Submitted:
19 May 2023
Posted:
22 May 2023
You are already at the latest version
Abstract
Plants have a natural protective process of altering their genetic molecules in response to changing environments. To uncover the genetic potential of plants, it is crucial to understand how they adapt to adverse conditions by analyzing their genetic molecules. In the study, we focused on understanding the responsive genes of tomatoes under drought conditions. We analyzed RNASeq data from different Tomato genotypes, tissue types, and different drought durations. We used a time series scale to identify early and late drought-responsive gene modules and applied a machine learning method to identify the best responsive genes. We found six candidate genes of Tomato (ASCT, FLA2, BAG5, DCL2b, NFP7.3, and ADC1) that were responsive to drought. We further constructed their protein-protein interaction network to identify their potential interactors and found them drought responsive proteins. The candidate genes can help to explore the adaptation of tomato plants under drought conditions. The identification of these candidate genes and modules can have far-reaching implications for molecular breeding and genome editing in Tomato, providing insights into the molecular mechanisms that underlie drought adaptation. This research underscores the importance of the genetic basis of plant adaptation, particularly in changing climates and growing populations.
Keywords:
Abiotic stresses
; Molecular breeding
; Machine learning
; Responsive genes
; Adapted crops
1. Introduction:
The growing global population and the worsening effects of climate change present a formidable challenge to the field of agriculture. To meet the increasing demand for food, agricultural scientists must find ways to increase production despite the adverse effects of climate change. One of the major factors that can reduce crop yields is water scarcity, which is a common problem in many regions, such as South Asia, where prolonged summers lead to low water availability and moisture content in the soil (Hayat et al., 2008). When crops are exposed to water stress, their growth and development are negatively affected, as water is essential for many physiological processes in plants, including photosynthesis. The prolonged stress can severely diminish plant growth and productivity, and cause the accumulation of reactive oxygen species (ROS) in the plant. Lower carbon fixation due to stomata closure can lead to reduced NADP+ regeneration through the Calvin cycle, which, when coupled with changes in chlorophyll production, can result in an increased production of ROS in water-stressed plants (Biehler et al., 1996). The accumulation of ROS can lead to a cascade of harmful effects, including peroxidation of lipids, protein oxidation, enzymatic activity inhibition, oxidative damage to nucleic acids, and ultimately, cell death. These consequences can have serious implications for agriculture, as they can ultimately lead to decreased crop yields and food shortages. Therefore, finding effective ways to mitigate the impact of water scarcity on crops is crucial to ensure food security in the face of climate change.
Tomatoes are a widely cultivated crop throughout the world, originating in southern America as a cultivated solanaceous plant known as Lycopersicon esculentum. Despite its origins, tomatoes are now produced and consumed worldwide, with demand slightly outstripping production (Bai et al., 2007). Due to their relatively short life cycle, easy cultivation, and simple genetics with lower gene duplication, tomatoes are considered to be a standard model of fruit (Bergougnoux, 2014). Molecular studies of tomatoes can also provide valuable insights into other related species (Kimura & Sinha, 2008). While tomatoes are economically important, there is still much to learn about their molecular responses to various abiotic stresses, such as water stress.
Next-generation sequencing technologies have rapidly become the preferred method for characterizing and quantifying entire genomes. One powerful application of these high-throughput sequencing methods is RNA sequencing (RNA-Seq), which allows researchers to gain insight into the transcriptome of a cell. Compared to previous approaches such as Sanger sequencing and microarray-based methods, RNA-Seq provides higher resolution and greater sensitivity for characterizing the dynamic nature of the transcriptome. In addition to quantifying gene expression, RNA-Seq data can facilitate the discovery of novel transcripts, identification of alternative splicing events, and detection of allele-specific expression. Recent advances in the RNA-Seq workflow, including improvements in sample preparation, library construction, and data analysis, have enabled researchers to uncover even more of the functional complexity of transcription. Furthermore, RNA-Seq can be used to investigate different populations of RNA, including total RNA, pre-mRNA, and noncoding RNA, such as microRNA and long noncoding RNA (Kukurba & Montgomery, 2015).
The phytohormone Abscisic acid (ABA) plays a crucial role in coordinating a plant's response to reduced water availability by modulating the expression of drought-responsive genes. In recent years, microRNAs (miRNAs) have been identified as key regulators of drought tolerance, post-transcriptionally regulating drought-responsive genes, including those controlled by ABA signaling pathways. An example of this intricate regulatory network is miR159 in Arabidopsis germinating seeds, which is induced by ABA and drought treatments, promoting transcript cleavage of the ABA positive regulators MYB33 and MYB101 transcription factors, thereby playing a critical role in ABA response (Lopez et al., 2020). Previous studies have investigated the molecular response of plants to reduced water availability, either focusing on specific tissues or the entire plant (Zhou et al., 2007; Kosmala et al., 2012). In a recent study on tomatoes, differentially expressed genes and corresponding enriched Gene Ontology (GO) categories were identified after long-term drought stress and rehydration. The study revealed that GOs enriched in down-regulated genes after drought stress included photosynthesis and cell proliferation, while upregulated genes belonged to GO categories more directly connected to stress responses (Iovieno et al., 2016). Additionally, proteomic studies have highlighted specific changes in components involved in transcription/translation machinery and/or in structural elements regulating cytoplasm hydration (Alam et al., 2010; Kosová et al., 2011).
In this study, we aim to investigate the responsive genes under prolonged water deficit in tomatoes using machine-learning techniques
2. Methods and Materials
2.1. RNA Seq data
For drought stress, RNA Seq data of tomatoes were extracted from Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/). The data was collected using the following keywords in GEO: "Solanum lycopersicum"[Organism] OR Tomato[All Fields]) AND ("drought response"[MeSH Terms] OR Drought stress[All Fields]). RNA Seq data were downloaded by SRAToolkit. There were both paired-end and single-end reads. Each treatment has its corresponding controls. Data were sorted according to time series points of 3, 6, 48, 72, 96, and 120 hours of treatment respectively. There was also data for recovery samples (Supplementary 1).
2.2. Mapping
Reads were mapped against the coding sequence (CDS) of the Heinz 1706 genome. The latest annotation (ITAG4.1, https://solgenomics.net/) of the Heinz 1706 genome was used. Kallisto was used for mapping. 34688 genes were found after mapping. The abundance file contained transcript id, length, effective length, estimated counts, and TPM (Transcript per million). Transcript id estimated counts and TPM values of all samples were gathered in one tab-separated value file for further analysis. The estimated counts of all samples per time point were averaged and labeled as a single time point.
