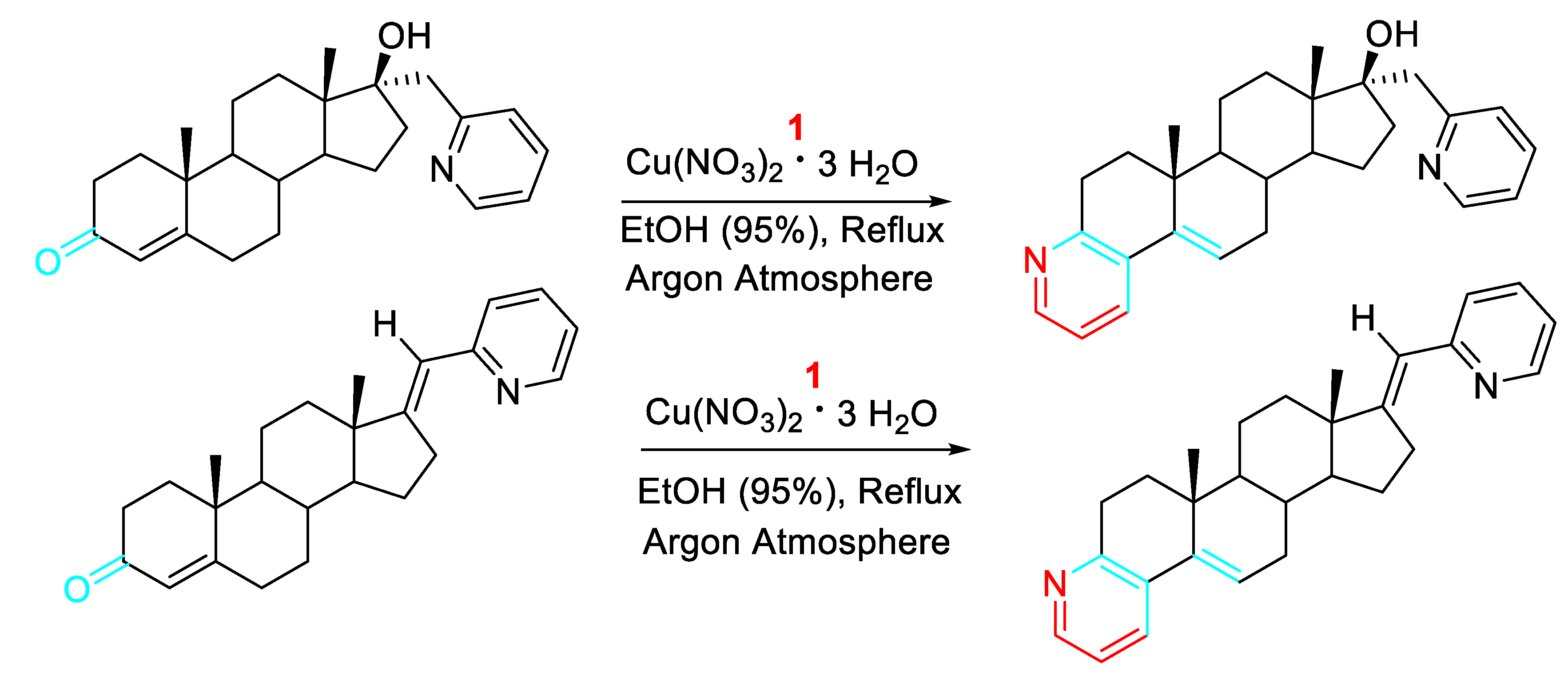
Optimization of the synthesis of relevant steroidal pyridines.
Scheme 9.
Gold-catalyzed synthesis of the carbacycloamine analog 11.
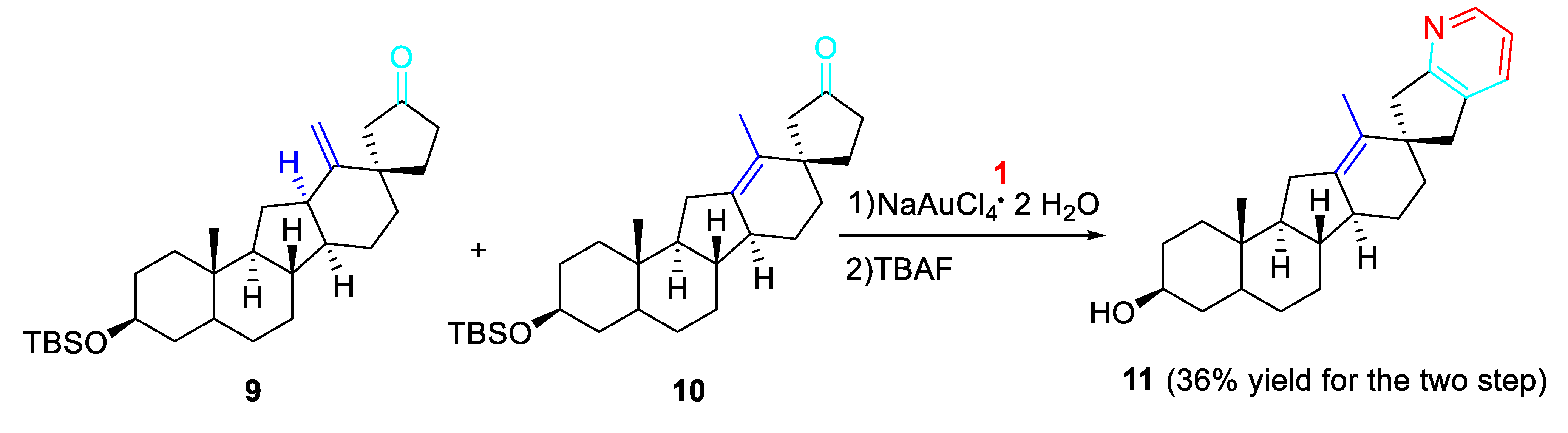
Scheme 10.
Gold-catalyzed synthesis of the potassium channel modulator 12.
Scheme 11.
Synthesis of Wnt signal path inhibitors.
Scheme 12.
Gold-catalyzed synthesis of BAY-298.
Scheme 13.
Synthesis of BMS-846372.
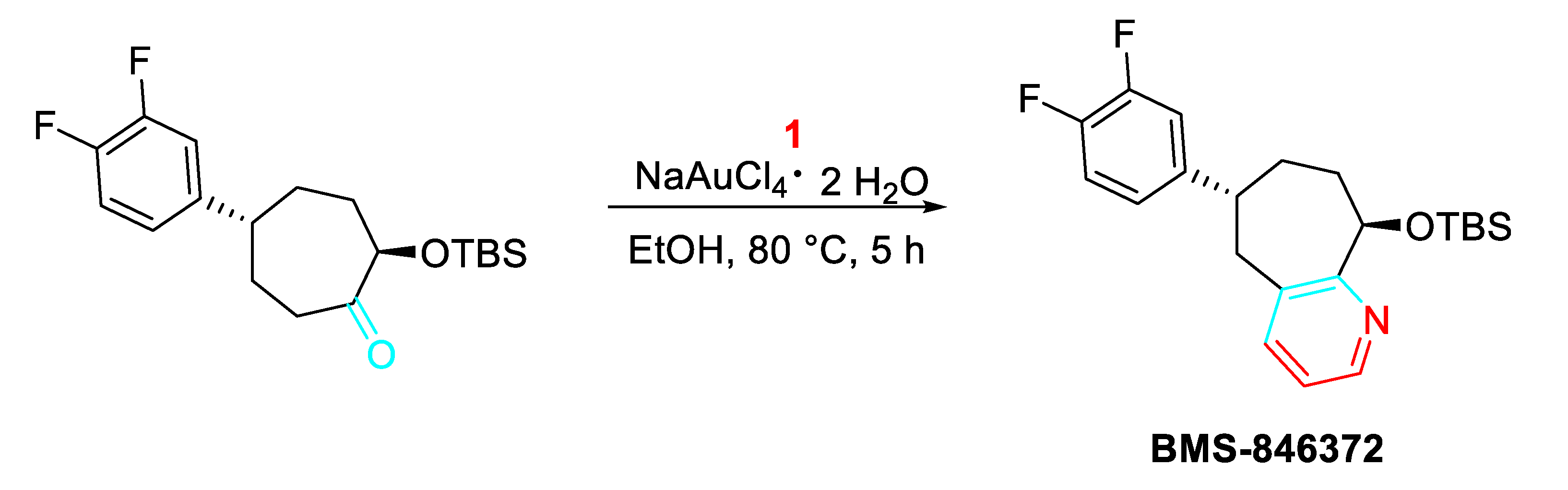
Scheme 14.
Gold-catalyzed synthesis of the benzomorphane derivative 13.
Scheme 15.
Gold-catalyzed synthesis of the tropane-related scaffold 14.
Scheme 16.
Synthesis of 11β-hydroxysteroid dehydrogenase inhibitors.
Scheme 17.
Gold-catalyzed synthesis of the ligand 17.
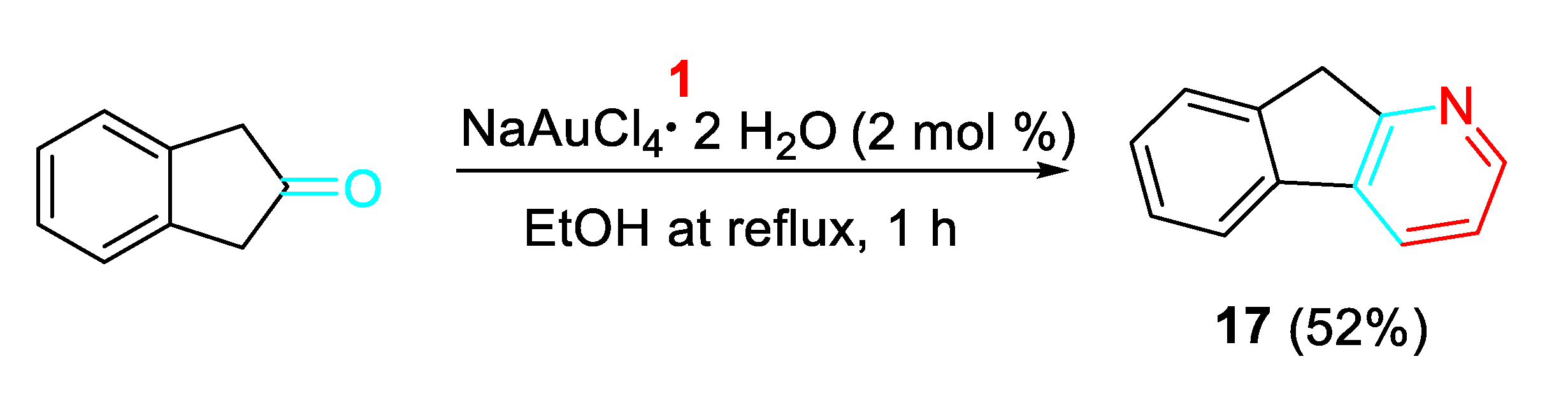
Scheme 18.
Gold-catalyzed synthesis of 2,5-dihydropyridines 18.
Scheme 19.
Synthesis of pyridinium salts.
Scheme 20.
Metal-free synthesis of the hetero-anthracene derivative 20.
Scheme 21.
Synthesis of the dihydrophenanthroline 21.
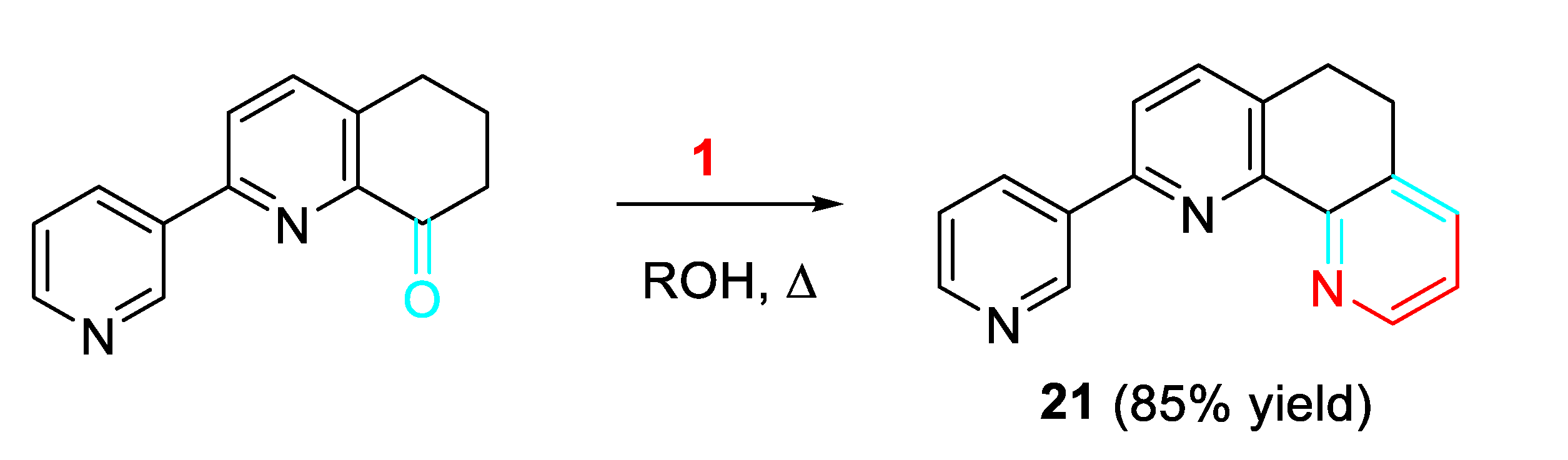
Scheme 22.
