
Submitted:
23 May 2023
Posted:
24 May 2023
You are already at the latest version
Abstract
Our recent studies showed that the treatment of human ovarian tumor cells with NCX4040 resulted in significant depletions of cellular glutathione, formation of reactive oxygen/nitrogen species and cell death. NCX4040 is also cytotoxic to several human colorectal cancer cells (CRC) in vitro and in vivo. Here, we have examined ferroptosis-dependent mechanism(s) of cytotoxicity of NCX4040 in HT-29 and K-RAS mutant HCT116 colon cell lines. Ferroptosis is characterized by accumulation of reactive oxygen species (ROS) within the cell, leading to an iron-dependent oxidative stress-mediated cell death. However, its relevance in the mechanism of NCX4040 cytotoxicity in CRC is not known. We found that NCX4040 generated ROS in CRC cells without any depletion of cellular GSH. Combinations of NCX4040 with erastin (ER) or RSL3 (RAS-selective lethal 3), known inducers of ferroptosis, enhanced CRC death. In contrast, ferrostatin-1, an inhibitor of ferroptosis, significantly inhibited NCX404-induced cell death. Treatment of CRC cells with NCX4040 resulted in induction of lipid peroxidation in a dose- and time-dependent manner. NCX4040 treatment induced several genes related to ferroptosis (e.g., CHAC1, GPX4 and NOX4) in both cell lines. Metabolomic studies also indicated significant increases in both lipid and energy metabolism following the drug treatment in HT-29 and HCT116 cells. These observations strongly suggest that NCX4040 causes ferroptosis-mediated cell death of CRC cells. Furthermore, combinations of NCX4040 and ER or RSL3 may contribute significantly for the treatment of CRC, including those that are difficult to treat due to the presence of Ras mutation in the clinic. NCX4040 induced ferroptosis may also be a dynamic form of cell death for the treatment of other cancers.
Keywords:
Ferroptosis
; NCX4040
; Erastin
; Ferrostatin-1
; RSL3
; Colon Cancer
1. Introduction
Cancer is one of the leading causes of death worldwide. While breast and lung cancers are most prevalent, colorectal cancer (CRC) represents third in morbidity and second in mortality of the total reported cancer deaths [1,2]. The incidence of CRC appears to be on the rise [3]. Current treatments for CRC include surgery, chemotherapy, radiotherapy, immunotherapy, and targeted therapy. Studies have shown that early diagnosis of CRC significantly improves the survival rate of patients. Unfortunately, CRC usually are detected at the advanced stages, leaving chemotherapy the primary choice with a poor survival [4]. This failure appears to result from the development of chemotherapy resistance with undesirable drug side effects. Therefore, development of newer drugs, selectively effective against CRC is urgently needed.
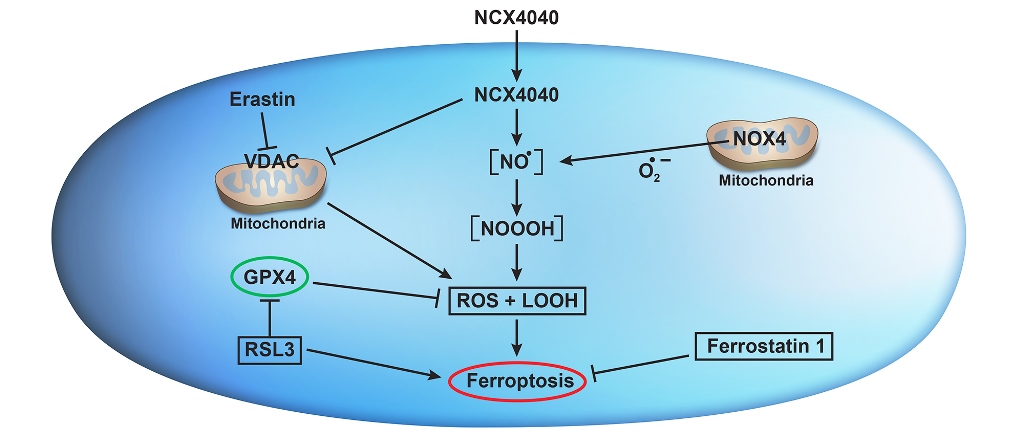
The non-steroidal nitric oxide donor, NCX4040 originally synthesized as an anti-carcinogenic compound, is highly cytotoxic to various tumors including human CRC and ovarian cells [5,6]. Because NCX4040 showed no significant toxicity in vivo [5], development of analogs of NCX4040 is highly desirable. Therefore, understanding of precise molecular mechanisms of cytotoxicity of NCX4040 is essential for the future development of more selective and effective NCX4040 analogs. Studies suggest that NCX4040 induces its cytotoxicity by releasing ●NO following hydrolysis by tumor esterases, depleting cellular glutathione and causing oxidative stress [7,8,9]. Our recent studies have also shown a significant depletion of GSH and formation of ROS by NCX4040 in human ovarian tumor cells [6]. Furthermore, our studies indicated that NCX4040 treatment resulted in induction of both CHAC1 and NOX4 genes in ovarian tumor cells [6]. Recently, CHAC1 and NOX4 along with GPx4 have been suggested to be hallmarks of ferroptosis in tumor cells [10].
Ferroptosis is a non-apoptotic iron-dependent form of cell death, resulting from the formation of ROS/RNS in cells followed by induction of cellular lipid peroxidation and membrane damage [11,12]. Ferroptosis, therefore, results from oxidative damage caused by the formation of ROS/RNS and the presence of ferrous iron and H2O2 (the Fenton reaction), or lipid peroxidation mediated by iron-containing lipoxygenases [13,14,15]. While free ferrous iron is not accumulated in cells, it can be generated by proteins involved in cellular iron metabolism, such as transferrin receptor 1, ferritin, and ferroportin. The antioxidant defenses involving glutathione peroxidase 4 (GPX4) which utilizes glutathione as the cofactor reduces hydroperoxide lipids, inhibiting ferroptosis-mediated cell death [16,17].
NCX4040 has been shown to induce apoptosis in a variety of tumor cells, resulting from the formation of both ROS and RNS [6,7,18]. However, so far, ferroptosis has not been investigated in the mechanism of NCX4040-dependent tumor cell death. As ferroptosis emerges as a promising approach for cancer therapy [19,20], and CRC tumor cells undergo facile ferroptosis [21], we have the examined role of ferroptosis in the mechanism of NCX4040 in cell death in human HT-29 and HCT116 CRC cells. Studies presented here show that NCX4040 indeed induces ferroptosis in both HT-29 and HCT116 tumor cells. Furthermore, combinations of NCX4040 with ER or RSL3 may provide a better treatment modality for the treatment of CRC in the clinic.
2. Methods and Materials
Material: NCX4040 was purchased from Sigma Chemicals (St. Lois, MO) and was dissolved in DMSO. Erastin (2-[1-[4-[2-(4-Chlorophenoxy)acetyl]-1-piperazinyl]ethyl]-3-(2-ethoxyphenyl)-4(3H)-quinazolinone), RSL3 and Ferrostatin-1 were purchased from Cayman Chemicals (Ann Arbor, MI) and were dissolved in DMSO. Stock solutions were stored at -800 C. Fresh drug solutions, prepared from the stock solutions, were used in all experiments. Antibodies to GPX4, CHAC1, NOX4 and β-actin were purchased from Abcam (Waltham, MA).
2.1. Cell culture
Authenticated human colon tumor cells, HT-29 and HCT116 cells were obtained from ATCC (Manassas, VA) and were grown in Phenol Red-free RPMI 1640 media supplemented with 10% fetal bovine serum and antibiotics. Tumor cells were routinely used for 20-25 passages, after which the cells were discarded, and a new cell culture was started from the frozen stock.
2.2. Cytotoxicity studies
The cytotoxicity studies were carried out with both a cell growth inhibition assay and Trypan Exclusion methods. Briefly, 50,000-60,000 cells/well were seeded onto a 6-well plate (in duplicate) and allowed to attach for 18h. Various concentrations of drugs (NCX4040 or combinations of NCX4040, and minimally cytotoxic concentrations of ER, ferrostatin-1, (FeS), or RSL3 were added to cells (HT-29 or HCT116) in fresh complete media (2 ML) and were incubated for either 24, 48 or 72h. When used, ER, FeS or RSL3 were preincubated with cells for 1-2 h before the addition of NCX4040. DMSO (0.01-0.1%) was included as the vehicle control when used. Following trypsinization, surviving cells were collected and counted in a cell counter (Beckman, Brea, CA). For the trypan blue exclusion assay 15 L of cell mixtures were combined with 15 µL of trypan blue and counted in T20 automatic cell counter (Bio-Rad, Hercules, CA).
2.3. Flow Cytometric Analysis of mitochondrial ROS
Analysis of mitochondrial ROS was determined by loading the cells with MitoSox Red (5 uM final concentration; Life Technologies, Carlsbad, CA) for 30 minutes at 37oC, 7% CO2 atmosphere before the addition of the drug. Cells were examined at 2h intervals with the addition of Sytox Blue as a vital dye by flow cytometry. A LSRFortessa flow cytometer (Benton Dickinson, San Jose, CA), equipped with FACSDiVa software, was used to analyze all samples. MitoSox and Sytox Blue were excited using a 561 nm and 405 nm laser and detected using a 610/20 nm and 450/50 nm filter, respectively. For each sample, 10, 000 cells were analyzed using FACSDiVa software.
2.4. Flow Cytometric Analysis for intracellular glutathione
Intracellular glutathione was determined as previously described [6]. Briefly, monochlorobimane dye (mBCl,10 µM final concentration; Life Technologies, Carlsbad, CA) was added to each sample for 15 minutes at 37oC, 5% CO2 atmosphere prior to examination. Propidium iodide (PI) was added (final concentration of 5 ug/ml) to samples before flow cytometric analysis using a LSRFortessa flow cytometer (Benton Dickinson, San Jose, CA) equipped with FACSDiVa software. mBCl and PI were excited using a 405 nm and 561 nm laser and detected using a 530/30 nm and 582/15 nm filter, respectively. For each sample, 10,000 cells were analyzed using FACSDiVa software.
2.5. Lipid Peroxidation Assay
Assay for the peroxidation of cellular lipids was carried out by measuring the formation of malondialdehyde (MDA) using 2-thiobarbituric acid as previously published [22,23]. Briefly, about 2-3 x 106 cells (HT-29 or HCT-116) were incubated with various concentrations of NCX4040 for 2-4h at 37oC. Following incubations, the reactions were stopped by adding 2% trichloroacetic acid and the mixtures were centrifuged (5 min at 1000g). Aliquots (1.0 ml) of the supernatant fractions were then reacted with 2.0 ml of 1% 2-thiobarbituric acid and the chromophore was developed at 90oC for 10 min. After the samples were cooled, the absorbance at 532 nm was determined.
2.6. Real Time RT-PCR
The expression levels of selected transcripts were examined by real-time polymerase chain reaction (RT-PCR) using absolute SYBR green ROX Mix (ThermoFisher Scientific, Rochester, NY) as previously described [6]. Total RNA was isolated using Trizol following treatment with NCX4040 (5µM) for 4 and 24h and purified. Data were analyzed using ΔΔCt method of relative quantification in which cycle times were normalized to β-actin (or GADPH) from the same sample. Primers for the selected genes were designed using Primer Express 1.0 software and in some cases were synthesized (Integrated DNA technologies, CA) from published literature or were purchased from Origene (Gaithersburg, MD). All real-time fluorescence detection was carried out on an iCycler (Bio-Rad, Hercules, CA). Experiments were carried out three different times and results are expressed as the mean ± SEM. Analyses were performed using unpaired Student’s t-test and considered significant when p ≤0.05.
