
Submitted:
26 May 2023
Posted:
29 May 2023
You are already at the latest version
Abstract
A high perovskite activity is sought for use in magnetic applications. In this paper, we present the simple synthesis of (2.5 and 5%) Tellurium-impregnated-LaCoO3 (Te-LCO), Te and LaCoO3 (LCO) by using a ball mill, chemical reduction, and hydrothermal synthesis, respectively. We also explored the structure stability along with the magnetic properties of Te-LCO. Te has a rhombohedral crystal structure, whereas Te-LCO has a hexagonal crystal system. The reconstructed Te was imbued with LCO that was produced by hydrothermal synthesis; as the concentration of the imbuing agent grew, the material became magnetically preferred. According to the X-ray photoelectron spectra, the oxidation state of the cobaltite is one that is magnetically advantageous. As a result of the fact that the creation of oxygen-deficient perovskites has been shown to influence the mixed (Te4+/2-) valence state of the incorporated samples, it is abundantly obvious that this process is of utmost significance. The TEM image confirms the inclusion of Te in LCO. The samples start out in a paramagnetic state (LCO), but when Te is added to the mixture, the magnetic state shifts to a weak ferromagnetic one. It is at this point that hysteresis occurs due to the presence of Te. Despite being doped with Mn in our prior study, rhombohedral LCO retains its paramagnetic characteristic at room temperature (RT). As a result, the purpose of this study was to determine the impacts of RT field dependency of magnetization (M-H) for Te-impregnated LCO in order to improve the magnetic properties of RT because it is a low-cost material for advanced multi-functional and energy applications.
Keywords:
Rhombohedral
; Hdtrothermal synthesis
; chemical reduction
; Oxygen deficiency
; Low spin state
; Ferromagnetism
1. Introduction
Perovskites are one of the most fascinating types of solid materials, exhibiting a wide range of physical events and characteristics. Extensive research has been conducted on ABO3-type perovskites with the general formula Ln1-xAxMO3 or LnBxM1-xO3(Ln-Lanthanides and M-dopant). Researchers have been intrigued by nano crystallite magnetic cobaltite due to its outstanding magnetic and electric capabilities for decades.Single-phase LaCoO3 perovskite is a good example of ceramic materials and is utilised in essential applications like ZrB2+10 wt.%SiC for leading edges and nose cones in hypersonic vehicles and LaCoO3 for solid oxide fuel cell cathodes [1]. A wide variety of practical applications are made possible by many intrinsic perovskite materials features due to the continuous interaction between structure and properties. Ferroelectricity [2,3,4], semi-conductivity [5,6], superconductivity [5,6], piezoelectricity [7,8], thermoelectricity [9], colossal magnetoresistance, ferromagnetism [10], half-metallic transport [11,12] are just some of the fascinating physical and chemical properties of perovskites. These oxides are increasingly being used in electronic and magnetic materials, automotive exhaust, water splitting catalysts, fuel cells, battery electrode materials [13], gas sensors, humidity sensors, microwave devices, high-density data storage, magnetic ferrofluids, magnetic switches, MRI, high-frequency, and power devices are among the applications for these materials [2]. As demonstrated by the discovery of superconductivity in Na0.3CoO2.1.3H2O, the fact that cobalt cations can assume multiple oxidation and spin states is the root cause of the wide range of observable physical features of cobaltites [14]. Specifically, the LCO perovskite is a classic example of thermally aided spin state transitions of trivalent cobalt [15,16]. Integration of divalent/trivalent or magnetic/nonmagnetic dopant ions into a lattice result in a drastic alteration of the structure and other properties defined by cation distribution. Dopant choice is needed to get the desired improvement in the unaltered perovskite cobaltite. In particular, the shift from paramagnetic (PM) to ferromagnetic (FM) at the Curie temperature (TC) and the accompanying insulator–metal transition (TIM) in the case of manganite and cobaltite have been known for some time. These include high conductivity, magnetic properties, excellent performance as a cathode or an anode, a high Seebeck coefficient, and various forms of oxygen vacancy order. The LaCoO3 perovskite, in instance, is a famous example of thermally aided trivalent cobalt spin state transitions. It has a nonmagnetic insulator ground state with only low-spin (LS) Co3+, but its magnetic susceptibility rises with temperature up to 100 K due to a transition from the LS to the intermediate-spin (IS) state. A second spin-state transition is then inferred from a shift in transport characteristics above 500 K from an activated regime (0.1 eV at 100 K) to a metallic regime (~1 mΩ cm), which corresponds to a conversion to high-spin (HS). The energetic proximity of the various Co3+ spin-states (LS, IS, and HS), as demonstrated by these two spin-state transitions, is also highlighted by the dramatic effect of a small amount of doping on physical attributes [17].
Ferroelastic materials having rhombohedral lattices are particularly fascinating. Stretching along one of the perovskite unit cell's four body diagonals distorts the parent cubic structure, causing ferro elasticity [18]. As previously reported, LCO is ferromagnetic at low temperatures [19], and B site incorporation increases the magnetic properties. The complicated interplay between the interatomic exchange interaction energy (Δex) and the crystal field splitting energy (Δcf) controls the active spin crossover of Co ion’s low and high spin states. A modest structural disruption brought on by strain can have a considerable effect since Δcf is particularly sensitive to changes in O-Co bond length and Co-O-Co bond angle [20,21].