Synthesis of polyfunctionalised pyridines from the reaction of α,ß-unsaturated carbonyl compounds with propargylic amines.
Scheme 22.
Synthesis of polyfunctionalised pyridines from the reaction of α,ß-unsaturated carbonyl compounds with propargylic amines.
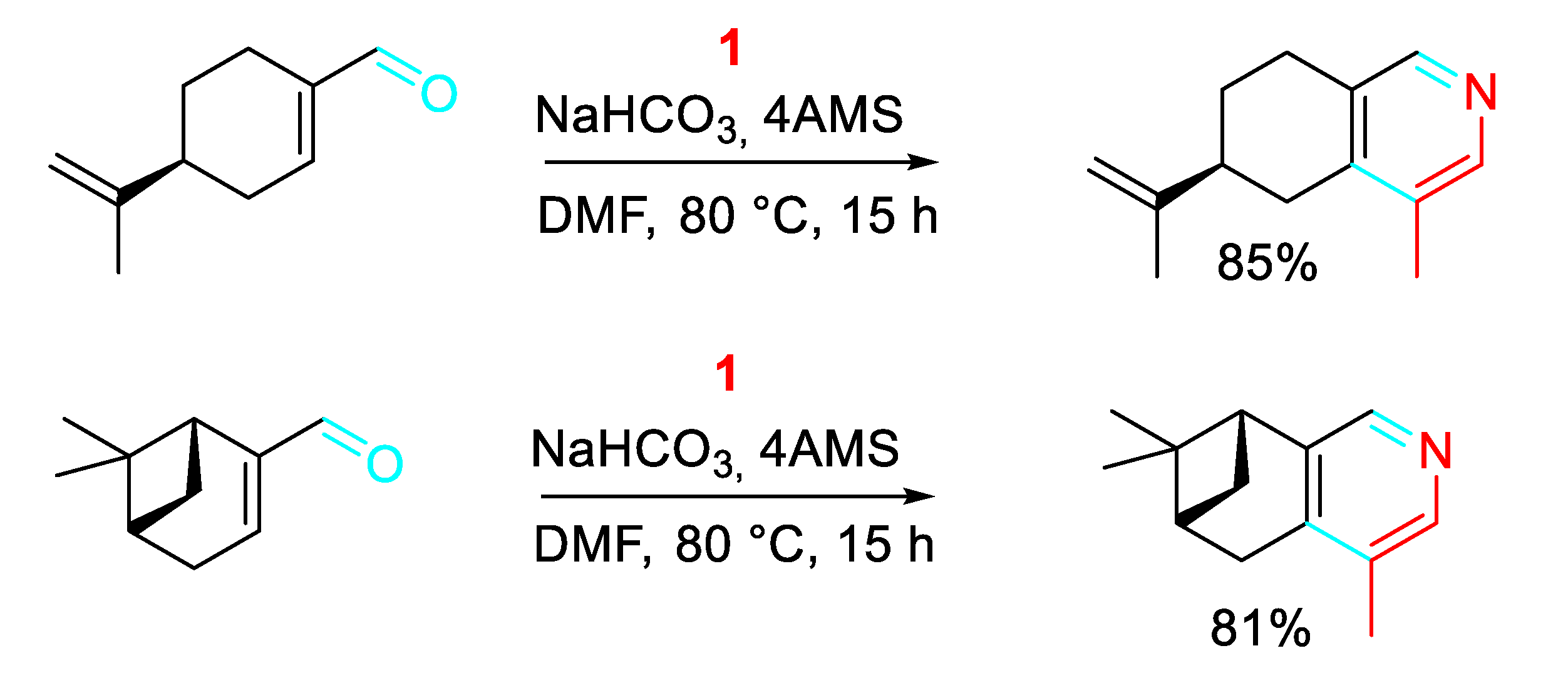
Scheme 23.
One-step synthesis of natural products.
Scheme 24.
Construction of pyridines starting from (S)-(-)-perillaldehyde and(1R)-myrtenal.
Scheme 25.
Retrosynthetic pathway of Suaveoline alkaloids.
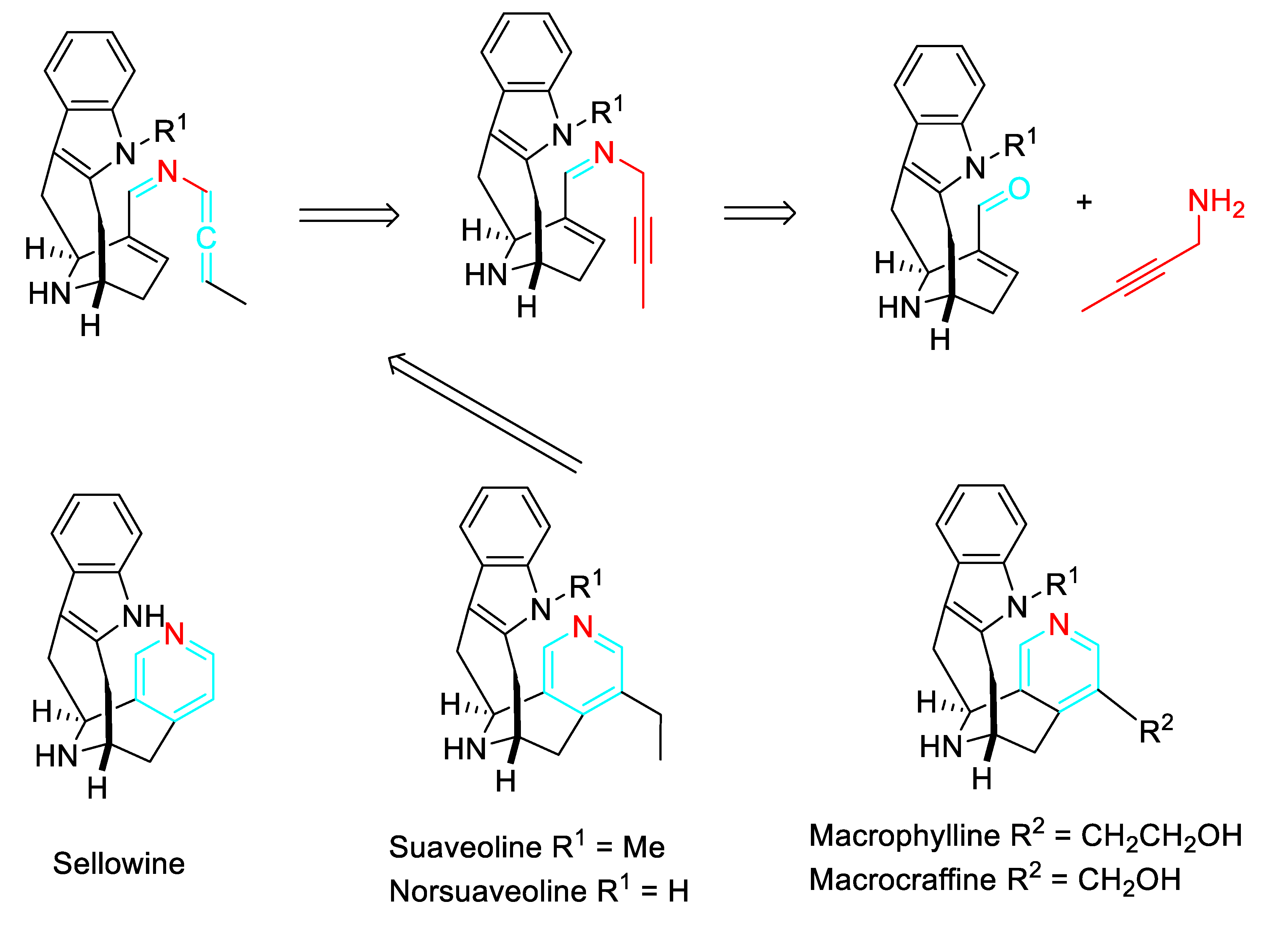
Scheme 26.
Pyridine synthesis by a 6π-3-azatriene electrocyclization.
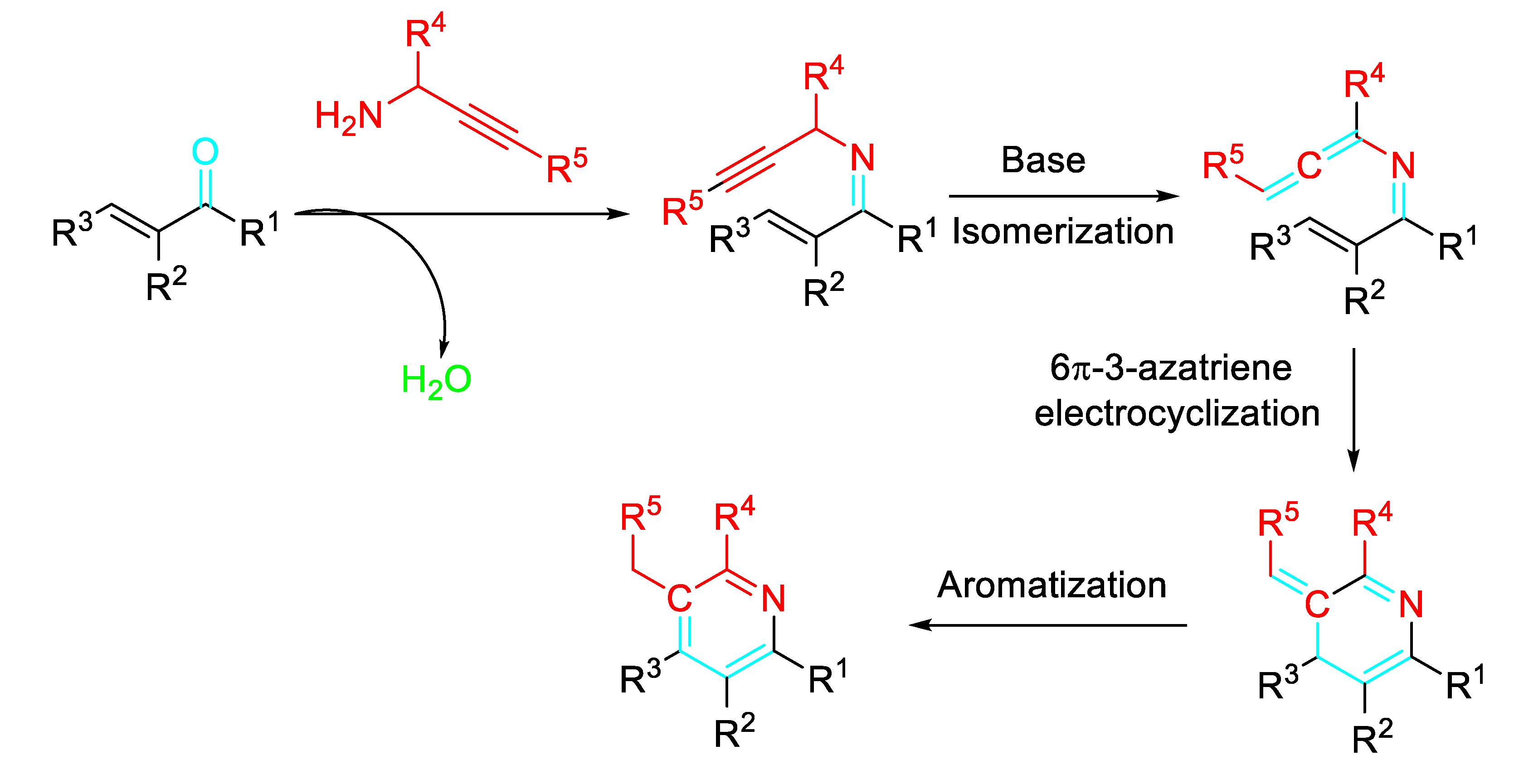
Scheme 27.
Synthesis of β- and γ-carbolines from indole aldehydes and substituted propargylic amines.
Scheme 27.
Synthesis of β- and γ-carbolines from indole aldehydes and substituted propargylic amines.