2.7. Western Blot Assay
The Western Blot analyses for various proteins were carried out following treatment with NCX4040 (5µM) for 4 and 24h using standard methods. Samples (5–20 μg of total protein) were electrophoresed under reducing conditions on 4–8% Tris-acetate gels (Novex, Life Technologies, Carlsbad, CA) for 50 minutes at 200 volts. After electrophoresis, proteins were transferred onto nitrocellulose membranes and probed with anti-NOX4, CHAC1, GPx4 and anti-β-actin antibodies. An Odssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE) was used to acquire images.
2.8. Metabolomics Studies
Cell culture extracts were analyzed using untargeted metabolomics, specifically ultra-high performance liquid chromatography (UHPLC) – high resolution tandem mass spectrometry (HRMS/MS). MS data were acquired from samples (n=1 injections) for comparison and statistical analysis. MS/MS data, used in the annotation of untargeted metabolomics features, were acquired from a pooled quality control sample analyzed multiple times via the AcquireX deep scan approach. Compound Discoverer 3.3.0.550 was used to process raw files to provide a tabulated output of features (i.e. unique descriptors of m/z and retention time) and corresponding annotations (MS/MS database matching). Outputs were formatted and further processed using in-house R scripts (via Jupyter Notebooks) to clean data prior to statistical analysis. Additional details are available in the Supplementary Information. The data supporting the annotated chemicals displayed in the figures were manually reviewed for MS, MS/MS, and retention time matching (when available), see SI Table 1.
2.9. Statistical Analysis
The results are expressed as mean ± SEM of minimum of 3 independent experiments (n = 3). One-way analysis of variance (ANOVA) was used for statistical analysis using Graph Pad Prism (GraphPad Software, Inc, La Jolla, CA). For multiple comparisons, the Tukey Multiple comparison’s test was utilized and were considered statistically significant when p < 0.05.
3. Results
3.1. Cytotoxicity studies with NCX4040
Our previous studies have shown that NCX4040 is cytotoxic to human ovarian tumor cells [6]. In this study, we found NCX4040 to be significantly cytotoxic to both human colon HT-29 and HCT116 cells (Figure 1A,B) and there were no significant differences in cytotoxicity as IC50 were very similar. (Figure 1C).
3.2. Measurements of mitochondrial ROS
Since NCX4040 generates ROS in various tumor cells which lead to tumor cell death, we investigated whether NCX4040 also forms ROS in these colon cancer cells. We used Mitosox for the detection of mitochondrial ROS as previously described [6]. While use of Mitosox has remained controversial [24], we [6] as well as others [7,25] have utilized Mitosox successfully to detect ROS in cells. Our results (Figure 2) clearly show that NCX4040 generates ROS in a dose-dependent manner in both cells. At lower concentrations of NCX4040 (e.g., 10 µM), more ROS were detected in HCT116 cells than in HT-29 cells.
3.3. Lipid Peroxidation in HT-29 and HCT116 Cells
Examination for peroxidation of HT-29 and HCT cellular lipids indicated that MDA formation increased in both cells over controls in the presence of NCX4040 in time- and dose-dependent manners. While increases in MDA formation were small, however, these increases were statistically significant (Figure 3).
3.4. RT-PCR Studies
3.4.1. NCX4040 induces Oxidative Stress Genes in HT-29 and HCT116 Cells
Our previous studies have shown that NCX4040 treatment results in modulations of various genes related to oxidative stress, inflammation and DNA-response in human ovarian tumor cells [6,26]. Therefore, we evaluated oxidative stress related genes in HT-29 and HCT-116 cells following NCX4040 treatment. Studies show that oxidative stress genes are significantly modulated by NCX4040 (Figure 4A,B). NCX4040 treatment resulted in a rapid (4h) induction of HMOX1/OX1 gene (6-fold) in HT-29 cells and more than 12-fold in HCT116 cells. HMOX1/OX1 gene remained elevated at 24h in HCT116 cells (3-fold), however, it decreased to the control values in HT-29 cells. HMOX1/OX1 gene is considered to be an important biomarker of oxidative stress and is induced by number of free radical producing drugs and chemicals [27,28]. Ferroptosis markers, GPX4, NOX4 and CHAC1 were also significantly induced by NCX4040 in both cell lines at 24h. However, CHAC1, which causes cleavage of glutathione, resulting in depletion of GSH [29], was rapidly induced (2-3-fold) in both HT-29 and HCT116 cells at 4h and remained elevated in both cells at 24h; 5-6 folds in HT-29 and 35-40-folds in HCT116 cells, respectively. SOD2, which catalyzes the decomposition of superoxide radical anion (O2●-) formed from NOX4, was induced by NCX4040 only in HCT116 cells. Furthermore, OGG1, which is responsible for repair of oxidative damage in DNA caused by ROS, was elevated in both HT-29 and HCT116 cells at 24h (Figure 4).
3.4.2. NCX4040 Induces Inflammatory Response Genes in HT-29 and HCT116 Cells
NCX4040 treatment of HT-29 and HCT116 colon cells also induced various inflammatory response genes (Figure 5A,B) as we found previously in ovarian tumor cells [6]. NCX4040 significantly induced VEGF in HCT116 cells at both 4 and 24 h while it was only induced at HT-29 cells at 24h. COX2 was significantly induced in HCT116 cells while NCX4040 had only a small effect in HT29 cells. It is also interesting that NCX4040 significantly decreased expression of NF-kB in HT-29 at 4h while it had no effects in HCT116 cells (Figure 5A).
3.5. Ferrostatin-1 Inhibits NCX4040 Cytotoxicity in HT-29 and HCT cells
Our RT-PCR results indicated significant modulation of GPX4, NOX4 and CHAC1 genes by NCX4040 in HT-29 and HCT116 cells. Since these genes are indicators of ferroptosis, we examined effects of Ferrostatin-1 (FES) on the cytotoxicity of NCX4040 in both HT-29 and HCT-116 cells. FES is a known inhibitor of ferroptosis in tumor cells [30,31,32]. Results shown in Figure 6A,B indicated FES (2 µM, minimally cytotoxic dose) significantly inhibited cytotoxicity of NCX4040 in both HT-29 and HCT cells, suggesting that NCX4040 induces ferroptosis in these CRC tumor cells. Furthermore, FES was more effective in inhibiting NCX4040-induced cytotoxicity in HCT116 cells than in HT-29 cells (Figure 7B; compare 10-5 M induced cell killing).
3.6. Erastin Enhances NCX4040 Cytotoxicity in HT-29 and HCT116 Cells
Erastin (ER) is a well-known inducer of ferroptosis in tumor cells and has been utilized extensively to decipher mechanism of ferroptosis [33,34,35]. We used various concentrations of ER to examine its effects on NCX4040 cytotoxicity in CRC cells. Our results, shown in Figure 7A,B, clearly indicated that ER was effective in enhancing NCX4040-mediating CRC cell death. Again, HCT116 cells were more sensitive to ER-dependent enhancement of NCX4040 cytotoxicity (Figure 7B).
3.7. RSL3 Enhances NCX4040 Cytotoxicity in HT-29 and HCT116 Cells
RSL3 (RAS-selective lethal 3) is a ferroptosis triggering agent that has been utilized extensively to induce ferroptosis in various tumor cells [17,36,37]. Therefore, effects of RSL3 on NCX4040 cytotoxicity was investigated and we found that it significantly enhanced NCX4040 cytotoxicity in both cells (Figure 8A,B). Again, HCT116 cells were more sensitive to RSL3-induced NCX4040-mediated cytotoxicity.
3.8. Metabolomic Studies in HT-29 and HCT116 cells
In order to further understand the mechanisms of NCX4040-induced ferroptosis in CRC cells we used untargeted metabolomics and examined effects of NCX4040 on cellular glutathione levels, lipid metabolism, and differential energy metabolism in HT-29 and HCT116 cells.
3.8.1. NCX4040 Increases Glutathione in HT-29 and HCT116 Cells
Our metabolomic studies show significant increases in glutathione (GSH) following NCX4040 treatment of CRC cells (Figure 9). GSH levels were elevated at 24h in both cells (Figure 10). The ratio of GSH to glutathione disulfide (GSSG), the oxidized form, remained similar indicative of the cellular ability to maintain redox potential. Further, taurine, a reported antioxidant and anti-inflammatory mediator was increased after 24h (Figure 9C), indicating NCX4040-induced oxidative stress in CRC cells.
3.9.2. NCX4040 Enhances Lipid Metabolism in HT-29 and HCT116 Cells
Lipid metabolism is a complex biochemical process that is involved in the regulation of cell survival and death, including ferroptosis [38]. Metabolomic studies indicated that NCX4040 treatment significantly increased lipid metabolism in these CRC cells (Figure 11). Acylcarnitines were observed to be statistically different between control and 24h samples in both cells. The acyl chain composition is reflective of energy metabolism patterns, including fatty acid oxidation which may result in formation of lipid radicals during beta oxidation, leading to increased cells death via ferroptosis.
The short-chain acylcarnitines (Figure 10A–C) increased after 24h in HT-29 and HCT116 cells. Increased levels suggest that energy production from glucose, amino acids, or fatty acid degradation is increased after treatment. The dicarboxylic acid conjugate, succinyl-carnitine (Figure 10D), reflects the selective patterns observed in carnitines. In contrast, the long-chain acylcarnitines (Figure 10E–H) decreased in 24h samples. The observed decrease may reflect alteration in fatty acid metabolism as the mitochondria are the primary location of synthesis and metabolism of long-chain acylcarnitines.
3.9.3. NCX4040 Enhances Energy Metabolism in HT-29 and HCT116 Cells
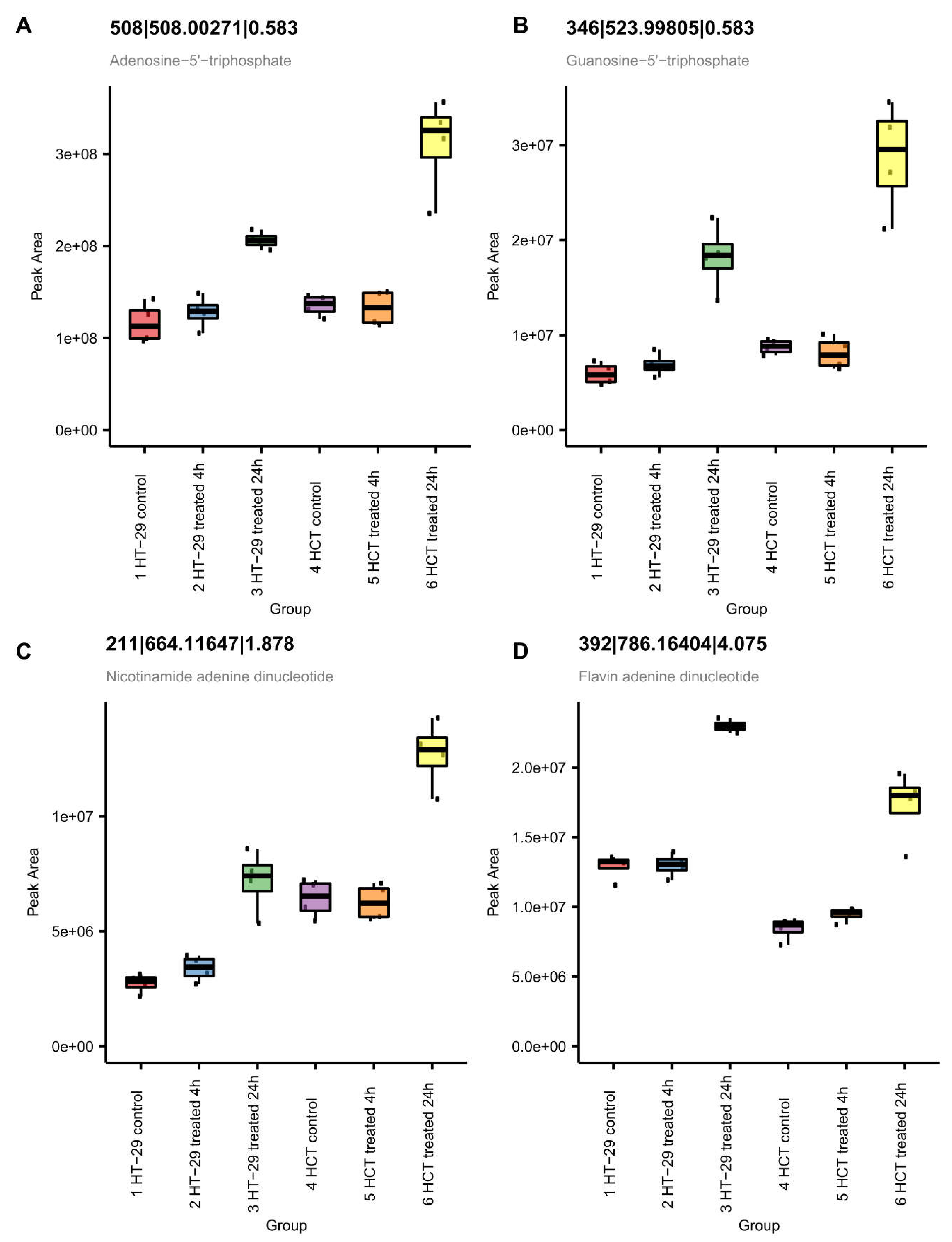
Metabolomic studies also show significant increases in ATP and cellular respiration co-factors, NAD+ and FAD+ (Figure 13) in both cells at 24 h. This observation suggest increases in energy production related to cell survival. The observed increase in energy production may reflect increased DNA/RNA repair of oxidative damage induced by NCX404 as supported by the observed increase in GTP, a building block of RNA and DNA (Figure 12).
4. Discussion
Ferroptosis is induced via iron-dependent lipid peroxidation following cellular formation of ROS [11,12]. While the mechanism of ferroptosis is under active investigation, it is different than those of other cell death processes, such as necrosis, autophagy, and apoptosis. The cellular death resulting from ferroptosis has been shown to arise from the inhibition of glutathione peroxidase 4 (GPX4) and accumulation of intracellular lipid hydroperoxides (LOOH), resulting in damage to cellular membranes (lipid peroxidation) in the presence of iron [11,12,16]. The damaging species is the reactive ●OH, formed from the reaction of H2O2 with Fe2+ (the Fenton reaction). Several small molecules, e.g., ER and RSL3, have been reported to induce ferroptosis in tumor cells [33,34,36,37]. Several pathways are now been reported for ER-induced ferroptosis, including inhibition of the system XC– (glutamate/cystine antiporter) [35], inhibition of the mitochondria bound voltage-dependent anion channel (VDAC) [33], and modulation of the tumor suppressor p53 gene [40,41]. System XC– is a transmembrane cystine–glutamate antiporter that specifically imports extracellular L-cystine into cells in exchange for glutamate. Cystine, disulfide form of cysteine, is imported into the cell by system XC– and is reduced to cysteine, which is the key intermediate for the synthesis of glutathione (GSH), an important cellular antioxidant. Thus, the inhibition of the system XC– by ER results in the depletion of the cellular GSH, leading to oxidative stress and ferroptosis-mediated cell death [12,42]. RSL3 is a potent ferroptosis-triggering agent which inhibits GPX4, thereby promoting ferroptosis, including in CRC cells [36]. RSL3 can kill RAS mutant cancer cells and activate iron-dependent, nonapoptotic cell death of RAS mutant cancer cells [43]. Increase in GPX4 in tumor cells has been shown to mediate suppression of ferroptosis [16,44].
However, ferroptosis has not been investigated in the treatment of CRC in the clinic. Therefore, exploitation of ferroptosis for killing of CRC cancer cells in response to NCX4040 needs elucidation for therapy. Herein, we have investigated the role of ferroptosis in the mechanism of NCX4040-dependent cell death in HT-29 and HCT116, a Ras mutated (codon 13) colon tumor cells. We utilized various inducers and inhibitors of ferroptosis to decipher the mechanisms of cell death. Our study showed that NCX4040 was equally cytotoxic to both CRC cells. Previous studies have shown that NCX4040 induces significant depletion of cellular GSH and generates ROS formation in tumor cells [6,7]. In this study, we found NCX4040 also generated ROS in HT-29 and HCT116 cells in both time- and dose-dependent manner. Less ROS was formed and detected at 2h than at 4h drug of treatment (not shown). Our studies also show that NCX4040 significantly increased lipid peroxidation in both HT-29 and HCT116 cells in a time- and dose-dependent manner. These observations suggest that NCX4040-generated ROS must have reacted with cellular lipids to form lipid peroxides in these tumor cells.
It should be noted that both the formation of ROS and increases in lipid peroxidation were independent of cellular GSH as we found no significant depletion of GSH in HT-29 and HCT116 cells by our mCBI flow cytometric detection method. In contrast, metabolomic studies indicated that treatment of CRC cells with NCX4040 results in increases in GSH following 24h drug treatment (Figure 11). We found that treatment with ER also failed to deplete GSH in these cells (data not shown, manuscript in preparation). These observations suggest that neither NCX4040 nor ER inhibits Xc-anti-transporter in HT-29 and HCT116 cells, indicating that other biochemical and metabolic changes besides cellular depletion of GSH are responsible for the induction of ferroptosis by NCX4040 in CRC cells.
Examination of effects of ER on the cytotoxicity of NCX4040 in HT-29 and HCT116 cells showed that ER is highly effective in enhancing NCX4040-dependent cell death in both CRC cells. More interestingly, we found that HCT116 cells are more sensitive to combinations of NCX4040 and ER (Figure 7). Similarly, RSL3 also significantly enhanced NCX4040-mediated cell death in both HT-29 and HCT116 cells. Again, HCT116 tumor cells are more sensitive to RSL3-NCX4040 combinations than HT-29 cells (Figure 8). In contrast, FES, a strong inhibitor of ferroptosis, significantly inhibited NCX4040-induced cells death in both HT-29 and HCT116 tumor cells (Figure 6). Again, we found that FES was more effective in inhibiting NCX4040-dependent cell killing in HCT116 cells. These observations, e.g., inhibition of NCX4040-dependent killing by FES and enhanced or synergistic killing of HT-29 and HCT116 cells by ER-NCX4040 and RSL3-NCX4040 combinations clearly suggest that NCX4040 induces ferroptosis in HT-29 and HCT116 cells. Furthermore, HCT-116 cells are more sensitive to ferroptosis-dependent cell death than HT-29 cells. While the reasons for this is not clear, this may be due to the fact HCT116 cells contain a KRAS-mutation and KRAS-mutant cells are more sensitive to both ER- and RSL3-induced ferroptosis [45]. Furthermore, HCT116 cells contain WTp53 while HT-29 cells contain mutantp53. Ferroptosis has been suggested to require WTp53 activity [41]. In addition, recent reports suggest the degree of FAM193A expression is a widespread enabler of p53 activity in cell death [46] which may relate to differential ferroptosis sensitivity of HT-29 and HCT116.
While mechanisms of NCX4040-induced ferroptosis is not completely clear, however, some reasonable conclusions can be made from our studies. First, NCX4040-induced ferroptosis-mediated tumor cell death is ROS-dependent as there was significant increase in ROS formation in both HT-29 and HCT116 cells. Formation of ROS in these cells are also confirmed by our RT-PCR studies showing significant increases in HMOX1/OX1, a hallmark of oxidative stress in cells. Furthermore, both SOD2 and NOX4 were induced indicating superoxide anion radical (O2-●) are formed in these cells. Our studies also show that lipid peroxides are generated in these cells as lipid peroxidation was enhanced in the presence of NCX4040. Lipid peroxidation has been suggested to trigger ferroptosis. Lipid peroxides/lipid hydroperoxides (L-OOH) are known to cause damage to the cellular plasma membrane due to the accelerated oxidation of the membrane lipids. Furthermore, increases in the concentrations of lipid peroxides can induce damage to nucleic acids and proteins from toxic aldehydes formed from the oxidation of lipids, generating additional toxicity and inducing cell death by ferroptosis [15].
Our studies showed that combined treatment of NCX4040 and RSL3 increased the death of both HT-29 and HCT116 CRC cells, due to the accumulation of cytotoxic lipid peroxidation products. RSL3, an inhibitor of GPX4 [16,17,36], enhanced cell death further confirms that lipid peroxides are responsible for cell death in HT-29 and HCT116 cells. Our study also shows that GPX4 transcripts were significantly enhanced in both HT-29 and HCT116 cells. GPX4 utilizes GSH as a cofactor to eliminate hydroperoxides and our metabolomic studies showed that cellular GSH was increased by NCX4040 in these cells.
It should be noted that CHAC1 gene (and proteins) was significantly enhanced in both cells, albeit significantly more in HCT116 cells. CHAC1, in addition to NOX4 and GPX4, is considered to be a hallmark of ferroptosis and has been reported to be involved in tumor cell death via induction of ferroptosis [12,47,48,49]. While CHAC1 is known to hydrolyze glutathione, leading to depletion of GSH in tumor cells, our study did not show depletion of GSH in either cell lines. However, CHAC1 is also known to induce ER stress via ATF4-CHOP-CHAC1 pathway, leading to ferroptosis [10,48].
Proteins levels of ferroptosis-associated CHAC1, GPX4 and NOX4 genes were evaluated using the Western blot methods (data not shown). CHAC1 protein was induced following NCX4040 in both cell lines, significantly more in HCT116 cells. Similarly, GPX4 proteins levels were also induced by NCX4040; however, protein levels of NOX4 remained unchanged in both HT-29 and HCT116 cells. While proteins expressions did show increases following treatment with NCX4040, there were no accord between transcript expressions with protein expressions. This may be due to differences in stability/half-life of transcripts compared to proteins. It is possible then that the half-lives of proteins are shorter due to rapid they degradation or turnover. Furthermore, it is also possible that these proteins are S-nitrosylated by ●NO/●NO-derived species formed from NCX4040 in cells, resulting in post-translation modifications, including decreased stability of the proteins as reported by us as well as others [50,51,52].
Our studies also show that various inflammatory response genes, e.g., COX2, VEGF and NF-kB were also modulated by NCX4040 in these CRC cells. These observations are consistent with our findings with NCX4040 in human ovarian cells [6]. While the exact roles of these genes in NCX4040-induced ferroptosis is not known at this time, several studies indicate that COX2 and VEGF genes are involved in the process of ferroptosis [53,54,55,56]. COX2 has been found to be increased during ferroptosis and has been suggested to be a biomarker as inhibitors of COX2 enzyme failed to modulate ferroptosis [57].
Finally, the inhibition of the mitochondria bound voltage-dependent anion channel (VDAC) has been suggested to play an important role in the mechanism of ferroptosis. Impairment of mitochondrial functions can increase sensitivity of anticancer drug of cancer cells. VDAC is an ion channel located in the outer mitochondrial membrane where it mediates and controls molecular and ion exchange between the mitochondria and the cytoplasm. The permeability of VDAC can be altered by drugs, causing mitochondrial metabolic disfunction, ROS production, and oxidative stress-mediated death. Yagoda et al [33] reported that ER can change the permeability of the mitochondrial outer membrane and that VDAC is the target of ER. ER was shown to reverse tubulin’s inhibition of VDAC in vitro and in vivo, allowing VDAC to open [33]. Opening of VDAC leads to various biological effects, including an increase of mitochondrial metabolism (the increase of Δψ), a decrease in glycolysis (anti-Warburg effect) and an increase in ROS production and oxidative stress [58]. The anti-Warburg action can lead to damage to cancer cells and decrease in cell proliferation. In addition, ER can hyperpolarize mitochondria in cancer cells, which is followed by rapid depolarization, resulting in mitochondrial dysfunction. ●NO, delivered via NO-donors, has been reported to directly inhibit VDAC functions [59]. While effects of NCX4040 on VDAC were not examined here, it is very likely that ●NO formed from NCX4040 inhibits/interferes with VDAC functions of CRC cells, generating ROS (as observed here) without effecting cellular GSH status. Increased/synergistic CRC cell death by the combinations of NCX4040 and ER may result from this combined effect of both agents on VDAC.
