Submitted:
18 May 2023
Posted:
19 May 2023
You are already at the latest version
Abstract
Gamma-aminobutyric acid (GABA), a non-protein-producing amino acid, is extensively found in microorganisms, plants and vertebrates, and is abundantly expressed in the spinal cord and brain. To date, GABA is considered to be the major inhibitory neurotransmitter in the central nervous system. Its physiological effects are related to the regulation of synaptic transmission, the promotion of neuronal development and relaxation, and the prevention of insomnia and depres-sion. Mediated through its specific receptors, it plays a pivotal role in the control of neuronal ex-citability, which can serve as anovel target for developing analgesics for pain management. This review provides an update on the accumulating evidence of specific GABA receptors and their subtypes in the involvement of pain analgesia.
Keywords:
Gamma-aminobutyric acid
; subtype
; analgesic effect
; mechanism
1. Introduction
Chronic pain impacts more people than heart disease, cancer, and diabetes combined [1]. Chronic pain includes many different forms with different causes and neuropathic pain is one of the most neurological disorders owing to the deficiency of effective clinical treatment [2]. Neuropathic pain refers to a chronic pain induced by an injury or disease regarding the peripheral or central nervous system [3].It affects 7-10% of the global population and has a serious influence on the life quality of patients [4].The condition is under-recognized and under-diagnosed, and its clinical management is a real challenge for physicians. The currently available treatment options focus on clinical symptoms, which usually have limited efficacy[5,6] and are often related to adverse side effects[4,7].
Neuronal synapses mediate the pain signal transduction from presynaptic neurons to postsynaptic neurons through the synaptic gap. Neuronal activity is heavily dependent on the dynamic equilibrium between inhibitory and excitatory functions [8]. The firing patterns of neuron in the brain are strongly regulated by the inhibitory neurotransmitter gamma-aminobutyric acid (GABA) [9].
GABA is the main inhibitory neurotransmitter existed in the central nervous system (CNS) and acts through two different types of receptors, the ionotropic GABAA and GABAC receptors and the metabotropic GABAB receptors [10]. In general, GABA is synthesized in presynaptic nerve cells from L-glutamate, stored in synaptic vesicles, and then catalyzed by glutamic acid decarboxylase. GABA is released into the synapse when the presynaptic neuron receives a pain signal, i.e. when the neuron is excited. GABA is important in human health, where it is involved in a variety of physiological roles and has a wide range of applications in pharmaceutical and food industries.
2. The role of GABA and its receptors in pain transduction and modulation
As stated above, there are three types of GABA receptors: alpha or A, beta or B, and gamma or C, which recognize and bind GABA; these receptors are primarily located on the postsynaptic membranes. GABAA and GABAC receptors belong to ligand-gated ion channels, while GABAB receptors pertain to G protein-coupled receptors. GABAA receptors mediate fast synaptic transmission, while GABAB receptors mediate slow synaptic transmission. Evidence shows that GABAA receptors are involved in epilepsy, resistance to injury, anxiety, and panic, while GABAB receptors are involved in memory, mood and pain modulation. Although GABAC receptors have been identified, their physiological function is not yet fully understood [11].
2.1. The role of GABA in pain transduction and regulation
GABA is widely distributed in the neuraxis and, according to previous studies, plays a vital role in regulating most central nervous system functions due to its widespread presence and relatively high levels in the spinal cord and brain. The agonists and antagonists of GABA receptor exhibit an extensive range of pharmacological activities including anxiolysis, hypnosis, amnesia, muscle relaxation, cognitive enhancement, euphoria and anticonvulsant activity [12].
As a neurotransmitter, GABA is released by one neuron to transmit information to another neuron. It is stored in membrane vesicles at the end of axons. There are thousands of GABA molecules stored in each vesicle. The vesicles are fused to the nerve cell membrane and the GABA molecules are released by the neuron and then by the cytosol. The GABA molecules subsequently are released into the synaptic gap and diffuse through the synaptic gap to the postsynaptic neuron. In humans, GABA functions at inhibitory synapses via binding to GABA receptors, which leads to the opening of ion channels, thus allowing potassium ions to flow out of the cell and chloride ions to flow in [13]. This action leads to a negative variation of transmembrane potential, which causes hyperpolarization and reduces neuronal excitability, presenting an analgesic effect. The released GABA in the synaptic gap that is not bound to the receptor is either degraded by enzymes or returned to the presynaptic axon terminal via active transport pattern by transporters or reuptake pumps [11].
An increase of GABA levels has been found to be correlated with the nocifensive behaviors in formalin test, suggesting the important role of GABA in pain sensations [14]. This probably is not surprising as it has long been proposed in the gate control theory of pain that the GABAergic neurotransmission in the dorsal horn of spinal cord plays a crucial role in controlling the transmission of pain signals. Braz et al [15] provided further evidence in supporting this theory by intraspinal transplantation of cortical precursors of GABAergic interneurons from the embryonic medial ganglionic eminence (MGE) to restore spinal cord GABAergic signaling. They demonstrated that MGE cell transplants reversed the mechanical allodynia and thermal hyperalgesia in nerve injury- and chemotherapy (paclitaxel)-induced models of neuropathic pain in mice. In contrast, the transplantation of cells with a deletion of the vesicular GABA transporter, which is required for the storage of synthesized GABA, has no effect on the pain-like behaviors, indicating that GABA released from these cells are crucial for the transmission of pain sensation[15,16]. These studies demonstrate that GABA plays important roles in pain transduction and regulation.
2.2. The role of GABAA receptors in pain transduction and modulation
The relationship between GABAA and analgesia has been becoming a hot pursuit in pain research in recent years. GABAA receptors are the most important of the three subtypes of receptors and are currently the most intensively studied. GABAA receptors belong to pentameric ligand-gated ion channels mediating most of the rapid inhibitory synaptic transmission in central nervous system, and GABAA receptor dysfunction is associated with many psychiatric and neurological disorders, such as anxiety, epilepsy and substance use disorders [9]. The GABAA receptor is comprised of five distinct subunits whose expression varies according to brain region, cell type and subcellular structural domain, as well as their function. To date, at least 19 receptor subunits are found, which are divided into subclasses according to homologous amino acid sequences: α1, α2, α3, α4, α5, α6, β1, β2, β3, and γ1, γ2, γ3, δ, ε, π, θ, and ρ1, ρ2, ρ3 [17]. Although the receptor subunits vary in combination and function, not all subunits assemble efficiently into functional receptor subtypes. Most GABAA receptors include two α-, two β- and one γ- subunits[18,19], which form an integrated negative ion channel that is permeable to chloride and bicarbonate ions. GABAA receptors are primarily involved in antinociceptive effects at the supraspinal level, modulating GABA-induced inhibition of short duration monosynapses. When GABA combines with GABAA receptors, chloride ion permeability on the nerve membrane increases, causing an anion-selective channel to open. This channel mainly gates chloride ions which allows them to flow inside neurons via ion concentration gradients, leading to hyperpolarization of the cell membrane, thereby inhibiting the neural excitability of GABAergic interneurons. As a result, this reduces pain signal transduction and further produces analgesic effect (see Figure 1) [20].