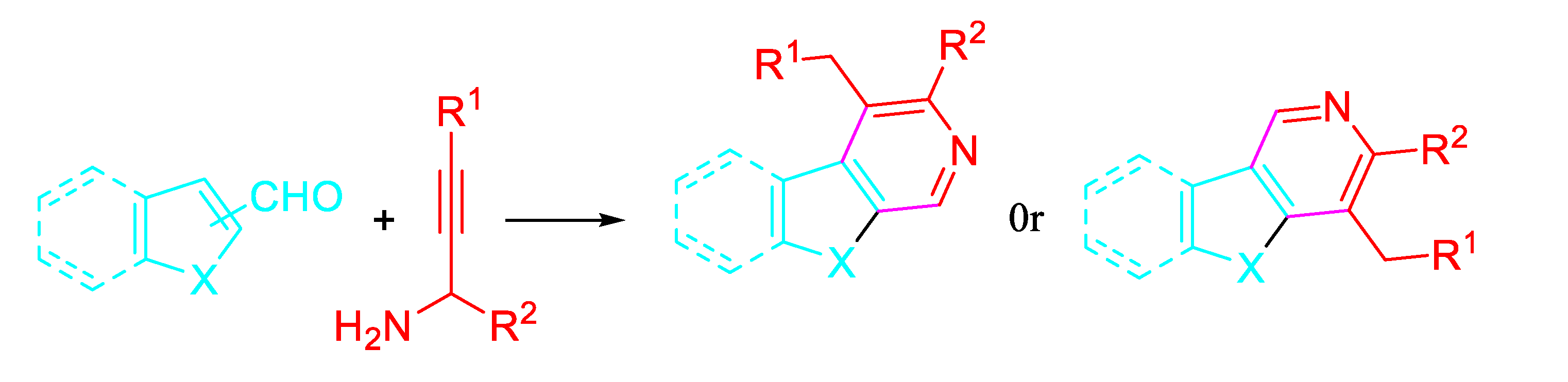
Scheme 28.
Synthesis of 4-benzylated carbolines 22.
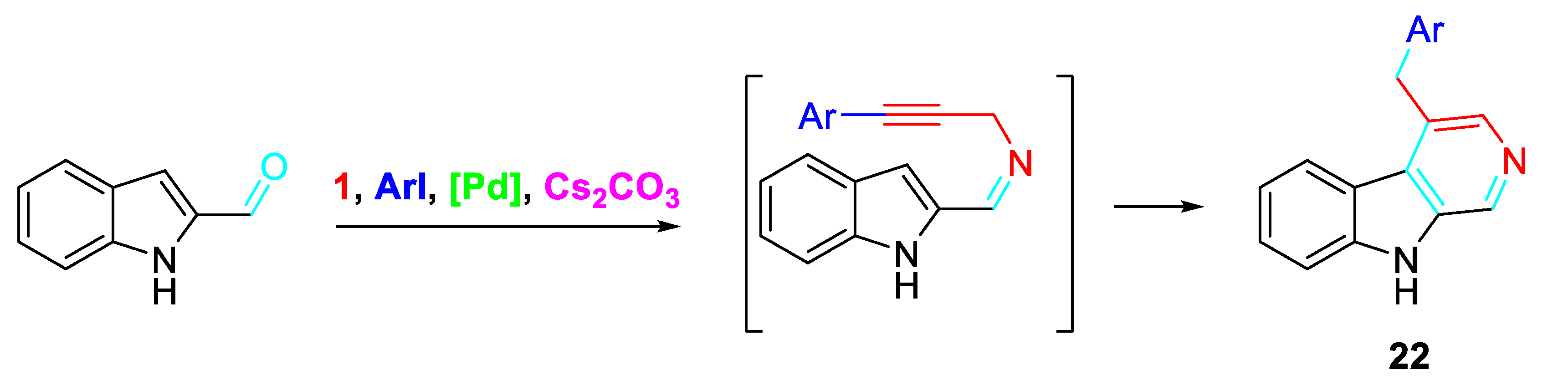
Scheme 29.
Alternative synthesis of polysubstituted pyridine derivatives from α,β-unsaturated carbonyl compound and propargylamine hydrochloride.
Scheme 29.
Alternative synthesis of polysubstituted pyridine derivatives from α,β-unsaturated carbonyl compound and propargylamine hydrochloride.
Scheme 30.
Total synthesis of onychine 23.
Scheme 31.
Mechanism for the formation of chromenopyridines 25.
Scheme 32.
Sequential reaction of 2-(diprop-2-ynylamino)benzaldehyde 26 with the propargylamine amine.
Scheme 32.
Sequential reaction of 2-(diprop-2-ynylamino)benzaldehyde 26 with the propargylamine amine.
Scheme 33.
Synthesis of naphthyridines 32.
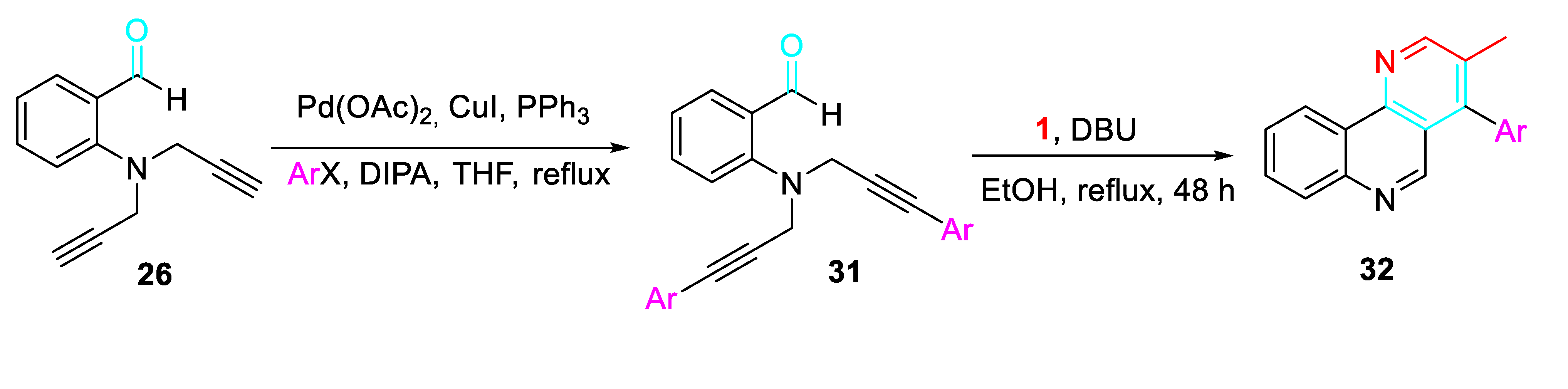
Scheme 34.
Synthesis of the chromenopyrazinones 33.
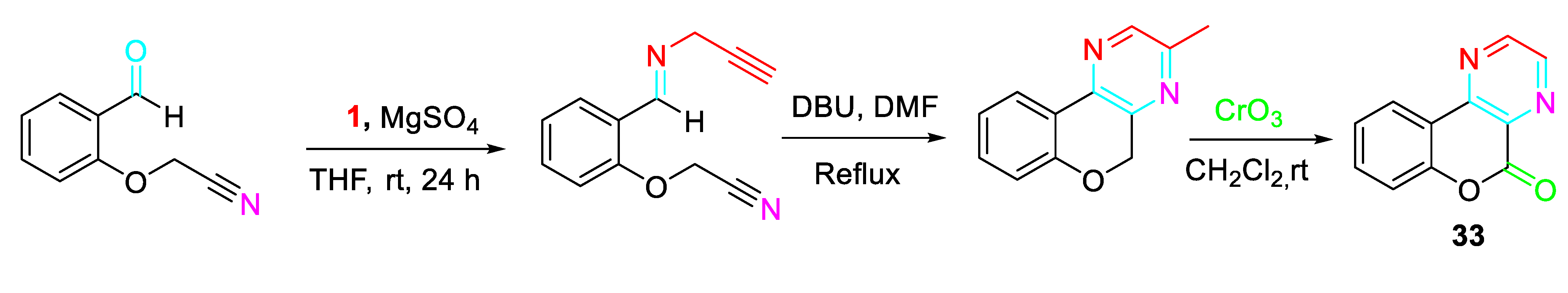
Scheme 35.
Gold-catalyzed synthesis of pyrazines 34 from the reaction of propargylamine with aldehydes.
Scheme 35.
Gold-catalyzed synthesis of pyrazines 34 from the reaction of propargylamine with aldehydes.
Scheme 36.
Proposed mechanism for the Au-catalyzed formation of pyrazines 34.
Scheme 37.
Sequential reaction of propargylamine with aldehydes catalyzed by MCM-41-immobilized phosphine gold(I) complex [MCM-41-PPh3-AuNTf2].
Scheme 37.
Sequential reaction of propargylamine with aldehydes catalyzed by MCM-41-immobilized phosphine gold(I) complex [MCM-41-PPh3-AuNTf2].
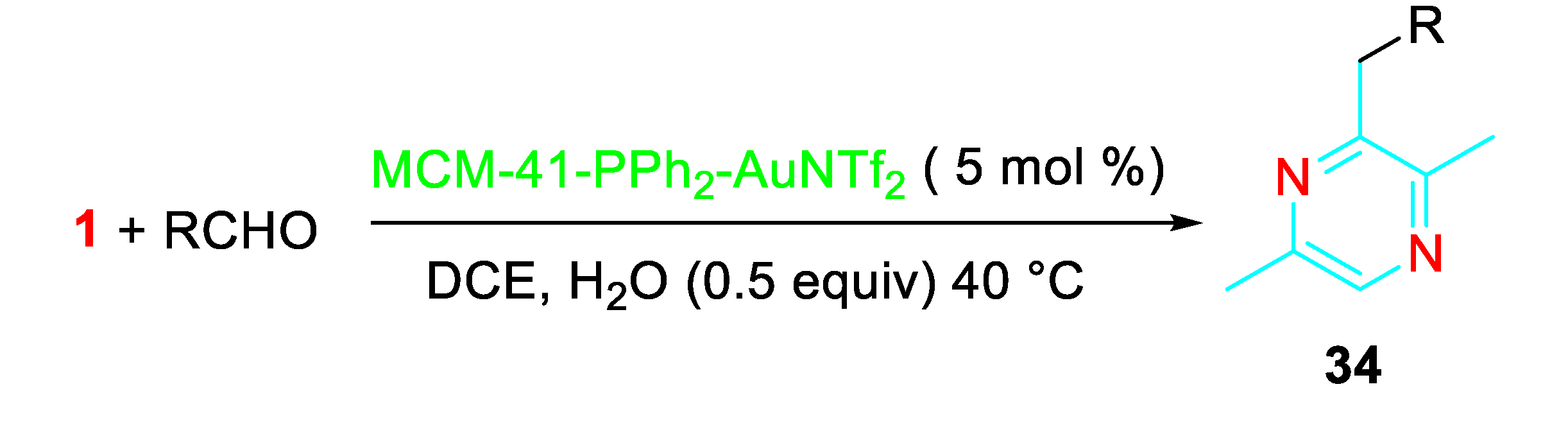
Scheme 38.
Synthesis of 1,2,3-triazolo-1,4-benzodiazepines 35.
Scheme 39.
Diversely substituted 1,2,3-triazolo 1,4-benzodiazepines 36.
Scheme 40.
Indium-catalyzed multicomponent synthesis of 9H-benzo[f]imidazo[1,2-d][1,2,3]triazolo[1,5-a][1,4]diazepines 37.
Scheme 41.
Indium-catalyzed synthesis of β-(N-indolyl)-α,β-unsaturated esters 40.
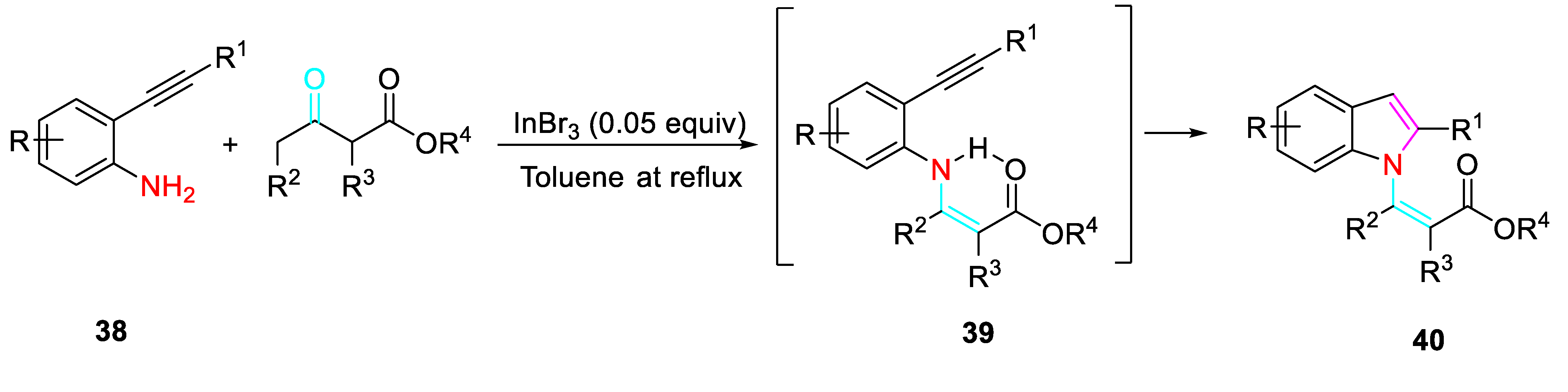
Scheme 42.
