
Submitted:
18 May 2023
Posted:
19 May 2023
You are already at the latest version
Abstract
We have shown that lipid-associated loci discovered by genome-wide association studies (GWAS) have pleiotropic effects on lipid metabolism, carotid intima-media thickness (CIMT), and CAD risk. Here, we investigated the impact of lipid-associated GWAS loci on the efficacy of rosuvastatin therapy in terms of changes in plasma lipid levels and CIMT. The study comprised 116 CAD patients with hypercholesterolemia. CIMT, total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and triglycerides (TG) were measured at baseline and after 6 and 12 months of follow-up, respectively. Genotyping of fifteen lipid-associated GWAS loci was performed by the MassArray-4 System. Linear regression analysis adjusted for sex, age, body mass index, and rosuvastatin dose was used to estimate the phenotypic effects of polymorphisms, and P-values were calculated through adaptive permutation tests. Over one-year therapy, a decrease in CIMT was linked to rs1689800, rs4846914, rs12328675, rs55730499, rs9987289, rs11220463, rs16942887, and rs881844 polymorphisms (Pperm<0.05). TC change was associated with rs55730499, rs11220463, and rs6065906; LDL-C change was linked to the rs55730499, rs1689800, and rs16942887 polymorphisms; and TG change was linked to polymorphisms rs838880 and rs1883025 (Pperm<0.05). In conclusion, the polymorphisms rs1689800, rs55730499, rs11220463, and rs16942887 were found to be predictive markers for multiple antiatherogenic effects of rosuvastatin in CAD patients.
Keywords:
coronary artery disease
; plasma lipids
; carotid intima-media thickness
; pharmacogenetics
; personalized medicine
; single nucleotide polymorphisms
; lipid-lowering therapy
; rosuvastatin.
1. Introduction
Atherosclerosis is a complex disease, which pathogenesis involving numerous pathological processes increased plasma cholesterol, its deposition in the arterial wall, endothelial dysfunction and vascular remodeling. These processes are influenced both by environmental and genetic factors [1,2]. It is widely agreed that dyslipidemia plays a crucial role in the development of atherosclerosis and coronary artery disease (CAD) [2]. Lipid metabolism abnormalities and coronary heart disease are determined by substantial effects of genetic factors [3,4,5]. According to the GWAS Catalog (https://www.ebi.ac.uk/gwas/home), approximately 1700 genome-wide association studies (GWAS) on over 450 cardiovascular phenotypes have been conducted, yielding substantial insights into the genetic etiology of cardiovascular disease and related traits [6]. Numerous GWAS including large-scale studies of the Global Lipids Genetics Consortium have discovered loci associated with plasma lipids [7,8,9,10,11,12]. GWAS were also conducted to find genetic determinants of statin pharmacogenetics, including JUPITER, the largest study of lipid-lowering effects of rosuvastatin [13,14,15]. Statins are the first line of lipid-lowering drugs widely used in both primary and secondary prevention of CAD. For one of the most popular statins, rosuvastatin, it was shown that it could reduce both blood lipid levels, including total cholesterol (TC), LDL-C (low-density lipoprotein cholesterol), triglycerides (TG), and carotid intima-media thickness (CIMT) [16].
Carotid intima-media thickness is a surrogate marker for the presence and progression of atherosclerosis, useful for evaluating risk and incidence of cardiovascular disease [17]. A causal relationship between LDL-C and CIMT has been observed [18], and CIMT is being considered as a marker of preclinical atherosclerosis [18,19]. A growing body of evidence has been provided to consider CIMT as a predictive marker for coronary artery atherosclerosis, its severity, and the extent of plaque burden [20,21,22,23]. Importantly, rosuvastatin may reduce not only CIMT but also atherosclerotic plaque growth [24,25]. It is impossible to claim definitely whether hypolipidemic effects of rosuvastatin are solely responsible for the regression of atherosclerosis, because pleiotropic effects of the drug may contribute to in arterial wall changes through inflammation reduction [25,26]. The mechanisms by which rosuvastatin is responsible for vascular wall changes may be unraveled by pharmacogenetic studies; however, a majority of such studies have investigated only lipid-lowering effects of the drug [13,14,27,28]. Our research team has conducted several studies on the effect of gene polymorphisms on plasma lipids and CIMT, as well as the dynamics of their changes during rosuvastatin lipid-lowering therapy [29,30,31]. It has been discovered that some lipid-associated GWAS loci contribute to lipid-lowering effects associated with rosuvastatin therapy and determine CIMT regression. Without a doubt, CIMT seems to be a surrogate marker of atherosclerosis in CAD patients, despite it correlates with disease severity [20,21,22,23]. However, in terms of clinical application, CIMT regression during rosuvastatin therapy may mirror vascular wall changes and be indicative of drug efficacy. The aim of our pharmacogenetic study was to identify the effects of fifteen lipid-associated GWAS loci such as rs4846914 (GALNT2), rs11220463 (ST3GAL4), rs881844 (STARD3), rs1689800 (ZNF648), rs12328675 (COBLL1), rs9987289 (PPP1R3B), rs55730499 (LPA), rs3136441 (F2), rs6065906 (PLTP), rs838880 (SCARB1), rs386000 (LILRA3), rs1883025 (ABCA1), rs3764261(CETP), rs217406 (NPC1L1), and rs16942887 (PSKH1) on plasma lipids and CIMT in CAD patients taking rosuvastatin. These loci have recently been observed to be associated with CAD susceptibility and CIMT [32].
2. Materials and Methods
Study design. The present research was designed as a prospective study, in which the participants received rosuvastatin at an initial dose of 5 mg. Then, in 4 weeks, we controlled the levels of LDL-C. In cases where the levels were below the target level (1.8 mmol/l), patients continued taking the dose, which let them attain the target level. If it was not attained, the dose of rosuvastatin was gradually increased to 10, 20, and finally the maximum dose of 40 mg. The control of LDL-C levels was performed every 4 weeks after the dose increase. For the association analysis, we used the change (delta, Δ) in TC, LDL-C, TG, and CIMT values over the first 6 and 12 months of the study.
Study participants. The patient cohort comprised 116 Russian patients with the diagnosis of CAD, stable angina pectoris grade II–III. The study participants were described in detail in our previous work [31]. In brief, the cohort included 85 men (73%) and 31 postmenopausal women (27%), with the mean age of 61.0 ± 7.25 years. Approximately 58% of CAD patients experienced a myocardial infarction in the past, and 97.5% of study participants had arterial hypertension. Diagnosis of CAD was confirmed by qualified cardiologists according to the Canadian Cardiovascular Society, ECG stress tests (treadmill test), and 24-h Holter’s ECG monitoring. TC levels higher than 4.0 mmol/l and LDL-C levels higher than 1.8 mmol/l were considered as criteria for dyslipidemia.
Biochemical and ultrasound investigations were described in our previous paper [31]. Plasma lipid levels, except LDL-C, were detected using an automatic laboratory analyzer. LDL-C were calculated using Friedewald’s equation. CIMT measurement was performed in B-mode using the standard method at the distal third of the common carotid artery at a distance of 1–1.5 cm proximal to the bifurcation along the posterior wall [33]. The following two parameters of carotid intima-media thickness were used in the study: mean CIMT (mean value of all measurements on the right and left sides) and maximal CIMT (the maximal value obtained from measurements on both sides with the further assessment of its change on the side where it was the highest).