GABAA receptors are the most prevalent receptors of inhibitory neurotransmitter in the CNS, and primary afferent neurons (PAD) are principally depolarized by GABA through the action of the cationic chloride transporter protein (NKCC1) [21]. NKCC1 helps maintain a high intracellular concentration of chloride ion (Cl-) [22], and the binding of GABA to GABAA receptors leads to anion efflux to depolarize PAD. The subsequent shunting of afferent action potentials leads to a decrease in excitatory transmitters released from injurious terminals. This process is known as presynaptic inhibition and plays a crucial role in controlling the hyperexcitability of neurons in the dorsal horn of the spinal cord [23]. However, there are exceptions. For example, certain types of injury reportedly can enhance PAD such that spiking potentials can be sequentially evoked at primary afferent terminals, thus leading to the change in the PAD from a normal inhibitory process to an excitatory one [24].
2.2.1. Mechanism of analgesic action of alpha(α) subtype receptors
Studies have shown that the cerebral cortex, hippocampus, and caudate nucleus exhibit complex expression patterns of multiple GABAA receptor subtype combinations. Subunits are expressed in the hypothalamus, striatal-nigral fibers and cerebellar stellate/basket cells, and are also extensively existed in the spinal cord. Alpha (α) subunits are important determining factors for localization and function of receptors. The two main isoforms of α, α1 and α2, are widely distributed in the brain. Especially, the most abundant subunit, α1, is almost ubiquitously present in the brain [25]. The α1 subunit in the spinal cord is concentrated around the central canal. Although the α2 subunit is widely expressed, the regional expression patterns of the α1 and α2 subunits in the brain are negatively correlated. The α2 subunit is highly present in the cerebellum and forebrain, including the amygdala, hippocampus, olfactory bulb, lateral septum and granule cells of the medial preoptic area of the hypothalamus, and is most abundant in the superficial dorsal horn of the spinal cord. The α3 subunit expression is localized in the olfactory bulb, cerebral cortex and brainstem nuclei, and is also distributed throughout the dorsal horn of the spinal cord and around the central canal. The α4 subunit expression is limited to the hippocampus, thalamus and striatum. The α5 subunit is abundant in the hippocampus and is also expressed in the granule cells of the olfactory bulb, neocortex and hypothalamus, but is less abundant [26]. It is also weakly expressed in the spinal cord. The expression pattern of the α6 subunit is the most restricted of the α subunits, existing in cochlear nucleus granule cells and cerebellar granule cells [27]. Taken together, α1, α2 and α3 are mainly concentrated at synaptic sites, whereas α4, α5 and α6 are usually located extrasynaptically [28].

Table 1.
Main distribution of the alpha subunit of the GABAA receptor.
| Subunit | Main distribution areas |
|---|---|
| α1 | Hippocampus, cerebral cortex, pericentral canal of spinal cord |
| α2 | Cerebellum, forebrain, superficial dorsal horn of spinal cord |
| α3 | Cortical, dorsal horn of the spinal cord and pericentral canal |
| α4 | Striatum, thalamus |
| α5 | Olfactory bulb, hippocampus |
| α6 | Cochlear nucleus granule cells, Cerebellar granule cells |
The involvement of the GABAA receptor subunit in analgesia has been well documented. Among the GABAA receptor binding sites, we focus on benzodiazepine receptors, which are often used in chronic pain patients to facilitate their sleeping. However, benzodiazepines do not have clear analgesic efficacy, particularly when they are administered systemically. The high affinity of the benzodiazepine receptor binding site needs the existence of a histidine residue at a conserved site in the N-terminal structural domain of the α subunit, which is present in the α1, α2, α3, α5 subunits but not in the α4 and α6 subunits. Mutation of histidine residues to arginine effectively reduces the affinity of GABAA receptors for benzodiazepine receptor binding sites without altering their response to GABA. McKernan et al. [29] reported that the sedative effect of benzodiazepines is primarily mediated through the α1-GABAA receptor as a significant reduction in the sedative effect of benzodiazepines was observed in mice carrying the α1 subunit H→R point mutation. In contrast, one study by Crestani et al. [30] found that point mutations in the α2 subunit could lead to a loss of anxiolytic effects of benzodiazepines. Through the study of benzodiazepine-mediated effects in mice with various α-subunit point mutations [31], it is generally believed that GABAA receptors containing α1 subtype mediate sedative effects, whereas receptors containing α2/α3 are associated with anxiety and those containing α5 are associated with memory function.
