
Preprint
Review
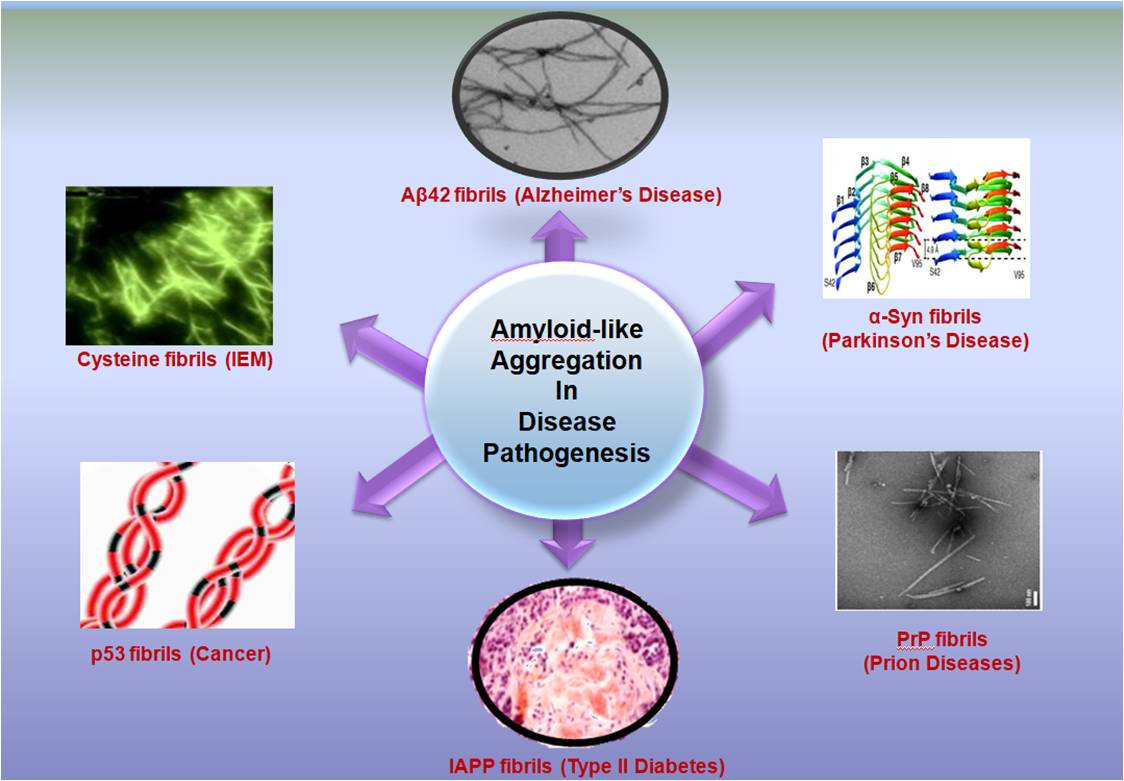
Realization of Amyloid like Aggregation as a Common Cause for the Pathogenesis in Diseases
This version is not peer-reviewed.


Submitted:
22 May 2023
Posted:
23 May 2023
You are already at the latest version
A peer-reviewed article of this preprint also exists.
Abstract
Amyloids were conventionally referred to as extracellular and intracellular accumulation of Aβ42 peptide which causes the formation of plaques and neurofibrillary tangles inside the brain leading to the pathogenesis in Alzheimer’s disease. Subsequently, amyloid-like deposition was found in the etiology of Prion diseases, Parkinson disease, Type II Diabetes and Cancer which was attributed to the aggregation of Prion protein, α-Synuclein, Islet Amyloid Polypeptide Protein and p53 protein respectively. Hence, traditionally amyloids were considered as aggregates formed by the proteins or peptides exclusively. However, since the last decade it has been discovered that other metabolites like single amino acids, nucleobases, lipids, glucose derivatives etc. have propensity to form amyloid-like toxic assemblies. Several studies suggest direct implications of these metabolite assemblies in the patho-physiology of various Inborn errors of metabolisms like Phenylketonuria, Tyrosinemia, Cystinuria and Gaucher’s disease to name a few. In this review, we present a comprehensive literature overview which suggests amyloid-like structure formation as a common phenomenon for the disease progression and pathogenesis in multiple syndromes. The review on this topic is urgently required to create awareness about the understanding of fundamental molecular mechanism behind the origin of diseases from an amyloid perspective and possibly look for a common therapeutic strategy for the treatment of these maladies by designing generic amyloid inhibitors.
Keywords:
Amyloid
; Aggregation
; Self-assembly
; Gene mutation
; Disease Pathogenesis
; Inborn errors of metabolism
1. Introduction
In 1838, German Botanist Matthias Schleiden for the very first time introduced the term “amyloid” to scientific literature for representing the amylaceous constituent of plants [1]. Later in 1854, Rudolph Virchow coined the term “amyloid” to describe the tissue abnormality which displayed an iodine staining reaction [2]. Further, in 1907, German psychiatrist Aloysius (Alois) Alzheimer referred plaques and neurofibrillary tangles present in the brain of patients with senile dementia as “amyloid” [3,4]. These abnormal aggregates were formed by the aggregation of Aβ42 and Aβ40 peptides and the disease was later referred as Alzheimer’s disease (AD) [5]. Subsequently, aggregation of Tau oligomers was also considered a hallmark in the progression of AD [6]. Similar protein aggregation was also discovered in Parkinson’s disease (PD) wherein α-synuclein (αSyn) protein aggregates inside the brain leading to pathogenesis like dementia and memory loss [7]. Further, it was also discovered that Islet Amyloid Polypeptide (IAPP) aggregates to fibrillar structures leading to Type II diabetes [8]. Research on studying the progression of plethora of prion -diseases suggests formation of infectious prion protein (PrP) with a tendency to cross seed aggregation in other proteins. Bovine spongiform encephalopathy (BSE or "mad cow" disease) in cattle, Creutzfeldt-Jakob disease (CJD) and variant CJD in humans, scrapie in sheep, and Chronic Wasting disease (CWD) in deer, elk, mouse and reindeer are some of the diseases associated with PrP aggregation [9]. A similar aggregation as observed for PrP protein was also noted for p53 protein in the patho-physiology of Cancer [10].
Initially, it was thought that amyloids are formed exclusively by the aggregation of proteins and peptides. However, several literature reports in last decade implicate single amino acids and non-proteinaceous metabolites also exhibit tendency to aggregate and form amyloid like structures [11-15]. Interestingly, the accumulation of metabolites reveals a similar aggregation pathway as that exhibited by conventional amyloidogenic proteins and peptides such as Aβ42, PrP and αSyn [16]. Hence, it may be surmised that the patho-physiology of rare Inborn errors of metabolisms (IEMs) may have a common etiology to amyloid associated diseases [16]. The research group of Gazit and coworkers has worked extensively on the metabolite assemblies research and reported amyloid-like toxic aggregates formed by single amino acids like phenylalanine, tyrosine, tryptophan and non-proteinaceous metabolites like uracil, orotic acid, adenine and oxalic acid and proposed a “Generic Amyloid Hypothesis” which implicate amyloid like structure formation as a general phenomenon in patho-physiology of diseases [11,14].
In this review, we will discuss amyloid-like structure formation which occurs in various diseases like AD, PD, PrP diseases, Type II diabetes, Cancer and IEMs. It is envisaged that the literature overview presented on this topic will be helpful in understanding the fundamental molecular mechanism behind the origin of several diseases like IEMs, Cancer, Type 2 diabetes and will enlighten the readers about their common etiology with conventional amyloid associated diseases caused by protein misfolding like Alzheimer’s, Parkinson’s and range of Prion-diseases.
2. General characteristics of Amyloid structures
The aggregates which are formed by amyloid have some common characteristics like binding to amyloid specific dyes Congo red (CR) and Thioflavin T (ThT) [17, 18]. Typically, amyloids are rich in β-sheets and hence produce a characteristic negative band around 190 nm in circular dichroism (CD) [19,20]. The morphology of amyloid like aggregates have been studied by microscopic techniques like Transmission Electron Microscopy (TEM), Scanning Electron Microscopy (SEM), Atomic Force Microscopy (AFM), and light microscopy [12-15,21,22]. Amyloid like aggregation is commonly associated with the formation of varying morphologies (polymorphs) during the course of disease progression [13]. The early aggregates are usually spherulite like structures which gradually transform to fibrillar assemblies. The early aggregates i.e., the pre-fibrillar aggregates of Aβ-peptide, huntingtin, α-synuclein and transthyretin are the most toxic and infectious one [14,23]. These early aggregates impair cellular functions by interacting with cell membranes and causing oxidative stress. Furthermore, pre-fibrillar aggregates increase free Ca2+ ion concentration which eventually leads to apoptotic or neural cell death [24]. Several studies also suggest that not only the pre-fibrillar aggregates but the soluble oligomers of many proteins or peptides are also toxic in major amyloid diseases for instance, spongiform encephalopathies, Huntington disease, Type-2 diabetes, AD and PD [25]. Recently, metabolite assemblies have also been characterized using similar methodologies which insinuate their amyloid character [16].
3. Aβ42 and Aβ40 aggregation in Alzheimer’s disease (AD)
Alzheimer’s disease (AD) is associated with the formation of senile plaques and neurofibrillary tangles in the brain which causes dementia and memory loss [4]. The plaques are the extracellular deposits of Amyloid β (Aβ) protein while the neurofibrillary tangles are intracellular accumulation [26]. Aβ42 is a peptide formed by 42 amino acids and is the main constituent of the lesions found in the brain of patients with AD whereas Aβ40 is the most abundant isoform constituted by 40 amino acid sequence [27,28]. Astrocytes and neurons produce Aβ peptides in the brain by the proteolytic processing of β-amyloid precursor protein (APP) mediated by enzymes such as β-secretase and γ-secretase [29]. It is well-documented that the astrocytes affected by AD express high levels of APP, β-secretase and γ-secretase, the three main components required for amyloid production [29]. Proteolysis of APP by α-secretase and β-secretase leads to secretion of sAPPα and sAPPβ respectively [30]. The secreted sAPPα or sAPPβ have C-terminal fragments which can be cleaved by γ-secretase extracellularly to release the peptides Aβ42 and Aβ40 [30]. Zhao et al. studied the effect of cytokines on the production of amyloid in the astrocytes of brain in AD affected patients. Their study suggests enhanced production of cytokines stimulate secretion of β-site APP cleaving enzyme 1 (BACE1) in C57BL/6J astrocytes. Furthermore, cytokines strengthen activity of β-secretase enzyme in the patients suffering from Swedish familial AD mutation [31]. In another study Calhoun et al. reported overexpression of APP in the mouse model of cerebrovascular disease, Cerebral Amyloid Angiopathy (CAA) and their research suggests enhanced production of Aβ leads to neuronal loss, microglial activation, synaptic abnormalities and microhemorrhage in the patho-physiology of CAA [32]. Brothers and others also discovered that secretion of Aβ42 is not only limited to neuronal/astrocyte cells of brain but other non-neural tissue such as skin, skeletal muscle and intestinal epithelium can also secrete Aβ42 [33]. It was also discovered that aggregation of Aβ42 and Aβ40 peptides is caused by mutation in the gene producing Aβ peptide [4,34]. Mutations in βAPP (β-amyloid precursor protein) gene and two presenilin genes PS1 and PS2 lead to production of abnormal Aβ42 peptides which rapidly aggregates and get deposited as extracellular plaques and tangles in the brain of patients suffering from Familial AD [4,34].
Yankner et al. investigated the neurotoxicity produced by Aβ fibrils on hippocampal neuronal cell lines by co-incubation studies under different time periods. Their study suggests both concentration and period of co-incubation of Aβ40 fibrils with the neuronal cells play a crucial role in predicting the trophic and toxic response of Aβ40 and its effect on neuronal differentiation [35]. In a recent study, Antonino et al. reported amyloidogenic processing of APP by the oligomers and fibrils of Aβ. It was noted that exacerbated and intracellular accumulation of Aβ42 is caused due to colocalization and physical interaction of APP and BACE1. It was also noted that cells overexpressing the mutant forms of APP, which cannot bind to Aβ could not increase colocalization of APP with BACE1 indicating crucial role of physical binding of Aβ to APP/ BACE1 in causing amyloidogenic deposits. Further in this study, it was noted that gallein prevents Aβ-dependent interaction of APP and BACE1 in endosomes and hence can serve as good therapeutic target [36]. A comparative analysis of Aβ42 and Aβ40 indicates that Aβ42 has more tendency of oligomerization as well as a more hydrophobic and fibrillogenic nature as compared to that of Aβ40 due to which Aβ42 cause more cytotoxic response, hence implicating crucial role of oligomerization in the pathogenesis of neurodegenerative diseases [25].
In 1963, Kidd first reported the structure of amyloid fibrils present in the cerebral cortex of AD patient with the help of electron microscopy (EM). The micrograph revealed amyloid fibrils exist as paired helical filament coiled in squash racket shape [37]. EM of Aβ42 and Aβ40 mature fibers suggest long straight fibrillar morphology having diameter in the range of 70-80 Å [38]. Negative stain EM of the protofibrils of Aβ42 and Aβ40 revealed flexible fibers having diameter in the range of 60-100 Å. The structures of the Aβ42 and Aβ40 protofibrils were also analyzed by AFM which revealed the diameter of Aβ40 protofibrils to be around 3.1±0.9 nm and the diameter of Aβ40 long fibrillar species as 7.8±0.45 nm [39]. The diameter of Aβ42 protofibrils was found to be 4.2±0.58 nm while the diameter of bifurcated type-1 and type-2 fibrils of Aβ42 was found to be 7.3±0.53 nm and 3.8±0.43 nm respectively. Kollmer et.al., further reported the Cryogenic Electronic Microscopy (Cryo-EM) structure of Aβ amyloid fibrils from meningeal Alzheimer’s brain tissue. Their studies suggest Aβ amyloid fibrils have polymorphic nature and their morphology is right-hand twisted [40]. The X-ray diffraction (XRD) analysis of amyloid fibers reveals a strong 4.8 Å reflection on the meridian which corresponds to the hydrogen bonding distance between β-strands. The 10-11 Å reflection on equator, on the other hand corresponded to the inter sheet distance of 10.7 Å [41]. Griffin and his team reported atomic resolution structure of monomorphic Aβ42 amyloid fibrils. The structure shows that the core of fibril consists of a dimer of Aβ42 molecules, each containing four β-strands in a S-shaped amyloid fold and arranged in a manner that generates two hydrophobic cores that are capped at the end of the chain by a salt bridge [42]. Riek and coworkers presented the 3D structure of Aβ42 fibril which reveal residues 15-42 in Aβ42 form double horseshoe like cross β-sheet structure [43]. Circular dichroism (CD) analysis of aqueous solutions of Aβ42 and Aβ40 suggests the aggregates have mostly β-sheet structure [38]. The studies by Soto et al, discussed how α-sheet changes to β-sheet in mutated Aβ42 and characterized this morphological transition using amide shift [44]. A characteristic apple-green color birefringence could also be observed when Aβ42 and Aβ40 aggregates are stained with CR while an abrupt increase in fluorescence intensity of ThT may be noted when these fibrils were co-incubated with ThT [45].