2.3. Gene expression analysis
Differentially expressed genes were analyzed using the Bioconductor edgeR package in R. Estimated counts value of all samples were taken to analyze differentially expressed genes (DEGs). Data were normalized with library size. Dispersions of samples were calculated with glm approaches of the edgeR package by comparing the treatments with their respective controls. Exact tests were carried out to verify the significance of change gene expression and their log fold change values. Multiple testing corrections were done with the topTags function. It ranks genes by p-value. The threshold of considering differentially expressed genes was False discovery rate (FDR)<0.01. For upregulated genes, the logFC threshold was >1, and for downregulated genes, it was < -1. Visualizing of gene expression was done with heatmap using the Complexheatmap Bioconductor package of R. Gene expression was shown with genotype-specific, tissue-specific, stress-specific, heat level-specific, and sample-specific using row Annotation, bottom Annotation, and top Annotation functions of Complex heatmap package in R. Intersection and total set of genes of samples was visualized with an upset plot using R for both upregulated genes and downregulated genes. Some selected transcription factors from diversified families were collected in literature which are said to be drought-responsive genes. The expressions of that transcription factor were also analyzed across time series scales by comparing them with their respective controls.
2.4. Gene ontology analysis
Gene ontology enrichment analysis of significant genes of samples was carried out with the topGO package in R. The genome annotation format (GAF, ITAG4.1) file of tomato was collected from (Carolyn, 2021, https://doi.org/10.25739/zh2v-4p15) and a map file was created with the bash script. The map file contained two columns with tabs separated where the first column was GO terms and the second column was genes to respective GO terms with space-separated. The functions were considered only those who were involved in biological processes. Fisher's classic test was done to find significant levels of GO terms and their functions. The top 15 functions were selected as thresholds.
2.5. Candidate selection
We used a tree model with gradient boosting, and XGBoost R implementation, to train and test the models. For each species, we split the data into training (80%) and testing (20%) sets. We used five-fold internal cross-validation to select the optimized hyperparameters. We tuned “nrounds” (number of trees), “colsample_bytree” (the proportion of features for constructing each tree), “subsamples” (the portion of training data samples for training each additional tree), and “eta” (shrinkage of feature weights to make the boosting process more conservative and prevent overfitting) in an XGBoost: classification model. We used the XGBoost-generated feature importance score that indicates how useful each feature was in the construction of the model.
2.6. Protein-protein interaction
To find out the likely interactions of candidate genes the STRING ((https://string-db.org) tool was used (Karimizadeh et al., 2019). For the search interface, parameters were set to full network type, confidence score >0.4, and more than 10 interactors. In the organism interface, it was restricted to Solanum lycopersicum. In the networks graph, the colored nodes display the proteins and the edges represent the interactions.
3. Results
3.1. Differentially expressed gene patterns:
The expression of genes can vary over time in response to different stimuli, and the identification of DEGs can provide insights into the underlying biological processes. In this study, the expression of genes in a time series scale was analyzed, and the results were visualized in Figure 1.
The DEGs were compared with their individual controls, and the analysis revealed interesting patterns (Figure 2 and Figure 3). Specifically, it was observed that the most upregulated genes were found in the later stages of stress, specifically at 120 hours post-treatment (Figure 2). This finding suggests that the cellular stress response becomes more pronounced as the duration of the stress increases. Interestingly, some genes were also found to be upregulated in the early stages of stress and overlapped with those found in the later stages. This observation implies that the cellular stress response may involve immediate and delayed mechanisms. And in the medium stages of water deficit, a few genes were found to be significantly upregulated. Although the number of DEGs was lower in this stage, it is important to note that these genes may play important roles in the early stages of the stress response. This study provides valuable insights into the temporal dynamics of gene expression in response to stress. The identification of DEGs at different time points highlights the importance of considering the duration of stress when studying cellular responses. Additionally, the overlapping DEGs observed across different stages suggest that the cellular stress response involves a complex interplay of immediate and delayed mechanisms.
3.2. Gene ontology enrichment analysis:
Gene ontology enrichment analysis is a powerful tool used to interpret the biological function of differentially expressed genes (DEGs) in a high-throughput manner. In this study, the functions of DEGs were predicted from GO and the results were visualized in Figure 4. The analysis revealed that a large proportion of DEGs (approximately 600 genes) were involved in organic substance biosynthetic processes and organonitrogen compounds. This finding suggests that the cellular stress response involves the production and modification of organic compounds, which play important roles in cellular metabolism and signaling pathways. Moreover, approximately 500 genes were found to be related to stress, indicating that the cellular stress response involves the activation of various stress response pathways. This finding is in agreement with previous studies that have shown that stress response genes are upregulated in response to various environmental stimuli. Additionally, more than 400 genes were found to respond to chemicals, highlighting the importance of chemical signaling in cellular responses. These genes may be involved in the synthesis, transport, and degradation of chemical signals, or they may play roles in downstream signaling pathways that are activated by chemical stimuli. Furthermore, approximately 200 genes were found to be related to translation and responded to osmotic stress. This finding suggests that osmotic stress may affect translation processes, which play important roles in the synthesis of proteins that are involved in cellular responses to stress. In summary, the gene ontology enrichment analysis conducted in this study provides valuable insights into the biological function of DEGs in response to stress. The identification of genes involved in various metabolic, signaling, and stress response pathways highlights the complexity of the cellular stress response and emphasizes the importance of considering multiple biological processes when studying stress responses.
3.3. Machine learning performance:
In this study, we utilized gene expression values as features to train classification models using the XGBoost algorithm. XGBoost is a popular implementation of the gradient boosting algorithm, which combines multiple weak learners, such as shallow trees, into a strong one. To evaluate the performance of our models, we split our dataset into training and test sets. The accuracy of the models was measured using the area under the curve (“auc”) value, which is a commonly used metric for evaluating classification models. The results of our analysis, shown in Figure 5, demonstrate that the accuracy of the training and test sets are quite close to each other, indicating that our models are well-fitted. Furthermore, we observed that the accuracy of the models increased over iterations, suggesting that the models learned from the data and were able to improve their performance as they were trained with more data. The use of the XGBoost algorithm in our study is particularly beneficial due to its ability to handle high-dimensional datasets, such as those generated from gene expression studies. By incorporating multiple weak learners, the algorithm can effectively capture the complex relationships between gene expression values and biological outcomes, such as disease states or cellular responses to stress. In summary, our results demonstrate the effectiveness of XGBoost in classifying biological samples based on gene expression values. The close agreement between the accuracies of the training and test sets suggests that our models are robust and generalizable. The use of XGBoost has enabled us to extract meaningful insights from high-dimensional gene expression datasets, and its flexibility makes it a valuable tool for future studies in this area.
3.4. Feature importance:
In this study, we employed a robust approach to determine the most influential genes that contribute to the classification of samples based on their gene expression values. Following the training and validation of our classification model using the XGBoost algorithm, we extracted the features with their corresponding importance scores. The importance score of a feature reflects its contribution to the classification of samples and thus provides valuable insights into the underlying biological mechanisms. Among the top six genes identified as candidate genes, we found that FLA2 gene had the highest importance score (Figure. 6). This observation suggests that the expression of FLA2 gene is a critical factor in determining the classification of samples in our study. FLA2 gene encodes for fasciclin-like arabinogalactan protein 2, which is a cell surface protein involved in various cellular processes such as cell adhesion, growth, and differentiation. The high importance score of FLA2 gene in our analysis may indicate its involvement in the cellular stress response and thus may have potential implications in the development of stress-tolerant crops. It is worth noting that the other candidate genes identified in our analysis also play important roles in various cellular processes. Further investigation of these genes could provide valuable insights into the underlying biological mechanisms involved in stress responses. In conclusion, the identification of FLA2 gene as the most important gene in our study highlights its potential significance in stress responses. The use of the XGBoost algorithm has enabled us to extract meaningful insights from high-dimensional gene expression datasets, and its flexibility makes it a valuable tool for future studies in this area.
3.5. Protein-protein network:
To gain a better understanding of the biological functions and interactions of the candidate genes identified in our study, we constructed protein-protein interaction networks for each gene using publicly available databases. The resulting network for each candidate gene is shown in Figure 7, with nodes representing genes and edges representing interactions between genes. In these networks, we observed that the candidate genes identified in our study were represented by the color red, indicating their high importance in abiotic stress response. We also observed that the targets of these candidate genes showed abiotic stress-related functions, suggesting their involvement in the cellular stress response. The protein-protein interaction networks provide valuable insights into the potential pathways and mechanisms involved in the cellular stress response. By identifying the interactions between genes, we can better understand the complex interactions and functions of genes in the network. This information could be further used to develop targeted interventions for improving stress tolerance in crops.
Overall, the protein-protein interaction networks of the candidate genes identified in our study highlight their potential significance in stress responses and provide a foundation for further investigation into their roles in stress tolerance (Supplementary 2).
3.6. Candidate genes overlapped with drought QTLs of Tomato
In this study, we aimed to explore the potential role of three candidate genes, FLA2, ASCT, and NPF7.3, in the response of tomato plants to drought stress. To investigate this, we compared the location of these genes with the previously identified QTLs associated with drought-related traits in tomato by Diouf et al. (2018).
Our analysis revealed that all three candidate genes overlapped with QTLs that were previously associated with drought stress, suggesting their potential involvement in the plant's response to water deficit conditions (Table 1). Specifically, the QTLs RIP, SSC, NFr, and FW were identified under drought stress for the traits of time to ripe, soluble solid content, number of fruit, and fruit weight, respectively.
The importance of our findings lies in the potential for identifying specific genes that contribute to the drought stress response of tomato plants. A better understanding of the genetic mechanisms involved in this response is crucial for developing crop varieties that can withstand environmental stressors such as water scarcity.
Our study suggests that FLA2, ASCT, and NPF7.3 could be promising candidate genes for further investigation in the context of drought stress in tomato plants. Future studies could focus on elucidating the specific molecular pathways through which these genes affect the plant's response to water deficit conditions. This could lead to the development of more targeted breeding programs that utilize these genes to develop drought-tolerant tomato varieties.
4. Discussion
The abstract of our study highlights the crucial importance of gaining a thorough understanding of the genetic composition of tomato plants to improve their ability to withstand drought stress, especially given the significant global demand for this staple crop. To achieve this objective, we employed state-of-the-art machine learning techniques to identify the most responsive genes associated with prolonged water scarcity in tomato plants. Our investigation uncovered six promising candidate genes, with FLA2 emerging as the top gene, indicating its potential as a critical target for enhancing drought resistance in tomatoes. Moreover, the gene ontology enrichment analysis conducted in our study demonstrated that the functions of the identified candidate genes were closely linked to drought stress. These findings are consistent with previous research (Iovieno, et al. 2016), reinforcing the notion that these candidate genes play a pivotal role in imparting drought tolerance in tomato plants. Our study significantly advances our knowledge of the molecular mechanisms underlying the response of tomato plants to drought stress. The identification of FLA2 and other candidate genes, coupled with the functional analysis of their gene ontology, lays a robust foundation for future research focused on developing drought-resistant tomato varieties. Such research can help mitigate the negative impact of water scarcity on global food production, making our study highly pertinent and timely.
The findings of this study demonstrate that specific genes were upregulated in later stages of stress, while others overlapped with early stages, aligning with previous research on plant response to drought stress (Atkinson et al., 2013). These results underscore the complexity of the molecular mechanisms involved in plant responses to stress and emphasize the need for a comprehensive understanding of these mechanisms that encompass multiple genetic pathways. Moreover, our detection of motif repeats in the six selected genes provides insights into the potential regulatory elements involved in drought stress response. Additionally, the identification of these genes on different chromosomes suggests that they may have distinct modes of action in responding to stress, underscoring the significance of investigating multiple genes and their interactions to gain a deeper understanding of the fundamental molecular processes. By shedding light on the intricate mechanisms that govern plant response to drought stress, this study contributes to advancing our knowledge in the field and lays a foundation for future research aimed at developing drought-tolerant plant varieties.
This article emphasizes the critical importance of identifying candidate genes involved in the response to abiotic stress, particularly drought stress, and understanding their interactions using protein-protein interaction networks. Our study focused on six genes (FLA2, ASCT, DCL2b, ADC1, NPF7.3, and BAG5), and the constructed protein-protein interaction networks offer valuable insights into the potential pathways and mechanisms involved in drought responses. This research provides a foundation for developing targeted interventions to enhance stress tolerance in crops. The study highlights the significant role of the identified candidate genes in the response to abiotic stress, especially drought stress, with these genes exhibiting functions related to the cellular stress response. Our findings are consistent with previous research (Muhammad et al., 2019), which identified various genes, including SlMAPK3, involved in drought stress responses in tomatoes. The overexpression of SlMAPK3 improved the tolerance of tomato plants to drought stress by regulating the expression of genes involved in drought stress responses. Therefore, our study makes a valuable contribution to ongoing research on drought stress tolerance in tomatoes. The identification of candidate genes involved in drought stress response and the construction of protein-protein interaction networks lay a foundation for further investigation into the molecular mechanisms involved in stress tolerance. These findings could be leveraged to develop new strategies for improving crop productivity and sustainability in the face of drought stress and other environmental stressors, ultimately benefiting global food security.
The identification of candidate genes that potentially play a role in the response of tomato plants to drought stress is a significant breakthrough in the field of agriculture. The study has provided compelling evidence that FLA2, ASCT, and NPF7.3 genes overlap with QTLs associated with drought-related traits in tomato plants. In a recent study by Verinico et al. (2022), FLA2 was identified as drought-responsive, which further confirms the importance of this gene in the plant's response to water deficit conditions.
Developing crops that are more resilient to environmental stressors like water scarcity has become an urgent priority, given the growing global population and climate change challenges facing agriculture. By understanding the molecular pathways through which these candidate genes affect the plant's response to drought stress, researchers could develop targeted breeding programs to develop drought-tolerant tomato varieties.
The potential of genetic research to develop more resilient crops is underscored by this study's findings. The identification of candidate genes such as FLA2, ASCT, and NPF7.3 provides a solid foundation for further research and breeding programs aimed at improving the drought tolerance of tomato plants. It is critical to continue exploring the molecular mechanisms underlying the response to water deficit conditions to develop crops that can withstand environmental stressors effectively.
In conclusion, this study provides a promising starting point for developing crops that are more resilient to drought stress, with the potential to enhance global food security. The identification of candidate genes like FLA2, ASCT, and NPF7.3 offers valuable insights into the underlying molecular processes involved in plant response to drought stress, paving the way for future breeding programs and research.
The integration of bioinformatics and computational methods, particularly machine learning, has greatly advanced the field of plant stress response research. The study presented in this paper is a remarkable example of how such approaches can provide novel insights into the genetic mechanisms underlying plant responses to environmental stress (Auer et al., 2010). By identifying candidate genes and regulatory elements involved in stress response, machine learning can help develop crop breeding programs that enhance stress tolerance, ultimately contributing to addressing food security challenges (Rico-Chávez et al., 2022).
The use of machine learning allows for the analysis of large amounts of genomic data and the identification of complex gene networks involved in stress response. This approach has enabled researchers to identify critical genetic targets and pathways that can be manipulated to enhance stress tolerance in crops. Such insights can be leveraged to develop crop varieties that can withstand harsh environmental conditions, including drought stress, which is one of the most significant challenges facing global food production (Rico-Chávez et al., 2022).
Furthermore, machine learning can also help identify and prioritize the most promising candidate genes and regulatory elements for further experimental validation. This approach can significantly accelerate the breeding process by reducing the time and resources required to develop new crop varieties. Therefore, the integration of bioinformatics and computational methods, particularly machine learning, represents a powerful approach to identifying the genetic mechanisms underlying plant responses to environmental stress.
Such approaches have the potential to make significant contributions to addressing food security challenges, particularly in areas where drought stress is a significant constraint on crop production. The use of such methods can ultimately lead to the development of drought-resistant crop varieties, which is critical for ensuring global food security in the face of climate change and other environmental challenges. Therefore, the integration of bioinformatics and computational methods is a promising tool for future research aimed at developing crops that can withstand environmental stressors and ultimately improve global food production.
5. Conclusion
The findings of this study shed light on the genetic makeup of tomato plants and their response to prolonged water deficit, and environmental stress that can significantly impact crop yield and quality. The results showed that some genes were significantly upregulated in later stages of stress, while some genes were also found in the early stages and overlapped with later stages. This suggests that different genes may play distinct roles in the plant's response to drought stress and that a comprehensive understanding of these genes is crucial for developing strategies to improve crop resistance to stress. Furthermore, the machine learning approach used in this study appears to be highly effective in identifying responsive genes under prolonged water deficit, with a well-fitted accuracy of the model. The identification of six candidate genes, with FLA2 having the highest importance score, provides a starting point for further research into the specific functions and mechanisms of the genes in the context of drought stress. The gene ontology enrichment analysis predicted that the functions of candidate genes are related to drought stress, indicating that they may play crucial roles in the plant's ability to cope with environmental stress. Our study suggests that FLA2, ASCT, and NPF7.3 could be promising candidate genes for further investigation in the context of drought stress in tomato plants. Overall, this study highlights the importance of understanding the genetic makeup of tomato plants to develop strategies to increase stress resistance and fruit quality.
Authors Contribution: Mehede Hasan Rubel and Rabiul Haq Chowdhury conceived and designed the study. Fatiha Sultana Eti did the data analysis. Rabiul Haq Chowdhury and Fatiha Sultana Eti wrote the manuscript. Md. Atiqur Rahman Bhuiyan and Shipan Das Gupta edited the manuscript. Mehede Hassan Rubel and Fatiha Sultana Eti reviewed the manuscript. All authors read and approved the final draft of the manuscript.
Conflict of interest: The authors declared that there is no conflict of interest
Acknowledgments
The study was partially supported by the Noakhali Science and Technology University Research Cell.
References
- Bergougnoux, V. 2014. The history of tomatoes: from domestication to biopharming. Biotechnology advances. 32, 170-189. [CrossRef]
- Kosová, K., Vítámvás, P., Prášil, I. T., Renaut, J. 2011. Plant proteome changes under abiotic stress—contribution of proteomics studies to understanding plant stress response. Journal of proteomics. 74, 1301-1322. [CrossRef]
- Alam, I., Sharmin, S. A., Kim, K. H., Yang, J. K., Choi, M. S., Lee, B. H. 2010. Proteome analysis of soybean roots subjected to short-term drought stress. Plant and Soil. 333, 491-505. [CrossRef]
- Iovieno, P., Punzo, P., Guida, G., Mistretta, C., Van Oosten, M. J., Nurcato, R., Grillo, S. 2016. Transcriptomic changes drive physiological responses to progressive drought stress and rehydration in tomato. Frontiers in plant science. 7, 371. [CrossRef]
- Kosmala, A., Perlikowski, D., Pawłowicz, I., Rapacz, M. 2012. Changes in the chloroplast proteome following water deficit and subsequent watering in a high-and a low-drought-tolerant genotype of Festuca arundinacea. Journal of experimental botany. 63, 6161-6172. [CrossRef]
- Rico-Chávez, A.K.; Franco, J.A.; Fernandez-Jaramillo, A.A.; Contreras-Medina, L.M.; Guevara-González, R.G.; Hernandez-Escobedo, Q. 2022. Machine Learning for Plant Stress Modeling: A Perspective towards Hormesis Management. Plants. 11, 970. [CrossRef]
- Zhou, J., Wang, X., Jiao, Y., Qin, Y., Liu, X., He, K., Deng, X. W. 2007. Global genome expression analysis of rice in response to drought and high-salinity stresses in shoot, flag leaf, and panicle. Plant molecular biology. 63, 591-608. [CrossRef]
- Kimura, S., Sinha, N. 2008. Tomato (Solanum lycopersicum): a model fruit-bearing crop. Cold Spring Harbor Protocols. 2008, pdb-emo105.
- Bai, Y., Lindhout, P. 2007. Domestication and breeding of tomatoes: what have we gained and what can we gain in the future?. Annals of botany. 100, 1085-1094. [CrossRef]
- Bradford, K. J., Hsiao, T. C. 1982. Physiological responses to moderate water stress. In Physiological plant ecology II (pp. 263-324). Springer, Berlin, Heidelberg.
- Karimizadeh, E., Sharifi-Zarchi, A., Nikaein, H., Salehi, S., Salamatian, B., Elmi, N., Mahmoudi, M. 2019. Analysis of gene expression profiles and protein-protein interaction networks in multiple tissues of systemic sclerosis. BMC medical genomics. 121, 1-12. [CrossRef]
- Osakabe, Y. , Osakabe, K., Shinozaki, K., Tran, L. S. P. 2014. Response of plants to water stress. Frontiers in plant science. 5, 86. [CrossRef]
- Atkinson, N. J. , Lilley, C. J., Urwin, P. E. 2013. Identification of genes involved in the response of Arabidopsis to simultaneous biotic and abiotic stresses. Plant physiology. 162, 2028–2041. [CrossRef]
- Auer, Paul. L., Doerge, R, W., 2010. Statistical Design and Analysis of RNA Sequencing Data. In Genetics, 185, 405-416. [CrossRef]
- Kukurba, K. R. Montgomery, S. B. 2015. RNA Sequencing and Analysis. Cold Spring Harbor protocols, 2015. 951–969. [CrossRef]
- Hayat, S., Hasan, S. A., Fariduddin, Q., Ahmad, A. 2008. Growth of tomato (Lycopersicon esculentum) in response to salicylic acid under water stress. Journal of Plant Interactions. 3(4), 297-304. [CrossRef]
- Biehler, K., Fock, H. 1996. Evidence for the contribution of the Mehler-peroxidase reaction in dissipating excess electrons in drought-stressed wheat. Plant physiology. 112, 265-272. [CrossRef]
- López-Galiano, M. J., García-Robles, I., González-Hernández, A. I., Camañes, G., Vicedo, B., Real, M. D., Rausell, C. 2019. Expression of miR159 is altered in tomato plants undergoing drought stress. Plants. 8, 201. [CrossRef]
- Diouf, I. A., Derivot, L., Bitton, F., Pascual, L., & Causse, M. (2018). Water deficit and salinity stress reveal many specific QTL for plant growth and fruit quality traits in tomato. Frontiers in Plant Science, 9, 279.
- VMuhammad, T., Zhang, J., Ma, Y., Li, Y., Zhang, F., Zhang, Y., & Liang, Y. 2019. Overexpression of a mitogen-activated protein kinase SlMAPK3 positively regulates tomato tolerance to cadmium and drought stress. Molecules, 24(3), 556.
- Veronico, P., Rosso, L. C., Melillo, M. T., Fanelli, E., De Luca, F., Ciancio, A., ... & Pentimone, I. 2022. Water stress differentially modulates the expression of tomato cell wall metabolism-related genes in meloidogyne incognita feeding sites. Frontiers in Plant Science, 13, 776. [CrossRef]
Figure 1.
Heatmap showing differentially expressed genes under drought stress in different time series data.
Figure 1.
Heatmap showing differentially expressed genes under drought stress in different time series data.
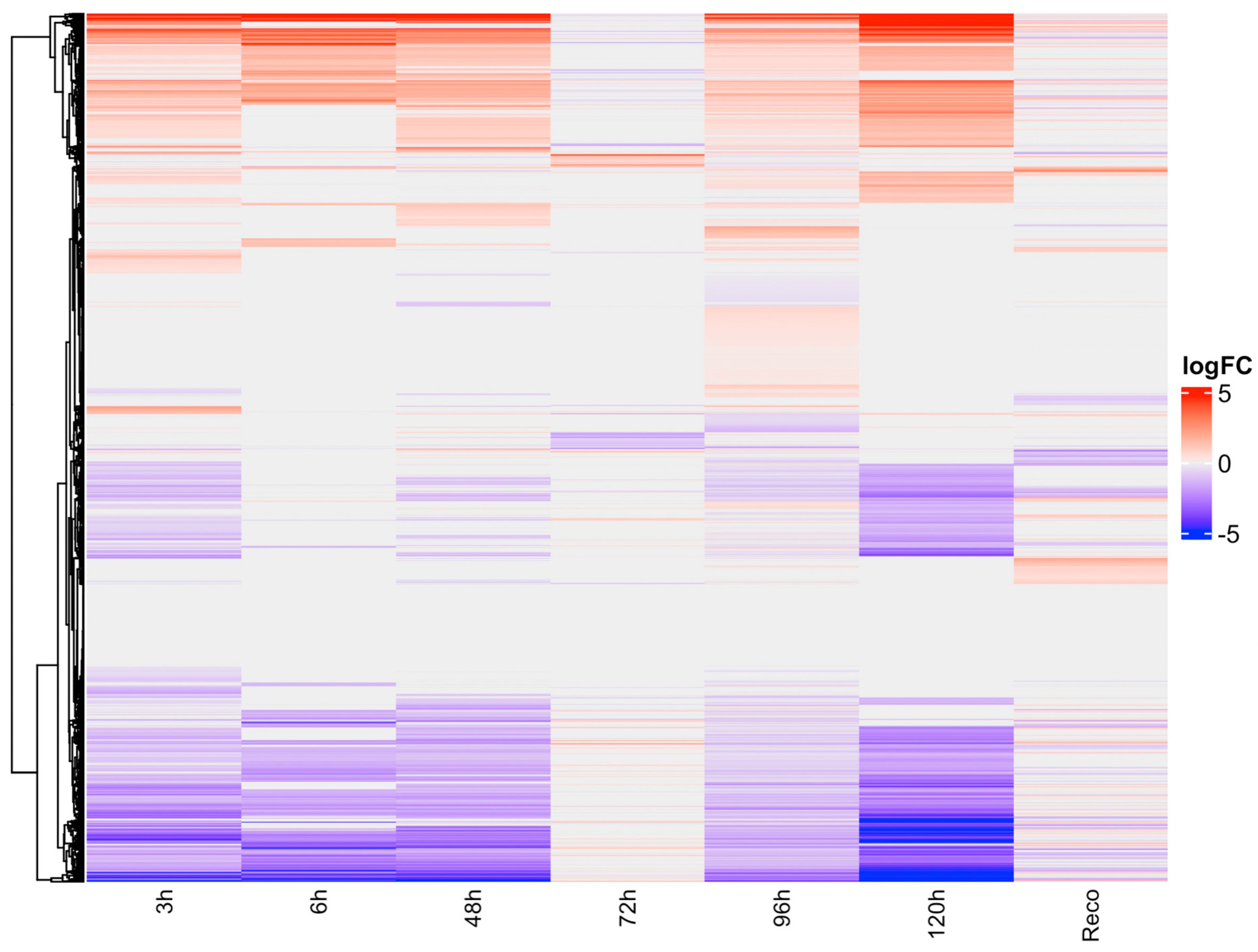
Figure 2.
Upset plot showing differentially expressed upregulated genes under drought stress in different time series data.
Figure 2.
Upset plot showing differentially expressed upregulated genes under drought stress in different time series data.
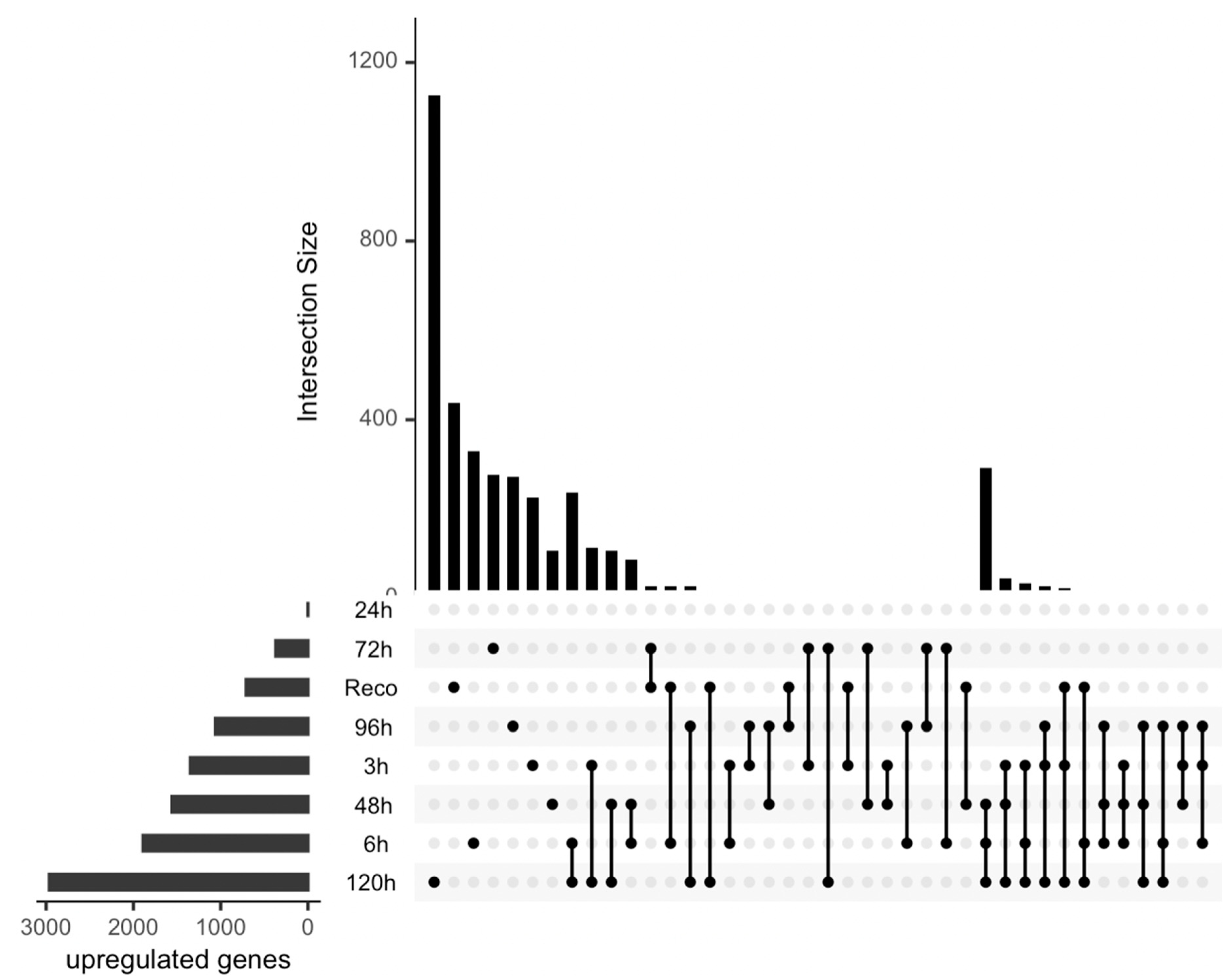
Figure 3.
Upset plot showing differentially expressed downregulated genes under drought stress in different time series data.
Figure 3.
Upset plot showing differentially expressed downregulated genes under drought stress in different time series data.
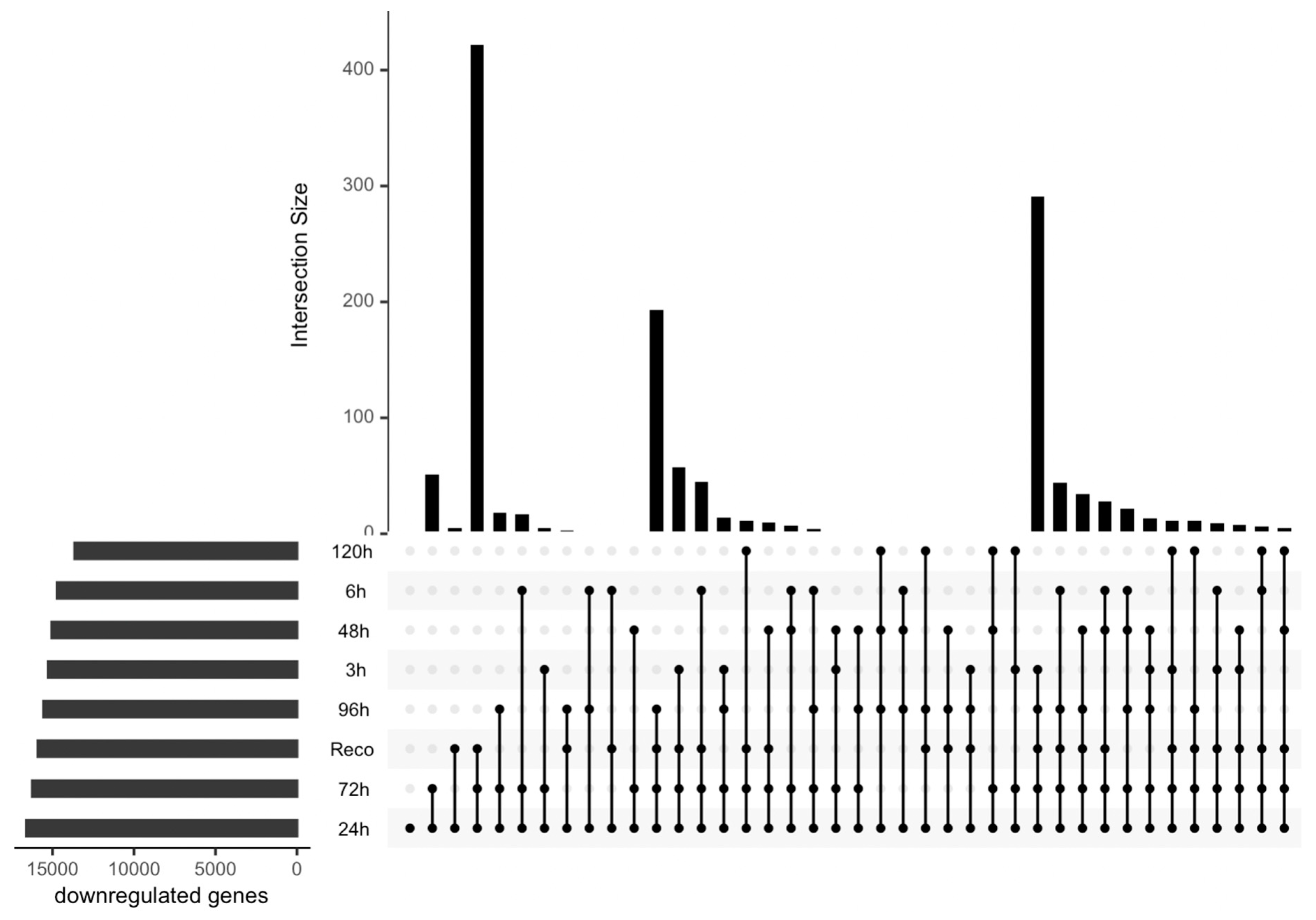
Figure 4.
Functions of differentially expressed genes predicted from GO term of Tomato.
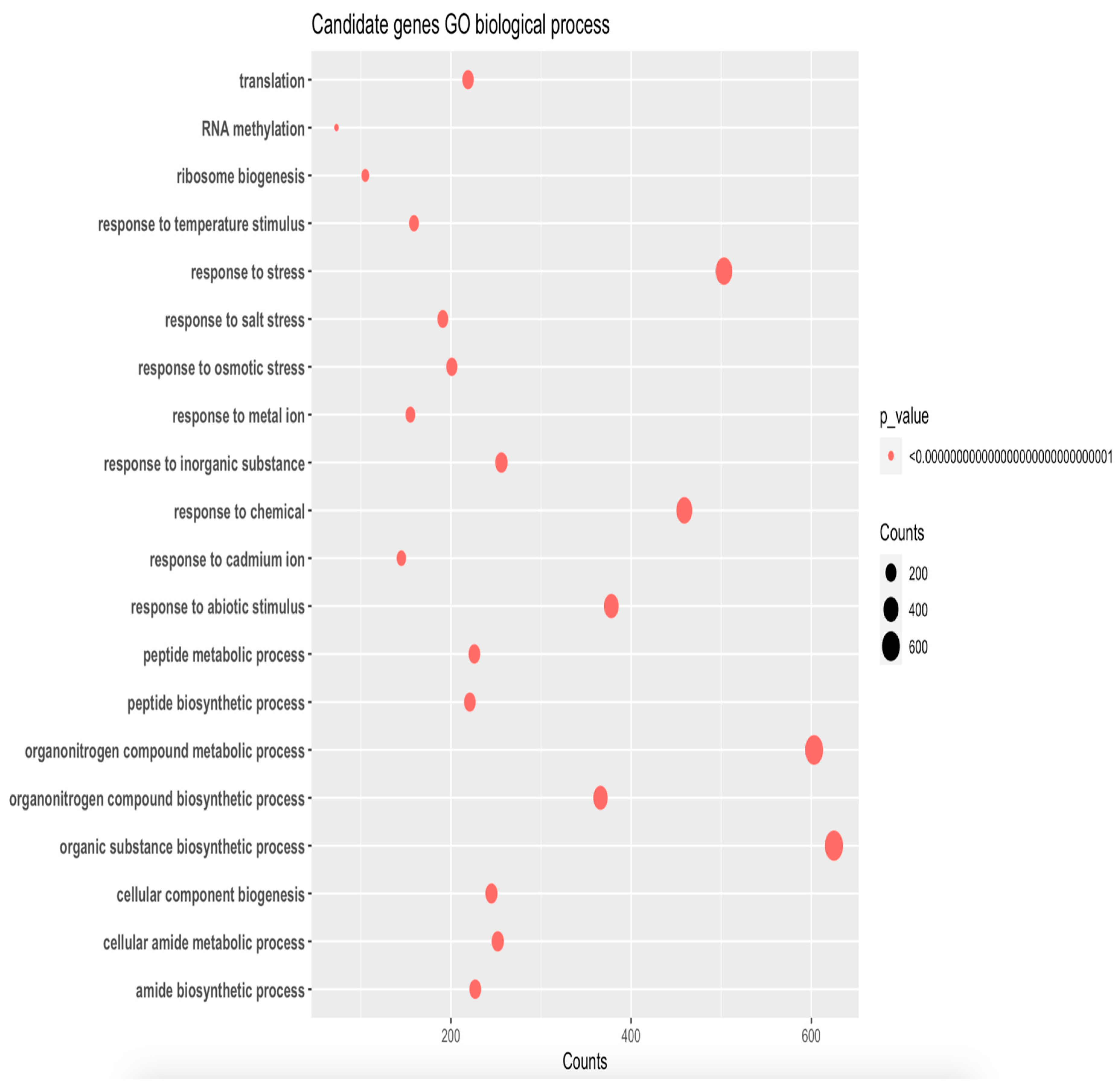
Figure 5.
Plot showing the “auc” value for training data and test data sets over iterations.