Knockout mice proves to be a powerful tool to dissect the specific involvement of different α subtypes in pain sensation. In a series of studies, specific point mutant mice were created, i.e. knocking out one of four alpha subtype: α1H→R(RHHH), α2H→R(HRHH), α3H→R(HHRH), α5H→R(HHHR) [32]. Mouse models of inflammatory pain and neuropathic pain were then established by injecting formalin into the hind paw or performing chronic constriction injury surgery (CCI). After intrathecal injection of diazepam into these mice, respectively, the antinociceptive/hyperalgesic effect was found to be significantly reduced in α2 (HRHH) and α3 (HHRH) point-mutant mice, suggesting that GABAA receptors containing α2 and α3 subtypes in the spinal cord mediated the analgesic effect of intrathecal diazepam administration and were the main contributors [32]. In addition, a minimal effect of spinal GABAA receptors containing α5 was found in the α5 (HHHR) point-mutant mice in a model of inflammatory pain. Compound NS11394 is a partial agonist with functional selectivity for α5>α3>α2>α1 GABAA receptors [33]. In rodent central sensitization-related pain models, NS11394 can increase mechanical pain and thermal pain thresholds and effectively alleviate arousal behavior in rats. After treated with flumazenil, the effect of NS11394 was reversed, thus demonstrating that NS11394 can produce antinociceptive effects by binding to GABAA receptors in vivo [34]. NS11394 was then made a comparison with other known positive allosteric modulators. Knabl et al. [32] selected an anxiolytic and selective GABAA receptor partial agonist, L-838417, which positively modulates α2, α3 and α5 GABAA receptors (43%, 43%, and 39%), but lacks α1 GABAA receptor activity (1.5%) [35]. L-838417 not only is an α2, α3 and α5 partial agonist, but also is an α1 antagonist. Since α2, α3 and α5 subunits are the major α subunits in the spinal cord, and α1 subunits are mainly expressed in the periaqueductal gray (PAG). L-838417 may positively modulate spinal antinociceptive alloreceptors while blocking central prenociceptive GABAA receptors. Both actions contribute to its antinociceptive effects, suggesting that L-838417 has analgesic effects in inflammatory and neuropathic pain models in wild-type rats. These findings suggest that α2/α3 selective or α2/α3/α5 selective agonists may be a new class of analgesics that can be used alone or in combination with existing analgesics for disorders associated with inflammatory pain or neuropathic pain. This demonstrates that, in addition to selectivity per se, a minimal level of positive regulation of individual α subunits (α2, α3 and possibly α5) may be a key determinant of antinociceptive hyperalgesia. On the other hand, TPA023 also activates α2 and α3 GABAA receptors. Although TPA023 is not as potent as L-838417, it also lacks activity at α1 GABAA receptors and is significantly less potent at α5 GABAA receptors [36]. It is an α2/α3 selective partial agonist, but the analgesic effect is not significant, probably due to insufficient α2/α3 efficacy [37]. Similarly, compound HZ166, an imidazolobenzodiazepine with selective partial agonism for receptors containing α2 and α3 subunits, showed reversible hyperalgesic effects of flumazenil in mice with yeast polysaccharide A-induced inflammation and CCI-induced mechanical pain hypersensitivity [38], which demonstrated its analgesic effects in mouse models of neuropathic and inflammatory pain. Taken together, NS11394, L-838417, and HZ166 are all potent agonists (positive allosteric modulators) at α2/α3-GABAA receptors with in vivo analgesic efficacies in preclinical studies.
Ralvenius et al. [39] also bred four point mutant mice with only one GABAA receptor subtype remaining benzodiazepine-sensitive, HRRR, RHRR, RRHR, RRRH (mice only with α1 or α2 or α3 or α5-GABAA receptors are sensitive to benzodiazepines), and tested them with diazepam. Although diazepam is a classic non-selective benzodiazepine, its activity was only restricted to a single GABAA subtype in these triple point mutant mice. The results showed that the order of action of the subtypes was α2>α5>α3 for anti-mechanical hyperalgesia, and α2>α3>α5 for anti-thermal hyperalgesia and chemical injury. These results further support the critical role of α2-GABAA receptor as an antihyperalgesic target. The lack of consistent analgesic effect of benzodiazepines such as diazepam is likely due to their broad pharmacological actions which overshadow the bona fide analgesic efficacy. These studies facilitate the enthusiasm of developing subtype-selective (i.e., α2 and α3-specific) benzodiazepines for pain management.
2.2.2. Mechanism of analgesic action of delta(δ) subtype receptors
GABAA receptors can be divided into synaptic receptors and extrasynaptic receptors based on their distribution within neurons. The two receptor groups also have distinct molecular structural features: synaptic receptors typically contain the γ subtype, while most extrasynaptic receptors are composed of the delta (δ) subtype. Unlike the α subtype GABAA receptors which are regulated by benzodiazepines through the benzodiazepine receptor binding site, the extrasynaptic delta-subtype-containing GABAA receptors are insensitive to positive allosteric modulation by benzodiazepines [40]. Instead, they are highly sensitive to the modulation of neurosteroids.
Peng et al. [41] demonstrated the protein expression of δ-GABAA receptors in the isolated spinal cord by Western blotting. In another study [42], the presence of δ-GABAA receptor mRNA was found by reverse transcription-polymerase chain reaction analysis of lamina tissue. These results suggest that δ-GABAA receptors are present in the spinal cord, By electrophysiological recording, neurons in the spinal cord laminae were found to display a δ-GABAA receptor-mediated tonic inhibitory current [43]. One known δ-GABAA receptor-preferring agonist with analgesic properties is THIP. Studies showd that THIP's enhancement of δ-GABAA receptor activity reduced the excitability of spinal neurons in wild-type mice, but not in Gabrd-/- mice (δ-GABAA receptor-deficient mice). Compared to GABA, THIP excites greater currents from GABAA receptors containing the δ subunit than from GABAA receptors lacking the δ subunit [44]. The δ-GABAA receptor produced low-amplitude tonic inhibitory currents on spinal lamina II neurons, suggesting its potential as a pharmacological target for reducing acute injurious sensations by modulating central sensitization [45]. Another compound with well-characterized analgesic efficacy was flupirtine, whose analgesic activity has been demonstrated in various animal models and in humans [46]. Flupirtine has long been thought of as a selective neuronal potassium (K+) channel opener [47]. However, Klinger et al. [48] found that flupirtine has different effects on synaptic and extrasynaptic GABAA receptors and preferentially acts on extrasynaptic GABAA receptors. This suggests that δ-containing GABAA receptors may be an important target of flupirtine and its analgesic activities may be at least partially mediated through δ-containing GABAA receptors. In summary, there are some evidence in the literature suggesting that δ-GABAA receptors may also be involved in pain modulation and warrants further examination.