It has been found that Aβ42 and Tau oligomers both are responsible for AD. Several studies have also reported formation of oligomers as key factors for memory loss and anatomical pathology [46]. The binding studies with Oligomeric specific antibodies proved that an increased level of tau oligomers are found in human AD affected brain compare to control [6]. A diagrammatic representation of amyloid formation in Alzheimer’s Disease is illustrated in Figure 1 [42,47].
4. α-Synuclein Aggregation and Parkinson’s disease
Parkinson’s disease (PD) was reported for the very first time, by James Parkinson in 1817. The main pathological characteristics of PD are formation of Lewy bodies inside the brain [48]. The key component present in the Lewy bodies are misfolded α-Syn peptides [49]. Jiang et.al., through their studies reported mutation in Synuclein Alpha (SNCA) gene as the main reason behind abnormal aggregation of α-Syn in PD (Figure: 2) [50]. The amyloidosis process of α-syn peptide includes the formation of soluble oligomers which further converts into insoluble amyloid fibrillar morphology by self-association [51]. These fibrils are the pathological emblem of PD and other Synucleinopathies are formed due to protein misfolding and aggregation of α-Syn, a 140 amino acid long protein which is normally present at high levels in the brain and is embroiled in crucial synaptic processes in the neurons [52,53]. It is basically a disorganized protein but adopts a partial α-helix motif upon binding with membranes [54]. The presence of the amphipathic region enables the α-Syn protein to bind with membranes [55]. The amphipathic region of α-Syn is followed by hydrophobic or non-amyloid component (NAC) region which is mainly responsible for inducing its abnormal aggregation [56]. It was found that β and γ-synuclein, are the two isoforms of α-Syn [57]. Dysregulation of cellular processes and toxic gain or losses of function are amongst the major molecular mechanisms that may account for the neurotoxicity associated with α-Syn aggregation [58]. Volles et.al, demonstrated toxicity of α-Syn aggregation through their experiment with synthetic membranes. The results obtained from this study suggest α-Syn protofibrillar aggregates cause cell death through the induction of transient cell permeability by annihilation of vesicle membranes. It is apparent that this transient cell permealization may persuade unregulated Ca2+ influx, mitochondrial depolarization and leakage of dopamine in cytoplasm [59]. Desplats and coworkers through their experiments reported α-Syn forms Lewy bodies-like inclusions when it transmits through neuron, ultimately leading to apoptosis and cellular death [60]. Araki et.al. proposed PD as a type of amyloidosis. Through immunostaining and XRD studies, they revealed Lewy Bodies (LBs) present in the PD affected brains contain cross β-sheet structure [61]. Musteikyte et.al, reported the interaction of α-Syn oligomers with the lipid membrane and its implications in the pathogenesis of PD [62]. Dean and Lee identified a link between melanoma and PD, since their experiments suggest that aggregation of Premelanosome Protein 17 (Pmel17) in the melanosome was ameliorated by α-Syn amyloids [63]. Research study by Maji and coworkers reported the crucial role of glycosaminoglycans (GAGs) in α-Syn amyloid formation. Their experiments suggest interactions between GAGs and α-Syn promoted amyloid fibrillation and aggregation was enhanced in a range of proteins by increasing GAG concentration [64]. A recent study by Reyes et.al., revealed that α-Syn peptide can accumulate in the liver via originating from the brain in Lewy Body Diseases. Liver might help to remove the pathological α-Syn aggregates as part of the liver’s detoxification and clearance process [65]. Yamaguchi et al. reported polyphosphates (polyPs) can persuade α-Syn amyloid formation at both low and high concentrations under neutral pH. At low concentration, polyPs diminish α-Syn solubility via charge-charge interactions with positively charged N-terminal KTKEGV repeats whereas at high concentration polyPs diminish α-Syn solubility via Hofmeister salting out effects [66].
Tuttle et al. characterized α-Syn fibrils by using TEM analysis which revealed α-Syn fibrils have a width of 4.6±0.4 nm. The X-ray diffraction analysis showed archetypal meridional diffraction pattern at 4.8 Å which corresponds to a cross β-sheet structure of α-Syn fibrils. The high-resolution 3D structure of a single untwisted α-Syn amyloid fibril in substantia nigra of the brains of people having PD reveals a diameter of around 5 nm as assessed through EM. The structure of a pathogenic fibril of full-length human α-Syn was studied through solid-state NMR and was validated by EM and XRD. These studies suggest α-Syn fibrils exhibit typical amyloid features which includes parallel, in-register β-sheets and hydrophobic-core residues with substantial complexity arising from diverse structural features like an intermolecular salt bridge, a glutamine ladder, close backbone interactions involving small residues, and several steric zippers stabilizing a new orthogonal Greek-key topology. These characteristics contribute to the robust propagation of this fibril form, as supported by the structural similarity of early-onset-PD mutants [67]. Stahlberg and coworkers also elucidated the Cryo-EM structure of α-Syn amyloid fibrils (residues 1-121) at a resolution of 3.4Å which reveals, two protofilaments are intertwined in left-handed helix and the protofilaments offer Greek-like topology [68]. In yet another study, Flynn et.al, characterized the α-Syn amyloid fibril structure with the help of Raman spectroscopy. Using this spectroscopy, an initial disordered conformation was characterized by obtaining amide-I bond for α-Syn. Narrowing amide-I band during aggregation indicates β-sheet structure formation [69]. Kumari et al. proposed a structural insight into α-Syn monomer fibrils. They showed how transient electrostatic interactions drive α-Syn monomer fibril binding with the help of nuclear magnetic and electron paramagnetic resonance spectroscopy. Their experimental data presented how intramolecular unfolding of α-Syn leads to secondary nucleation process which causes formation α-Syn amyloid fibrils [70]. With the help of AFM-IR, Zhou et.al, were able to characterize individual α-Syn oligomers. The AFM-IR results revealed that oligomers contain α-helix/random coil, parallel and antiparallel β-sheet structures in the early stage of aggregation, whereas in the late stage parallel β-sheet structure predominates [71]. The CD spectra display a local minimum of 195 to 220 nm indicating a change in the secondary structure of α-Syn from a mix of α-helix and random coil elements to β-sheet enriched structure. FTIR study also revealed a clear shift of α-sheet to β-sheet structure for α-Syn oligomers [72]. α-Syn like Aβ also binds ThT which can be noted by an enhanced fluorescence intensity of ThT at 480 nm [57]. A diagrammatic representation of amyloid formation in PD is illustrated in Figure 2 [73-76].
5. Aggregation of Prion protein (PrP) and the associated diseases
Missense, insertion and point mutations in the Prion Protein gene (PRNP) causes abnormal production of Prion protein (PrP) which has a tendency to self-aggregate and accumulate as insoluble fibrils and plaques (Figure 3) [77]. Prusiner et.al, for the first time reported the amyloidogenic nature of prion protein [78]. The main conformational change in the brain during the amyloidosis of PrP is from the cellular form of PrP (PrPC) into the disease-causing isoform i.e., the scrapie form (PrPSc) [79]. Accumulation of Prion protein (PrP) amyloids in the brain causes fatal degenerative disorders such as Gerstmann Straussler-Scheinker disease (GSS), prion protein cerebral amyloid angiopathy (PrP-CAA), scrapie of sheep and goat, spongiform encephalopathy of cattle, kuru, Creutzfeldt-Jakob disease (CJD) and fatal familial insomnia (FFI). PrPSc is involved with Kuru and Scrapie disease [77]. PRNP gene mutation can be octapeptide repeat insertional mutation [80]. The most common associated mutation of PRNP is Glu200Lys [81]. Sanz-Hernández et al. provided a molecular insight into misfolding of human PrPC which occurred due to the pathological mutation in T183A [82]. Wang et al. also recently described the E196K mutation in wild type PrP amyloid fibrils [83]. Goldferb and others clinically investigated about the two repeat octapeptide insertions in the patients with CJD disease [84]. Cochran et al. studied the five octapeptide insertional effect in familial CJD patients [85]. Goldfarb et.al., carried out research about the transmissible familial CJD disease with five, six, eight, nine and more extra octapeptide repeat insertion (OPRI) [86]. Lee and coworkers performed two experiments of PrP variants with octapeptide and without octapeptide insertion. Furthermore, they found that octapeptide repeat insertions effect both folded and misfolded Prion Proteins. Intriguingly, it was also observed that deletion of octapeptide forms fewer twisted fibrils and insertion makes silk-like fibers but insertion of octapeptide does not increase cytotoxicity whereas deletion helps to weaken cytotoxicity [87]. Areškevičiūtė et.al., have recently discussed the 8-OPRI in PRNP gene resulting prion diseases in a Danish Family [88]. Different PrP amyloid fibrils can be generated under same selected experimental conditions [89]. Different types of structures of PrP amyloid fibrils are temperature dependent and this type of temperature dependency may be due to the size variability in initial PrP molecules [90]. Honda through in vitro study showed that Aβ persuades PrP amyloid formation at sub micromolar concentrations [91]. Artikis et al. examined the accommodation of N-glycans on both ends in each monomer of an octameric parallel in-register intermolecular β-sheet (PIRIBS) protofibril. Through in silico molecular dynamics studies they proved that the triantennary glycans can be sterically accommodated in register on both N-linked glycosylation sites of each monomer [92]. Takahashi and coworkers reported that PrP accumulating plaques are associated with Aβ oligomers and appear prior to AD in aged human brains [93]. Sakaguchi and Hara described how N-terminal region regulates the normal function of PrPC, the conversion of PrPC to PrPSc and neurotoxicity of PrPSc. Non-structural, flexible N-terminal domain including polybasic region, OR region, post-OR regions etc., regulates the conversion of PrPC to PrPSc and neurotoxicity of PrPSc. This specific domain has a role in maintaining normal function of PrPC [94]. It has been suggested that high temperature and pressure dissociate PrP fibrils at the molecular level [95]. High temperature and pressure mediated PrP amyloid fibrils dissociation implies that the density of interactions and packing in the amyloid fold is lower than that of native protein [95]. Purro et.al, discussed therapeutic strategies for the interaction between Aβ and PrPC [96]. PrP:Aβ interaction has a crucial role in Aβ-mediated toxicity. It was hypothesized that a fraction of Aβ toxicity is determined by PrPC which can be therapeutically useful in AD [96].
Molecular mechanisms of PrP seeded amyloid fibril formation have been deciphered with the help of CD, mass spectrometer, ultracentrifugation and chemical cross linking [97]. TEM study revealed that PrP rods are 10-20 nm in diameter and 100 to 200 nm in length in negative staining. By rotating with tungsten, the individual rods are 25 nm in diameter. Cryo-EM analysis evinces that each PrP fibril is composed of two protofibrils intertwined in a left-handed helix and the fibril core diameter is of ~14 nm and width of ~25 nm [98]. From solid State NMR study, it is evident that the PrPSc fibrils have a parallel intermolecular β-sheet architectures [99]. Like other amyloids, prion aggregates too reveal characteristic amyloid dye binding properties as after binding with ThT, PrP amyloid fibrils exhibited an enhanced fluorescence at 485 nm and revealed apple green birefringence under cross polarized light after staining of fibrils with CR [100]. A diagrammatic representation of amyloid formation in prion diseases is illustrated in Figure 3 [101-103].
6. IAPP aggregation and Type 2 diabetes
Islet Amyloid Polypeptide (IAPP) or Amylin is a pancreatic hormone produced by pancreatic β-cells [104]. Amylin is a 32 amino acid-based peptide. Mutation in the Islet Amyloid Polypeptide (IAPP) gene results in the formation of Amylin amyloid fibrils which are primarily observed in late onset type 2 diabetes (T2D) [105]. In 1993, O’Brien et.al, discussed the biological role of IAPP and its function in T2D [106]. Lorenzo and coworkers investigated the toxicity of IAPP fibrils to the β-cells and from their experiments; it was evident that human IAPP self-assemblies are toxic to islet β-cells [107]. Howard described the deposition of amylin toxic aggregates in primates and cats and correlated those toxic assemblies with T2D in these mammals [108]. Among these mammals, only rats could not produce amyloids and as a result they do not generate T2D [109]. Mirzabekov et al. analyzed that human amylin can form ion channel in lipid bilayers whereas rat amylin cannot form ion channels [110]. Raimundo et al. depicted the role of IAPP amyloids in the T2D and AD disease pathology. They explained IAPP-centered drug development strategies against AD as the result of ‘’diabetes brain phenotype’’ [111]. Mucibabic and others demonstrated cross seeding of α-syn and IAPP fibrils in vitro to assess the mutual interaction of two amyloidogenic fibrils on T2D . Their findings state that IAPP amyloid formation is diminished in mice due to the deficiency in endogenous α-Syn However, when α-Syn is present, β-Cells take α-Syn in an IAPP and glucose dependent manner in vitro. Further, it was also noted that tail vein injected α-Syn promotes islet amyloid formation in vivo in mice [112]. Mukherjee et.al, proposed that the IAPP amyloid aggregates are just like prion protein aggregates. Their results imply that some of the clinical and pathological changes in T2D might be contagious through a similar manner at which prion proteins disseminate in prion diseases [113]. Kakinen and others demonstrated the coaggregation of IAPP with its primary and secondary amyloidogenic fragments 19-29 S20G and 8-20. They examined that instead of protofilaments, mature fibrils are elongated in coaggregation [114].