Divergent sequential gold-catalyzed cyclisation/alkenylation reaction of 2-alkynylanilines with 1,3-dicarbonyl compounds.
Scheme 42.
Divergent sequential gold-catalyzed cyclisation/alkenylation reaction of 2-alkynylanilines with 1,3-dicarbonyl compounds.
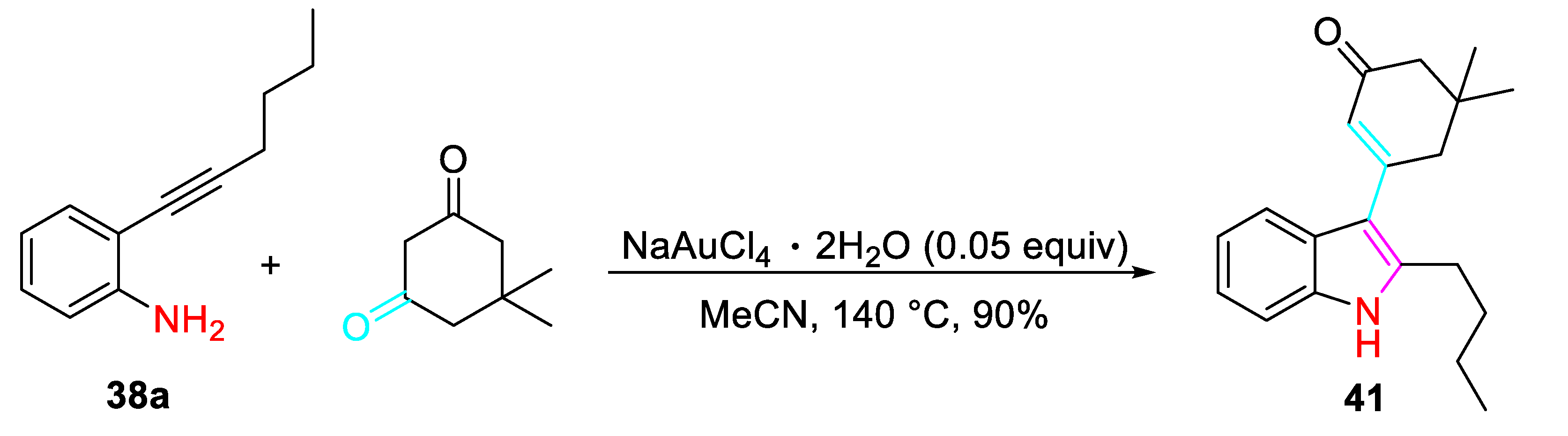
Scheme 43.
p-TsOH promoted synthesis of 4-alkyl-2,3-disubstituted quinolines 42.
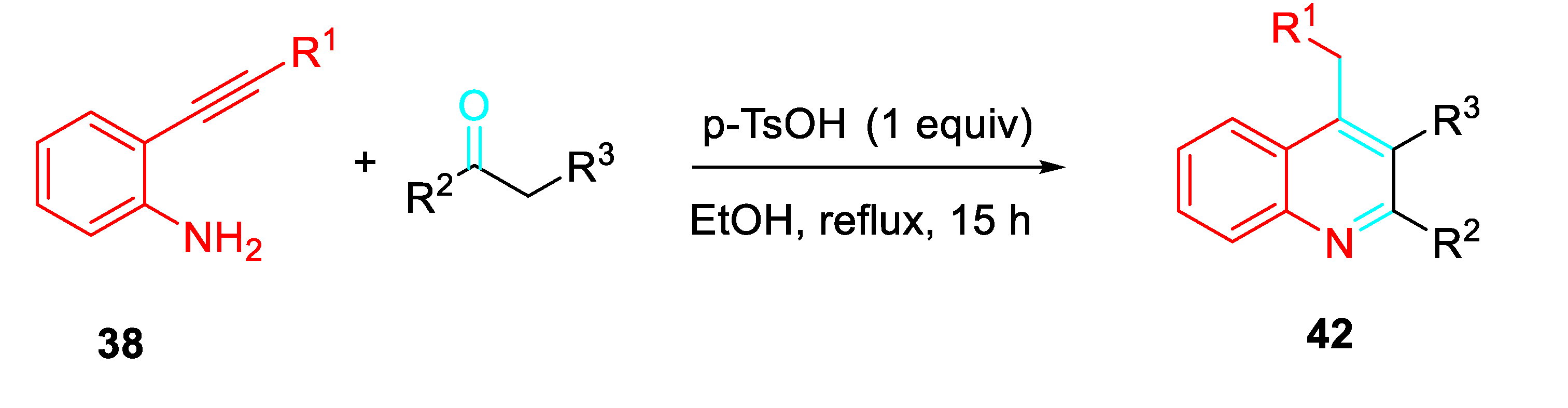
Scheme 44.
Synthesis of dimeric quinolines 43.
Scheme 45.
Iron(III)-catalyzed sequential condensation, cyclization and aromatization of 1,3-diketones with 2-ethynylaniline to afford 4-methyl-2,3-disubstituted quinolines 44.
Scheme 45.
Iron(III)-catalyzed sequential condensation, cyclization and aromatization of 1,3-diketones with 2-ethynylaniline to afford 4-methyl-2,3-disubstituted quinolines 44.
Scheme 46.
Sequential reaction of levulinic acid with 2-ethynylaniline under solventless conditions.
Scheme 46.
Sequential reaction of levulinic acid with 2-ethynylaniline under solventless conditions.
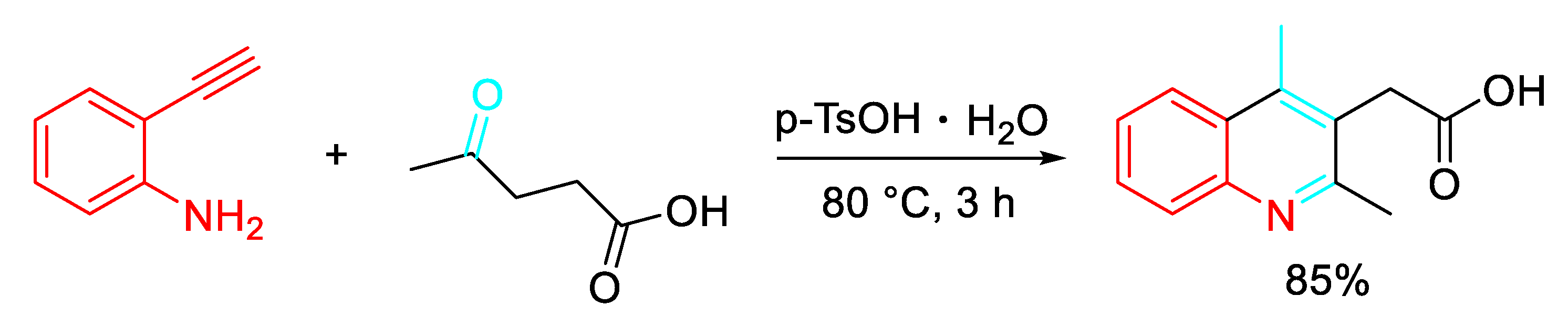
Scheme 47.
Copper(I)-catalyzed synthesis of 2-acylquinolines.
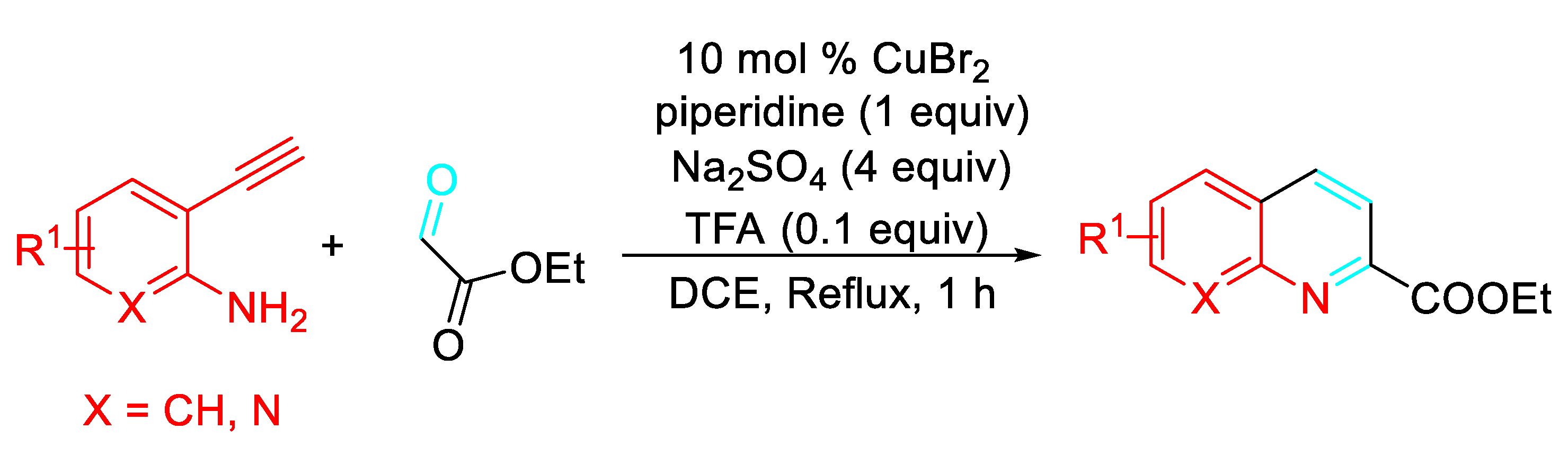
Scheme 48.
Copper-catalyzed synthesis of quinolines 45 from ethynylaniline and ethyl glyoxylate.
Scheme 49.
Synthesis of 4-alkoxy-2-arylquinolines 46.
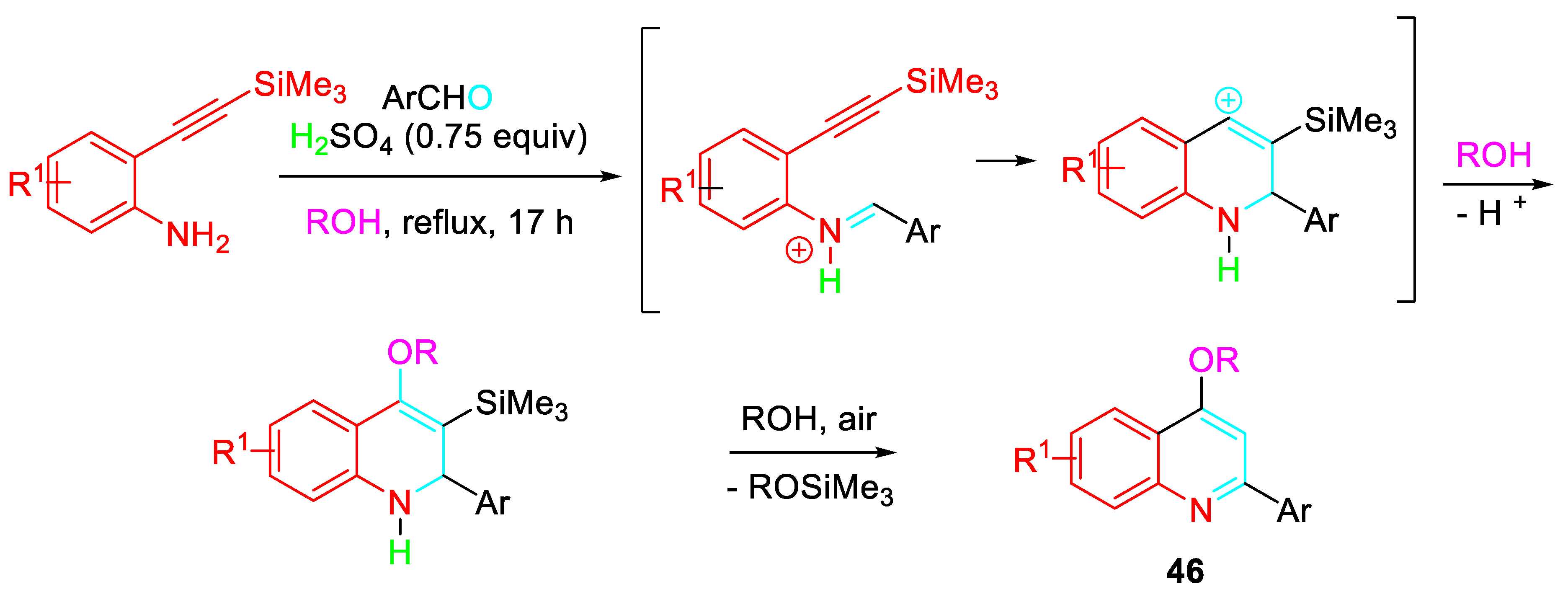
Scheme 50.