Genetic analysis. Single nucleotide polymorphisms (SNPs) selected for this study have previously been found to be associated with plasma lipids in several genome-wide association studies [7,8,9,10,11,12] (GWAS Catalog (https://www.ebi.ac.uk/gwas, accessed date 13 March 2023). As mentioned above, some of the SNPs have been recently found to be associated with CAD susceptibility and CIMT [32], and we are interested in investigating whether these variants determine the lipid- and CIMT-lowering effects of rosuvastatin in CAD patients. The investigated SNPs included rs4846914 (GALNT2), rs11220463 (ST3GAL4), rs881844 (STARD3), rs1689800 (ZNF648), rs12328675 (COBLL1), rs9987289 (PPP1R3B), rs55730499 (LPA), rs3136441 (F2), rs6065906 (PLTP), rs838880 (SCARB1), rs386000 (LILRA3), rs1883025 (ABCA1), rs3764261 (CETP), rs217406 (NPC1L1), and rs16942887 (PSKH1).
Extraction of DNA was performed from venous blood samples using phenol-chloroform method and precipitation with ethanol. Genotyping was performed using iPLEX technology on the MassARRAY 4 system (Agena Bioscience, San-Diego, CA, USA). Software MassARRAY Assay Design Suite was used to select a primer set and to design a multiplex panel for SNP genotyping. The sequences of primers are available on request.
Statistical analysis. The normality of the distribution of lipid and CIMT values was determined using the Kolmogorov-Smirnov and Shapiro-Wilk tests. As the trait distributions deviated from the normal one, the values were expressed as median and interquartile range (Me, Q1; Q3). Statistical package STATISTICA v13.0 (Statsoft, Tulsa, OK, USA) was utilized for descriptive statistics, distribution analysis, and determining the significance of lipid and CIMT changes. The significance of lipid and CIMT change during rosuvastatin therapy was tested by the Wilcoxon’s matched pairs test. The distribution of genotype frequencies according to Hardy–Weinberg equilibrium was assessed with Fisher’s exact test. For the association analysis, we used linear regression with adjustments for sex, age, body mass index (calculated as body mass in kilograms divided by height in meters in a square), and rosuvastatin dose, which allowed the participants to attain the target LDL-C level. Change in lipid levels was calculated as the difference between natural log-transformed on- and off-treatment levels divided by the natural log-transformed off-treatment level as described by Postmus et al. [13]. We applied the same approach for the CIMT change, but before the logarithmic transformation, the CIMT data were multiplied by 10. Empirical P-values (Pperm) were calculated through the adaptive permutation procedure. We tested three genetic models for SNP-phenotype associations, such as additive, dominant, and recessive. The PLINK v1.92 software [34] was used for all genetic calculations, including estimation of minor allele frequencies (MAF), tests for Hardy-Weinberg equilibrium (HWE), multiple regression analysis, and permutation procedures. When the p-value was less than 0.05, all of the results were declared statistically significant.
3. Results
The majority of the patients (N=110, 94,8 %) attained their target LDL-C level during rosuvastatin therapy. The change in lipid and CIMT values was assessed during 6 and 12 months of observation. The reduction of all lipid parameters (p< 1×106 for TC, LDL-C, and TG change during 6- and 12-month periods) was significant, except HDL-C levels (P=0.16 and 0.87, respectively). Reductions in the maximum (p< 1×106) and mean CIMT (P=0.025 and < 1×105 for the change in 6 and 12 months, respectively) were also significant (Supplementary Table 1).
3.1. Associations of the SNPs with lipid and CIMT reduction during the 6-month therapy by rosuvastatin.
The distribution of genotypes for the majority of SNPs (except for rs16942887) was in Hardy-Weinberg equilibrium. The observed effects of SNPs on the lipid-lowering effect and CIMT regression in CAD patients taking rosuvastatin for the 6-month period are shown in Table 1. We found that plasma TC reduction was associated with two SNPs, such as rs55730499 of LPA (beta = 0.323, Pperm=0.0022, recessive effect) and rs6065906 of PLTP (beta = 0.140, Pperm=0.0135, recessive effect). Based on the beta coefficients, it can be estimated that the carriage of homozygous genotypes for the minor alleles of the above polymorphisms is associated with 32.3% and 14.0 % of TC reduction, respectively, compared with the carriers of alternative genotypes (the positive beta indicates a worse lipid reduction and consequently a worse statin response). LDL-C reduction was found to be associated with rs55730499 of the LPA gene (Pperm=0.0224, recessive effect), and rs1689800 of the ZNF648 gene (Pperm=0.0493, additive effect). The rs1689800G allele is associated with a 4.6 percent reduction in plasma LDL-C levels after 6 months of rosuvastatin therapy. The effect size for the rs55730499 polymorphism of LPA was greater in the TT homozygotes and characterized by a 50.4% decrease in LDL-C concentration than in carriers of alternative genotypes.
As it can be seen from Table 1, four lipid-associated GWAS loci were associated with both maximum and mean CIMT change: rs4846914 in GALNT2 (Pperm=0.0133 and 0.0344 for the association with maximum and mean CIMT change, respectively), rs11220463 in ST3GAL4 (Pperm=0.0159 and 0.0243, respectively), rs16942887 in PSKH1 (Pperm=0.0421 and 0.0483, respectively), and rs881844 in STARD3 (Pperm=0.0086 and 0.0033, respectively). SNP rs1689800 in the ZNF648 gene (Pperm=0.0234) was associated with a change in the maximum CIMT. The most significant association with TC and LDL-C reduction during 6 months of rosuvastatin therapy was found for SNP rs55730499 in LPA, whereas the rs881844 polymorphism of the STARD3 gene showed the most substantial association with the regression of CIMT in CAD patients.
Table 2 shows associations between the SNPs and plasma triglycerides reduction. It has been revealed that the only SNP rs838880 of the SCARB1 gene was associated with TG reduction (beta = 1.736, Pperm=0.0478, recessive effect) after 6 months of lipid-lowering therapy.
3.2. Associations of SNPs with lipid and CIMT reduction during 12-month therapy by rosuvastatin
Table 3 shows associations of studied SNPs with the 12-month lipid-lowering effect and carotid intima-media thickness change in CAD patients receiving rosuvastatin.
We observed that two polymorphisms contributed to TC reduction: rs55730499 in LPA (beta = 0.364, Pperm=0.0001, recessive effect) and rs11220463 in ST3GAL4 (beta = -0.181, Pperm=0.0273, recessive effect) after one year of lipid-lowering therapy. For the rs11220463 polymorphism, the homozygous genotype for the minor allele T was associated with an 18.1% more pronounced TC-lowering effect than in carriers of alternative genotypes. The reduction in plasma LDL-C levels was associated with rs55730499 of LPA (Pperm=0.0415, recessive effect) and rs16942887 of PSKH1 (Pperm=0.0175, dominant effect).
Three SNPs were associated with both maximum and mean CIMT change: rs1689800 of ZNF648 (better statin response; Pperm=0.0105 and 0.0282 for the association with maximum and mean CIMT change, respectively), rs12328675 of COBLL1 (worse statin response; Pperm=0.0213 and 0.0056, respectively), and rs9987289 of PPP1R3B (worse statin response; Pperm=0.0359 and 0.0109, respectively). Two polymorphisms, such as rs55730499 of LPA (Pperm=0.0146) and rs881844 of STARD3 (Pperm=0.0223) were associated with the mean CIMT change.
During 12 months of lipid-lowering therapy with rosuvastatin, the most significant effect on the TC reduction was found for SNP rs55730499 of LPA, whereas the most significant effects of rs16942887 of PSKH1 and rs12328675 of COBLL1 were found on the reduction of LDL-C and CIMT, respectively. Associations of TG reduction (Table 2) with genotype in a 12-month period were found for rs1883025 of the ABCA1 gene (beta = -0.7246, Pperm= 0.0160, dominant effect) and for rs11220463 in ST3GAL4 (beta = 3.624, Pperm=0.05, recessive effect).