Westermark et al. for the first time investigated the amyloid nature of IAPP aggregates using optical microscopy in T2D [115]. EM, AFM and X-ray fiber diffraction techniques showed that the structures of IAPP aggregates resemble twisted protofilaments with distinct cross-β sheet. EM of IAPP amyloid fibrils present that the fibrils are long, unbranched and exhibit diameter of about 100 Å. X-ray diffraction pattern depicts that the peak positions of the IAPP fibril occur at 4.7 Å meridional and 10 Å equatorial reflections corresponding to a cross-β pattern. CD and FTIR studies further implicate that the IAPP aggregates contain β sheets predominantly. The Cryo-EM analysis of full-length IAPP fibrils in ice, revealed a strong reflection at 4.7 Å which is similar to strong meridional signal at 4.7 Å observed for Aβ (11–25) fibrils, supporting this cryo-EM feature as a common characteristic present in amyloid fibrils [116]. Cooper et.al, monitored the growth, bidirectionality and morphological change of individual Amylin fibrils using time lapse AFM. During growth of a protofibril, it is elongated at both ends indicating bidirectionality of protofibril growth [117]. The structure of protofibril of amylin suggest about 2.6 human amylin molecules are packed in 1nm protofibril [118]. Roeder and coworkers described the similarities of IAPP fibrils with Amyloid-β fibrils with the help of cryo-EM [119]. Their atomic model reveals two S-shaped fold intertwined protofilaments of IAPP in the main polymorph resembles S-fold of Aβ fibrils in AD [120]. In the solid-state NMR (ssNMR) study, β-hairpin structures are observed in IAPP amyloid fibrils and this β-hairpin structure consists of two β-strands [120]. Like other amyloids, after staining with CR, apple green birefringence was observed under cross polarized light and enhancement in fluorescence intensity of ThT was noted after binding of IAPP aggregates [116]. A diagrammatic representation of amyloid formation in Type-II diabetes is illustrated in Figure 4 [121].
7. Amyloid like aggregation in the patho-physiology of Cancer.
p53 protein acts as a global transcription factor to maintain the integrity of the cells under different stress [122]. p53 consists of 393 amino acid residues. and abnormality in its biological function leads to Cancer. The mutation in Tumor Suppressor gene (TP53) causes formation of abnormal p53 protein which has tendency to self-aggregate and form amyloidogenic structures [123, 124]. Interestingly the aggregation propensity of p53 protein is remarkably similar to PrP protein and likewise PrP, p53 aggregates are also infectious and cross seed aggregation in other proteins [124]. Maji and his team through their studies demonstrated that aggregation of p53 protein results in impairment of its regular functions leading to production of cells with leaky membranes a characteristic feature of Cancer pathogenesis [125]. Several research findings around the globe suggest almost half of the human cancers are associated with p53 gene mutation [123, 125]. The studies by De Oliveira and others suggest p53 DNA binding domain (DBD) is more prone to pressure induced unfolding and aggregation due to poorer backbone hydrogen bond formation between p53 and DBD [126]. [126]. Maji and coworkers examined the contribution of wild type p53 aggregates in formation of tumors and hypothesize its implications as potential therapeutic targets [127]. Maritschnegg et al. detected p53 aggregates using the ELISA system (enzyme linked immunosorbent-assay) and further enhanced the understanding of the influence of p53 misfolding in Cancer. Seprion-ELISA system binds with high molecular p53 aggregates whereas tetramers of p53 do not bind with Seprion-ELISA system [128]. Farmer et.al., reported the formation of p53 oligomers and fibrils in human AD brains. Furthermore, their studies implied that in the AD, impairment of DNA may occur due to the interaction of p53 oligomers with tau oligomers [129]. Maji and his group carried out circular dichroism (CD) analysis of p53 amyloids which revealed β-sheet rich character of p53 aggregates with negligible fraction of α-helix. From the TEM study both fibrillar and non-fibrillar structures of p53 could be observed. Further like other amyloid p53 aggregates could also bind ThT and CR dyes [125]. Solution state NMR evinces disruption of salt bridge between monomers during p53 tetramerization as the reason which leads to oligomerization and amyloid like fibril formation by p53 [130]. A diagrammatic representation of amyloid formation by p53 protein in Cancer is illustrated in Figure 5 [131].
8. Metabolites assemblies as a surprising extension to Generic amyloid Hypothesis.
8.1. Amyloid like aggregation of Single amino acids and its implications in IEMs
Gazit and coworkers for the very first time delineated a common etiology between rare IEM Phenylketonuria (PKU) and amyloid associated diseases. In their pathbreaking research the group reported amyloid like fibrils formed by self-assembly of phenylalanine (Phe). Mutation in the Phenylalanine Hydroxylase (PAH) gene causes deficiency in Phenylalanine Hydroxylase (PAH) enzyme. Blood concentration of Phe is elevated due to the deficient activity of PAH enzyme. Elevated Phe concentration is responsible for autosomal recessive disorder PKU. The study revealed Phe fibrils just like conventional amyloid could bind CR and ThT dye. The MTT assay suggest Phe fibrils are cytotoxic to both hepatic as well as neural cell lines. The immune-histochemical analysis connotes formation of antibodies against Phe fibrils. Hence, the results illustrated in this work suggest a common etiology between PKU and amyloid associated diseases and implicates pathogenesis in PKU is associated with the formation of toxic fibrillar assemblies by phenylalanine [11]. Concomitantly, the same research group investigated the morphological, structural and mechanical properties of Phe nano fibrils in both hydrated and dehydrated states by using AFM and electron microscopy techniques. Their report described that the Young Modulus of Phe fibrils may show a value up to 30 GPa which is higher than any other biological fibrillar structures hence, revealing their exceptional mechanical strength [132].
Subsequent to Phe fibril discovery, Gazit and his team explained the abrupt the formation of amyloid-like supramolecular nanostructures by the self-assembly of Tyrosine (Tyr) [14]. Rare IEM disorder Tyrosinemia occurs due to the accumulation of Tyr in body which is also caused by a mutation in gene expressing enzyme tyrosine hydroxylase (TyH) which catabolize Tyr [133]. Ménard-Moyon et.al, encountered that the Tyr amyloid nano fibrillar structures accelerate the aggregation of globular proteins and aromatic metabolites and this type of amyloid cross seeding results in creation of lethal aggregation trap of proteins. For instance, Tyrosine can self-assemble into a variety of structures such as nanoribbons, dendritic structures, fiber-like structures etc [134]. Tyr amyloid structures display apple green color after staining with CR and an enhanced fluorescence intensity after binding with ThT. The cytotoxicity analysis of Tyr assemblies by XTT assay suggest on increasing the concentrations of Tyr from 0.2 to 4 mg/ml, the cell viability was decreased from 80% to 40% [14]. Further, Gazit and coworkers developed anti-Tyr antibodies to inhibit the formation of toxic Tyr amyloid fibrils. Their study revealed pre-incubation of Tyr assemblies with anti-Tyr antibodies resulted in almost 80% cell viability in SH-SY5Y cells suggesting a possible therapeutic cure of tyrosinemia by these antibodies [135]. Further experimental studies by Kar and coworkers elucidate ability of Tyr assemblies to cross-seed amyloid like aggregation in lysozyme, BSA and myoglobin proteins under physiological conditions [136]. Subsequently Gazit and coworkers also reported toxic amyloid like fibrils formed by aggregation of Tryptophan (Trp) which induces apoptosis in neuronal cells and have implication in Hypertryptophanemia and Hartnup diseases [137]. MD simulation experiments for assessing the mechanism of cytotoxicity caused by Trp and Phe fibrils suggest these metabolite assemblies can penetrate inside membrane leading to its disruption and ultimately inducing apoptosis [138].
Although, the aggregation of aromatic amino acids
to amyloid like morphologies was well studied by Gazit and coworkers, however,
amyloid like aggregation propensities for non-aromatic amino acids were
unknown. In this context, our research group for the very first-time reported
amyloid-like structures formed by non-aromatic amino acids Cysteine (Cys) and
Methionine (Met). The amyloid like aggregation of Cys and Met was validated
through microscopic techniques like SEM and TEM and spectroscopy tools like
solid state NMR, FTIR, TGA and XRD along with preliminary MD simulation
studies. The MTT assay also revealed both Cys and Met fibrils are cytotoxic and
decreased cell viability both in neural and kidney cells, suggesting in the
role of amyloidogenic pathway in diseases like cystinuria and
hypermethioninemia [12]. Further, our group
reported unusual aggregates formed by proline, hydroxyproline and lysine [13]. Currently, we ae also studying the aggregation
properties of non-aromatic polar amino acids and our initial experiments
suggest glutamine (Gln), aspartic acid (Asp) and glutamic acid (Glu) may
aggregate to amyloid like structures [139].
Self-assembly of Glycine (Gly) crystalline fern-like architects is also
reported [140]. Gazit and coworkers have also
previously studied the self-assembly of Cystine to amyloid-like fibrillar
morphologies by using TEM, Confocal fluorescence microscopy and ThT binding
assay. It was also discovered that Cystine aggregates are cytotoxic and induced
cell death up to 62% as indicated by Annexin V and Propidium iodide (PI)
apoptosis assay, [14]. Gazit and coworkers
illustrated well-organized supramolecular β-sheet architects of all naturally
occurring amino acids at the nano level. Hence, this study suggests
a potential of self-assembly in all naturally occurring amino acids which need
to be deciphered in future studies [141]. A
diagrammatic representation of amyloid like structure formation by single amino
acids is illustrated in Figure 6 [11-13,
135, 137]. Plasma concentrations of all amino acids are also mentioned in Table 2.
8.2. Non-proteinaceous metabolite assemblies and its implications in IEMs
Gazit and coworkers have recently reported the research on the association of homocysteine amyloid toxic fibrils with the Alzheimer's disease pathogenesis. They characterized the homocysteine crystals and its amyloid fibrils. Furthermore, they demonstrated the inhibition methodology of homocysteine fibrils by polyphenolic compounds [142]. Zaguri et.al., examined the self-assembly process of oxalate, which results in supramolecular nanofibrils without accumulation of calcium. They provided a mechanistic insight to explain the inconsistency between impaired retinal function and lack of crystal deposition by examining self-assembly of oxalate into supramolecular nano architects. Using TEM analysis, they displayed the existence of elongated oxalate nanofibrillar structures. They also obtained the crystals of oxalate by using a supersaturated solution containing ~70mM of calcium oxalate in Phosphate Buffer Saline (PBS) [143]. Quinolinic acid (QA) is a neurometabolite in the kynurenine pathway, the biosynthetic pathway of tryptophan, which is associated with deadly neurodegenerative diseases. Tavassoly et.al, first demonstrated the self-assembled nanostructures formed by QA. They showed the QA self-assembly by dissolving QA in PBS at 90°C to obtain a homogenous solution followed by gradual cooling of the solution followed by the characterization of the morphologies formed by TEM. From TEM micrographs, it was observed that the fibrillar aggregates are homogeneous in length and width and having diameter in between 3-6 nm. They further studied the cytotoxicity and apoptotic behavior of QA fibrils on human neuroblastoma cells. Surprisingly, it was observed that QA assemblies can result in up to 100% cell death. Further, it was noted that QA nanofibrils can be disrupted by EGCG. To examine the cross-seeding effect of QA fibrils into the α-Syn self-aggregation, they co-incubated α-Syn with QA seeds and after a couple of hours, it was found that those QA seeds induced α-Syn aggregation [144].
Gazit and his team delineated the cytotoxicity of Adenine, Orotic acid and Uracil self-assemblies. Cytotoxicity of adenine was carried out using XTT assay in SH-SY5Y cells and it was found that as the concentration of Adenine was increased to 2mg/ml, cell viability decreases around 50% which is similar to that of Orotic acid. Uracil assemblies on the other hand displayed 90% cell viability at the same concentration. From the cytotoxicity analysis, it was surmised that Adenine and Orotic acid self-assemblies exhibit more cytotoxicity as compared to Uracil aggregates. On the other hand, the apoptotic behavior of Adenine, Orotic acid and Uracil assemblies was analysed using Annexin V and Propidium iodide (PI) assays. This suggests that Adenine and Uracil assemblies cause 40 to 50% cell apoptosis, whereas Orotic acid exhibits about 30% cell apoptosis [14]. Gazit and coworkers also illustrated the Adenine fibril formation and its accumulation in a yeast model [145]
Gazit and coworkers recently reported robust twisted ribbon like structures formed by the self-assembly of Glucosylceramide (GlcCer). Notably, a deficiency of lysosomal enzyme Glucocerebrosidase results in the accumulation of GlcCer and causes Gaucher’s Disease [16]. The cytotoxicity analysis of GlcCer aggregates along with other characteristic amyloidogenic traits suggest etiology of Gaucher’s Disease is also associated to amyloidosis [146]. Kar and coworkers discussed amyloid mimicking self-assemblies formed by artificial sweetener Aspartame through conventional microscopy analysis, turbidimetry, ThT binding assay cytotoxicity and haemolysis analysis along with MD simulation experiments [147]. Further they also recently reported amyloid like structures formed by dopamine using similar methodologies [148]. In our very recent research, we have extended generic amyloid hypothesis to assemblies formed by the metabolites of urea cycle and uric acid pathway [149]. A diagrammatic representation of amyloid like structure formation by non-proteinaceous metabolites is illustrated in Figure 7 [14,142,143].
9. Critical analysis and future outlook
Various research in disease progression suggests amyloid-like structure formation as a common cause for pathogenesis in diseases. The studies illustrated in this review provide a comprehensive literature survey which implicates validity of Generic amyloid hypothesis in the etiology of plethora of diseases like AD, PD, prion diseases, T2D, cancer and IEMs. Several literature reports also suggest common therapeutic remedies for these diseases can be envisaged by designing drugs which could act as generic amyloid inhibitors. However, it is apparent that the diseases like AD which was first discovered to be caused by amyloid like aggregation still does not have cure. The clinical trials suggest most of drugs which could inhibit aggregation and hinder amyloid like aggregation have not been found effective in the treatment of this disease. This is surprising and the reason of inefficacy of these drugs is still unknown. Hence, if the generic amyloid hypothesis is considered a common cause for pathogenesis in other diseases including the most recently discovered association with IEMs, its significance in unraveling the therapeutic remedy for these diseases is still questionable, since the drug which inhibit such amyloid like progression should have been clinically successful. However, limited success of such drug in AD implies that there might be another pathway through which the disease may progress. In future, it is indeed very important to study aggregation of other metabolites, the excess of which causes rare diseases to decipher if an amyloid like etiology could be precisely understood. There should be a lot more clinical studies for assessing the effect of generic amyloid inhibitors like polyphenols, tannic acid, quercetin, flavonoids as for a common therapeutic drug for the diseases mentioned in review to unravel the role of amyloid hypothesis in their etiology. Further the studies should also be conducted on various morphological transitions which occur in amyloid formation and the effect of toxicity of aggregates present in different stages right from nucleation to prefibrillar aggregation and fibril formation. It might be possible that the drugs which inhibit amyloid do not work as the aggregates which are formed in preliminary nucleation step might be more lethal/toxic. Moreover, most of the studies wherein generic amyloid hypothesis have been proposed are performed only at in vitro stage. Still, there is an urgent need for extensive in vivo studies which may pave the ways for clinical trials which appear a far-reaching goal at present. Table 3 summarizes the characteristic features of different types of amyloids associated with the pathogenesis

Table 2.