Condensative cyclization of 6-amino-5-[(trimethylsilyl)ethynyl]-2H-chromen-2-one 47.
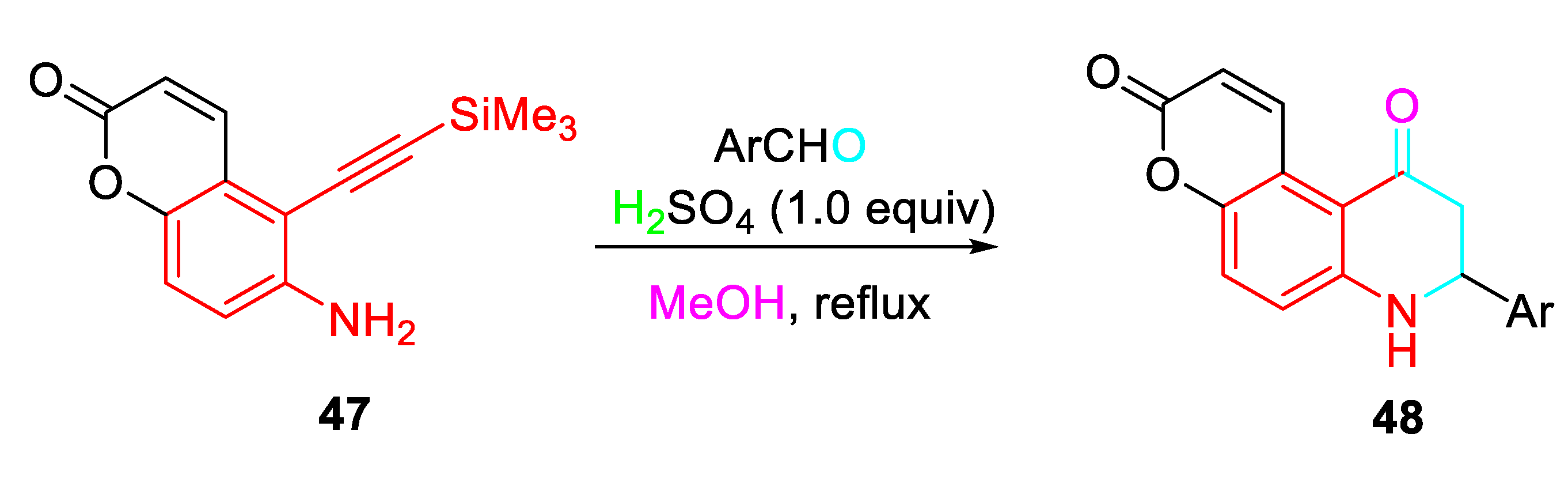
Scheme 51.
Synergistic effect of Pd(II) and acid catalysts on the synthesis of ring-fused quinolines.
Scheme 51.
Synergistic effect of Pd(II) and acid catalysts on the synthesis of ring-fused quinolines.
Scheme 52.
Sc(OTf)3-catalyzed bicyclization of 2-alkynylanilines with aldehydes.
Scheme 53.
Gold-catalyzed synthesis of polycyclic frameworks 54.
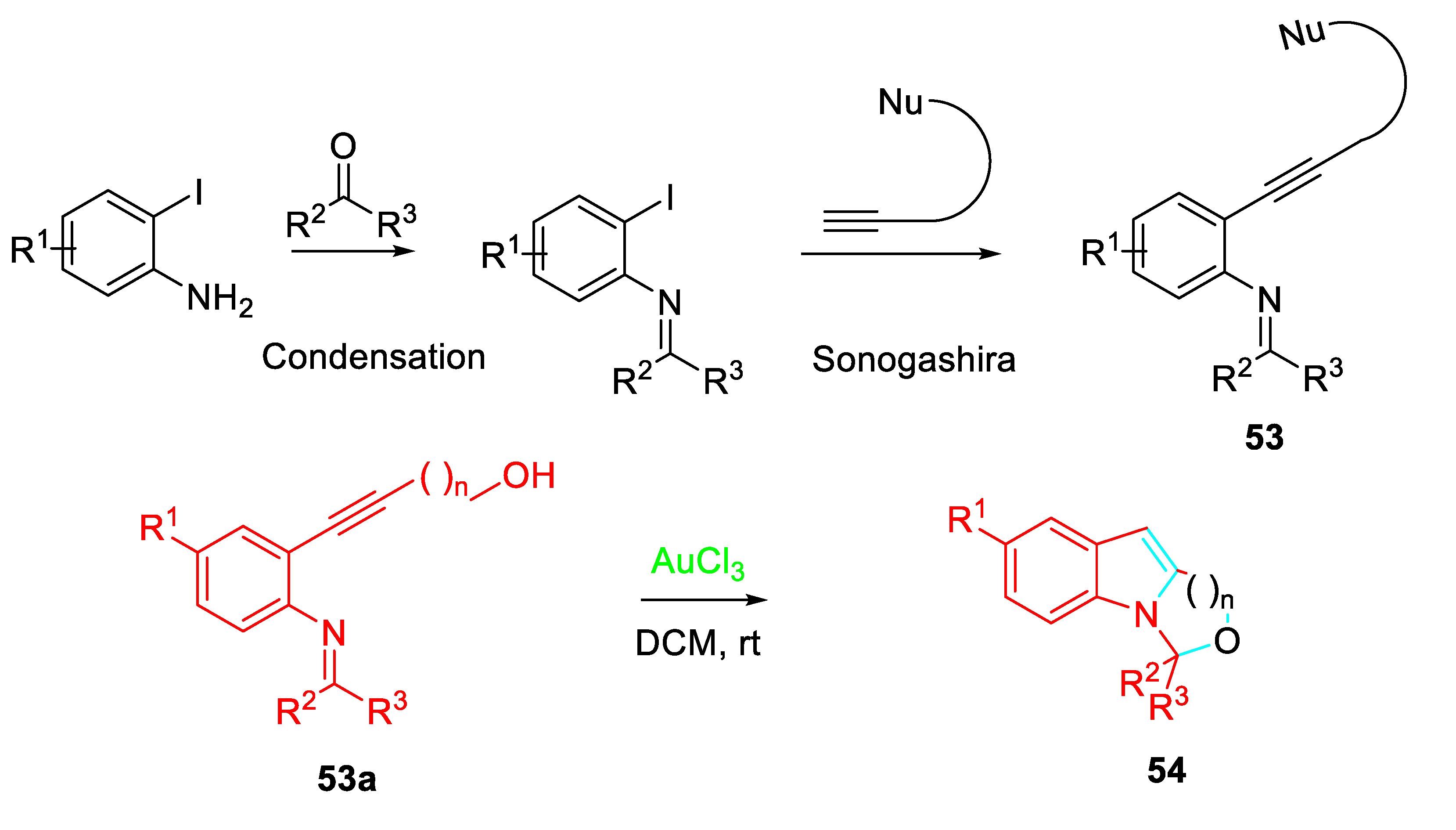
Scheme 54.
Iodonium induced tandem cyclization of N-(2-alkynylphenyl) imines 55.
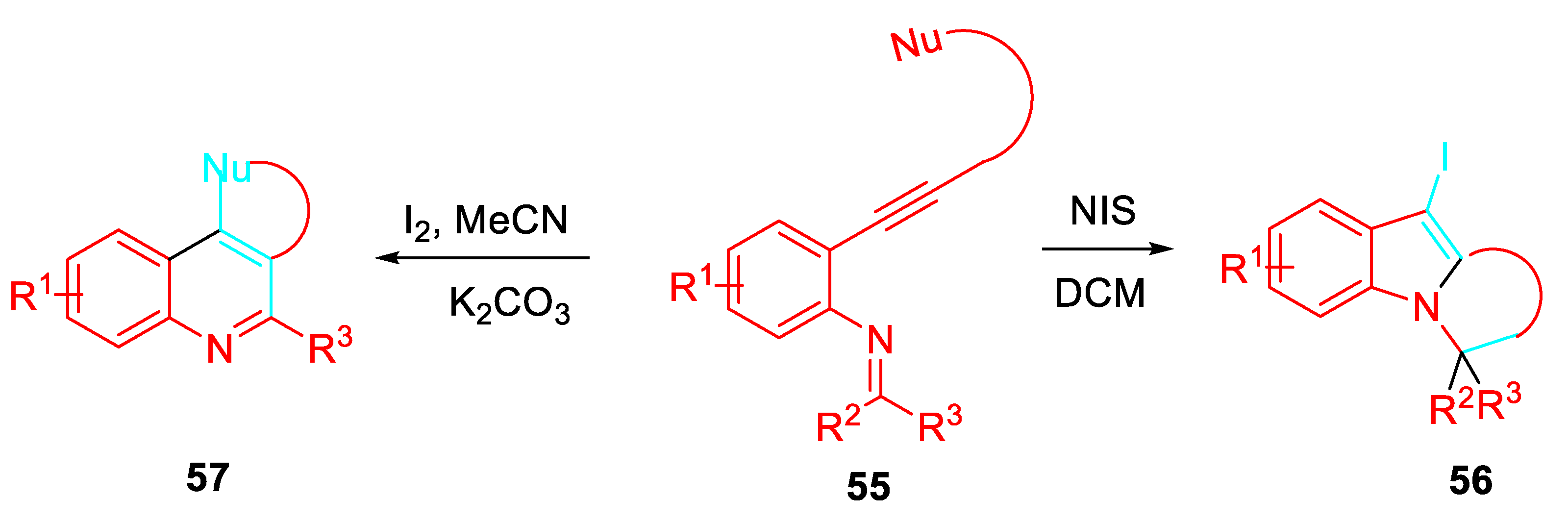
Scheme 55.
One-pot synthesis of 2,2’-disubstituted diindolylmethanes 58.
Scheme 56.
Gold-catalyzed synthesis of 1-substituted 2-tosyl-2,3,4,5-tetrahydropyrido[4,3-b]indoles 63.
Scheme 56.
Gold-catalyzed synthesis of 1-substituted 2-tosyl-2,3,4,5-tetrahydropyrido[4,3-b]indoles 63.
Scheme 57.
Cu(OTf)2-catalyzed synthesis of quinoline-annulated polyheterocyclic frameworks 66.
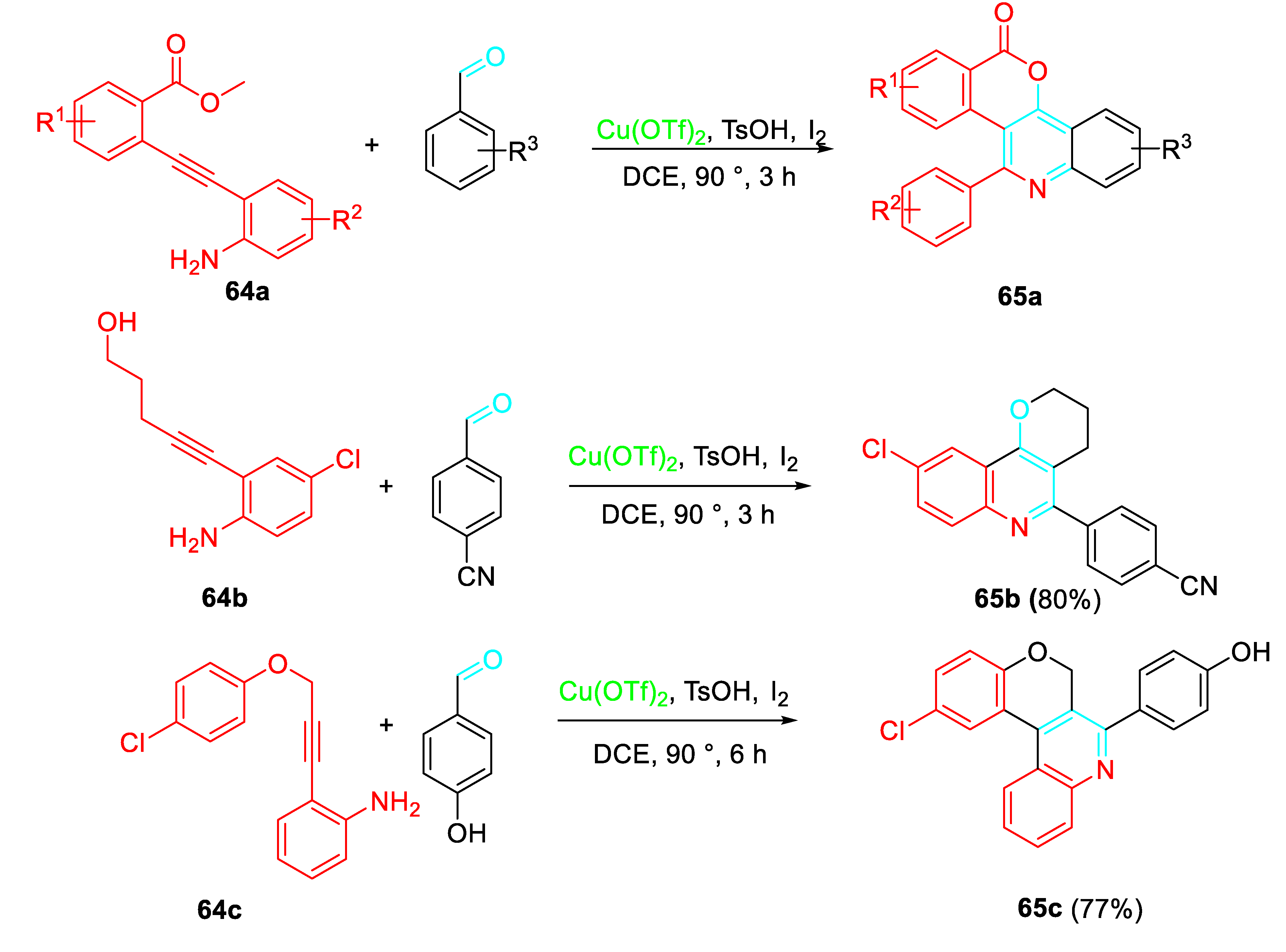
Scheme 58.