Microwave heating, chemical co-precipitation, sol-gel auto-combustion [22], micro- and nano-emulsions, hydrothermal techniques, high-temperature breakdown, and reverse micelle have all been used to create cobaltite magnetic nanoparticles (NPs) [23,24,25]. The best way to produce LaCoO3 is through hydrothermal synthesis because it is easy, cheap, and safe for the environment. One of the most important aspects of our study was the hydrothermal method, which allowed us to produce superior perovskite precipitates with the required stoichiometry and microstructure. Hydrothermal synthesis of LCO by L. Tepech-Carrillo.et.al., followed by a study of LCO structure at different calcination temperatures is elaborated. However, they did not look into LCO or Te-LCO composites' structures or magnetic properties. Since Te(2.5% and 5%) in LaCoO3 modifies the magnetic properties, we are attracted in studying the magnetic properties and structural analysis of this material [26].Unlike our prior work, in which we discovered no change in paramagnetic states at RT when doping Mn into LCO, we found that the Te inclusion in LCO has impacted the RT magnetic characteristics of LCO crystals. Therefore, we set out to improve LCO's magnetic characteristics when it was exposed to RT. After discovering LT ferromagnetism in Mn-doped LCO, we set out to investigate whether or not the same phenomenon would occur in Te-incorporated LCO.
2. Materials and Methods
2.1. Materials
2.1.1. Materials for the preparation of Te (Method 1).
The precursor materials used for the preparation of Te are Tellurium dioxide (TeO2) [Sigma Aldrich], and Sodium borohydride (NaBH4) [Merck].
2.1.2. Materials for the preparation of LaCoO 3 (Method 2).
The precursor materials used for the preparation of LCO are Lanthanum (III) nitrate hexahydrate (La(NO3)3⋅6H2O) [Merck], Cobalt (II) Nitrate Hexahydrate (Co(NO3)2⋅6H2O) [Merck], Ammonia Solution About 25% (NH4OH) [Merck], and Sodium Hydroxide (NaOH) [Merck].
2.1.3. Materials for the preparation of Te (2.5%) impregnated LaCoO3 and Te (5%) impregnated LaCoO3(Method 3).
The materials obtained after preparation of method 1 and method 2 are Te and LaCoO3. These two materials are used to prepare Te impregnated LaCoO3.All of the chemicals are reagent grade with high purity (99.9%) and can be used straight out of the package.
2.2. Methods
2.2.1. Method 1: The preparation of Te.
From room temperature (RT) to boiling point (BP), 100 ml of C2H6O2 and TeO2 (2 g wt.) were homogenously mixed using a magnetic stirrer in a silicon oil bath to maintain a temperature of 180 ºC in a double-neck round-bottomed flask equipped with a reflux cooler for 2 hours or longer. The ratio of salt to reductant was maintained at 1:4. After 2 h, 8 g of NaBH4 was added slowly into the mixture with the evolution of gases. While introducing NaBH4, regular personnel protective equipment (PPE) precautions and other safety measures were taken. After adding the reducing agent, the reaction mixtures were stirred for two hours and cooled to RT naturally. The ultimate precipitate was centrifuged and repeatedly washed with DI-water, acetone, and ethanol before being dried at RT Equation depicts the reaction of the sodium borohydride and tellurium dioxide (1).
TeO (OH-) is formed in two steps (Eq. 1), involving reduction by hydrogenation. Initially, TeO2 was reduced to equivalent ions and hydrogenated oxygen (H+O) was collected at the surface. The evolved H2 may dissociate into H atoms or ions during the reduction process.
2.2.2. Method 2: The preparation of LaCoO3.
We have briefly discussed the hydrothermal synthesis of LCO in our previous report [18].
2.2.3. Method 3: The preparation of Te impregnated LaCoO3.
The third method is ball milling of the samples obtained from the first two methods. The ball-milling of Te and LCO is as follows: 2.5 and 5% Te are mixed with 97.5 and 95% LCO by weight respectively and well ground for 10 hours with intervals of 30 minutes to avoid settling of powder on the sides of the ball-mill container. After the ball milling, the material is taken for further studies.
2.3. Characterisation Technique
To investigate the phase transformations at ambient temperatures, a CuK-alpha radiation was utilised in an Empyrean Malvern Panalytical X-ray Powder Diffractometer. A field emission scanning electron microscope (FESEM) equipped with energy dispersive X-Ray spectroscopy (EDAX) was used to investigate the surface topography and composition of the microstructure. The vibrational modes of the materials are studied using a confocal micro-Raman microscope (WiTec Alpha 300, Germany) equipped with AFM imaging and a He-Ne laser as the excitation source (λ-exc = 532 nm) in a backscattering setup. In order to investigate the chemical composition of the surface, we used X-ray photo-electron spectroscopy (XPS) with Al K-alpha X-rays (Thermo Scientific, UK) was utilised. A JEOL JEM 2100 HRTEM was utilised for microscopic examination and evaluation. The effect of a magnetic field strength of 5 T on the material's magnetization (M-H) at room temperature was investigated using SQUID-MPMS (Quantum Design, USA).