Amyloids and its characteristics:.
| Type of Amyloids (Disease caused) | Aβ42/40 (AD) | α-Syn (PD) | PrP (prion diseases) | IAPP (Type 2 Diabetes) | p53 (Cancer) | Metabolite Amyloid (IEMs) |
|---|---|---|---|---|---|---|
| Characteristics | 1. Aβ42 – 42 amino acids based-peptide. Aβ40 – 40 amino acids based-peptide. 2.βAPP, PSN1 and PSN2 genes mutations lead to an accumulation of Aβ42 and Aβ40 peptides. 3. Aβ42 accumulation causes AD. 4. Aβ42/40 disrupts cell membrane. |
1. α-Syn is a 140 amino acid based long protein. 2. SNCA gene encodes for α-syn. Mutation in this gene leads to accumulation of α-syn. 3. Accretion of α-Syn causes PD. 4.α-synuclein amyloid fibrils are the major component of Lewy bodies. 5. α-Syn amyloid disrupts cellular membrane. |
1. PrP is 208 amino acids-based protein. 2.Two isomeric forms of PrP are as follows: Cellular form: PrPC Scrapie form: PrPSc. 3. PRNP gene mutation. Mutations in PRNP gene can be missense, insertion or point mutations 4. Prion protein accumulation is known to be associated with the prion diseases. 5. PrP amyloid disrupts cell membrane |
1. It is 37 amino acids-based -polypeptide. 2. It is a regulatory peptide in the islets and it inhibits insulin and glucagon secretion. 3. The human plasma IAPP concentration is only 1-2% of that of insulin. 4.Mutation in the IAPP gene is the main cause. 5. This amyloid causes type 2 diabetes. 6. IAPP amyloids disrupt cell membrane. |
1. It is 393 amino acids based Protein. 2.TP53 gene mutation is responsible for p53 accumulation. 3. Accumulation of p53 is associated with Cancer. 3. It is a tumor suppressor protein. 4.p53 amyloid disrupts cell membrane. |
1. Few amino acids and non- proteinaceous metabolites exhibit amyloid. 2. Mutation in specific genes is the main reason behind accumulation of metabolites. 3.These amyloids can induce cross-seeding. 4.These amyloids can act as functional amyloids. 5. It causes rare Inborn Errors of Metabolism. 6.These amyloids interact with cell membrane leading to disruption of membrane structure. |
| Secondary Structure | β-sheet | β-sheet | β-sheet | β-sheet | β-sheet | β-sheet |
| X-ray | 1. 4.8Å reflection on the meridian. 2. 10-11 Å reflection on equator. |
Meridional diffraction pattern was found at 4.8 Å whereas equatorial diffraction was at 10 Å |
1. A strong meridional diffraction signal at 4.6-4.7 Å. 2. A second strong reflection at equator of 8.5-9.7 Å. |
4.7Å meridional and 10 Å equatorial reflections. | 4.7Å meridional and 10 Å equatorial reflections. | Detects crystalline or non-crystalline nature in assembly process. |
| CD | Aqueous solution of Aβ40 indicates majority portions are β-sheet structure whereas Aβ42 contains only β-sheet structure. |
A mix of α-sheet and random coil to β-sheet enriched structure. | Highly ordered β-sheet conformation. | IAPP aggregates contain mostly β-sheet. | CD spectra shows that both fibrillar and ordered p53 aggregates have β-sheet rich profile. | CD spectra shows β-sheet formation. |
| CR | Apple green birefringence color after staining with CR. | Apple green birefringence color after staining with CR. | Apple green birefringence color after staining with CR. | Apple green birefringence color after staining with CR. | Apple green birefringence color after staining with CR. | It might display Apple green birefringence color after staining with CR. |
| ThT | ThT fluorescence displays an enhanced intensity at 480 nm. | ThT fluorescence displays an enhanced intensity at 480 nm. | ThT fluorescence displays an enhanced intensity at 485 nm. | ThT fluorescence displays an enhanced intensity at 480 nm. | ThT fluorescence displays an enhanced intensity at 480 nm. | It may display an enhanced intensity at 480 nm due to ThT fluorescence. |
| Location | Brain | Brain | Brain | Pancreatic β-cells | Cancer Tissues | Brain and other organs |
10. Conclusions
Overall, in this review we have tried to present a common etiology for diseases like Alzheimer’s, Parkinson’s, prion diseases, type II diabetes, Cancer and IEM from an amyloid perspective. The studies illustrate that there is a common origin of these diseases i.e., at first mutation in gene causes enzyme dysfunction which further leads to accumulation and subsequent aggregation of proteins, peptides and metabolites inside the body. Hence mutations in βAPP, PS1 and PS2 genes leads to Alzheimer’s; SNCA gene mutation causes Parkinson’s; mutated PRNP gene leads to prion protein aggregation and causes prion diseases; IAPP gene mutation leads to Type-II diabetes, TP53 gene mutates and causes accumulation of p53 protein leading to pathogenesis in cancer while mutations in various genes responsible for production of enzymes which catabolize metabolites thus causing accumulation and amyloid-like aggregation of metabolite assemblies leading to plethora of IEMs. Hence, in this review we have tried to explain the importance of “Generic amyloid hypothesis” for the origin of wide range of diseases. We believe in future more extensive research will be pursued to unravel the role of amyloidosis in pathogenesis of diseases. Such studies can be useful in paving the way for identifying a generic therapeutic module for treatment of these diseases.
Author Contributions
Both the authors have read and agreed to the published version of the manuscript. Both authors contributed equally.
Funding
This study was supported by the SERB research grant SPG/2021/00521.
Acknowledgments
We thank Basil Wilson, Bharti Koshti, Vivekshinh Kshtriya and Monisha Patel for help in Figures and useful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
AFM –-Atomic force microscopy
APP - Amyloid precursor protein
AD -Alzheimer’s disease
AKU - Alkaptonuria
Cryo-EM – Cryogenic Electron Microscopy
CR - Congo red
CD - Circular dichroism
CJD - Creutzfeldt-Jakob disease
CWD – Chronic Wasting Disease
Cys - Cysteine
EM - Electron microscopy
EGCG - Epigallocatechin gallae
FE-SEM – Field-emission scanning electron microscopy
FTIR - Fourier-transform infrared spectroscopy
FFI - Fatal familial insomnia
GlcCer – Glucosylceramide
GAG – Glycosaminoglycans
GSS - Gerstmann Straussler-Scheinker
Hcys - Homocysteine
Htt – Huntingtin
Hyp – Hydroxyproline
IEMs - Inborn errors of metabolism
LNAA - Large neutral amino acids
Lys – Lysine
Met – Methionine
MD simulation - Molecular dynamics simulations
NMR - Nuclear magnetic resonance
OPRI - Octapeptide repeat insertion
PKU - Phenylketonuria
Phe - Phenylalanine
Pro – Proline
Polyps – Polyphosphates
PH – Primary Hyperoxaluria
PD - Parkinson’s disease
PBS – Phosphate Buffer Saline
QA – Quinolinic acid
SEM -Scanning electron microscopy
SH – Secondary Hyperoxaluria
TEM - Transmission electron microscopy
ThT - Thioflavin T
TGA - Thermogravimetric analysis
TA - Tannic acid
Tyr - Tyrosine
Trp – Tryptophane
Type II Diabetes – T2D
XRD- X-ray diffraction
References
- Kyle, R.A. Amyloidosis: A convoluted story. British journal of haematology 2001, 114, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer's 1907 paper," Uber eine eigenartige Erkankung der Hirnrinde". Clinical anatomy (New York, NY) 1995, 8, 429–431. [Google Scholar]
- Plascencia-Villa, G.; Perry, G. Neuropathologic Changes Provide Insights into Key Mechanisms of Alzheimer Disease and Related Dementia. Am. J. Pathol. 2022, 192, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Guo, Z. Alzheimer's Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. Journal of neurochemistry 2013, 126, 305–311. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. The FASEB Journal 2012, 26, 1946. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.-H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res. 2015, 2016, 1–18. [Google Scholar] [CrossRef]
- Wille, H.; Requena, J.R. The structure of PrPSc prions. Pathogens 2018, 7, 20. [Google Scholar] [CrossRef]
- Rangel, L.P.; Costa, D.C.; Vieira, T.C.; Silva, J.L. The aggregation of mutant p53 produces prion-like properties in cancer. Prion 2014, 8, 75–84. [Google Scholar] [CrossRef]
- Adler-Abramovich, L.; Vaks, L.; Carny, O.; Trudler, D.; Magno, A.; Caflisch, A.; Frenkel, D.; Gazit, E. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol. 2012, 8, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Gour, N.; Kanth P, C.; Koshti, B.; Kshtriya, V.; Shah, D.; Patel, S.; Agrawal-Rajput, R.; Pandey, M.K. Amyloid-like structures formed by single amino acid self-assemblies of cysteine and methionine. ACS chemical neuroscience 2018, 10, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Koshti, B.; Kshtriya, V.; Singh, R.; Walia, S.; Bhatia, D.; Joshi, K.B.; Gour, N. Unusual Aggregates Formed by the Self-Assembly of Proline, Hydroxyproline, and Lysine. ACS Chem. Neurosci. 2021, 12, 3237–3249. [Google Scholar] [CrossRef] [PubMed]
- Shaham-Niv, S.; Adler-Abramovich, L.; Schnaider, L.; Gazit, E. Extension of the generic amyloid hypothesis to nonproteinaceous metabolite assemblies. Sci. Adv. 2015, 1, e1500137–e1500137. [Google Scholar] [CrossRef]
- Koshti, B.; Kshtriya, V.; Naskar, S.; Narode, H.; Gour, N. Controlled aggregation properties of single amino acids modified with protecting groups. New J. Chem. 2022, 46, 4746–4755. [Google Scholar] [CrossRef]
- Gour, N.; Gazit, E. Metabolite assemblies: A surprising extension to the amyloid hypothesis. Curr. Opin. Chem. Biol. 2021, 64, 154–164. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Khurana, R.; Coleman, C.; Ionescu-Zanetti, C.; Carter, S.A.; Krishna, V.; Grover, R.K.; Roy, R.; Singh, S. Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 2005, 151, 229–238. [Google Scholar] [CrossRef]
- Zaman, M.; Khan, A.N.; Wahiduzzaman; Zakariya, S. M.; Khan, R.H. Protein misfolding, aggregation and mechanism of amyloid cytotoxicity: An overview and therapeutic strategies to inhibit aggregation. Int. J. Biol. Macromol. 2019, 134, 1022–1037. [Google Scholar] [CrossRef]
- Amdursky, N.; Stevens, M.M. Circular Dichroism of Amino Acids: Following the Structural Formation of Phenylalanine. Chemphyschem 2015, 16, 2768–2774. [Google Scholar] [CrossRef]
- Carulla, N.; Zhou, M.; Arimon, M.; Gairí, M.; Giralt, E.; Robinson, C.V.; Dobson, C.M. Experimental characterization of disordered and ordered aggregates populated during the process of amyloid fibril formation. Proc. Natl. Acad. Sci. 2009, 106, 7828–7833. [Google Scholar] [CrossRef]
- Adamcik, J.; Mezzenga, R. Study of amyloid fibrils via atomic force microscopy. Curr. Opin. Colloid Interface Sci. 2012, 17, 369–376. [Google Scholar] [CrossRef]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef]
- Arispe, N.; Pollard, H.B.; Rojas, E. The Ability of Amyloid β-Protein [AβP (1–40)] to Form Ca2+ Channels Provides a Mechanism for Neuronal Death in Alzheimer's Disease. Annals of the New York Academy of Sciences 1994, 747, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.; Brennan, S.; Keon, M.; Ooi, L. The role of amyloid oligomers in neurodegenerative pathologies. Int. J. Biol. Macromol. 2021, 181, 582–604. [Google Scholar] [CrossRef]
- Villegas, S.; Roda, A.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar] [CrossRef]
- Ghiso, J.; Frangione, B. Amyloidosis and Alzheimer’s disease. Advanced drug delivery reviews 2002, 54, 1539–1551. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, W.; Sun, Y.; Dong, X. Insights into the cross-amyloid aggregation of Aβ40 and its N-terminal truncated peptide Aβ11-40 affected by epigallocatechin gallate. Chin. J. Chem. Eng. 2021, 45, 284–293. [Google Scholar] [CrossRef]
- Baazaoui, N.; Iqbal, K. Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound. Biomolecules 2022, 12, 1409. [Google Scholar] [CrossRef]
- Parkin, E.T.; Sultan, F. The Amyloid Precursor Protein Plays Differential Roles in the UVA Resistance and Proliferation of Human Retinal Pigment Epithelial Cells. Protein Pept. Lett. 2022, 29, 313–327. [Google Scholar] [CrossRef]
- Zhao, J.; O'Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer's disease pathogenesis. Journal of neuroinflammation 2011, 8, 1–17. [Google Scholar] [CrossRef]
- Calhoun, M.E.; Burgermeister, P.; Phinney, A.L.; Stalder, M.; Tolnay, M.; Wiederhold, K.-H.; Abramowski, D.; Sturchler-Pierrat, C.; Sommer, B.; Staufenbiel, M.; et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc. Natl. Acad. Sci. 1999, 96, 14088–14093. [Google Scholar] [CrossRef]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer's disease. Frontiers in aging neuroscience 2018, 118. [Google Scholar] [CrossRef]
- Sehar, U.; Rawat, P.; Reddy, A.P.; Kopel, J.; Reddy, P.H. Amyloid beta in aging and Alzheimer’s disease. International journal of molecular sciences 2022, 23, 12924. [Google Scholar] [CrossRef]
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and Neurotoxic Effects of Amyloid β Protein: Reversal by Tachykinin Neuropeptides. Science 1990, 250, 279–282. [Google Scholar] [CrossRef]
- Antonino, M.; Marmo, P.; Freites, C.L.; Quassollo, G.E.; Sánchez, M.F.; Lorenzo, A.; Bignante, E.A. Aβ Assemblies Promote Amyloidogenic Processing of APP and Intracellular Accumulation of Aβ42 Through Go/Gβγ Signaling. Frontiers in Cell and Developmental Biology 2022, 638. [Google Scholar] [CrossRef]
- Kidd, M. Paired helical filaments in electron microscopy of Alzheimer's disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2000, 1502, 16–30. [Google Scholar] [CrossRef]
- Harper, J.D.; Wong, S.S.; Lieber, C.M.; Lansbury Jr, P.T. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chemistry & biology 1997, 4, 119–125. [Google Scholar]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Morris, K.L.; Serpell, L.C. X-ray fibre diffraction studies of amyloid fibrils. Amyloid proteins: Methods and protocols 2012, 121–135.
