
Submitted:
16 May 2023
Posted:
17 May 2023
You are already at the latest version
Abstract
Nonalcoholic fatty liver disease (NAFLD) has become a widely studied subject due to its increasing prevalence and links to diseases such as type 2 diabetes and obesity. It has severe complications, including nonalcoholic steatohepatitis, cirrhosis, hepatocellular carcinoma, and portal hypertension that can lead to liver transplantation in some cases. To better prevent and treat this pathology, it is important to understand its underlying physiology. Here, we identify three main factors that play a crucial role in the physiopathology of NAFLD: oxidative stress, insulin resistance, and the key role of carcinoembryonic antigen related cell adhesion molecule 1 (CEACAM1). We discuss the pathophysiology linking these factors to NAFLD pathophysiology.
Keywords:
NAFLD
; NASH
; insulin resistance
; reactive oxygen species
; CEACAM1
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is a serious disease that affects 25% of the world's population [1]. The prevalence of NAFLD has also been increasing for several years due to the epidemic of overweight and obesity [2]. This is an important pathology to understand because it can be complicated by advanced hepatic fibrosis (nonalcoholic steatohepatitis, NASH) or even hepatocellular carcinoma [3]. Due to the prevalence and associated complications of NAFLD, it is essential to analyze the diverse physiological mechanisms for targeted and effective treatment. This study will focus on two significant processes that contribute to the pathogenesis of NAFLD, namely oxidative stress and carcinoembryonic antigen related cell adhesion molecule 1 (CEACAM1). CEACAM1 is a transmembrane protein that plays a crucial role in insulin degradation and excretion, as well as regulating insulin homeostasis in the liver. The redox equilibrium must remain within physiological parameters in order to be considered safe. However, if exposed to certain triggers, a redox imbalance may arise, leading to an increase of oxidative stress and ultimately contributing to the pathogenesis of NAFLD.
2. Method
A literature review was realized using PubMed, Google Scholar and Web of Science including several studies from 1986 until 2023, but mostly from recent years, which were linked to the association between NAFLD (or MAFLD), CEACAM1 and Oxidative stress. Medical Subject Headings terms such as “Nonalcoholic fatty liver disease”, “Liver disease”, “NASH”, “Steatohepatitis”, “Steatosis”, “Hepatic Lipogenesis” were associated with “CEACAM1”, “Insulin clearance” and “Oxidative Stress”. The different articles were analyzed and selected according to their abstract relevance. Similar articles suggested by the research sites were also taken into consideration and selected. In total, this review was based on the study of 94 different articles. The analyzed articles were all restricted to English language.
2.1. NAFLD: Definition and prevalence, Diagnosis, Physiopathology and Therapeutical approach
2.1.1. Definition and prevalence
NAFLD is a spectrum of diseases ranging from simple hepatic steatosis to nonalcoholic steatohepatitis and hepatic fibrosis. It is a very common disease and the most common hepatic disease worldwide with an estimated prevalence of 25% [1]. It is defined by hepatic steatosis (at least 5%) and the absence of a secondary cause of hepatopathy such as excessive alcohol consumption, chronic infection with viral hepatitis, and other chronic liver diseases [4,5,6]. Liver biopsy is the gold standard for NAFLD diagnosis, as well as for NASH and hepatic fibrosis [7]. In histological terms, Nonalcoholic steatohepatitis (NASH), which is a subcategory of NAFLD, is characterized by the presence of hepatic steatosis, liver inflammation, and hepatocellular ballooning on biopsy in the absence of any chronic liver disease or alcohol consumption [7,8]. NASH prevalence in the general population is estimated to range from 1.5% to 6.4% [1].
2.1.2. Risk factors
NAFLD prevalence increases dramatically in diabetic and obese patients [9]. It has been estimated that approximately 70% of individuals suffering from Type 2 Diabetes Mellitus (T2DM) have NAFLD, with an additional 25% presenting with a more severe manifestation known as Nonalcoholic Steatohepatitis (NASH) [10,11]. Although evidence is limited, these patients appear to be at a higher risk of developing advanced fibrosis and hepatocellular carcinoma [12]. The prevalence of NAFLD in individuals with obesity has been estimated to range from 60-95% [13,14,15]. The presence of sleep apnea syndrome is independently associated with NAFLD, degree of steatosis, inflammation, and fibrosis [16]. NAFLD is also associated with consumption of western foods, including soft drinks, red meat, processed foods, and refined grains [17,18].
2.1.3. Diagnosis of NAFLD
The diagnosis of NAFLD can be confirmed through histological examinations post-biopsy or through imaging modalities such as magnetic resonance imaging (MRI) or ultrasound. Liver biopsy remains the gold standard for identifying and categorizing histological changes in NAFLD [19,20]. Histologically, NAFLD is defined by the presence of at least 5% of hepatic fat content. NASH within the context of NAFLD is clinically characterized by hepatocellular injury, lobular inflammation, and hepatocellular ballooning [21].
Given the potential risks associated with liver biopsy, imaging is generally the favored method for detecting NAFLD. Computed tomography (CT), MRI, and ultrasonography are common techniques employed to quantify hepatic fat in the body; however, these conventional imaging modalities can be limited due to a lack of sensitivity and specificity (in CT and ultrasonography), subjectivity (in MRI and ultrasonography), potential radiation-related hazards (in CT), and other confounding factors [22]. Differential diagnosis, particularly hepatic glycogenesis and glycogenic hepatopathy, are among the major confusing factors [23]. However, current developments in imaging, such as multi-parametric MRI, can aid in more effectively detecting hepatic fat. Indeed, multi-parametric MRI, particularly with the proton density fat fraction, has been developed. As a result, it has evolved into a virtual liver biopsy method that may be used to monitor patients during treatment and prevent needless biopsies [22]. This new imaging technique may be essential given the high and rising frequency of NAFLD.