3. Results and Discussion
3.1. Powder X-Ray Diffraction (P-XRD) of Te Incorporated LaCoO3
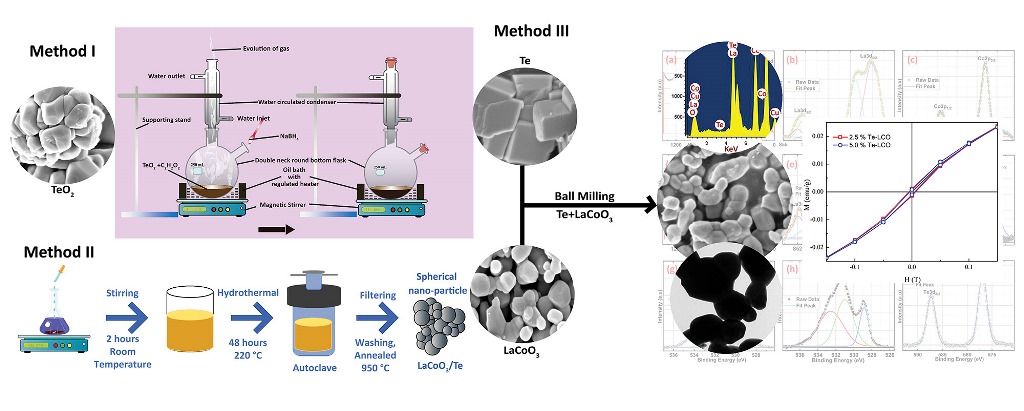
For parent LCO and two different concentrations of 2.5% and 5%Te-LCO, the powder XRD patterns were taken. The standard single-phase crystallite perovskite structure of LCO (JCPDS 48-0123) can be used to assign all of the diffraction peaks of the prepared samples, proving that the perovskite structures are well preserved after Te in-corporation as shown in Figure 1. There were no peaks that could be attributed to impurities. Furthermore, an enlarged scale of the higher intensity diffraction peaks of the prepared samples (2θ range: 30-38°) is shown at the outset of Figure 1 and reveals a slight shift towards higher 2θ as the concentration of Te2- (ionic radius 207 pm) increases, which has been attributed to Te's larger ionic radius compared with that of Te. The crystal planes of rhombohedral LCO are marked in Figure 1 with reference to standard JCPDS Card No. 48-0123. To understand the reduction in TeO2, we took XRD for both parent and reduced Te, which is shown in Figure 2. TeO2 is successfully reduced to Te, as shown by the XRD data, which is supported by the ICSD collection codes 34-422 and 65-3048 for TeO2 and Te, respectively. The crystal structure transforms from tetragonal to hexagonal during the reduction of TeO2 to Te, as reported in the reference data sheet. Table 1 illustrates the lattice parameter, crystal system, space group, and crystallite size of TeO2, Te, LCO, 2.5% Te-LCO, and 5% Te-LCO. The average crystal size was determined using the Debye-Scherrer formula, and the lattice parameters were determined using unit cell software.
3.2. Raman Spectrum analysis of Te Incorporated LaCoO3
Hydrothermally produced LCO, chemically reduced Te, and Te-impregnated LCO are analysed for their chemical distribution using Raman spectroscopy. In Figure 3, Raman scattering measurements of LCO, Te, 2.5% Te-LCO, and 5% Te-LCO are displayed. The primeval LCO has weak Raman signals compared to Te, 2.5 percentage Te–LCO, and 5 percentage Te-LCO, whereas the 2.5 and 5 percentage Te in cooperated LCO have good Te signals, which match the Te and have a slight shift and have a significant Raman characteristic peak. The Co-O-Co stretching mode in strongly deformed CoO6 may be related to the newly widened band at 516 cm-1 [27]. The Eg symmetry, which may be related to exterior mode (La-O) vibration, is responsible for the 116 and 140 cm-1 peaks [28,29]. Te exhibits strong Raman-active phonon modes as a result of its high atomic number and electronic polarizability. Three atoms make up each tellurium unit cell, which is arranged in an unending chain parallel to the c-axis. One A1 mode and two degenerate E modes, which are identified by rigid-chain rotation across the a- and b-axes and which fit into the Te lattice's D3 symmetry group, are indicated by the Raman spectra. The week bands at 103 cm-1 are attributed to the E(1) modes, which are responsible for the Raman spectra's E(1) bands. The E(2) mode, which is primarily distinguished by asymmetries along the c-axis, corresponds to the bands at 141 cm-1. The broad bands are of the second order at 268.9 cm-1[30,31,32]. Tellurium’s characteristic peaks at 122 and 141 cm-1 can be seen in both 2.5% Te-LCO and 5% Te-LCO which corresponds to the A1 bond-stretching mode and two degenerate E bond-stretching modes, respectively [33]. There is a slight red shift for the high intensity Te peak in Te-LCO, and increasing the Te conc. broadens the peaks.
3.3. SEM and Energy-Dispersive X-Ray (EDS) Analysis of Te Incorporated LaCoO3
Te-LCO composites are created by impregnating tellurium into the pores of LCO, which successfully preserves the material's irregular spherical shape. Figure 4 (a), (b), (c), (d), and (e) present the surface morphology of TeO2, Te, LCO, 2.5% Te-LCO, and 5% Te-LCO, respectively. TeO2 and Te show (Figure 4 a & b) that the reduction leads to the uniformly distributed plated nanostructure from mono-dispersed irregular microcrystals of about a few micrometres to nearly 500 nm. The hydrothermal synthesis of LCO (Figure 4 c) leads to a well dispersed spherical crystallite structure and the impregnating of Te in LCO (Figure 4 d & e) has impacted slightly on the microstructure. The Te impregnated LCO is well segregated, which is visible in the SEM micrograph. The EDAX spectrum of TeO2, Te, LCO, 2.5% Te-LCO, and 5% Te-LCO is revealed in Figure 4, which confirms the occurrence of constituent elements.