4. Discussion
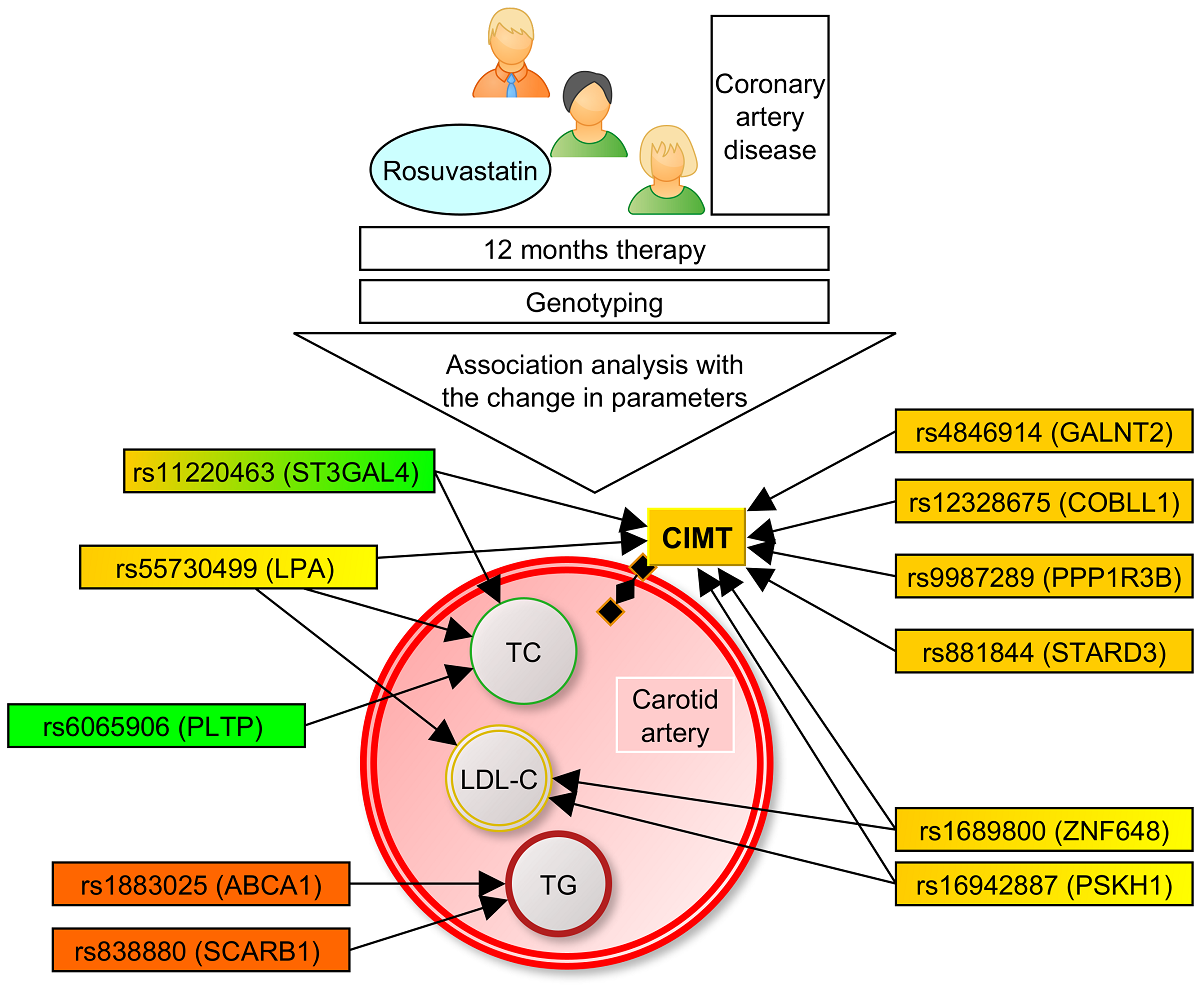
The present study demonstrated, for the first time, the impact of lipid-associated GWAS loci on CIMT (Figure 1) and plasma levels of TC, LDL-C, and TG in CAD patients during rosuvastatin therapy (Figure 2).
In particular, we found associations of SNPs located at ZNF648, LPA, ST3GAL4, PSKH1, GALNT2, COBLL1, PPP1R3B, and STARD3 genes with reduction of CIMT. Pharmacogenetic associations of the variants in ZNF648, LPA, ST3GAL4, PSKH1, PLTP, SCARB1, and ABCA1 with the change in lipid levels were also found for the first time. Thus, lipid-associated GWAS loci are associated not only with lipids and the risk of coronary artery disease [32], but are also responsible for the antiatherogenic effects of rosuvastatin after 6 and 12 months of lipid-lowering therapy.
Potential mechanisms by which polymorphic variants of the investigated lipid metabolism genes can impact the antiatherogenic effects of rosuvastatin therapy are of interest. The ZNF648 gene encodes zinc finger protein 648, which may be involved in the DNA-templated transcription [35]. In the present study rs1689800 polymorphism was associated with CIMT change. The possible mechanism of the effect of the variant on vascular wall may include the influence on oxidative stress (GLUL catabolizes glutamate, an amino acid required for glutathione synthesis), because such a mechanism is described for the nearest SNP rs10911021 located in the same genomic region [36].
The GALNT2 gene encodes N-acetylgalactosaminyltransferase 2, an enzyme involved in O-linked glycosylation of the substrates, including those regulating lipid metabolism such as apolipoprotein C-III (APOC-III), angiopoietin-related protein 3 (ANGPTL3) and phospholipid transfer protein (PLTP); in these ways it influences HDL-C and TG metabolism [37,38]. The rs4846914G allele has been linked to atherogenic changes in plasma lipid metabolism, such as an increase in TG and a decrease in HDL-C levels [9,10]. We found no associations of this variant with changes in any plasma lipid, but this SNP was significantly associated with a better CIMT regression during a 6-month period of rosuvastatin therapy. The presence of the link with CIMT change and absence of any association with lipid level change suggest that the pharmacogenetic effect on CIMT change might be mediated through non-lipid-related mechanisms, taking into account that SNP rs4846914 is known to be associated with endothelial function, serum levels of insulin and glucose [39], as well as hypertension [40]. In particular, the latter study demonstrated higher promoter methylation of the GALNT2 gene, higher levels of ApoB, and lower levels of ApoA1 in hypertensives with the GG genotype [40].
The COBLL1 gene encodes a cordon-bleu WH2 repeat protein like 1, having actin monomer and cadherin binding activity [41]. In the present study, the variant rs12328675 was associated with worse CIMT regression (for the carriers of the C-allele, dominant effect) during 12 months of observation. This association was the most significant among all studied variants over a 12-month period. There were no associations with lipid levels change. The link between this polymorphism and CIMT change can be explained by the ability of the risk allele C, associated with the higher CAD risk, to form transcription factor binding sites for the factors involved in the processes of vascular inflammation regulation and angiogenesis (AP-1 (syn. JUN), SMAD2, SMAD3, SMAD4, and E2F8) [42].
The LPA gene encodes lipoprotein (a), which is a well-known and independent risk factor for coronary artery disease [11]. The studied rs55730499 variant is associated with Lp(a) concentrations [11], coronary artery disease risk [8], myocardial infarction risk [43], and stroke risk [44]. The T-allele is associated with higher TC levels out of therapy [43], and in our study, the TT genotype (homozygous for the minor allele) was associated with reduced drug response in terms of TC, LDL-C, and CIMT change. The association with TC change had the highest significance among all associations with TC change, and moreover, rs55730499 was the only variant studied that was associated with both TC, LDL-C, and CIMT change in plasma at the same period of observation (12 months). Based on these findings, the studied LPA variant can be considered the first of all studied variants to be used as a predictor for rosuvastatin therapy personalization.