2.1.4. Physiopathology
The progression of NAFLD is caused by a number of underlying factors, including accumulation of intrahepatic fat, adipose tissue malfunction, and de novo intrahepatic lipogenesis. Ultimately, this process is mainly attributed to insulin resistance [24]. Additionally, some lipid intermediates involved in NAFLD development, such as diacylglycerols and ceramides, have a higher propensity than others to lead to hepatic insulin resistance, feeding a vicious loop that results in a rise in NAFLD [25]. In fact, increasing levels of free fatty acids (FFA) in the blood and ectopic lipid buildup in the liver are related to insulin resistance, which can further encourage inflammation and endoplasmic reticulum stress, contributing to the vicious cycle of the insulin resistant state [26]. With inflammatory mediators like cytokines and adipokines having a fundamental role not only in inflammation but also in metabolic energy balance and immunological response, inflammation appears to have a significant role in both insulin resistance and NAFLD [27]. The increased presence of intracellular reactive oxygen species (ROS), which cause oxidative stress, is a major factor in the emergence of NAFLD. The primary ROS generators are NADPH Oxidase (NOX) enzymes, and it has been demonstrated that NAFLD and insulin resistance are associated with increased NOX activity because of hepatic lipid overload [28,29]. Additionally, it is well-known that obesity and poor eating practices increase the formation of ROS by disrupting the balance between their production and clearance, which contributes further to the emergence of insulin resistance and liver tissue damage, perpetuating the vicious cycle [28]. Since adiponectin is known to have anti-inflammatory properties and enhance hepatic insulin sensitivity, subjects with NAFLD often have lower plasma adiponectin concentrations than those without NAFLD [30].
2.1.5. Therapeutical approach
Lifestyle management is the foundation of NAFLD treatment, and mostly consists in improving diet and increasing exercise. Studies have shown that weight reduction of 7-10% is associated with a decrease in NAFLD-related inflammation [31]. A study has shown that bariatric surgery in the setting of weight loss results in histologic improvement of steatosis, steatohepatitis, and liver fibrosis in the majority of patients with NAFLD [32]. Given the importance of insulin resistance in the physiopathology of NAFLD, insulin-sensitizing drugs have been evaluated. Metformin has not shown convincing results in patients with NAFLD [33]. Statins are commonly taken by patients with NAFLD, but there are no studies demonstrating their efficacy in NAFLD. As for antioxidant therapies, vitamin E therapy tends to have a good results, but its long-term efficacy is still not well known [34]. Glucagon-like peptide-1 receptor agonists are a type of medication that can cause dose-dependent weight loss. Additionally, these drugs have been demonstrated to improve insulin resistance in the liver and adipose tissue, reduce de novo lipogenesis and lipolysis in fat tissue, and reduce oxidative stress [35,36,37,38,39]. In a randomized, double-blind, placebo-controlled, multicenter study lasting 48 weeks, liraglutide (a glucagon-like peptide-1 receptor agonist) was found to effectively and safely treat nonalcoholic steatohepatitis (NASH), leading to histological improvement [38].
2.2. Oxidative stress
2.2.1. Definition
Oxidative stress refers to several harmful processes that result from an imbalance between excessive formation of prooxidants and inadequate antioxidant defenses [42]. This imbalance leads to cell death and tissue damage. Classic prooxidants are the reactive oxygen species (superoxide, hydrogen peroxide, hydroxyl radical) and reactive nitrogen species (oxide nitrate, peroxynitrite). Under physiological conditions, ROS continue to be formed but are eliminated by cell scavenging systems (catalase, superoxide dismutase, glutathione). Studies have identified several circulating biomarkers of lipid peroxidation in patients with NAFLD/NASH [43,44,45,46] and disease severity was found to be at high levels of these markers [47]. More than 90% of patients with NAFLD were found to have an imbalance of oxygen reduction and decreased plasma antioxidant capacity [48].
ROS are generated at various sites, such as the mitochondrial respiratory chain, cytochrome P450, oxidative enzymes, and some heme proteins. ROS can also be caused by radiation, certain drugs, or UV radiation. Oxidation of fatty acids in the liver is a reaction that is one of the main sources of ROS. Beta-oxidation occurs in the mitochondria as well as in the peroxisome, whereas omega-oxidation occurs in the endoplasmic reticulum by cytochrome p450 [49].
2.2.2. Oxidative stress in NAFLD development
Although poorly understood, the progression of hepatic steatosis to NASH was initially explained by the two-hits hypothesis. The first cause is hepatic steatosis resulting from insulin resistance, which leads to lipogenesis in the liver and less than optimal export of fatty acids [50,51] and hormonal disorders such as the alteration of adiponectin and leptin [52,53]. When insulin resistance first occurs in muscle and adipose tissue, less glucose is delivered to these tissues, creating a catabolic state that results in the release of fatty acids into the circulation through peripheral fat. To compensate for hyperglycemia, the pancreas secretes more insulin and hyperinsulinemia results. Since the liver is relatively more sensitive to insulin, it enters an anabolic state and secretes and stores lipids [54]. The second hit is triggered by two types of factors, those that increase inflammatory cytokines that activate stellate cells and cause fibrosis and endoplasmic reticulum stress, and those that increase oxidative stress. [55,56,57,58,59]. Endoplasmic reticulum stress is involved in the development of steatosis and NASH [60]. In fact, ceramides, but also sphingolipids synthesized from saturated fatty acids, have been shown to be synthesized when saturated fatty acid content is increased. Ceramides induce hepatocyte apoptosis through endoplasmic reticulum stress and may induce mitochondrial fragmentation through interaction with mitochondrial fragmentation factor [61]. In addition, a large stream of free fatty acids, folded and unfolded proteins accumulate, leading to a process called unfolded protein response. The latter leads to a stress of the endoplasmic reticulum and thus to the production of ROS. [62].
ROS can attack molecules such as polyunsaturated fatty acids in the cell, producing aldehyde by-products such as 4-hydroxy-2-ninenal and malondialdehyde [63,64]. Since the mitochondrial membrane is composed of polyunsaturated fatty acids for the proper assembly of respiratory complexes, the production of these two aldehyde products impairs mitochondrial function. A study has shown that ROS can directly attack hepatic mitochondrial DNA, leading to a decrease in the synthesis of proteins important for the respiratory chain. It is worth noting that a new theory of "multiple hits" has more recently emerged (Figure 2). It hypothesizes that in addition to the two steps described above, there are genetic and epigenetic mutations and factors from the gut microbiota that would play a role in the pathogenic development of NAFLD [65]. Finally, one study found that production of ROS, lipid peroxidation, alteration of the mitochondrial respiratory chain, and changes in mitochondrial membrane composition are present in all patients with hepatic steatosis and tend to progress to NAFLD [66].
At the same time, nutrition also plays an important role. Chronic malnutrition, especially based on high fat and low glycemic index carbohydrates, can stimulate intracellular liver pathways leading to the development of oxidative stress. One study has shown that saturated fat consumption plays an important role in steatosis and NAFLD as a mechanism. Another study found that the antioxidant components of the diet were significantly reduced in patients with NASH compared to healthy people [67]. There are mechanisms to balance the accumulation of triglycerides and fatty acids in patients with a high-fat diet. Thus, there is an increase in mitochondrial palmitoyl transfer carnitine and mitochondrial uncoupling proteins, which lower plasma levels of ROS and thus play a protective role in the development of NAFLD [61].