3.4. X-Ray Photoelectron Spectroscopy (XPS) studies of Te Incorporated LaCoO3
X-ray photoelectron spectroscopy (XPS)is utilised to investigate the valence state of pure LCO and Te in-cooperated LCO with a Te content of 5 wt.%. The XPS survey spectra in Figure 5(a) and (d) demonstrate the presence of Co, O, La, and Te (5 wt.% Te in-cooperated LCO). Core-level XPS spectra verify the real valence states of the individual components. Co 2p core level spectra, both pure and Te in-cooperated, are shown in Figures 5 c and f, respectively. LCO's two asymmetric peaks are located at 780.18 and 795.45 eV and are most similar to those of Co 2p3/2 and Co 2p1/2. Paramagnetic Co2+ is formed at the surface, accounting for the visible satellite peaks above the primary photo peaks [34]. Because of the presence of numerous excitations and the coexistence of Co3+ and Co2+ states, the Co 2p peak expanded and shifted toward higher energy when Te was present in conjunction with LCO. The existence of both high-spin Co2+ and low-spin Co3+ ions were further validated by the appearance of the Co2p3/2 peaks at 781.23 eV with decreased satellite peaks [35,36]. The Co 2p photoelectron spectra of Te-LCO are chemically shifted sufficiently to permit chemical identification. The Co 2p3/2 and Co 2p1/2 lines for Te-LCO were chemically shifted to 1.05 eV and 1.14 eV, respectively, to higher binding energies than for LCO. The Co 2p3/2 has diminished satellite peak may be due to well exposure of LCO when we mechanically ball mill with Te, which leads to further oxidation of the samples towards the surface. According to research by Frost and colleagues [34,37], photoelectron spectra of high-spin Co2+ compounds show robust satellites, while those of low-spin Co2+ compounds either show weak satellites or none at all. Te-LCO has a core photoemission peak separation of 15.36 eV, while LCO's is 15.27 eV. These values are fairly close to the CoO and Co3O4 values that have previously been published [34,38] as depicted in Figure 5. As anticipated, the La 3d line shows the four component peaks (b and e). According to our prior research [19], the La 3d5/2 and La 3d3/2 are associated to the double peaks of LCO, which emerge at 834.7 and 838.34 eV, respectively. These peaks can be attributed to the La3+ state and represent charge transfer between the La2O3, O2p and La 4f orbits [36,37,38] or substantial electronic configuration final state mixing [42]. Te in-corporation causes a shift toward greater binding energies, indicating that the lattice structure is being destroyed. Because of Te's multivalence state, the additive peak with the highest intensity at 837.34 eV could be attributable to the synthesis of La sub-oxide (La-Ox). The observed core-level Te3d spectra have been deconvoluted into single 3d3/2 and 3d5/2 spin-state peaks at about 587 and 576 eV, respectively, to help explain it better.The chemical shift seen at Te 3d5/2 of approximately 3 eV towards higher binding energy from Te2- is characteristics of NaBH4 reduced TeO2, which may form some surface oxidation layers (Te0). Figure 5 (i) shows the two asymmetric peaks of high-resolution Te 3d XPS spectroscopy, which reveals the Te4+/Te2-/Te0 oxidation state for the NaBH4 reduced TeO2.The binding energy difference between 3d3/2 and 3d5/2 is 10.4 eV, which agrees with the literature [43,44,45]. As was previously mentioned, the presence of Te at high oxidation states reconstructs the crystal structure and produces more Co2+ and Co3+ at various spin states, which leads to additional defects. The perovskite's A and B sites are both affected by the equally distributed Te4+, which results in an increase in the amount of La2+ in the A site and an anisotropic Co2+ in the B site to balance the charge. As a result, the magnetic state of the perovskite LCO was affected by the discrete La 3d and Co 2p photoemission peaks that the 5% Te in-coordinated LCO displayed at various spin states [46,47]. The same O1s peaks associated with the LCO and 5% Te-LCO provide evidence that Te played a part in the incorrect perovskite oxide's formation. As demonstrated in Figure 5, both the pristine and Te-LCO O1s peaks had three unique peaks (h & g). O1s peaks observed at 528.8, 530.9, and 532.6 eV on the LCO surface. This major signal at 528.8 eV can be attributable to either bulk oxide or to lattice oxygen (O2-), which is in line with the findings from earlier studies [41]. When looking at the oxide system, the broad peaks that have greater binding energies are the ones that are the most challenging to interpret. The value of 530.9 eV can be explained by the presence of chemisorbed oxygen (O-) or adsorbed H2O/OH- species, both of which create a vacancy at the surface for oxygen to occupy [48].O 1s XPS spectra of the Te impregnated LCO surface show that superoxide (O2-) formation occurs at a peak energy of 532.6 eV, while the other two peaks can be attributed to lattice oxygen, chemically adsorbed oxygen species on the oxygen vacancies, and physically adsorbed oxygen species on the surface. Three peaks with centres at BE = 529.5, 531.6, and 532.7 eV were revealed by deconvolution of the Te-impregnated LCO asymmetric O 1s spectra (Figure 5 g) [49]. These peaks correspond to lattice oxygen (such as O2), chemically adsorbed oxygen (such as O-), and physically adsorbed oxygen (H2O, O2), respectively. This encourages the conception of more peroxide/superoxide ions, which results in the acquisition of stronger peaks at 532.7 eV. Te4+ is in higher oxidation states in the Te in-cooperated LCO as a result of the exchange of oxygen species that occurs as oxygen vacancies form between bulk and surface oxides [40].
3.4. Magnetic Properties of Te Incorporated LaCoO3
At 300 K, the field dependency of magnetization (M-H) for pristine LCO, 2.5% Te-LCO, and 5% Te-LCO was measured for a field change from -2.5 T to 2.5 T. The M-H loop at 300 K (Figure 6 a) suggests that the samples are originally in a paramagnetic state (LCO) and that the magnetic state changes to a weak ferromagnetic with the addition of Te, as shown in Figure 6 a, where the hysteresis develops with Te impregnation of LCO. When compared to the parent LCO, the overall magnetization value of the incubated samples is lower. The samples have a very low magnetic saturation (MS) value after incorporation with Te. However, some previous studies relate the magnetic state to the anti-ferromagnetic exchange interaction in the samples [50], which arises from the antiferromagnetically ordered localized high spin states present in these systems with the incorporation of Te [51]. The values of magnetization (MS), MR and HC extracted from the magnetic data (Figure 6) are given in Table 2. With the initial incorporation of 2.5% Te, it induces the coercivity and it decreases for higher concentrations of Te (5%). The anti-symmetric exchange interaction, which arises from the interaction of low spin Co (III) with the excited high spin Co (III). This further influences the magneto-crystallite anisotropy and reduces the coercive field [52]. In our previous study of Mn doped LCO, the RT M-H shows only a paramagnetic state even after incorporation with Mn at the Co site [19]. From the XPS study, it can be understood that the different valence states of Te may occupy La/Co sites, which enhances the magnetic property in the present LCO system.