The PPP1R3B gene encodes protein phosphatase 1 regulatory subunit 3B, a key protein in hepatic glycogen metabolism [12]. The rs9987289 variant is associated with plasma TC and LDL-C levels [9,12], but was not tested for carotid atherosclerosis traits or the pharmacogenetics of rosuvastatin. In our study, the presence of the minor A-allele was associated with a worse CIMT response to rosuvastatin. Such an effect could be explained by the association of the studied SNP with inflammatory markers, such as C-reactive protein [45], known for its influence on cardiovascular risk and atherosclerosis progression [46], and also by the link of the variant with metabolic syndrome [47] and glucose levels [48], taking into account the known role of hyperglycemia in the dysfunction of the endothelium [49,50].
The ABCA1 gene encodes phospholipid-transporting ATPase ABCA1, which provides the efflux of intracellular cholesterol to apolipoproteins and the formation of nascent high-density lipoproteins [51]. The minor T-allele of the rs1883025 SNP is associated in GWAS with an anti-atherogenic phenotype: lower TC [9], LDL-C [52], TG [53], and HDL-C levels [52], and with a better response to statin therapy ([54], the particular drug and lipid are not specified). In the present study, we confirmed a better response to statin therapy in terms of triglyceride reduction in carriers of the minor T-allele (the dominant effect of the SNP).
The ST3GAL4 gene encodes CMP-N-acetylneuraminate-beta-galactosamide-alpha-2,3-sialyltransferase 4, an enzyme involved in the terminal sialylation of glycoproteins and glycolipids. A beta-galactoside alpha2-3 sialyltransferase takes part in hemostasis (sialylation of plasma von Willebrand factor) and the inflammatory process (selectin-mediated rolling and adhesion of leukocytes during extravasation) [55]. The studied SNP in this gene, rs11220463, is associated with both blood lipids (TC and LDL-C) [53] and inflammation (CRP levels) [56]. In the present study, we have found pharmacogenetic associations of the studied variant with TC and CIMT reduction during therapy. The association with CIMT can be possibly explained by the involvement of the product of ST3GAL4 in inflammatory processes, taking into account the known association of systemic inflammation (assessed by the systemic immune-inflammatory index) with CIMT [57]. For rs11220463 the association with inflammation was found in terms of CRP levels [56], and CRP is known to be associated with CIMT [58,59], but this association is not proven to be causal [58].
The PSKH1 gene encodes serine/threonine-protein kinase H1, involved in intracellular protein trafficking and pre-mRNA processing [60]. For the rs16942887 variant, we found associations with LDL-C and CIMT changes on rosuvastatin therapy. There are no reported associations in the literature between this variation and changes in LDL-C and CIMT when taking rosuvastatin. The possible mechanism of the effect of SNP is difficult to predict because of the lack of information on the mechanisms of lipid and vascular influence of PSKH1.
The STARD3 gene encodes StAR-related lipid transfer protein 3, which mediates cholesterol transport from the endoplasmic reticulum to endosomes [61]. In the literature, the rs881844 polymorphism in STARD3 was known to be associated with plasma lipids, including TC and HDL-C [43,53]. In the present study we didn’t find any associations with changes in plasma lipid levels on the rosuvastatin therapy, however, this SNP showed the strongest effects on CIMT changes in both 6- and 12-month periods. The effect on the vascular wall can be explained by the involvement of STARD3 in the regulation of cholesterol-dependent inflammation and sensitivity to proinflammatory cytokines [62].
The PLTP gene encodes phospholipid transfer protein, which is involved in the transfer of phospholipids and free cholesterol from LDLs and VLDLs into HDLs [63], as well as the uptake of cholesterol from peripheral cells [64]. Taking into account the function of the gene product, it was rather logical to find the influence of rs6065906 in PLTP on the lipid-lowering effect of rosuvastatin in terms of TC reduction in the present study, which was not reported before.
Thus, the molecular mechanisms underlying the effects of the studied loci in terms of lipid and CIMT reduction remains unknown, but taking into account the biological functions of the proteins, these genes possess pleiotropic effects on biological and pathological processes including lipid metabolism, inflammation, leukocyte adhesion to the endothelium, endothelial dysfunction, and glucose metabolism. Further experimental studies are needed to explain the functional effects of polymorphisms. Many of the studied loci have been associated with HDL-C levels in GWAS. However, because of non-significant HDL-C change on the therapy, observed in the present study, we didn’t test the SNPs for associations with HDL-C change. This is the limitation of our study, but we can’t say it’s a big omission, because one of the main phenotypes studied here was CIMT, which is known to have a causal relationship with LDL-C and with other factors but not with HDL-C itself [18].
5. Conclusions
In the present study, associations of both TC (and/or LDL-C) and CIMT changes at the same period of follow-up (6 or 12 months) were observed for the polymorphisms rs55730499 of LPA (after 12 months of therapy) and rs1689800 of ZNF648 (after 6 months of therapy). In general (including 6- and 12-month periods), four polymorphisms namely rs1689800 in ZNF648 (LDL-C and CIMT), rs55730499 in LPA (TC, LDL-C, and CIMT), rs11220463 in ST3GAL4 (TC and CIMT), and rs16942887 in PSKH1 (LDL-C and CIMT) were associated with a decrease in atherogenic lipids and CIMT. There were no TG change-associated loci, that were also associated with CIMT change. A half of eight GWAS-associated lipid loci, such as rs4846914 of GALNT2, rs12328675 of COBLL1, rs9987289 of PPP1R3B, and rs881844 of STARD3, that were linked to CIMT changes in our study were not associated with any changes in plasma lipids during rosuvastatin therapy. Polymorphisms rs881844 of STARD3 and rs12328675 of COBLL1 were most strongly associated with CIMT regression, whereas rs55730499 of LPA – with the reduction of plasma TC and rs16942887 of PSKH1 – with LDL-C change. The observed associations should be replicated in independent pharmacogenetic studies with a larger number of hyperlipidemic patients. The successful validation of these associations may provide novel genetic markers for the personalization of rosuvastatin therapy in the monitoring of the drug effectiveness in patients with atherogenic changes in lipid metabolism.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Change in lipid and carotid intima-media thickness values during rosuvastatin therapy. Supplementary Table 1. Change in lipid and carotid intima-media thickness values during rosuvastatin therapy.
Author Contributions
Conceptualization, A.P.; methodology, A.P., S.K., M.A., M.C.; software, S.K. and A.P.; validation, S.K.,A.P.; formal analysis, S.K., M.A.; genetic investigation, Iu.A., E.K., and M.B.; resources, A.P.; data curation, S.K. and A.P.; writing—original draft preparation, S.K.; writing—review and editing, A.P.; visualization, S.K.; supervision, A.P.; project administration, A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received funding from Kursk State Medical University.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Ethics Committee of Kursk State Medical University (11 May, 2015).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data supporting reported results are available upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gupta, RM; Schnitzler, G; Fang, S; Lee-Kim, VS; Barry, A. Multiomic Analysis and CRISPR Perturbation Screens Identify Endothelial Cell Programs and Novel Therapeutic Targets for Coronary Artery Disease. Arterioscler Thromb Vasc Biol. 2023. [CrossRef]
- Blackburn, H. Invited commentary: 30-year perspective on the seven countries study. Am J Epidemiol. 2017, 185, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R; Tybjaerg-Hansen, A. Genetics of Coronary Artery Disease. Circ Res. 2016, 118, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Churilin, M.I.; Kononov, S.I.; Mal, G.S.; Polonikov, A.V.; Lazarenko, V.A. Lipid metabolism genes and predisposition to ischemic heart disease. Medical News of North Caucasus, 2019, 14, 401–407. [Google Scholar] [CrossRef]
- Mutlu, AS; Duffy, J; Wang, MC. Lipid metabolism and lipid signals in aging and longevity. Dev Cell. 2021, 56, 1394–1407. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Jurgens, S.J.; Erdmann, J. , Bezzina, C.R. Genome-wide association studies of cardiovascular disease. Physiol Rev. [CrossRef]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [PubMed]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Kathiresan, S.; Melander, O.; Guiducci, C.; Surti, A.; Burtt, N.P.; Rieder, M.J.; Cooper, G.M.; Roos, C.; Voight, B.F.; Havulinna, A.S.; et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 2008, 40, 189–197. [Google Scholar] [CrossRef]
- Mack, S.; Coassin, S.; Rueedi, R.; Yousri, N.A.; Seppälä, I.; Gieger, C.; Schönherr, S.; Forer, L.; Erhart, G.; Marques-Vidal, P.; et al. A genome-wide association meta-analysis on lipoprotein (a) concentrations adjusted for apolipoprotein (a) isoforms. J. Lipid Res. 2017, 58, 1834–1844. [Google Scholar] [CrossRef]
- Zhang, Y.; Gan, W.; Tian, C.; Li, H.; Lin, X.; Chen, Y. Association of PPP1R3B polymorphisms with blood lipid and C-reactive protein levels in a Chinese population. J. Diabetes. 2013, 5, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Postmus, I; Trompet, S; Deshmukh, H. A.; Arsenault B.J.; Avery C.L.; Bis J.C.; Chasman D.I.; de Keyser C.E.; Deshmukh H.A.; Evans D.S.; et al. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat. Commun. 2014, 5, 5068. [Google Scholar] [CrossRef] [PubMed]
- Chasman, D.I.; Giulianini, F.; MacFadyen, J. , Barratt B.J., Nyberg F., Ridker P.M. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ. Cardiovasc. Genet. 2012, 5, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Chasman, D.I. , Giulianini, F., Demler, O.V., Udler, M.S. Pleiotropy-Based Decomposition of Genetic Risk Scores: Association and Interaction Analysis for Type 2 Diabetes and CAD. Am J Hum Genet. 2020, 106, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.R. 3rd, Raichlen, J.S., Riley, W.A.; Evans G.W.; Palmer M.K.;, O'Leary D.H.; Grobbee D.E.; Bots M.L.; METEOR Study Group. Effect of rosuvastatin on progression of carotid intima-media thickness in low-risk individuals with subclinical atherosclerosis: the METEOR Trial. JAMA. 2007, 297, 1344–1353. [Google Scholar] [PubMed]
- Nezu, T; Hosomi, N; Aoki, S; Matsumoto M. Carotid Intima-Media Thickness for Atherosclerosis. J Atheroscler Thromb. 2016, 23, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Shah, S; Casas, J. P., Drenos, F.; Whittaker, J.; Deanfield, J.; Swerdlow, D.I.; Holmes, M.V.; Kivimaki, M.; Langenberg, C.; Wareham, N.; et al. Causal relevance of blood lipid fractions in the development of carotid atherosclerosis: mendelian randomization analysis. Circ. Cardiovasc Genet. 2013, 6, 63–72. [Google Scholar] [CrossRef]
- Vargas, JD; Manichaikul, A; Wang, X. Q.; Rich, S.S.; Rotter, J.I.; Post, W.S.; Polak, J.F.; Budoff, M.J., Bluemke, D.A. Detailed analysis of association between common single nucleotide polymorphisms and subclinical atherosclerosis: The Multi-ethnic Study of Atherosclerosis. Data Brief. 2016, 7, 229–242. [Google Scholar] [CrossRef]
- Guaricci, A.I.; Arcadi, T.; Brunetti, ND; Maffei, E. ; Montrone, D.; Martini, C.; De Luca, M.; De Rosa, F.; Cocco, D.; Midiri, M.; et al. Carotid intima media thickness and coronary atherosclerosis linkage in symptomatic intermediate risk patients evaluated by coronary computed tomography angiography. Int J Cardiol. 2014, 176, 988–993. [Google Scholar] [CrossRef]
- Amato, M.; Montorsi, P.; Ravani, P. , et al. Carotid intima-media thickness by B-mode ultrasound as surrogate of coronary atherosclerosis: correlation with quantitative coronary angiography and coronary intravascular ultrasound findings. Eur Heart J. 2007, 28, 2094–2101. [Google Scholar] [CrossRef]
- Graner M, Varpula M, Kahri J et al. Association of carotid intima-media thickness with angiographic severity and extent of coronary artery disease. Am. J. Cardiol. 2006, 97, 624–629. [Google Scholar] [CrossRef]
- Matsushima Y, Kawano H, Koide Y, Baba T, Toda G, Seto S, Yano K. Relationship of carotid intima-media thickness, pulse wave velocity, and ankle brachial index to the severity of coronary artery atherosclerosis. Clin Cardiol. 2004, 27, 629–634. [Google Scholar] [CrossRef]
- Ballantyne CM, Raichlen JS, Nicholls SJ et al. Effect of rosuvastatin therapy on coronary artery stenoses assessed by quantitative coronary angiography: a study to evaluate the effect of rosuvastatin on intravascular ultrasound-derived coronary atheroma burden. Circulation 2008, 117, 2458–2466. [Google Scholar] [CrossRef]
- Puri R, Libby P, Nissen SE et al. Long-term effects of maximally intensive statin therapy on changes in coronary atheroma composition: insights from SATURN. Eur. Heart J. Cardiovasc. Imaging. 2014, 15, 380–388. [Google Scholar] [CrossRef]
- Rumyantsev NA, Kukes VG, Kazakov RE, Rumyantsev AA, Sychev DA. Ispol'zovanie farmakogeneticheskogo testirovaniia dlia predotvrashcheniia nezhelatel'nykh lekarstvennykh reaktsiĭ pri terapii statinami [Use of pharmacogenetic testing to prevent adverse drug reactions during statin therapy]. Ter Arkh. 2017, 89, 82–87. Russian. [Google Scholar] [CrossRef]
- Soko ND, Masimirembwa C, Dandara C. Pharmacogenomics of Rosuvastatin: A Glocal (Global + Local) African Perspective and Expert Review on a Statin Drug. OMICS 2016, 20, 498–509. [Google Scholar] [CrossRef]
- Alfonsi JE, Hegele RA, Gryn SE. Pharmacogenetics of lipid-lowering agents: precision or indecision medicine? Curr. Atheroscler. Rep. 2016, 18, 24. [Google Scholar] [CrossRef]
- Churilin, M.I. , Kononov, S.I., Luneva, Y.V.,...Solodilova, M.A., Polonikov, A.V. Polymorphisms of Intracellular Cholesterol Transporters Genes: Relationship to Blood Lipid Levels, Carotid Intima-Media Thickness, and the Development of Coronary Heart Disease. Russian Journal of Genetics 2020, 56, 234–241. [Google Scholar] [CrossRef]
- Churilin, M.I. , Kononov, S.I., Luneva, Y.V.,...Polonikov, A.V., Kazanov, V.A. Apolipoprotein E gene polymorphisms: A relationship with the risk of coronary artery disease and the effectiveness of lipid-lowering therapy with rosuvastatin. Cardiovascular Therapy and Prevention (Russian Federation), 2020b, 19, pp. 17–23.
- Kononov S, Mal G, Azarova I, Klyosova E, Bykanova M, Churnosov M, Polonikov A. Pharmacogenetic loci for rosuvastatin are associated with intima-media thickness change and coronary artery disease risk. Pharmacogenomics. 2022 Jan;23, 15-34. [CrossRef]
- Lazarenko V, Churilin M, Azarova I, Klyosova E, Bykanova M, Ob'edkova N, Churnosov M, Bushueva O, Mal G, Povetkin S, Kononov S, Luneva Y, Zhabin S, Polonikova A, Gavrilenko A, Saraev I, Solodilova M, Polonikov A. Comprehensive Statistical and Bioinformatics Analysis in the Deciphering of Putative Mechanisms by Which Lipid-Associated GWAS Loci Contribute to Coronary Artery Disease. Biomedicines. 2022 Jan 25;10, 259. [CrossRef]
- Pignoli P, Tremoli E, Poli A, Oreste P, Paoletti R. Intimal plus medial thickness of the arterial wall: a direct measurement with ultrasound imaging. Circulation 74, 1399–1406 (1986).
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007 Sep;81, 559–575. [CrossRef]
- Gaudet P, Livstone MS, Lewis SE, Thomas PD. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief Bioinform. 2011 Sep;12, 449–462. [CrossRef]
- Shahid SU, Shabana, Humphries S. The SNP rs10911021 is associated with oxidative stress in coronary heart disease patients from Pakistan. Lipids Health Dis. 2018 Jan 5;17, 6. [CrossRef]
- Khetarpal SA, Schjoldager KT, Christoffersen C, Raghavan A, Edmondson AC, Reutter HM, Ahmed B, Ouazzani R, et al. Loss of Function of GALNT2 Lowers High-Density Lipoproteins in Humans, Nonhuman Primates, and Rodents. Cell Metab. 2016 Aug 9;24, 234–245. [CrossRef]
- Zilmer M, Edmondson AC, Khetarpal SA, Alesi V, Zaki MS, Rostasy K, Madsen CG, et al. Novel congenital disorder of O-linked glycosylation caused by GALNT2 loss of function. Brain. 2020 Apr 1;143, 1114-1126. [CrossRef]
- Chen J, Guan L, Liu H, Liu Q, Fan P, Bai H. GALNT2 Gene Variant rs4846914 Is Associated with Insulin and Insulin Resistance Depending on BMI in PCOS Patients: a Case-Control Study. Reprod Sci. 2021 Apr;28, 1122-1132. [CrossRef]
- Zhang X, Zhao H, Zhang J, Han D, Zheng Y, Guo X, He D, Guo J, Wang Y. Gene environment interaction of GALNT2 and APOE gene with hypertension in the Chinese Han Population. Biomed Mater Eng. 2015;26 Suppl 1:S1977-83. [CrossRef]
- Guo Z, Neilson LJ, Zhong H, Murray PS, Zanivan S, Zaidel-Bar R. E-cadherin interactome complexity and robustness resolved by quantitative proteomics. Sci Signal. 2014 Dec 2;7, rs7. [CrossRef]
- Churilin, MI. Association of RS12328675 COBLL1 polymorphism with coronary heart disease and intermediate phenotypes of atherosclerosis: validation study in Central Russia. Research Results in Biomedicine. 2020;6, 209-218. Russian. [CrossRef]
- Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, Narita A, Konuma T, Yamamoto K, Akiyama M, Ishigaki K, Suzuki A, Suzuki K, Obara W, Yamaji K, Takahashi K, Asai S, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021 Oct;53, 1415-1424. [CrossRef]
- Mishra A, Malik R, Hachiya T, Jürgenson T, Namba S, Posner DC, Kamanu FK, Koido M, Le Grand Q, Shi M, He Y, Georgakis MK, Caro I, Krebs K, Liaw YC, Vaura FC, Lin K, et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature. 2022 Nov;611, 115-123. Erratum in: Nature 2022, 612, E7. [CrossRef]
- Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, Highland HM, Patel YM, Sorokin EP, Avery CL, Belbin GM, Bien SA, Cheng I, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019 Jun;570, 514-518. [CrossRef]
- Sethwala AM, Goh I, Amerena JV. Combating Inflammation in Cardiovascular Disease. Heart Lung Circ. 2021 Feb;30, 197-206. [CrossRef]
- Lind, L. Genome-Wide Association Study of the Metabolic Syndrome in UK Biobank. Metab Syndr Relat Disord. 2019 Dec;17, 505-511. [CrossRef]
- Chen J, Spracklen CN, Marenne G, Varshney A, Corbin LJ, Luan J, Willems SM, Wu Y, Zhang X, Horikoshi M, Boutin TS, Mägi R, Waage J, Li-Gao R, Chan KHK, Yao J, Anasanti MD, Chu AY, et al. The trans-ancestral genomic architecture of glycemic traits. Nat Genet. 2021 Jun;53, 840-860. [CrossRef]
- Meza CA, La Favor JD, Kim DH, Hickner RC. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int J Mol Sci. 2019 Aug 2;20, 3775. [CrossRef]
- Clyne, AM. Endothelial response to glucose: dysfunction, metabolism, and transport. Biochem Soc Trans. 2021 Feb 26;49, 313-325. [CrossRef]
- Krimbou L, Denis M, Haidar B, Carrier M, Marcil M, Genest J Jr. Molecular interactions between apoE and ABCA1: impact on apoE lipidation. J Lipid Res. 2004 May;45, 839–848. [CrossRef]
- Huang QQ, Sallah N, Dunca D, Trivedi B, Hunt KA, Hodgson S, Lambert SA, Arciero E, Wright J, Griffiths C, Trembath RC, Hemingway H, et al. Transferability of genetic loci and polygenic scores for cardiometabolic traits in British Pakistani and Bangladeshi individuals. Nat Commun. 2022 Aug 9;13, 4664. [CrossRef]
- Hoffmann TJ, Theusch E, Haldar T, Ranatunga DK, Jorgenson E, Medina MW, Kvale MN, Kwok PY, Schaefer C, Krauss RM, Iribarren C, Risch N. A large electronic-health-record-based genome-wide study of serum lipids. Nat Genet. 2018 Mar;50, 401-413. [CrossRef]
- Wu Y, Byrne EM, Zheng Z, Kemper KE, Yengo L, Mallett AJ, Yang J, Visscher PM, Wray NR. Genome-wide association study of medication-use and associated disease in the UK Biobank. Nat Commun. 2019 Apr 23;10, 1891. [CrossRef]
- Mondal N, Buffone A Jr, Stolfa G, Antonopoulos A, Lau JT, Haslam SM, Dell A, Neelamegham S. ST3Gal-4 is the primary sialyltransferase regulating the synthesis of E-, P-, and L-selectin ligands on human myeloid leukocytes. Blood. 2015 Jan 22;125, 687–696. [CrossRef]
- Ligthart S, Vaez A, Hsu YH; Inflammation Working Group of the CHARGE Consortium; PMI-WG-XCP; LifeLines Cohort Study; Stolk R, Uitterlinden AG, Hofman A, Alizadeh BZ, Franco OH, Dehghan A. Bivariate genome-wide association study identifies novel pleiotropic loci for lipids and inflammation. BMC Genomics. 2016 Jun 10;17:443. [CrossRef]
- Ozbeyaz NB, Gokalp G, Algul E, Kilic P, Saricam O, Aydinyilmaz F, Guliyev I. Could Systemic Inflammation in Healthy Individuals With Obesity Indicate Subclinical Atherosclerosis? Angiology. 2022 Apr 28:33197221089375. [CrossRef]
- Kivimäki M, Lawlor DA, Smith GD, Kumari M, Donald A, Britton A, Casas JP, Shah T, Brunner E, Timpson NJ, Halcox JP, Miller MA, Humphries SE, Deanfield J, Marmot MG, Hingorani AD. Does high C-reactive protein concentration increase atherosclerosis? The Whitehall II Study. PLoS One. 2008 Aug 20;3, e3013. [CrossRef]
- Moran CA, Sheth AN, Mehta CC, Hanna DB, Gustafson DR, Plankey MW, Mack WJ, Tien PC, French AL, Golub ET, Quyyumi A, Kaplan RC, Ofotokun I. The association of C-reactive protein with subclinical cardiovascular disease in HIV-infected and HIV-uninfected women. AIDS. 2018 ;32, 999-1006. 15 May. [CrossRef]
- Brede G, Solheim J, Prydz H. PSKH1, a novel splice factor compartment-associated serine kinase. Nucleic Acids Res. 2002 Dec 1;30, 5301–5309. [CrossRef]
- Wilhelm LP, Wendling C, Védie B, Kobayashi T, Chenard MP, Tomasetto C, Drin G, Alpy F. STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 2017 ;36, 1412-1433. 15 May. [CrossRef]
- Li L, Liu Y, Liu X, Zheng N, Gu Y, Song Y, Wang X. Regulatory roles of external cholesterol in human airway epithelial mitochondrial function through STARD3 signalling. Clin Transl Med. 2022 Jun;12, e902. [CrossRef]
- Zhang M, Zhai X, Li J, Albers JJ, Vuletic S, Ren G. Structural basis of the lipid transfer mechanism of phospholipid transfer protein (PLTP). Biochim Biophys Acta Mol Cell Biol Lipids. 2018 Sep;1863, 1082-1094. [CrossRef]
- Albers JJ, Vuletic S, Cheung MC. Role of plasma phospholipid transfer protein in lipid and lipoprotein metabolism. Biochim Biophys Acta. 2012 Mar;1821, 345–357. [CrossRef]
Figure 1.