5. Conclusions
Studies presented here show that NCX4040 induces formation of ROS in HT-29 and HCT116 colon cancer cells without significantly effecting cellular GSH. NCX4040 treatment of these tumor cells resulted in increases in lipid peroxidation. Combinations of ferroptosis inducers, erastin and ESL3, significantly enhanced NCX4040 cytotoxicity while ferrostatin-1, an inhibitor of ferroptosis, significantly inhibited NCX4040 cytotoxicity in CRC cells. Our studies also showed that treatment of HT-29 and HCT116 cells resulted in significant modulations of CHAC1, GPX4, NOX4 and COX2 genes, biomarkers of ferroptosis. These events, taken together, strongly suggest that NCX4040 induces ferroptosis in CRC cells. Combinations of erastin or RSL3 with NCX4040 may provide better treatment modality for CRC in the clinic.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Table S1.
Author Contributions
Conceptualization and Design, Birandra K. Sinha, Methodology, BKS, CDB, AKJ, EJT, CM, XW, HW and REC, Writing—original draft preparation, BKS, Editing, BKS, AKJ, and REC.
Funding
This research was supported by the intramural research program (grant numbers ZIA E505013922, ZIA BC 0011476, and ZIC ES103363-03) of the National Institute of Environmental Health Sciences, NIH. Statements contained herein do not necessarily represent the statements, opinions, or conclusions of NIEHS, NIH, or the US Government.
Acknowledgments
We thank Drs. Michael Fessler and B. Alex Merrick for critical review of the manuscript.
Conflicts of Interest
The authors declare no actual or potential conflicts of interest.
Abbreviations
NO, nitric oxide; NCX4040, 2-(acetyl)benzoic acid 4-(nitroxy; ethyl) phenyl ester; ROS, reactive oxygen species; RNS, reactive nitrogen species CRC, Colorectal Cancer; FES, Ferrostatin-1; ER, Erastin, 2-[1-[4-[2-(4-Chloroph; noxy)acetyl]-1-piperazinyl]ethyl]-3-(2-ethoxyphenyl)-4(3H)-quinazolinone; RSL3, RAS-selective lethal 3; MDA, Malondialdehyde; OGG1, 8-oxoguanine DNA glycosylase1; HMOX1/OX1; Heme Oxygenase 1; CHAC1, Glutathione Specific Gamma-Glutamylcyclotransferase 1; NOX4, NADPH Oxidase 4; GPX4, Glutathione Peroxidase 4; COX2, Cyclooxygenase 2; RT-PCR, real-time polymerase chain reaction.
References
- Averboukh, F.; Ziv, Y.; Kariv, Y.; Zmora, O.; Dotan, I.; Klausner, J.M.; Rabau, M.; Tulchinsky, H. Colorectal carcinoma in inflammatory bowel disease: a comparison between Crohn’s and ulcerative colitis. Colorectal Dis 2011, 13, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Z.; Sun, W.; Zhang, J.; Xu, Q.; Zhou, X.; Mao, L. Ferroptosis in colorectal cancer: Potential mechanisms and effective therapeutic targets. Biomed Pharmacother 2022, 153, 113524. [Google Scholar] [CrossRef]
- Rigas, B.; Kashfi, K. Nitric-oxide-donating NSAIDs as agents for cancer prevention. Trends Mol Med 2004, 10, 324–330. [Google Scholar] [CrossRef]
- Sinha, B.K.; Tokar, E.J.; Bortner, C.D. Molecular Mechanisms of Cytotoxicity of NCX4040, the Non-Steroidal Anti-Inflammatory NO-Donor, in Human Ovarian Cancer Cells. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Gao, J.; Liu, X.; Rigas, B. Nitric oxide-donating aspirin induces apoptosis in human colon cancer cells through induction of oxidative stress. Proc Natl Acad Sci U S A 2005, 102, 17207–17212. [Google Scholar] [CrossRef] [PubMed]
- Bratasz, A.; Selvendiran, K.; Wasowicz, T.; Bobko, A.; Khramtsov, V.V.; Ignarro, L.J.; Kuppusamy, P. NCX-4040, a nitric oxide-releasing aspirin, sensitizes drug-resistant human ovarian xenograft tumors to cisplatin by depletion of cellular thiols. J Transl Med 2008, 6, 9. [Google Scholar] [CrossRef]
- Dunlap, T.; Chandrasena, R.E.; Wang, Z.; Sinha, V.; Wang, Z.; Thatcher, G.R. Quinone formation as a chemoprevention strategy for hybrid drugs: balancing cytotoxicity and cytoprotection. Chemical research in toxicology 2007, 20, 1903–1912. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, N.; Chen, C.; Xu, X.; Luo, A.; Yan, Y.; Lu, Y.; Liu, J.; Ou, X.; Tan, Y.; et al. Sevoflurane Induces Ferroptosis of Glioma Cells Through Activating the ATF4-CHAC1 Pathway. Front Oncol 2022, 12, 859621. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim Biophys Acta Mol Cell Res 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem Biophys Res Commun 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol 2018, 9, 1371. [Google Scholar] [CrossRef]
- Tesei, A.; Zoli, W.; Fabbri, F.; Leonetti, C.; Rosetti, M.; Bolla, M.; Amadori, D.; Silvestrini, R. NCX 4040, an NO-donating acetylsalicylic acid derivative: efficacy and mechanisms of action in cancer cells. Nitric Oxide 2008, 19, 225–236. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: past, present and future. Cell Death Dis 2020, 11, 88. [Google Scholar] [CrossRef]
- Delvaux, M.; Hague, P.; Craciun, L.; Wozniak, A.; Demetter, P.; Schoffski, P.; Erneux, C.; Vanderwinden, J.M. Ferroptosis Induction and YAP Inhibition as New Therapeutic Targets in Gastrointestinal Stromal Tumors (GISTs). Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Wang, D.; Tang, L.; Zhang, Y.; Ge, G.; Jiang, X.; Mo, Y.; Wu, P.; Deng, X.; Li, L.; Zuo, S.; et al. Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis 2022, 13, 544. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Trush, M.A.; Gram, T.E. Stimulation by adriamycin of rat heart and liver microsomal NADPH-dependent lipid peroxidation. Biochemical pharmacology 1981, 30, 2797–2804. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Kennedy, K.A.; Trush, M.A.; Sinha, B.K. Adriamycin-enhanced membrane lipid peroxidation in isolated rat nuclei. Cancer research 1985, 45, 3296–3304. [Google Scholar]
- Zielonka, J.; Kalyanaraman, B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic Biol Med 2010, 48, 983–1001. [Google Scholar] [CrossRef]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React Oxyg Species (Apex) 2016, 2, 361–370. [Google Scholar] [CrossRef]
- Sinha, B.K.; Tokar, E.J.; Li, J.; Bushel, P.R. Gene Expression Profiling Elucidates Cellular Responses to NCX4040 in Human Ovarian Tumor Cells: Implications in the Mechanisms of Action of NCX4040. Cancers (Basel) 2022, 15. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, Y.; Zheng, T.; Yang, G.; Zhang, X.; Sun, Z.; Shi, C.; Zhao, S. Inhibition of heme oxygenase-1 enhances anti-cancer effects of arsenic trioxide on glioma cells. J Neurooncol 2011, 104, 449–458. [Google Scholar] [CrossRef]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol 2015, 2, e1054549. [Google Scholar] [CrossRef]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J Biol Chem 2015, 290, 15878–15891. [Google Scholar] [CrossRef]
- Chu, J.; Liu, C.X.; Song, R.; Li, Q.L. Ferrostatin-1 protects HT-22 cells from oxidative toxicity. Neural Regen Res 2020, 15, 528–536. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vuckovic, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol 2020, 28, 101328. [Google Scholar] [CrossRef]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett 2020, 25, 10. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Li, S.; He, Y.; Chen, K.; Sun, J.; Zhang, L.; He, Y.; Yu, H.; Li, Q. RSL3 Drives Ferroptosis through NF-kappaB Pathway Activation and GPX4 Depletion in Glioblastoma. Oxid Med Cell Longev 2021, 2021, 2915019. [Google Scholar] [CrossRef]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther 2020, 5, 108. [Google Scholar] [CrossRef]
- Lee, J.Y.; Nam, M.; Son, H.Y.; Hyun, K.; Jang, S.Y.; Kim, J.W.; Kim, M.W.; Jung, Y.; Jang, E.; Yoon, S.J.; et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc Natl Acad Sci U S A 2020, 117, 32433–32442. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med 2019, 133, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis 2021, 12, 1079. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol Life Sci 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Szwarc, M.M.; Guarnieri, A.L.; Joshi, M.; Duc, H.N.; Laird, M.C.; Pandey, A.; Khanal, S.; Dohm, E.; Bui, A.K.; Sullivan, K.D.; et al. FAM193A is a positive regulator of p53 activity. Cell Rep 2023, 42, 112230. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Wang, S.F.; Hsu, C.Y.; Yin, P.H.; Yeh, T.S.; Lee, H.C.; Tseng, L.M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef]
- Wang, N.; Zeng, G.Z.; Yin, J.L.; Bian, Z.X. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s Lymphoma. Biochem Biophys Res Commun 2019, 519, 533–539. [Google Scholar] [CrossRef]
- Xiao, R.; Wang, S.; Guo, J.; Liu, S.; Ding, A.; Wang, G.; Li, W.; Zhang, Y.; Bian, X.; Zhao, S.; et al. Ferroptosis-related gene NOX4, CHAC1 and HIF1A are valid biomarkers for stomach adenocarcinoma. J Cell Mol Med 2022, 26, 1183–1193. [Google Scholar] [CrossRef]
- Iwakiri, Y. S-nitrosylation of proteins: a new insight into endothelial cell function regulated by eNOS-derived NO. Nitric Oxide 2011, 25, 95–101. [Google Scholar] [CrossRef]
- Martinez-Ruiz, A.; Araujo, I.M.; Izquierdo-Alvarez, A.; Hernansanz-Agustin, P.; Lamas, S.; Serrador, J.M. Specificity in S-nitrosylation: a short-range mechanism for NO signaling? Antioxid Redox Signal 2013, 19, 1220–1235. [Google Scholar] [CrossRef]
- Sharma, N.K.; Kumar, A.; Kumari, A.; Tokar, E.J.; Waalkes, M.P.; Bortner, C.D.; Williams, J.; Ehrenshaft, M.; Mason, R.P.; Sinha, B.K. Nitric Oxide Down-Regulates Topoisomerase I and Induces Camptothecin Resistance in Human Breast MCF-7 Tumor Cells. PloS one 2015, 10, e0141897. [Google Scholar] [CrossRef]
- Tan, H.; Chen, J.; Li, Y.; Li, Y.; Zhong, Y.; Li, G.; Liu, L.; Li, Y. Glabridin, a bioactive component of licorice, ameliorates diabetic nephropathy by regulating ferroptosis and the VEGF/Akt/ERK pathways. Mol Med 2022, 28, 58. [Google Scholar] [CrossRef]
- Zuo, Y.; Xie, J.; Li, X.; Li, Y.; Thirupathi, A.; Zhang, J.; Yu, P.; Gao, G.; Chang, Y.; Shi, Z. Ferritinophagy-Mediated Ferroptosis Involved in Paraquat-Induced Neurotoxicity of Dopaminergic Neurons: Implication for Neurotoxicity in PD. Oxid Med Cell Longev 2021, 2021, 9961628. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Gao, M.; Liang, J.; Chen, Y.; Wang, Y.; Wang, Y.; Xiao, Y.; Zhao, Z.; Wan, X.; Jiang, M.; et al. SLC7A11 Reduces Laser-Induced Choroidal Neovascularization by Inhibiting RPE Ferroptosis and VEGF Production. Front Cell Dev Biol 2021, 9, 639851. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, Y.; Zhao, H. Identification of ferroptosis-related genes and predicted overall survival in patients with burns. Front Surg 2022, 9, 1060036. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, J.; Wang, Y.; Liu, Z.; Wu, Y. Ferroptosis drives photoreceptor degeneration in mice with defects in all-trans-retinal clearance. J Biol Chem 2021, 296, 100187. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front Oncol 2017, 7, 154. [Google Scholar] [CrossRef]
- Cheng, Q.; Sedlic, F.; Pravdic, D.; Bosnjak, Z.J.; Kwok, W.M. Biphasic effect of nitric oxide on the cardiac voltage-dependent anion channel. FEBS Lett 2011, 585, 328–334. [Google Scholar] [CrossRef]
Figure 1.