3.5. Transmission analysis of Te Incorporated LaCoO3
Furthermore, TEM was used to verify the nanoparticle size and material structure. LCO Figure 7 (a and b), 2.5 wt.% Te-LCO (d and e), and 5 wt.% Te-LCO (g and h) demonstrate an evident ellipsoid structure with a 100-200 nm distribution in Figure 7. Due of the low Te concentration in LCO, lattice plane separation cannot be observed. In addition, Figure 7 shows the 5% Te-LCO’s selected area electron diffraction (SAED) patterns. The composite reflected a single crystal structure with a few irregular brilliant spots, showing that the addition of Te affected the original structure of LCO [53]. Figure 7 depicts the interplanar spacing (dhkl) measured from the SAED patterns.
4. Conclusion
The findings provide a systematic examination of Te impregnation in LCO perovskite oxide at various concentrations. As-prepared perovskite oxides have been analysed for their magnetic properties and crystal structures. The results showed that the samples exhibited an imperfect rhombohedral crystal structure. Adding Te to LCO produces a perovskite with a high multi-valence state of Te4+/2- and three distinct oxygen species where creates oxygen deficiency. Raman spectroscopy and XPS analysis both show that the presence of Te4+ prevents the formation of defective cobalt oxide. After being subjected to impact and iteration in a ball mill, the SEM micrograph of LCO and Te-impregnated LCO demonstrates structural similarity between the parent and included samples. However, XRD analysis revealed that Te-LCO was successfully reduced and incorporated into the mixture as a result of the ball mill's influence on the crystallite structure. The structural integrity of the perovskite samples has also been verified using transmission electron microscopy investigation. The presence of weak ferromagnetic order at ambient temperature after impregnation implies its potential application in magnetic devices and hypersonic vehicles.
Data Availability
All the data used in the manuscript are within the manuscript.
Acknowledgments
The authors would like to thank the National Research Foundation of Korea (NRF) grant, funded by the Korean government (MIST) (No. 2022R1C1C1006414) and (No.2021R1A4A1032207). The author SEM acknowledges the funding agency of SERB-TARE fellowship (TAR/2021/000097).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- N Fist, J Dinan, R Stadelmannand N Orlovskaya, (2012) “In situ three point bending device for measurements of vibrational response of ceramics under stress by microRaman spectroscopy,” Advances in Applied Ceramics, vol. 111, no. 7, pp. 433-439, 2012. [CrossRef]
- C.B Samantaray, H Sim, and H Hwang, “Electronic structure and optical properties of barium strontium titanate (BaxSr1−xTiO3) using first-principles method,”Physica B: Condensed Matter, vol. 351, no. 1-2, pp. 158-162, 2004. [CrossRef]
- C.B. Samantaray, H. Sim, and H. Hwang, “The electronic structures and optical properties of BaTiO3 and SrTiO3 using first-principles calculations,” Microelectronics Journal, vol. 36, no. 8, pp.725-728, 2005. [CrossRef]
- J. G. Bednorz, and K. A. Müller, “Sr1−xCaxTiO3: An XY Quantum Ferroelectric with Transition to Randomness,” Physical Review Letters, vol. 52, pp. 2289-2292, 1984. [CrossRef]
- H. P. R. Frederikse, W. R. Thurber, and W. R. Hosler, “Electronic Transport in Strontium Titanate,” Physical Review Journals Archive, vol. 134, pp. A442-A445, 1964. [CrossRef]
- C. S. Koonce, M. L. Cohen, J. F. Schooley, W. R. Hosler, and E. R. Pfeiffer, “Superconducting Transition Temperatures of Semiconducting SrTiO3,”Physical Review Journals Archive, vol. 163, pp. 380-390, 1967. [CrossRef]
- H. Wang, B. Wang, Q. Li, Z. Zhu, R. Wang, and C. H. Woo, “First-principles study of the cubic perovskites BiMO3 (M=Al, Ga, In, and Sc),” Physical Review B,vol. 75, pp. 245209, 2007. [CrossRef]
- P. Baettig, C.F. Schelle, R. LeSar, U. V. Waghmare, and N.A. Spaldin, “Theoretical prediction of new high-performance lead-free Piezoelectrics,” Chemistry of Materials, vol. 17, pp. 1376–1380, 2005. [CrossRef]
- H. Muta, K. Kurosaki, and S. Yamanaka, “Thermoelectric properties of rare earth doped SrTiO3,” Journal of Alloys and Compounds, vol. 350, no. 1-2, pp. 292-295, 2003. [CrossRef]