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S. Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. Journal of the American Chemical Society 2016, 138, 9663–9674. [Google Scholar] [CrossRef]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ (1–42) amyloid fibril. Proceedings of the National Academy of Sciences 2016, 113, E4976–E4984. [Google Scholar] [CrossRef]
- Soto, C.; Castaño, E.M.; Frangione, B.; Inestrosa, N.C. The α-Helical to β-Strand Transition in the Amino-terminal Fragment of the Amyloid β-Peptide Modulates Amyloid Formation∗. Journal of Biological Chemistry 1995, 270, 3063–3067. [Google Scholar] [CrossRef]
- Habiba, U.; Merlin, S.; Lim, J.K.; Wong, V.H.; Nguyen, C.T.; Morley, J.W.; Bui, B.V.; Tayebi, M. Age-Specific Retinal and Cerebral Immunodetection of Amyloid-β Plaques and Oligomers in a Rodent Model of Alzheimer’s Disease. J. Alzheimer's Dis. 2020, 76, 1135–1150. [Google Scholar] [CrossRef]
- Zhang, T.; Xia, Y.; Hu, L.; Chen, D.; Gan, C.-L.; Wang, L.; Mei, Y.; Lan, G.; Shui, X.; Tian, Y. Death-associated protein kinase 1 mediates Aβ42 aggregation-induced neuronal apoptosis and tau dysregulation in Alzheimer's disease. International Journal of Biological Sciences 2022, 18, 693. [Google Scholar] [CrossRef]
- Tiiman, A.; Krishtal, J.; Palumaa, P.; Tõugu, V. In vitro fibrillization of Alzheimer’s amyloid-β peptide (1-42). Aip Advances 2015, 5, 092401. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R. Mutation in the α-synuclein gene identified in families with Parkinson's disease. science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Jiang, Z.; Huang, Y.; Zhang, P.; Han, C.; Lu, Y.; Mo, Z.; Zhang, Z.; Li, X.; Zhao, S.; Cai, F.; et al. Characterization of a pathogenic variant in GBA for Parkinson’s disease with mild cognitive impairment patients. Mol. Brain 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Choi, M.L.; Gandhi, S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer's and Parkinson's diseases. The FEBS journal 2018, 285, 3631–3644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.-Q.; Jia, C.; Lim, Y.-J.; Feng, G.; Xu, E.; Long, H.; Kimura, Y.; Tao, Y.; Zhao, C. Mechanistic basis for receptor-mediated pathological α-synuclein fibril cell-to-cell transmission in Parkinson's disease. Proceedings of the National Academy of Sciences 2021, 118, e2011196118. [Google Scholar] [CrossRef]
- Clayton, D.F.; George, J.M. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254. [Google Scholar] [CrossRef]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed]
- Meena, V.K.; Kumar, V.; Karalia, S.; Dangi, R.S.; Sundd, M. Structural and mechanistic insights into modulation of α-Synuclein fibril formation by aloin and emodin. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2022, 1866, 130151. [Google Scholar] [CrossRef]
- George, J.M. The synucleins. Genome biology 2001, 3, 1–6. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2012, 14, 38–48. [Google Scholar] [CrossRef]
- Volles, M.J.; Lee, S.-J.; Rochet, J.-C.; Shtilerman, M.D.; Ding, T.T.; Kessler, J.C.; Lansbury, P.T. Vesicle permeabilization by protofibrillar α-synuclein: Implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry 2001, 40, 7812–7819. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.-J.; Bae, E.-J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.-J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef]
- Araki, K.; Yagi, N.; Aoyama, K.; Choong, C.-J.; Hayakawa, H.; Fujimura, H.; Nagai, Y.; Goto, Y.; Mochizuki, H. Parkinson’s disease is a type of amyloidosis featuring accumulation of amyloid fibrils of α-synuclein. Proc. Natl. Acad. Sci. 2019, 116, 17963–17969. [Google Scholar] [CrossRef]
- Musteikytė, G.; Jayaram, A.K.; Xu, C.K.; Vendruscolo, M.; Krainer, G.; Knowles, T.P.J. Interactions of α-synuclein oligomers with lipid membranes. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183536. [Google Scholar] [CrossRef]
- Dean, D.N.; Lee, J.C. Linking Parkinson's Disease and Melanoma: Interplay Between α-Synuclein and Pmel17 Amyloid Formation. Movement Disorders 2021, 36, 1489–1498. [Google Scholar] [CrossRef]
- Mehra, S.; Ghosh, D.; Kumar, R.; Mondal, M.; Gadhe, L.G.; Das, S.; Anoop, A.; Jha, N.N.; Jacob, R.S.; Chatterjee, D.; et al. Glycosaminoglycans have variable effects on α-synuclein aggregation and differentially affect the activities of the resulting amyloid fibrils. J. Biol. Chem. 2018, 293, 12975–12991. [Google Scholar] [CrossRef]
- Reyes, J.F.; Ekmark-Léwen, S.; Perdiki, M.; Klingstedt, T.; Hoffmann, A.; Wiechec, E.; Nilsson, P.; Nilsson, K.P.R.; Alafuzoff, I.; Ingelsson, M. Accumulation of alpha-synuclein within the liver, potential role in the clearance of brain pathology associated with Parkinson’s disease. Acta neuropathologica communications 2021, 9, 1–20. [Google Scholar] [CrossRef]
- Yamaguchi, K.; So, M.; Aguirre, C.; Ikenaka, K.; Mochizuki, H.; Kawata, Y.; Goto, Y. Polyphosphates induce amyloid fibril formation of α-synuclein in concentration-dependent distinct manners. J. Biol. Chem. 2021, 296, 100510. [Google Scholar] [CrossRef]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. elife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Flynn, J.D.; McGlinchey, R.P.; Walker, R.L.; Lee, J.C. Structural features of α-synuclein amyloid fibrils revealed by Raman spectroscopy. J. Biol. Chem. 2018, 293, 767–776. [Google Scholar] [CrossRef]
- Kumari, P.; Ghosh, D.; Vanas, A.; Fleischmann, Y.; Wiegand, T.; Jeschke, G.; Riek, R.; Eichmann, C. Structural insights into α-synuclein monomer–fibril interactions. Proceedings of the National Academy of Sciences 2021, 118, e2012171118. [Google Scholar] [CrossRef]
- Zhou, L.; Kurouski, D. Structural Characterization of Individual α-Synuclein Oligomers Formed at Different Stages of Protein Aggregation by Atomic Force Microscopy-Infrared Spectroscopy. Anal. Chem. 2020, 92, 6806–6810. [Google Scholar] [CrossRef]
- Hong, D.-P.; Fink, A.L.; Uversky, V.N. Structural Characteristics of α-Synuclein Oligomers Stabilized by the Flavonoid Baicalein. J. Mol. Biol. 2008, 383, 214–223. [Google Scholar] [CrossRef]
- Faustini, G.; Longhena, F.; Bruno, A.; Bono, F.; Grigoletto, J.; La Via, L.; Barbon, A.; Casiraghi, A.; Straniero, V.; Valoti, E.; et al. Alpha-synuclein/synapsin III pathological interplay boosts the motor response to methylphenidate. Neurobiol. Dis. 2020, 138, 104789. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Kumari, R.; Kumar, S.; Jangir, D.K.; Maiti, T.K. Extracellular α-Synuclein Disrupts Membrane Nanostructure and Promotes S-Nitrosylation-Induced Neuronal Cell Death. Biomacromolecules 2018, 19, 1118–1129. [Google Scholar] [CrossRef]
- Reis, R.A.d.M.; Freitas, H.R.; de Mello, F.G. Cell Calcium Imaging as a Reliable Method to Study Neuron–Glial Circuits. Front. Neurosci. 2020, 14, 569361. [Google Scholar] [CrossRef] [PubMed]
- Bit-Ivan, E.N.; Bigio, E.H. Neuropathology of neurodegenerative disorders. Progressive Cognitive Impairment and its Neuropathologic Correlates 2016, 1–16. [Google Scholar]
- Ghetti, B.; Piccardo, P.; Frangione, B.; Bugiani, O.; Giaccone, G.; Young, K.; Prelli, F.; Farlow, M.R.; Dlouhy, S.R.; Tagliavini, F. Prion Protein Amyloidosis. Brain Pathol. 1996, 6, 127–145. [Google Scholar] [CrossRef]
- Prusiner, S.B.; McKinley, M.P.; Bowman, K.A.; Bolton, D.C.; Bendheim, P.E.; Groth, D.F.; Glenner, G.G. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 1983, 35, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Kamali-Jamil, R.; Vázquez-Fernández, E.; Tancowny, B.; Rathod, V.; Amidian, S.; Wang, X.; Tang, X.; Fang, A.; Senatore, A.; Hornemann, S.; et al. The ultrastructure of infectious L-type bovine spongiform encephalopathy prions constrains molecular models. PLOS Pathog. 2021, 17, e1009628. [Google Scholar] [CrossRef]
- Owen, F.; Poulter, M.; Shah, T.; Collinge, J.; Lofthouse, R.; Baker, H.; Ridley, R.; McVey, J.; Crow, T.J. An in-frame insertion in the prion protein gene in familial Creutzfeldt-Jakob disease. Mol. Brain Res. 1990, 7, 273–276. [Google Scholar] [CrossRef]
- Mastrianni, J.A. Genetics of Prion Disease. Prions and Diseases 2023, 375–424. [Google Scholar]
- Sanz-Hernández, M.; Barritt, J.D.; Sobek, J.; Hornemann, S.; Aguzzi, A.; De Simone, A. Mechanism of misfolding of the human prion protein revealed by a pathological mutation. Proceedings of the National Academy of Sciences 2021, 118, e2019631118. [Google Scholar] [CrossRef]
- Wang, L.-Q.; Zhao, K.; Yuan, H.-Y.; Li, X.-N.; Dang, H.-B.; Ma, Y.; Wang, Q.; Wang, C.; Sun, Y.; Chen, J.; et al. Genetic prion disease–related mutation E196K displays a novel amyloid fibril structure revealed by cryo-EM. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, L.; Brown, P.; Little, B.; Cervenakova, L.; Kenney, K.; Gibbs, C.; Gajdusek, D. A new (two-repeat) octapeptide coding insert mutation in Creutzfeldt-Jakob disease. Neurology 1993, 43, 2392–2392. [Google Scholar] [CrossRef] [PubMed]
- Cochran, E.J.; Bennett, D.A.; Cervenakova, L.; Kenney, K.; Bernard, B.; Foster, N.L.; Benson, D.F.; Goldfarb, L.G.; Brown, P. Familial Creutzfeldt-Jakob disease with a five-repeat octapeptide insert mutation. Neurology 1996, 47, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, L.G.; Brown, P.; McCombie, W.R.; Goldgaber, D.; Swergold, G.D.; Wills, P.R.; Cervenakova, L.; Baron, H.; Gibbs Jr, C.; Gajdusek, D.C. Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proceedings of the National Academy of Sciences 1991, 88, 10926–10930. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.-H.; Huang, M.-Y.; Lee, Y.-R.; Lin, Y.-K.; Chen, H.-R.; Lee, C.-I. The Effect of Octapeptide Repeats on Prion Folding and Misfolding. Int. J. Mol. Sci. 2021, 22, 1800. [Google Scholar] [CrossRef] [PubMed]
- Areškevičiūtė, A.; Høgh, P.; Bartoletti-Stella, A.; Melchior, L.C.; Nielsen, P.R.; Parchi, P.; Capellari, S.; Broholm, H.; Scheie, D.; Lund, E.L. A Novel Eight Octapeptide Repeat Insertion in PRNP Causing Prion Disease in a Danish Family. J. Neuropathol. Exp. Neurol. 2019, 78, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Ziaunys, M.; Sneideris, T.; Smirnovas, V. Formation of distinct prion protein amyloid fibrils under identical experimental conditions. Sci. Rep. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Ziaunys, M.; Sakalauskas, A.; Mikalauskaite, K.; Snieckute, R.; Smirnovas, V. Temperature-Dependent Structural Variability of Prion Protein Amyloid Fibrils. Int. J. Mol. Sci. 2021, 22, 5075. [Google Scholar] [CrossRef]
- Honda, R. Amyloid-β Peptide Induces Prion Protein Amyloid Formation: Evidence for Its Widespread Amyloidogenic Effect. Angew. Chem. Int. Ed. 2018, 57, 6086–6089. [Google Scholar] [CrossRef]
- Artikis, E.; Roy, A.; Verli, H.; Cordeiro, Y.; Caughey, B. Accommodation of In-Register N-Linked Glycans on Prion Protein Amyloid Cores. ACS Chem. Neurosci. 2020, 11, 4092–4097. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Yokotsuka, M.; Tobiume, M.; Sato, Y.; Hasegawa, H.; Nagao, T.; Gouras, G.K. Accumulation of cellular prion protein within β-amyloid oligomer plaques in aged human brains. Brain Pathol. 2021, 31, e12941. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Sakaguchi, S. N-Terminal Regions of Prion Protein: Functions and Roles in Prion Diseases. Int. J. Mol. Sci. 2020, 21, 6233. [Google Scholar] [CrossRef] [PubMed]
- Torrent, J.; Martin, D.; Noinville, S.; Yin, Y.; Doumic, M.; Moudjou, M.; Béringue, V.; Rezaei, H. Pressure Reveals Unique Conformational Features in Prion Protein Fibril Diversity. Sci. Rep. 2019, 9, 2802. [Google Scholar] [CrossRef] [PubMed]
- Purro, S.A.; Nicoll, A.J.; Collinge, J. Prion Protein as a Toxic Acceptor of Amyloid-β Oligomers. Biol. Psychiatry 2017, 83, 358–368. [Google Scholar] [CrossRef]
- Stöhr, J.; Weinmann, N.; Wille, H.; Kaimann, T.; Nagel-Steger, L.; Birkmann, E.; Panza, G.; Prusiner, S.B.; Eigen, M.; Riesner, D. Mechanisms of prion protein assembly into amyloid. Proceedings of the National Academy of Sciences 2008, 105, 2409–2414. [Google Scholar] [CrossRef]
- Wang, L.Q.; Zhao, K.; Yuan, H.Y.; Wang, Q.; Guan, Z.; Tao, J.; Li, X.N.; Sun, Y.; Yi, C.W.; Chen, J.; et al. Cryo-EM structure of an amyloid fibril formed by full-length human prion protein. Nat. Struct. Mol. Biol. 2020, 27, 598–602. [Google Scholar] [CrossRef]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef]
- Yamaguchi, K.-I.; Kuwata, K. Formation and properties of amyloid fibrils of prion protein. Biophys. Rev. 2017, 10, 517–525. [Google Scholar] [CrossRef]
- Soto, C.; Satani, N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 2011, 17, 14–24. [Google Scholar] [CrossRef]
- Matos, C.O.; Passos, Y.M.; do Amaral, M.J.; Macedo, B.; Tempone, M.H.; Bezerra, O.C.L.; Moraes, M.O.; Almeida, M.S.; Weber, G.; Missailidis, S.; et al. Liquid-liquid phase separation and fibrillation of the prion protein modulated by a high-affinity DNA aptamer. FASEB J. 2020, 34, 365–385. [Google Scholar] [CrossRef]
- Ciuperca, I.S.; Hingant, E.; Palade, L.I.; Pujo-Menjouet, L. Fragmentation and monomer lengthening of rod-like polymers, a relevant model for prion proliferation. arXiv preprint arXiv:1112.4342 2011. arXiv:1112.4342 2011.