2.3. The role of GABAB receptors in pain transduction and modulation
GABAB receptors, first identified by Dr. Norman Bowery in 1979, belong to the G protein-coupled receptor superfamily with seven transmembrane segments. The functional GABAB receptor is a heterodimer composed of GABAB1 and GABAB2 subunits. GABAB1 subunits possesses the ligand-binding site mediating the interaction between the receptor and the ligand, while GABAB2 subunit mediates the downstream signaling via associated G proteins and modulates the affinity of GABAB1 subunit to GABA. Both GABAB1 subunit and GABAB2 subunit have an intracellular C-terminal domain, an extracellular N-terminal domain, and a core transmembrane domain. GABAB1 subunit has several splice variants with GABAB1a and GABAB1b as two most common varaints. The presence of two sushi domains on the GABAB1a subunit makes it distinct from the GABAB1b subunit, which affects the transportation of the receptor [49]. Upon activation, GABAB receptor mediates slow and prolonged inhibitory effect via the activation of the associated Gi/o-protein. On the pre-synaptic side, the dissociated Gαi subunit usually inhibits the activity of adenylyl cyclase and reduces the production of cAMP, leading to the reduced neurotransmitter release, while the Gβγ segment can inhibit the voltage-gated calcium channel, decreasing the vesicle fusion and transmitter release. On the post-synaptic side, GABAB receptor activates the inwardly rectifying potassium channels, hyperpolarizing the membrane potential, decreasing the excitability. The inhibitory effects of GABAB receptor make it a promising therapeutic target in pain management [50]. The schematic diagram for GABAB receptor-mediated analgesic effect are shown in Figure 2.
GABAB receptors are widely expressed in the central and peripheral nervous system exhibiting a development- and region-specific manner, including the thalamus, brainstem nuclei, and spinal cord and play a variety of important roles, such as synaptic development and memory [51]. The extensive expression of GABAB receptors in the pain sensing neurons in the superficial layers of the dorsal horn in the spinal cord and trigeminal ganglia suggests that it could be targetable for the modulation of analgesia [52]. For example, GABAB receptors in dorsal reticular nucleus in the spinal cord are involved in inflammatory pain [14]. It has also been shown that GABAB receptor expression in neurons, astrocytes and microglia in the spinal cord dynamically changed in cancer-induced bone pain (CIBP) rat model, providing evidence that down-regulation of GABAB receptor contributes to the development and maintenance of CIBP [53]. It has also been demonstrated that spinal GABAB receptor normalizes the N-methyl-D-aspartate receptor expression via the cAMP response element-binding protein, contributing to the diabetic neuropathic pain [54]. These studies further support the development of novel analgesics by targeting the GABAB receptor signaling.
In fact, it has been demonstrated as early as 1990s that systemic administration of (±) baclofen produced significant analgesic and antinociceptive effects in acute and chronic pain models, which can be blocked by the intrathecal GABAB receptor antagonists, but neither the GABAA receptor antagonist bicuculline nor the opioid receptor antagonist naloxone can block its effect, suggesting the involvement of GABAB receptor in this process [55]. In addition, intrathecal application of baclofen significantly attenuated CIBP-induced mechanical pain [53]. Consistent with the antinociceptive effect of GABAB receptor, the GABAB1 knockout mice lacking GABA binding sites and GABAB2 knockout mice lacking the functional GABAB receptors have been shown to exhibit hyperalgesia[56,57]. Thus, it is possible to develop GABAB receptor-specific analgesics, although their safety in long-term use is unclear.
3. Conclusion
In summary, GABA and its receptors are critically implicated in the transmission and regulation of pain. Among them, GABAA receptors play a role in analgesia mainly through two subtypes, α2 and α3, although the δ subtype may also be involved in pain modulation. In addition, GABAB receptors also seem to play a certain role in analgesia, which warrants further exploration. These evidence suggests the potential of targeting GABAA or GABAB receptor subtypes for the development of pain analgesics. Given the large number of receptor subtypes and interacting proteins that GABA interacts with, more fundamental research is needed to dissect the specific physiological and pharmacological roles of the targets of GABA. The emerging and promising evidence suggest that targeting GABAergic system could be a fruitful endeavor in the development of new efficacious analgesics in the clinical management of various painful conditions.