Cytotoxicity of NCX4040 in HT-29 (A) and HCT116 (B) following 48 and 72h drug exposures, respectively. Plot of cytotoxicity curves of NCX4040 in HT-29 and HCT116 showing similar cytotoxicity(C). ** p values < 0.005 compared to untreated control.
Figure 1.
Cytotoxicity of NCX4040 in HT-29 (A) and HCT116 (B) following 48 and 72h drug exposures, respectively. Plot of cytotoxicity curves of NCX4040 in HT-29 and HCT116 showing similar cytotoxicity(C). ** p values < 0.005 compared to untreated control.
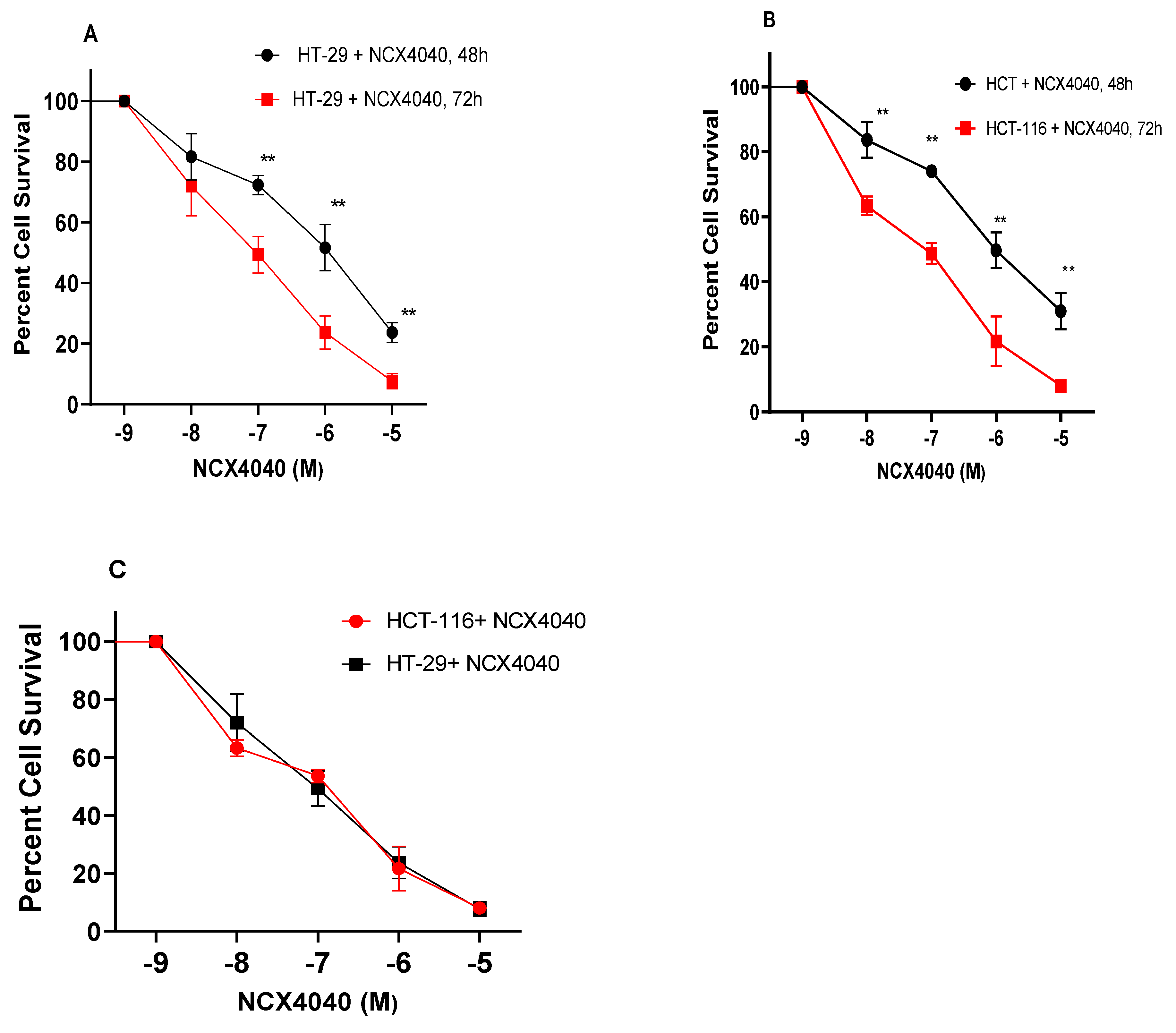
Figure 2.
Dose-dependent ROS formation as detected by mitosox from NCX4040 in HT-29 and HCT116 cells following 4h treatment. *, and *** p values < 0.05, and 0.001 respectively compared to untreated control.
Figure 2.
Dose-dependent ROS formation as detected by mitosox from NCX4040 in HT-29 and HCT116 cells following 4h treatment. *, and *** p values < 0.05, and 0.001 respectively compared to untreated control.
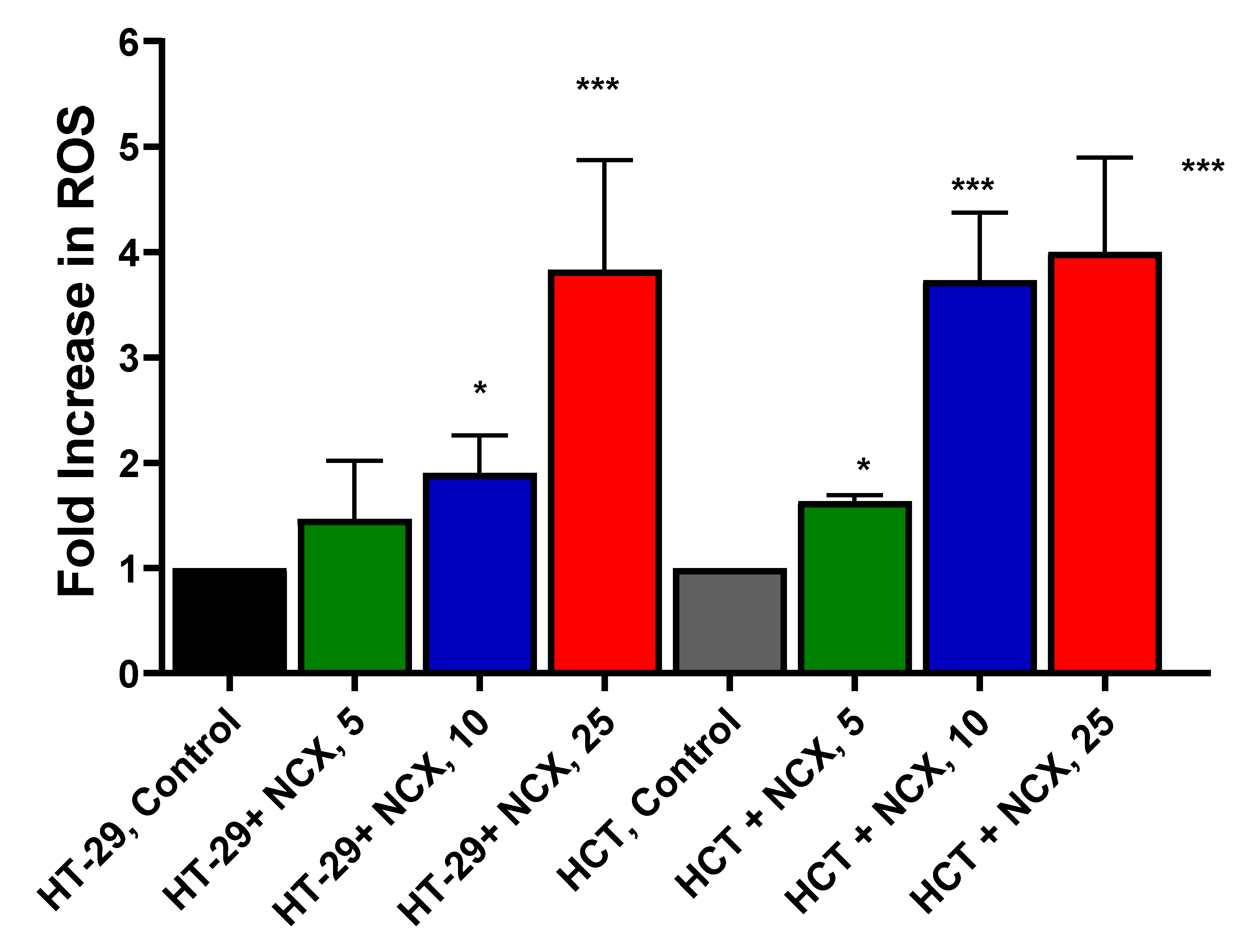
Figure 3.
Dose- and time-dependence of NCX4040-induced lipid peroxidation in HT-29 (A) and HCT116 (B). MDA formation was measured at 532mM. *, ** and *** p values < 0.05, 0.005 and 0.001 respectively compared to untreated control.
Figure 3.
Dose- and time-dependence of NCX4040-induced lipid peroxidation in HT-29 (A) and HCT116 (B). MDA formation was measured at 532mM. *, ** and *** p values < 0.05, 0.005 and 0.001 respectively compared to untreated control.
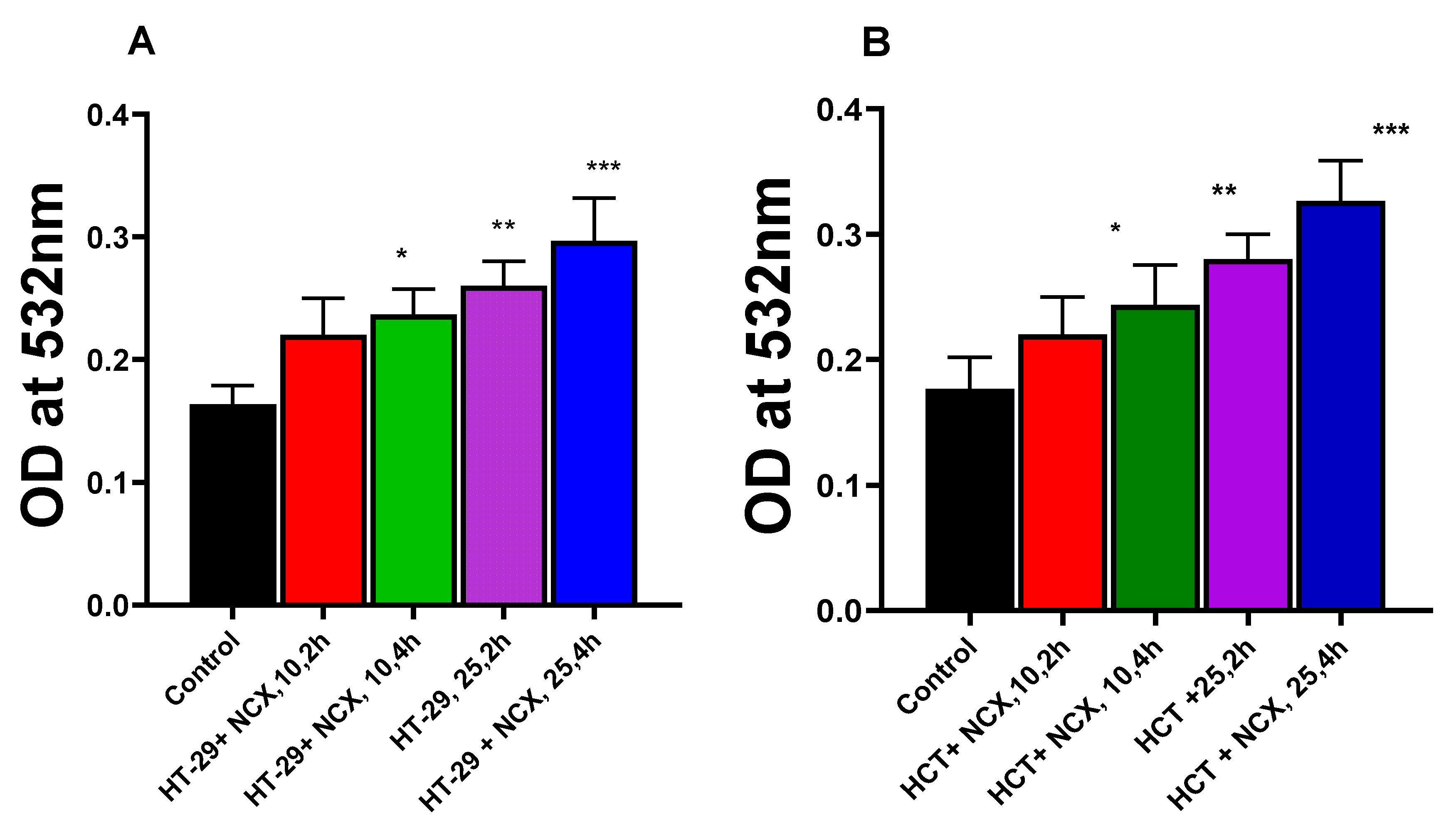
Figure 4.
Effects of NCX4040 (5µM) on various Oxidative stress related genes in HT-29 (A) and HCT116 (B) cells following treatment with NCX4040 for 4 and 24h. *, ** and *** p values < 0.05, 0.005 and 0.001 respectively compared to control (β-Actin at 4h and 24h, respectively) .
Figure 4.
Effects of NCX4040 (5µM) on various Oxidative stress related genes in HT-29 (A) and HCT116 (B) cells following treatment with NCX4040 for 4 and 24h. *, ** and *** p values < 0.05, 0.005 and 0.001 respectively compared to control (β-Actin at 4h and 24h, respectively) .
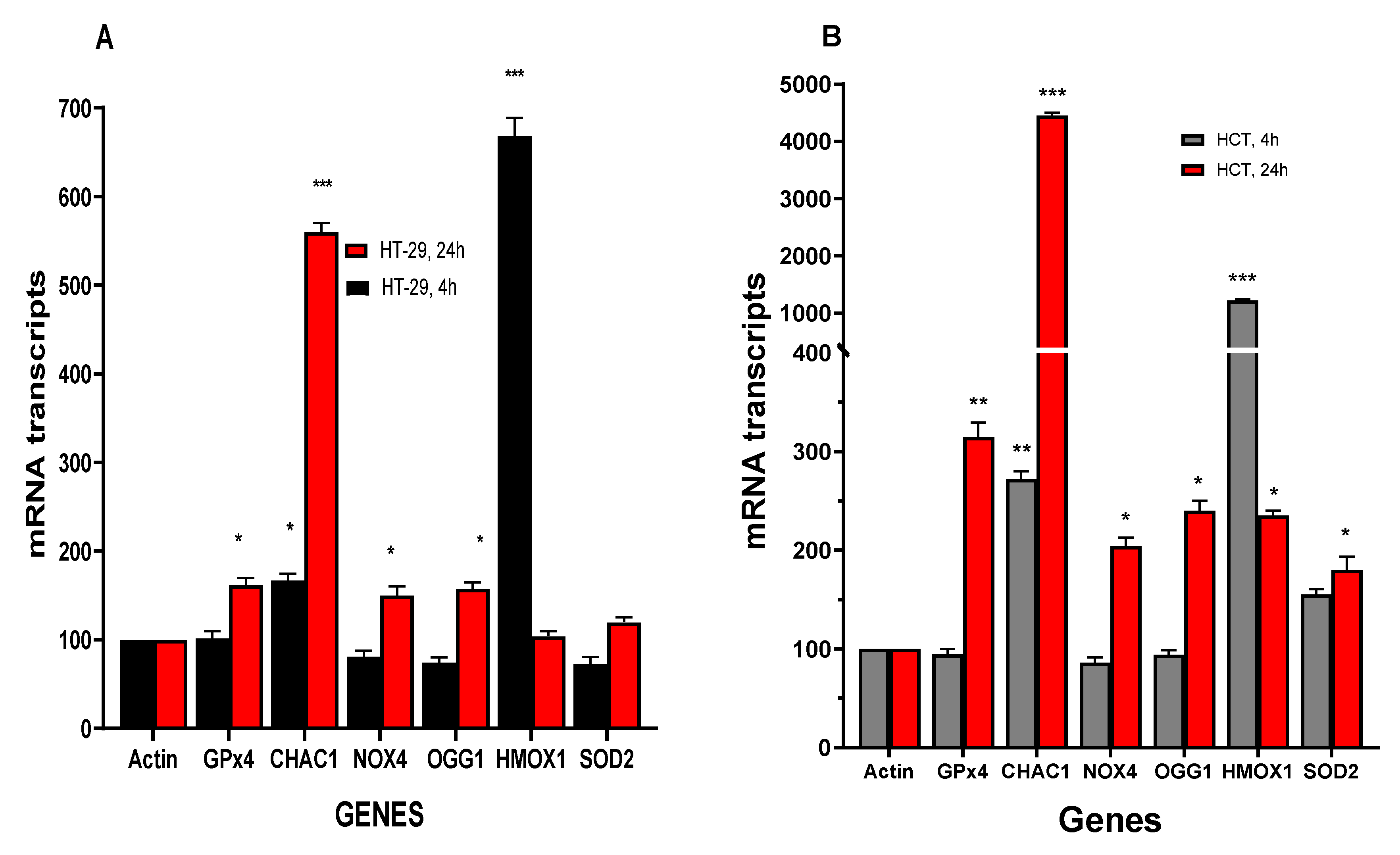
Figure 5.
Effects of NCX4040 (5µM) on various inflammatory response genes in HT-29 (A) and HCT116 (B) cells following treatment with NCX4040 for 4 and 24h. *, ** and *** p values < 0.05, 0.005 and 0.001 respectively compared to control (β-Actin at 4h and 24h, respectively).
Figure 5.
Effects of NCX4040 (5µM) on various inflammatory response genes in HT-29 (A) and HCT116 (B) cells following treatment with NCX4040 for 4 and 24h. *, ** and *** p values < 0.05, 0.005 and 0.001 respectively compared to control (β-Actin at 4h and 24h, respectively).
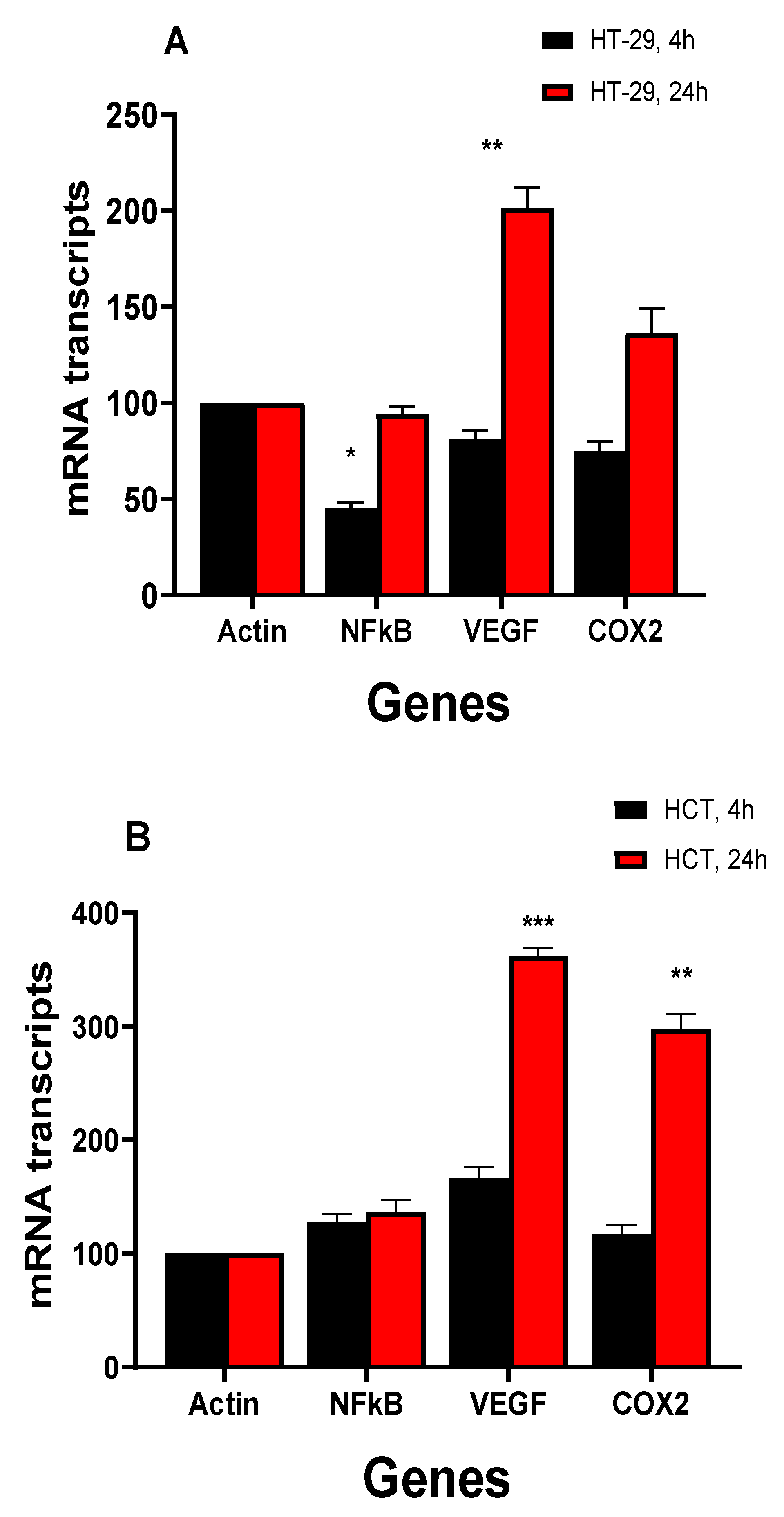
Figure 6.
Effects of ferrostatin-1 (2µM) on the cytotoxicity of NCX400 in HT-29 (A) and HCT116 (B) following 48h drug treatment. FES was pre-incubated with the cells for 1-2h before adding NCX4040. *, and ** p values < 0.05, 0.005, respectively compared treatment matched samples.
Figure 6.
Effects of ferrostatin-1 (2µM) on the cytotoxicity of NCX400 in HT-29 (A) and HCT116 (B) following 48h drug treatment. FES was pre-incubated with the cells for 1-2h before adding NCX4040. *, and ** p values < 0.05, 0.005, respectively compared treatment matched samples.
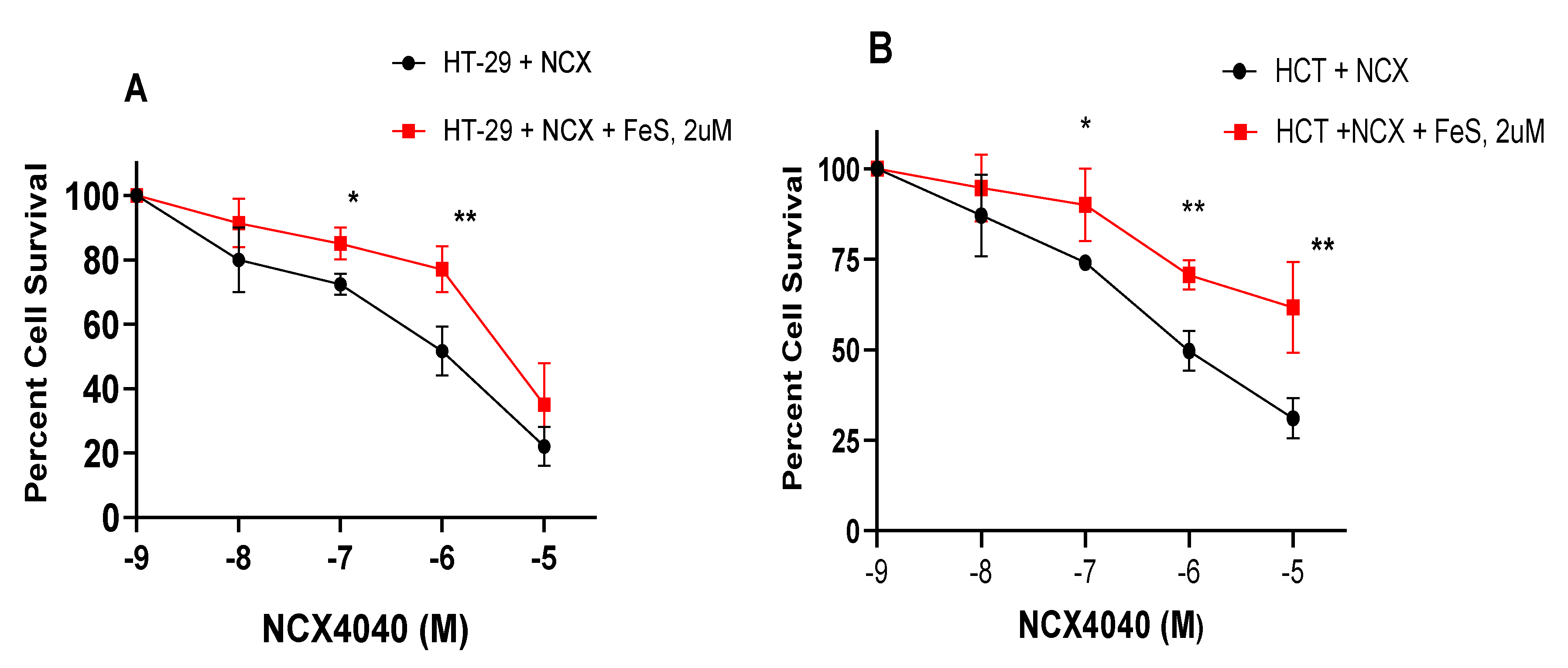
Figure 7.
Erastin-dependent enhancement of NCX4040 (10-6 M and 10-5 M) cytotoxicity in HT-29 (A) and HCT116 (B) tumor cells following 24h drug treatments. ER was pre-incubated with cells for 1-2 h before adding NCX4040. *, and $ (p values < 0.05) compared to the control alone. **, and $$ (p values < 0.005), compared to the control and 10-5 M NCX4040. $$$ (p values < 0.001) compared to 10-5 M NCX4040.
Figure 7.
Erastin-dependent enhancement of NCX4040 (10-6 M and 10-5 M) cytotoxicity in HT-29 (A) and HCT116 (B) tumor cells following 24h drug treatments. ER was pre-incubated with cells for 1-2 h before adding NCX4040. *, and $ (p values < 0.05) compared to the control alone. **, and $$ (p values < 0.005), compared to the control and 10-5 M NCX4040. $$$ (p values < 0.001) compared to 10-5 M NCX4040.
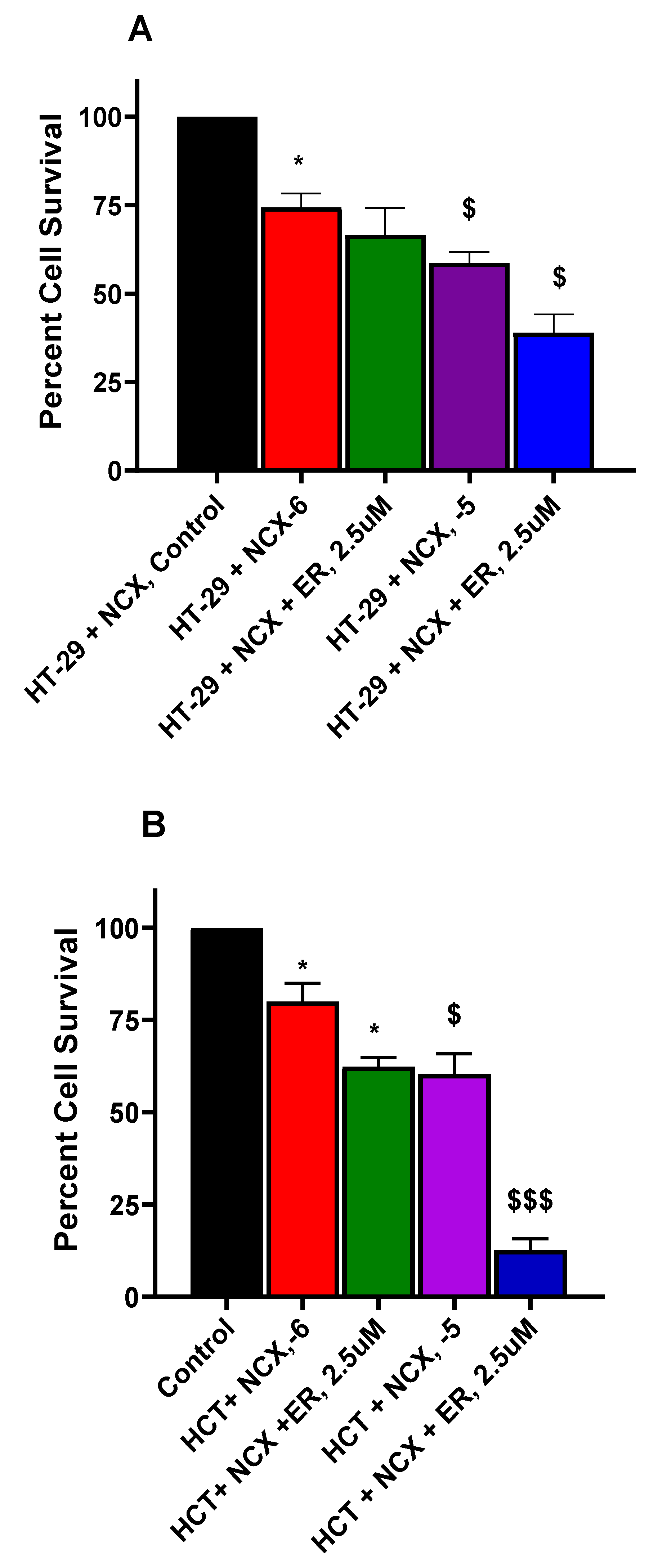
Figure 8.
RSL3-induced enhancement of NCX4040 ( 10-6 and 10-5 M) cytotoxicity in HT-29 (A) and HCT116 (B) tumor cells following 24h drug exposures. RSL3 was pre-incubated with cells for 1-2 h before adding NCX4040. * and $ (p values < 0.05) compared to the control and 10-6 M NCX4040. $$ (p values < 0.005) compared to 10-5 M NCX4040.
Figure 8.
RSL3-induced enhancement of NCX4040 ( 10-6 and 10-5 M) cytotoxicity in HT-29 (A) and HCT116 (B) tumor cells following 24h drug exposures. RSL3 was pre-incubated with cells for 1-2 h before adding NCX4040. * and $ (p values < 0.05) compared to the control and 10-6 M NCX4040. $$ (p values < 0.005) compared to 10-5 M NCX4040.
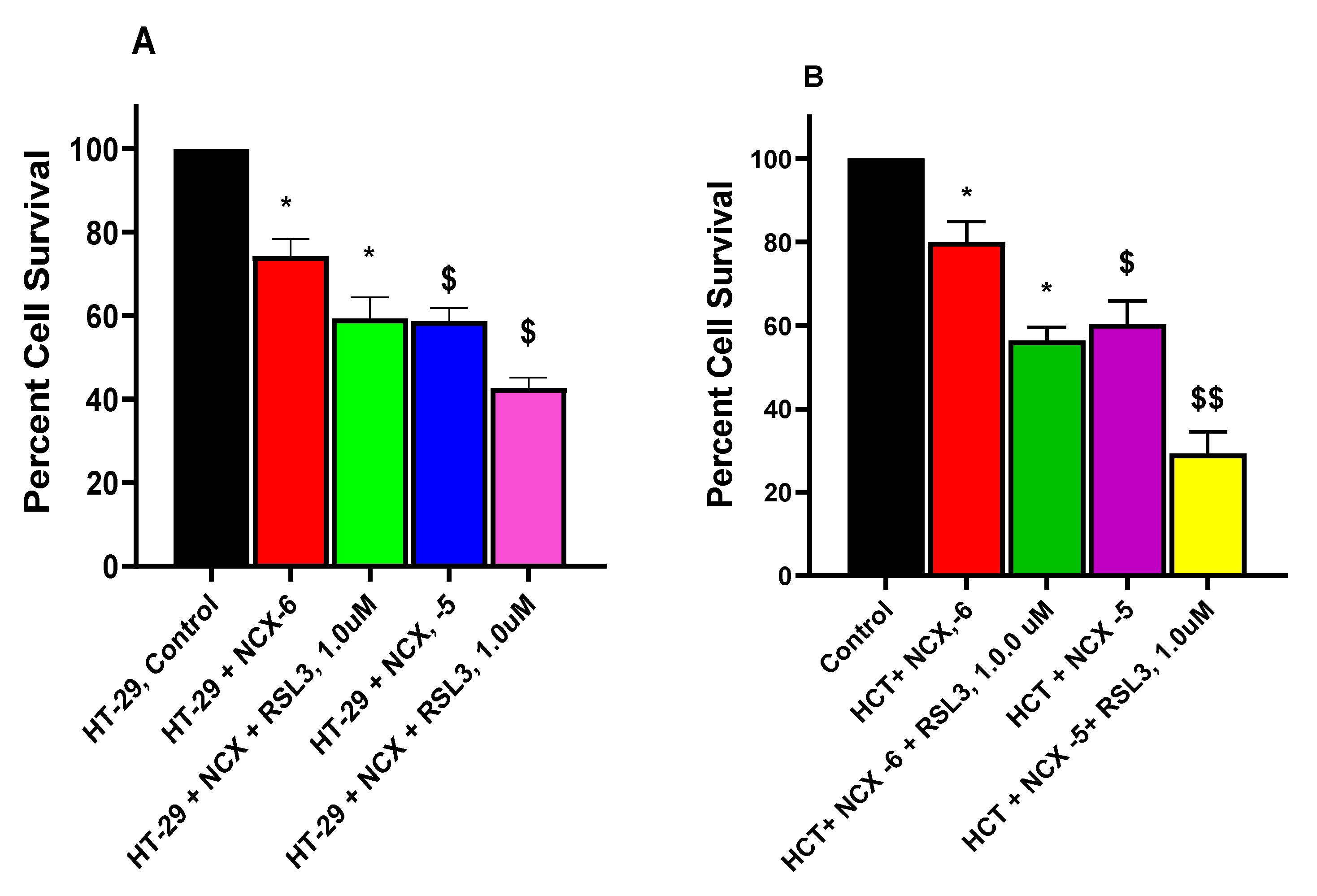
Figure 9.
Effects of NCX4040 on glutathione (A), ratio of GSH to GSSG (B), and taurine (C) in HT-29 and HCT116 cells with time.
Figure 9.
Effects of NCX4040 on glutathione (A), ratio of GSH to GSSG (B), and taurine (C) in HT-29 and HCT116 cells with time.