- Ferreira, S.; Raimundo, A.F.; Menezes, R.; Martins, I.C. Islet amyloid polypeptide & amyloid beta peptide roles in Alzheimer’s disease: Two triggers, one disease. Neural Regeneration Research 2021, 16, 1127. [Google Scholar] [PubMed]
- Novials, A.; Rojas, I.; Casamitjana, R.; Usac, E.; Gomis, R. A novel mutation in islet amyloid polypeptide (IAPP) gene promoter is associated with Type II diabetes mellitus. Diabetologia 2001, 44, 1064–1065. [Google Scholar] [PubMed]
- O'Brien, T.D.; Butler, P.C.; Westermark, P.; Johnson, K.H. Islet Amyloid Polypeptide: A Review of Its Biology and Potential Roles in the Pathogenesis of Diabetes Mellitus. Veter- Pathol. 1993, 30, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756–760. [Google Scholar] [CrossRef]
- Howard Jr, C.F. Insular amyloidosis and diabetes mellitus in Macaca nigra. Diabetes 1978, 27, 357–364. [Google Scholar] [CrossRef]
- Betsholtz, C.; Christmansson, L.; Engström, U.; Rorsman, F.; Svensson, V.; Johnson, K.H.; Westermark, P.; Mietlicki-Baase, E.G.; Serrano, A.L.; Lomont, J.P.; et al. Sequence divergence in a specific region of islet amyloid polypeptide (IAPP) explains differences in islet amyloid formation between species. FEBS Lett. 1989, 251, 261–264. [Google Scholar] [CrossRef]
- Mirzabekov, T.A.; Lin, M.-C.; Kagan, B.L. Pore Formation by the Cytotoxic Islet Amyloid Peptide Amylin. J. Biol. Chem. 1996, 271, 1988–1992. [Google Scholar] [CrossRef]
- Raimundo, A.F.; Ferreira, S.; Martins, I.C.; Menezes, R. Islet Amyloid Polypeptide: A Partner in Crime With Aβ in the Pathology of Alzheimer's Disease. Frontiers in Molecular Neuroscience 2020, 13, 35. [Google Scholar] [CrossRef]
- Mucibabic, M.; Steneberg, P.; Lidh, E.; Straseviciene, J.; Ziolkowska, A.; Dahl, U.; Lindahl, E.; Edlund, H. α-Synuclein promotes IAPP fibril formation in vitro and β-cell amyloid formation in vivo in mice. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morales-Scheihing, D.; Salvadores, N.; Moreno-Gonzalez, I.; Gonzalez, C.; Taylor-Presse, K.; Mendez, N.; Shahnawaz, M.; Gaber, A.O.; Sabek, O.M.; et al. Induction of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. J. Exp. Med. 2017, 214, 2591–2610. [Google Scholar] [CrossRef] [PubMed]
- Kakinen, A.; Xing, Y.; Arachchi, N.H.; Javed, I.; Feng, L.; Faridi, A.; Douek, A.M.; Sun, Y.; Kaslin, J.; Davis, T.P.; et al. Single-Molecular Heteroamyloidosis of Human Islet Amyloid Polypeptide. Nano Lett. 2019, 19, 6535–6546. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P. Quantitative Studies of Amyloid in the Islets of Langerhans. Upsala J. Med. Sci. 1972, 77, 91–94. [Google Scholar] [CrossRef]
- Makin, O.S.; Serpell, L.C. Structures for amyloid fibrils. The FEBS journal 2005, 272, 5950–5961. [Google Scholar] [CrossRef] [PubMed]
- Goldsbury, C.; Kistler, J.; Aebi, U.; Arvinte, T.; Cooper, G.J. Watching amyloid fibrils grow by time-lapse atomic force microscopy. J. Mol. Biol. 1999, 285, 33–39. [Google Scholar] [CrossRef]
- Goldsbury, C.S.; Cooper, G.J.; Goldie, K.N.; Müller, S.A.; Saafi, E.L.; Gruijters, W.; Misur, M.P.; Engel, A.; Aebi, U.; Kistler, J. Polymorphic Fibrillar Assembly of Human Amylin. J. Struct. Biol. 1997, 119, 17–27. [Google Scholar] [CrossRef]
- Röder, C.; Kupreichyk, T.; Gremer, L.; Schäfer, L.U.; Pothula, K.R.; Ravelli, R.B.G.; Willbold, D.; Hoyer, W.; Schröder, G.F. Cryo-EM structure of islet amyloid polypeptide fibrils reveals similarities with amyloid-β fibrils. Nat. Struct. Mol. Biol. 2020, 27, 660–667. [Google Scholar] [CrossRef]
- Asthana, S.; Mallick, B.; Alexandrescu, A.T.; Jha, S. IAPP in type II diabetes: Basic research on structure, molecular interactions, and disease mechanisms suggests potential intervention strategies. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1765–1782. [Google Scholar] [CrossRef]
- Tomita, T. Apoptosis in pancreatic β-islet cells in Type 2 diabetes. Bosn. J. Basic Med Sci. 2016, 16, 162–179. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and Drug Targeting Insights on Mutant p53. Cancers 2021, 13, 3344. [Google Scholar] [CrossRef]
- Billant, O.; Friocourt, G.; Roux, P.; Voisset, C. , p53, A Victim of the Prion Fashion. Cancers 2021, 13, 269. 124. Bom, A.P.A.; Rangel, L.P.; Costa, D.C.; de Oliveira, G.A.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.; Cepeda, A.O.; Stumbo, A.C. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. Journal of Biological Chemistry 2012, 287, 28152–28162. [Google Scholar]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. p53 amyloid formation leading to its loss of function: implications in cancer pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, G.A.P.; Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Vieira, T.C.R.G.; Cino, E.A.; Silva, J.L. The Status of p53 Oligomeric and Aggregation States in Cancer. Biomolecules 2020, 10, 548. [Google Scholar] [CrossRef] [PubMed]
- Navalkar, A.; Pandey, S.; Singh, N.; Patel, K.; Datta, D.; Mohanty, B.; Jadhav, S.; Chaudhari, P.; Maji, S.K. Direct evidence of cellular transformation by prion-like p53 amyloid infection. J. Cell Sci. 2021, 134. [Google Scholar] [CrossRef] [PubMed]
- Maritschnegg, E.; Heinzl, N.; Wilson, S.; Deycmar, S.; Niebuhr, M.; Klameth, L.; Holzer, B.M.; Koziel, K.; Concin, N.; Zeillinger, R. Polymer-Ligand-Based ELISA for Robust, High-Throughput, Quantitative Detection of p53 Aggregates. Anal. Chem. 2018, 90, 13273–13279. [Google Scholar] [CrossRef] [PubMed]
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathologica Communications 2020, 8, 1–21. [Google Scholar] [CrossRef]
- Galea, C.; Bowman, P.; Kriwacki, R.W. Disruption of an intermonomer salt bridge in the p53 tetramerization domain results in an increased propensity to form amyloid fibrils. Protein Sci. 2005, 14, 2993–3003. [Google Scholar] [CrossRef]
- Rajan, R.; Ahmed, S.; Sharma, N.; Kumar, N.; Debas, A.; Matsumura, K. Review of the current state of protein aggregation inhibition from a materials chemistry perspective: special focus on polymeric materials. Mater. Adv. 2021, 2, 1139–1176. [Google Scholar] [CrossRef]
- Zaguri, D.; Shaham-Niv, S.; Chakraborty, P.; Arnon, Z.A.; Makam, P.; Bera, S.; Rencus-Lazar, S.; Stoddart, P.R.; Gazit, E.; Reynolds, N.P. Nanomechanical Properties and Phase Behavior of Phenylalanine Amyloid Ribbon Assemblies and Amorphous Self-Healing Hydrogels. ACS Appl. Mater. Interfaces 2020, 12, 21992–22001. [Google Scholar] [CrossRef]
- Aliu, E.; Kanungo, S.; Arnold, G.L. Amino acid disorders. Annals of translational medicine 2018, 6. [Google Scholar] [CrossRef]
- Ménard-Moyon, C.; Venkatesh, V.; Krishna, K.V.; Bonachera, F.; Verma, S.; Bianco, A. Self-Assembly of Tyrosine into Controlled Supramolecular Nanostructures. Chem. – A Eur. J. 2015, 21, 11681–11686. [Google Scholar] [CrossRef] [PubMed]
- Zaguri, D.; Kreiser, T.; Shaham-Niv, S.; Gazit, E. Antibodies towards Tyrosine Amyloid-Like Fibrils Allow Toxicity Modulation and Cellular Imaging of the Assemblies. Molecules 2018, 23, 1273. [Google Scholar] [CrossRef] [PubMed]
- Anand, B.G.; Prajapati, K.P.; Shekhawat, D.S.; Kar, K. Tyrosine-Generated Nanostructures Initiate Amyloid Cross-Seeding in Proteins Leading to a Lethal Aggregation Trap. Biochemistry 2018, 57, 5202–5209. [Google Scholar] [CrossRef] [PubMed]
- Shaham-Niv, S.; Rehak, P.; Vuković, L.; Adler-Abramovich, L.; Král, P.; Gazit, E. Formation of Apoptosis-Inducing Amyloid Fibrils by Tryptophan. Isr. J. Chem. 2016, 57, 729–737. [Google Scholar] [CrossRef]
- Shaham-Niv, S.; Rehak, P.; Zaguri, D.; Kolusheva, S.; Král, P.; Gazit, E. Metabolite amyloid-like fibrils interact with model membranes. Chem. Commun. 2018, 54, 4561–4564. [Google Scholar] [CrossRef]
- Gour, N.; Jaiswal, A.; Patel, M.; Revi, N.; Rengan, A. Aggregation characteristics of non-aromatic polar amino acids and its association to amyloids. ChemRxiv. Cambridge: Cambridge Open Engage; 2023; This content is a preprint and has not been peer-reviewed.
- Singh, P.; Pandey, S.K.; Grover, A.; Sharma, R.K.; Wangoo, N. Understanding the self-ordering of amino acids into supramolecular architectures: co-assembly-based modulation of phenylalanine nanofibrils. Mater. Chem. Front. 2020, 5, 1971–1981. [Google Scholar] [CrossRef]
- Bera, S.; Mondal, S.; Rencus-Lazar, S.; Gazit, E. Organization of Amino Acids into Layered Supramolecular Secondary Structures. Accounts Chem. Res. 2018, 51, 2187–2197. [Google Scholar] [CrossRef]
- Yazdi, D.S.; Bar-Yosef, D.L.; Adsi, H.; Kreiser, T.; Sigal, S.; Bera, S.; Zaguri, D.; Shaham-Niv, S.; Oluwatoba, D.S.; Levy, D.; et al. Homocysteine fibrillar assemblies display cross-talk with Alzheimer’s disease β-amyloid polypeptide. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef]
- Zaguri, D.; Shaham-Niv, S.; Naaman, E.; Mimouni, M.; Magen, D.; Pollack, S.; Kreiser, T.; Leibu, R.; Rencus-Lazar, S.; Adler-Abramovich, L.; et al. Induction of retinopathy by fibrillar oxalate assemblies. Commun. Chem. 2020, 3, 1–9. [Google Scholar] [CrossRef]
- Tavassoly, O.; Sade, D.; Bera, S.; Shaham-Niv, S.; Vocadlo, D.J.; Gazit, E. Quinolinic Acid Amyloid-like Fibrillar Assemblies Seed α-Synuclein Aggregation. J. Mol. Biol. 2018, 430, 3847–3862. [Google Scholar] [CrossRef]
- Laor, D.; Sade, D.; Shaham-Niv, S.; Zaguri, D.; Gartner, M.; Basavalingappa, V.; Raveh, A.; Pichinuk, E.; Engel, H.; Iwasaki, K.; et al. Fibril formation and therapeutic targeting of amyloid-like structures in a yeast model of adenine accumulation. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Jacoby, G.; Bar-Yosef, D.L.; Beck, R.; Gazit, E.; Segal, D. Glucosylceramide Associated with Gaucher Disease Forms Amyloid-like Twisted Ribbon Fibrils That Induce α-Synuclein Aggregation. ACS Nano 2021, 15, 11854–11868. [Google Scholar] [CrossRef] [PubMed]
- Anand, B.G.; Prajapati, K.P.; Dubey, K.; Ahamad, N.; Shekhawat, D.S.; Rath, P.C.; Joseph, G.K.; Kar, K. Self-Assembly of Artificial Sweetener Aspartame Yields Amyloid-like Cytotoxic Nanostructures. ACS Nano 2019, 13, 6033–6049. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, K.P.; Anand, B.G.; Ansari, M.; Temgire, M.; Tiku, A.B.; Kar, K. Amyloid-mimicking toxic nanofibers generated via self-assembly of dopamine. Nanoscale 2022, 14, 8649–8662. [Google Scholar] [CrossRef] [PubMed]
- Gour N, Patel M, Gupta S, Wilson B. Metabolite assemblies of urea cycle and uric acid pathway. ChemRxiv. Cambridge: Cambridge Open Engage; 2022; This content is a preprint and has not been peer-reviewed.
Figure 1.
A diagrammatic representation of Alzheimer’s disease formation in human brain. Middle: TEM image of Aβ42 fibers [47]. Bottom right: The Griffin group’s ribbon representation of the Aβ42 fibril structure [42].