Author Contributions
Conceptualization, Q.Z.; methodology, X.Q. and X.Z.; software, X.Q. and X.Z.; validation, J.-L.L., J.-X.L., and Q.Z.; information retrieval, X.Q., X.Z., L.Y., Y.Y., X.-D.Z., and L.W.; writing—original draft preparation, X.Q. and X.Z.; writing—review and editing, J.-L.L., J.-X.L., and Q.Z.; project administration, Q.Z.; funding acquisition, J.-L.L., J.-X.L., and Q.Z.. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China [grant number 82071238, 82070600, 81971243], the Natural Science Foundation of Jiangsu Province [grant number BK20181459], and the Postgraduate Research & Practice Innovation Program of Jiangsu Province [grant number: KYCX21_3131, KYCX21_3096]
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Institute Of Medicine Us Committee On Advancing Pain Research, C. A. Education. In Relieving pain in america: A blueprint for transforming prevention, care, education, and research; National Academies Press (US): Washington, DC, USA, 2011. [Google Scholar]
- Jensen, T.S.; Baron R; Haanpaa, M. ; Kalso, E.; Loeser, J.D.; Rice, A.; Treede, R.D. A new definition of neuropathic pain. Pain. 2011, 152, 2204–2205. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic pain: From mechanisms to treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef]
- van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain. 2014, 155, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; Mcnicol, E.; Baron R; Dworkin, R. H.; Gilron, I.; Haanpaa, M.; Hansson, P.; Jensen, T.S.; Kamerman, P.R.; Lund, K.; Moore, A.; Raja, S.N.; Rice, A.S.; Rowbotham, M.; Sena, E.; Siddall, P.; Smith, B.H.; Wallace, M. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [PubMed]
- Kalso, E.; Aldington, D.J.; Moore, R.A. Drugs for neuropathic pain. BMJ. 2013, 347, f7339. [Google Scholar] [CrossRef] [PubMed]
- Baron R; Binder, A. ; Wasner, G. Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010, 9, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Barker, J.S.; Hines, R.M. Regulation of GABA(A) receptor subunit expression in substance use disorders. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Frolund, B.; Ebert, B.; Kristiansen, U.; Liljefors, T.; Krogsgaard-Larsen, P. GABA(A) receptor ligands and their therapeutic potentials. Curr. Top. Med. Chem. 2002, 2, 817–832. [Google Scholar] [CrossRef]
- Sarasa, S.B.; Mahendran, R.; Muthusamy, G.; Thankappan, B.; Selta, D.; Angayarkanni, J. A brief review on the non-protein amino acid, gamma-amino butyric acid (GABA): Its production and role in microbes. Curr. Microbiol. 2020, 77, 534–544. [Google Scholar] [CrossRef]
- Enna, S.J.; Mccarson, K.E. The role of GABA in the mediation and perception of pain. Adv Pharmacol. 2006, 54, 1–27. [Google Scholar] [PubMed]
- Cellot, G.; Cherubini, E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front Pediatr. 2014, 2, 70. [Google Scholar] [CrossRef]
- Martins, I.; Carvalho, P.; de Vries, M.G.; Teixeira-Pinto, A.; Wilson, S.P.; Westerink, B.; Tavares, I. GABA acting on GABAB receptors located in a medullary pain facilitatory area enhances nociceptive behaviors evoked by intraplantar formalin injection. Pain. 2015, 156, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Braz, J.M.; Wang, X.; Guan, Z.; Rubenstein, J.L.; Basbaum, A.I. Transplant-mediated enhancement of spinal cord GABAergic inhibition reverses paclitaxel-induced mechanical and heat hypersensitivity. Pain. 2015, 156, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Braz, J.M.; Sharif-Naeini, R.; Vogt, D.; Kriegstein, A.; Alvarez-Buylla, A.; Rubenstein, J.L.; Basbaum, A.I. Forebrain GABAergic neuron precursors integrate into adult spinal cord and reduce injury-induced neuropathic pain. Neuron. 2012, 74, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Steiger, J.L.; Russek, S.J. GABAA receptors: Building the bridge between subunit mRNAs, their promoters, and cognate transcription factors. Pharmacol Ther. 2004, 101, 259–281. [Google Scholar] [CrossRef] [PubMed]
