
Submitted:
16 May 2023
Posted:
18 May 2023
You are already at the latest version
Abstract
Traumatic brain injury (TBI) affects millions of people annually, however, our knowledge of the diffuse pathologies associated with TBI is limited. As diffuse pathologies, including axonal injury and neuroinflammatory changes, are difficult to visualize in the clinical population, animal models are used. In the current study we used the central fluid percussion injury (CFPI) model in a micro pig to study the potential scalability of these diffuse pathologies in a gyrencephalic brain of a species with inflammatory systems very similar to humans. We found that both axonal injury and microglia activation within the thalamus and corpus callosum are positively correlated to the weight-normalized pressure pulse, while subtle changes in blood gas and mean arterial blood pressure are not. We also found that the majority of tissue generated up to 10 years previously is viable for immunofluorescent labeling after long-term refrigeration storage. This study indicates that a micro pig CFPI model could allow for specific investigations of various degree of diffuse pathological burdens following TBI.
Keywords:
Traumatic brain injury
; Axonal injury
; Micro pig
; Diffuse pathology
; Microglia
; Aged tissue
1. Introduction
Traumatic brain injury (TBI) is a major health care concern that carries significant personal and societal burdens[1,2,3,4]. There are approximately 2 million reported cases of TBI annually in the United States with global cases reaching 50 million each year with the understanding that these numbers are likely higher due to the vast majority of TBIs being mild and going unreported[4,5,6,7]. Although our knowledge of the complex pathologies associated with TBI has progressed, understanding of diffuse pathologies, which are associated with morbidity in the human population[8,9,10], is still limited.
Diffuse traumatic axonal injury (TAI) is one of the pathological hallmarks of TBI and is commonly used in experimental mild TBI studies to convey the degree of injury[11,12,13,14,15,16,17,18]. Neuroinflammation has become another key pathological feature of brain injury in both human and animal studies[19,20,21,22]. Many recent studies have demonstrated the impact of inflammatory cascades in regulating behavioral morbidities, general pathology, and neuronal function in both the normal brain and in various disease states, including TBI[23,24,25,26]. Studies have demonstrated neuroinflammation in the human population following TBI in various brain regions, with consistent involvement of the thalamic domain[20,22,27,28]. Microglia, the innate immune cells of the brain, are critical mediators of these TBI-induced neuroinflammatory processes[29,30,31,32,33,34,35,36]. Astrocytes are key regulators of various processes, including maintenance of the blood brain barrier, water movement, synaptic activity regulation, glucose storage, and neuroinflammatory processes[33,37,38,39]. Serum levels of glial fibrillary acidic protein (GFAP), a component of the astrocyte cytoskeleton, are also correlated with negative outcomes following TBI and is one of two currently FDA approved biomarkers for TBI[40,41,42].
We currently don’t have the technology to specifically investigate TBI-induced diffuse pathological progression on an individual cellular level in the living human brain; therefore, animal models are used to tease out potential targets for further study. Many therapeutics have shown promising results in rodent models of TBI, unfortunately, this efficacy has been limited when translated to humans[43,44,45]. The high reliance on lower-order species for preclinical TBI research has been implicated in this translational failure in that some processes and mechanisms found in rodents don’t occur in humans, and vice versa[44,46]. Therefore, there have recently been calls for more models using higher order mammals with inflammatory responses and brain cytoarchitecture more similar to humans, such as pigs to better evaluate potential therapeutics prior to human translation[45,47,48,49,50,51,52].
With this goal in mind, we began characterization of a central fluid percussion injury (CFPI) model of mild diffuse TBI in the adult micro pig[53,54,55,56]. We found that CFPI resulted in diffusely dispersed injured axons and activated microglia in various brain regions without producing focal lesions or gross tissue damage[54]. We further found that this model recapitulated diffuse pathological and serum biomarker profiles of the human population[53,54,55]. As human injury occurs on a spectrum, the current investigation aimed to evaluate the potential for graded levels of injury-induced pathology in the micro pig model of CFPI. Additionally, due to the expense of generating large animal TBI models, we sought to determine the duration that our previously generated micro pig brain tissue would maintain viability for immunolabeling.
2. Materials and Methods
2.1. Animals
Data from animals used in this investigation have been published previously[54,55]. We used tissue from a total of twenty-one ~6-month-old adult male Yukatan micro pigs (n=3 sham; n=18 injured), weighing 15-25kg at time of injury. Prior to injury induction, animals were housed in pairs on a 12h light–dark cycle in pens with free access to food and water. Pens had epoxy flooring with a drain in the back of the pen and a mixture of solid cinderblock wall and fencing. Following injury, pigs were single housed in a separate pen within the same room. All experimental protocols were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee, an AAALAC international accredited and USDA registered organization. The Virginia Commonwealth University Institutional Animal Care and Use Committee adheres to all regulations outlined in the “Guide for the Care and Use of Laboratory Animals: 8th Edition” (National Research Council[57]).
2.2. Surgical preparation and injury induction
As previously published[54], micro pigs were initially sedated with an intramuscular injection in the rear flank of 100mg/ml Xylazine (2.2mg/kg; AnaSed Injection, Shenondoah, IA, USA),100mg/ml Telazol (2.0mg/kg; Tiletamine HCL and Zolazepam HCL; Pfizer, New York, NY, USA), and 0.01mg/kg Glycopyrrolate. After initial sedatives took full effect (5-10min post injection), sodium pentobarbital (60mg/kg; Sigma-Aldrich, St. Louis, MO, USA) was administered intravenously through a superficial ear vein. Loss of the palpebral reflex, in which the canthus of the palpebral fissure of the eye was touched gently to elicit a reflexive eye twitch/closing, and jaw tone were used to verify the plane of anesthesia. Once 2-3 gentle touches to the canthus of the palpebral fissure did not elicit a response and the jaw was easily manipulated, the micro pig was intubated with a size 6-7 endotracheal tube and ventilated with 1-2% isoflurane mixed in 100% oxygen throughout the experiment. Ophthalmic lubricant was applied to avoid damage or drying of the eye during surgery or recovery. Body temperature was monitored with a rectal thermometer and maintained at 37°C by adjusting the level of the self-heating surgical table. Chux under pads were placed between the stainless-steel surface of the surgical table and the pig to reduce chances of burns. The head and inner thigh were shaved with electric clippers and sanitized with betadine and 70% ethanol wipes. Heparin-coated catheters were inserted within the right femoral artery and vein and sutured into place for continuous monitoring of mean arterial blood pressure (MABP; via the arterial line), assessment of blood gases via the arterial line, and replacement of fluids with Lactated Ringer’s (Hospira, Lake Forest, IL, USA) via the venous line to maintain hydration. Following the canula placement, the animal was placed on its sternum and the animal’s head was shaved, sterilized with betadine and 70% ethanol wipes, and draped. A midline incision was made from the supraorbital process to the nuchal crest and a 16mm diameter circular craniotomy was trephined along the sagittal suture with a manual surgical trephine (Medicon 57-63-16), positioning the center of the craniotomy 14mm anterior to lambda (on the nuchal crest) and leaving the Dura intact (Figure 1A). A custom stainless-steel threaded hub (Custom Design and Fabrication, Richmond, VA, USA) with an outer diameter of 17mm and an inner diameter of 14mm (Figure 1B) was screwed into the craniotomy site to a depth of ~4mm. Dental acrylic (methyl-methacrylate; Hygenic Corp., Akron, OH) was applied around the hub to insure hub stability. The hub was filled with sterile saline and wet gauze was placed over the dental acrylic to serve as a heat sink for the exothermic reaction that occurs during hardening.
The induction of the central fluid percussion injury (cFPI) was done as described previously[53,54]. Following verification that the dental acrylic was fully hardened, anesthetized micro pigs were connected to the cFPI device that had been fitted with an L-shaped stainless steel adaptor onto which the injury hub locked (Figure 1B,C). This adaptor allowed for the redirection of the fluid pulse downward through the injury hub on the ventral surface of the pig’s skull. Following connection and verification that bubbles hadn’t been introduced to the closed fluid-filled system, micro pigs were injured with a fluid pressure pulse at a magnitude of 1.39-1.83 atmospheres and duration of 24-32msec (Supplementary Table S1) measured by a transducer affixed to the injury device and displayed on an oscilloscope (Tektronix, Beaverton, OR, USA). Immediately after injury induction, animals were disconnected from the injury device, the set screws were unscrewed from the skull and the dental acrylic, hub, and screws were removed as a unit. The hub was then removed from the surrounding dental acrylic for sterilization and reuse. While there was some sub-arachnoid bleeding following injury, the cFPI did not result in any breach of the Dura mater or induce subdural hematoma formation. Gel foam was placed over the craniotomy/injury site to alleviate small amounts of bone bleeding and the scalp was sutured without replacing the bone to the craniotomy site.
2.3. Physiology
Systemic physiology was monitored and recorded throughout the duration of anesthesia, both prior to cFPI and throughout the post-injury monitoring period. Mean arterial blood pressure was measured through the canulated femoral artery, hemoglobin oxygen saturation was monitored via a pulse oximeter, and a rectal thermometer was used to measure body temperature. All physiological readings were measured and visualized by a Cardell® MAX-12HD (Sharn veterinary, inc., Chicago, IL, USA) system. The partial pressures of oxygen and carbon dioxide in arterial blood, PaO2 and PaCO2, respectively, hematocrit (Hct), bicarbonate (HCO3), hemoglobin (Hb) and pH values were assessed on blood draws from the femoral arterial cannula using a Stat Profile pHOx blood gas machine (NOVA Biomedical, Waltham, MA, USA). Summarized physiological results for these animals were previously published [54]. However, this manuscript investigates individual animals pre and post-injury physiological readouts (Supplementary Table S1).
2.4. Tissue processing
At terminal endpoints of 6h post-cFPI micro pigs were overdosed with 3ml euthasol euthanasia-III solution (Henry Schein, Dublin, OH; USA) and immediately transcardially perfused with 6L of 0.9% saline followed by 8L of 4% paraformaldehyde/0.2% gluteraldehyde in Millonig’s buffer (136 mM sodium phosphate monobasic/109mM sodium hydroxide), as previously published[54]. After transcardial perfusion, the brains were removed from the skull and post-fixed in 4% paraformaldehyde/0.2% gluteraldehyde/Millonig’s fixation buffer for an additional 48-72h at 4°C. Postfixed brains were blocked into 5mm coronal segments throughout the rostral-caudal extent using a small pig brain slicer matrix (Cat. # PBMPBS050-1 Zivic Instruments, Pittsburgh, PA, USA). Segments containing the thalamus or corpus callosum were bisected at the midline and the left side was further processed. All segmented brain tissue not used in active studies was cataloged, cryoprotected in 30% sucrose and frozen in tissue tek Optimal Cutting Temperature (O.C.T.) compound (Tissue-Tek #4583; Sakura Finetek; Torrance, CA, USA) prior to freezing and storage at -80°C for future use. The 5mm coronal segments containing the left hemi thalamus or corpus callosum were never frozen, rather, they were embedded in agarose and coronally sectioned in 0.1M phosphate buffer with a vibratome (Leica, Banockburn, IL, USA) at a thickness of 40µm. Sections were collected serially in 6-well plates (240μm between sections in each well) and stored in Millonig’s buffer at 4°C. Thalamic tissue treated in this manner was sotred in the refrigerator for multiple years prior to investigations of viability for immunobiological analysis. Buffer was changes and/or added every few years to avoid dehydration.
2.5. Immunohistochemistry
2.5.1. Previous labeling of thalamic and corpus callosal tissue for axonal injury and microglial activity index assessments. All immunohistological labeling and analyses that was used to determine injury model scalability was done on freshly generated tissue during the conduct of the studies published previously Lafrenaye et al., 2015[54]. Mean thalamic data for injured and sham groups was previously published in Lafrenaye et al., 2015[54], however, corpus callosum data was not reported. For immunohistological labeling, a random well (1–6) was selected using a random number generator and six pieces of tissue containing the thalamic or twelve sections containing the corpus callosum were taken for immunolabeling. Tissue from all animals was processed concomitantly to reduce variability between animals.
To visualize injured axons, fluorescent immunohistochemistry against amyloid precursor protein (APP) was used to detect axonal transport issues indicative of axonal injury[13,14,58]. In this procedure sections were blocked and permeabilized at room temperature in 5% normal goat serum (NGS), 2% bovine serum albumin (BSA) and 1.5% triton in phosphate buffered saline for 2hr followed by overnight incubation with a rabbit antibody against the C-terminus of β-APP (1:700; Cat.# 51-2700, Life Technologies, Carlsbad, CA, USA) at 4C° in 5% NGS/2% BSA. Tissue was washed with 1%NGS/1%BSA in PBS at least six times prior to secondary antibody incubation with Alexa Fluor 568-conjugated goat anti-rabbit IgG (1:700; Cat.# A-11011, Life Technologies, Carlsbad, CA, USA) in 1%NGS/1%BSA/PBS at room temperature for 1hr. Tissue was washed in 1%NGS/1%BSA in PBS at least six times. Tissue was mounted on slides using Vectashield hardset mounting medium with Dapi (Cat.#H-1500; Vector Laboratories, Burlingame, CA, USA).
For the visualization of microglia, chromatic immunohistochemistry against ionized calcium-binding adaptor molecule 1 (Iba-1) was done. Endogenous peroxidases were quenched with hydrogen peroxide prior to chromatic immunolabeling. Tissue was blocked and permeabilized in 1.5% triton/10% NGS/PBS for 2hrs followed by incubation with a rabbit antibody against Iba-1 (1:1000; Cat.#019-19741 Wako; Richmond, VA, USA) in 10% NGS/PBS at 4C°. Tissue was washed with 1%NGS/1%BSA in PBS at least six times prior to secondary antibody incubation with biotinylated goat anti-rabbit IgG (1:1000; Cat.# BA-1000, Vector Laboratories, Burlingame, CA, USA). To enhance the chromatic signal, tissue was incubated in avidin biotinylated enzyme complex using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA). The substrate used for the chromatic reaction to visualize microglia was 0.05% diaminobenzidene/ 0.01% H2O2/0.3% imidazole/PBS. Following cessation of the reaction tissue was mounted, dehydrated in a series of alcohols, and cover-slipped with Permount mounting media (Fisher Scientific, Cat#SP15).
2.5.2. Labeling of stored thalamic tissue for immunohistochemistry efficacy assessments.
Archived thalamic tissue from 19 different animals (generated between 7/25/2013-7/21/2015) that had been perfusion fixed with 4% paraformaldehyde/0.2% glutaraldehyde followed by at least 3 days of post-fixation and stored at to be used for immunohistological labeling. Data from these animals have been published previously[54]. Fixed tissue used in this study was sectioned in 0.1M dibasic sodium phosphate buffer within 3 months of being generated and stored long term at 4° C in Millonigs buffer (136 mmol/L sodium phosphate monobasic/109 mmol/L sodium hydroxide). Glial fibrillary acidic protein (GFAP) was used to label astrocytic soma and processes, and Iba-1 was used to label microglial soma and processes. Tissue was sequentially co-labeled for both markers, with the majority of samples (n=13) being initially labeled for GFAP followed by Iba-1 and the remaining samples (n=6) being labeled for Iba-1 first followed by GFAP labeling. For this, tissue was washed in PBS 3 times for 5 mins, blocked and permeabilized for 1hr at room temperature in 5% NGS/2% BSA/1.5% Triton/PBS, and incubated overnight at 4° C with either mouse anti-GFAP primary antibody (1:1000; Cat #MAB360; Millipore) or rabbit anti-Iba-1 primary antibody (1:1000; Waco). Tissue was then washed 6 times for 5 mins in 1% NGS/1% BSA/PBS prior to 1hr incubation at room temperature with either goat anti-mouse Alexa 568 (1:500, Cat# ; ) for GFAP visualization, or goat anti-Rabbit Alexa 488 (1:500, Cat# ; ) for Iba-1 visualization. Tissue was washed again 4 times for 5 mins in PBS. The single-labeled tissue was then blocked again for 1 hr at room temperature in 5% NGS/2% BSA/PBS and immunolabeled with the other primary antibody, either rabbit anti-Iba-1 or mouse anti-GFAP followed by secondary antibody either goat anti-rabbit Alexa 488 for Iba-1 visualization or goat anti-mouse Alexa 568 for GFAP visualization, as above. Tissue was mounted with Vectashield Hardset mounting medium with DAPI prior to imaging.
2.6. Image analysis
Imaging of APP and microglia used for the correlation matrices was done prior to 2015 using a Nikon Eclipse 800 microscope (Nikon, Tokyo, Japan) equipped with an Olympus DP71 camera (Olympus, Center Valley, PA, USA). Image acquisition settings were held consistent for all animals and the regions of interest were restricted to the thalamus and corpus callosum using anatomical landmarks. Imaging was done by an investigator blinded to animal identity.
2.6.1. Quantitative image analysis of diffuse axonal injury
For the APP assessments in the thalamus a total of 60 images (10 images in each of the 6 sections assessed) were taken at 10X magnification (0.72mm2 field) in a systematically random fashion starting at the dorsal lateral aspect of the thalamus. For the corpus callosum a total of 24 images (2 images in each of 12 sections) were taken at 10X magnification, which covered the majority of the region of interest. Nuclear DAPI label was used for field advancement and to verify focus as well as restriction within the region of interest. Thresholded masks of APP intensity were generated in ImageJ software (NIH, Bethesda, MD). The particle analysis tool in ImageJ was then used to assess the number of APP+ axonal swellings in each image (particle analysis settings circularity=0.2-1, size= 20-infinity). The number of APP+ swellings per unit area was quantified for each image and averaged for each animal.
2.6.2. Quantitative image analysis of microglial activity index
Assessment of microglia activation for the thalamus was published previously[54]. For this the entire thalamus or corpus callosum was assessed for each of the sections selected for each animal (6 sections for the thalamus and 12 sections for the corpus callosum). Identification of microglia activation was based on specific morphological criteria. Microglia with highly ramified fine process networks that were lightly labeled with Iba-1 were considered non-reactive, while microglia with thicker, shorter, or absent processes and darker Iba-1 labeling were identified as active/reactive [59,60,61]. The degree of microglia activation was assessed using an index from 0-5 in which 0= no microglial activation observed, 1= ramified microglia with thicker processes and darker Iba-1 labeling observed in ~5% of the region of interest, 2= activated microglia observed in ~5-10% of the region of interest, 3= activated microglia observed in ~10<25% of the region of interest, 4= activated microglia observed in ~25<50% of the region of interest and 5= activated microglia observed in >50% of the region of interest. Two blinded investigators analyzed all sections independently and their scores were averaged for each animal.
2.6.3. Qualitative analysis of glial labeling in old pig tissue
Immunofluorescent labeling of microglia and astrocytes in pig tissue generated >7 years prior and stored at 4°C was qualitatively assessed by investigators blind to age of tissue. Images were taken with a Keyence BZ-X800 microscope (Keyence Corporation of America, Itasca, IL, USA) at 40x magnification with all settings held constant for each sample. Experimenters randomly selected 6 areas of each section within the thalamic domain to evaluate the degree of labeling of Iba-1+ microglia and GFAP+ astrocytes. A high-degree label was associated with widely distributed and clearly visible cell soma and processes, a medium-degree label was associated with sporadically distributed and visible cell soma and/or processes, and a low/no-degree label was associated with no visible cell soma or processes. Age of tissue was assessed for each labeling category (high, medium and low/no) to gain insight regarding the potential viability of tissue for immunohistochemistry over time.
2.7. Statistical analysis
Two-tailed unpaired T-test with equal variance not assumed were performed to assess corpus callosal axonal injury and microglial activation index between sham and cFPI animals. Individual animal correlation data was analyzed with a Spearman’s Rho Correlation analysis due to its lack of reliance on the assumption of data normality. Kruskal Wallis with a Dunn’s post hoc were done for assessment of the age of tissue for each labeling category. Data are presented as mean ± standard error of the mean (SEM) or median ± quartiles as indicated in the figure legends. Statistical significance was set at a p value <0.05.
3. Results
3.1. Central fluid percussion injury induces significant diffuse axonal injury in the thalamus and the corpus callosum that correlate with microglial activation.
In our previous study[54], we reported TAI in various brain regions, including the thalamus and corpus callosum. We focused our previous assessments for that study on the thalamus, finding a significant increase in the degree of axonal injury within the thalamic domain (t18=4.46, p=1.4X10-5) as well as a significant increase in the microglial activation index (t4=0.51, p=0.004)[54]. For this study, we extended our evaluation of the degree of axonal injury and microglia activation to the corpus callosum, another brain region sustaining consistent TAI at 6h following a central fluid percussion injury (CFPI) in our micro pig model[54]. We found that the degree of TAI, indicated as amyloid precursor protein (APP) accumulations at the proximal axonal swelling, within the corpus callosum was significantly higher at 6h following CFPI (n=18, 50.89±19.26) compared to sham (n=3, 0.64±0.3.7; t17=1.44, p=0.018; Figure 2A). Microglial activation, signified as an increase in the microglial activation index, was also significantly increased in the corpus callosum at 6h following CFPI in our micro pig model (sham=1.17±0.0.13 CFPI=3.96±0.15; t9=2.51, p=1.57X10-7; Figure 2B), suggesting that the corpus callosum also sustains significant TAI and microglial activation within 6h of CFPI.
3.2. Central fluid percussion injury generates scalable diffuse pathology that does not significantly impact systemic physiology.
To investigate the potential of the micro pig cFPI model to scale the degree of pathology while maintaining a diffuse brain injury in which no cortical contusion was generated, we correlated the degree of TAI and microglial activation index within both the thalamus and the corpus callosum to the atmospheric pressure (ATM) induced in each individual animal generated in our 2015 study[54] (Figure 3). As the male micro pigs generated for this study varied in terms of weight from 14.9kg to 29.8kg (Supplementary Table S1), we first normalized the ATM pressure to body weight. This normalized ATM pressure (ATM/kg) was significantly correlated to the raw ATM pressure transduced through the fluid percussion device (Rho=0.96 p<0.0001, Figure 3A), while allowing for specific weight-based refinement of the injury intensity.
The body weight normalized ATM pressure positively correlated to TAI within both the thalamus (Spearman Rho=0.45 p=0.04) and the corpus callosum (Spearman Rho=0.60 p=0.004; Figure 3B). The degree of axonal injury within the corpus callosum also significantly correlated with the degree of TAI found in the thalamus (Spearman Rho=0.71, p=0.0003; Figure 3B), indicating that both regions scale with the intensity of the fluid pressure pulse. As previously published, thalamic TAI significantly correlates with the microglia activation index within the thalamus (Spearman Rho=0.76 p=<0.0001)[54], indicating an interplay between microglial activation and TAI in this region. Microglial activation within the corpus callosum also significantly and positively correlated with TAI within the region (Spearman Rho=0.64 p=0.0018), indicating that this association is not thalamus specific.
The microglial activation index within the thalamus was found to be positively correlated with the microglial activation index within the corpus callosum (Spearman Rho=0.54 p=0.019; Figure 3B), suggesting that microglial activation might also scale with injury intensity in these two regions. While the microglial activation index within the thalamus was not correlated with the weight-normalized fluid pressure pulse (Spearman Rho=0.29 p=0.20), there was a significant positive correlation between ATM/kg and the microglial activation index within the corpus callosum (Spearman Rho=0.68 p=0.0008; Figure 3B).
Post-injury blood gases were also assessed to investigate the potential that the intensity of cFPI alters systemic physiology. As we previously published, neither the CFPI nor sham groups had blood gases that were outside of the normal range (Supplementary Table S1). None of the individual animal’s blood measurements, including Oxygen saturation (O2%), partial pressure of O2 (PaO2), partial pressure of CO2 (PaCO2), mean arterial blood pressure (MABP), blood pH, hematocrit (Hct), bicarbonate in the blood (HCO3), or Hemoglobin (Hb) were correlated with weight normalized pressure induction (Figure 4). This indicates that while the diffuse pathology is correlated with the injury intensity within mild ranges, the overall systemic physiology reflected in the blood gases are not altered.
We did find significant positive correlations between diffuse pathology and subtle blood gas changes. The degree of TAI within the corpus callosum correlated with PaO2 (Spearman Rho=0.51 p=0.018), HCO3 (Spearman Rho=0.47 p=0.034), and blood pH (Spearman Rho=0.46 p=0.037. Thalamic TAI only correlated with blood pH (Spearman Rho=0.44 p=0.047) and thalamic microglia activation correlated with HCO3 (Spearman Rho=0.44 p=0.045). However, the lack of correlation between injury intensity and blood gases suggests that blood gas readouts are not a good metric of injury intensity in this model.
3.3. Fixed pig tissue maintains the capacity for immunohistochemical labeling for years after refrigerated storage.
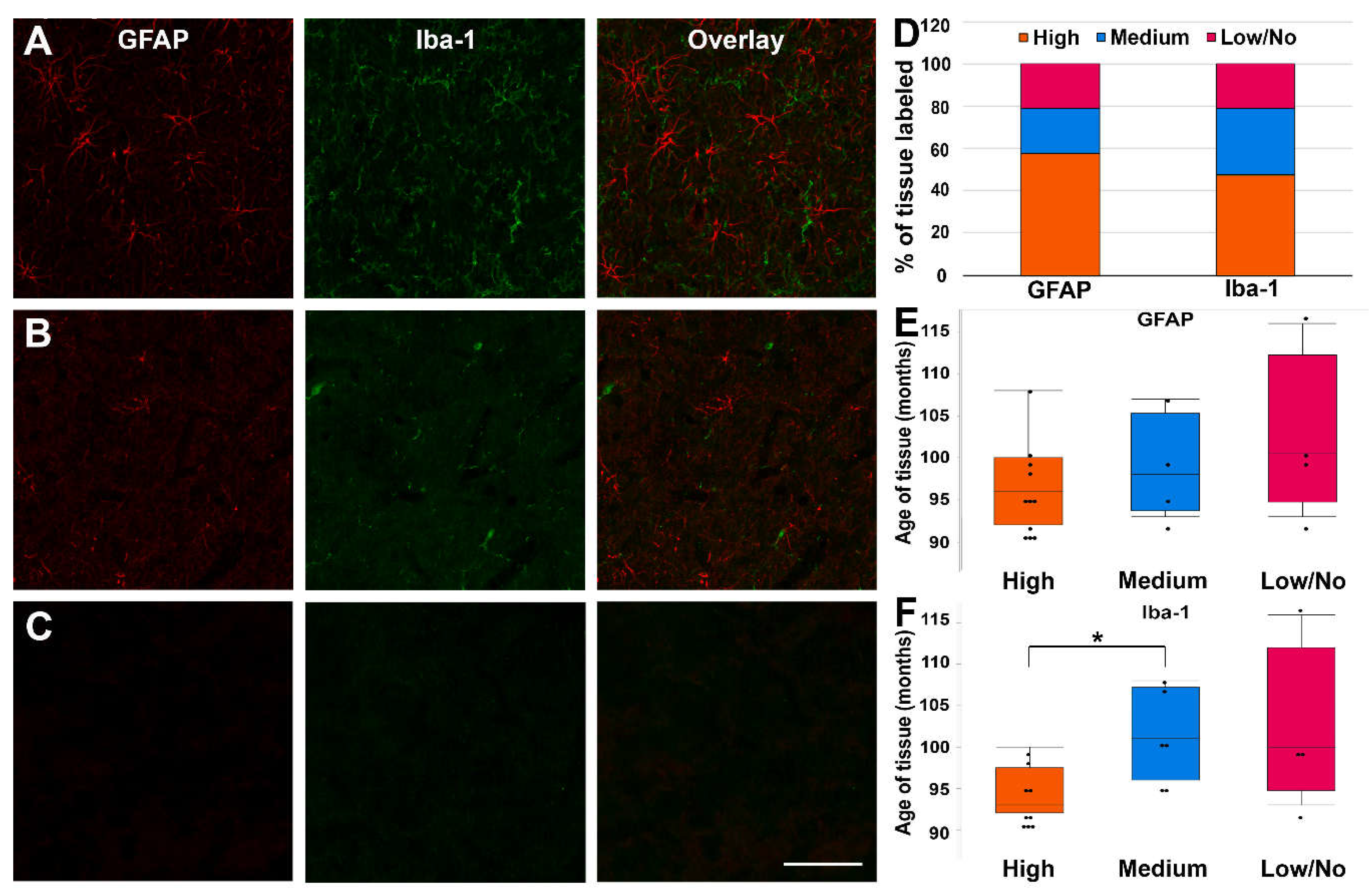
Fixed thalamic micropig tissue following long-term refrigerated storage was immunolabeled for astrocytes using GFAP and microglia using Iba-1 to determine the tissue’s viability for use in immunofluorescent microscopy. The degree of labeling was distinguished by the visibility of cell soma and processes, as well as the density of labeling within the images that were assessed. Samples were categorized as having a high-degree of labeling (densely labeled cell soma and processes), a medium degree of labeling (sporadically labeled cell soma or processes), or a low/no-degree of labeling (non-labeled cell soma and processes; Figure 5A-C). Most of the cases assessed had a high-degree of labeling for both GFAP (58%) and Iba-1 (47%; Figure 5D). Medium-degree labelling was more prevalent in Iba-1 labelling (32%) versus in GFAP (21%; Figure 5D). Only 21% of tissue did not label for GFAP or Iba-1, indicating that this tissue maintains some degree of immunofluorescent labeling capacity.
As there was a difference in the efficacy of immunolabeling for GFAP and Iba-1, we further assessed the potential that the degree of immunolabeling was associated with the age of the tissue for each marker. The age of tissue did not significantly change when stratified by labeling degree for GFAP (X2(2)= 2.10, p=0.35; Figure 5E). In contrast, the age of the tissue was significantly different when stratified by the degree of labeling for Iba-1 (X2(2)=7.55, p=.023; Figure 5F). Specifically, the age of tissue with a high-degree of Iba-1 labeling was lower (94.8±1.03 months) than tissue with a medium degree of Iba-1 labeling (101.5±2.1 months; p=0.011) and trended toward being lower than tissue with low/no Iba-1 labeling (102.3±4.9; p=0.065), suggesting that the age of the tissue impacts the labeling efficacy of Iba-1.
4. Discussion
Mild TBI is associated with diffuse pathological progressions including, but not limited to, TAI and neuroinflammation, which are associated with negative outcomes and morbidities in the human population[62,63,64,65]. Although classified as mild clinically based on their Glasgow comma score, mild TBI injuries occur on a spectrum, which is still not fully understood, but involves changes in the degree of diffuse pathology. As diffuse axonal injury as a clinical diagnosis can only be confirmed using histological assessment in postmortem tissue[17,18,66], detailed investigations into the spectrum of mild injury requires animal modeling. The cytoarchitecture, inflammatory system, and metabolism of pigs are highly consistent with human[47,50,67], therefore, we utilized a pig model to investigate the spectrum of diffuse injury produced by a CFPI model of mild TBI.
In our initial study we found significant axonal injury and microglia activation within the thalamic domain, which is consistent with findings in the human population[22,54,68,69,70]. We had noted TAI within the corpus callosum in that study as well, however, we had not previously quantitatively assessed the burden of TAI within the corpus callosum. In this study we found that our model of CFPI in the micro pig produced significant TAI and microglial activation within the corpus callosum (Figure 2). This is consistent with what is seen in the human population with microscopic damage to the corpus callosum being one of the primary indicators of grade 1 diffuse axonal injury in the Adams diffuse axonal injury classification[71].
The burden of axonal injury within the corpus callosum and thalamus were significantly correlated to one another (Figure 3), demonstrating that in the pig CFPI model of mild TBI both grey and white matter regions are impacted to a similar degree allowing versatility for pathological assessments in this model. Axonal injury within both regions also demonstrated significant positive correlations to microglia activation following CFPI, indicating that microglia activation likely maps to areas of axonal injury. While we previously showed this correlation between TAI and microglia activation to be due to microglia process convergence onto injured axonal swellings in the thalamus in the pig following CFPI[53,54], further studies are needed to investigate the physical relationship between TAI and microglia activation in the corpus callosum.
Importantly, there was a significant correlation between the intensity of the fluid pulse and the degree of TAI within both the thalamus and corpus callosum, showing that this pig model of mild TBI allows assessment of subtle changes in both grey and white matter areas along the spectrum of mild TBI. This finding is consistent with clinical investigations in which the degree of diffuse injury has been associated with severity of injury and progressive morbidity following TBI in experimental and clinical studies[9,20,62].
While the degree of TAI in both the corpus callosum and thalamus significantly correlated with fluid pulse intensity, only corpus callosal microglia activation was significantly correlated to the pressure pulse (Figure 3). As the microglia activation index within the thalamus and corpus callosum were found to be significantly correlated, the lack of significance of thalamic microglia activation with injury intensity is likely an artifact of the assessment strategy. The assessment within the corpus callosum involved twice as many sections for analysis for each animal since the corpus callosum occupies a much smaller area than the thalamus. The lower degree of sampling within the thalamus of each animal could have introduced a higher degree of inter-animal variability. This difference between the correlation of the corpus callosum and thalamic microglia activation index with injury intensity could also be due to the more homogenous fiber orientations found within the corpus callosum compared to the thalamus which has a larger overall area and more heterogeneous axonal orientations.
Interestingly, while the injury intensity was not significantly correlated with blood gas readouts and all blood gas readouts remained within normal ranges, diffuse pathology was found to be correlated with some blood gas readouts. Specifically, the degree of TAI within the corpus callosum significantly correlated with blood gas readouts, including paO2, pH, and HCO3, and the degree of thalamic TAI correlated with blood pH. However, this might not indicate a specific relationship between the brain pathology and subtle blood gas changes. Axonal injury and these subtle blood gas changes could both be symptoms of mild TBI that occur together but do not necessarily drive one another. Rather, the changes in blood gases could indicate subtle changes in systemic organs following a mild TBI. Studies have recently shown that brain injury can induce changes within systemic organs systems, including both the kidney and lung, both regions involved in regulating the HCO3, pH, and PaO2 of the blood. Kidney and lung pathology has been shown to occur in rodent models of mild TBI[72,73,74,75,76]. Retrospective clinical studies also found that either acute kidney injury[77] or acute lung injury[78] was associated with worse outcomes in TBI patients, indicating that systemic organ changes could be occurring in the human population as well. While these are intriguing possibilities that require further investigation, due to the blood gas readouts all being within normal ranges and the lack of correlation between any blood gas readout and the pressure pulse, it does not appear that CFPI substantially changes the blood gas readouts. Therefore, it is likely that whatever potential changes might be occurring systemically, they are not extreme enough to break homeostasis.
In terms of the ability to label tissue years following generation, most of the tissue stored in long-term (7.5-9.6 years) refrigeration express a high degree of immunolabeling of both astrocytic GFAP and microglial IBA1 markers. This data suggests that the efficacy of GFAP labeling is not altered as the age of tissue increases. Iba-1, on the contrary, demonstrates more medium and low/no degree of labeling in older tissues, suggesting that the specific antibody is less effective in older tissues. The Wako Iba-1 antibody has been used in various specifies to identify microglia with a high degree of efficacy[79,80,81,82]. The foundational studies using this Iba-1antibody all appeared to use transcardial perfusion with 4% paraformaldehyde followed by post-fixation[80,81,82]. Our prior published studies with the same tissue indicates that the Iba-1 antibody did not suffer decreased labeling in newly generated tissues as it did with tissues from long-term storage. This may suggest that the Wako IBA-1 antibody loses.
5. Conclusions
Overall, the current study shows that a mild model of central fluid percussion injury can produce scalable diffuse injury within multiple brain regions without significantly altering blood gases. Additionally, we found that the majority of tissue fixed with 4% paraformaldehyde/0.2% glutaraldehyde and stored in Millings buffer under refrigeration conditions is usable for immunofluorescent analysis up to 8 years following generation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Full data set.
Author Contributions
Conceptualization, A.D.L. methodology, A.D.L.; formal analysis, M.P. and A.D.L..; investigation, A.D.L. and M.P; writing—original draft preparation, M.P. and A.D.L. writing—review and editing, M.P. and A.D.L.; supervision, A.D.L.; funding acquisition, A.D.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the US Army grant W81XWH-10-1-0623 and NINDS grants R56NS128104, R21NS126611
Data Availability Statement
Full data for each animals can be found in Supplementary Table S1.
Acknowledgments
We would like to thank Martina Hernandez, Karen Gorse, and Amanda Logan-Wesley for technical assistance and scientific discussion.
Conflicts of Interest
The authors declare no conflict of interest.
References
- James, S.L.; Theadom, A.; Ellenbogen, R.G.; Bannick, M.S.; Montjoy-Venning, W.; Lucchesi, L.R.; Abbasi, N.; Abdulkader, R.; Abraha, H.N.; Adsuar, J.C.; et al. Global, Regional, and National Burden of Traumatic Brain Injury and Spinal Cord Injury, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. The Lancet Neurology 2019, 18, 56–87. [Google Scholar] [CrossRef]
- Ortiz-Prado, E.; Mascialino, G.; Paz, C.; Rodriguez-Lorenzana, A.; Gómez-Barreno, L.; Simbaña-Rivera, K.; Diaz, A.M.; Coral-Almeida, M.; Espinosa, P.S. A Nationwide Study of Incidence and Mortality Due to Traumatic Brain Injury in Ecuador (2004-2016). Neuroepidemiology 2019, 1–12. [Google Scholar] [CrossRef]
- Brau, R.H.; Acevedo-Salas, Y.; Giovannetti, K. Epidemiological Trends of Traumatic Brain and Spinal Cord Injury in Puerto Rico from November 10th, 2006, through May 24th, 2011. Puerto Rico Health Sciences Journal 2018, 37, 67–77. [Google Scholar]
- Faul, M.; Coronado, V. Chapter 1 - Epidemiology of Traumatic Brain Injury. In Handbook of Clinical Neurology; Traumatic Brain Injury, Part I; Grafman, J., Salazar, A.M., Eds.; Elsevier, 2015; Vol. 127, pp. 3–13. [Google Scholar]
- Maas, A.I.; Roozenbeek, B.; Manley, G.T. Clinical Trials in Traumatic Brain Injury: Past Experience and Current Developments. Neurotherapeutics 2009, 7, 115–126. [Google Scholar] [CrossRef]
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic Brain Injury–Related Emergency Department Visits, Hospitalizations, and Deaths — United States, 2007 and 2013. MMWR. Surveillance Summaries 2017, 66, 1–16. [Google Scholar] [CrossRef]
- Gardner, R.C.; Yaffe, K. Epidemiology of Mild Traumatic Brain Injury and Neurodegenerative Disease. Molecular and Cellular Neuroscience 2015, 66, 75–80. [Google Scholar] [CrossRef]
- Coughlin, J.M.; Wang, Y.; Munro, C.A.; Ma, S.; Yue, C.; Chen, S.; Airan, R.; Kim, P.K.; Adams, A.V.; Garcia, C.; et al. Neuroinflammation and Brain Atrophy in Former NFL Players: An in Vivo Multimodal Imaging Pilot Study. Neurobiology of Disease 2015, 74, 58–65. [Google Scholar] [CrossRef]
- Grossman, E.J.; Inglese, M. The Role of Thalamic Damage in Mild Traumatic Brain Injury. Journal of Neurotrauma 2016, 33, 163–167. [Google Scholar] [CrossRef]
- Little, D.M.; Kraus, M.F.; Joseph, J.; Geary, E.K.; Susmaras, T.; Zhou, X.J.; Pliskin, N.; Gorelick, P.B. Thalamic Integrity Underlies Executive Dysfunction in Traumatic Brain Injury. Neurology 2010, 74, 558–564. [Google Scholar] [CrossRef]
- Gennarelli, T.A.; Thibault, L.E.; Adams, J.H.; Graham, D.I.; Thompson, C.J.; Marcincin, R.P. Diffuse Axonal Injury and Traumatic Coma in the Primate. Annals of neurology 1982, 12, 564–574. [Google Scholar] [CrossRef]
- Povlishock, J.T. Pathobiology of Traumatically Induced Axonal Injury in Animals and Man. Ann Emerg Med 1993, 22, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Sherriff, F.E.; Bridges, L.R.; Sivaloganathan, S. Early Detection of Axonal Injury after Human Head Trauma Using Immunocytochemistry for Beta-Amyloid Precursor Protein. Acta neuropathologica 1994, 87, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, S.M.; Nash, M.J.; Sweeting, C.J.; Graham, D.I.; Roberts, G.W. β-Amyloid Precursor Protein (ΒAPP) as a Marker for Axonal Injury after Head Injury. Neuroscience Letters 1993, 160, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.H.; Meaney, D.F. Axonal Damage in Traumatic Brain Injury. The Neuroscientist 2000, 6, 483–495. [Google Scholar] [CrossRef]
- Scheid, R.; Walther, K.; Guthke, T.; Preul, C.; von Cramon, D.Y. Cognitive Sequelae of Diffuse Axonal Injury. Archives of neurology 2006, 63, 418–424. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, X.; Xu, L.; Zhang, R.; Li, Z.; Yue, X.; Qiao, D. Traumatic Axonal Injury: Neuropathological Features, Postmortem Diagnostic Methods, and Strategies. Forensic Sci Med Pathol 2022, 18, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Mckee, A.C.; Daneshvar, D.H. The Neuropathology of Traumatic Brain Injury. In Handbook of Clinical Neurology; Elsevier B.V., 2015; Vol. 127, pp. 45–66.
- Velázquez, A.; Ortega, M.; Rojas, S.; González-Oliván, F.J.; Rodríguez-Baeza, A. Widespread Microglial Activation in Patients Deceased from Traumatic Brain Injury. Brain Injury 2015, 29, 1126–1133. [Google Scholar] [CrossRef]
- Coughlin, J.M.; Wang, Y.; Munro, C.A.; Ma, S.; Yue, C.; Chen, S.; Airan, R.; Kim, P.K.; Adams, A. V.; Garcia, C.; et al. Neuroinflammation and Brain Atrophy in Former NFL Players: An in Vivo Multimodal Imaging Pilot Study. Neurobiology of Disease 2015, 74, 58–65. [Google Scholar] [CrossRef]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R. a; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after Trauma: Microglial Activation and Traumatic Brain Injury. Annals of neurology 2011, 70, 374–383. [Google Scholar] [CrossRef]
- Zhou, Y.; Lui, Y.W.; Zuo, X.-N.; Milham, M.P.; Reaume, J.; Grossman, R.I.; Ge, Y. Characterization of Thalamocortical Association Using Amplitude and Connectivity of FMRI in Mild Traumatic Brain Injury. J Magn Reson Imaging 2014, 39, 1558–1568. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Satgunaseelan, L.; Bye, N.; Kossmann, T. Modulation of Immune Response by Head Injury. Injury 2007, 38, 1392–1400. [Google Scholar] [CrossRef]
- Nizamutdinov, D.; Shapiro, L.A. Overview of Traumatic Brain Injury: An Immunological Context. Brain Sciences 2017, 7. [Google Scholar] [CrossRef]
- Kelley, B.J.; Lifshitz, J.; Povlishock, J.T. Neuroinflammatory Responses after Experimental Diffuse Traumatic Brain Injury. J Neuropathol Exp Neurol 2007, 66, 989–1001. [Google Scholar] [CrossRef]
- Das, M.; Mohapatra, S.; Mohapatra, S.S. New Perspectives on Central and Peripheral Immune Responses to Acute Traumatic Brain Injury. Journal of Neuroinflammation 2012, 9, 236. [Google Scholar] [CrossRef]
- Velázquez, A.; Ortega, M.; Rojas, S.; González-Oliván, F.J.; Rodríguez-Baeza, A. Widespread Microglial Activation in Patients Deceased from Traumatic Brain Injury. Brain Injury 2015, 29, 1126–1133. [Google Scholar] [CrossRef]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R. a; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after Trauma: Microglial Activation and Traumatic Brain Injury. Annals of neurology 2011, 70, 374–383. [Google Scholar] [CrossRef]
- Mannix, R.C.; Whalen, M.J. Traumatic Brain Injury, Microglia, and Beta Amyloid. International journal of Alzheimer’s disease 2012, 2012, 608732. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M.R.; Franklin, R.J.M. Debris Clearance by Microglia: An Essential Link between Degeneration and Regeneration. Brain : a journal of neurology 2009, 132, 288–295. [Google Scholar] [CrossRef]
- Smith, C. Review: The Long-Term Consequences of Microglial Activation Following Acute Traumatic Brain Injury. Neuropathology and Applied Neurobiology 2013, 39, 35–44. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial Physiology: Unique Stimuli, Specialized Responses. Annual review of immunology 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The Contribution of Astrocytes and Microglia to Traumatic Brain Injury. British Journal of Pharmacology 2015. [Google Scholar] [CrossRef]
- Tay, T.L.; Hagemeyer, N.; Prinz, M. The Force Awakens: Insights into the Origin and Formation of Microglia. Current Opinion in Neurobiology 2016, 39, 30–37. [Google Scholar] [CrossRef]
- Kigerl, K. a; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of Two Distinct Macrophage Subsets with Divergent Effects Causing Either Neurotoxicity or Regeneration in the Injured Mouse Spinal Cord. The Journal of neuroscience : the official journal of the Society for Neuroscience 2009, 29, 13435–13444. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A. Microglia in the TBI Brain: The Good, the Bad, and the Dysregulated. Experimental Neurology 2016, 275, 316–327. [Google Scholar] [CrossRef]
- Chen, Y.; Swanson, R.A. Astrocytes and Brain Injury. Journal of Cerebral Blood Flow and Metabolism 2003, 23, 137–149. [Google Scholar] [CrossRef]
- Lafrenaye, A.D.; Simard, J.M. Bursting at the Seams: Molecular Mechanisms Mediating Astrocyte Swelling. International journal of molecular sciences 2019. [Google Scholar] [CrossRef]
- Laird, M.D.; Vender, J.R.; Dhandapani, K.M. Opposing Roles for Reactive Astrocytes Following Traumatic Brain Injury. NeuroSignals 2008, 16, 154–164. [Google Scholar] [CrossRef]
- Mondello, S.; Muller, U.; Jeromin, A.; Streeter, J.; Hayes, R.L.; Wang, K.K. Blood-Based Diagnostics of Traumatic Brain Injuries. Expert review of molecular diagnostics 2011, 11, 65–78. [Google Scholar] [CrossRef]
- Czeiter, E.; Mondello, S.; Kovacs, N.; Sandor, J.; Gabrielli, A.; Schmid, K.; Tortella, F.; Wang, K.K.W.; Hayes, R.L.; Barzo, P.; et al. Brain Injury Biomarkers May Improve the Predictive Power of the IMPACT Outcome Calculator. Journal of neurotrauma 2012, 29, 1770–1778. [Google Scholar] [CrossRef]
- Papa, L. Potential Blood-Based Biomarkers for Concussion. Sports Med Arthrosc 24, 115. [CrossRef]
- Schumacher, M.; Denier, C.; Oudinet, J.P.; Adams, D.; Guennoun, R. Progesterone Neuroprotection: The Background of Clinical Trial Failure. Journal of Steroid Biochemistry and Molecular Biology 2016, 160, 53–66. [Google Scholar] [CrossRef]
- Dai, J.X.; Ma, Y.B.; Le, N.Y.; Cao, J.; Wang, Y. Large Animal Models of Traumatic Brain Injury. International Journal of Neuroscience 2018, 128, 243–254. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Jackson, T.C.; Jha, R.M.; Clark, R.S.B.; Okonkwo, D.O.; Bayır, H.; Poloyac, S.M.; Wagner, A.K.; Empey, P.E.; Conley, Y.P.; et al. Paths to Successful Translation of New Therapies for Severe Traumatic Brain Injury in the Golden Age of Traumatic Brain Injury Research: A Pittsburgh Vision. Journal of Neurotrauma 2019, neu.2018.6203. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic Responses in Mouse Models Poorly Mimic Human Inflammatory Diseases. Proceedings of the National Academy of Sciences of the United States of America 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Sauleau, P.; Lapouble, E.; Val-Laillet, D.; Malbert, C.H. The Pig Model in Brain Imaging and Neurosurgery. Animal 2009, 3, 1138–1151. [Google Scholar] [CrossRef]
- Wernersson, R.; Schierup, M.H.; Jørgensen, F.G.; Gorodkin, J.; Panitz, F.; Staerfeldt, H.-H.; Christensen, O.F.; Mailund, T.; Hornshøj, H.; Klein, A.; et al. Pigs in Sequence Space: A 0.66X Coverage Pig Genome Survey Based on Shotgun Sequencing. BMC genomics 2005, 6, 70. [Google Scholar] [CrossRef]
- Fairbairn, L.; Kapetanovic, R.; Sester, D.P.; Hume, D. a The Mononuclear Phagocyte System of the Pig as a Model for Understanding Human Innate Immunity and Disease. Journal of leukocyte biology 2011, 89, 855–871. [Google Scholar] [CrossRef]
- Lind, N.M.; Moustgaard, A.; Jelsing, J.; Vajta, G.; Cumming, P.; Hansen, A.K. The Use of Pigs in Neuroscience: Modeling Brain Disorders. Neuroscience and Biobehavioral Reviews 2007, 31, 728–751. [Google Scholar] [CrossRef]
- Marklund, N.; Hillered, L. Animal Modelling of Traumatic Brain Injury in Preclinical Drug Development: Where Do We Go from Here? British Journal of Pharmacology 2011, 164, 1207–1229. [Google Scholar] [CrossRef]
- Statler, K.D.; Jenkins, L.W.; Dixon, C.E.; Clark, R.S.; Marion, D.W.; Kochanek, P.M. The Simple Model versus the Super Model: Translating Experimental Traumatic Brain Injury Research to the Bedside. J Neurotrauma 2001, 18, 1195–1206. [Google Scholar] [CrossRef]
- Gorse, K.M.; Lafrenaye, A.D. The Importance of Inter-Species Variation in Traumatic Brain Injury-Induced Alterations of Microglial-Axonal Interactions. Frontiers in Neurology 2018, 9. [Google Scholar] [CrossRef]
- Lafrenaye, A.D.; Todani, M.; Walker, S.A.; Povlishock, J.T. Microglia Processes Associate with Diffusely Injured Axons Following Mild Traumatic Brain Injury in the Micro Pig. Journal of Neuroinflammation 2015, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Lafrenaye, A.D.; Mondello, S.; Wang, K.K.; Yang, Z.; Povlishock, J.T.; Gorse, K.; Walker, S.; Hayes, R.L.; Kochanek, P.M. Circulating GFAP and Iba-1 Levels Are Associated with Pathophysiological Sequelae in the Thalamus in a Pig Model of Mild TBI. Scientific Reports 2020, 10, 13369. [Google Scholar] [CrossRef] [PubMed]
- Lafrenaye, A.; Mondello, S.; Povlishock, J.; Gorse, K.; Walker, S.; Hayes, R.; Wang, K.; Kochanek, P.M. Operation Brain Trauma Therapy: An Exploratory Study of Levetiracetam Treatment Following Mild Traumatic Brain Injury in the Micro Pig. Frontiers in Neurology 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- National Research Council GUIDE LABORATORY ANIMALS FOR THE CARE AND USE OF Eighth Edition Committee for the Update of the Guide for the Care and Use of Laboratory Animals Institute for Laboratory Animal Research Division on Earth and Life Studies; 2011; ISBN 978-0-309-15400-0.
- Bramlett, H.M.; Kraydieh, S.; Green, E.J.; Dietrich, W.D. Temporal and Regional Patterns of Axonal Damage Following Traumatic Brain Injury: A Beta-Amyloid Precursor Protein Immunocytochemical Study in Rats. Journal of Neuropathology and Experimental Neurology 1997, 56, 1132–1141. [Google Scholar] [CrossRef]
- Taetzsch, T.; Levesque, S.; McGraw, C.; Brookins, S.; Luqa, R.; Bonini, M.G.; Mason, R.P.; Oh, U.; Block, M.L. Redox Regulation of NF-ΚB P50 and M1 Polarization in Microglia. Glia 2015, 63, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, K.R.; Loane, D.J.; Stoica, B. a; Zhang, J.; Faden, A.I. Delayed MGluR5 Activation Limits Neuroinflammation and Neurodegeneration after Traumatic Brain Injury. Journal of Neuroinflammation 2012, 9, 43. [Google Scholar] [CrossRef]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.-B.; Julius, D. The P2Y12 Receptor Regulates Microglial Activation by Extracellular Nucleotides. Nature neuroscience 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- van Eijck, M.M.; Schoonman, G.G.; van der Naalt, J.; de Vries, J.; Roks, G. Diffuse Axonal Injury after Traumatic Brain Injury Is a Prognostic Factor for Functional Outcome: A Systematic Review and Meta-Analysis. Brain Injury 2018, 32, 395–402. [Google Scholar] [CrossRef]
- Davceva, N.; Sivevski, A.; Basheska, N. Traumatic Axonal Injury, a Clinical-Pathological Correlation. Journal of Forensic and Legal Medicine 2017, 48, 35–40. [Google Scholar] [CrossRef]
- Vieira, R. de C.A.; Paiva, W.S.; de Oliveira, D.V.; Teixeira, M.J.; de Andrade, A.F.; de Sousa, R.M.C. Diffuse Axonal Injury: Epidemiology, Outcome and Associated Risk Factors. Front Neurol 2016, 7, 178. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.S.N.; Jolly, A.; Zimmerman, K.; Bourke, N.J.; Scott, G.; Cole, J.H.; Schott, J.M.; Sharp, D.J. Diffuse Axonal Injury Predicts Neurodegeneration after Moderate–Severe Traumatic Brain Injury. Brain 2020, 143, 3685–3698. [Google Scholar] [CrossRef]
- Mesfin, F.B.; Gupta, N.; Hays Shapshak, A.; Taylor, R.S. Diffuse Axonal Injury. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023. [Google Scholar]
- Hoffe, B.; Holahan, M.R. The Use of Pigs as a Translational Model for Studying Neurodegenerative Diseases. Frontiers in Physiology 2019, 10, 838. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sinha, S.; Suri, V.; Agarwal, D.; Bisht, A.; Garg, K.; Gupta, D.; Kakkar, A.; Kale, S.; Lalwani, S.; et al. Histological Changes in Thalamus in Short Term Survivors Following Traumatic Brain Injury: An Autopsy Study. Neurology India 2013, 61, 599. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Parker, D.; Whyte, J.; Hart, T.; Pluta, J.; Ingalhalikar, M.; Coslett, H.B.; Verma, R. Disrupted Structural Connectome Is Associated with Both Psychometric and Real-World Neuropsychological Impairment in Diffuse Traumatic Brain Injury. Journal of the International Neuropsychological Society 2014, 20, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Banks, S.D.; Coronado, R.A.; Clemons, L.R.; Abraham, C.M.; Pruthi, S.; Conrad, B.N.; Morgan, V.L.; Guillamondegui, O.D.; Archer, K.R. Thalamic Functional Connectivity in Mild Traumatic Brain Injury: Longitudinal Associations With Patient-Reported Outcomes and Neuropsychological Tests. Archives of Physical Medicine and Rehabilitation 2016, 97, 1254–1261. [Google Scholar] [CrossRef]
- Adams, J.; Doyle, D.; Ford, I.; Gennarelli, T.; Graham, D.; McLellan, D. Diffuse Axonal Injury in Head Injury: Definition, Diagnosis and Grading. Histopathology 1989, 15, 49–59. [Google Scholar] [CrossRef]
- Prus, R.; Pokotylo, P.; Logash, M.; Zvir, T. Morphological Particularities and Morphometry of Rats’ Kidneys under the Effect of Experimental Mild Traumatic Brain Injury. Folia Morphologica 2021, 80, 310–316. [Google Scholar] [CrossRef]
- Tekin Neijmann, Ş.; Kural, A.; Sever, N.; Dogan, H.; Sarıkaya, S. Evaluation of Renal Function in Rats with Moderate and Mild Brain Trauma. Ulus Travma Acil Cerrahi Derg 2022, 28, 1–7. [Google Scholar] [CrossRef]
- Ruan, F.; Chen, J.; Yang, J.; Wang, G. MILD TRAUMATIC BRAIN INJURY ATTENUATES PNEUMONIA-INDUCED LUNG INJURY BY MODULATIONS OF ALVEOLAR MACROPHAGE BACTERICIDAL ACTIVITY AND M1 POLARIZATION. Shock 2022, 58, 400. [Google Scholar] [CrossRef]
- Vermeij, J.-D.; Aslami, H.; Fluiter, K.; Roelofs, J.J.; van den Bergh, W.M.; Juffermans, N.P.; Schultz, M.J.; Van der Sluijs, K.; van de Beek, D.; van Westerloo, D.J. Traumatic Brain Injury in Rats Induces Lung Injury and Systemic Immune Suppression. Journal of Neurotrauma 2013, 30, 2073–2079. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Jung, H.; Youn, D.H.; Kim, T.Y.; Han, S.W.; Kim, B.J.; Lee, J.J.; Jeon, J.P. Mild Traumatic Brain Injury and Subsequent Acute Pulmonary Inflammatory Response. J Korean Neurosurg Soc 2022, 65, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhao, W.-G.; Zhang, W.-F. Acute Kidney Injury in Patients with Severe Traumatic Brain Injury: Implementation of the Acute Kidney Injury Network Stage System. Neurocrit Care 2011, 14, 377–381. [Google Scholar] [CrossRef]
- Holland, M.C.; Mackersie, R.C.; Morabito, D.; Campbell, A.R.; Kivett, V.A.; Patel, R.; Erickson, V.R.; Pittet, J.-F. The Development of Acute Lung Injury Is Associated with Worse Neurologic Outcome in Patients with Severe Traumatic Brain Injury. Journal of Trauma and Acute Care Surgery 2003, 55, 106. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-Specific Localisation of a Novel Calcium Binding Protein, Iba1. Molecular Brain Research 1998, 57, 1–9. [Google Scholar] [CrossRef]
- Rodriguez-Callejas, J.D.; Fuchs, E.; Perez-Cruz, C. Evidence of Tau Hyperphosphorylation and Dystrophic Microglia in the Common Marmoset. Frontiers in Aging Neuroscience 2016, 8. [Google Scholar] [CrossRef]
- Ahn, J.-H.; Choi, J.-H.; Park, J.-H.; Yan, B.-C.; Kim, I.-H.; Lee, J.-C.; Lee, D.-H.; Kim, J.-S.; Shin, H.-C.; Won, M.-H. Comparison of Alpha-Synuclein Immunoreactivity in the Spinal Cord between the Adult and Aged Beagle Dog. Lab Anim Res 2012, 28, 165–170. [Google Scholar] [CrossRef]
- Gaigé, S.; Bonnet, M.S.; Tardivel, C.; Pinton, P.; Trouslard, J.; Jean, A.; Guzylack, L.; Troadec, J.-D.; Dallaporta, M. C-Fos Immunoreactivity in the Pig Brain Following Deoxynivalenol Intoxication: Focus on NUCB2/Nesfatin-1 Expressing Neurons. NeuroToxicology 2013, 34, 135–149. [Google Scholar] [CrossRef]
Figure 1.
Adult male micro pigs were subjected to either sham injury or a mild central fluid percussion injury (CFPI). Representative drawings of (A) dorsal and (B) lateral view of the pig skull with the injury hub screwed into the skull along the sagittal suture. (C) Picture of the fluid percussion device used to induce CFPI in the micro pig. The injury hub was screwed into a 16mm craniectomy placed 14mm anterior to the nuchal crest. The hub was filled with sterile saline then connected to the fluid percussion device L-shaped adaptor. Following connection to the device the hammer was released producing a fluid pressure pulse that transduced through the dura to the cerebrospinal fluid.
Figure 1.
Adult male micro pigs were subjected to either sham injury or a mild central fluid percussion injury (CFPI). Representative drawings of (A) dorsal and (B) lateral view of the pig skull with the injury hub screwed into the skull along the sagittal suture. (C) Picture of the fluid percussion device used to induce CFPI in the micro pig. The injury hub was screwed into a 16mm craniectomy placed 14mm anterior to the nuchal crest. The hub was filled with sterile saline then connected to the fluid percussion device L-shaped adaptor. Following connection to the device the hammer was released producing a fluid pressure pulse that transduced through the dura to the cerebrospinal fluid.
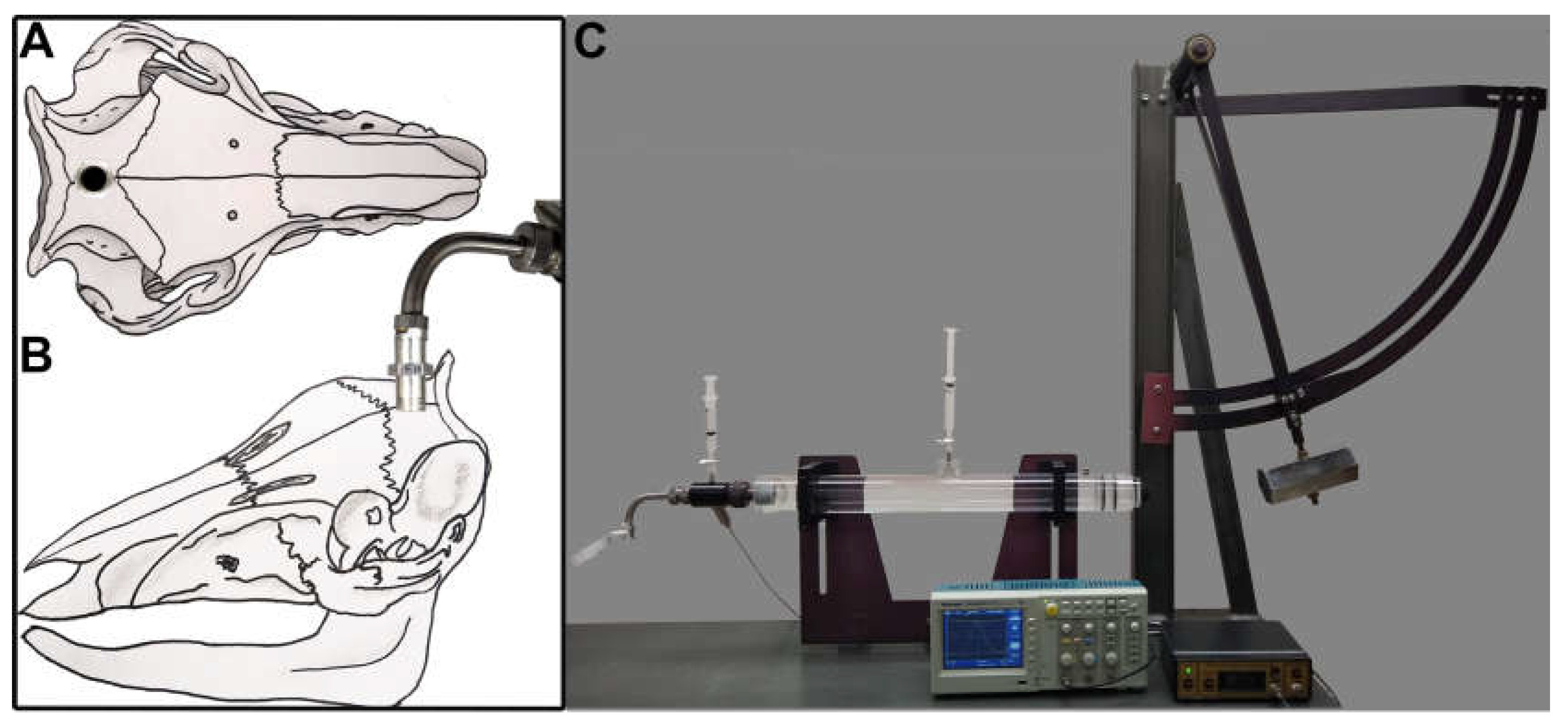
Figure 2.
Central fluid percussion injury results in significant axonal injury and microglial activation within the corpus callosum. Box plots of (A) APP+ Axonal swellings indicative of diffuse traumatic axonal injury and (B) microglial activation index at 6hr following sham (n=3, grey boxes) or cFPI (n=18, yellow boxes). Each black circle indicates an individual animal. Graphs depict the median ± quartile. *p<0.05.
Figure 2.
Central fluid percussion injury results in significant axonal injury and microglial activation within the corpus callosum. Box plots of (A) APP+ Axonal swellings indicative of diffuse traumatic axonal injury and (B) microglial activation index at 6hr following sham (n=3, grey boxes) or cFPI (n=18, yellow boxes). Each black circle indicates an individual animal. Graphs depict the median ± quartile. *p<0.05.
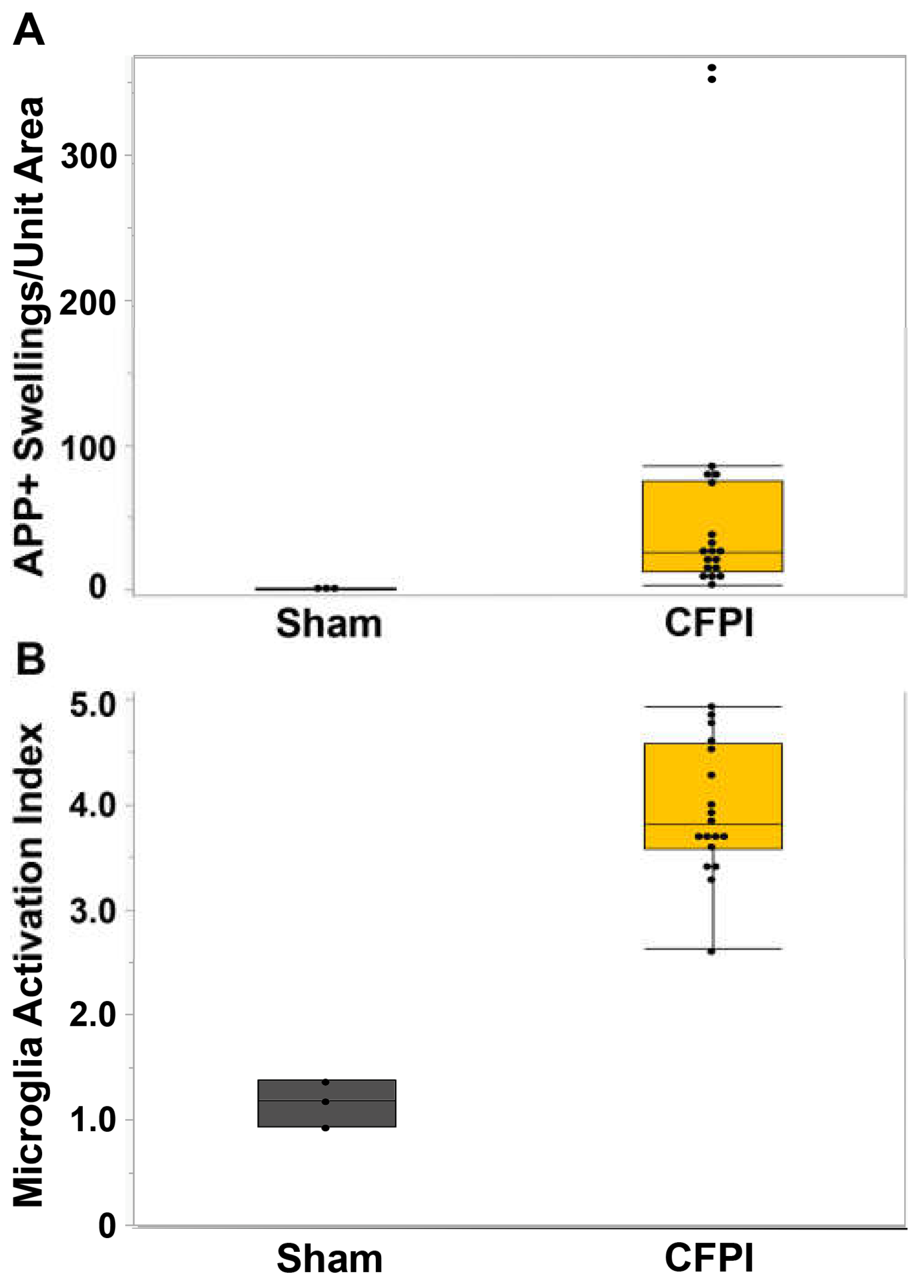
Figure 3.
Weight normalized central fluid percussion pressure pulse positively correlates with diffuse traumatic axonal injury and microglia activation in the thalamus and corpus callosum. (A) Scatter plot of total fluid percussion ATM pressure and the ATM pressure normalized to kg weight. (B) Correlation matrix depicting the Spearman Rho correlation for the weight normalized injury intensity (ATM/kg) with the degree of axonal injury in the corpus callosum (APP in CC) or thalamus (APP in Thalamus) as well as the degree of microglia activation in the corpus callosum (Microglia index in CC) or thalamus (Microglia index in Thalamus). Each pair of assessment values on the y and x axis is colored with red indicating a positive correlation, or blue indicating a negative correlation, with the correlation strength demonstrated by the circle’s size with larger circles representing lower p values. The lower part of the correlation matrix shows the correlation curves for each x and y comparison with each black dot representing data from an individual animal. Note that degree of axonal injury and microglial index values show positive correlations with ATM/kg, suggesting scalability between the pressure pulse inflicted and the degree of injury as well as the level of neuroinflammation in both regions.
Figure 3.
Weight normalized central fluid percussion pressure pulse positively correlates with diffuse traumatic axonal injury and microglia activation in the thalamus and corpus callosum. (A) Scatter plot of total fluid percussion ATM pressure and the ATM pressure normalized to kg weight. (B) Correlation matrix depicting the Spearman Rho correlation for the weight normalized injury intensity (ATM/kg) with the degree of axonal injury in the corpus callosum (APP in CC) or thalamus (APP in Thalamus) as well as the degree of microglia activation in the corpus callosum (Microglia index in CC) or thalamus (Microglia index in Thalamus). Each pair of assessment values on the y and x axis is colored with red indicating a positive correlation, or blue indicating a negative correlation, with the correlation strength demonstrated by the circle’s size with larger circles representing lower p values. The lower part of the correlation matrix shows the correlation curves for each x and y comparison with each black dot representing data from an individual animal. Note that degree of axonal injury and microglial index values show positive correlations with ATM/kg, suggesting scalability between the pressure pulse inflicted and the degree of injury as well as the level of neuroinflammation in both regions.
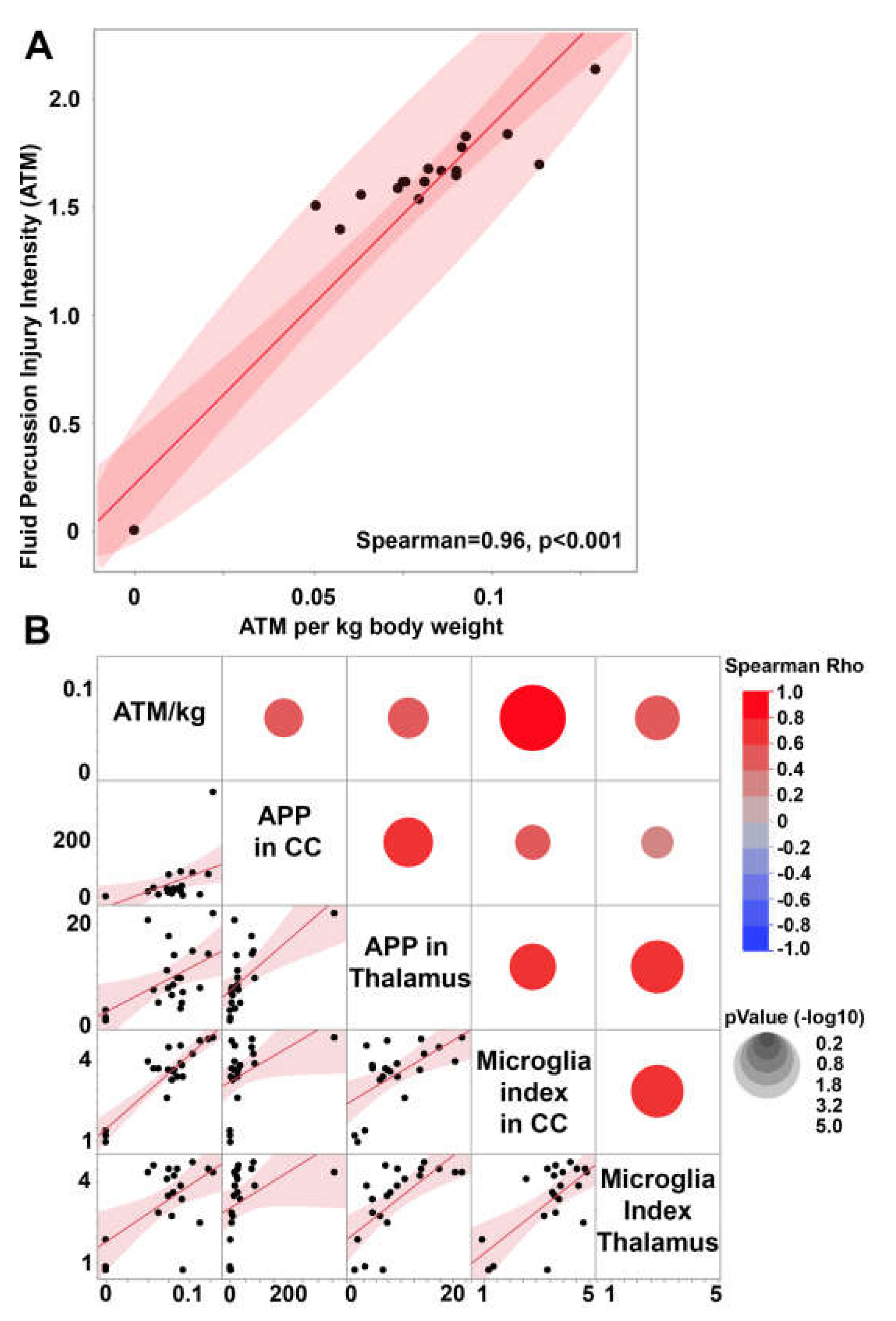
Figure 4.
Blood gas readings do not correlate with cFPI injury intensity (ATM/kg) in micro pigs. Correlation matrixes depicting the Spearman Rho correlation for the weight normalized injury intensity (ATM/kg) with the various blood gas measurements and the mean arterial blood pressure (MABP) 6hr post-CFPI. Each pair of assessment values on the y and x axis is colored with red indicating a positive correlation, or blue indicating a negative correlation, with the correlation strength demonstrated by the circle’s size. The lower part of the correlation matrix shows the correlation curves for each x and y comparison with each black dot representing data from an individual animal.
Figure 4.
Blood gas readings do not correlate with cFPI injury intensity (ATM/kg) in micro pigs. Correlation matrixes depicting the Spearman Rho correlation for the weight normalized injury intensity (ATM/kg) with the various blood gas measurements and the mean arterial blood pressure (MABP) 6hr post-CFPI. Each pair of assessment values on the y and x axis is colored with red indicating a positive correlation, or blue indicating a negative correlation, with the correlation strength demonstrated by the circle’s size. The lower part of the correlation matrix shows the correlation curves for each x and y comparison with each black dot representing data from an individual animal.
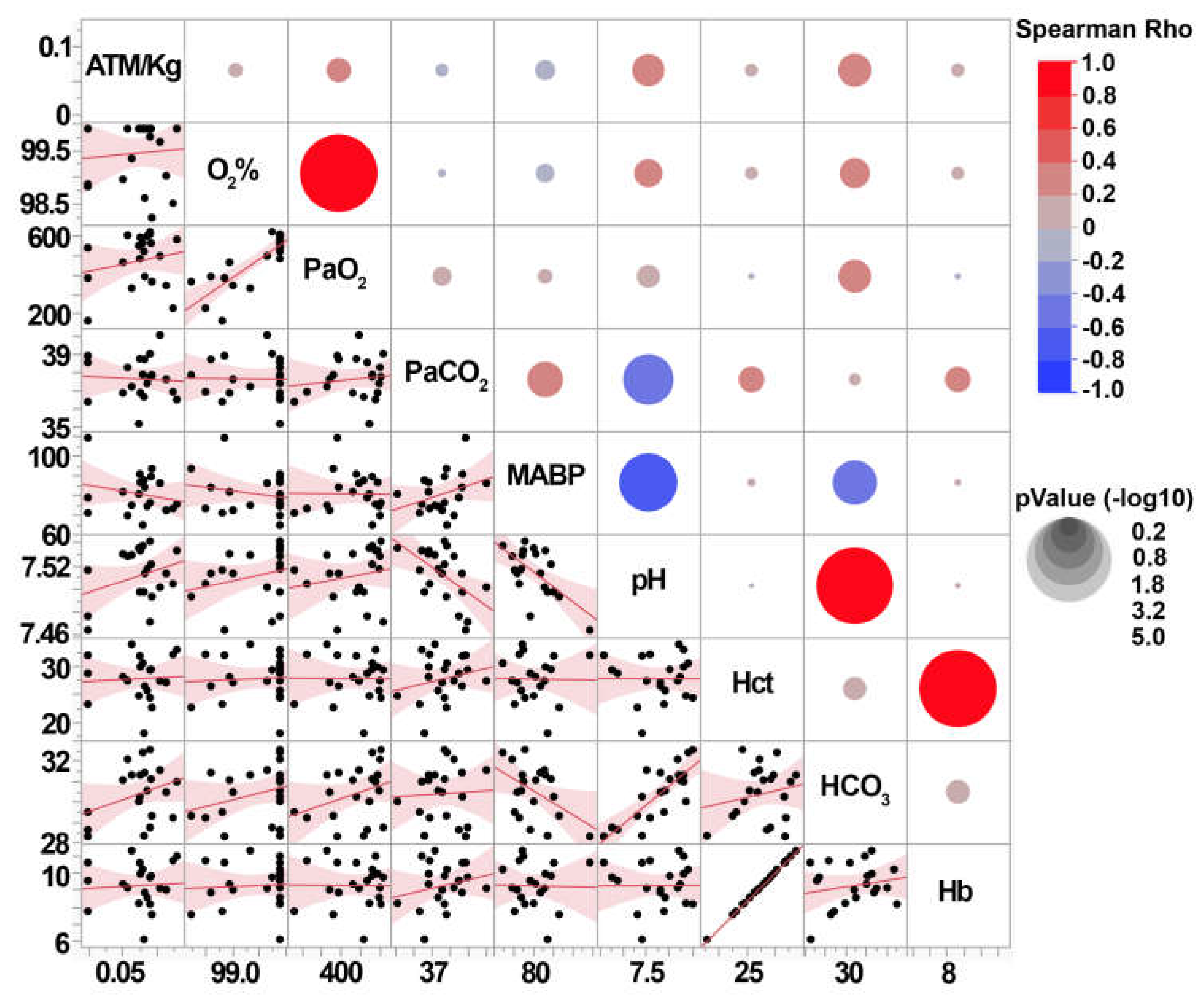
Figure 5.
The majority of labeled tissues exhibited high-degree labeling of both GFAP and Iba-1.(A-C) Representative epifluorescent micrographs show astrocytes labeled with GFAP (first column; red) and microglia with Iba-1 (second column; green). The third column shows an overlay of GFAP and Iba-1. Each row shows representative images for the degree of labeling, with (A) high-degree, (B) medium-degree, and (C) low/no-degree labeling of GFAP and IBA1. Scale bar: 50μm. (D) Stacked bar graphs show the proportional number of tissues that were categorized into high-degree, medium-degree, and low/no-degree labeling for both GFAP and IBA1. (E-F) Box plots depicting the interquartile range and median age of tissue of each animal labeled with (E) GFAP or (F) IBA1 are categorized by degree of labeling. Note that the mean ages of GFAP-labeled tissue are less variable between labeling degrees, while the mean age of IBA1 tissue increases significantly from high-degree to medium-degree labeled tissue. Graphs depict the median ± quartile. Black dots denote individual tissue samples. *p < 0.05.
Figure 5.
The majority of labeled tissues exhibited high-degree labeling of both GFAP and Iba-1.(A-C) Representative epifluorescent micrographs show astrocytes labeled with GFAP (first column; red) and microglia with Iba-1 (second column; green). The third column shows an overlay of GFAP and Iba-1. Each row shows representative images for the degree of labeling, with (A) high-degree, (B) medium-degree, and (C) low/no-degree labeling of GFAP and IBA1. Scale bar: 50μm. (D) Stacked bar graphs show the proportional number of tissues that were categorized into high-degree, medium-degree, and low/no-degree labeling for both GFAP and IBA1. (E-F) Box plots depicting the interquartile range and median age of tissue of each animal labeled with (E) GFAP or (F) IBA1 are categorized by degree of labeling. Note that the mean ages of GFAP-labeled tissue are less variable between labeling degrees, while the mean age of IBA1 tissue increases significantly from high-degree to medium-degree labeled tissue. Graphs depict the median ± quartile. Black dots denote individual tissue samples. *p < 0.05.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