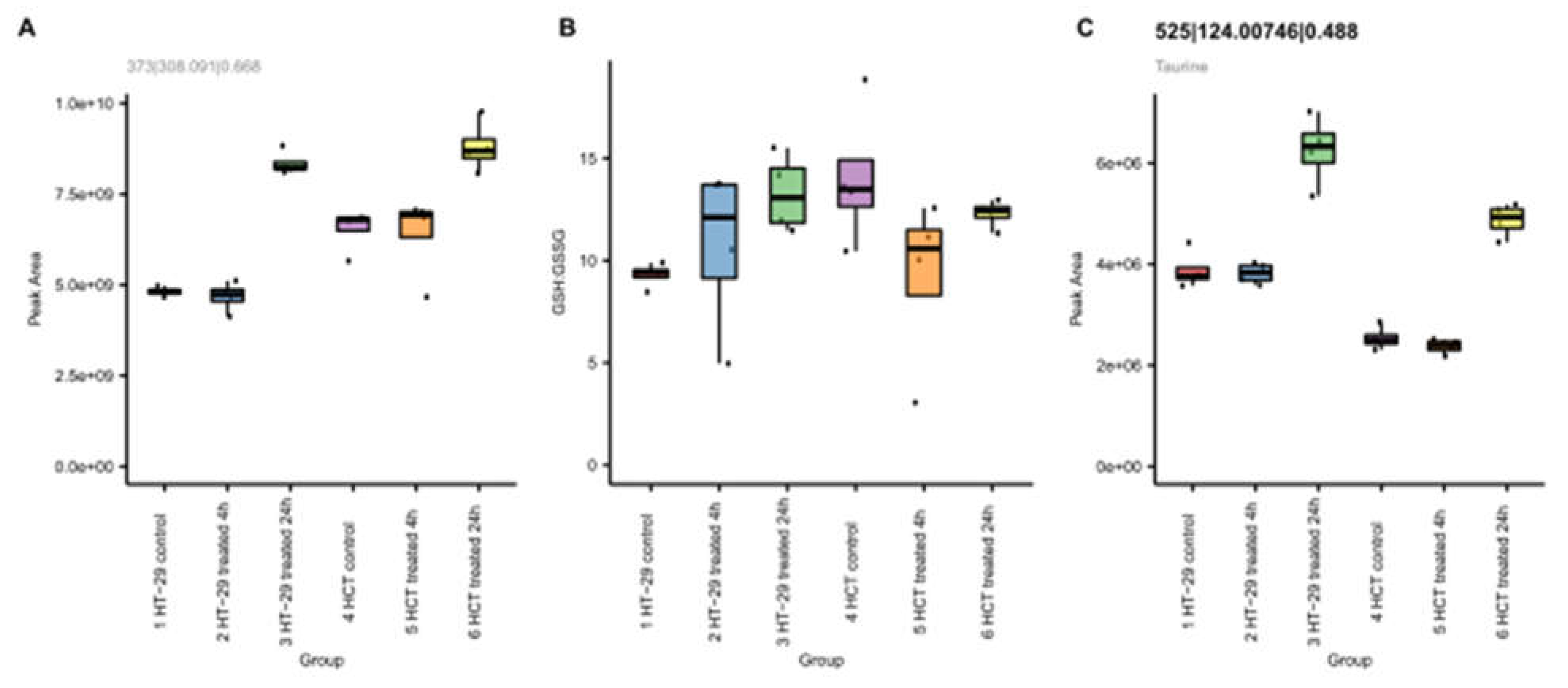
Figure 10.
Positive ionization mode data. Acylcarnitines with illustrative short and long acyl chains formation following NCX4040 treatment.
Figure 10.
Positive ionization mode data. Acylcarnitines with illustrative short and long acyl chains formation following NCX4040 treatment.
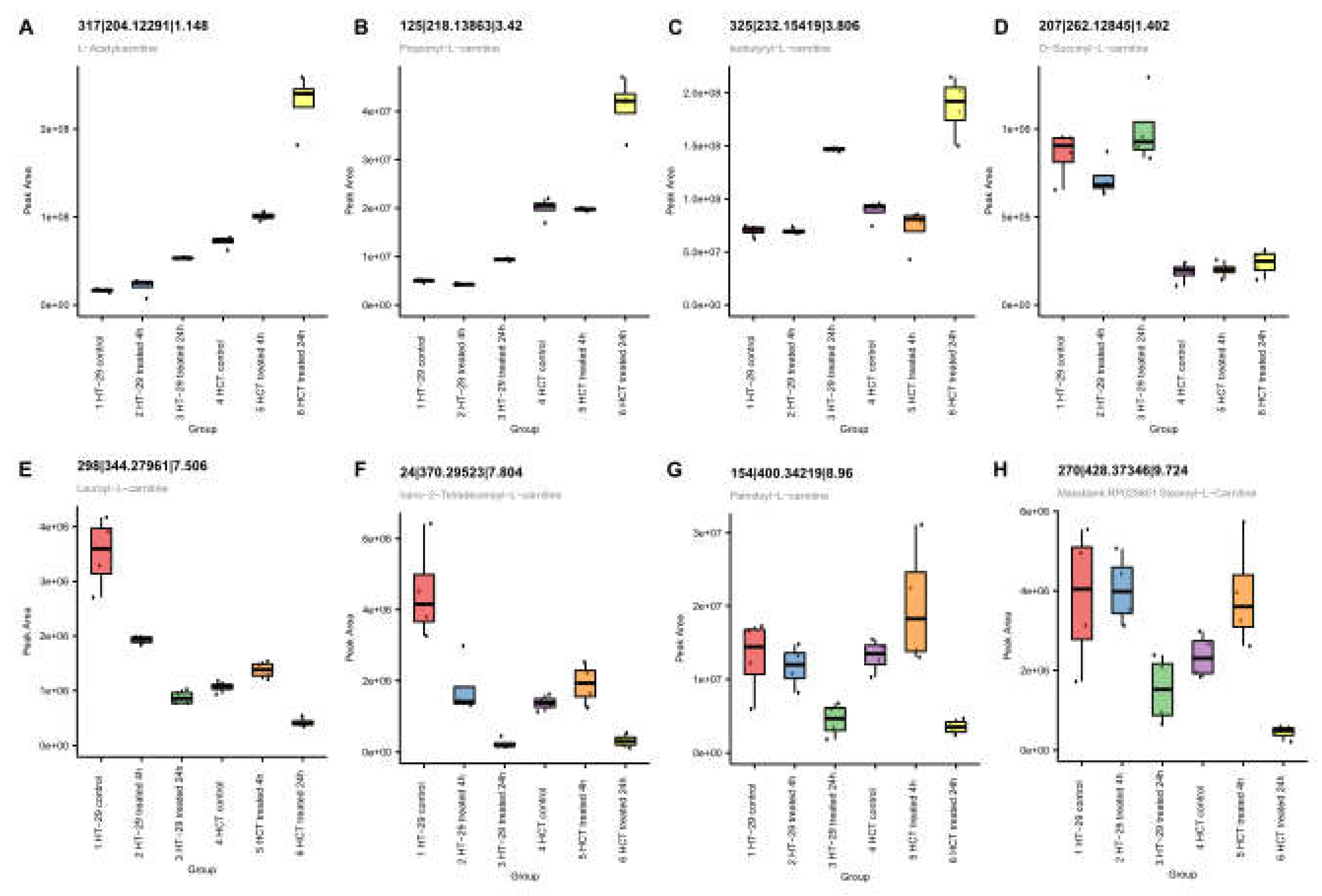
Figure 11.
Feature 147|303.23339|8.722 (negative mode data) is putatively annotated as arachidonic acid present in HT-29 and HCT116 cells following treatment with NCX4040.
Figure 11.
Feature 147|303.23339|8.722 (negative mode data) is putatively annotated as arachidonic acid present in HT-29 and HCT116 cells following treatment with NCX4040.
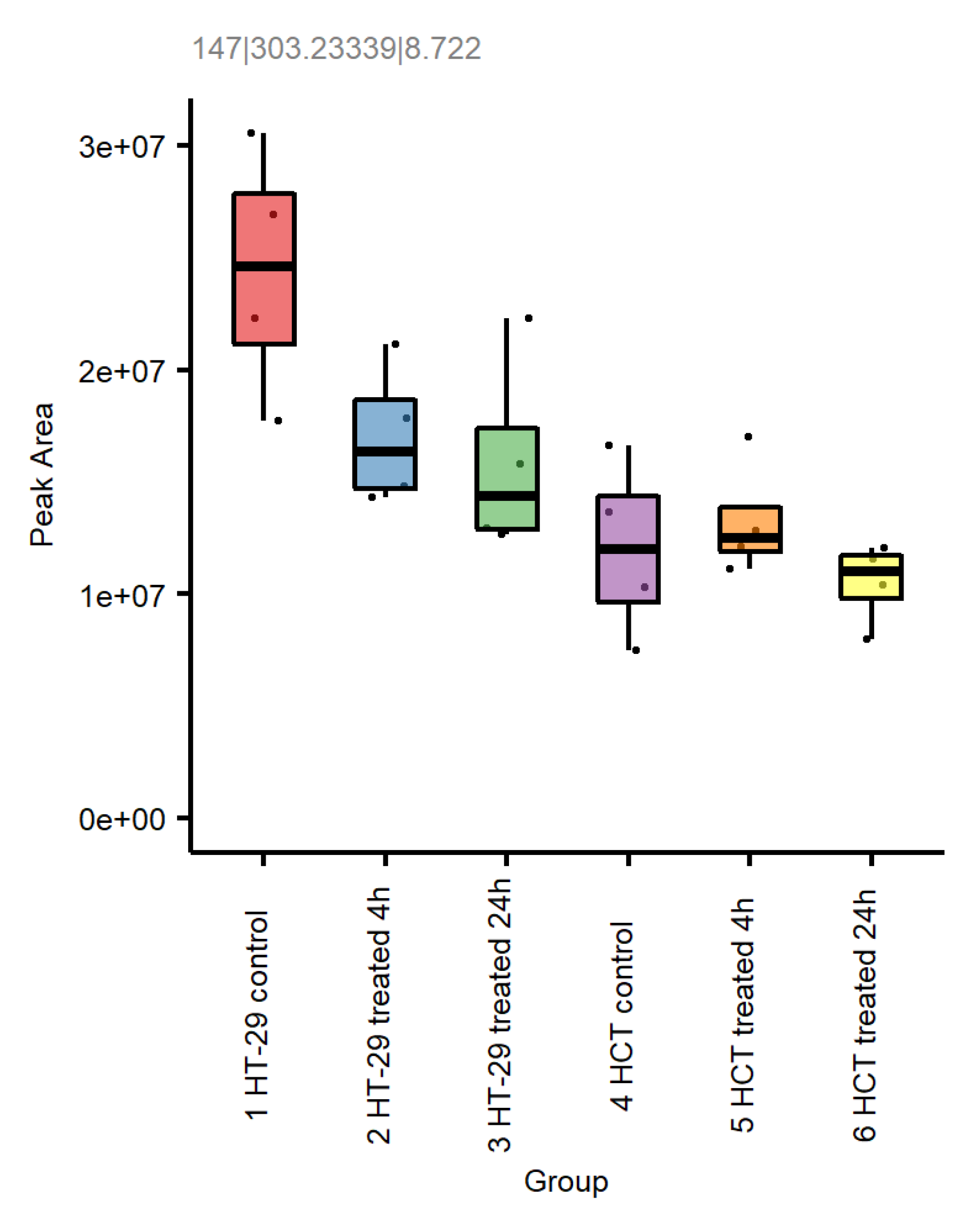
Figure 12.
Metabolomic measurement of ATP (A), NAD+ (B), and FAD+ (C) and (D) GTP in HT-29 and HCT116 cells following NCX4040 treatment.
Figure 12.
Metabolomic measurement of ATP (A), NAD+ (B), and FAD+ (C) and (D) GTP in HT-29 and HCT116 cells following NCX4040 treatment.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