Plausible reaction mechanism.
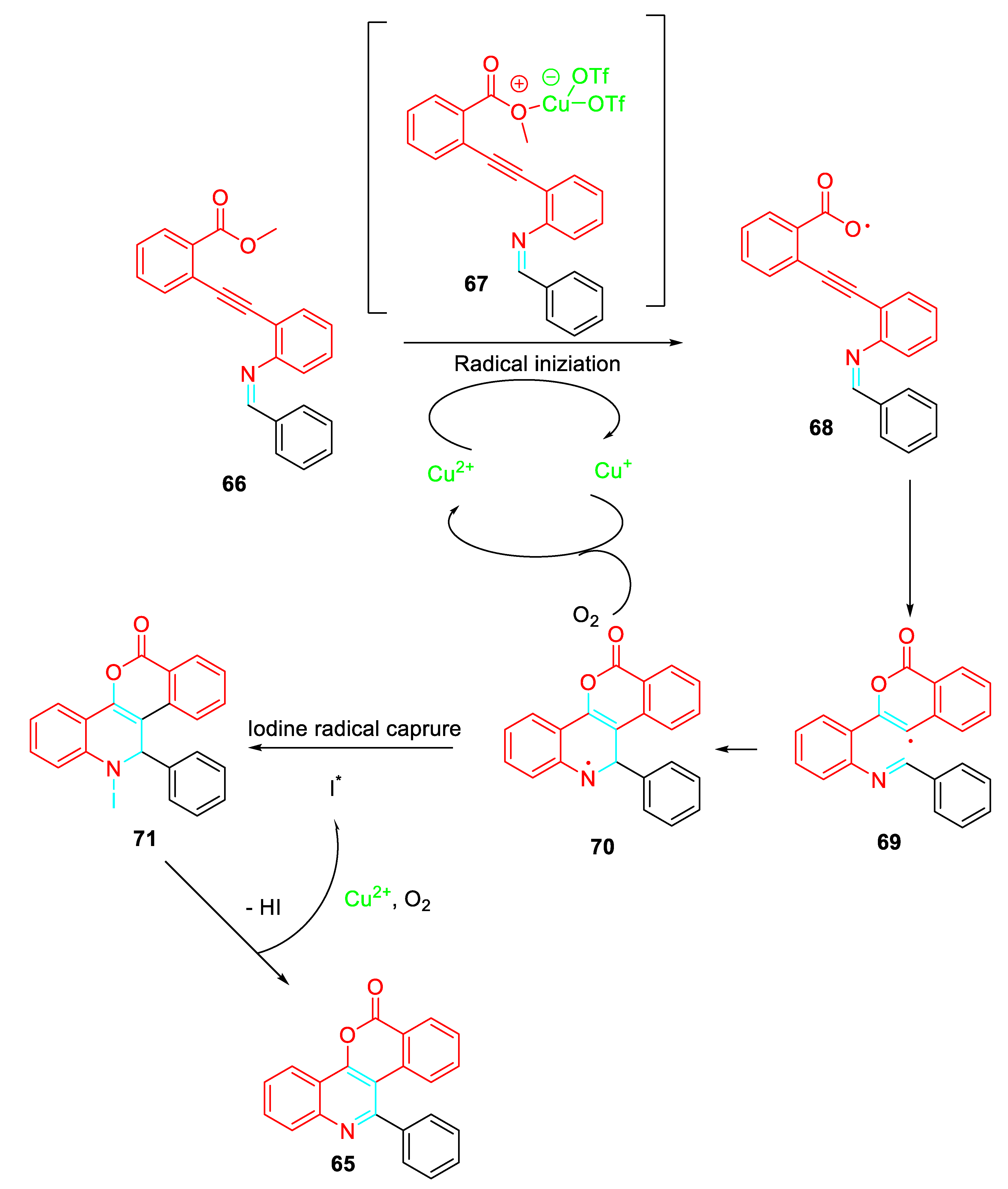
Scheme 59.
Palladium-catalyzed synthesis of 1-benzoazepine carbonitriles 72.
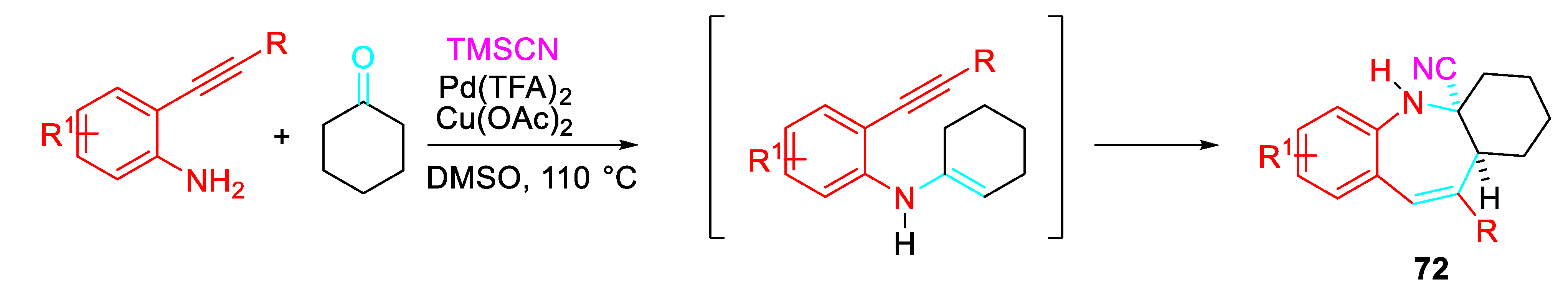
Scheme 60.
Product selectivity control of the sequential reaction of 2-alkynylaniline with ketones.
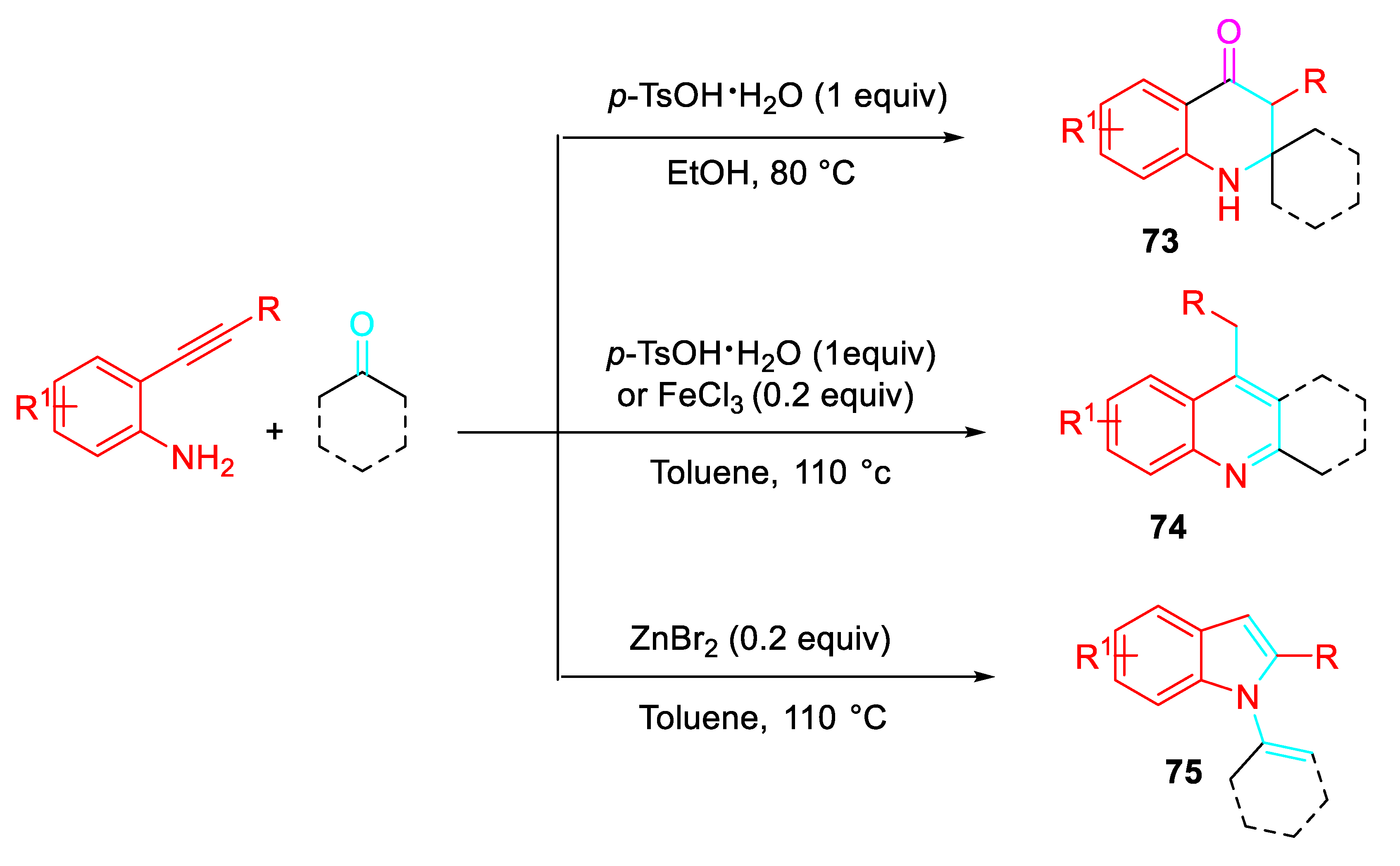
Scheme 61.
Proposed mechanism for the synthesis of quinolinones 73.
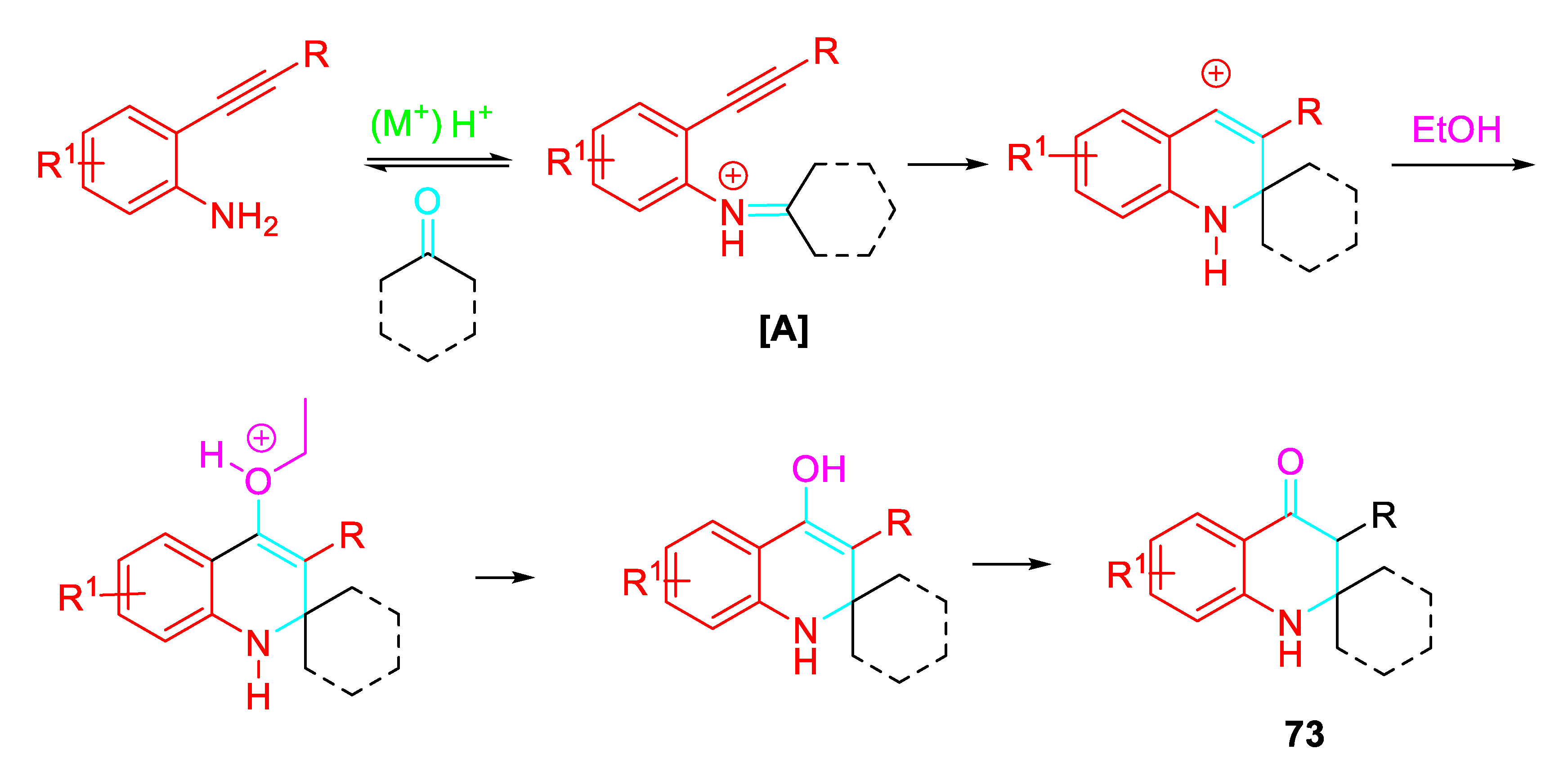
Scheme 62.