- Sieghart, W.; Fuchs, K.; Tretter, V.; Ebert, V.; Jechlinger, M.; Hoger, H.; Adamiker, D. Structure and subunit composition of GABA(A) receptors. Neurochem. Int. 1999, 34, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Backus, K.H.; Arigoni, M.; Drescher, U.; Scheurer, L.; Malherbe, P.; Mohler, H.; Benson, J.A. Stoichiometry of a recombinant GABAA receptor deduced from mutation-induced rectification. Neuroreport. 1993, 5, 285–288. [Google Scholar] [CrossRef]
- Luo, Y.; Kusay, A.S.; Jiang, T.; Chebib, M.; Balle, T. Delta-containing GABA(A) receptors in pain management: Promising targets for novel analgesics. Neuropharmacology. 2021, 195, 108675. [Google Scholar] [CrossRef]
- Price, T.J.; Cervero, F.; de Koninck, Y. Role of cation-chloride-cotransporters (CCC) in pain and hyperalgesia. Curr. Top. Med. Chem. 2005, 5, 547–555. [Google Scholar] [CrossRef]
- Kahle, K.T.; Staley, K.J.; Nahed, B.V.; Gamba, G.; Hebert, S.C.; Lifton, R.P.; Mount, D.B. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol. 2008, 4, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Willis, W.D. John Eccles' studies of spinal cord presynaptic inhibition. Prog. Neurobiol. 2006, 78, 189–214. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Nicas, E.; Laird, J.M.; Cervero, F. GABAA-Receptor blockade reverses the injury-induced sensitization of nociceptor-specific (NS) neurons in the spinal dorsal horn of the rat. J. Neurophysiol. 2006, 96, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Pirker, S.; Schwarzer, C.; Wieselthaler, A.; Sieghart, W.; Sperk, G. GABA(A) receptors: Immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000, 101, 815–850. [Google Scholar] [CrossRef] [PubMed]
- Hortnagl, H.; Tasan, R.O.; Wieselthaler, A.; Kirchmair, E.; Sieghart, W.; Sperk, G. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience. 2013, 236, 345–372. [Google Scholar] [CrossRef] [PubMed]
- Uusi-Oukari, M.; Korpi, E.R. Regulation of GABA(A) receptor subunit expression by pharmacological agents. Pharmacol. Rev. 2010, 62, 97–135. [Google Scholar] [CrossRef]
- Wu, X.; Wu, Z.; Ning, G.; Guo, Y.; Ali, R.; Macdonald, R.L.; De Blas, A.L.; Luscher, B.; Chen, G. Gamma-Aminobutyric acid type a (GABAA) receptor alpha subunits play a direct role in synaptic versus extrasynaptic targeting. J. Biol. Chem. 2012, 287, 27417–27430. [Google Scholar] [CrossRef] [PubMed]
- Mckernan, R.M.; Rosahl, T.W.; Reynolds, D.S.; Sur, C.; Wafford, K.A.; Atack, J.R.; Farrar, S.; Myers, J.; Cook, G.; Ferris, P.; Garrett, L.; Bristow, L.; Marshall, G.; Macaulay, A.; Brown, N.; Howell, O.; Moore, K.W.; Carling, R.W.; Street, L.J.; Castro, J.L.; Ragan, C.I.; Dawson, G.R.; Whiting, P.J. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat. Neurosci. 2000, 3, 587–592. [Google Scholar] [CrossRef]
- Crestani, F.; Low, K.; Keist, R.; Mandelli, M.; Mohler, H.; Rudolph, U. Molecular targets for the myorelaxant action of diazepam. Mol. Pharmacol. 2001, 59, 442–445. [Google Scholar] [CrossRef]
- Munro, G.; Ahring, P.K.; Mirza, N.R. Developing analgesics by enhancing spinal inhibition after injury: GABAA receptor subtypes as novel targets. Trends Pharmacol. Sci. 2009, 30, 453–459. [Google Scholar] [CrossRef]
- Knabl, J.; Witschi, R.; Hosl, K.; Reinold, H.; Zeilhofer, U.B.; Ahmadi, S.; Brockhaus, J.; Sergejeva, M.; Hess, A.; Brune, K.; Fritschy, J.M.; Rudolph, U.; Mohler, H.; Zeilhofer, H.U. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. 2008, 451, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Mirza, N.R.; Larsen, J.S.; Mathiasen, C.; Jacobsen, T.A.; Munro, G.; Erichsen, H.K.; Nielsen, A.N.; Troelsen, K.B.; Nielsen, E.O.; Ahring, P.K. NS11394 [3'- [5-(1-hydroxy-1-methyl-ethyl)-benzoimidazol-1-yl]-biphenyl-2-carbonitrile], a unique subtype-selective GABAA receptor positive allosteric modulator: In vitro actions, pharmacokinetic properties and in vivo anxiolytic efficacy. J. Pharmacol. Exp. Ther. 2008, 327, 954–968. [Google Scholar] [CrossRef] [PubMed]
- Munro, G.; Lopez-Garcia, J.A.; Rivera-Arconada, I.; Erichsen, H.K.; Nielsen, E.O.; Larsen, J.S.; Ahring, P.K.; Mirza, N.R. Comparison of the novel subtype-selective GABAA receptor-positive allosteric modulator NS11394 [3'- [5-(1-hydroxy-1-methyl-ethyl)-benzoimidazol-1-yl]-biphenyl-2-carbonitrile] with diazepam, zolpidem, bretazenil, and gaboxadol in rat models of inflammatory and neuropathic pain. J. Pharmacol. Exp. Ther. 2008, 327, 969–981. [Google Scholar] [PubMed]
- Scott-Stevens, P.; Atack, J.R.; Sohal, B.; Worboys, P. Rodent pharmacokinetics and receptor occupancy of the GABAA receptor subtype selective benzodiazepine site ligand L-838417. Biopharm. Drug Dispos. 2005, 26, 13–20. [Google Scholar] [CrossRef]