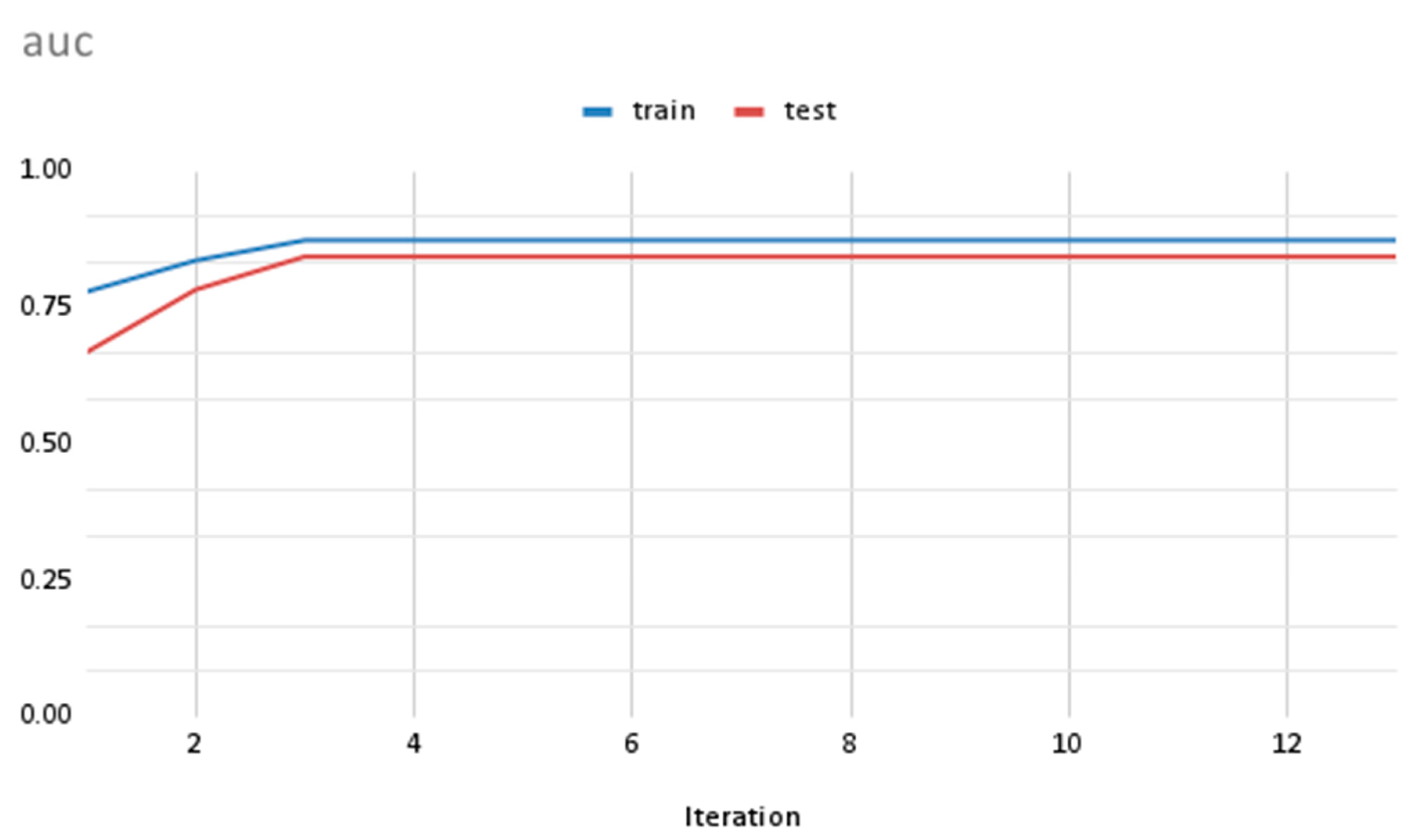
Figure 6.
Candidate genes and their scores for drought response.
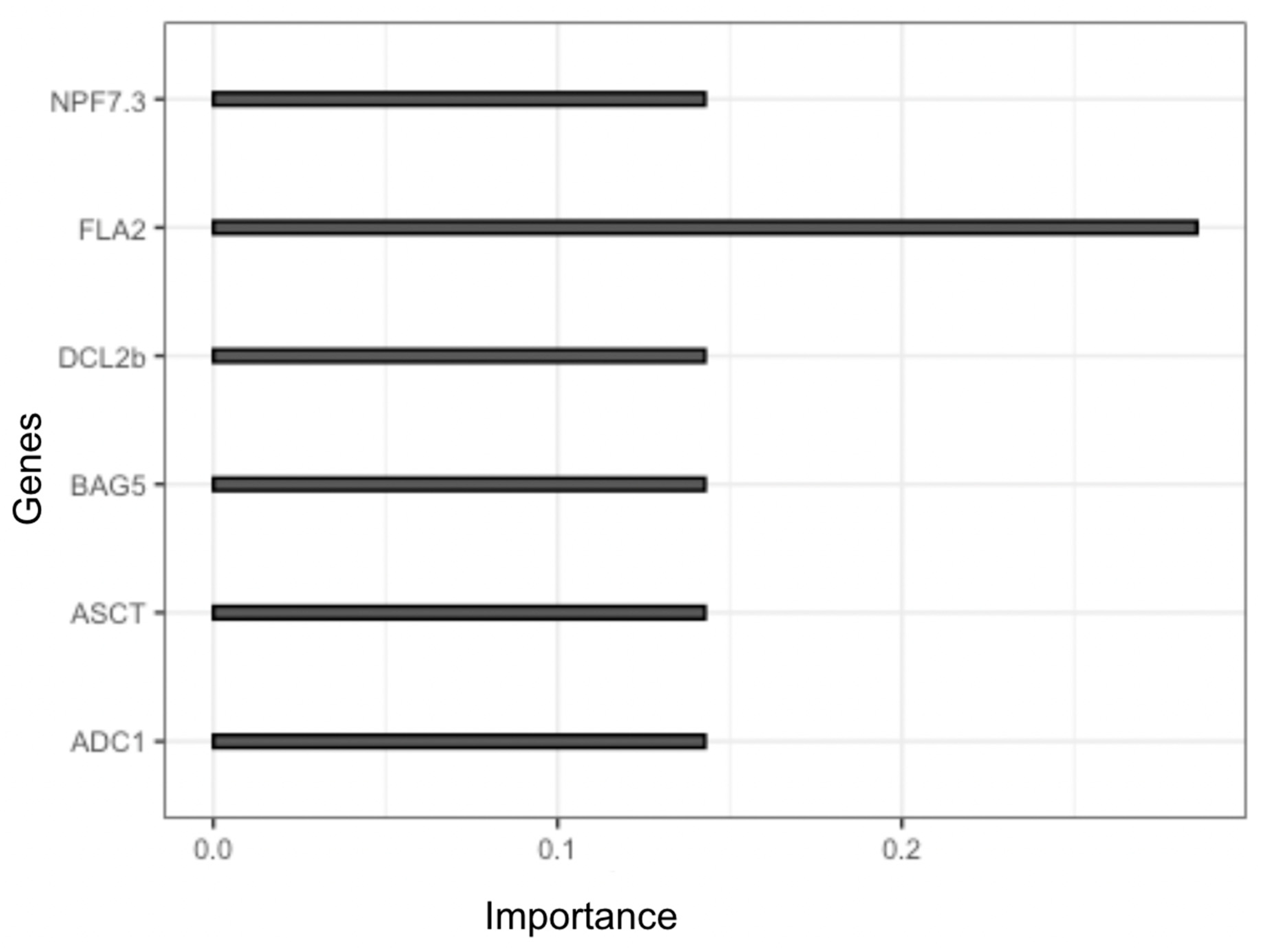
Figure 7.
Protein-protein interaction network for candidate genes.


Table 1.
Drought stress QTLs and overlapped candidate genes.
| Transcripts | Gene | Physical Map(Mbp) | QTLs |
|---|---|---|---|
| Solyc07g045440.1.1 | FLA2 | 58.6651 | RIP3.1, SSC4.1, RIP1.1, NFr1.1 |
| Solyc03g078150.3.1 | ASCT | 51.555 | FW2.2, NFr2.2, SSC11.1, RIP3.1 |
| Solyc01g080870.3.1 | NPF7.3 | 80.0395 | SSC1.1, NFr1.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