Proposed mechanism for the synthesis of quinolines 74 and N-alkenylindoles 75.
Scheme 63.
Alternative mechanism for the synthesis of quinolines 74.
Scheme 64.
Sequential amination/annulation/aromatization reactions of β-(2-aminophenyl)-α,β-ynones 76 with enolisable ketones.
Scheme 64.
Sequential amination/annulation/aromatization reactions of β-(2-aminophenyl)-α,β-ynones 76 with enolisable ketones.
Scheme 65.
Product selectivity control in the synthesis of polycyclic steroidal quinolines 77.
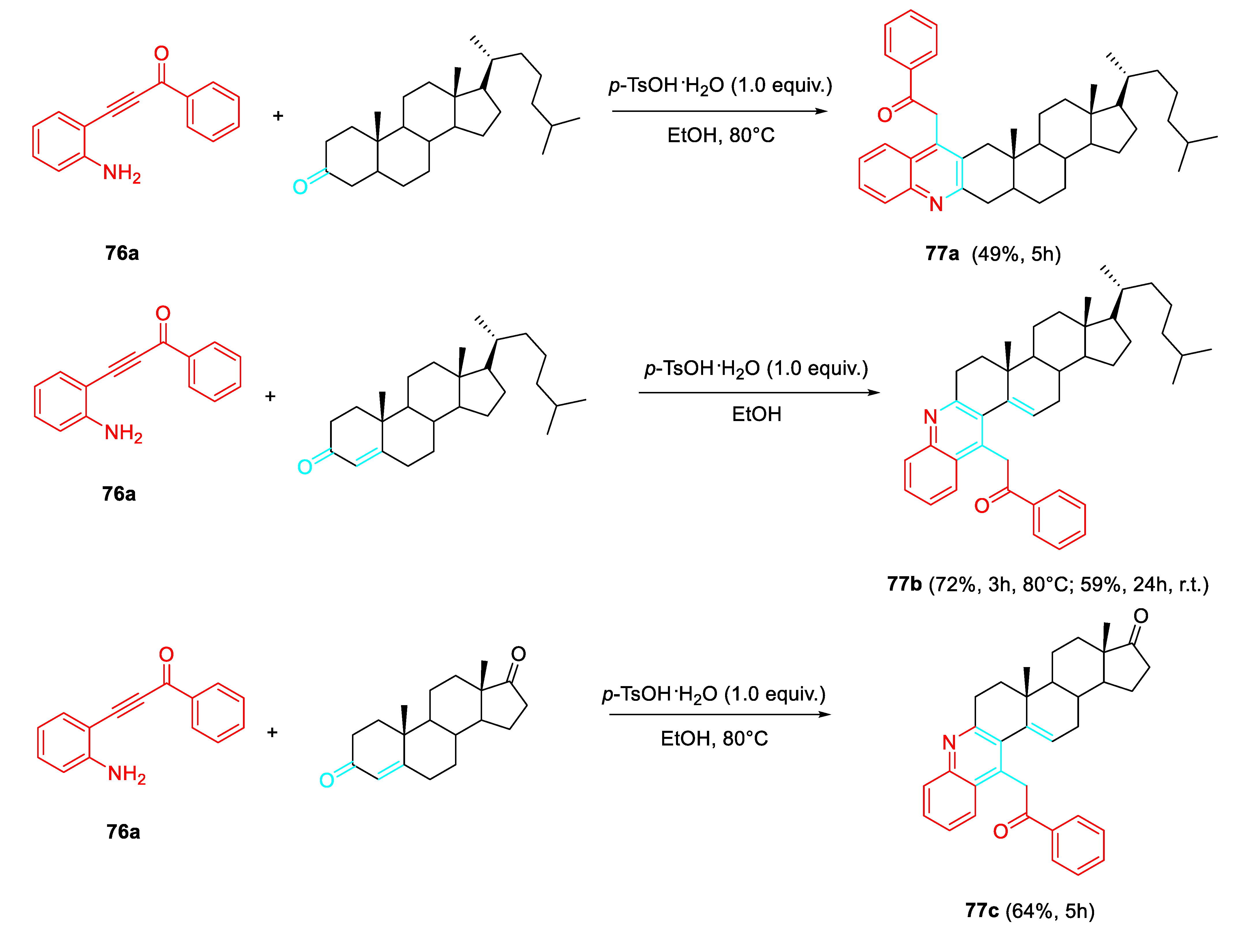
Scheme 66.
Domino reactions of β-(2-aminophenyl) α,β-ynones with 1,3-dicarbonyls.
Scheme 67.
Sequential aminopalladation of β-amino alkynes 80.
Scheme 68.
Retrosyntetic assembly of 11H-indolo[3,2-c]quinolines.
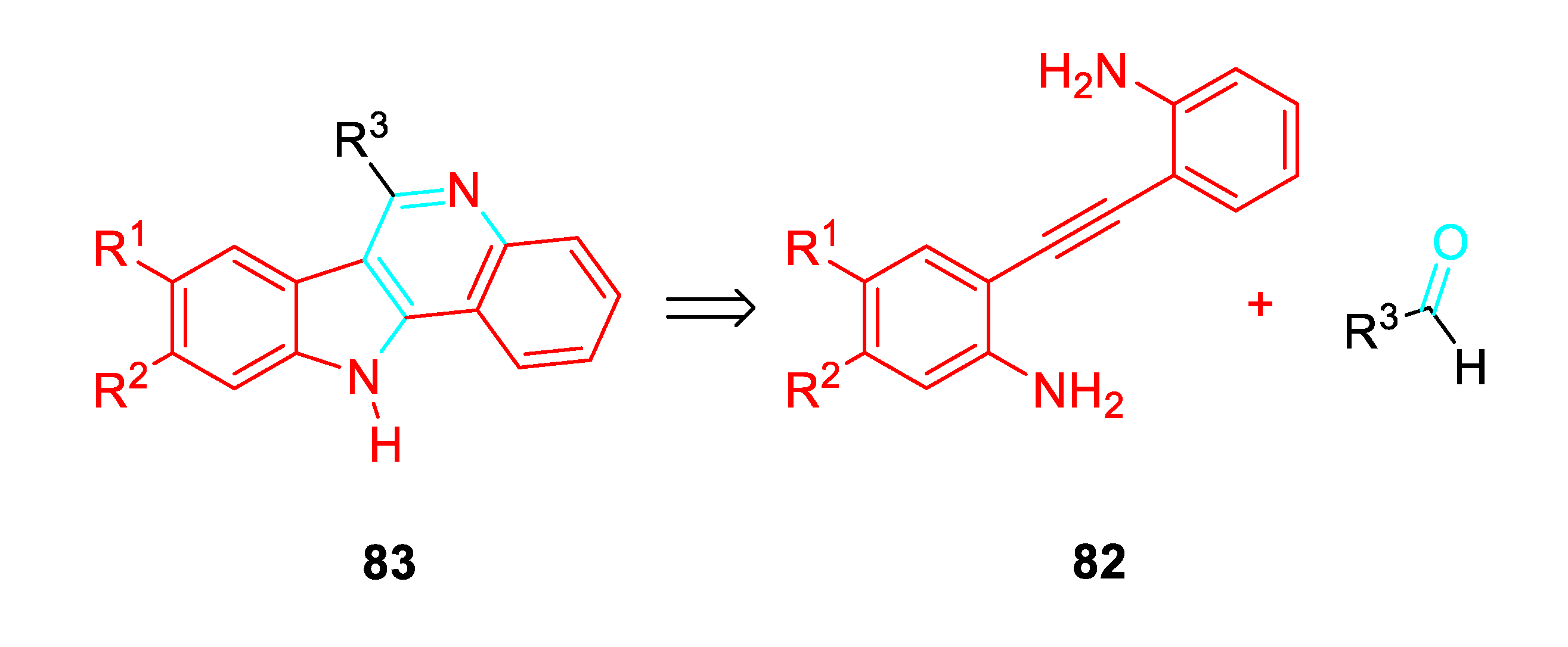
Scheme 69.
Gold-catalyzed synthesis of 11H-Indolo[3,2-c]quinoline 83.
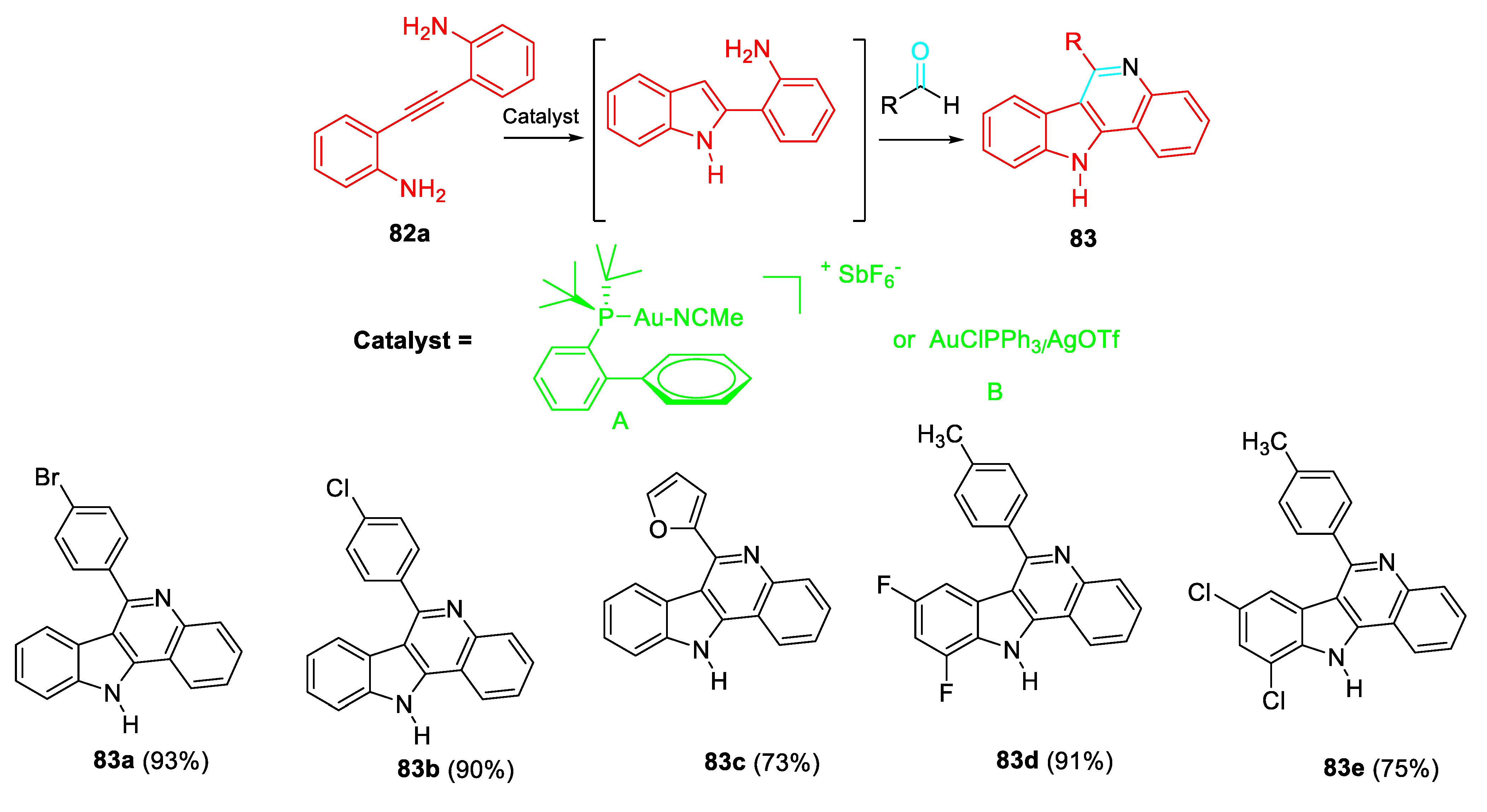
Scheme 70.