A decrease in the antioxidant activity of catalase and superoxide dismutase has also been observed [68]. Indeed, ROS, produced in NAFLD, can degrade antioxidant molecules and inhibit antioxidant enzymes [69]. In addition, nuclear factor-2, a transcriptional regulator of antioxidant proteins, is completely degraded, further worsening the redox balance [70].
2.3. Carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1)
2.3.1. Definition and Generalities
Carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) is a transmembrane glycoprotein, a member of the immunoglobulin superfamily and more precisely it belongs to the carcinoembryonic antigen (CEA) family [71]. In the liver, CEACAM1 is essentially expressed on epithelial cells and endothelial cells participating in the conservation of the epithelial polarity in hepatocytes during their differentiation. Originally, in humans the gene encoding CEACAM1 protein is located on the long arm of the chromosome 19 [72].
When it comes to splicing, human CEACAM1 gene generates twelve splice variants [73]. Given this diversity in splicing, it is not a surprise that it has a lot of different roles which can differ according to its location and its stimulus. Its roles include arrangement of tissue structure, angiogenesis, apoptosis, tumor suppression, metastasis and the regulation of immune response [71]. It is important to note that the major CEACAM1 isoforms are made of 1 transmembrane domain with 3 or 4 extracellular domains, including the N-terminal IgV-like domain, and a cytoplasmic tail, which can be short or long. To resume, there are the CEACAM1-4L and the CEACAM1-4S, which both have 4 extracellular domains but with a long cytoplasmic tail and a short one respectively, and the CEACAM1-3L and the CEACAM1-3S, which have 3 extracellular domains with a long cytoplasmic tail and a short one respectively [71].
2.3.2. CEACAM1 role in metabolic balance and NAFLD/NASH
Here, we focus on the role of CEACAM1 in the development of nonalcoholic fatty liver disease, including NAFLD, NASH and liver fibrosis on the molecular aspect.
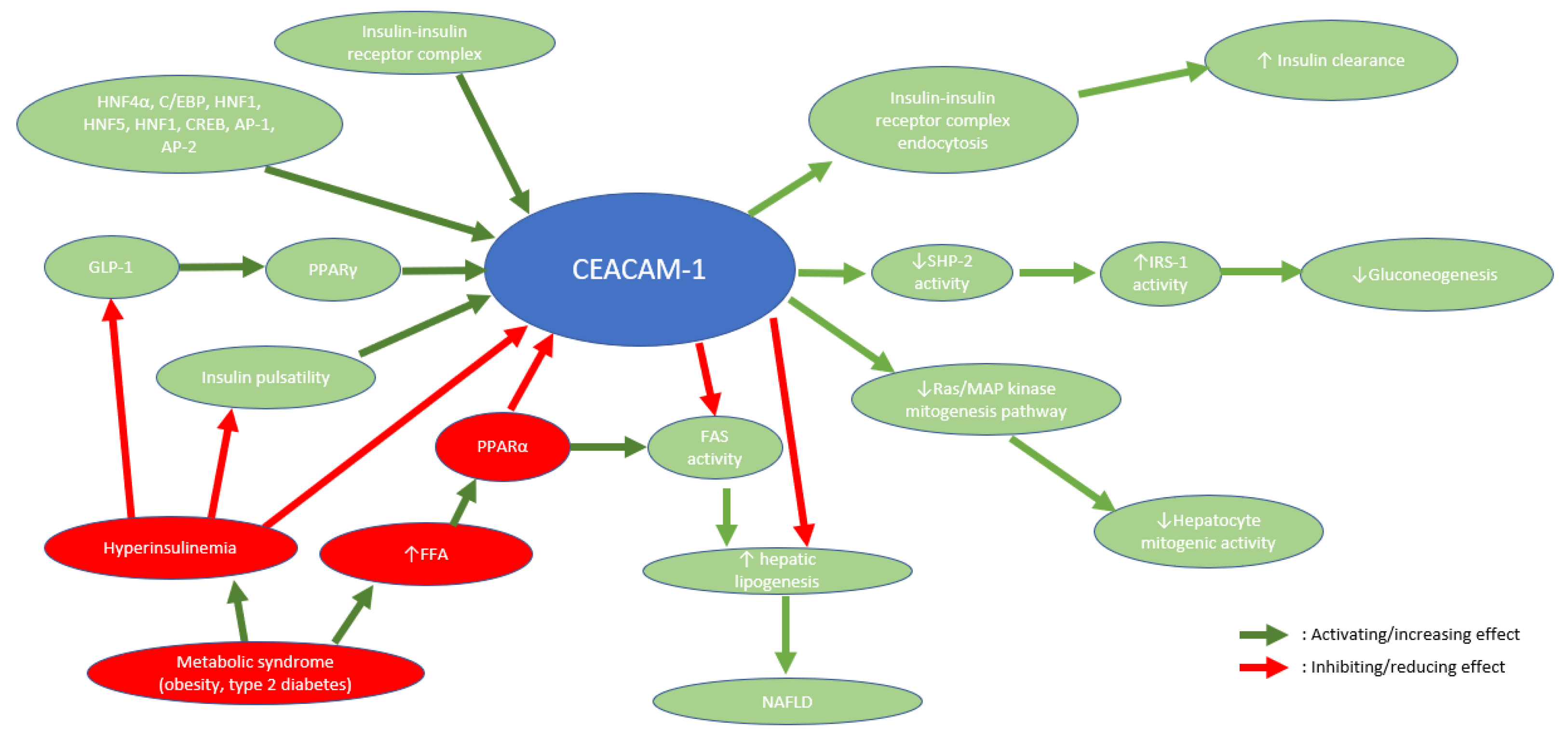
First, this protein is known to have a key role in hepatocyte differentiation, especially in early life where CEACAM1 is more expressed in hepatocytes. A major role in hepatic regeneration has also been noticed in rat livers after partial hepatectomy where CEACAM1 expression was analyzed through indirect fluorescence [74]. On the genetic aspect, CEACAM1 expression is essentially upregulated by the hepatocyte nuclear factor 4α (HNF4α) and CEACAM1’s promoter studies in rats have shown to have binding sites for factors including hepatonuclear factor 5 (HNF5), CCAAT/enhancer binding protein (C/EBP), hepatonuclear factor 1 (HNF1), glucocorticoids, cAMP-response element binding protein (CREB) and activator protein 1 and 2 (AP-1 and AP-2) [75]. CEACAM1 acts as a tumor suppressor, in tissue organization and in hepatocyte differentiation. These different functions have been known for a long time but CEACAM1’s role in metabolic processes, although suspected, has only recently been studied and explored.
Indeed, CEACAM1 plays a central function in insulin transduction since it has been reported as a substrate of the insulin receptor specifically in liver, in opposition to the other insulin-sensitive tissues (adipose tissue and skeletal muscle) [76].
Once insulin reaches the hepatocyte, it enhances CEACAM1 action on the hepatocyte mitogenic activity by downregulating it. In fact, the action of CEACAM1, phosphorylated by the insulin receptor, is allowed through its binding to SH2-containg adapter protein (Shc), which becomes sequestered by CEACAM1. Since Shc is a coupler protein, this has the effect of reducing the coupling of the growth factor receptor-bound protein 2 (Grb2) to the insulin receptor and down-regulates the Ras/MAP kinase mitogenesis pathway resulting in a reduced mitogenic activity. Another consequence of the Shc sequestering by the CEACAM1 is the reduced activity of the phosphoinositide 3’kinase (PI3K) and protein kinase B (AKT). This pathway is usually made possible through the binding between insulin receptor substrate 1 (IRS-1) and PI3k after IRS-1 is phosphorylated by the insulin receptor and leads to cell proliferation. CEACAM1’s binding to Shc competes with IRS-1 phosphorylation by the insulin receptor, hence downregulating cell proliferation in hepatocytes [77].
Regarding insulin clearance, CEACAM1-L plays a central role, orchestrating multiple intracellular reactions initiated by insulin binding to its receptor. This binding causes CEACAM1-L phosphorylation, allowing its indirect attachment to the insulin receptor in order to enhance the endocytosis, degradation and clearance of the insulin-insulin receptor complex [78].
SHP-2 is a tyrosine phosphatase which is a protein that dephosphorylates and inhibits insulin receptor substrate 1 (IRS-1) action. CEACAM1-L attaches itself to the SHP-2 tyrosine phosphatase, preventing its negative action on IRS-1, which results in a sustained action of IRS-1 in the hepatocyte. IRS-1 sustained action is also favored by the insulin-insulin receptor complex endocytosis because of insulin receptor tyrosine kinase prolonged exposure to IRS-1 [78,79].
These mechanisms point out the major role of CEACAM1 in insulin degradation and in insulin clearance in hepatocytes since hepatic insulin removal is responsible for clearing approximatively 80 % of the total insulin synthetized by the pancreatic β cells [80]. Mutation on the phosphorylation site in CEACAM1-L has been shown to cause hyperinsulinemia, glucose resistance and leads ultimately to chronic fatty hepatopathy in mouse models [81].
The role and impact of CEACAM1 protein in the development of chronic NAFLD is intimately linked to its function as an insulin clearer. In non-mutated individuals, it has been hypothesized that chronic hyperinsulinemia and impaired insulin secretion pulsatility reduce CEACAM1 phosphorylation and action, creating a vicious cycle [82,83]. There is clear evidence that hyperinsulinemia in patients with NAFLD correlates much more with impaired insulin clearance than with increased insulin secretion [84], implying that CEACAM1 is one of the major key factors linking insulin resistance, hyperinsulinemia, and fatty liver disease.
Intrahepatic lipogenesis is essentially stimulated by insulin by increasing sterol regulatory element-binding proteins (SREBPs) in hepatocytes, which promotes cholesterol, free fatty acids, triglycerides and phospholipids synthesis and uptake [85]. SREBPs are also essential for enzymes expression, for example glucokinase, liver-type pyruvate kinase (LPK), fatty acid synthase (FAS), and acetyl-CoA-carboxylase (ACC) which are required for lipogenesis [86]. Insulin concentration is much higher in the portal vein than in the peripheral blood circulation [80], therefore the liver seems to be more likely increase de novo lipogenesis through insulin action. However, despite the liver expressing more lipogenic genes, the activity of the fatty acid synthase (FAS) in normal insulinemic individuals is nearly imperceptible and it is mainly due to insulin release pulsatility [83]. This allows an acute and fast action of insulin on the hepatocytes, phosphorylating CEACAM1-L protein, permitting insulin clearance through endocytosis of the insulin-insulin receptor complex. As a reminder, insulin is usually secreted by the β cells in two phases, a first secretion peak phase following glucose intake, then a second deferred phase that helps bring glycemia back to normal blood concentration [87]. In individuals with chronic hyperinsulinemia, this mechanism is impaired and insulin secretion pulsatility is progressively diminished. Since CEACAM1 is the main factor for insulin clearance, and impaired insulin clearance correlates more with hyperinsulinemia than with insulin secretion [84], it is safe to deduce that CEACAM1 impairment leads to a hyperinsulinemic state.
Interestingly, in patients with Type 1 Diabetes (T1D), the only treatment relies on exogenous insulin administration in the peripheral circulation. Not only is it unlikely that all the administrated insulin reaches the liver, but also this causes altered dynamic of insulin delivery, in opposition to endogenous insulin production [84,88]. This altered hepatic exposure to insulin is congruent with the loss of the insulin pulsatility effect, described above. Knowing the importance of the insulin pulsatility effect on the proper functioning of CEACAM1, patients with T1D are probably more likely to have CEACAM1 impairment [82].
CEACAM1 also plays a critical role in hepatic lipogenesis. As explained, CEACAM1-L in hepatocytes is activated through its phosphorylation mediated by the insulin-insulin receptor complex. Phosphorylated CEACAM1-L binds to the fatty acid synthase (FAS) and suppresses its activity by sequestering it. FAS is an enzymatic complex essential for enhancing fatty acids synthesis, and its inhibition by the activated CEACAM1-L leads to decreased liver lipogenesis [89].
Obese individuals have excessive white adipose tissue and, hence, high levels of circulating FFA [90]. FFA action on hepatocytes will promote and activate peroxisome proliferator-activated receptor α (PPARα) [91], which is a nuclear receptor acting as a ligand-activated transcriptional factor regulating the expression of crucial genes participating in fatty acid beta-oxidation [92]. PPARα reduces one of CEACAM1 promoter activity, thus downregulating the CEACAM1 protein transcription by decreasing CEACAM1-mRNA action [91]. Individuals with metabolic syndrome tend to have more FFA and thus have a much lower CEACAM1 activity. Whenever CEACAM1 activity is reduced by 50% or more, its sequestration of FAS is greatly diminished, and insulin clearance is also impaired inducing hyperinsulinemia, then leading to insulin resistance and to increased FAS activity [76]. This ultimately leads to steatosis and to NAFLD development and puts forward the crucial importance of CEACAM1 protein in the metabolic balance and in the buildup of chronic fatty liver disease.
CEACAM1 protein synthesis in hepatocytes is promoted by the peroxisome proliferator-activated receptor γ (PPARγ) [93]. PPARγ expression and action is induced and upregulated by incretin hormones, such as glucagon-like peptide 1 (GLP-1) [80,93], the activity of which has been shown to be reduced in patients with chronic hyperinsulinemia [94]. This suggests that the reduced activity of GLP-1 implies a downregulation and, thus, a reduced activity of CEACAM1, but also implies that a treatment with GLP-1 receptor agonists could be a possible therapy to increase CEACAM1 protective activity in liver.
3. Discussion
NAFLD has become a rising subject of interest during recent years. It is known now that there are several complex biological mechanisms leading to NAFLD. Understanding these mechanisms is key to help further research towards potential therapeutic targets.
Oxidative stress is a factor in the development of NAFLD and occurs when there is an imbalance between pro-oxidants and antioxidants. This can lead to cellular damage due to ROS. Treatments targeting oxidative stress, such as vitamins C and E in combination with statins, induce improved redox status in NAFLD patients. Coffee may help protect against NAFLD by regulating metabolism, fat accumulation, and providing polyphenol antioxidants. Metformin appears to slow NAFLD progression by altering the gut microbiota and promoting ROS clearance through SOD. GLP-1 receptor agonists appear to have antioxidant effects by activating antioxidant genes. However, there are still multiple therapeutic approaches to be studied given the multi-mechanical roles of ROS in NAFLD physiopathology [61].
CEACAM1, a transmembrane protein, is critical for many different types of processes, including tissue organization, metastasis, immune response control, and, more significantly here, metabolic homeostasis. It has been shown that its main effect regarding the liver is rather protective and its actions prevent fat deposition in the liver [80]. CEACAM1 is downregulated in individuals with metabolic syndrome and its downregulation enhances a vicious circle leading to the worsening of NAFLD. Since CEACAM1 plays a key role in the control of the metabolic aspect in NAFLD, its actions and the pathways it enhances could be interesting targets for therapeutic approaches. A stimulation of CEACAM1 through the known mechanisms (Figure 1) or through a new molecule is yet to be tested in NAFLD in order to assess its healing potential [61].
Despite all the current research performed in the field, there is no highly specific blood marker for NAFLD put in place yet. A blood marker or a constellation of blood markers correlated to CEACAM1 activity could potentially be interesting to help assess the NAFLD presence or severity.
4. Conclusion
ROS and CEACAM1 activity are important factors to understand NAFLD development in individuals suffering from the metabolic syndrome (obesity, insulin resistance, T2D). Their understanding at the molecular level is crucial to unravel potential therapies targeting these different molecular pathways.
Limiting ROS activation via antioxidant therapy or via appropriate diet has shown promising results, giving hope for other molecular alternatives that reduce ROS activation. There is strong evidence that CEACAM1 upregulation reduces fat deposition in the liver and optimizes insulin clearance, limiting the hyperinsulinemic state. To that matter, some potential molecules such as GLP-1 receptor agonists, used to treat T2D, or other molecules have yet to be further studied in order to judge their efficiency on NAFLD/NASH.
Understanding these molecular mechanisms involved in NAFLD development open new therapeutic fields of interest, especially since NAFLD remains the most frequent chronic liver disease in the general population [1].
References
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Powell EE, Wong VWS, Rinella M. Nonalcoholic fatty liver disease. The Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Younossi ZM, Gramlich T, Matteoni CA, Boparai N, McCullough AJ. Nonalcoholic fatty liver disease in patients with type 2 diabetes. Clin Gastroenterol Hepatol. 2004, 2, 262–265. [Google Scholar] [CrossRef]
- Berzigotti A, Tsochatzis E, Boursier J, Castera L, Cazzagon N, Friedrich-Rust M, et al. EASL Clinical Practice Guidelines on non-invasive tests for evaluation of liver disease severity and prognosis – 2021 update. J Hepatol. 2021, 75, 659–689. [Google Scholar] [CrossRef] [PubMed]
- Marjot T, Moolla A, Cobbold JF, Hodson L, Tomlinson JW. Nonalcoholic Fatty Liver Disease in Adults: Current Concepts in Etiology, Outcomes, and Management. Endocr Rev. 2020, 41, 66–117. [Google Scholar] [CrossRef] [PubMed]
- Zhou JH, Cai JJ, She ZG, Li HL. Noninvasive evaluation of nonalcoholic fatty liver disease: Current evidence and practice. World J Gastroenterol. 2019, 25, 1307–1326. [Google Scholar] [CrossRef]
- Brunt EM, Janney CG, Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef]
- Rich NE, Oji S, Mufti AR, Browning JD, Parikh ND, Odewole M, et al. Racial and Ethnic Disparities in Nonalcoholic Fatty Liver Disease Prevalence, Severity, and Outcomes in the United States: A Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol. 2018, 16, 198–210. [Google Scholar] [CrossRef]
- Bril F, Cusi K. Nonalcoholic Fatty Liver Disease. Endocrinol Metab Clin North Am. 2016, 45, 765–781. [Google Scholar] [CrossRef]
- Williamson RM, Price JF, Glancy S, Perry E, Nee LD, Hayes PC, et al. Prevalence of and Risk Factors for Hepatic Steatosis and Nonalcoholic Fatty Liver Disease in People With Type 2 Diabetes: the Edinburgh Type 2 Diabetes Study. Diabetes Care. 2011, 34, 1139–1144. [Google Scholar] [CrossRef]
- El-serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004, 126, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, Ahmed A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol. 2017, 23, 8263–8276. [Google Scholar] [CrossRef] [PubMed]
- Pallayova M, Taheri S. Nonalcoholic fatty liver disease in obese adults: clinical aspects and current management strategies: Nonalcoholic fatty liver disease in obese adults. Clin Obes. 2014, 4, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli L, Beaugrand M. Transient elastography in nonalcoholic fatty liver disease. Ann Hepatol. 2012, 11, 172–178. [Google Scholar] [CrossRef]
- Jin S, Jiang S, Hu A. Association between obstructive sleep apnea and nonalcoholic fatty liver disease: a systematic review and meta-analysis. Sleep Breath 2018, 22, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Kalafati IP, Borsa D, Dimitriou M, Revenas K, Kokkinos A, Dedoussis GV. Dietary patterns and nonalcoholic fatty liver disease in a Greek case–control study. Nutrition. 2019, 61, 105–110. [Google Scholar] [CrossRef]
- Yang CQ, Shu L, Wang S, Wang JJ, Zhou Y, Xuan YJ, et al. Dietary Patterns Modulate the Risk of Nonalcoholic Fatty Liver Disease in Chinese Adults. Nutrients. 2015, 7, 4778–4791. [Google Scholar] [CrossRef]
- de Vries M, Westerink J, Kaasjager KHAH, de Valk HW. Prevalence of Nonalcoholic Fatty Liver Disease (NAFLD) in Patients With Type 1 Diabetes Mellitus: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab. 2020, 105, 3842–3853. [Google Scholar] [CrossRef]
- Cotter TG, Rinella M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Mertens J, Van Gaal LF, Francque SM, De Block C. NAFLD in type 1 diabetes: overrated or underappreciated? Ther Adv Endocrinol Metab. 2021, 12, 204201882110555. [Google Scholar] [CrossRef] [PubMed]
- Kinner S, Reeder SB, Yokoo T. Quantitative Imaging Biomarkers of NAFLD. Dig Dis Sci. 2016, 61, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Mertens J, De Block C, Spinhoven M, Driessen A, Francque SM, Kwanten WJ. Hepatopathy Associated With Type 1 Diabetes: Distinguishing Nonalcoholic Fatty Liver Disease From Glycogenic Hepatopathy. Front Pharmacol. 2021, 12, 768576. [Google Scholar] [CrossRef] [PubMed]
- Gariani K, Philippe J, Jornayvaz FR. Nonalcoholic fatty liver disease and insulin resistance: From bench to bedside. Diabetes Metab. 2013, 39, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Gariani K, Jornayvaz FR. NAFLD: From Mechanisms to Therapeutic Approaches. Biomedicines 2022, 10, 1747. [Google Scholar]
- Asrih M, Jornayvaz FR. Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol Cell Endocrinol. 2015, 418, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Asrih M, Jornayvaz FR. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J Endocrinol. 2013, 218, R25–R36. [Google Scholar] [CrossRef]
- Nascè A, Gariani K, Jornayvaz FR, Szanto I. NADPH Oxidases Connecting Fatty Liver Disease, Insulin Resistance and Type 2 Diabetes: Current Knowledge and Therapeutic Outlook. Antioxidants 2022, 11, 1131. [Google Scholar] [CrossRef]
- Delli Bovi AP, Marciano F, Mandato C, Siano MA, Savoia M, Vajro P. Oxidative Stress in Nonalcoholic Fatty Liver Disease. An Updated Mini Review. Front Med. 2021, 8, 595371. [Google Scholar] [CrossRef]
- Shabalala SC, Dludla PV, Mabasa L, Kappo AP, Basson AK, Pheiffer C, et al. The effect of adiponectin in the pathogenesis of nonalcoholic fatty liver disease (NAFLD) and the potential role of polyphenols in the modulation of adiponectin signaling. Biomed Pharmacother. 2020, 131, 110785. [Google Scholar]
- Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Mummadi RR, Kasturi KS, Chennareddygari S, Sood GK. Effect of Bariatric Surgery on Nonalcoholic Fatty Liver Disease: Systematic Review and Meta-Analysis. Clin Gastroenterol Hepatol. 2008, 6, 1396–402. [Google Scholar] [CrossRef] [PubMed]
- Li Y, Liu L, Wang B, Wang J, Chen D. Metformin in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Biomed Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N Engl J Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Gupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, et al. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef]
- Armstrong MJ, Hull D, Guo K, Barton D, Hazlehurst JM, Gathercole LL, et al. Glucagon-like peptide 1 decreases lipotoxicity in nonalcoholic steatohepatitis. J Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef]
- Gastaldelli A, Gaggini M, Daniele G, Ciociaro D, Cersosimo E, Tripathy D, et al. Exenatide improves both hepatic and adipose tissue insulin resistance: A dynamic positron emission tomography study. Hepatology 2016, 64, 2028–2037. [Google Scholar] [CrossRef]
- Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with nonalcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. The Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N Engl J Med. 2021, 384, 989–1002. [Google Scholar] [CrossRef]
- Newsome PN, Allison ME, Andrews PA, Auzinger G, Day CP, Ferguson JW, et al. Guidelines for liver transplantation for patients with nonalcoholic steatohepatitis. Gut. 2012, 61, 484–500. [Google Scholar] [CrossRef]
- Taylor RS, Taylor RJ, Bayliss S, Hagström H, Nasr P, Schattenberg JM, et al. Association Between Fibrosis Stage and Outcomes of Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 2020, 158, 1611–1625. [Google Scholar] [CrossRef] [PubMed]
- Robertson G, Leclercq I, Farrell GC. II. Cytochrome P -450 enzymes and oxidative stress. Am J Physiol-Gastrointest Liver Physiol. 2001, 281, G1135–G1139. [Google Scholar] [CrossRef] [PubMed]
- Świderska M, Maciejczyk M, Zalewska A, Pogorzelska J, Flisiak R, Chabowski A. Oxidative stress biomarkers in the serum and plasma of patients with nonalcoholic fatty liver disease (NAFLD). Can plasma AGE be a marker of NAFLD? Oxidative stress biomarkers in NAFLD patients. Free Radic Res. 2019, 53, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Irie M, Sohda T, Iwata K, Kunimoto H, Fukunaga A, Kuno S, et al. Levels of the Oxidative Stress Marker γ-Glutamyltranspeptidase at Different Stages of Nonalcoholic Fatty Liver Disease. J Int Med Res. 2012, 40, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Yesilova Z, Yaman H, Oktenli C, Ozcan A, Uygun A, Cakir E, et al. Systemic Markers of Lipid Peroxidation and Antioxidants in Patients with Nonalcoholic Fatty Liver Disease. Am J Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Videla LA, Rodrigo R, Araya J, Poniachik J. Oxidative stress and depletion of hepatic long-chain polyunsaturated fatty acids may contribute to nonalcoholic fatty liver disease. Free Radic Biol Med. 2004, 37, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Liu S, Shi W, Li G, Jin B, Chen Y, Hu H, et al. Plasma reactive carbonyl species levels and risk of nonalcoholic fatty liver disease: Reactive carbonyl species and NAFLD. J Gastroenterol Hepatol. 2011, 26, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Loguercio C, De Girolamo V, de Sio I, Tuccillo C, Ascione A, Baldi F, et al. Nonalcoholic fatty liver disease in an area of southern Italy: main clinical, histological, and pathophysiological aspects. J Hepatol. 2001, 35, 568–574.
- Gambino R, Musso G, Cassader M. Redox Balance in the Pathogenesis of Nonalcoholic Fatty Liver Disease: Mechanisms and Therapeutic Opportunities. Antioxid Redox Signal. 2011, 15, 1325–1365. [Google Scholar] [CrossRef]
- Lavoie JM, Gauthier MS. Regulation of fat metabolism in the liver: link to nonalcoholic hepatic steatosis and impact of physical exercise. Cell Mol Life Sci. 2006, 63, 1393–1409. [Google Scholar] [CrossRef]
- Tessari P, Coracina A, Cosma A, Tiengo A. Hepatic lipid metabolism and nonalcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2009, 19, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Parola M, Marra F. Adipokines and Redox Signaling: Impact on Fatty Liver Disease. Antioxid Redox Signal. 2011, 15, 461–483. [Google Scholar] [CrossRef] [PubMed]
- Larter CZ, Chitturi S, Heydet D, Farrell GC. A fresh look at NASH pathogenesis. Part 1: The metabolic movers. J Gastroenterol Hepatol. 2010, 25, 672–690. [Google Scholar] [CrossRef] [PubMed]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Sanyal AJ, Campbell–Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Chitturi S, Farrell GC. Etiopathogenesis of Nonalcoholic Steatohepatitis. Semin Liver Dis. 2001, 21, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, Wakasa K. In situ detection of lipid peroxidation and oxidative DNA damage in nonalcoholic fatty liver diseases. J Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Marchesini G, Marzocchi R, Agostini F, Bugianesi E. Nonalcoholic fatty liver disease and the metabolic syndrome. Curr Opin Lipidol. 2005, 16, 421–427. [Google Scholar] [CrossRef]
- Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol-Gastrointest Liver Physiol. 2006, 290, G1339–G1346.
- Pagliassotti, MJ. Endoplasmic Reticulum Stress in Nonalcoholic Fatty Liver Disease. Annu Rev Nutr. 2012, 32, 17–33. [Google Scholar] [CrossRef]
- Smirne C, Croce E, Di Benedetto D, Cantaluppi V, Comi C, Sainaghi PP, et al. Oxidative Stress in Nonalcoholic Fatty Liver Disease. Livers 2022, 2, 30–76. [Google Scholar] [CrossRef]
- Sozio M, Liangpunsakul S, Crabb D. The Role of Lipid Metabolism in the Pathogenesis of Alcoholic and Nonalcoholic Hepatic Steatosis. Semin Liver Dis. 2010, 30, 378–90. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991, 11, 81–128. [Google Scholar] [CrossRef] [PubMed]
- Gardner, HW. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic Biol Med. 1989, 7, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Takaki A, Kawai D, Yamamoto K. Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Nonalcoholic Steatohepatitis (NASH). Int J Mol Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed]
- Meex RCR, Blaak EE. Mitochondrial Dysfunction is a Key Pathway that Links Saturated Fat Intake to the Development and Progression of NAFLD. Mol Nutr Food Res. 2021, 65, 1900942. [Google Scholar] [CrossRef]
- Erhardt A, Stahl W, Sies H, Lirussi F, Donner A, Häussinger D. Plasma levels of vitamin E and carotenoids are decreased in patients with nonalcoholic steatohepatitis (NASH). Eur J Med Res. 2011, 16, 76. [Google Scholar] [CrossRef]
- Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Nonalcoholic fatty liver disease. Free Radic Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. 2020, 152, 116–41. [Google Scholar] [CrossRef]
- Reccia I, Kumar J, Akladios C, Virdis F, Pai M, Habib N, et al. Nonalcoholic fatty liver disease: A sign of systemic disease. Metabolism. 2017, 72, 94–108. [Google Scholar] [CrossRef]
- Horst A, Najjar S, Wagener C, Tiegs G. CEACAM1 in Liver Injury, Metabolic and Immune Regulation. Int J Mol Sci. 2018, 19, 3110. [Google Scholar] [CrossRef] [PubMed]
- Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, et al. Ensembl 2020. Nucleic Acids Res. 2019, gkz966. [Google Scholar]
- Kammerer R, Zimmermann W. Coevolution of activating and inhibitory receptors within mammalian carcinoembryonic antigen families. BMC Biol. 2010, 8, 12. [Google Scholar]
- Odin P, Öbrink B. Dynamic expression of the cell adhesion molecule cell-CAM 105 in fetal and regenerating rat liver. Exp Cell Res. 1986, 164, 103–14. [Google Scholar] [CrossRef] [PubMed]
- Najjar SM, Boisclair YR, Nabih ZT, Philippe N, Imai Y, Suzuki Y, et al. Cloning and Characterization of a Functional Promoter of the Rat pp120 Gene, Encoding a Substrate of the Insulin Receptor Tyrosine Kinase. J Biol Chem. 1996, 271, 8809–17. [Google Scholar] [CrossRef] [PubMed]
- Najjar SM, Caprio S, Gastaldelli A. Insulin Clearance in Health and Disease. Annu Rev Physiol. 2023, 85, 363–81. [Google Scholar] [CrossRef] [PubMed]
- Poy MN, Ruch RJ, Fernström MA, Okabayashi Y, Najjar SM. Shc and CEACAM1 Interact to Regulate the Mitogenic Action of Insulin. J Biol Chem. 2002, 277, 1076–84. [Google Scholar] [CrossRef]
- Boucher J, Kleinridders A, Kahn CR. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb Perspect Biol. 2014, 6, a009191–a009191. [Google Scholar] [CrossRef]
- Yousef A, Behiry E, Abd Allah W, Hussien A, Abdelmoneam A, Imam M, et al. IRS-1 genetic polymorphism (r.2963G>A) in type 2 diabetes mellitus patients associated with insulin resistance. Appl Clin Genet. 2018, 11, 99–106. [Google Scholar] [CrossRef]
- Najjar SM, Perdomo G. Hepatic Insulin Clearance: Mechanism and Physiology. Physiology 2019, 34, 198–215. [Google Scholar] [CrossRef]
- Poy MN, Yang Y, Rezaei K, Fernström MA, Lee AD, Kido Y, et al. CEACAM1 regulates insulin clearance in liver. Nat Genet. 2002, 30, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Memaj P, Jornayvaz FR. Nonalcoholic fatty liver disease in type 1 diabetes: Prevalence and pathophysiology. Front Endocrinol. 2022, 13, 1031633. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko AV, Liuwantara D, Gurlo T, Kirakossian D, Dalla Man C, Cobelli C, et al. Pulsatile Portal Vein Insulin Delivery Enhances Hepatic Insulin Action and Signaling. Diabetes 2012, 61, 2269–2279. [Google Scholar] [CrossRef]
- Bril F, Lomonaco R, Orsak B, Ortiz-Lopez C, Webb A, Tio F, et al. Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology 2014, 59, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Dif N, Euthine V, Gonnet E, Laville M, Vidal H, Lefai E. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem. J. 2006, 400, 179–188. [Google Scholar] [CrossRef]
- DeBose-Boyd RA, Ye J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Laurenti MC, Matveyenko A, Vella A. Measurement of Pulsatile Insulin Secretion: Rationale and Methodology. Metabolites 2021, 11, 409. [Google Scholar] [CrossRef]
- Rojano-Toimil A, Rivera-Esteban J, Manzano-Nuñez R, Bañares J, Martinez Selva D, Gabriel-Medina P, et al. When Sugar Reaches the Liver: Phenotypes of Patients with Diabetes and NAFLD. J Clin Med. 2022, 11, 3286. [Google Scholar] [CrossRef]
- Najjar SM, Yang Y, Fernström MA, Lee SJ, DeAngelis AM, Rjaily GAA, et al. Insulin acutely decreases hepatic fatty acid synthase activity. Cell Metab. 2005, 2, 43–53. [Google Scholar] [CrossRef]
- Camastra S, Ferrannini E. Role of anatomical location, cellular phenotype and perfusion of adipose tissue in intermediary metabolism: A narrative review. Rev Endocr Metab Disord. 2022, 23, 43–50. [Google Scholar] [CrossRef]
- Ramakrishnan SK, Khuder SS, Al-Share QY, Russo L, Abdallah SL, Patel PR, et al. PPARα (Peroxisome Proliferator-activated Receptor α) Activation Reduces Hepatic CEACAM1 Protein Expression to Regulate Fatty Acid Oxidation during Fasting-refeeding Transition. J Biol Chem. 2016, 291, 8121–8129. [Google Scholar] [CrossRef] [PubMed]
- van Raalte DH, Li M, Pritchard PH, Wasan KM. Peroxisome Proliferator-Activated Receptor (PPAR)- : A Pharmacological Target with a Promising Future. Pharm Res. 2004, 21, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Ghadieh HE, Muturi HT, Russo L, Marino CC, Ghanem SS, Khuder SS, et al. Exenatide induces carcinoembryonic antigen-related cell adhesion molecule 1 expression to prevent hepatic steatosis. Hepatol Commun. 2018, 2, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Salehi M, Aulinger B, Prigeon RL, D’Alessio DA. Effect of Endogenous GLP-1 on Insulin Secretion in Type 2 Diabetes. Diabetes 2010, 59, 1330–1337. [Google Scholar] [CrossRef]
Figure 2.
Multiple-hits hypothesis of NAFLD pathogenesis.
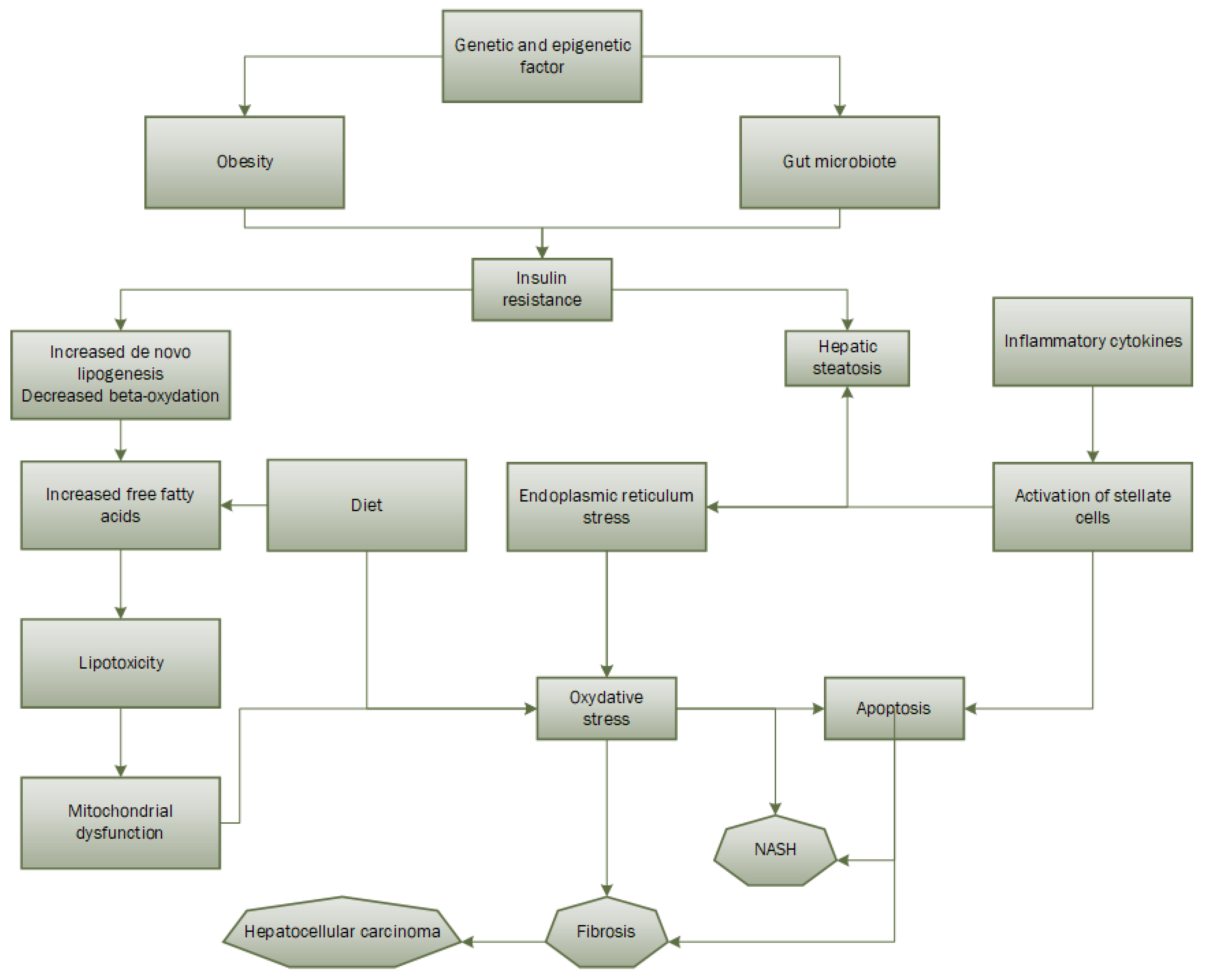
Figure 1.
Roles and actions of CEACAM1 as well as the factors influencing the functioning of CEACAM1.
Figure 1.
Roles and actions of CEACAM1 as well as the factors influencing the functioning of CEACAM1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