- Atack, J.R.; Wafford, K.A.; Tye, S.J.; Cook, S.M.; Sohal, B.; Pike, A.; Sur, C.; Melillo, D.; Bristow, L.; Bromidge, F.; Ragan, I.; Kerby, J.; Street, L.; Carling, R.; Castro, J.L.; Whiting, P.; Dawson, G.R.; Mckernan, R.M. TPA023 [7-(1,1-dimethylethyl)-6-(2-ethyl-2H-1,2,4-triazol-3-ylmethoxy)-3-(2-fluorophenyl)-1,2,4-triazolo [4,3-b]pyridazine], an agonist selective for alpha2- and alpha3-containing GABAA receptors, is a nonsedating anxiolytic in rodents and primates. J. Pharmacol. Exp. Ther. 2006, 316, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Nickolls, S.; Mace, H.; Fish, R.; Edye, M.; Gurrell, R.; Ivarsson, M.; Pitcher, T.; Tanimoto-Mori, S.; Richardson, D.; Sweatman, C.; Nicholson, J.; Ward, C.; Jinks, J.; Bell, C.; Young, K.; Rees, H.; Moss, A.; Kinloch, R.; Mcmurray, G. A comparison of the alpha2/3/5 selective positive allosteric modulators l-838,417 and TPA023 in preclinical models of inflammatory and neuropathic pain. Adv Pharmacol Sci. 2011, 2011, 608912. [Google Scholar]
- Di Lio, A.; Benke, D.; Besson, M.; Desmeules, J.; Daali, Y.; Wang, Z.J.; Edwankar, R.; Cook, J.M.; Zeilhofer, H.U. HZ166, a novel GABAA receptor subtype-selective benzodiazepine site ligand, is antihyperalgesic in mouse models of inflammatory and neuropathic pain. Neuropharmacology. 2011, 60, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Ralvenius, W.T.; Benke, D.; Acuna, M.A.; Rudolph, U.; Zeilhofer, H.U. Analgesia and unwanted benzodiazepine effects in point-mutated mice expressing only one benzodiazepine-sensitive GABAA receptor subtype. Nat. Commun. 2015, 6, 6803. [Google Scholar] [CrossRef]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: Phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 2005, 6, 215–229. [Google Scholar] [CrossRef]
- Peng, H.Y.; Chen, G.D.; Lee, S.D.; Lai, C.Y.; Chiu, C.H.; Cheng, C.L.; Chang, Y.S.; Hsieh, M.C.; Tung, K.C.; Lin, T.B. Neuroactive steroids inhibit spinal reflex potentiation by selectively enhancing specific spinal GABA(A) receptor subtypes. Pain. 2009, 143, 12–20. [Google Scholar] [CrossRef]
- Iura, A.; Takahashi, A.; Hakata, S.; Mashimo, T.; Fujino, Y. Reductions in tonic GABAergic current in substantia gelatinosa neurons and GABA(A) receptor delta subunit expression after chronic constriction injury of the sciatic nerve in mice. Eur. J. Pain. 2016, 20, 1678–1688. [Google Scholar] [CrossRef] [PubMed]
- Ataka, T.; Gu, J.G. Relationship between tonic inhibitory currents and phasic inhibitory activity in the spinal cord lamina II region of adult mice. Mol. Pain. 2006, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.; Kerby, J.; Bonnert, T.P.; Whiting, P.J.; Wafford, K.A. Pharmacological characterization of a novel cell line expressing human alpha(4)beta(3)delta GABA(A) receptors. Br J Pharmacol. 2002, 136, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Bonin, R.P.; Labrakakis, C.; Eng, D.G.; Whissell, P.D.; De Koninck, Y.; Orser, B.A. Pharmacological enhancement of delta-subunit-containing GABA(A) receptors that generate a tonic inhibitory conductance in spinal neurons attenuates acute nociception in mice. Pain. 2011, 152, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Szelenyi, I. Flupirtine, a re-discovered drug, revisited. Inflamm. Res. 2013, 62, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Kornhuber, J.; Bleich, S.; Wiltfang, J.; Maler, M.; Parsons, C.G. Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. Rapid communication. J Neural Transm (Vienna). 1999, 106, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Klinger, F.; Bajric, M.; Salzer, I.; Dorostkar, M.M.; Khan, D.; Pollak, D.D.; Kubista, H.; Boehm, S.; Koenig, X. Delta Subunit-containing GABAA receptors are preferred targets for the centrally acting analgesic flupirtine. Br J Pharmacol. 2015, 172, 4946–4958. [Google Scholar] [CrossRef]
- Benke, D. GABA(B) receptors and pain. Curr Top Behav Neurosci. 2022, 52, 213–239. [Google Scholar]
- Terunuma, M. Diversity of structure and function of GABA(B) receptors: A complexity of GABA(B)-mediated signaling. Proc Jpn Acad Ser B Phys Biol Sci. 2018, 94, 390–411. [Google Scholar] [CrossRef]
- Malcangio, M. GABA(B) receptors and pain. Neuropharmacology. 2018, 136, 102–105. [Google Scholar] [CrossRef]
- Antonopoulos, S.R.; Durham, P.L. Grape seed extract suppresses calcitonin gene-related peptide secretion and upregulates expression of GAD 65/67 and GABAB receptor in primary trigeminal ganglion cultures. IBRO Neurosci Rep. 2022, 13, 187–197. [Google Scholar] [CrossRef]
- Zhou, Y.Q.; Chen, S.P.; Liu, D.Q.; Manyande, A.; Zhang, W.; Yang, S.B.; Xiong, B.R.; Fu, Q.C.; Song, Z.P.; Rittner, H.; Ye, D.W.; Tian, Y.K. The role of spinal GABAB receptors in Cancer-Induced bone pain in rats. J. Pain. 2017, 18, 933–946. [Google Scholar] [CrossRef]
- Bai, H.P.; Liu, P.; Wu, Y.M.; Guo, W.Y.; Guo, Y.X.; Wang, X.L. Activation of spinal GABAB receptors normalizes N-methyl-D-aspartate receptor in diabetic neuropathy. J. Neurol. Sci. 2014, 341, 68–72. [Google Scholar] [CrossRef]
- Malcangio, M.; Ghelardini, C.; Giotti, A.; Malmberg-Aiello, P.; Bartolini, A. CGP 35348, a new GABAB antagonist, prevents antinociception and muscle-relaxant effect induced by baclofen. Br J Pharmacol. 1991, 103, 1303–1308. [Google Scholar] [CrossRef]
- Schuler, V.; Luscher, C.; Blanchet, C.; Klix, N.; Sansig, G.; Klebs, K.; Schmutz, M.; Heid, J.; Gentry, C.; Urban, L.; Fox, A.; Spooren, W.; Jaton, A.L.; Vigouret, J.; Pozza, M.; Kelly, P.H.; Mosbacher, J.; Froestl, W.; Kaslin, E.; Korn, R.; Bischoff, S.; Kaupmann, K.; van der Putten, H.; Bettler, B. Epilepsy, hyperalgesia, impaired memory, and loss of pre- and postsynaptic GABA(B) responses in mice lacking GABA(B(1)). Neuron. 2001, 31, 47–58. [Google Scholar] [CrossRef]
- Gassmann, M.; Shaban, H.; Vigot, R.; Sansig, G.; Haller, C.; Barbieri, S.; Humeau, Y.; Schuler, V.; Muller, M.; Kinzel, B.; Klebs, K.; Schmutz, M.; Froestl, W.; Heid, J.; Kelly, P.H.; Gentry, C.; Jaton, A.L.; Van der Putten, H.; Mombereau, C.; Lecourtier, L.; Mosbacher, J.; Cryan, J.F.; Fritschy, J.M.; Luthi, A.; Kaupmann, K.; Bettler, B. Redistribution of GABAB(1) protein and atypical GABAB responses in GABAB(2)-deficient mice. J. Neurosci. 2004, 24, 6086–6097. [Google Scholar] [CrossRef]
Figure 1.
GABAA receptor-mediated analgesic effect. Presynaptic GABAA receptors activation leads to Cl- efflux because of the high concentration of Cl- in the intracellular side of primary afferent neurons. The initial depolarization reduced the neurotransmitter release induced by noxious stimuli and pain signaling generation. Postsynaptic GABAA receptor activation leads to Cl- influx because of low concentration of intracellular Cl-, which hyperpolarizes the secondary sensory neurons and lowers their excitability reducing the paining sensation.
Figure 1.
GABAA receptor-mediated analgesic effect. Presynaptic GABAA receptors activation leads to Cl- efflux because of the high concentration of Cl- in the intracellular side of primary afferent neurons. The initial depolarization reduced the neurotransmitter release induced by noxious stimuli and pain signaling generation. Postsynaptic GABAA receptor activation leads to Cl- influx because of low concentration of intracellular Cl-, which hyperpolarizes the secondary sensory neurons and lowers their excitability reducing the paining sensation.
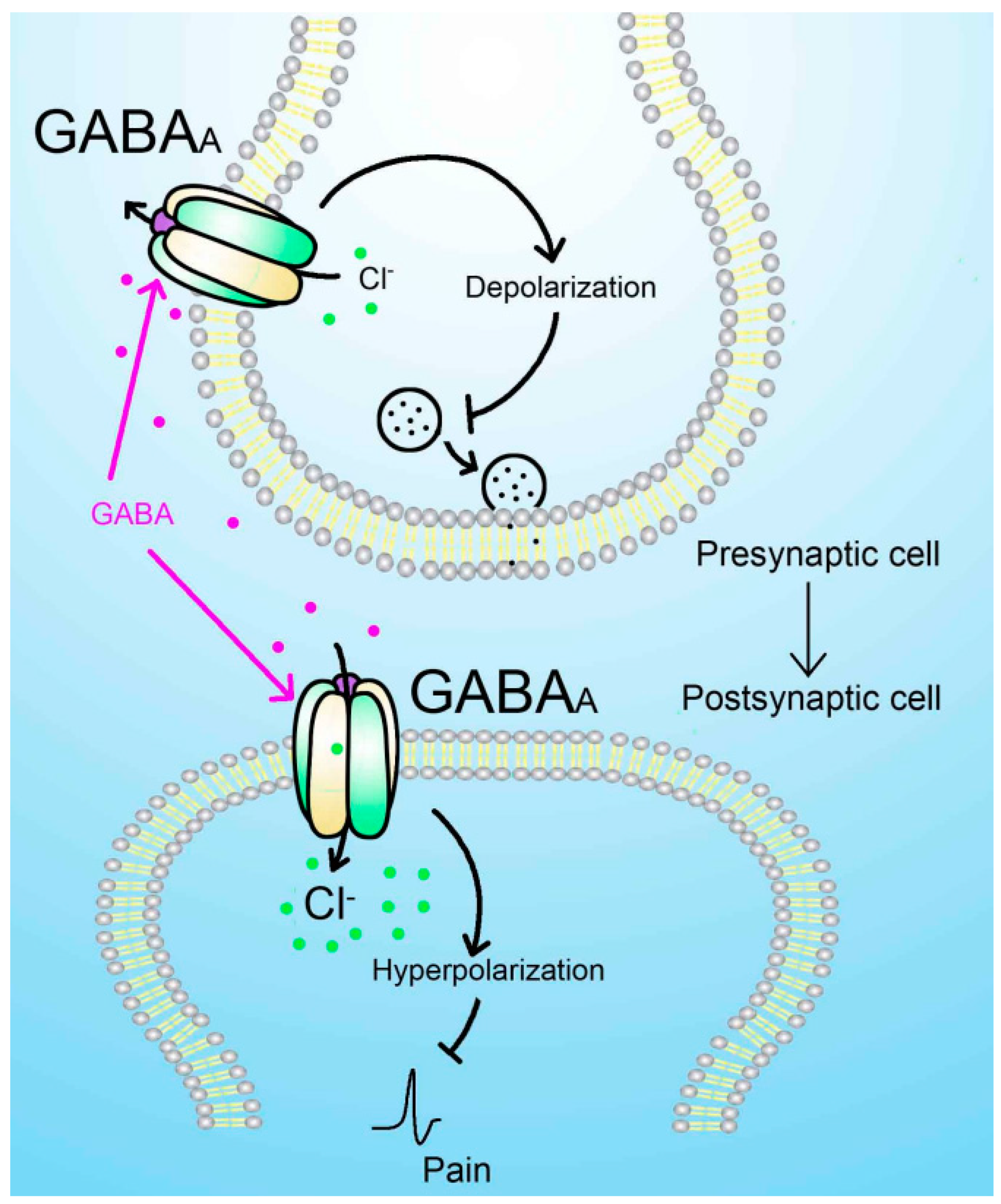
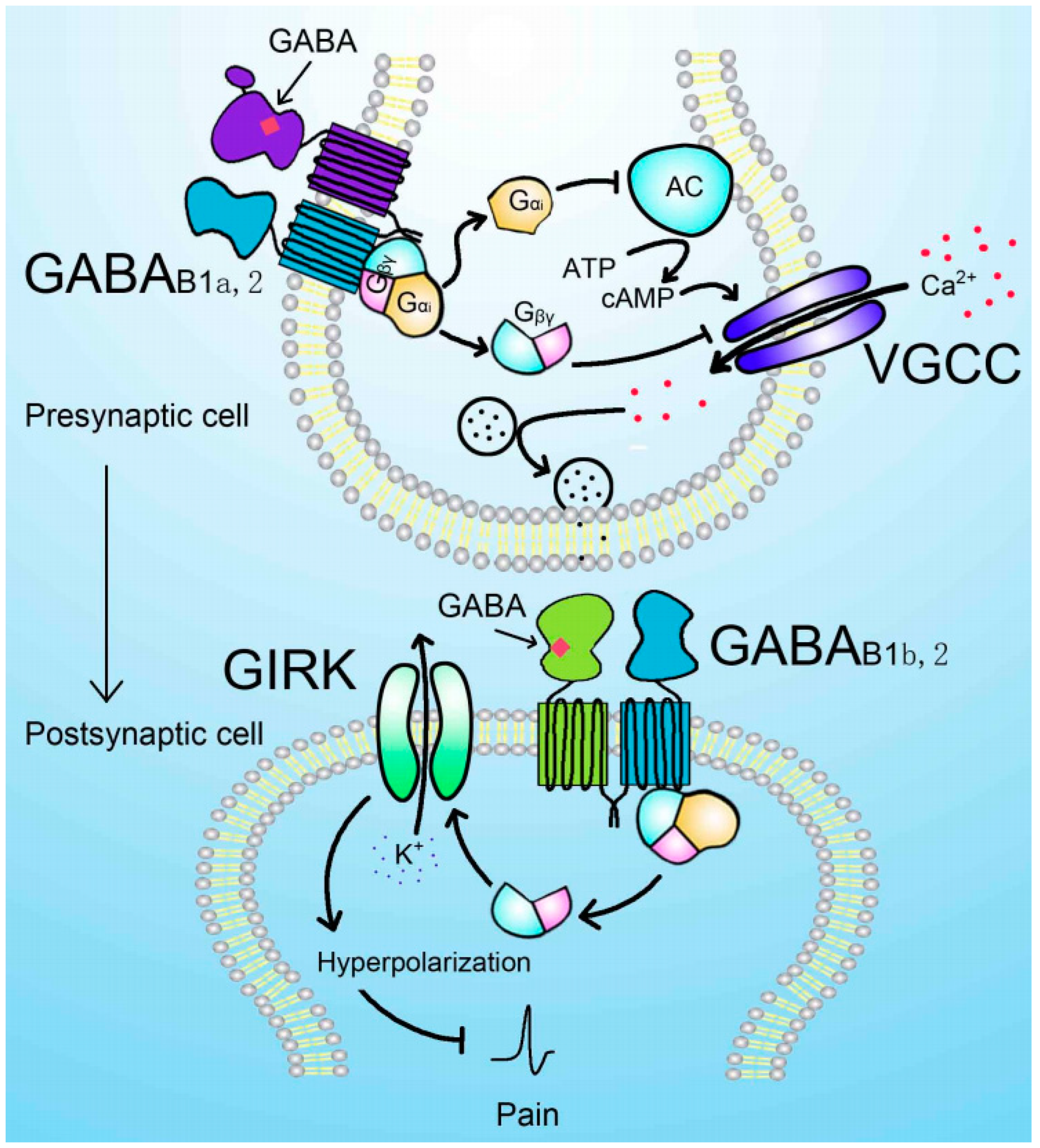
Figure 2.
GABAB receptor-mediated analgesic effect. Presynaptic B1a-containing GABAB receptor interaction with GABA promotes the activation of Gi protein. The Gαi inhibits the activity of adenylyl cyclase (AC) reducing the production of cAMP, which enhances the function of voltage-gated calcium channel (VGCC). The Gβγ can directly inhibit the function of VGCC. Both pathways contribute to the reduced activity of VGCC and neurotransmitter release. The postsynaptic B1b-containing GABAB receptor activation leads to the activation of G protein-gated inwardly rectifying potassium (GIRK) channels via Gβγ complex. GIRK channel opening promotes the efflux of K+ and hyperpolarization, which reduces the excitability of the postsynaptic cell and exerts analgesic effect.
Figure 2.
GABAB receptor-mediated analgesic effect. Presynaptic B1a-containing GABAB receptor interaction with GABA promotes the activation of Gi protein. The Gαi inhibits the activity of adenylyl cyclase (AC) reducing the production of cAMP, which enhances the function of voltage-gated calcium channel (VGCC). The Gβγ can directly inhibit the function of VGCC. Both pathways contribute to the reduced activity of VGCC and neurotransmitter release. The postsynaptic B1b-containing GABAB receptor activation leads to the activation of G protein-gated inwardly rectifying potassium (GIRK) channels via Gβγ complex. GIRK channel opening promotes the efflux of K+ and hyperpolarization, which reduces the excitability of the postsynaptic cell and exerts analgesic effect.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