Brønsted acid catalyzed synthesis of 2,2′-disubstituted 1H,1′H-3,3′-biindoles 84.
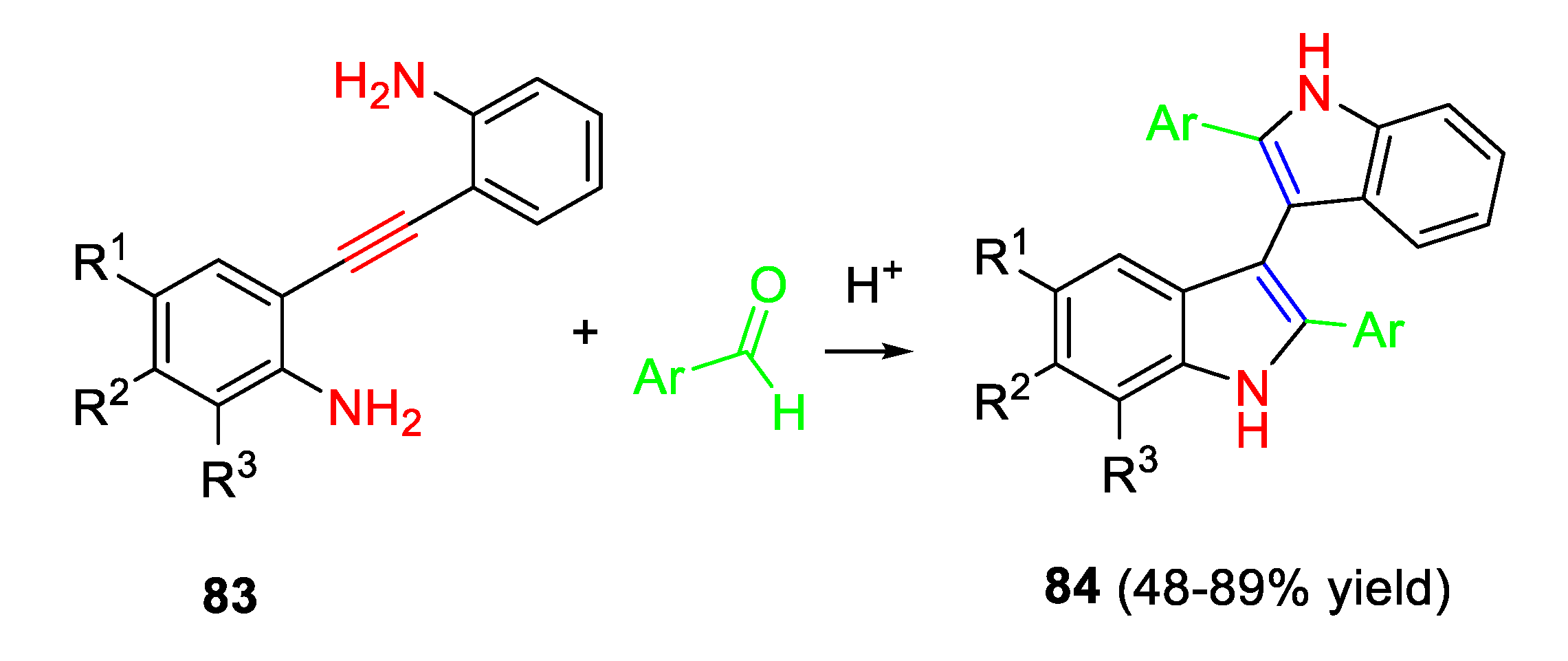
Scheme 71.
Proposed mechanism.
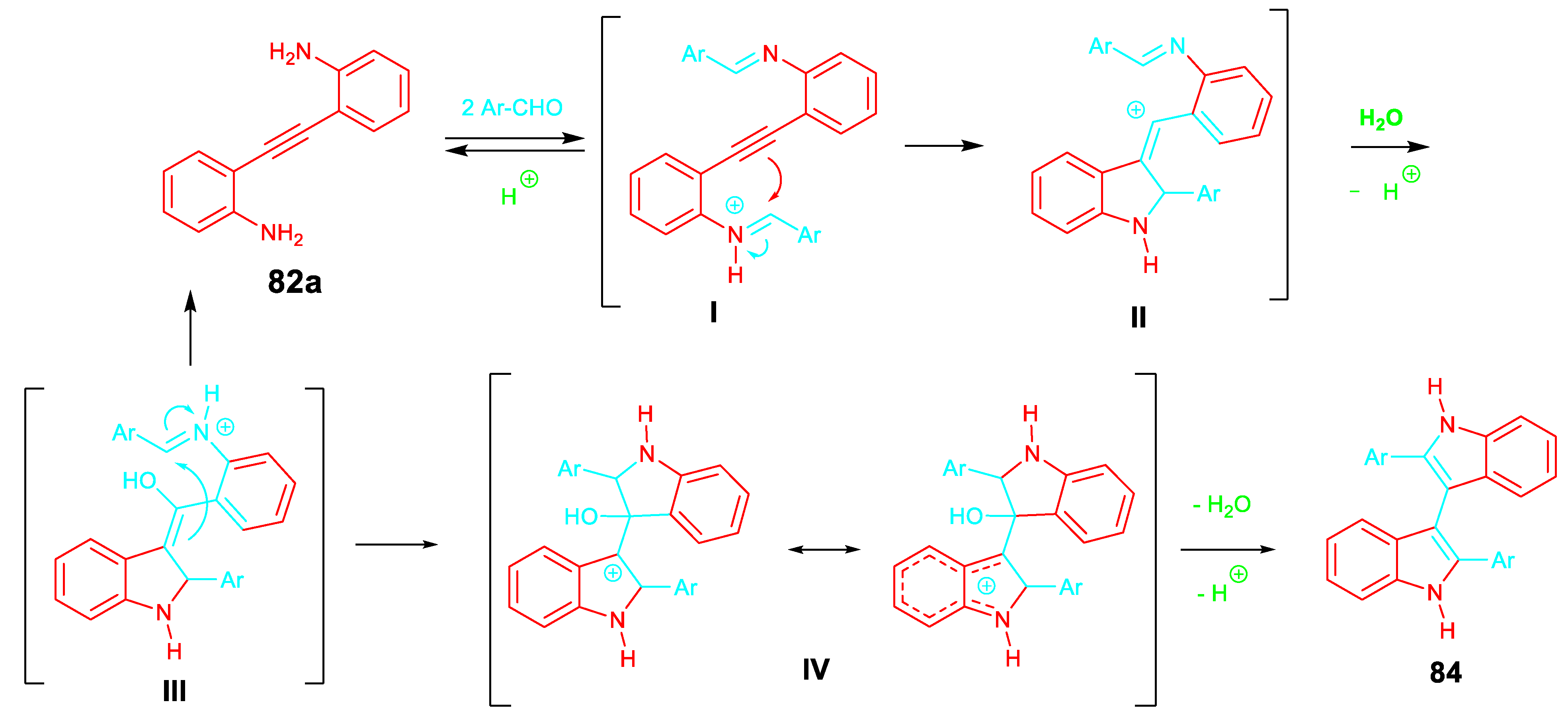
Scheme 72.
Gold-catalyzed reaction of 2-[(2-aminophenyl)ethynyl]phenylamines 83 with isatins and ketones.
Scheme 72.
Gold-catalyzed reaction of 2-[(2-aminophenyl)ethynyl]phenylamines 83 with isatins and ketones.
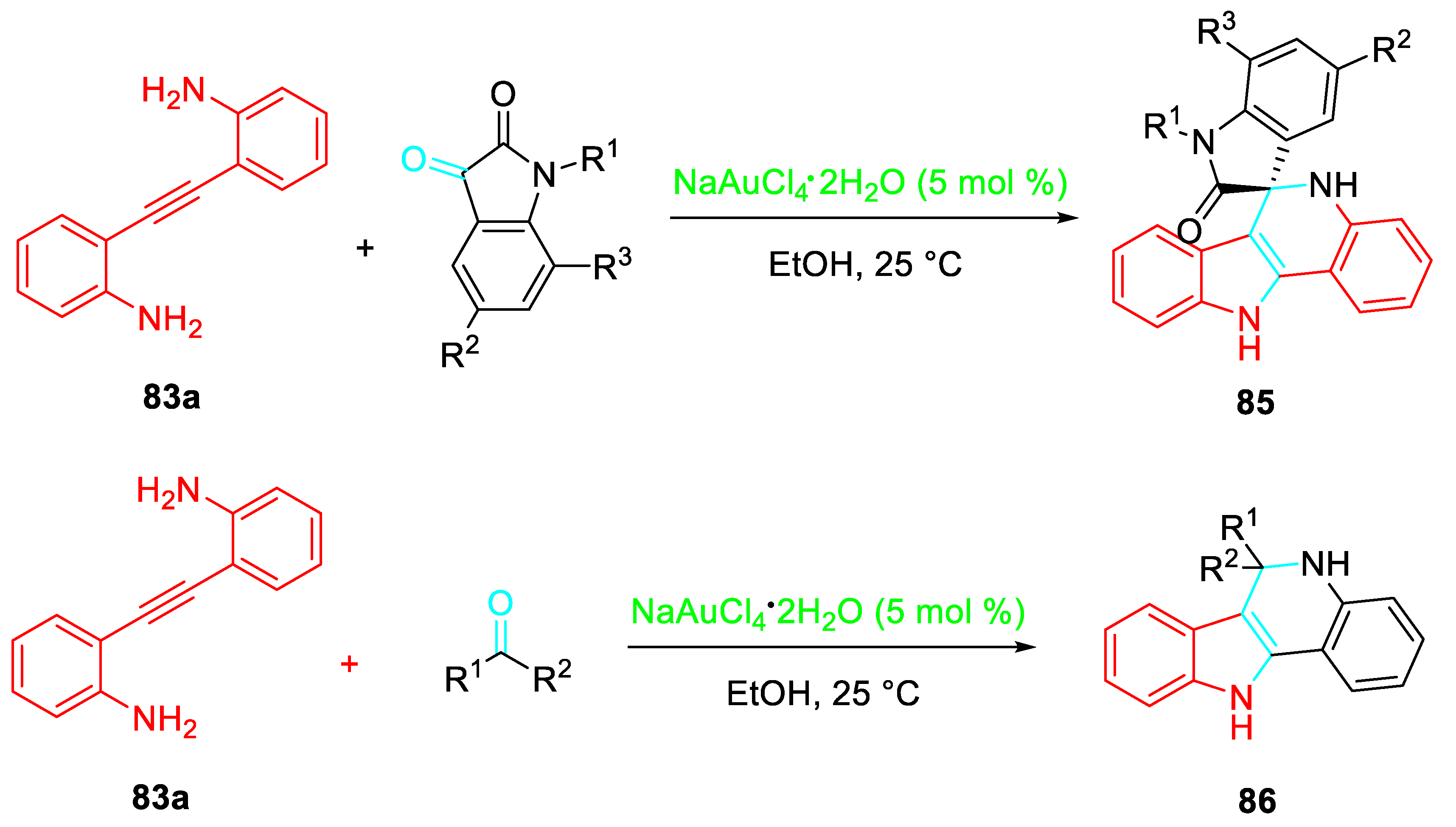
Scheme 73.
Indium(III)-catalyzed sequential reaction of 2-alkynylanilines with 2-alkynylbenzaldehydes.
Scheme 73.
Indium(III)-catalyzed sequential reaction of 2-alkynylanilines with 2-alkynylbenzaldehydes.
Scheme 74.
Synthesis of 2-alkoxyfuro[2,3-c]quinolones 88.
Scheme 75.
Proposed mechanism.
Scheme 76.
Sequential multicomponent approach to 2,2-disubstituted 3-methyleneindolines 94.
Scheme 77.
Synthesis of 1-tosyl-2,3,4,5-tetrahydro-1H-indeno[1,2-b]-pyridines 96.
Scheme 78.
Synthesis of pyrrolo[1,2-a]quinolines 99.
Scheme 79.
Platinum(II)-catalyzed synthesis of indolines 101.
Scheme 80.
Platinum(II)-catalyzed synthesis of tetrahydroquinolines 103.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated