
Submitted:
17 May 2023
Posted:
17 May 2023
You are already at the latest version
Abstract
The current classification of acute myeloid leukemia (AML) relies largely on genomic alterations. AML with mutated Nucleophosmin1 (NPM1-mut) is the largest of the genetically defined groups, involving about 30% of adult AMLs and is currently recognized as a distinct entity in the actual AML classifications. NPM1-mut AML usually occurs in de novo AML and is associated predominantly with a normal karyotype and relatively favorable prognosis. However, NPM1-mut AMLs are genetically, transcriptionally, and phenotypically heterogeneous. Furthermore, NPM1-mut is a clinically heterogenous group. Recent studies have in part clarified the consistent heterogeneities of these AMLs and have strongly supported the need for an additional stratification aiming to improve the therapeutic response of the different subgroups of NPM1-mut AML patients.
Keywords:
acute myeloid leukemia
; genetic classification
; mutational profiling
; transcriptome analysis
; prognostic stratification
; clonal evolution
1. Introduction
The development of the techniques for analysis of genome have greatly contributed to the development of a detailed molecular classification of acute myeloid leukemia. In 2016, it was proposed a first genomic classification of AMLs, that distinguishes 11 molecular subtypes, each with peculiar diagnostic molecular features [1]. More recently, according to the mutational profile and to cytogenetic analysis, this classification was updated and revised, supporting the existence of 16 molecular classes [2]. These molecular classifications have a relevant role in the diagnosis and in the treatment of AML patients and have been included in recent internationally accepted systems of leukemic classification or risk stratification, such as the 2022 World Health Organization (WHO) classification [3], the new International Consensus Classification (ICC) [4] and the European Leukemia Net (ELN) risk stratification [5]. All these classifications prioritize the role of genetic alterations to establish diagnosis and prognosis, to have criteria for definition and evaluation of minimal residual disease and, in some instances, also the indication of the optimal treatment.
Mutations at the level of the nucleophosmin 1 (NPM1) represent one of the most common gene mutations (25-30% of cases) observed in adult AML patients [6]. NPM1 encodes a multifunctional protein, prominently localized at the level of the nucleolus, that shutles between the nucleus and cytoplasm; the mutant NPM1 protein is delocalized at the level of the cytoplasm [7]. The nuclear export is mediated by the interaction of mutant NPM1 protein with exportin 1 (XPO1), a nuclear transporter acting as a direct carrier mediating the export of proteins containing a nuclear export signal into the cytoplasm. The biochemical and functional properties of normal and mutant NPM1 protein have been recently reviewed by Falini et al. [7]. Usually, NPM1-mutant AMLs at diagnosis display high percentages of blasts, high white cell counts, increased extramedullary involvement and are commonly associated with a normal karyotype [7]. Only about 15% of these patients display an abnormal karyotype, with the most frequent chromosomal abnormalities being represented by +8, +4, del 9q and +21) [7]. The screening of a large cohort of 2426 NPM1-mut AML patients negative for FLT3-ITD or with low FLT3-ITD allelic ratio (NPM1-mut/FLT3-ITDneg/low) showed that 17.6% of these patients displayed an abnormal karyotype: 13.6% of patients with intermediate-risk and 3.4% with adverse-risk chromosomal abnormalities [8]. Overall survival and event-free survival were significantly reduced in patients with adverse-risk chromosomal abnormalities [8].
NPM1 mutations are heterogeneous and mostly localized at the level of exon 12 of the NPM1 gene [2]. NPM1 mutations are always heterozygous and are caused by 4-bp insertions inducing a frameshift mutation at C-terminus of NPM1 protein resulting in a loss of a tryptophan residues (w288 and W290 or W290 alone) and gain of a new nuclear export signal (NES) determining a disruption of the folded helix structure with loss of the nucleolar localization signal (NoLS): all these changes determine a shift towards nuclear export and cytoplasmic localization of NPM1 protein [2]. These two tryptophan resudues are responsible for the nucleolar localization signal and interaction with ribosomal DNA. According to the different types of NPM1 mutations, NPM1-mut AMLs are subdivided into three main subgroups: type A, characterized an insertion of TCTG between nucleotides 860 and 863 (69% of cases); type B, characterized by insertion of CATG between nucleotides 863 and 864 (11% of cases); type D, characterized by insertion of CCTG between nucleotides 863 and 864 [9]. Type A NPM1-mut AMLs were characterized by high frequency of DNMT3A mutations [9].
The effect of mutant NPM1 is dominant over the normal NPM1 allele, a phenomenon due to the formation of heterodimers between mutant and normal NPM1, delocalized at the level of cytoplasm [2]. The dominance of the mutant allele over the normal NPM1 allele is reinforced also through a preferential transcription of the mutant allele [10].
The mechanisms through which NPM1-mut causes leukemic transformation remain largely undetermined. In this context, a key role seems to be played by the dysregulation of developmental and stem cell-associated genes such as HOXA cluster genes and MEIS1, highly expressed in NPM1-mut AMLs. The high expression of HOX genes in NPM1-mut AML cells requires the presence of the delocalized NPM1 mutant protein; in fact, pharmacological inhibition of XPO1 relocalized NPM1 mutant protein in the nucleus, resulting in immediated downregulation of HOX gene expression, differentiation of AML cells and prolongation of the survival of NPM1-mut leukemic mice [11]. Two recent studies have clarified the mechanism through which NPM1-mut directly upregulates the expression of target genes. Thus, Uckelmann et al. have shown that NPM1-mut directly binds at the level of specific chromating gene targets, co-occupied by the histone methyltransferase KMT2A (MLL1); targeted degradation of NPM1 determines a rapid decrease in gene expression and of activating histone modifications at the level of target genes [12]. Wang et al. showed that NPM1-mut binds at the level of active gene promoters in NPM1-mut AML cells, including HOXA/B gene clusters and MEIS1; NPM1-mut sustains the transcriptional activation of these genes by inhibiting the activity of histone deacetylases [13]. Studies based on mouse leukemogenesis models have shown that NPM1 mutations favor leukemic transformation through a double mechanism: hyperctivation of NPM1 target genes MEIS1 and HOXA genes and induction of a condition of haploinsufficiency of NPM1-WT determing insuffcient level of normal NPM1 protein at the level of the nucleus and nucleolus [14].
2. The mutational landscape of NPM1-mutant AMLs
Given their high frequency, NPM1-mutant AMLs have been characterized in detail for their mutational profile, showing that these AMLs display in most of cases one or more than one associated co-mutations. Some of these co-mutations play a key role in NPM1-mut AML development and are prognostically relevant.
Figure 1.
Most recurrent co-mutations observed in adult AML patients. These co-mutations involve genes pertaining to DNA methylation, activated signaling and cohesin complex. The data are issued from Ivey et al [15].
Figure 1.
Most recurrent co-mutations observed in adult AML patients. These co-mutations involve genes pertaining to DNA methylation, activated signaling and cohesin complex. The data are issued from Ivey et al [15].
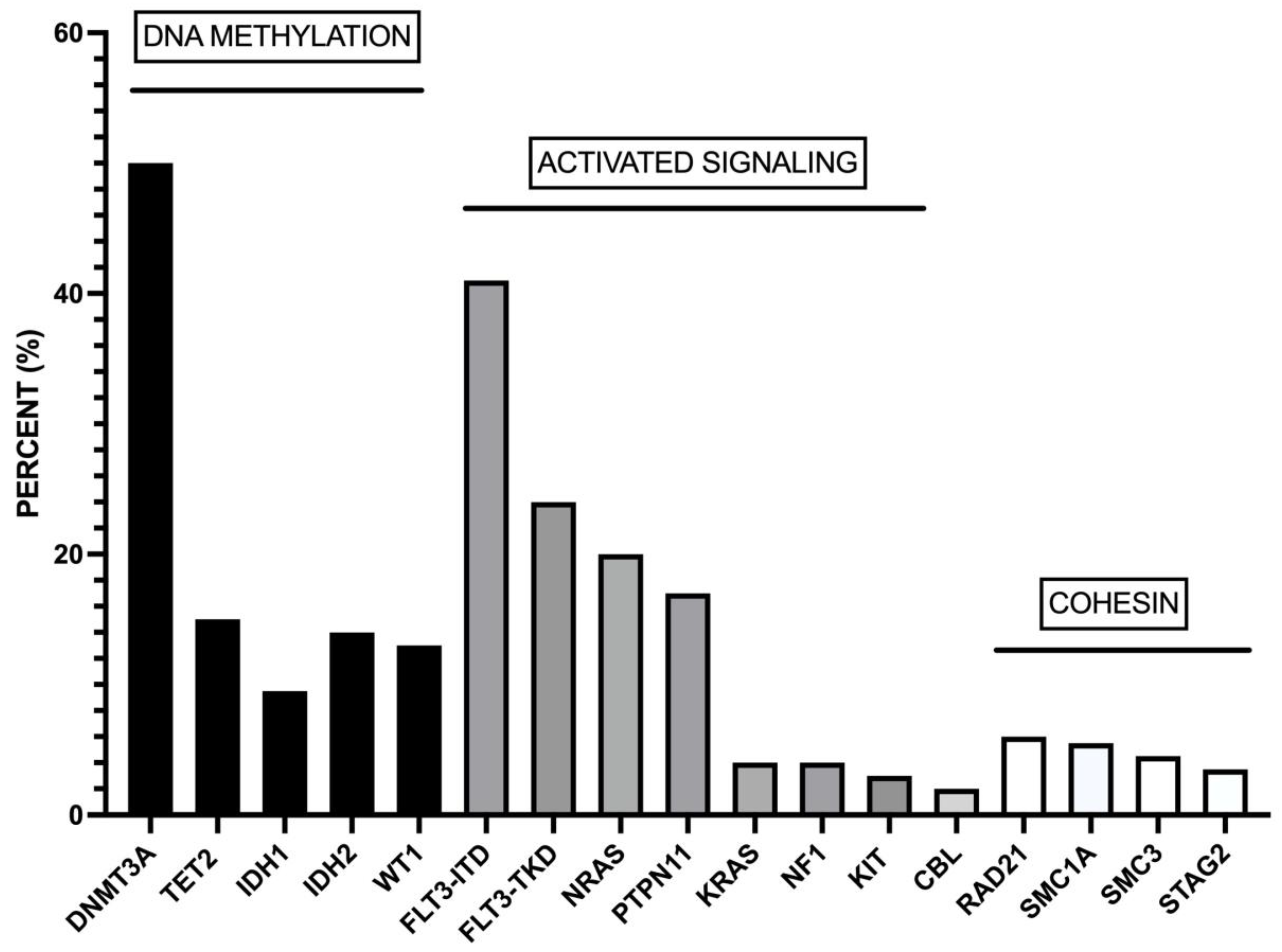
Studies on large cohorts of NPM1-mut AMLs showed recurrent mutations of genes involved in DNA methylation (DNMT3A (51%), TET2 (15.5%), IDH1 (12%), IDH2 (14%) and WT1 (8%)) and activated signaling (FLT3-ITD (39.5), NRAS (19%), FLT3-TKD (17.5%), PTPN11 (16%), KRAS (4%)) [15,16]. More than 95% of NPM1-mut AMLs display co-mutations of at least one of these genes [15,16]. The analysis of individual NPM1-mut AMLs showed that the large majority of cases with DNMT3A mutations display concomitant mutations of one or more than one gene of DNA methylation or activated signalling pathways: particularly frequent is the co-association with FLT3-ITD, FLT3-TKD, TET2, IDH1, IDH2, WT1 and NRAS mutations [15]. In other cases, TET2, IDH1, IDH2, WT1, FLT3-ITD, FLT3-TKD and NRAS mutations are not associated with DNMT3A mutations; in these cases, FLT3-ITD mutations are frequently associated with IDH2, WT1 and TET2 mutations, while FLT3-TKD mutations are frequently associated with IDH1 mutations [16].
A recent study reported the results of the mutational profiling of 2856 AML cases, including 640 NPM1-mut AMLs [17]. The most relevant results of this extensive analysis showed that NPM1 mutations were: (i) significantly co-mutated with FLT3 and DNMT3A mutations; (ii) highly associated with IDH1 mutations; (iii) exclusive with RUNX1, SRSF2, ASXL1 and IDH2-R172 mutations [17]. NPM1 mutations were associated with DNA methylation genes and activation of signaling genes but exclusive with myeloid transcription factors, spliceosome genes and chromatin-modifying gene mutations [17].
The favorable prognostic impact of NPM1 mutations decreases with increasing age of AML patients treated with standard treatments. This finding supported the study of the mutational profile of older NPM1-mut AML patients. Thus, one study reported in older NPM1-mut AML patients (≥75 years) a significant enrichment of TET2, SRSF2 and IDH2 mutations, with a reduced frequency of DNMT3A mutations, compared with what observed in younger NMP1-mut AML patients (45% vs 16%, 22% vs 3.5%, 28% vs 12% and 27% vs 52%, respectively) [18]. Similar observations were made by Lachowietz et al. reporting a higher frequency of TET2 and a lower frequency of DNMT3A mutations in ≥65 year NPM1-mut AMLs compared to those of patients with ≤65 year [19]. An extensive analysis carried out on 533 NPM1-mutated AML patients showed some notable differences in the mutational profile of ≤65 vs ≥65 year: TET2 (13% vs 27%), NRAS (13% vs 7%), SRSF2 (5% vs 15%), WT1 (10% vs 4%), ASXL1 (1% vs 7%) [20].
Therapy-related AMLs (t-AML) are a heterogeneous group of aggressive myeloid neoplasms occurring in patients with cytotoxic chemotherapy or ionizing radiation. The majority of AMLs display concomitant chromosomal abnormalities and TP53 alterations; a minority of t-AMLs have a normal karyotype (NK). About 35% of NK t-AMLs have NPM1 mutations. The mutational spectrum of NK t-AMLs was similar to that observed for NK de novo AMLs, although the frequency of some mutations show some significant differences: NPM1 (35% vs 49%, respectively), FLT3 (23% vs 36%, respectively), KRAS (12% vs 5%, respectively) and GATA2 (9% vs 2%, respectively) [21]. 7-9% of patients developing t-AML display NPM1 mutations. T-NPM1 AMLs exhibit unique features compared to t-AML non-NPM1-mut. In fact, t-NPM1-AMLs are similar to de novo NPM1-mut AMLs but different from the rest of t-AMLs: tNPM1-mut AMLs have a normal karyotype more frequently than t-AMLs (88% vs 28%, respectively); t-NPM1-mut AMLs are more frequently associated with DNMT3A-mut and TET2-mut than t-AMLs (43% vs 14% and 40% vs 10%, respectively); t-NPM1-mut AMLs are less frequently associated with TP53 mutations than t-AMLs (3% vs 35%, respectively) [22].
3. Cell differentiation heterogeneity of NPM1-mut AMLs
NPM1-mut AMLs, in addition to genetic heterogeneity, exhibit a consistent degree of phenotypic heterogeneity, with a subset showing monocytic differentiation and another subset lacking monocytic differentiation and showing a promyelocyte-like CD34-/HLA-DR- immunophenotype [2].
Mason et al. have explored a possible link between phenotypic and genotypic heterogeneities in a group of 239 NPM1-mut AMLs; 41% of these AMLs dispalyed monocytic differentiation and the remaining 59% of cases were subdivided into two subgroups, one lacking HLA-DR and CD34 expression (double negative, 30% of cases) and the orther defined as myeloid (29% of cases) [23]. These three phenotypic subtypes differed for some genotypic features: TET2 and IDH1-2 mutations are more frequent in DN cases (96% of positivity) than in myeloid (44%) or monocytic (48%) subtypes; DNMT3A mutations are significantly less frequent in DN AMls (27%) than in myeloid (44%) or monocytic cases (54%) [23]. These three phenotypic groups showed also significant differences in their outcome in that the DN-NPM1 displayed a DFS and OS (64.7 and 66.7 months, respectively) longer than monocytic NPM1 (20.6 and 44.3 months, respectively) and myeloid NPM1 (8.4 and 20.2 months, respectively) [23].
Using a machine learning approach for the analysis of gene expression profiles, Mer et al. have idnetiifed two different subtypes within NPM1-mut AML patients, one labeled as primitive and the other one as committed, based on the respective presence or absence of a stem cell signature [24]. FLT3-ITD mutations were significantly more frequent in primitive than committed NPM1 subtypes (62% vs 28%, respectively), while DNMT3A mutations were less common in primitive than in committed NPM1 subtype (38% vs 56%, respectively) [24]. The primitive subtype was associated with a significantly worse survival than the committed subtype; furthermore, the primitive subtype was more sensitive to kinase inhibitors [24].
The analysis of gene expression profile of NPM1-mut AMLs further supported their heterogeneity. Thus, Cheng et al. have explored the gene expression profiles by RNA sequencing and somatic genomic alterations by targeted or whole-exome sequencing of 655 AML patients and, based on enhanced consensus clustering, identified eigth stable gene expression subgroups (G1 to G8) [25]. NPM1-mut AMLs clustered into three different subgroups (G6, G7 and G8), showing high expression of HOXA/B genes and various differentiation stages, from hematopoietic stem/progenitor cells down to monocyte, and specifically HOX-primitive (G7), HOX-mixed (G8) and HOX-committed (G6); in the G6 and G8 subgroups clustered also KMT2A fusions and in the G7 subgroup NUP98 fusions [25]. NPM1-mut AMLs present in the G6-G8 subgroups show relevant differences at the level of their co-mutation profile: (i) DNMT3A represent the most frequent co-mutations in the G6 and G8 subgroups, while NPM1-mut present in the G7 subgroup rarely associate with DNMT3A mutations but frequently associate with IDH1 and IDH2 mutations; (ii) the frequency of triple mutated NPM1/DNMT3A/FLT3-ITD was higher in the G8 subgroup (with a percentage of 4.5%, 6% and 22.2% in G6, G7 and G8,respectively); (iii) the frequency of triple-mutated NPM1/FLT3-ITD/TET2 or NPM1/FLT3-ITD/IDH2 was more recurrent in the G7 subgroup compared to the two other subgroups (with 0%, 28.4% and 7.6% in G6, G7 and G8 , respectively) [25]. Concerning the differentiation state, G6 and G7 exhibited a more differentiated monocytic phenotype and stem cell phenotype, respectively [25]. The comparison of the prognostic profile of these three sungroups showed that patients in G8 (HOX-mixed) have the poorest prognosis, in terms of both OS and EFS, compared to those in G6 (HOX-committed) and G7 (HOX-primitive) [25].
4. Clonal architecture and clonal evolution of NPM1-mutant AMLs
The study of NPM1-mut AMLs is one of the best models to explore the mechanisms of leukemic clonal evolution. The analysis of allelic burden (VAF) of NPM1 mutations and of the associated co-mutations allowed to define a mutational clonal hierarchy of NPM1-mut at diagnosis. This analysis showed that in the majority of patients a higher VAF was detected for some co-mutations than for NPM1, including DNMT3A, IDH1, IDH2, SRSF2 and TET2, thus suggesting that mutations in those genes represent first hits and occur at an early phase of leukemic development; on the contrary, other co-mutations such as FLT3, NRAS and WT1 showed a significantly lower VAF than NPM1-mut, thus indicating that these are second hit mutations [26]. According to the VAF, Cappelli et al. distinguished co-mutations occurring in NPM1-mut AMLs and distinguished the mutations in CHIP-like, including DNMT3A, TET2, ASXL1, ID1, IDH2, SRFSF2 and STAG2, and CHOP-like, including FLT3, GATA2, NRAS, PTPN11, WT1, TP53 and RUNX1 [27]. The persistence or the acquisition of CHOP-like mutations was associated with an inferior outcome [27].
The study of the clonal architecture of NPM1-mut AMLs at single cell level showed that these leukemias are usually organized following simple clonal architectures with one to six subclones and branching; in all cases studied, NPM1 mutations are secondary or subclonal to other driver mutations; in a part of these leukemias, it was postulated, through the analysis of single CD34+/CCD33- cells, the exixtence of pre-leukemic cells bearing one or more driver mutations, lacking NPM1 mutations [28]. Importantly, after transplantation in immunodeficient mice, the dominant regenerative clone in vivo was a NPM1-mut subclone, even when NPM1-mut was minoritary at sublonal level in the diagnostic leukemic cells [28]. According to these findings, it was proposed a model in which NPM1-mut AMLs develop from pre-existing clonal hematopoiesis [28]. Additional studies supported this clonal evolution model of NPM1-mut AMLs. Thus, Desai et al. reported the longitudinal history of an AML patient with IDH2-mut clonal hematopoiesis who develop AML one year after the acquisition of a NPM1 mutations [29]. NPM1-mut AML patients with concomitant DNMT3A mutations, responding opimally to standard induction chemotherapy, despite the persistence of the DNMT3A mutations, achieved a long-term response [30,31].
Single-cell mutation analysis provided a fundamental tool to analyze AML clonal evolution. A study by Miles et al, exploring clonal evolution in 123 AML samples, provided evidence that AML development is characterized by a small number of mutant clones, frequently harbouring co-occurring mutations in epigenetic regulators [32]. In NPM1-mut AMLs with concomitant FLT3-ITD mutations, the size of double-mutant NPM1/FLT3 clones was significantly greater than those of NPM1 or FLT3 mutant single clones; in contrast, in NPM1-mut AMLs with concomitant RAS mutations, there is evidence of cooperativity between these mutations with respect to single-mutant RAS clones, but not to single-mutant NPM1 clones [32]. This observation indicates that in NPM1-mut AMLs different combinations of mutants differ in their capacity to promote clonal expansion [32].
The mutational dynamics at clonal level was explored in NPM1-mut AMLs at diagnosis and at relapse. Kronke et al. reported the evaluation of 53 relapsing NPM1-mut AMLs and showed that in about 90% of cases the recurrence was related to the original NPM1-mut clone at relapse [33]. The relapsed AMLs were characterized by an increased mutational complexity; some mutations, such as DNMT3A and IDH2 mutations were almost completely stable at relapse, while other mutations, such as FLT3-ITD and RAS mutations, showed low stability; recurrent genetic alterations acquired at relapse often involved ETV6, TP53, NF1, WT1 genes [33]. In larger analyses on relapsing NPM1-mut AML patients it was reported that 9-14% of patients relapsed with NPM1-WT AMLs [34,35]. At diagnosis, FLT3-ITD mutations were more frequent in patients with NPM1-mut at relapse, while DNMT3A mutations were more frequent in those relapsing with NPM1-WT AML [34]. According to the results of exome sequencing studies, it was proposed that: in NPM1-mut persistent patients an NPM1-mut clone survived to chemotherapy, showed additional mutational evolution and subsequently acquired a growth advantage, causing relapse; in NPM1-mut patients relapsing with NPM1-WT AML, the initial NPM1-mut clone is eradicated by chemotherapy and the relapse is ensured by a surviving clone with preleukemic mutations acquiring new mutations and leukemic properties [34,35].
5. Prognostic heterogeneity of NPM1-mut AMLs
The analysis of the overall survival on NPM1-mut AML patients showed a marked heterogeneity, with a part of patients showing a good OS, with an observed risk hazard distributed between favorable and intermediate ELN risk groups and another part of patients showing a poor OS with an observed risk hazard distributed between intermediate and adverse ELN risk groups [2].
The European HARMONY Alliance retrospectively analyzed a large cohort of 1011 NPM1-mut patients for their mutational profile and their response to standard therapy showing that: (i) the triple mutation group NPM1/DNMT3A/FLT3-ITDhigh identified a subgroup with adverse prognosis (2-year OS of 25%, similar to that observed for NPM1/TP53 double mutant AMLs); (ii) the double mutation groups FLT3-ITDlow/DNMT3A or FLT3-ITDhigh/DNMT3A-WT exhibited an intermediate prognosis (2-year OS of 45% and 53%, respectively); (iii) NRAS, KRAS, PTPN11 or RAD21 mutations were associated with a better OS (however, these mutations did not affect prognosis in the presence of the triple mutations NPM1/DNMT3A/FLT3-ITD) [36]. Using this large database, a machine learning algorithm was developed, allowing the idnetification of combinations of up to 4 comutations with prognostic significance [37]. This algorithm allowed to stratify NPM1-mut AML patients into four groups with increasing prognostic adversity: favorable, intyermediate-1, intermediate-2 and adevrse (Figure 2). Particularly, the triple combination NPM1-mut/FLT3-ITD/DNMT3A-mut idfentified a subgroup with adverse prognosis (2-year OS of 33%), similar to that observed for the small subgroup (1.5% of total NPM1-mut AMLs) (Figure 2). Two subgroups were identified in the favorable group: a first subgroup, involving TP53-WT/FLT3-WT/DNMT3A-mut and NRAS or KRAS or PTPN11 or RAD21-mutated AMLs; a second subgroup, involving TP53-WT/DNMT3A-WT/IDH-mut patients. The intermediate-1 group involves trwo subgroups: one composed by AMLs with a TP53-WT/FLT3-WTR/DNMT3A-WT mutationalk profile; the other one involves TP53-WT/FLçT3-WT/DNMT3A-mut/IDH-mut and NRAS/KRAS/PTPN11/RAD21-WT cases (Figure 2). The intermediate-2 group involves trwo subgroups: one subgroup implies AMLs TP53-WT/FLT3-WT/DNMT3A-mut/IDH-mut; the other sungroup is composed by AMLs TP53-WT/FLT3-ITD/DNMT3A-WT/IDH-WT (Figure 2). The 3-year OS of these groups was 78%, 63%, 48% and 29% for favorable, intermediate-1, intermediate-2 and adverse groups respectively [37]. The prognostic predictive capacity of this algorithm was evlauated in other datasets of NPM1-mut AML patients with availbale genetic and clinical information [37].
Mrozek et al. reported in 1637 adult AML patients the evaluation of the 2022 ELN stratification risk system [38]. The NPM1-mut/FLT3-ITD-negative was included in the favorable group; the outcome of these patients harboring myelodysplasia-related mutations was worse than the outcome of the patients without myelodysplasia-related mutations (CR rates: 67% vs 81%, respectively; PFS: 30% vs 43%, respectively; 5-year OS: 32% vs 42%, respectively) [38]. The outcome of the patients was similar to that of patients classified as intermediate-risk patients following ELN 2022 [38]. According to the ELN 2022 guidelines for AML, NPM1-mut AML patients with adverse cytogenetics are classified as adverse risk patients [5]. The evaluation of 13 of these patients showed that they have a shorter 5-year OS and DFS compared to that observed in NPM1-mut patients without adverse risk cytogenetics (23% vs 41% and 38% vs 41%, respectively) [38]. A comparison with other AML groups with intermediate or adverse risk shows that these NPM1-mut AML patients with adverse cytogenetics are more similar to patients with intermediate risk than to those with adverse risk [38].
Angenendt et al. re-evaluated the data on chromosomal abnormalities in 2426 patients with NPM1-mut AMLs, upgrading the risk category of risk based on chromosomal abnormalities evaluated following ELN 2022 [8,39]. In these patients, adverse cytogenetics according to ELN 2022 was associated with lower complete remission rates (87%, 85% and 66% for normal, aberrant-intermediate and adverse karyotypes, respectively) and inferior overall survival (40%, 36% and 16%, for normal, intermediate and adverse karyotypes, respectively) [39].
Several studies have explored a possible prognostic impact of NPM1-mut VAF, generating conflicting results. NPM1-mut VAF was shown to positively correlate with leukemic cellularity at diagnosis (WBC), the percentage of leukemic blasts in peripheral blood and to negatively correlate with platelet counts [40,41,42]. Three studies have explored a possible link between NPM1-mut VAF and outcomes. Patel et al. explored 109 patients with de novo NPM1-mut VAF (≥0.44) correlated with shortened OS and EFS compared to the rest of NPM1-mut AMLs; high NPM1-mut VAF had a particularly negative prognostic impact in NPM1-mut patients treated with stem cell transplantation in first remission and in patients with mutated DNMT3A [42]. In a second study, the same authors showed that high NPM1-mut VAF correlates with minimal residual disease (MRD) at first remission; both NPM1-mut VAF and MRD at first remission predicted a shortened EFS [44].
Abbas et al. reached a different conclusion in their evaluation of 147 NPM1-mut AML patients treated with induction chemotherapy. First, they observed a significantly higher NPM1-mut VAF in patients with FLT3-ITD compared to those FLT3-WT (42.7% vs 39.1%, respectively); however, NPM1-mut VAF did not correlate with DNMT3A muttional status or with the presence of cytogenetic abnormalities [41]. No any significant correlation was observed between the level of NPM1-mut VAF and either OS or EFS in the entire cohort of patients or in any subgroup [40]. It was suggested that differences between this study and the previous study could be related to differences in induction chemotherapy regimens used in these two studies [41].
Rothenberg-Thurley et al. reported the study of 417 NPM1-mut patients and the analysis of their NPM1-mut VAT showed that: the median NPM1-mut VAT was 0.43 and was higher in type A than in type B NPM1 mutations and was not associated with abnormal karyotype; patients with high NPM1-mut VAF more frequently had concomitant FLT3-ITD (47% vs 37%) and DNMT3A (63% vs 46%) mutations compared to those of patients with low NPM1-mut VAF; a high NPM1-mut VAF associated with shorter OS [42]. However, in multivariate analysis, after adjusting for FLT3-ITD allelic ratio and/or DNMT3A mutational status, only these genetic alterations but not NPM1-mut VAF remained associated with OS [42]. According to these results it was suggested that high NPM1-mut VAF may simply represent a marker of highly proliferative subsets of NPM1-mut AMLs, such as those with FLT3-ITD mutations, rather than an independent prognostic factor [42].
6. DNMT3A mutations in NPM1-mut AMLs
As above mentioned, DNMT3A mutations represent the mutations most frequently associated with NPM1 mutations in AMLs. DNMT3A mutations are frequently observed in aging individuals without overt leukemia and in association with clonal hematopoiesis of undetermined potential (CHIP). DNMT3A gene mutations precede NPM1 mutations, exerting a stimulatory effect on self-renewal of leukemic clones. In the ELN 2022 leukemia classification DNMT3A is not considered a high-risk mutation.
In AML patients the levels of DNMT3A mutations do not correlate with presenting clinical features or concurrent gene mutations and do not affect the OS; DNMT3A-mut expression persists in most AML patients achieving complete remission after induction chemotherapy, suggesting persistence of clonal hematopoiesis in hematological remission [31].
Cappelli et al. have retrospecively analyzed a large cohort of 1977 NPM1-mut AML patients [44]. In these patients, DNMT3A gene aws the most frequently co-mutated (45% of cases). The VAF of DNMT3A mutation was significantly higher than that of NPM1 mutations, thus indicating that they precede NPM1 mutations. DNMT3A mutations displayed a peculiar pattern according to age, being more frequent in younger than older (≥60 years) patients: 51% vs 40% respectively [43]. This pattern of DNMT3A mutational frequency was dependent on the type of mutations: in fact, DNMT3A-R882 mutations were more frequent in younger than older NPM1-mut patients (58% vs 42%, respectively), while non-R882 DNMT3A mutations were less frequent in young than in older patients (39% vs 61%, respectively) [44]. In contrast, other CHIP-related genes, such as ASXL1 and TET2 were less frequently mutated in younger than in older NPM1-mut AMLs (1% vs 4% and 17% vs 27%, respectively) [44]. Importantly, in NPM1-WT AMLs the frequency of DNMT3A mutations increased with age [44]. In addition to DNMT3A, other gene mutations were preferentially associated with younger age, such as WT1 (8% vs 3%), NRAS (25% vs 17%) and PTPN11 (8% vs 1%) [44]. The co-mutational pattern of NPM1-mut/DNMT3A-mut double mutated AMLs was significantly different compared to NPM1-mut/DNMT3A-WT: in fact, NPM1-mut/DNMT3A-mut was positively associated with FLT3-ITD, NRAS and PTPN11 mutations, and negatively associated with IDH2R140, STAG2 and SRFSF2 mutations [44]. The analysis of the survival according to DNMT3A mutational status showed that: in NPM1-mut patients DNMT3A mutations were not associated with survival, irrispectively of DNMT3A mutational subtype; the presence of FLT3-ITD mutations had a detrimental effect both in DNMT3A-mut and DNMT3A-WT NPM1-mut patients; particularly, the presence of DNMT3A-R882 mutation in association with FLT3-ITD was associaetd with worse outcomes [44].
In a more recent study, the same authors reported the study of 150 NPM1-mut AML patients achieving CR following induction chemotherapy: patients with CHIP mutations, such as DNMT3A, TET2, ASXL1, IDH1, IDH2 and SRSF2 have a frequency of relapse and a probability of OS comparable to that observed for NPM1-mut without co-mutations at remission; in contrast, patients with mutations not CHIP-related, such as FLT3-ITD, FLT3-TKD, GATA2, NRAS, PTPN11, WT1, TP53 and RUNX1, persistent at remission or acquired at relapse, have an increased probability of relapse and a poor prognosis [45]. These not-CHIP mutations were defined as CHOP mutations. Finally, this study showed that persistence of DNMT3A-R882 mutations is not associated with inferior survival [45].
Onate and coworkers have explored the prognostic impact of DMNT3A mutations in NPM1-mut AMLs subdivided into three subgroups according to FLT3 mutational status: DNMT3A-FLT3-WT, DNMT3A-FLT3-ITDlow, DNMT3A-FLT3-ITDhigh; patients with DNMT3A mutation have a delayed NPM1-mut clearance after induction chemotherapy but DNMT3A mutations do not modify the prognostic value of FLT3-ITD allelic ratio in NPM1-mut AMLs [46].
The characterization of DNMT3A-mut/NPM1-mut AMLs has led to the dientification of an AML subset chracterized by triple positivity for NPM1, DNMT3A and FLT3-ITD mutations. A part of these triple-positive AMLs display also either TET2 or WT1 mutations. An initial study by Loghavi and coworkers suggested that triple-positive NPM1/DNMT3A/FLT3-ITD may represent a peculiar subset of NPM1-mut AMLs associated with poor prognosis: in fact these AMLs displayed an OS shorter than that observed in double-positive NPM1-mut/FLT3-ITD AMLs [47]. The concomitant presence NPM1/DNMT3A/FLt3-ITD mutations was observed in about 6% of AMLs, characterized by high frequency of leukemia stem cells, aberrant immunophenotype (with low CD34 expression, associated with high CD56 expression) and high expression of hepatic leukemia factor(whose expression is required for the maintenance and the expansion of leukemic stem cells) [48].
Several studies have reported poor overall survival of tiple mutant NPM1/DNMT3A/FLT3-ITD patients: thus, Bezerra et al. reported an 5-year OS of only 4% for these patients, an increased risk of relapse and a lower disease-free survival [49]. In this study the analysis was limited to AML patients bearing R882-DNMT3A mutations, the only mutations of DNMT3A having biochemical changes [50] and consequences on clonal hematopoiesis [51].
Wakita and coworkers have retrospectively analyzed 605 Japanese patients with de novo AML (174 with NPM1-mut AML) [52]. The analysis of both NPM1-mut and NPM1-WT AML patients showed that the presence of DNMT3A-R882 mutations was associated with a reduced overall survival compared to the respective DNMT3A-WT patients; in both NPM1-mut/DNMT3A-WT and NPM1-mut/DNMT3A-R882 AMLs the co-occurrence of FLT3-ITD mutations, at both low and high allelic ratios, significantly reduced OS; triple-mutant NPM1/DNMT3A/FLT3-ITD patients showed a marked decline of OS [52].
7. FLT3 mutations in NPM1-mut AML patients
Two types of FLT3 mutations are observed in AMLs: internal tandem duplication of the juxta membrane domain (FLT3-ITD) and point mutations or deletion of tyrosine kinase domain (FLT3-TKD). FLT3 mutations are very frequent in NPM1-mut patients: FLT3-ITD (41%), FLT3-TKD (21%) and FLT3-ITD/FLT3-TKD (4.5%) [15]. FLT3-ITD mutations may occur at the level of the juxtamembrane domain (FLT3-ITD-JMD) or at the level of tyrosine kinase domain 1 (FLT3-ITD-TKD1) or in both these regions of FLT3 (FLt3-ITD-JMD-TKD1). In the RATIFY trial enrolling a large cohort of FLT3-ITD-mutated patients, it was reported that in NPM1-mut/FLT3-ITD patients 60.5% displayed FLT3-ITD-JMD mutations, 17% FLT3-ITD-TKD1 and 22.5% FLT3-ITD-JMD-TKD1 [53]. The FLT3-ITD-TKD1 is associated with a worse prognosis.
NPM1-mut/FLT3-ITD and NPM1-mut/FLT3-TKD have a similar overall survival when treated with intensive frontline therapy, while patients dispalying concomitant FLT3-ITD and FLT3-TKD mutations have a dismal overall survival [54].
The ELN 2017 classification supported the evaluation of FLT3-ITD allelic ratio as a prognostic parameter, classifying patients with a high FLT3-ITD ratio in a worse category group. FLT3-ITDhigh is associated with a higher WBC, higher blood and bone marrow blasts and with more frequent NPM1 mutations, while FLT3-ITDlow was associated with FLT3-TKD [55]. However, in spite these clinico-biologic differences, the outcomes of AML patients undergoing allogeneic HSC was similar for both FLT3low and FLT3high AML patients [55].
The evaluation of FLT3-ITD minimal residual disease by NGS in complete remission represents the best and more sensitive biomarker to predict the outcomes of these patients [56].
8. IDH1 and IDH2 mutations in NPM1-mut AMLs
About 25% of NPM1-mut AMLs have a mutation of IDH1 or IDH2 genes. IDH1 and IDH2 mutations occur in a part of cases in association also with DNMT3A mutations, while in other patients are co-mutated with FLT3-ITD or FLT3-TKD [15]. In NPM1-mut AMLs, the most frequent IDH1 mutations are represented by IDH1R132H, while IDH1R132C are less frequent; the most frequent IDH2 mutations are represented by IDH2R140Q, while IDH2R172K mutations are only rarely observed in NPM1-mut AMLs [56]. Importantly, the association of IDH1/IDH2 mutations with NPM1 mutations improved their prognostic impact, in comparison with the prognosis of AMLs with the same type of IDH1 or IDH2 mutations but in association with other co-mutations [57].
IDH1 mutations occur in about 7-8% of AML patients, mostly associated with a normal karyotype. NPM1 and DNMT3A gene mutations most frequently associated with IDH1 mutations. Particularly, 66% of IDH1-mut AMLs display NPM1-mut; IDH1-R132H was strongly associated with NPM1-mut (89% of cases), while IDH1-R132H was associated with NPM1-mut in 28.5% of cases; other more rare IDH1 mutations are also strongly associated with NPM1-mut (75% of cases) [58]. IDH2R140 mutations are associated with NPM1 mutations in about 50% of cases and in NPM1-mut AMLs are associated with frequent DNMT3A, FLT3-ITD and SRSF2 co-mutations [59]. These findings indicate that the association between IDH1/IDH2 and NPM1 mutations is stronger than the association between NPM1 and IDH1/IDH2 mutations.
Mason et al. have distinguished two subtypes of NPM1-mut AMLs according to their immunophenotypic features: the acute promyelocytic-like subtype, characterized by absence of CD34 and HLA-DR expression and strong myeloperoxidase expression, was highly enriched in IDH1, IDH2 or TET2 co-mutated cases [23]. This APL-like subtype is associated with longer relapse-free and overall survival, when compared with cases that were positive for CD34 and/or HLA-DR [23].
In conclusion, NPM1-mut AMLs with IDH1 or IDH2 co-mutations do not seem to have a worse prognosis compared to NPM1-mut AMLs without IDH1 or IDH2 mutations.
9. Cohesin complex gene mutations in NPM1-mut AMLs
Cohesin complex genes, STAG2, RAD21, SMC1A and SMC3 mutations, are observed in about 19% of NPM1-mut AMLs [15]. Complex cohesin genes were found to be mutated in about 11% of all AMLs [59]. Some of the cohesin genes, including RAD21, SMC1A and SMC3 dispaleyd the highest frequency of mutations in NPM1-mut AMLs [60].
STAG2 mutations occur in about 3% of NPM1-mut AMLs [15]; NPM1-mut AMLs represent 15% of all STAG2-mut AMLs [59]. NPM1 is less commonly mutated in STAG2-mut AMLs than in the rest of AMLs (15% vs 32%, respectively). NPM1-mut AMLs with STAG2 mutations frequently diplay also FLT3-ITD and NRAS mutations and, more rarely, DNMT3A mutations.
RAD21 mutations occur in about 6% of NPM1-mut AMLs [15]; NPM1-mut AMLs represent 57% of all RAD21-mut AMLs [59]. NPM1 is significantly more frequently mutated in RAD21-mut AMLs than in the rest of AMLs /57% vs 30%, respectively) [60]. Double-mutant NPM1-RAD21 AMLs display a pattern of associated mutations comparable to that observed in the whole group of NPM1-mut AMLs [61].
SMC3 is mutated in 4.5% of NPM1-mut AMLs [60].; NPM1-mut AMLs represent 65% of all SMC3-mut AMLs [59]. Double mutant NPM1/SMC3 frequently display additional mutations of NRAS [59].
SMC1A is mutated in about 5% of NPM1-mut AMLs [15]; NPM1-mut AMLs represent 40% of all SMC1A-mut AMLs [60].
Simonetti et al. have explored the metabolomic profile of AMLs and, through an integrated analysis of genomic-metabolic profiles defined two subgroups of NPM1-mut AMLs: one of these two subroups was enriched in cohesin/DNA damage-related genes and showed higher mutation load, transcriptomic signatures of reduced inflammatory state and better ex vivo response to EGFR and MET inhibition [61].
Benard et al. exploring the genomic data of 2829 AML patients reached the conclusion that clonal architecture represents a predictive parameter of clinical outcomes and drug sensitivity [62]. In some instances, the order of mutations in functional classes stratified survival: this is the case of patients with co-occurring mutations in NPM1 and chromatin/cohesin complex genes; in these patients, if a chromatin/cohesin mutation occurred before an NPM1 variant, there was a strong association with poor survival [62].
Studies in inducible mouse models of NPM1-mut/SMC3-mut have shown that cohesin gene mutations alter the transcriptome in the context of NPM1-mutant; particularly, it was shown that the Rac 1-2 exchange factor Dock1 is specifically upregulated in double mutant NPM1/SMC3 cells and could represent a therapeutic target in these leukemias [63].
10. RAS mutations in NPM1-mut AMLs
RAS genes are frequently mutated in NPM1-mut AMLs: NRAS in about 20% of cases and KRAS in about 4% of cases [15]. NRAS mutations in these patients are frequently associated with DNMT3A and PTPN11 mutations; NRAS is rarely co-mutated with FLT3-ITD or FLT3-TKD. NPM1 mutations preferentially associate with NRASG12/13 mutations but not with NRASQ21 mutations [1]. NRAS mutations do not affect the outcomes of NPM1-mut AMLs; NPM1/DNMT3A/NRAS triple-mutant AMLs are associated with a favorable prognosis [64].
Rivera et al. have explored 273 de novo AML patients treated with induction therapy and showed that in these patients favorable karyotype and concomitant NPM1 mutations were associated with higher CR, ORR and OS [65].
11. Myelodysplasia-related alterations in NPM1-mut AMLs
AML with myelodysplastic changes (AML-MRC) is a subgroup of AMLs, usually associated with poor prognosis. The diagnosis of AML-MRC englobes a variety of AMLs, based on three main criteria: history of a myelodysplatic syndrome or of myelodysplastic/myeloprolifetative neoplasm (AML-MRC-H); Presence of a MDS-defining cytogenetic abnormality (AML-MRC-C); morphological detection of multilineage dysplasia (AML-MRC-M) [66].
According to these criteria, four different AML-MRC subtypes can be identified: AML-MRC-C, AML-MRC-H, AML-MRC-M and AML-MRC-TS (this last subtype identiifes AMLs originated from previously treated MDS or MDS-MPN).
NPM1-mut MDS
At variance of AMLs in which NPM1 mutations are frequent, myelodysplatsic syndromes (MDS) only rarely display NPM1 mutations. In fact, the frequency of NPM1-mut in patients with a diagnosis of MDS or myelodysplastic/myeloproliferative neoplasm (MDS/MPN) is low, ranging from 0% to 9% [67,68,69]. NPM1-mut MDS or MDS/MPN exhibit an aggressive clinical course with a high rate of transformation to AML [69]. Forghieri et al. proposed that NPM1-mut MDS or MDS/MPN may be classified as AML, even in the presence of <20% bone marrow blasts [69].
Maurya et al. explored 111 MDS patients and reported a NPM1-mut frequency of 3.6%, with 50% of NPM1-mut patients showing a IPSS-low-risk and 50% a IPSS-high-risk and a reduced OS compared to MDS patients without NPM1 mutations [70].
However, the conclusions of these studies were based on the analysis of very small numbers of NPM1-mut MDS patients.
Montalban-Bravo et al. reported the analysis of 31 NPM1-mut MDS patients observed in a cohort of 1900 MDS patients [71]. This analysis included the largest series of NPM1-mut MDS patients reported thus far. These patients were predominantly classified as intermediate-risk and high-risk, with a median BM blast percentage of 10% and with a normal karyotype in 77% of cases; compared to the rest of MDS patients, NPM1-mut MDS patients were younger, had lower hemoglobin levels, had higher median BM blasts percentage at diagnosis and had higher frequency of normal karyotype [71]. 38% of these patients had a transformation to AML after a median of 14 months, maintaining in all cases the NPM1 mutation [70]. The analysis of the mutational profile of these patients showed a pattern of mutations similar to that observed in NPM1-mut AMLs: in fact, frequent NRAS (32%), DNMT3A (27%), TET2 (18%), WT1 (18%), PTPN11 (13%), FLT3 (13%) and IDH2 (10%) mutations were observed [71]. 32% of these patients received cytotoxic chemotherapy and 65% hypomethylating agents: patients treated with chemotherapy had higher complete resposne rates and longer overall survival compared to those treated with hypomethylating agents (90% vs 28% and not recahed vs 16 months, respectively) [71]. These observations, although based on a retrospective analysis, striongly support the treatment of NPM1-mut MDS patients with intensive chemotherapy, possibly followed by allogeneic SCT [71].
Another recent study showed a very low response rate of NPM1-mut MDS patients to hypomethylating agents, while NPM1-mut sAMLs treated with intensive chemotherapy showed a high rate of complete responses with 39% of patients undergoing allogeneic SCT [72]. These findings, together with previous studies, support the view that NPM1-mut MDSs are an aggressive clinicopathologic entitity requiring, if clinically suitable, treatment with intensive chemotherapy [72].
A second study reported the characetrization at clinical and molecular levels of a consistent number (45) of NPM1-mut MDS. NPM1-mut MDS compared to NPM1-WT MDS were associated with younger age, lower WBC and bone marro cellularity at daignosis [73]. NGS studies showed some remarkable differences between NPM1-mut and NPM1-WT MDS: IDH1, IDH2, ASXL1, RUNX1 and TP53 mutations are less frequent in NPM1-mut MDS than in NPM1-WT MDS; PTPN11 and DNMT3A mutations are more frequent in NPM1-mut than in NPM1-WT MDS [73]. (Figure 3) The frequency of patients with abnormal karyotype is markedly lower in NPM1-mut than in NPM1-WT MDS (12% vs 61%, respectively) [73]. For the mutational profile and for chromosomic abnormalities, NPM1-mut MDS are more similar to NPM1-mut AML than to NPM1-WT MDS [73].
On the basis of the findings of these studies Falini et al. have proposed that NPM1-mut MDS represent NPM1-mut AML diagnosed at early stage and that must be treated with intensive chamotherapy, followed by allogeneic SCT, as typical NPM1-mut AMLs [74].
NPM1-mut secondary AMLs
It is unclear if NPM1 mutations remain a positive prognostic indicator within secondary AMLs evolving from a prior myeloid neoplasm. Smith et al. have retrospectively analyzed a group of 54 NPM1-mut sAMLs evolving from MDS, CMML, MPN, atypical CML [81]. Compared to de novo NPM1-mut AMLs, NPM1-mut sAMLs are similar concerning their classification according to ELN 2017, karyotype abnormalities and occurrence of FLT3-ITD mutations; however, NPM1mut sAMLs, compared to NPM1-mut de novo AMLs, were more likely to have RUNX1 mutations (14% vs 3%, respectively), CBL mutations (95 vs 2%) and NRAS mutations (26% vs 11) [81]. The overall survival of NPM1-mut sAMLs was similar to that observed for NPM1-mut de novo AMLs (2-year survival: 46% for sAML and 56% for de novo AML) [81].
Another recent study provided preliminary evidence that NPM1-mut sAMLs are molecularly heterogeneous, with only a part of patients bearing sMut: patients with sMut exhibited a signifiant lower OS compared to those without sMut [82]. This property is not unique to NPM1-mut sAMLs but is observed also in NPM1-mut de novo AMLs [82].
12. TET2 mutations in IDH1-mut AML
TET2 mutations are observed in about 16% of de novo AMLs and are usually associated with clinical signs of hyperleukocytosis, high blast percentage, a normal karyotype and are mutually exclusive with IDH mutations [1]. In Npm1-mut AMLs about 15% of cases dipslay TET2 mutations [15]. A significant proportion of NPM1-mut/TET2-mut AMLs display additional co-mutations at the level of DNMT3A: in both DNMT3A-mut and DNMT3A-WT cases, FLT3-ITD and FLT3-TKD are also frequent co-mutational events.
Studies of molecular characterization of AML patients with hyperleukocytosis at diagnosis have shown frequent involvement of AMLs with normal karyotype [83,84]. Particularly, 75%, 73% and 45% of patients with normal karyotype and hyperleukocytosis at daignosis are NPM1-mut, FLT3-mut and TET2-mut, respectively; 25% of these patients have concomitant NPM1 and TET2 mutations and 21% concomitant NPM1, TET2 and FLT3 mutations [84]. A recent study showed that the combined mutation of NPM1/TET2/FLT3-ITD and DNMT3A observed in about 4% of NPM1-mut AMLs results in aggressive leukemia phenotype [85].
13. PTPN11 mutations in NPM1-mut AMLs
PTPN11 mutations are observed in about 7-10% of adult AML patients [88,89]. PTPN11 mutations are observed in about 17% of NPM1-mut AMNLs [15]; in PTP11-mut AMLs, NPM1-mut AMLs are very frequent (60-65% of cases) and represent the most frequent co-mutations, followed by DNMT3A, NRAS, FLT3-ITD, IDH2 and TET2 mutations [87,88]. PTPN11-mut AMLs can be subdivided into two subgroups according to the presence of NPM1 mutations: DNMT3A and FLT3-ITD mutations are more frequent in the PTPN11-mut/NPM1-mut subgroup than in the PTPN11-mut/NPM1-WT subgroup, while the contrary was observed for BCOR, RUNX1, ASXL1 and SF3B1 mutations [88,89].
PTPN11mut AMLs are most frequent in the AMLs classified in the favorable risk genetic group following the ELN risk classification and are associated with higher leukocyte counts [88,89].
The clinical impact of PTPN11 mutations in AML patients was recently explored. Most of these studies provided evidence that PTPN11 mutations had an adverse effect on overall survival and a negative prognostic effect on event-free survival [88,89,90,91]. However, Metzeler et al. failed to confirm these results and observed no negative clinical impact of PTPN11 mutations in a cohort of 116 newly diagnosed PTPN11-mut AML patients [92]. Finally, Fobare et al. in their analysis on 1725 newly diagnosed AML patients, including 140 PTPN11-mut AML patients, treated with intensive induction chemotherapy, showed that PTPN11 mutations did not affect outcomes of NPM1-mut patients, but had an adverse effect on NPM1-WT patients [89]. This differential sensitivity of PTPN11-mut/NPM1-mut vs PTPN11-mut/NPM1-WT patients seems to be related to the selective enrichment in PTPN11/NPM1-WT patients of co-mutations, such as BCOR, RUNX1, TP53, associated with adverse outcomes [89].
14. Therapy of NPM1-mut AMLs
The standard therapy for NPM1-mut AML patients includes “3+7”-induction chemotherapy and consolidation therapy. It was estimated in these patients a complete remission rate of about 80% and an overall survival rate of about 40%. However, more than 50% of NPM1-mut AML patients relapse; thus, for high-risk NPM1-mut patients allogeneic stem cell transplantation (allo-HASCT) and additional treatments (such as FLT3 inhibitors) are important therapeutic choices.
In a cohort of 1570 AML patients, NPM1-mut cases displayed a favorable prognosis, with a hazard ratio of death of 0.7 and a median OS of nearly 6 years compared to about 2 years in those with NPM1-WT AML [1]. The prognostic impact of NPM1 VAF of NPM1-mut AMLs is unclear and should be not used to stratify the risk status of NPM1-mut AML patients. This favorable prognostic index was much more pronounced in younger than in older patients: Mrozek et al. showed an OS of 10.5 years in patients with a median age of 44 years compared to 1.7 years in patients with a median age of 69 years [93].
Recent clinical studies have shown the efficacy of Venetoclax (an inhibitor of the anti-apoptotic Bcl-2 protein) in the treatment of older NPM1-mut AML patients when administered together with hypomethylating agents [94,95] or low-dose AraC [96,97] or intensive chemotherapy [98,99]. An updated analysis of the CAVEAT trial implying the treatment of elderly AML patients treated with intensive chemotherapy and ≥12 months of VEN-based therapy showed that 45% of patients reponding to therapy ceased treatment: >50% of these patients remained in remission after ceasing treatment (treatment-free remission, TFR) [100]. The majority of patients with TFR displayed NPM1 and/or IDH2 mutations at diagnosis [100]. A retrospective analysis compared outcomes of NPM1-mut AML patients treated with three different regimens (intensive chemotherapy, hypomethylating agents alone and venetoclax plus hypomethylating agents): venetoclax plus hypomethylating agents improved OS compared to hypomethylating agents alone or to intensive chemotherapy [100]. Particularly, in patients treated with Ven+HMA an OS of 80% after median 1-year follow-up was observed [101].
A recent study retrospectively explored the response to venetoclax-based regimens in a group of 206 relapsing/refractory NPM1-mut AML patients in comparison with a group of NPM1-WT AML patients: high-intensity but not low-intensity regimens were associated wit a higher rate of complete responses in NPM1-mut patients compared to NPM1-WT patients (63% vs 37%, respectively for high-intensity regimens); the addition of venetoclax to low-intesity regimens significantly improved the CR and OS rate in NPM1-mut but not in NPM1-WT patients (71% vs 32%, respectively, for CR; 14.7 months vs 5.9 months, respectively for OS) [102].
High CD33 expression in NPM1-mut AMLs provided a rationale supporting the evaluation of the drug-conjugated anti-CD33 antibody gentuzumab ozogamicin (GO) in this AML subtype. The prospective randomised AMLSG 09-09 phase >III study evaluated the efficacy of induction therapy with idarubicin, cytarabine, and all-trans retinoic acid with or without GO; the early death rate was higher in the GO arm compared with the standard arm, while the incidence of relapse in patients achieving complete remission was lower in the GO arm compared to the standard arm [103]. GO failed to improve event-free survival (EFS) rate; subgroup analysis showed an improvement of EFS in FLT3-ITD-negative patients induced by GO [103]. The analysis of MRD levels showed that GO addition reduced the levels of residual NPM1-mut transcript levels during all treatment cycles, leading to a significantly lower relapse rate [104]. An updated analysis of the results observed in the AMLSG 09-09 study confirmed the absence of a significant benefit of GO on EFS and OS [105]. Subgroup analysis showed a benefit of GO in terms of EFS for patients FLT3-ITD-WT and patients with DNMT3A mutations [105]. It is important to note that the final results of this trial confirmed that GO administration significantly reduces the cumulative incidence of relapse rate, thus indicating that the addition of GO might reduce the need for salvage therapy [106].
The NCRI AML 29 trial randomized 1475 patients with newly-diagnosed AML or high-risk MDS, with no-adverse cytogenetics, to receive FLG-Ida or DA (Danurubicin plus AraC); 1031 of these patients were also randomized to receive a single or a fractionated dose of GO [107]. Subgroup analysis showed a significant improvement in NPM1-mut AML patients treated with FLAG-Ida-GO compared to DA-GO (Os 82% vs 64% respectively at 3 years); concerning FLT3-ITD-mutated patients, a significant OS benefit was observed among NPM1-mut/FLT3-mut patients [107].
Several potential targeted therapies against NPM1-mut AMLs have been discovered, inhibiting with some relevant biochemical properties of NPM1 mutant protein, interfering with NPM1 oligomerization or with the abnormal trafic of NPM1 mutant protein (XPO1 inhibitors), inducing selective NPM1-mutant protein degradation (ATRA/ATO, deguelin, (-)-epigallocatechin-3 gallate) and targeting the integrity of the nucleolar structure (actnomycin D) [107]. The properties of these dufferent drugs and their potential therapeutic implications for NPM1-mut AMLs have been recently reviewed [107,108]. Here the analysis of these drugs was restricted to those in a more advanced stage of clinical evaluation.
Experimental studies have supported a possible efficacy of menin inhibitors in the treatment of NPM1-mut AMLs. Histone modifiers MLL1 and DOTL1 control HOX gene expression and FLT3 expression in NPM1-mut AMLs [109]. Menin-MLL1 tarteing inhibited preleukemia cells in a mouse model of NPM1-mut AML cells [110] Importantly, menin inhibition synergizes with ven etoclax in mediating the inhibition of NPM1-mut and FLT3-mut AML cells [111]. Similarly, the combination of menin inhibitors with FLT3 inhibitors resulted in an enhanced inhibitory effect on the proliferation and stimulatory induction of apoptosis of primary FLT3-mut leukemic blasts [112] and of leukemia cells in a murine model of leukemia promoted by NPM1-mut and FLT3-ITD [113]. A recent study showed that menin inhibitors synergize with drugs targeting chromatin regulation and DNA damage, as well as with drugs targeting apoptosis and cell cycle; particularly interesting was the observation of a synergistic interaction bewtween menin inhibitors and ATRA [114].
Fiskus et al have given an important contribution to the understanding of the biochemical mechanisms through which the menin inhibitor ziftomenib (KO-539) inhibits menin activity: this drug triggers menin protein degradation through the ubiquitin-proteasome with consequent marked decline of menin levels and of the expression of menin dependendent genes, such as BCL1, MEIS1, FLT3, CDK6 and MEF2C; these effects are associated with induction of leukemic cell differentiation and reduced cell viability [115].
Initial phase I studies have shown a good tolerance and therapeutic efficacy of two menin inhibitors, KO-539 [116] and SNDX-5613 [117]. Recently, the first clinical results of these two menin inhibitors were reported. Issa et al. reported the results of the AUGMENT-101 trial, the first-in-human phase I trial of the menin inhibitor SNDX-5613. (revumenib) in patients with relapsed/refractory AML KMT2A-rearranged (46 patients) and NPM1-mut (14 patients) patients; these patients were heavily pretreated [118]. In the whole population of 60 enrolled patients, the overall response rate was 53% with a CR rate of 38%, of which 78% were MRD-negative; 21% of the NPM1-mut AML patients displayed a complete response, with a 100% MRD-negativity in this responding population [118]. Patients with acquired resistance to menin inhibition displayed somatic mutations in MEN1 at the revumenib-menin interface [119].
The second study, the trial KOMET-001, involved the phase I evaluation of KO-539 (ziftomenib) in adult relapsing/refractory AML pastients: in a heavily pre-treated cohort of relapsed/refractory NPM1-mut AML patients an overall response rate of 40% and a complete response rate of 35% were observed [120].Differentiation syndrome wqas an adverse event observed in soime patients; the occurrence of differentiation syndrome was associoated with improved response [120]. Future clinical trials will involve the evaluation of the safety and efficacy of ziftomenib in combination with venetoclax plus azacitidine or the 7+3 chemotherapy regimen (trial KOMET-007) or the evaluation of revumenib in association with the chemotherapy regimen based on fludarabine and cytarabine (trial AUGMENT-102).
15. Conclusions
NPM1-mut AMLs represent the largest genetically defined group of AMLs. NPM1 mutations appear to be secondary events, being virtually absent in CHIP, and occurring after mutations in DNMT3A, IDH1 or NRAS during the development of AML. The key role of NPM1 gene in these AMLs is clear, but it equally evident that NPM1 mutations alone are not able to generate a full leukemic process and have to cooperate with other mutant driver genes. The diversity of these oncogenetic partners of NPM1 mutant gene generates a consistent degree of heterogeneity; thus, it was estimated that in these AMLs a number of five oncogenic mutations per patient. This implies that NPM1-mut AMLs must be explored by NGS for their mutational profile and consequently stratified in various risk groups. The presence of TP53 mutations, the triple mutational combination DNMT3A/NPM1/FLT3-ITD, the presence of MDS-related co-mutations and of high-risk chromosomic abnormalities are all conditions that shift NPM1-mut AML patients into an adverse-risk condition.
Author Contributions
For the analysis of literature data all the authors contributed equally; for writing the manuscript and for original draft preparation all the authors contributed equally. U.T. contributed to the final editing of the manuscript. All the authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analysed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, K.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Hauser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Tazi, Y.; Arango-Ossa, J.E.; Zhou, Y.; Bernard, E.; Thomas, I.; Gilkes, A.; Freeman, S.; Pradat, Y.; Johnson, S.J.; Hills, R.; et al. Unified classification and risk stratification in acute myeloid leukemia. Nature Commun 2022, 13, 4622. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of myeloid neoplasms and acute leukemia: integrating morphological, clinical and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla OAkkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan John, K.C.; Chen, W.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 ELN Recommendations from an International Expert Panel. Blood. 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-mutated acute myeloid leukemia: from bench to bedside. Blood 2020, 136, 1707–1720. [Google Scholar] [CrossRef]
- Angenendt, L.; Rollig, C.; Montesinos, P.; Martinez-Cuadròn, D.; Barragan, E.; Garcia, R.; Botella, C.; Martinez, P.; Ravandi, F.; Kadia, T.; et al. Chromosomal abnormalities and prognosis in NPM1-mutated acute myeloid leukemia: a pooled analysis of individual patient data from nine international cohorts. J Clin Oncol 2019, 37, 2632–2642. [Google Scholar] [CrossRef]
- Alpermann, T.; Schnittger, S.; Eder, C.; Dicker, F.; Meggendorfer, M.; Kern, W.; Schmid, C.; Aul, C.; Staib, P.; Wendtner, C.M.; Schmitz, N.; et al. Molecular subtypes of NPM1 mutations have different clinical profiles specific patterns of accompanying molecular mutations and varying outcome in intermediate risk acute myeloid leukemia. Haematologica 2016, 101, e55. [Google Scholar] [CrossRef]
- Bailey, G.D.; Doolan, L.; Baskar, A.; Smith, L.C.; Seedhouse, C.H. Preferential transcription of the mutated allele in NPM1 mutated acute myeloid leukemia. Scient Rep 2020, 10, 17695. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.S.; Ramabradan, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 maintains the elukemic state through HOX expression. Cancer Cell 2018, 34, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Uckelmann, H.J.; Haarer, E.L.; Takeda, R.; Wong, E.M.; Hatton, C.; Marinaccio, C.; Perner, F.; Rajput, M.; Antonissen, N.; Wen, Y.; et al. Mutant NPM1 directly regulates oncogenic transcription in acute myeloid leukemia. Cancer Discov 2023, 13, 746–765. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.D.; Fan, D.; Han, Q.; Liu, Y.; Miao, H.; Wang, X.; Li, Q.; Chen, D.; Gore, H.; Himadewi, P.; et al. Mutant NPM1 hijacks transcriptional hubs to maintain pathogenic gene programs in acute myeloid leukemia. Cancer Discov 2023, 13, 724–745. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, A.; Ammer, T.; Kechter, A.; Rawat, V.; Sinha, A.; Gonzalez-Menendiez, I.; Quintanilla-Martinez, L.; Azoitein, A.; Gunes, C.; Mupo, A.; et al. Npmi haploinsufficiency in collaboration with MEIS1 is sufficient to induce AML in mice. Blood Adv 2023, 7, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, M.A.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; Wall, K.; et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Sargas, C.; Ayala, R.; Larrayoz, M.J.; Chillon, M.C.; Carillo-Cruz, E.; Bialbao-Sieyro, C.; Prados de la Torre, E.; Martinez-Cadron, D.; Rodriguez-Veiga, R.; Boluda, B.; et al. Molecular landscape and validation of new genomic classification in 2668 adult AML patients: real life data from the PETHEMA group. Cancers 2023, 15, 438. [Google Scholar] [CrossRef]
- Petterson, L.; Holmgren, B.; Juliusson, G.; Lazaveric, V.; Ehinger, M. Mutational spectrum of de novo NPM1-mutated acute myeloid leukemia patients older than 75 years. Leuk & Lymphoma 2021, 62, 1958–1966. [Google Scholar]
- Lachowiez, C.A.; Loghavi, S.; Kadia, T.M.; Daver, N.; Borthakur, G.; Pemmaraju, N.; Naqvi, K.; Alvarado, Y.; Yilmaz, M.; Short, N.; et al. Outcomes of older patients with NPM1-mutated AML: current treatments and the promise of venetoclax-based regimens. Blood Adv 2020, 4, 1311–1320. [Google Scholar] [CrossRef]
- Khan, A.M.; Reddy, S.N.; Aly, M.; Dhillon, V.; Sbihi, A.A.; Kewan, T.; Bahaj, W.; Gurnari, C.; Al_Share, B.; Dyson, G.; et al. Comprehensive age-stratified impact of NPM1-Mutation in acute myeloid leukemia. Blood 2022, 140, 1433–1434. [Google Scholar] [CrossRef]
- Cantu, M.D.; Kanagal-Shamanna, R.; Wang, S.; Kadia, T.; Bueso-Ramos, C.E.; Patel, S.S.; Geyer, T.T.; Tam, W.; Madanat, Y.; et al. Clinicopathologic and molecular analysis of normal karyotype-related and de novo acute myeloid leukemia: a multi-institutional study by the bone marrow pathology group. JCO Precis Oncol 2023, 7, e2200400. [Google Scholar] [CrossRef] [PubMed]
- Ohtman, J.; Meggendorfer, M.; Tiacci, E.; Thiede, C.; Schlenk, R.; Dillon, R.; Stasik, S.; Venanzi, A.; Bertoli, S.; Delabesse, E.; et al. Overlapping features of therapy-related and de novo NPM1-mutated AML. Blood 2023, 141, 1846–1857. [Google Scholar]
- Mason, E.F.; Hasserjian, R.P.; Aggarwal, N.; Seegmiller, A.C.; Podznyakova, O. Blast phenotype and comutations in acute myeloid leukemia with mutated NPM1 influence disease biology and outcome. Blood Adv 2019, 3, 3322–3332. [Google Scholar] [CrossRef] [PubMed]
- Mer, A.S.; Haeth, E.M.; Tonekaboni, S.A.M.; Dogan-Artun, N.; Nair, S.K.; Murison, A.; Garcia-Prat, L.; Shlush, L.; Hurren, R.; Voisin, V.; et al. Biological and therapeutic implications of a unique subtype pf NPM1 mutated AML. Nat Commun 2021, 12, 1054. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.Y.; Li, J.F.; Zhu, Y.M.; Lin, X.J.; Wen, L.J.; Zhang, F.; Zhang, Y.L.; Zhao, M.; Fang, H.; Wang, S.Y.; et al. Transcriptome-based molecular subtypes and differentiation hierarchies improve the classification framework of acute myeloid leukemia. Proc Natl Acad Sci USA 2022, 119, e2211429119. [Google Scholar] [CrossRef]
- Cappelli, L.V.; Meggendorfer, M.; Baer, C.; Nadarajah, N.; Kern, W.; Haferlach, T.; Haferlach, C.; Hollein, A. NPM1 mutated AML is characterized by pre-leukemic mutations and the persistence and acquisition of co-mutations in molecular remission leads to inferior prognosis. Blood 2018, 132 (Suppl. 1), 996. [Google Scholar] [CrossRef]
- Cappelli, L.V.; Meggendorfer, M.; Baer, C.; Nadarajah, N.; Hutter, S.; Jeromin, S.; Dicker, F.; Kern, W.; Haferlach, T.; Haferlach, C.; Hollein, A. Indeterminate and oncogenic potential: CHIP vs CHOP mutations in AML with NPM1 alteration. Leukemia 2022, 36, 394–402. [Google Scholar] [CrossRef]
- Potter, N.; Miraki-Moud, F.; Ermini, L.; Titley, I.; Vijayaraghavan, G.; Papaemmanuil, E.; Campbell, P.; Gribben, J.; Taussig, D.; Graeves, M. Single cell analysis of clonal architecture in acute myeloid leukemia. Leukemia 2019, 33, 1113–1123. [Google Scholar] [CrossRef]
- Desai, P.; Marcia-Trichant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Ploen, G.G.; Nederby, L.; Guldberg, P.; Hansen, M.; Ebbesen, L.H.; Jensen, U.B.; Hokland, P.; Aggerholm, A. Persistence of DNMT3A mutations at long-term remission in adult patients with AML. Br j Haematol 2014, 167, 478–486. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Weber, D.; Paschka, P.; Kaumanns, A.; Krieger, S.; Carbacioglu, A.; Kronke, J.; Kapp-Schworer, S.; Kramer, D.; Horst, H.A.; et al. DNMT3A mutant transcriopt levels persist in remission and do not predict outcome in patients with acute myeloid leukemia. Leukemia 2018, 32, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Bullinger, L.; Teleanu, V.; Tschrtz, F.; Gaidzik, V.I.; Kuhn, M.; Rucker, F.G.; Holzmann, K.; Paschka, P.; Kapp-Schworer, S.; et al. Clonal evolution in relapsed NPM1-mutatted acute myeloid leukemia. Blood 2013, 122, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Hollein, A.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T. NPM1 mutated AML can relapse with wild-type NPM1: persistent clonal hematopoiesis can drive relapse. Blood Adv 2018, 2, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Cocciardi, S.; Dolnik, A.; Kapp-Schoerer, S.; Rucker, F.G.; Lux, S.; Blatte, T.J.; Skambraks, S.; Kronke, J.; Heidel, F.H.; Schnoder, T.M.; et al. Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nat Commun 2019, 10, 2031. [Google Scholar] [CrossRef]
- Hernandez-Sanchez, A.; Villaverde-Ramiro, A.; Elicegui, J.M.; Gonzalez, T.; Benner, A.; Strong, E.; Castellani, G.; Eckman, C.A.; Versluis, J.; Abaigar, M.; et al. NPM1 mutated AML: impact of co-mutational patterns. Results of the European HARMONY Alliance. HemaSphere 2022, 6, S3. [Google Scholar] [CrossRef]
- Hernandez-Sanchez, A.; Villaverde-Ramiro, A.; Strang, E.; Castellani, G.; Heckman, C.A.; Versluis, J.; Abaigar, M.; Sobas, M.A.; Melchor, R.A.; Benner, A.; et al. Machine learning allows the dientification on new co-mutationsl patterns with prognostic implications in NPM1-mutated AML: results of the European Hasrmiony Alliance. Blood 2022, 140 (Suppl. 1), 739–742. [Google Scholar] [CrossRef]
- Mrozek, K.; Kohlschmidt, J.; Blachly, J.S.; Nicolet, D.; Carroll, A.J.; Archer, K.J.; Mims, A.S.; Lerkin, K.T.; Orwick, S.; Oakes, C.C.; et al. Outcome prediction by the 2022 European Leukemia Net genetic-risk classification for adults with acute myeloid leukemia: an Alliance study. Leukemia 2023, 37, 788–798. [Google Scholar] [CrossRef]
- Angenendt, L.; Rollig, C.; Montesinos, P.; Ravandi, F.; Juliusson, G.; Récher, C.; Htzykson, R.; Racil, Z.; Wei, A.H.; Schliemann, C. Revisiting coexisting chromosomal abnormalities in NPM1-mutated AML in light of the revised ELN 2022 classification. Blood 2023, 141, 433–435. [Google Scholar] [CrossRef]
- Patel, S.S.; Kuo, F.C.; Gibson, C.J.; Steensma, D.P.; Soiffer, R.J.; Edwin III, A.; Chen, Y.B.A.; Fathi, A.T.; Graubert, T.A.; Brunner, A.M.; et al. High NPM1-mutant allele burden at diagnosis predicts unfavorable oucomes in de novo AML.
- Abbas, H.A.; Ravandi, F.; Loghavi, S.; Borthakur, G.; Kadia, T.M.; Jabbour, E.; Takahashi, K.; Cortes, J.; Issa, G.C.; Konopleva, M.; et al. NPM1 mutant variant allele frequency correlates with leukemia burden but does not provide prognostic information in NPM1-mutated AML. Am J Hematol 2019, 94, E158–E160. [Google Scholar] [CrossRef]
- Rothenberg-Thurley, M.; Herold, T.; Gorlich, D.; Sauerland, C.; Janke, H.; Prassek, V.V.; Konstandin, N.P.; Dufour, A.M.; Schneider, S.; Ksienzyk, B.; et al. NPM1 variant allele frequency and outcomes in AML. Blood, 132 (Suppl. 1), 1486.
- Patel, S.S.; Pinkus, G.S.; Ritterhouse, L.L.; Segal, J.P.; Dal Cin, P.; Restrepo, T.; Harris, M.H.; Stone, R.M.; Hasserjian, R.P.; Weinberg, O.K. High NPM1 mutant allele burden at diagnosis correlates with minimal residual disease at first remission in de novo acute myeloid leukemia. Am J Hematol 2019, 94, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.V.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Hutter, S.; Kern, W.; Haferlach, T.; Haferlach, C.; Hollen, A. DNAMT3A mutations are over-represented in younf adults with NPM1 mutated AML and prompt a distinct co-mutational pattern. Leukemia 2019, 33, 2741–2746. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.V.; Meggendorfer, M.; Baer, C.; Nadarejah, N.; Hatter, S.; Jeromin, S.; Dicker, F.; Kern, W.; Haferlach, T.; Haferlach, C.; et al. Indeterminate and oncogenic potential: CHIP vs CHOP mutations in AML with NPM1 alterations. Leukemia 2022, 36, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Onate, G.; Bataller, A.; Garrido, A.; Hoyos, M.; Vives, S.; Coll, R.; Tommo, M.; Sampol, A.; Escoda, L.; Salamero, O.; et al. Prognostic impact of DNMT3A mutation in acute myeloid leukemia with mutataed NPM1. Blood Adv 2022, 6, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Loghavi, S.; Zuo, Z.; Ravandi, F.; Kantajian, H.M.; Bueso-Ramos, C.; Zhang, L.; Singh, R.R.; Patel, K.P.; Medeiros, L.J.; Stingo, F.; et al. Clinical features of de novo acute myeloid leukemia with concurrent DNMT3A, FLT3 and NPM1 mutations. J Hematol Oncol 2014, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Reyes-Palomares, A.; He, L.; Bergeron, A.; Lavallée, V.P.; Lemieux, S.; Gendron, P.; Rohde, C.; Xia, J.; Jaghdane, P.; et al. Hepatic leukemia factor is a novel leukemic stem cell regulator in DNMT3A, NPM1, and FLT3-ITD triple-mutatedAML. Blood 2019, 134, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Beserra, M.F.; Lima, A.S.; Piqué-Borras, M.R.; Silveira, D.R.; Coelho-Silva, J.L.; Pereira-Martins, D.A.; Weinhauser, I.; Franca-Neto, P.L.; Quek, L.; Corby, A.; et al. Co-occurrence of DNMT3A, NPM1, FLT3 mutations identifies a subset of acute myeloid leukemia with adverse prognosis. Blood 2020, 135, 870–875. [Google Scholar] [CrossRef]
- Holz-Scheitinger, C.; Matie, D.M.; Reich, N.O. Mutations in Dna methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J Biol Chem 2012, 287, 30941–30951. [Google Scholar] [CrossRef]
- Genovese, G.; Kahler, A.K.; Hnadsaker, R.E. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequences. N Engl J Med 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Wakita, S.; Marumo, A.; Morita, K.; Kako, S.; Toya, T.; Najima, Y.; Doki, N.; Kanda, J.; Kuroda, J.; Mori, S.; et al. Mutational analysis on DNMT3A improves the prognostic stratification of patients with acute myeloid leukemia. Cancer Science 2013, 114, 1297–1308. [Google Scholar] [CrossRef]
- Rucker, F.G.; Luck, T.J.; Benner, A.; Krzykalla, J.; Gathmann, I.; Voso, M.T.; Amadori, S.; Prior, T.W.; Brandwein, J.M.; Appelbaum, F.R.; et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia 2022, 36, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.; Al Ali, N.; Ball, S.; Grenet, J.; Hana, C.; Deutsch, Y.F.; Tashkandi, H.; Zhang, L.; O Hussaini, M.; Yun, S.; et al. The prognostic impact of FLT3 in NPM1-mutated AML: co-occurrence of FLT3-ITD and FLT3-TKD cofers poor outcomes. Blood 2022, 140 (Suppl. 1), 3435–3437. [Google Scholar] [CrossRef]
- Jentsch, M.; Bischof, L.; Brauer, D.; Backaus, D.; Ussmann, J.; Franke, G.N.; Vucinic, V.; Platzbecker, U.; Schwind, S. Clinical implications of the FLT3-ITD allelic ratio in acute myeloid leukemia in the context of an allogeneic stem cell transplantation. Cancers 2023, 15, 1312. [Google Scholar] [CrossRef]
- Grob, T.; Sanders, M.A.; Vonk, C.M.; Kavelaars, F.G.; Rijken, M.; Hanekamp, D.; Gradowska, P.L.; Cloos, J.; Floisand, Y.; Kooy, M.M.; et al. Prognostic value of FLT3-internal tandem duplication residual disease in acute myeloid leukemia. J Clin Oncol 2023, 41, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Middeke, J.M.; Metzeler, K.H.; Rollig, C.; Kramer, M.; Eckardt, J.N.; Stasik, S.; Greif, P.A.; Spiekermann, K.; Rothenbrg-Thurley, M.; Krug, U.; et al. Differential impact of IDH1/2 mutational subclasses on outcome in adult AML: results from a large munlticenter study. Blood Adv 2022, 6, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Spinelli, O.; Meggendorfer, M.; Martelli, M.P.; Bigerna, B.; Ascani, S.; Stein, H.; Rambaldi, A.; Haferlach, T. IDH1-R132 changes vary according to NPM1 and other mutations status in AML. Leukemia 2019. [Google Scholar] [CrossRef] [PubMed]
- Meggendorfer, M.; Cappelli, L.V.; Walter, W.; Haferlach, C.; Kern, W.; Falini, B.; Haferlach, T. IDH1R132, IDH2R140 and IDH2R172 in AML: different genetic landscapes correlate with outcome and may influence targeted treatment strategies. Leukemia 2018, 32, 1249–1253. [Google Scholar] [CrossRef]
- Eckardt, J.N.; Stasik, S.; Rollig, C.; Sauer, T.; Scholl, S.; Hochaus, A.; Crysandt, M.; Bummendorf, T.; Naumann, R.; Steffen, B.; et al. Alterations of cohesin complex genes in acute myeloid leukemia: differential co-mutations, clinical presentation and impact on outcome. Blood Cancer J 2023, 13, 18. [Google Scholar] [CrossRef]
- Simonetti, G.; Mengucci, C.; Padella, A.; Fonzi, E.; Picone, G.; Delpino, C.; Nanni, J.; De Tommaso, R.; Franchini, E.; Papayannidis, C.; et al. Integrated genomic-metabolic classification of acute myeloid leukemia defines a subgroup with NPM1 and cohesin/DNA damage mutations. Leukemia 2021, 35, 2813–2826. [Google Scholar] [CrossRef]
- Benard, BA.; Leak, L.B.; Azizi, A.; Thomas, D.; Gentles, A.J.; Majeti, R. Clonal architecture predicts clinical outcomes and drug sensitivity in acute myeloid leukemia. Nat Commun 2021, 12, 7244. [Google Scholar] [CrossRef]
- Meyer, A.E.; Stelloh, C.; Pulakanti, K.; Burns, R.; Fisher, J.B.; Heimbruch, K.E.; Tarima, S.; Furumo, Q.; Brennan, J.; Zheng, Y.; et al. Cominatorial genetics reveals the Dock1-Rac2 axis as a potential target for the treatment of NPM1;Cohesin mutated AML. Leukemia 2022, 36, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, Z.; Li, T.; Li, Y.; Wang, W.; Hao, Q.; Xie, X.; Wan, D.; Jiang, Z.; Wang, C.; Liu, Y. Mutational sepctrum and prognosis in NRAS-mutated acute myeloid leukemia.
- Rivera, D.; Kim, K.; Kanagal-Shamanna, R.; Borthakur, G.; Montalban-Bravo, G.; Daver, N.; DiNardo, C.; Short, N.J.; Yilmaz, M.; Pemmaraju, N.; et al. Implications of RAS mutational status in subsets of patients with newly diagnosed acute myeloid leukemia across therapy subtypes. Am J Hematol 2022, 97, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Erba, H.P. Diagnosis and treatmentr of patients with acute myeloid leukemia with myelodysplastic changes (AML-MRC). Am J Clin Pathol 2020, 154, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Bains, A.; Luthra, R.; Medeiros, L.J.; Zuo, Z. FLT3 and NPM1 mutations in myelodysplastic syndromes: frequency and potential value for predicting progression to acute myeloid leukemia. Am J Clin Pathol 2011, 135, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biologica implicxations of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Forghieri, F.; Paolini, A.; Morselli, M.; Bigliardi, S.; Bonacorsi, G.; Leonardi, G.; Coluccio, V.; Maccaferri, M.; Fantuzzi, V.; Faglioni, L.; et al. NPM1 mutations may reveal acute myeloid leukemia in cases otherwise morphologically diagnosed as myelodysplastic syndromes or myelodysplatic/myeloproliferative neoplasms. Leuk Lymphoma 2015, 56, 3222–3226. [Google Scholar] [CrossRef]
- Maurya, N.; Mohanty, P.; Dhangar, S.; Panchal, P.; Jijina, F.; Mathan, L.P.; Shanmukhaiah, C.; Madkaikar, M.; Vudinti, B.R. Comprehensive analysis of genetic factors predicting overall survival in myelodysplastic syndromes. Scient Rep 2020. [Google Scholar] [CrossRef]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Sasaki, K.; Patel, K.; Ganan-Gomez, I.; Jabbour, E.; Kadia, T.; Ravandi, F.; DiNardo, C.; Borthakur, G.; Takahashi, K.; et al. NPM1 mutations define a sepcific subgroup of MDS and MDS/MPN patients with favorable outcomes with intensive chemotherapy. Blood Adv 2019, 3, 922–933. [Google Scholar] [CrossRef]
- Wang, C.; Chan, O.; Ali, A.; Kuykendall, A.; Padron, E.; Sweet, K.; Lancet, J.; Komrokji, R.S.; Sallam, D.A. Clinical characterization and outcomes of patients with NPM1-mutated myelodysplastic syndromes or chronic myelomonocytic leukemia. Blood 2022, 140, 6936–6937. [Google Scholar] [CrossRef]
- Patel, S.S.; Ptashkin, R.N.; Sudigh, S.; Bugg, A.; Geyger, J.T.; Xu, M.L.; Prebet, T.; Mason, E.F.; Seegmille, A.C.; et al. Clinicopathologic and genetic characterization of nonacute NPM1-mutated myeloid neoplasms. Blood Adv 2019, 3, 1540–1545. [Google Scholar] [CrossRef]
- Falini, B.; Martelli, M.P.; Brunetti, L.; Gjerten, B.T.; Andresen, V. The NPM1 defines AML irrespective of blasts count. Am J hematol 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Gardin, C.; Pautas, C.; Fournier, E.; Itzykson, R.; Lemasle, E.; Bourhis, J.H.; Adès, L.; Marolleau, J.P.; Malfuson, J.V.; Gastaud, L.; Raffoux, E. Added prognostic value of secondary AML-like hene mutations in ELN intermediate-risk older AML: ALFA-1200 study results. Blood Adv 2020, 4, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, I.; Lenk, M.; Haferlach, T.; Stengel, A.; Hutter, S.; Baer, C.; Meggendorfer, M.; Kern, W.; Haferlach, C. AML, NOS and AML-MRC as defined by multilineage dysplasia share a common mutation pattern which is distinct from AML-MRC as defiend by MDS-related cytogenetics. Leukemia 2022, 36, 1929–1942. [Google Scholar] [CrossRef]
- Tsai, X.; Sun, K.J.; Lo, N.Y.; Tien, F.M.; Kuo, Y.Y.; Tseng, M.H.; Peng, Y.L.; Chuang, Y.K.; Ko, B.S.; Tang, J.L.; et al. Poor prognostic implications of myelodysplastic-related mutations in both older anf younger patients with de novo AML. Blood Cancer J 2023, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.; Al Ali, N.; Tashkandi, H.; Ellis, A.; Ball, S.; Grenet, J.; Hana, C.; Deutsch, Y.E.; Zhang, L.; O Hussaini, M.; et al. Secondary AML mutations confer poor prognosis in aptients with ELN favorable risk NPPM1-mutated AML. Blood 2022, 140 (Suppl. 1), 3419–3421. [Google Scholar] [CrossRef]
- Wang, Y.; Quesada, A.E.; Zuo, Z.; Medeiros, L.J.; Yin, C.C.; Li, S.; Xu, J.; Borthakur, G.; Li, Y.; Yang, C.; et al. The impact of mutation of myelodysplasia-related genes in de novo acute myeloid leukemia carrying NPM1 mutation. Cancers 2023, 15, 198. [Google Scholar] [CrossRef]
- Smith, E.C.; Atenafu, E.G.; Bankar, A.; Chan, S.M.; Davidson, M.B.; Gupta, V.; Minden, M.D.; Richard-Carpentier, G.; Schimmer, A.D.; Schuh, A.C.; et al. Clinical outcomes in de novo versus secondary NPM1-mutated AML. Blood 2022, 140 (Suppl. 1), 8952–8953. [Google Scholar] [CrossRef]
- Zhao, D.; Zairf, M.; Eladl, E.; Capo-Chichi, J.M.; Smith, A.C.; Atenafu, E.G.; Tierens, A.; Minden, M.D.; Schuh, A.; Chang, H. NPM1-mutated AML-MRC diagnosed on the basis of history of MDS or MDS/MPN frequently harbours escondary-type mutations and confers inferior outcome compared to AML with mutated NPM1. Leuk Res 2022, 118, 106869. [Google Scholar] [CrossRef]
- Tien, F.M.; Hou, H.A.; Tsai, C.T.; Tang, J.L.; Chen, C.Y.; Kuo, Y.Y.; Li, C.C.; Lin, C.T.; Yao, M.; Huang, S.Y. Hyperleukocytosis is associated with ditsinct genetic alterations and is an independent poor-risk factor in de novo acute myeloid leukemia patients. Eur J Hematol 2018, 101, 86–94. [Google Scholar] [CrossRef]
- Pastore, F.; Pastore, A.; Rothenberg-Thurley, M.; Metzler, K.H.; Ksienzyk, B.; Schneider, S.; Bohlander, S.K.; Braess, J.; Sauerland, M.C.; Gorlich, D.; Berdel, W.E.; et al. Molecular profiling of patients with cytogeneticcally normal acute myeloid leukemia and hyperleukocytosis. Cancer 2022, 128, 4213–4222. [Google Scholar] [CrossRef] [PubMed]
- Ramdas, B.; Reddy, P.L.; Mali, R.S.; Pasupuleti, S.K.; Zhang, J.; Kelley, M.R.; Paczesny, S.; Zhang, C.; Kapur, R. Combined heterozygosity of FLT3ITD, TET2, and DNMT3A results in aggressive leukemia. JCI Insight 2022, 7, e162016. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.C.; Chou, S.C.; Liu, C.Y.; Chen, C.Y.; Hou, H.A.; Kuo, Y.Y.; Lee, M.C.; Ko, B.S.; Tang, J.L.; Yao, M.; et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 2011, 118, 3803–3810. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Xu, Y.; Yin, J.; Tian, H.; Chen, S.; Wu, D.; Sun, A. TET2 mutation is unfavorable prognostic factor in cytogenetically normal acute myeloid leukemia patients with NPM1+ and FLT3-ITD- mutations. Int J Hematol 2014, 100, 96–104. [Google Scholar] [CrossRef]
- Stasik, S.; Eckardt, J.N.; Kramer, M.; Rollig, C.; Kramer, A.; Scholl, S.; Hocchaus, A.; Crysandt, M.; Brummanford, T.H.; Naumann, R.; et al. Impact of PTPN11 mutations in clinical outcome analyzed in 1529 patients with acute myeloid leukemia. Blood Adv b 2011, 5, 3279–3289. [Google Scholar] [CrossRef] [PubMed]
- Fobare, S.; Kohlschmidt, J.; Ozer, H.G.; Mrozek, K.; Nicolet, D.; Mims, A.S.; Garzon, R.; Blachly, J.S.; Orwick, S.; Carroll, A.J.; et al. Molecular, clinical and prognostic implications of PTPN11 mutations in acute myeloid leukemia. Blood Adv 2022, 6, 1371–1380. [Google Scholar] [CrossRef]
- Alfayez, M.; Issa, G.C.; Patel, K.P.; Wang, F.; Wang, X.; Short, N.J.; Cortes, J.E.; Kadia, T.; Ravandi, F.; Pierce, S.; et al. The clinical impact of PTPN11 mutastions in adults with acute myeloid leukemia. Leukemia 2021, 35, 691–700. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, F.; Huo, L.; Cai, W.; Wang, Q.; Wen, L.; Yan, L.; Shen, H.; Xu, X.; Chen, S. Clinical characteristics and prognostic analysis of acute myeloiod leukemia patients with PTPN11 mutations. Hematology 2022, 27, 1184–1190. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Rotrhenberg-Thurley, M.; Gorlich, D.; Sauerland, M.C.; Dufour, A.M.; Schneider, S.; Subkelewe, M.; Braess, J.; Wormann, B.J.; Wormann, W.E.; et al. PTPN11 mutations and outcome in adult patients with acute myeloid leukemia. Blood 2020, 136 (Suppl. 1), 4–5. [Google Scholar] [CrossRef]
- Mrozek, K.; Marcucci, G.; Nicolet, D. Prognostic significance of the Uropean Leukemia Net standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol 2012, 30, 4515–4523. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; et al. Venetoclax combined with decitabine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullakat, V.; Thirman, M.J.; Garcia, J.S.; M Wei, A.H. Azacitidine and vcenetoclax in previously untreated acute myeloid leukemia. N Engl J Med 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Strickland, S.A.; Hou, J.Z.; Fiedler, J.W.; Lin, T.L.; Walter, R.B. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/Ii study. J Clin Oncol 2019, 37, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Montesinos, P.; Inanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K. Venetoclax plus Idac for newly diagnosed AML imnelegible for intensive chemotherapy: a phase 3 randomised placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.C.; Roberts, A.W.; Reynolds, I.; Fong, C.Y.; Ting, S.B.; Salmon, J.M. Chemotherapy and venetoclax in elderly acute myeloid leukemia trial (Caveat): a phase Ib dose-escalation study of venetoclax combined with modified intensive chemotherapy. J Clin Oncol 2020, 38, 3506–3517. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loighavi, S.; Xiao, L.; Kadia, T. Venetoclax combined with Fag-Ida induction and consolidation in newly diagnosed and relapsed or refractory acute myeloid leukemia. J Clin Oncol 2021, 39, 2768–2778. [Google Scholar] [CrossRef]
- Chua, C.C.; Hammond, D.; Kent, A.; Tiong, I.S.; Konopleva, M.; Pollyea, D.A.; DiNardo, C.D.; Wei, A.H. Treatment-free rbemission after ceasing venetoclax-based therapy in patients with acute myeloid leukemia. Blood Adv 2022, 6, 3879–3885. [Google Scholar] [CrossRef]
- Lachowiez, C.A.; Loghavi, S.; Kadia, T.M.; Daver, N.; M Barthakur, G.; Pemmaraju, N.; Naqvi, K.; Alvarado, Y.; Yilmaz, M.; Short, N.; et al. Oucomes of older patients with NPM1-mutated AML: current treatments and the rpomise of venetoclax-based regimens. Blood Adv 2020, 4, 1311–1320. [Google Scholar] [CrossRef]
- Issa, G.C.; Bidikian, A.; Venugopal, S.; Konopleva, M.; DiNardo, C.D.; Kadia, T.M.; Borthakur, G.; Jabbour, E.; Pemmaraju, N.; Yilmaz, M.; et al. Clinical outcomes associated with NPM1 mutations in patients with relapsed or refractory AML. Blood Adv 2023, 7, 933–942. [Google Scholar] [CrossRef]
- Schlenk, R.F.; Paschka, P.; Krzyalla, J.; Weber, D.; Kapp-Schwoerer, S.; Gaidzik, V.I.; Leis, C.; Fiedler, W.; Kindler, T.; Schroeder, T. Gentuzumab ozogamicin in NPM1-mutated acute myeloid leukemia: early results from the prospective. Randomized AMLSG-09-09. Phase III study. J Clin Oncol 2020, 38, 623–632. [Google Scholar] [CrossRef]
- Kapp-Schwoerer, S.; Weber, D.; Carbacioglu, A.; Gaidzik, V.I.; Pashka, P.; Kronke, J.; Theis, F.; Rucker, F.G.; Teleanu, M.V.; panina, E.; et al. Impact of gemtuzumab ozogamicin. On MRD and relapse risk in patients with NPM1-mutated AML: results from the AMLSG 09-09 trial. Blood 2020, 136, 3041–3050. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Weber, D.; Krzykalla, J.; Fiedler, W.; Kuhn, M.; Schroeder, T.; Mayer, K.; Lubbert, M.; Watted, M.; Gotze, K.; et al. Gemtuzumab ozogamicin plus intensive chemotherapy for patients. With NPM1-mutated acute myeloid leukemia. Blood 2022, 140 (Suppl. 1), 218. [Google Scholar] [CrossRef]
- Dohner, H.; Weber, D.; Krzykalla, J.; Fiedler, W.; Kuhn, M.; Schroeder, T.; Mayer, K.; Lubbert, M.; Watted, M.; Gotze, K.; et al. Intensive chemotherapy with or without gemtuzumab ozogamicin in patients with NPM1-mutated acute myeloid leukemia (AMLSG 09-09): a randomised, open-lebal, munlticentre, phase 3 trial. Lancet Hematol 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Russell, N.H.; Wilhelm-Benarizi, C.; Knapp, S.; Batten, L.M.; Canham, J.; Hinson, E.L.; Overgaad, U.M.; Gilkes, A.; Othman, J.; Potter, N.; et al. PLAG-Ida combined with gemtuzumab ozogamicin (GO) improves event free survival in younger patients with newly diagnosed acute myeloid leukemia (AML) and shows an overall survival benefit in NPM1 and FLT3 mutated subgroups. Results fgrom the UK NCRI AML 19 trial. Blood 2022, 140 (Suppl. 1), 526–528. [Google Scholar]
- Ranieri, R.; Pianigiani, G.; Sciabolacci, S.; Perriello, V.M.; Marra, A.; Cardinali, V.; Pierangeli, S.; Milano, F.; Gionfriddo, I.; Brunetti, L.; et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia 2022, 36, 2351–2367. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, P.; Chang, C.L.; Zhong, S.Z.; Zhu, H.H. Targetd therapy in NPM1-mutated AML: knowns and unknowns. Front Oncol 2022, 12, 972606. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.W.; Song, E.; Feng, Z.; Sinha, A.; Chen, C.W.; Deshpande, A.J.; et al. Targeting chromatin regulators inhibits leukemogenic gene expression in Npm1 mutant leukemia. Cancer Discovery 2016, 6, 1166–1181. [Google Scholar] [CrossRef]
- Uckelmann, H.J.; Kim, S.M.; Wong, E.M.; Hatton, C.; Giovinazzo, H.; Gadrey, J.Y. Therapeutic targeting of preleukemia cells in a mouse model of Npmn1 mutant acute myeloid leukemia. Science 2020, 367, 586–590. [Google Scholar] [CrossRef]
- Carter, B.Z.; Tao, W.; Mak, P.Y.; Ostermann, L.B.; Mak, D.; nMcGeehan, G. Menin inhibition decreases bcl-2 and synergizes with venetoclax in Npm1/Flt3-mutated AML. Blood 2021, 138, 1637–1641. [Google Scholar] [CrossRef]
- Dzama, M.M.; Steiner, M.; Rausch, J.; Sasca, D.; Schonfeld, J.; Kunz, K. Synergistic trargeting of Flt3 mutations in AML via combined menin-MLL and Flt3 inhibition. Blood 2020, 136, 2442–2456. [Google Scholar] [CrossRef]
- Miao, H.; Kim, E.; Chen, D.; Purohit, T.; Kempinska, K.; Ropa, J. Combinatoria treatment with menin and Flt3 inhibitors induces complete remission in AML models with activating Flt3 mutations. Blood 2020, 136, 2958–2963. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.; Dzama, M.M.; Dolgikh, N.; Stiller, H.L.; Bohl, S.R.; Lahrmann, C.; Kunz, K.; Kessler, L.; Echchannaoui, H.; Chen, C.W.; et al. Menin inhibitor ziftyomenib (KO-539) synergiozes with drugs targeting chromatin regulation or apoptosis and sisitizes acute myeloid leukemia with MLL rearrangement or NPM1 mutation to venetoclax. Haematologica 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Daver, N.; Boettcher, S.; Mill, C.P.; Sasaki, K.; Birdwell, C.E.; Davis, J.A.; Das, K.; Takahashi, K.; Kadia, T.M.; et al. Activity of menin inhibitor. Ziftomenib (KO-539) as monotherapy or in combinations against AML cells with MLL1 rearrangement or mutant NPM1. Leukemia 2022, 36, 2729–2733. [Google Scholar] [CrossRef] [PubMed]
- Wang, ER.S.; Altman, J.K.; Pettit, K.; De Botton, S.; Walter, R.P.; Fenaux, P. Preliminary data on a phase 1/2a first in human study of the menin-Kmt2A (MLL) inhibitor KO-539 in patients with relapsed or refractory acute myeloid leukemia. Blood 2020, 136 (Suppl. 1), 7–8. [Google Scholar] [CrossRef]
- Stein, E.M.; Aldoss, I.; DiPersio, J.F.; Stone, R.M.; Arellano, M.L.; Rosen, G. Safety and efficacy of menin inhibition in patients (Pts) with MLL-rearranged and NPM1 mutant acute acute leukemia: a phase (Ph) 1, first-in-human study of SNDX-5613 (Augment 101). Blood 2021, 138 (Suppl. 1), 699. [Google Scholar] [CrossRef]
- Issa, G.C.; Aldoss, I.; DiPersio, J.; Cugklievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Pater, M.R.; Dickens, D.S.; Shenoy, S.; et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukemia. Nature 2023, 615, 920–925. [Google Scholar] [CrossRef]
- Perner, F.; Stein, E.M.; Wenge, D.V.; Singh, S.; Kim, J.; Apazidis, A.; Rahnamoun, H.; Anand, D.; Marinaccio, C.; Hatton, C.; et al. MEN1 mutations mediate clinical resistance to menin inhibition. Nature 2023, 615, 9013–919. [Google Scholar] [CrossRef]
- Erba, H.P.; Fathi, A.T.; Issa, G.C.; Altman, J.K.; Montesinos, P.; Patnaik, M.M.; Foran, J.M.; De Botton, S.; Baer, M.R.; Schiller, G.J.; et al. Update of a phase 1-2 first-in-human study of the menin-KMT2A (MLL) inhibitor ziftomenib (KO-539) in patients with relapsed or refractory acute myeloid leukemia. Blood 2022, 140 (Suppl.1), 153–156. [Google Scholar] [CrossRef]
Figure 2.
Mutational profile of NPM-1-mut AMLs subdivided into four different prognostic groups (favorable, intermediate-1, intermediatye-2 and adverse), each comprising two different subgroups, whose mutational profile is shown. Within parenthesis it is shown the frequency of each subgroup with respect to total NPM1-mut AMLs. The Figure is freely adapted from data reported by Sanchez et al. [37].
Figure 2.
Mutational profile of NPM-1-mut AMLs subdivided into four different prognostic groups (favorable, intermediate-1, intermediatye-2 and adverse), each comprising two different subgroups, whose mutational profile is shown. Within parenthesis it is shown the frequency of each subgroup with respect to total NPM1-mut AMLs. The Figure is freely adapted from data reported by Sanchez et al. [37].
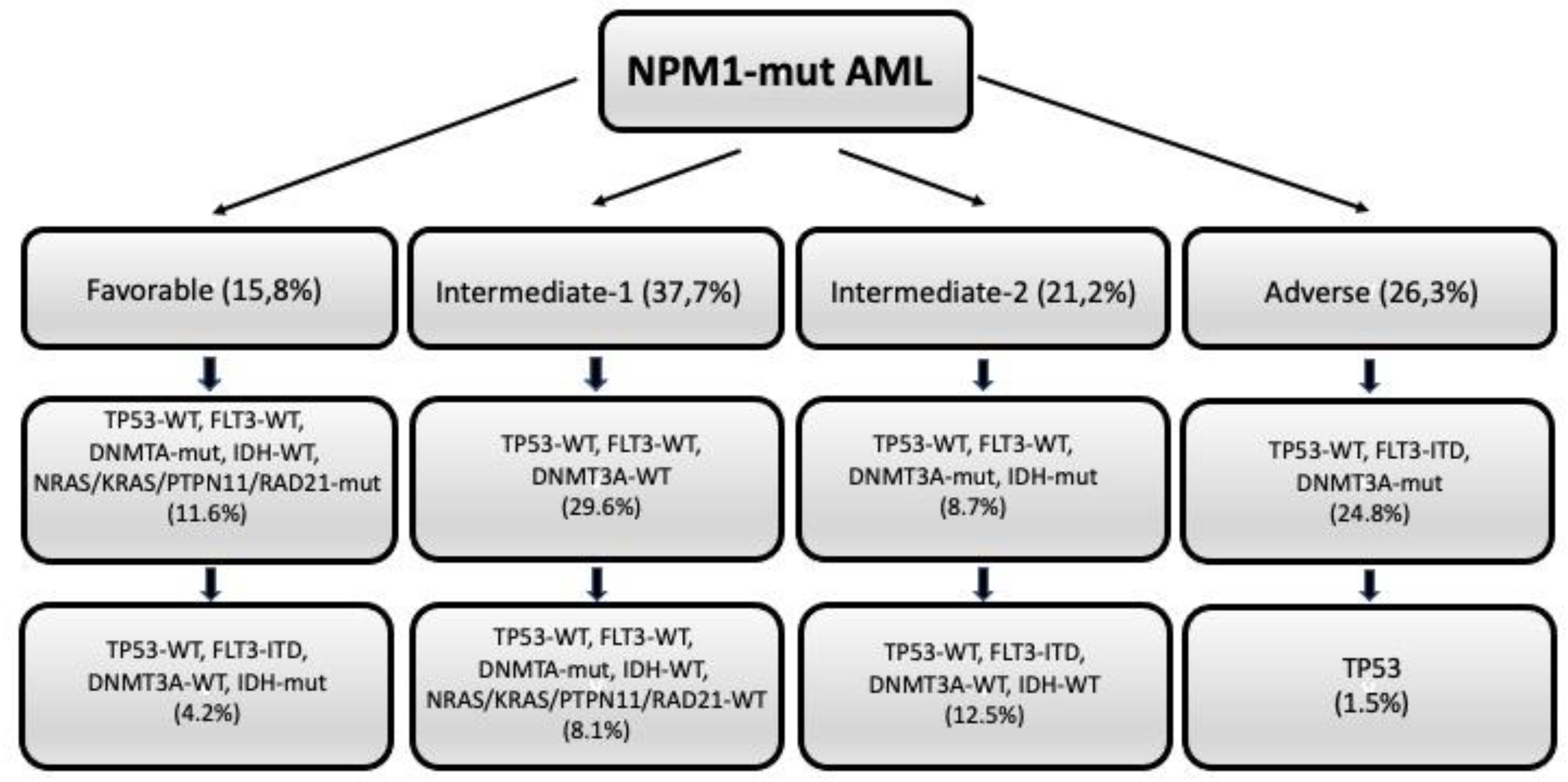
Figure 3.
Comparison of the mutational profile observed in MDS NPM1-mut, MDS NPM1-WT and AML NPM1-mut. The data are reported in Patel et al. 2020 [73].
Figure 3.
Comparison of the mutational profile observed in MDS NPM1-mut, MDS NPM1-WT and AML NPM1-mut. The data are reported in Patel et al. 2020 [73].
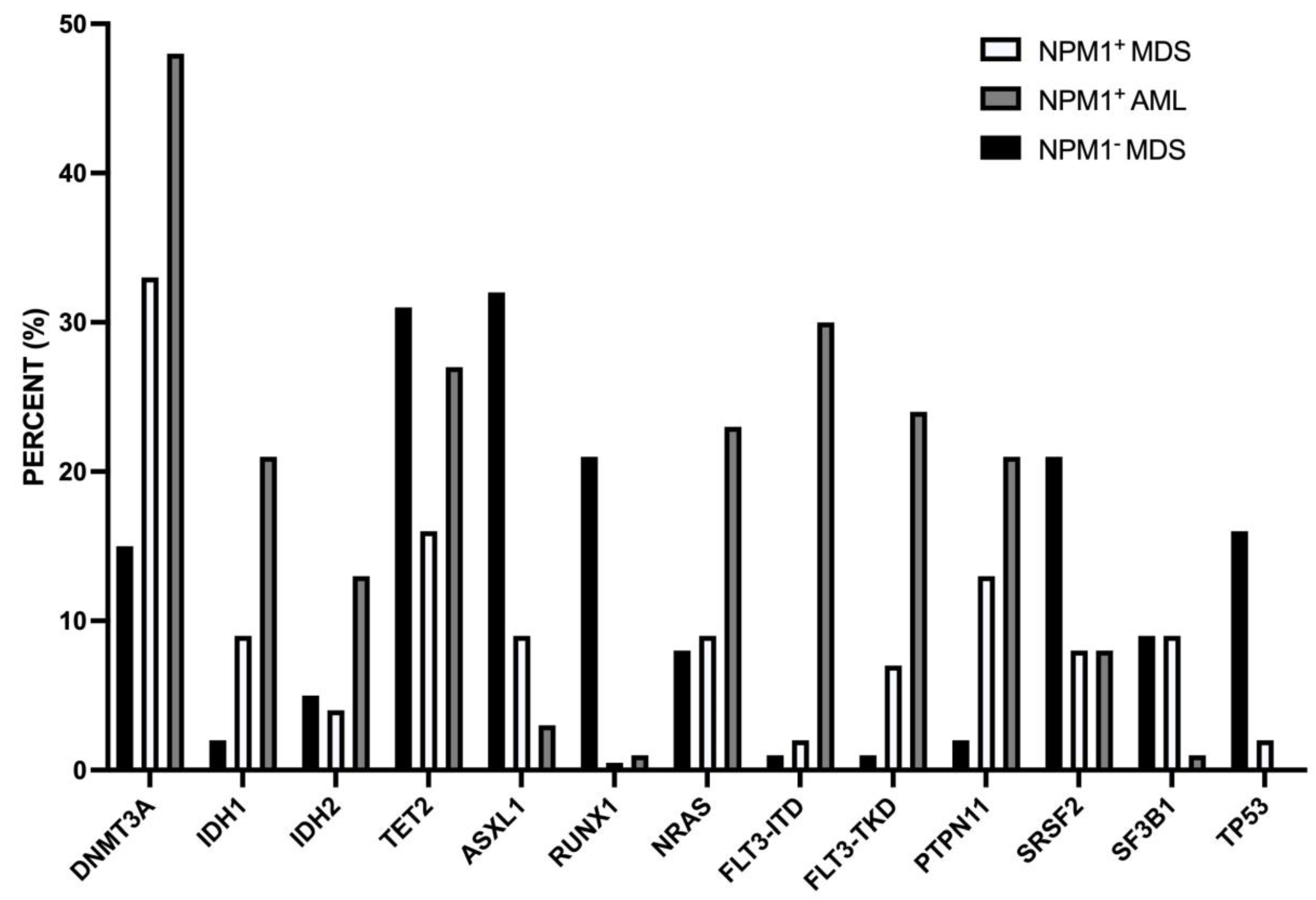
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