- P. Sivaprakash, S.Divya, R.Parameshwari,C. Saravanan, S.Sagadevan, S. Arumugam and S. Esakki Muthu,“Influence of Zn2+ doping towards the structural, magnetic, and dielectric properties of NiFe2O4 composite. Journal of Materials Science: Materials in Electronics,vol. 31, pp. 16369–16378, 2020. [CrossRef]
- J. Millis, Boris I. Shraiman, and R. Mueller, “Dynamic Jahn-Teller Effect and Colossal Magnetoresistance in La1-xSrxMnO3,” Physical Review Letters, vol. 77, 175-178, 1996. [CrossRef]
- S. Divya, P. Sivaprakash, S. Raja, S. Esakki Muthu, Emad M. Eed, S. Arumugam and Tae Hwan Oh, “Temperature-dependent dielectric and magnetic properties of NiFe2O4 nanoparticles,” Applied Nanoscience, 2021. [CrossRef]
- M. Dragan, S. Enache, M. Varlam, and K. Petrov, “Perovskite-Type Lanthanum Cobaltite LaCoO3: Aspects of Processing Route toward Practical Applications,” Cobalt Compounds and Applications, IntechOpen, 2019. [CrossRef]
- H. Hilgenkamp, Ariando, H.J.H. Smilde, D.H.A. Blank, G. Rijnders, H. Rogalla, J.R. Kirtley, and C.C. Tsuel, “Ordering and manipulation of the magnetic moments in large-scale superconducting π-loop arrays,” Nature, vol. 422, pp. 50–53, 2003. [CrossRef]
- P. M. Raccah, and J. B. Goodenough, “First-Order Localized-Electron ⇆ Collective-Electron Transition in LaCoO3,” Physical Review Journals Archive,vol. 155, pp. 932-943, 1967. [CrossRef]
- M.A. Señarís-Rodríguez, and J.B. Goodenough, “Magnetic and Transport Properties of the System La1-xSrxCoO3-δ (0 < x ≤ 0.50),” Journal of Solid State Chemistry,vol. 118, pp. 323–336, 1995. [CrossRef]
- Maignan, D. Flahaut, and S. Hebert, “Sign change of the thermoelectric power in LaCoO3,” The European Physical Journal B - Condensed Matter and Complex Systems, vol. 39, pp. 145–148, 2004. [CrossRef]
- E. J. Guo, R. Desautels, D.Keavney, M. A. Roldan, B. J. Kirby, D. Lee, Z. Liao, T. Charlton, A.Herklotz, T. Z. Ward, M. R. Fitzsimmons, andH. N. Lee, “Nanoscale ferroelastic twins formed in strained LaCoO3 films,” Science advances, vol. 5, pp. 1-5, 2019. [CrossRef]
- S. Jhelai, M. Radhakrishnan, N. Padmanathan, S. Esakki Muthu, P. Sivaprakash, and M. Kadiresan, “Effect of Mn substitution on magnetic behaviour of oxygen defective LaCoO3 perovskite oxide,” Materials Science and Engineering: B, vol. 284, pp. 115875, 2022. [CrossRef]
- J.S. Zhou, J.Q. Yan, and J.B. Goodenough, “Bulk modulus anomaly in RCoO3 (R=La, Pr, and Nd),” Physical Review B - Condensed Matter and Materials Physics, vol. 71, pp. 22010, 2005. [CrossRef]
- T. Vogt, J.A. Hriljac, N.C. Hyatt, and P. Woodward, “Pressure-induced intermediate-to-low spin state transition in LaCoO3,” Physical Review B- Condensed Matter and Materials Physics, vol. 67, pp. 140401, 2003. [CrossRef]
- S. Divya, P. Sivaprakash, S. Raja, S. Esakki Muthu, Ikhyun Kim, N. Renuka, S. Arumugam, and Tae Hwan Oh, “Impact of Zn doping on the dielectric and magnetic properties of CoFe2O4 nanoparticles,” Ceramics International, vol. 48,pp. 33208-33218, 2022. [CrossRef]
- Y. Xu, P. Zielke, N. van Nong, S. Pirou, R. Reolon, X. Si, S.B. Simonsen, P. Norby, H. Lühmann, W. Bensch, and R. Kiebach, “Hydrothermal Synthesis, Characterization, and Sintering Behavior of Core-Shell Particles: A PrincipleStudy on Lanthanum Strontium Cobaltite Coated with Nanosized Gadolinium Doped Ceria,” Ceramics. vol.1,pp. 246-260, 2018. [CrossRef]
- M. Ayyob, I. Ahmad, F. Hussain, M. Kashif Bangash, J.A. Awan, and J.N. Jaubert, “A new technique for the synthesis of lanthanum substituted nickel cobaltite nanocomposites for the photo catalytic degradation of organic dyes in wastewater,” Arabian Journal of Chemistry,vol. 13, pp. 6341–6347, 2020. [CrossRef]
- Deeksha, P. Kour, I. Ahmed, K. K.Haldar,and K. Yadav, “Tuning the Morphology of Lanthanum Cobaltite Using the Surfactant-Assisted Hydrothermal Approach for Enhancing Oxygen Evolution Catalysis,” Proceedings of the National Workshop on Recent Advances in Condensed Matter and High Energy Physics, Springer Proceedings in Physics, Springer, Singapore,vol. 278, pp. 15-24, 2022. [CrossRef]
- L. Tepech-Carrillo, A. Escobedo-Morales, A. Pérez-Centeno, E. Chigo-Anota, J.F. Sánchez-Ramírez, E. López-Apreza, and J. Gutiérrez-Gutiérrez, “Preparation of Nanosized LaCoO3 through Calcination of a Hydrothermally Synthesized Precursor,” Journal of Nanomaterials,vol. 2016, pp. 7, 2016.