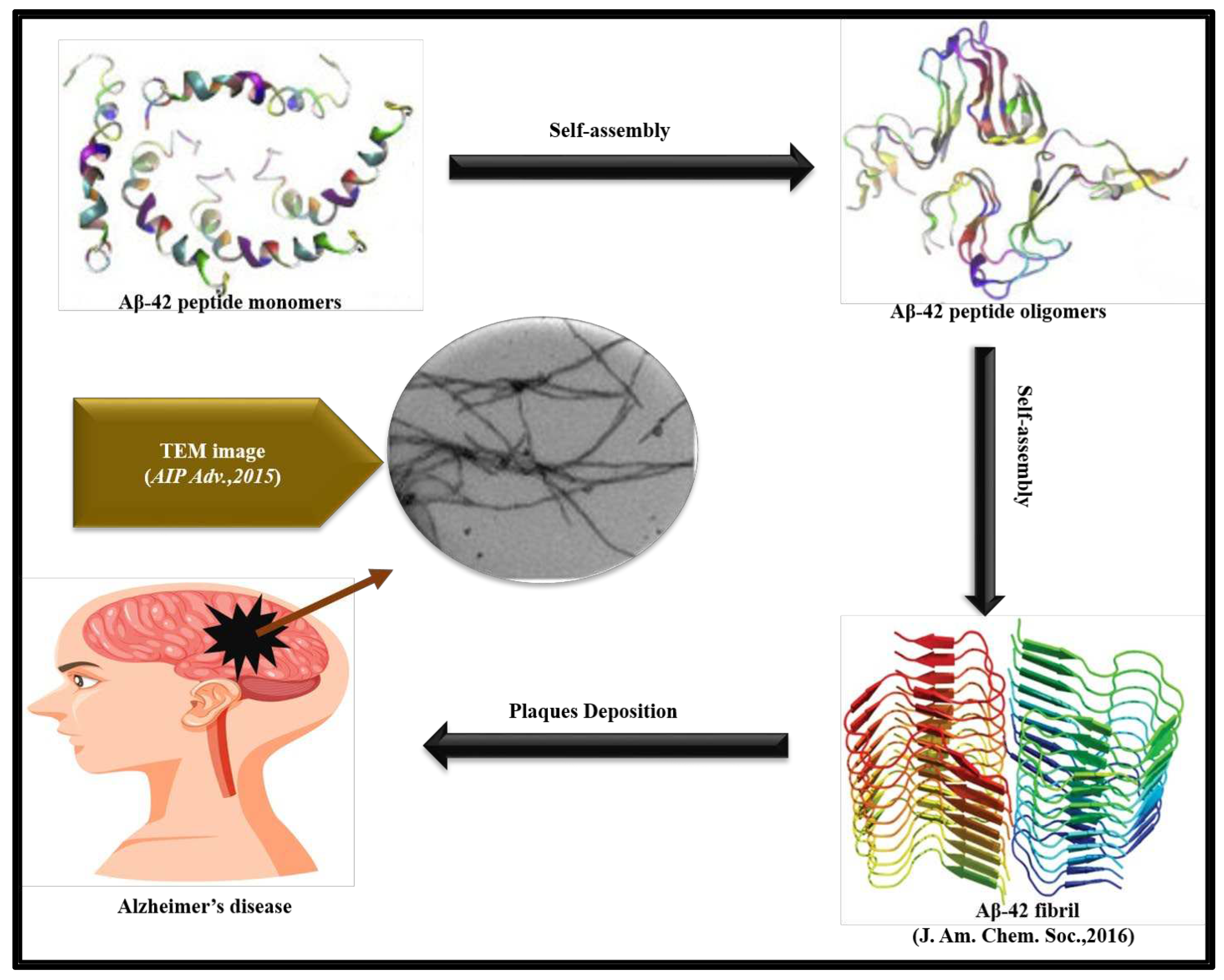
Figure 2.
Pathogenesis caused by the α-syn fibrils due to SNCA gene mutation. Top middle: The Griffin group’s ribbon representation of α-syn fibrils [73]. Bottom middle: Representation of α-syn fibrils bind with cellular membrane [74]. Bottom right: Ca2+ influx into neuron cell [75]. Top right: Optical microscopy images of Lewy body formed in Parkinson disease affected brain [76].
Figure 2.
Pathogenesis caused by the α-syn fibrils due to SNCA gene mutation. Top middle: The Griffin group’s ribbon representation of α-syn fibrils [73]. Bottom middle: Representation of α-syn fibrils bind with cellular membrane [74]. Bottom right: Ca2+ influx into neuron cell [75]. Top right: Optical microscopy images of Lewy body formed in Parkinson disease affected brain [76].
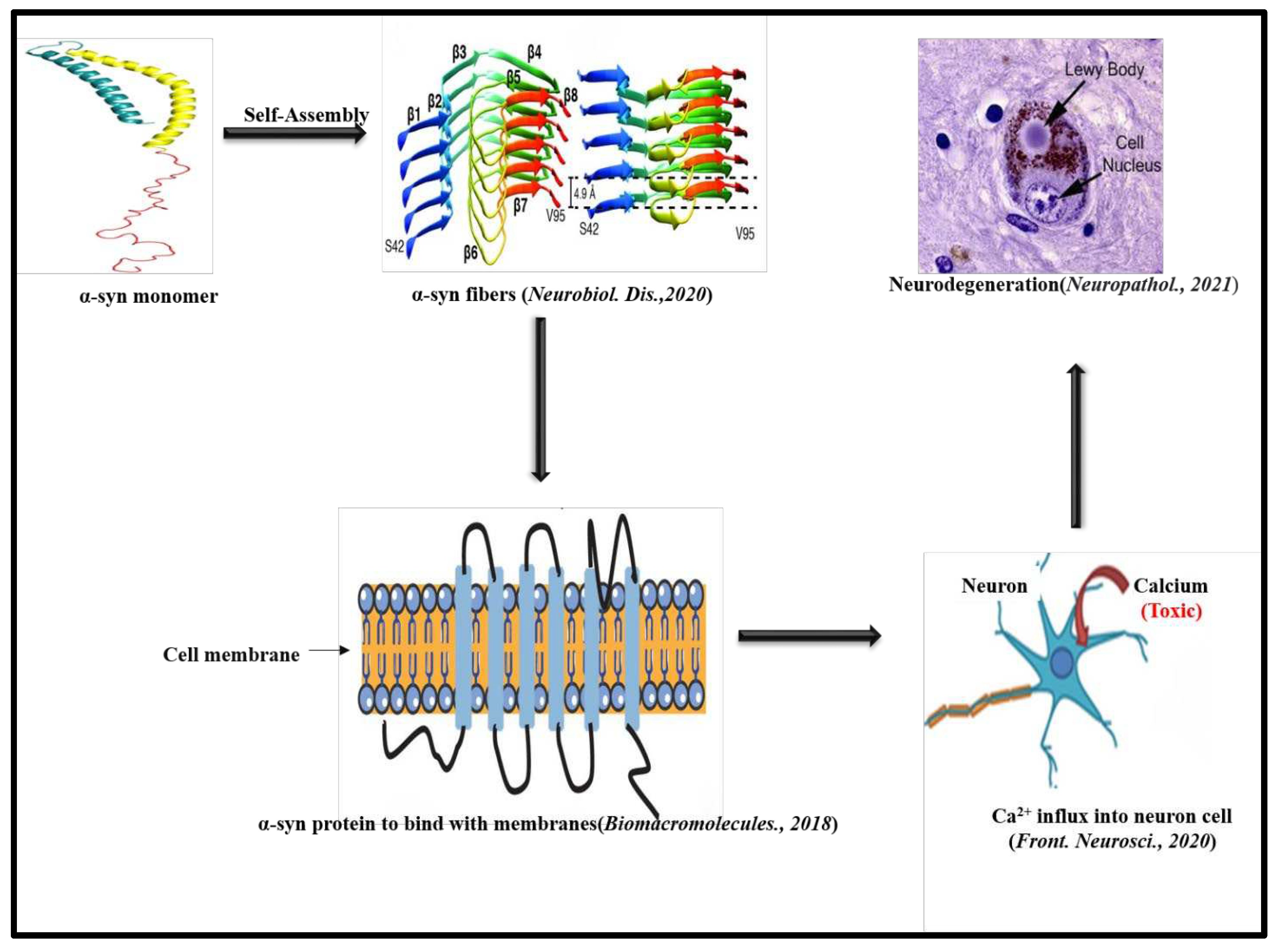
Figure 3.
Diagrammatic representation of prion fibril formation in human brain. Top: Optical microscopic images of Prion infected brain [101]. Left: TEM image of Misfolded prion protein [102]. Right: TEM image of Prion protein fibers [103].
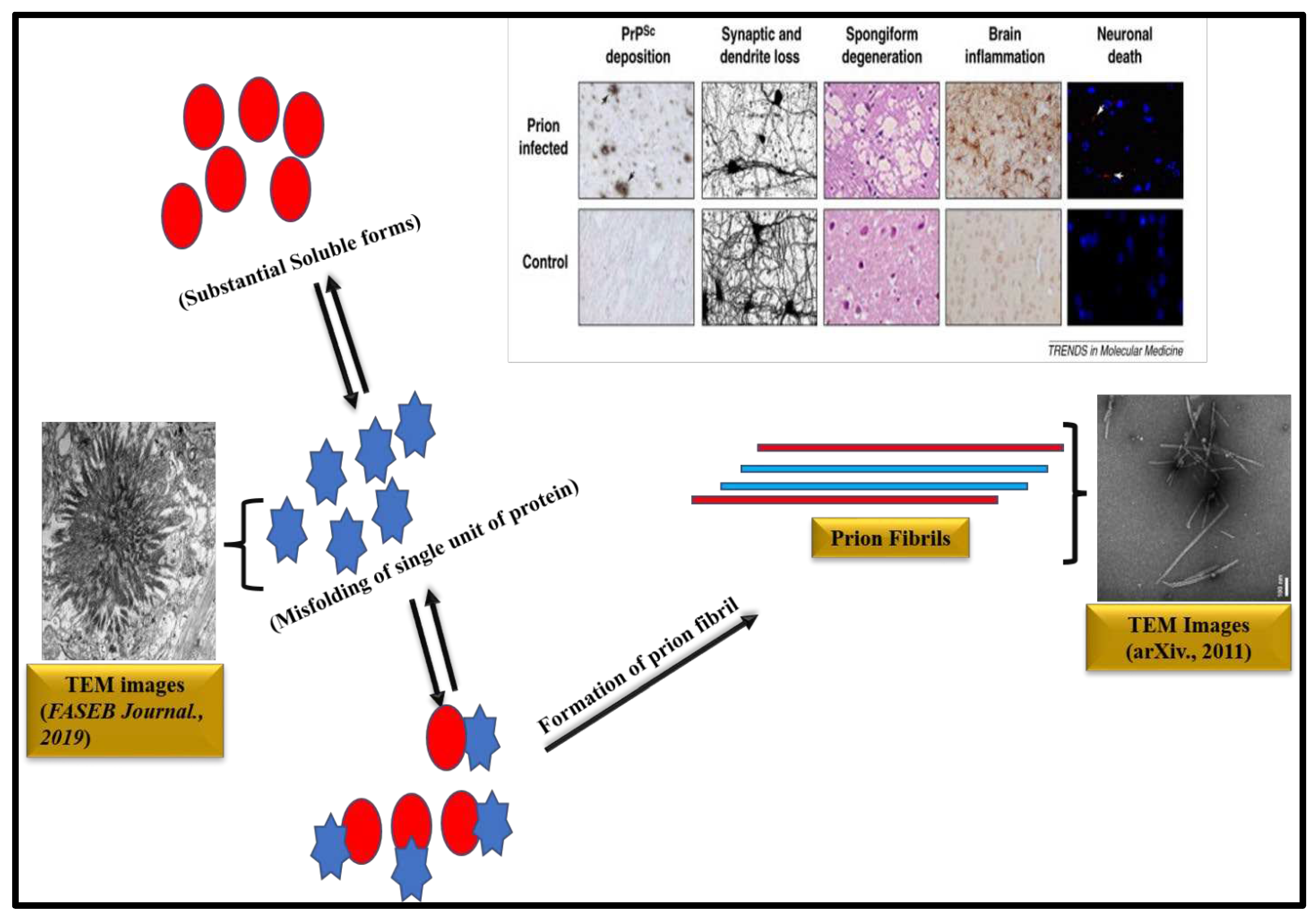
Figure 4.
Steps in IAPP aggregation and β-cell death. Right: Optical microscopy images of Islet β-cell (Congo red) [121]. Left: Optical microscopic images of Deposition of Amyloid and β-cell death (Congo red assay) [121].
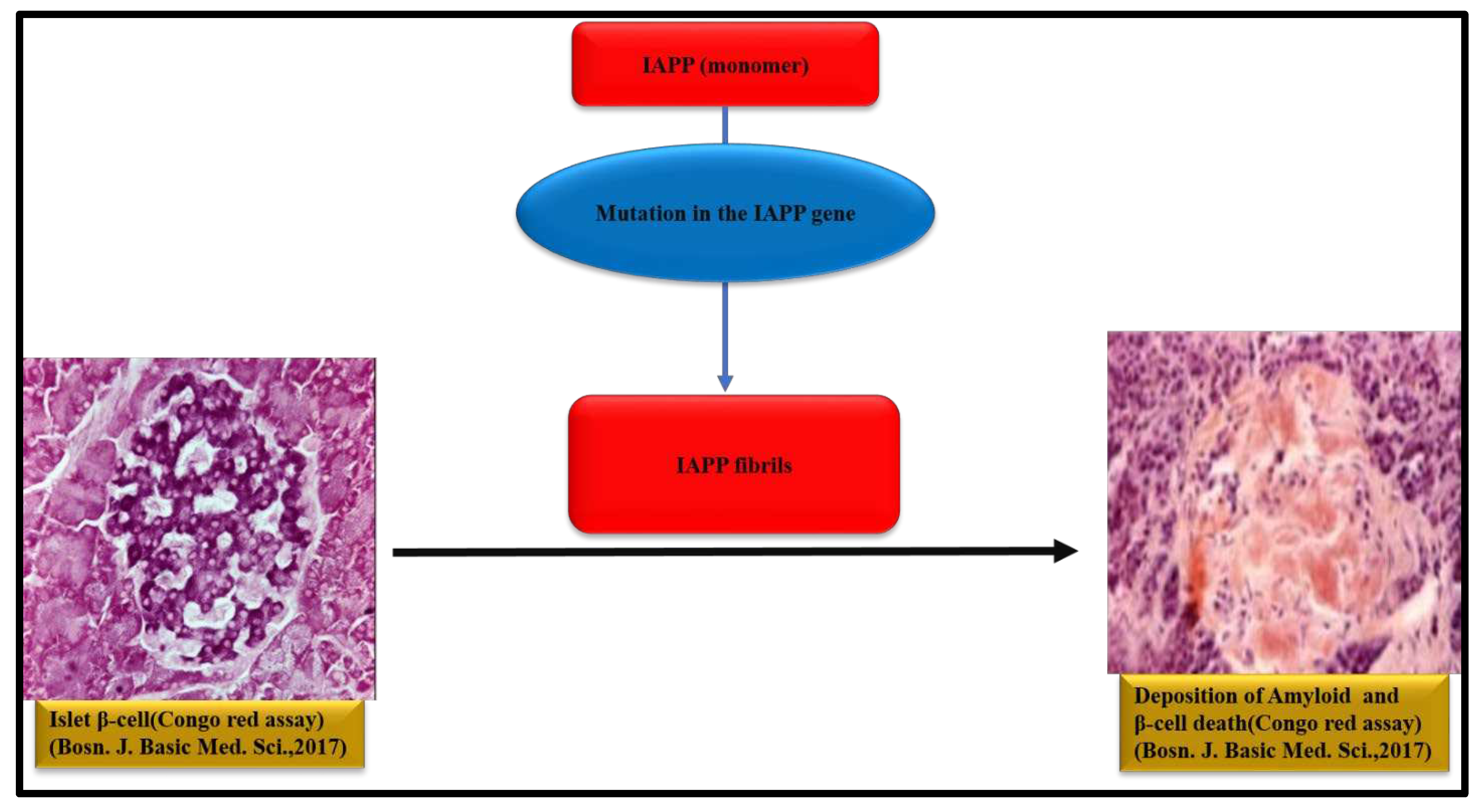
Figure 5.