Associations of single nucleotide polymorphisms with carotid intima-media thickness change on rosuvastatin at 6 and 12 months of therapy.
Figure 1.
Associations of single nucleotide polymorphisms with carotid intima-media thickness change on rosuvastatin at 6 and 12 months of therapy.

Figure 2.
Associations of single nucleotide polymorphisms with lipid- lowering effect of rosuvastatin at 6 and 12 months of therapy. TC, total cholesterol; LDL-C, low density lipoprotein cholesterol; TG, triglyceride levels.
Figure 2.
Associations of single nucleotide polymorphisms with lipid- lowering effect of rosuvastatin at 6 and 12 months of therapy. TC, total cholesterol; LDL-C, low density lipoprotein cholesterol; TG, triglyceride levels.


Table 1.
Associations of single nucleotide polymorphisms with the 6-month lipid-lowering effect (total cholesterol, low density lipoprotein cholesterol) and carotid intima-media thickness change in coronary artery disease patients taking rosuvastatin/.
Table 1.
Associations of single nucleotide polymorphisms with the 6-month lipid-lowering effect (total cholesterol, low density lipoprotein cholesterol) and carotid intima-media thickness change in coronary artery disease patients taking rosuvastatin/.
| Chr | Gene (SNP ID) | Effect allele | EAF | N | Total cholesterol | LDL-C | CIMT, maximum | CIMT, mean | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beta* | Pperm# | Beta* | Pperm# | Beta* | Pperm# | Beta* | Pperm# | |||||
| 1 | ZNF648 (rs1689800) | G | 0.392 | 115 | 0.020 | 0.1786 | 0.046 | 0.0493A | -0.084 | 0.0234R | -0.021 | 0.2308 |
| 1 | GALNT2 (rs4846914) | G | 0.388 | 115 | 0.004 | 0.7778 | -0.005 | 0.8571 | -0.045 | 0.0133A | -0.038 | 0.0344A |
| 2 | COBLL1 (rs12328675) | C | 0.170 | 111 | -0.003 | 0.8571 | -0.003 | 1.0000 | 0.035 | 0.2647 | 0.041 | 0.1000 |
| 6 | LPA (rs55730499) | T | 0.056 | 115 | 0.323 | 0.0022R | 0.504 | 0.0224R | -0.010 | 0.8571 | 0.028 | 0.8571 |
| 7 | NPC1L1 (rs217406) | G | 0.203 | 115 | 0.017 | 0.2308 | 0.033 | 0.3556 | -0.001 | 1.0000 | -0.003 | 1.0000 |
| 8 | PPP1R3B (rs9987289) | A | 0.086 | 115 | 0.019 | 0.3404 | -0.013 | 0.8571 | 0.041 | 0.3404 | 0.051 | 0.1550 |
| 9 | ABCA1 (rs1883025) | T | 0.263 | 115 | 0.001 | 1.0000 | -0.004 | 0.8571 | -0.024 | 0.2982 | -0.025 | 0.2535 |
| 11 | F2 (rs3136441) | C | 0.180 | 102 | 0.011 | 0.6429 | 0.025 | 0.7778 | 0.021 | 0.6923 | 0.023 | 0.2982 |
| 11 | ST3GAL4 (rs11220463) | T | 0.190 | 115 | -0.031 | 0.1280 | -0.068 | 0.0803 | 0.066 | 0.0159D | 0.061 | 0.0243A |
| 12 | SCARB1 (rs838880) | C | 0.336 | 115 | 0.002 | 1.0000 | 0.005 | 1.0000 | -0.014 | 0.7273 | -0.015 | 0.6923 |
| 16 | CETP (rs3764261) | A | 0.147 | 115 | 0.007 | 0.6250 | 0.027 | 0.8571 | -0.045 | 0.2466 | -0.042 | 0.1900 |
| 16 | PSKH1 (rs16942887) | A | 0.116 | 115 | 0.009 | 0.6429 | 0.025 | 0.4643 | 0.066 | 0.0421D | 0.068 | 0.0483D |
| 17 | STARD3 (rs881844) | C | 0.310 | 115 | 0.003 | 0.8571 | -0.038 | 0.1667 | 0.048 | 0.0086A | 0.057 | 0.0033A |
| 19 | LILRA3 (rs386000) | C | 0.203 | 115 | 0.006 | 0.5455 | -0.001 | 1.0000 | 0.015 | 0.7778 | 0.003 | 0.8571 |
| 20 | PLTP (rs6065906) | C | 0.160 | 115 | 0.140 | 0.0135R | -0.008 | 1.0000 | -0.022 | 0.6923 | -0.022 | 0.5455 |
| Chr, chromosome; SNP, single nucleotide polymorphism; EAF, effect allele frequency; LDL-C, low-density lipoprotein cholesterol; CIMT, carotid intima-media thickness. *Beta for difference between the natural log-transformed on- and off-treatment values adjusted for natural log-transformed off-treatment level. The beta reflects the fraction of differential lipid- or CIMT-lowering effect in carriers versus non-carriers of the minor allele (for additive model), in carriers of the genotype according to the model (dominant, recessive) versus the carriers of alternative genotypes; a negative beta indicates a better statin response (stronger lipid or CIMT reduction), a positive beta – a worse statin response. Betas were generated using linear regression analysis with age, sex, body mass index, and rosuvastatin dose as covariates. #p-values were generated using the permutation procedure. Superscript indicates genetic model: R, recessive; A, additive; D, dominant. | ||||||||||||
Table 2.
Associations of single nucleotide polymorphisms with triglyceride-lowering effect in coronary artery disease patients taking rosuvastatin.
Table 2.
Associations of single nucleotide polymorphisms with triglyceride-lowering effect in coronary artery disease patients taking rosuvastatin.
| Chr | Gene (SNP ID) | Effect allele | EAF | N | 6-month period | 12-month period | ||
|---|---|---|---|---|---|---|---|---|
| Beta* | Pperm# | Beta* | Pperm# | |||||
| 1 | ZNF648 (rs1689800) | G | 0.392 | 114 | -0.6495 | 0.1148 | -0.1946 | 0.4643 |
| 1 | GALNT2 (rs4846914) | G | 0.388 | 114 | 0.4816 | 0.1919 | 0.05046 | 0.7778 |
| 2 | COBLL1 (rs12328675) | C | 0.170 | 110 | -0.2948 | 0.6250 | -0.1431 | 0.7778 |
| 6 | LPA (rs55730499) | T | 0.056 | 114 | -0.5923 | 0.4242 | 0.1825 | 0.6923 |
| 7 | NPC1L1 (rs217406) | G | 0.203 | 114 | 0.4106 | 0.4118 | -0.1208 | 0.6429 |
| 8 | PPP1R3B (rs9987289) | A | 0.086 | 114 | -0.9197 | 0.1887 | -0.7977 | 0.0756 |
| 9 | ABCA1 (rs1883025) | T | 0.263 | 114 | -0.177 | 0.5789 | -0.7246 | 0.0160D |
| 11 | F2 (rs3136441) | C | 0.180 | 101 | -0.185 | 0.5789 | 0.02512 | 1.0000 |
| 11 | ST3GAL4 (rs11220463) | T | 0.190 | 114 | -0.774 | 0.1587 | 3.624 | 0.0503R |
| 12 | SCARB1 (rs838880) | C | 0.336 | 114 | 1.736 | 0.0478R | 0.1063 | 0.6429 |
| 16 | CETP (rs3764261) | A | 0.147 | 114 | 0.695 | 0.2931 | 0.02338 | 1.0000 |
| 16 | PSKH1 (rs16942887) | A | 0.116 | 114 | -0.6306 | 0.1439 | -0.2626 | 0.3947 |
| 17 | STARD3 (rs881844) | C | 0.310 | 114 | 0.4075 | 0.4815 | -0.2337 | 0.2647 |
| 19 | LILRA3 (rs386000) | C | 0.203 | 114 | -0.1847 | 0.8571 | -0.1448 | 0.6923 |
| 20 | PLTP (rs6065906) | C | 0.160 | 114 | -0.4654 | 0.5789 | -0.0718 | 0.8571 |
| Chr, chromosome; SNP, single nucleotide polymorphism; EAF, effect allele frequency. *Beta for difference between the natural log-transformed on- and off-treatment values adjusted for natural log-transformed off-treatment level. Betas were generated using linear regression analysis with age, sex, body mass index, and rosuvastatin dose as covariates. #p-values were generated using the permutation procedure. Superscript indicates genetic model: R, recessive; A, additive; D, dominant. | ||||||||
Table 3.
Associations of single nucleotide polymorphisms with the 12-month lipid-lowering effect and carotid intima-media thickness change in coronary artery disease patients taking rosuvastatin.
Table 3.
Associations of single nucleotide polymorphisms with the 12-month lipid-lowering effect and carotid intima-media thickness change in coronary artery disease patients taking rosuvastatin.
| Chr | Gene (SNP ID) | Effect allele | EAF | N | Total cholesterol | LDL-C | CIMT, maximum | CIMT, mean | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beta* | Pperm# | Beta* | Pperm# | Beta* | Pperm# | Beta* | Pperm# | |||||
| 1 | ZNF648 (rs1689800) | G | 0.392 | 113 | 0.010 | 0.3091 | 0.024 | 0.2043 | -0.093 | 0.0105R | -0.036 | 0.0282A |
| 1 | GALNT2 (rs4846914) | G | 0.388 | 113 | -0.004 | 0.7273 | -0.016 | 0.4118 | 0.002 | 0.8571 | -0.002 | 0.8571 |
| 2 | COBLL1 (rs12328675) | C | 0.170 | 109 | -0.011 | 0.4643 | -0.018 | 0.4643 | 0.059 | 0.0213A | 0.075 | 0.0056D |
| 6 | LPA (rs55730499) | T | 0.056 | 113 | 0.364 | 0.0001R | 0.367 | 0.0415R | 0.018 | 0.8571 | 0.414 | 0.0146R |
| 7 | NPC1L1 (rs217406) | G | 0.203 | 113 | -0.002 | 1.0000 | 0.003 | 0.2043 | 0.019 | 0.3636 | 0.018 | 0.4516 |
| 8 | PPP1R3B (rs9987289) | A | 0.086 | 113 | 0.004 | 0.8571 | -0.068 | 0.4118 | 0.072 | 0.0359D | 0.079 | 0.0109D |
| 9 | ABCA1 (rs1883025) | T | 0.263 | 113 | 0.010 | 0.3148 | 0.001 | 0.4643 | 0.011 | 0.8571 | 0.019 | 0.2687 |
| 11 | F2 (rs3136441) | C | 0.180 | 100 | 0.013 | 0.3478 | 0.018 | 0.2043 | -0.011 | 0.6429 | -0.005 | 1.0000 |
| 11 | ST3GAL4 (rs11220463) | T | 0.190 | 113 | -0.181 | 0.0273R | -0.027 | 0.4118 | 0.001 | 1.0000 | 0.032 | 0.1709 |
| 12 | SCARB1 (rs838880) | C | 0.336 | 113 | -0.001 | 1.0000 | -0.005 | 0.4643 | -0.003 | 1.0000 | -0.004 | 1.0000 |
| 16 | CETP (rs3764261) | A | 0.147 | 113 | 0.017 | 0.3478 | 0.021 | 0.2043 | -0.053 | 0.0833 | -0.043 | 0.0880 |
| 16 | PSKH1 (rs16942887) | A | 0.116 | 113 | 0.006 | 0.5789 | 0.086 | 0.0175D | -0.011 | 0.5217 | -0.004 | 0.7778 |
| 17 | STARD3 (rs881844) | C | 0.310 | 113 | 0.009 | 0.4375 | 0.010 | 0.8571 | 0.028 | 0.1852 | 0.040 | 0.0223A |
| 19 | LILRA3 (rs386000) | C | 0.203 | 113 | 0.019 | 0.2400 | 0.015 | 0.5200 | 0.026 | 0.2982 | 0.010 | 0.7778 |
| 20 | PLTP (rs6065906) | C | 0.160 | 113 | 0.013 | 0.4815 | 0.031 | 0.2571 | -0.029 | 0.3478 | -0.006 | 1.0000 |
| Chr, chromosome; SNP, single nucleotide polymorphism; EAF, effect allele frequency; LDL-C, low-density lipoprotein cholesterol; CIMT, carotid intima-media thickness. *Beta for difference between the natural log-transformed on- and off-treatment values adjusted for natural log-transformed off-treatment level. The beta reflects the fraction of differential lipid- or CIMT-lowering effect in carriers versus non-carriers of the minor allele (for additive model), in carriers of the genotype according to the model (dominant, recessive) versus the carriers of alternative genotypes; a negative beta indicates a better statin response (stronger lipid or CIMT reduction), a positive beta – a worse statin response. Betas were generated using linear regression analysis with age, sex, body mass index, and rosuvastatin dose as covariates. #p-values were generated using the permutation procedure. Superscript indicates genetic model: R, recessive; A, additive; D, dominant. | ||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