- M. Popa, J. Frantti, and M. Kakihana, “Characterization of LaMeO3 (Me: Mn, Co, Fe) perovskite powders obtained by polymerizable complex method,” Solid State Ionics. Vol. 154–155, pp. 135-141, 2002. [CrossRef]
- M.N. Iliev, and M. V Abrashev, “Raman phonons and Raman Jahn-Teller bands in perovskite-like manganites,” Journal of Raman Spectroscopy, vol. 32, no. 10, pp. 805–811, 2001. [CrossRef]
- N. Orlovskaya, D. Steinmetz, S. Yarmolenko, D. Pai, J. Sankar, and J. Goodenough, “Detection of temperature- and stress-induced modifications of LaCoO3 by micro-Raman spectroscopy,”Physical Review B - Condensed Matter and Materials Physics, vol. 72, pp. 014122, 2005. [CrossRef]
- R.M. Martin, G.I. R.M. Martin, G.I. Ucovsky, and K. Helliwell, “Intermolecnlar bonding and lattice dynamics of Se and Te,” Physical Review B - Condensed Matter and Materials Physics, vol. 13, pp. 1383, 1976. [CrossRef]
- S. Pine and G. Dresselhaus, “Raman Scattering in Paratellurite, TeO2,” Physical Review B - Condensed Matter and Materials Physics, vol. 5, pp. 4087, 1972. [CrossRef]
- . Marini, D. Chermisi, M. Lavagnini, D. Di Castro, C. Petrillo, L. Degiorgi, S. Scandolo, and P. Postorino, “High-pressure phases of crystallite tellurium: A combined Raman and ab initio study,”Physical Review B - Condensed Matter and Materials Physics, vol. 86. Pp. 064103, 2012. [CrossRef]
- J. He, W. Lv, Y. Chen, K. Wen, C. Xu, W. Zhang, Y. Li, W. Qin, and W. He, “Tellurium-Impregnated Porous Cobalt-Doped Carbon Polyhedra as Superior Cathodes for Lithium-Tellurium Batteries,” ACS Nano,vol. 11, pp. 8144–8152, 2017. [CrossRef]
- J. Haber, and L. Ungier, “On chemical shifts of ESCA and Auger lines in cobalt oxides,”Journal of Electron Spectroscopy and Related Phenomena, vol. 12,pp. 305-312, 1977. [CrossRef]
- N. S. McIntyre, D. D. Johnston, L. L.Coatsworth, R. D. Davidson, and J. R. Brown, “X-ray photoelectron spectroscopic studies of thin film oxides of cobalt and molybdenum,” Surface and Interface Analysis, vol. 15, no. 4, pp. 265-272, 1990. [CrossRef]
- H. Wang, W. Xu, S. Richins, K. Liaw, L. Yan, M. Zhou, and H. Luo, “Polymer-assisted approach to LaCo1-xNixO3 network nanostructures as bifunctional oxygen electrocatalysts,” Electrochimica Acta, vol. 296, pp. 945–953, 2019. [CrossRef]
- D.C. Frost, C.A. McDowell, and I.S. Woolsey, “Evidence for multiplet splitting of 2p photoelectron lines of transition metal complexes,” Chemical Physics Letters, vol. 17, pp. 320-323, 1972. [CrossRef]
- H. Seim, M. Nieminen, L. Niinistö, H. Fjellvåg, and L.S. Johansson, “Growth of LaCoO3 thin films from β-diketonate precursors,” Applied Surface Science, pp. 112, pp. 243–250, 1997. [CrossRef]
- C. V. Ramana, R.S. Vemuri, V. V. Kaichev, V.A. Kochubey, A.A. Saraev, and V. V. Atuchin, “X-ray photoelectron spectroscopy depth profiling of La2O3/Si thin films deposited by reactive magnetron sputtering,” ACS Applied Materialsand Interfaces, vol. 3, pp. 4370–4373, 2011. [CrossRef]
- X. Jiang, Y. Dong, Z. Zhang, J. Li, J. Qian, and D. Gao, “Cation substitution of B-site in LaCoO3 for bifunctional oxygen electrocatalytic activities,” Journalof Alloys and Compounds,vol. 878, pp. 160433, 2021. [CrossRef]
- L. Armelao, D. Barreca, G. Bottaro, A. Gasparotto, C. Maragno, and E. Tondello, “LaCoO3Nanosystems by a Hybrid CVD/Sol-Gel Route: An XPS Investigation,” Surface Science Spectra,vol. 10, pp. 143–149, 2003. [CrossRef]
- R. P. Vasquez, “X-ray photoemission measurements of La1-xCaxCoO3(x=0, 0.5),” Physical Review B - Condensed Matter and Materials Physics, vol. 54, pp. 14938, 1996. [CrossRef]
- A.J. Ricco, H.S. White, and M.S. Wrighton, “X-ray photoelectron and Auger electron spectroscopic study of the CdTe surface resulting from various surface pretreatments: Correlation of photoelectrochemical and capacitance-potential behavior with surface chemical composition,” Journal of Vacuum Science & Technology A: Vacuum, Surfaces, and Films, vol. 2, pp. 910–915, 1984. [CrossRef]
- A.B. Christie, I. Sutherland, and J.M. Walls, “Studies of the composition, ion-induced reduction and preferential sputtering of anodic oxide films on Hg0.8Cd0.2Te by XPS,” Surface Science, vol. 135, no. 1-3, pp. 225-242, 1983. [CrossRef]
- K. Hidetaka, and Y. Yoshihisa, “Ylide-Metal Complexes. XIV. An X-Ray Photoelectron Spectroscopic Study on Tellurium Complexes of Methylenetriphenylphosphorane,” Bulletin of the Chemical Society of Japan,vol. 61, no. 8, pp. 2990-2992, 1988. [CrossRef]
- W. Branford, M.A. Green, and D.A. Neumann, “Structure and ferromagnetism in Mn4+spinels: AM0.5Mn1.5O4 (A = Li, Cu; M = Ni, Mg),” Chemistry of Materials, vol. 14, no. 4, pp. 1649–1656, 2002. [CrossRef]
- K.P. Thummer, M.C. Chhantbar, K. Modi, and H. Joshi, Effect of Mn4+ substitution on magnetic behaviour of cobalt ferrite, Indian Journal of Physics, vol. 79, pp. 41–45, 2005.