Conversion of p53 protein monomers to p53 fibers and associated diseases [131].
Figure 5.
Conversion of p53 protein monomers to p53 fibers and associated diseases [131].
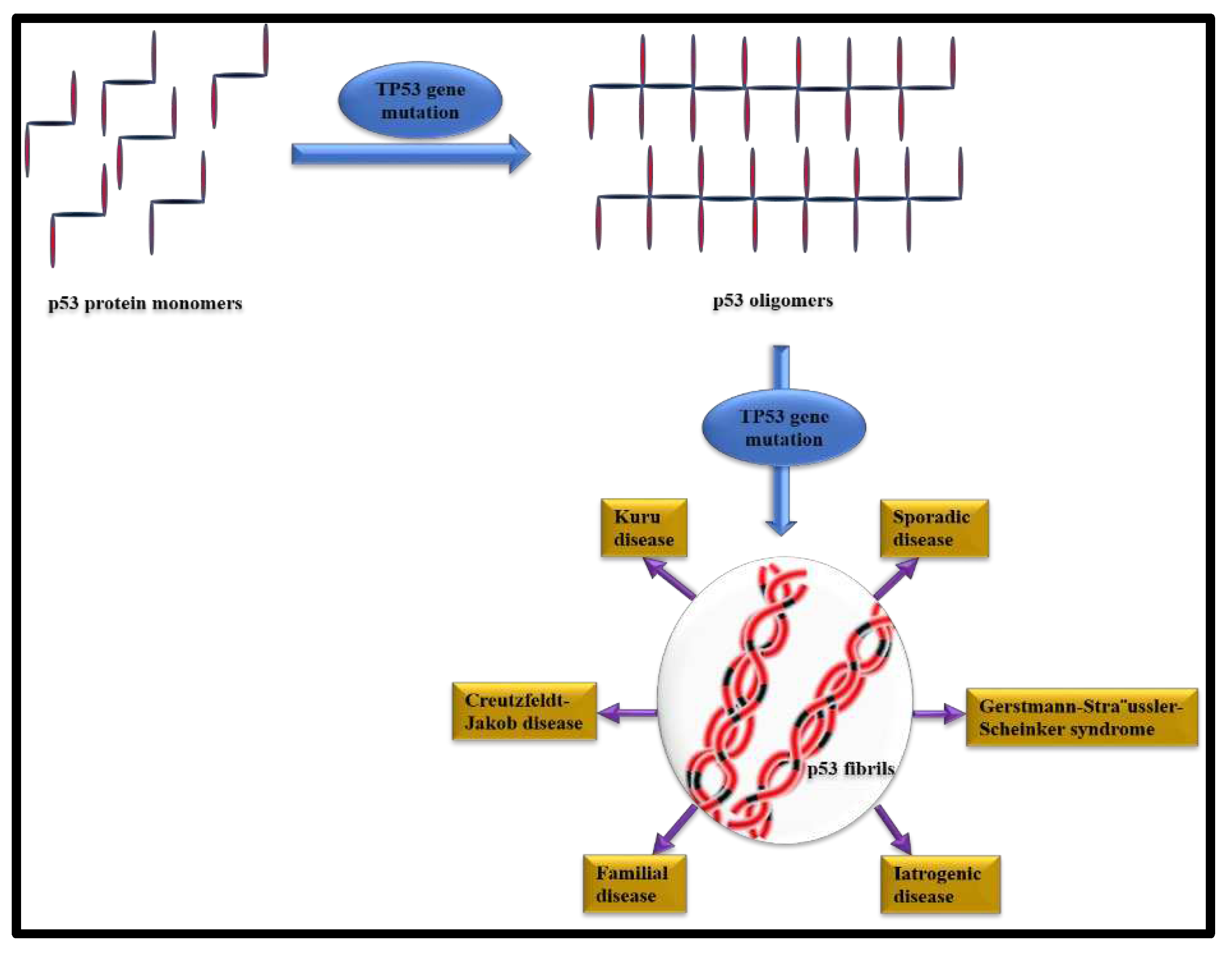
Figure 6.
A graphical summary of the different reports which illustrates amyloid-like structures formed by the self-assembly of single amino acids [11-13, 135, 137].
Figure 6.
A graphical summary of the different reports which illustrates amyloid-like structures formed by the self-assembly of single amino acids [11-13, 135, 137].
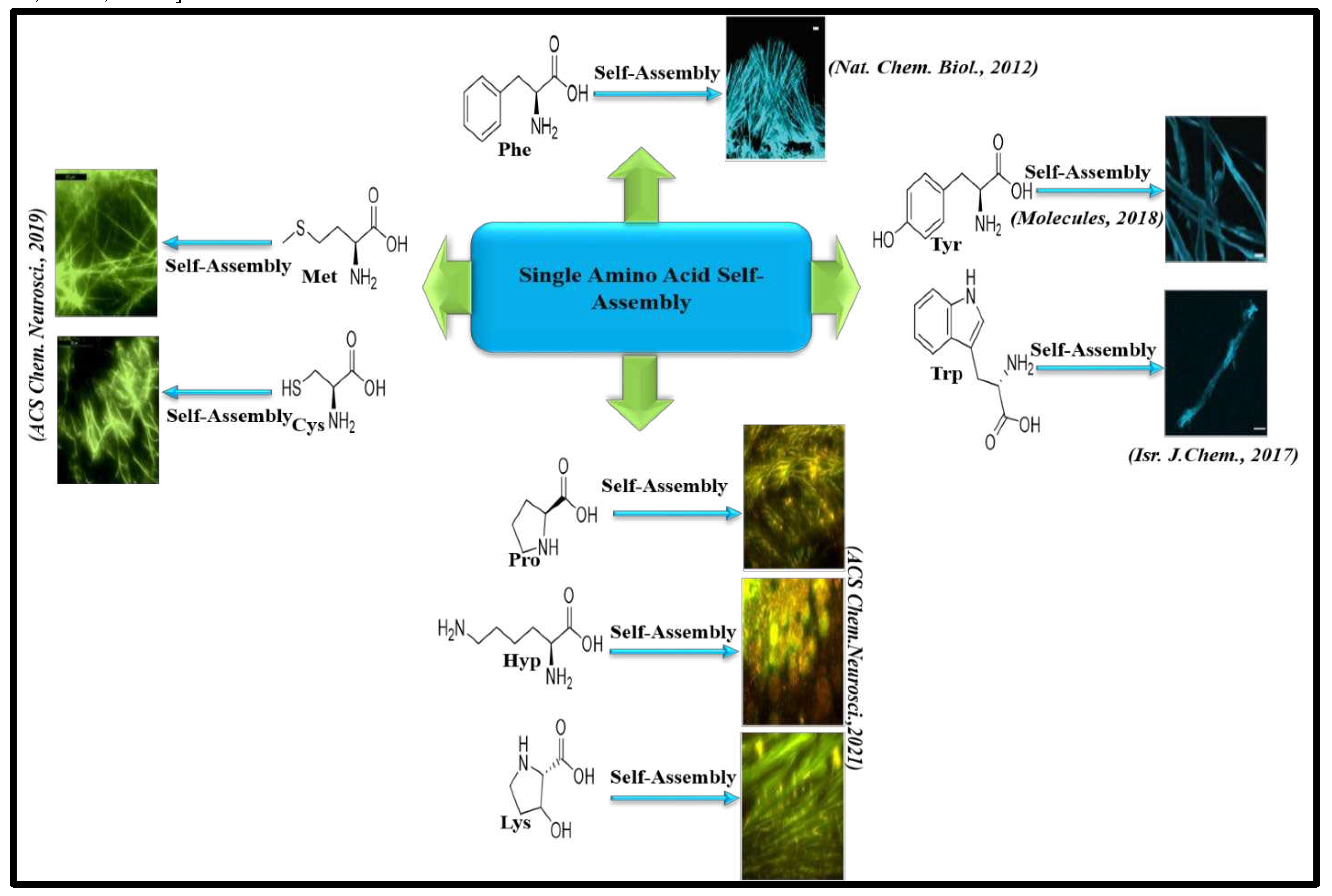
Figure 7.
A graphical summary of the different reports which illustrates amyloid-like structures formed by the self-assembly of non-proteinaceous metabolites [14,142,143].
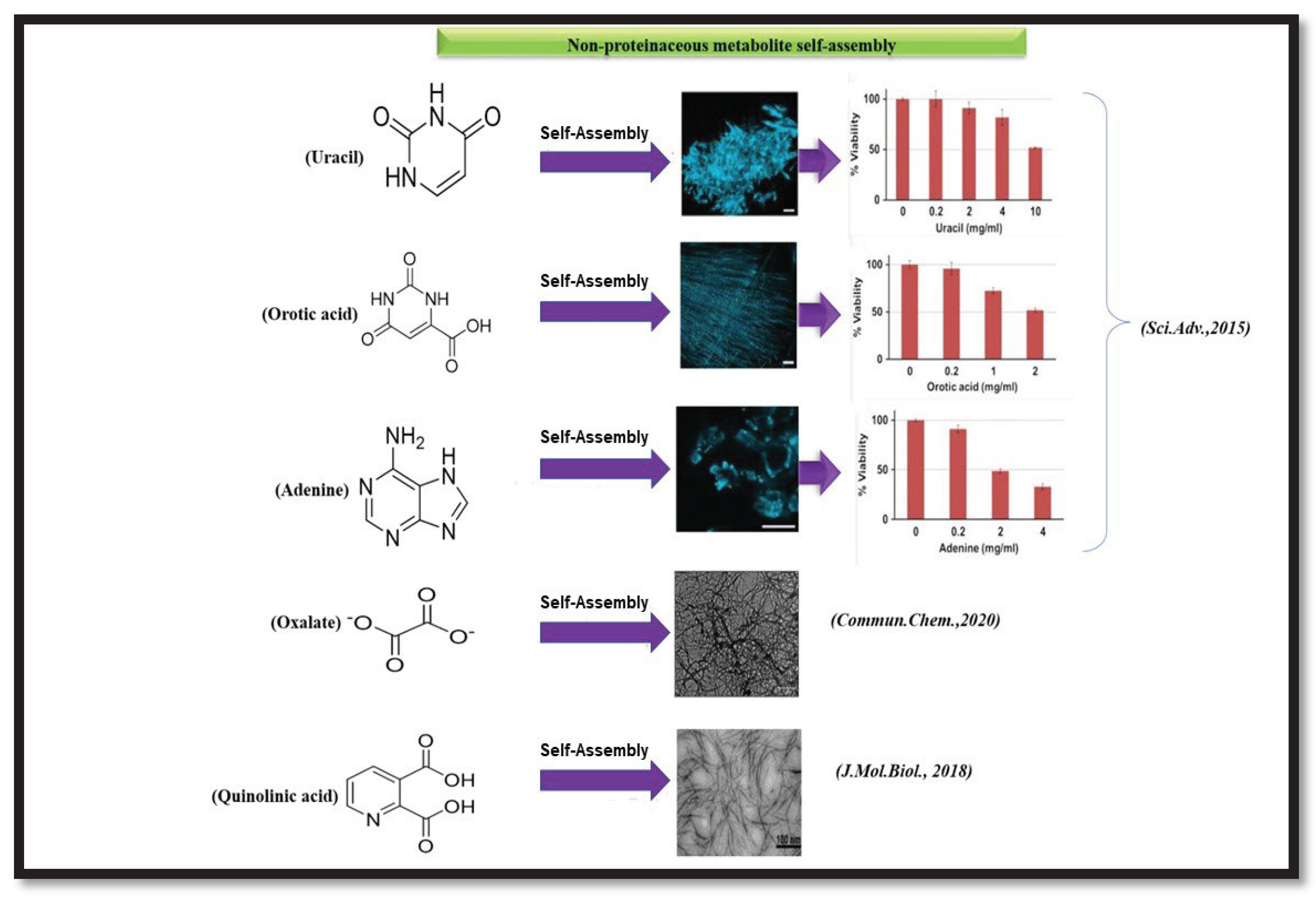
Table 2.
Amino acids and its plasma concentrations.
| Amino acids | Plasma concentration 5range (µmol/L) |
|---|---|
| Alanine | 564-644 |
| Arginine | 44-51 |
| Asparagine | 92-98 |
| Aspartate | 64-69 |
| Glutamate | 261-296 |
| Glutamine | 506-547 |
| Glycine | 390-452 |
| Histidine | 114-122 |
| Isoleucine | 96-102 |
| Leucine | 191-210 |
| Lysine | 210-242 |
| Methionine | 27-31 |
| Proline | 244-265 |
| Phenylalanine | 95-101 |
| Serine | 191-201 |
| Tyrosine | 73-83 |
| Threonine | 164-168 |
| Tryptophan | 65-72 |
| Valine | 217-233 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Downloads
324
Views
152
Comments
0
Subscription
Notify me about updates to this article or when a peer-reviewed version is published.



MDPI Initiatives
Important Links
© 2025 MDPI (Basel, Switzerland) unless otherwise stated