- H. Liang, Y. Hong, C. Zhu, S. Li, Y. Chen, Z. Liu, and D. Ye, “Influence of partial Mn-substitution on surface oxygen species of LaCoO3 catalysts,” Catalysis Today,vol. 201, pp. 98–102, 2013. [CrossRef]
- P. Xiao, J. Zhu, H. Li, W. Jiang, T. Wang, Y. Zhu, Y. Zhao, and J. Li, “Effect of textural structure on the catalytic performance of LaCoO3 for CO oxidation,”ChemCatChem, vol. 6, no. 6,pp. 1774–1781, 2014. [CrossRef]
- R. Schmidt, J. Wu, C. Leighton, and I. Terry, Dielectric response to the low-temperature magnetic defect structure and spin state transition in polycrystalline LaCoO3,” Physical Review B - Condensed Matter and Materials Physics,vol. 79, pp. 125105, 2009. [CrossRef]
- K. Tomiyasu, M. Sato, S.I. Koyama, T. Nojima, R. Kajimoto, S. Ji, and K. Iwasa, “Magnetic properties of electron-doped LaCoO3,” Journal of the Physical Society of Japan, vol. 86, no. 9, pp. 094706, 2017. [CrossRef]
- D. Gignoux, Etienne Du Tremolet De Lacheisserie, M. Schlenker, “Magnetism: Materials and Applications,” Springer. 0-387-23063-7 (2005).
- R. Wang, C. Ye, H. Wang, and F. Jiang, “Z-Scheme LaCoO3/g-C3N4 for Efficient Full-Spectrum Light-Simulated Solar Photocatalytic Hydrogen Generation,” ACS Omega. vol. 5, no. 47, pp. 30373–30382, 2020. [CrossRef]
Figure 1.
X-ray Diffraction of LCO, Te, & Te(2.5 & 5%)-LCO. Outset shows the magnified image of high intensity peaks of LCO, Te, & Te(2.5 & 5%)-LCO.
Figure 1.
X-ray Diffraction of LCO, Te, & Te(2.5 & 5%)-LCO. Outset shows the magnified image of high intensity peaks of LCO, Te, & Te(2.5 & 5%)-LCO.
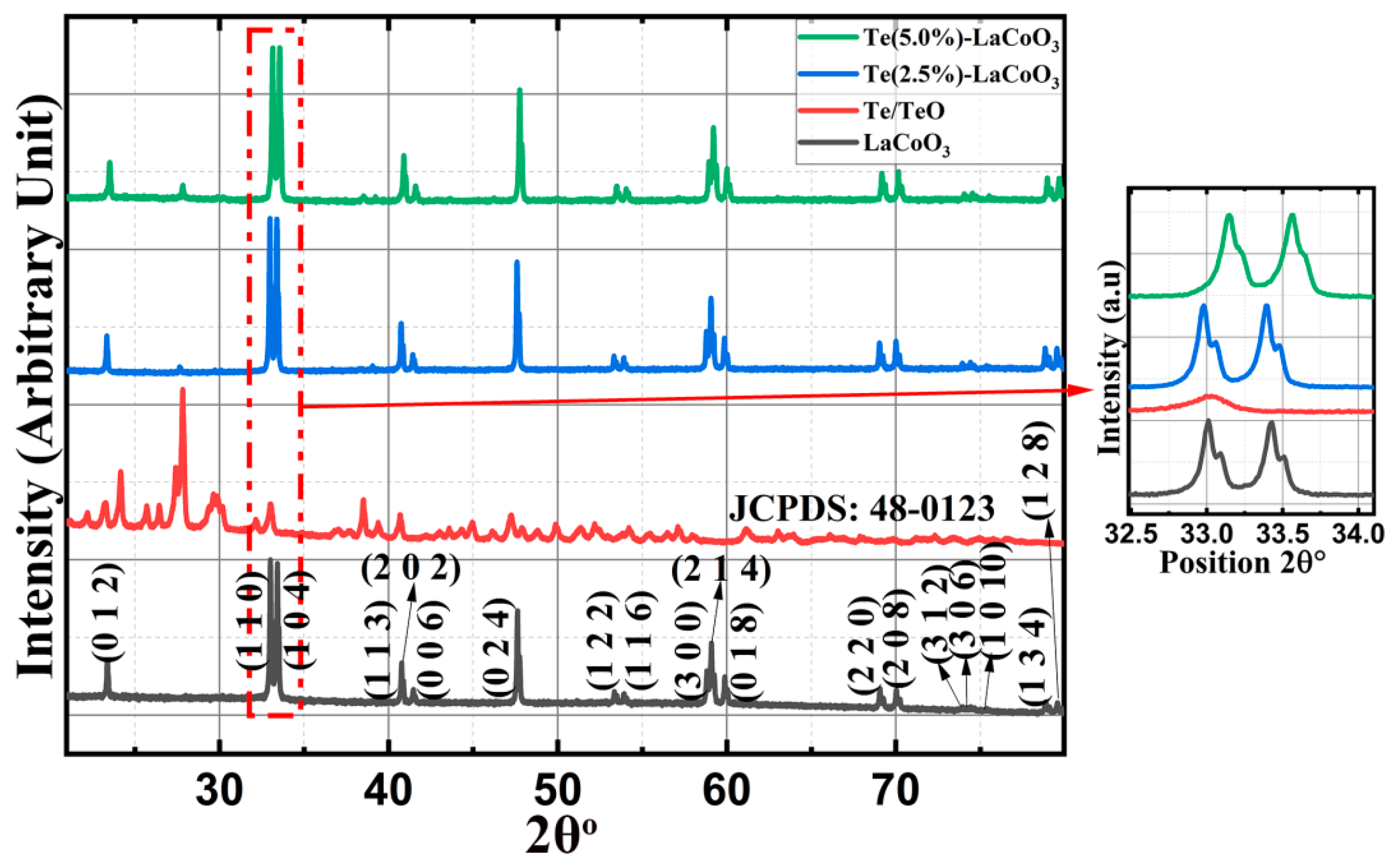
Figure 2.
X-ray Diffraction of TeO2 and Te.
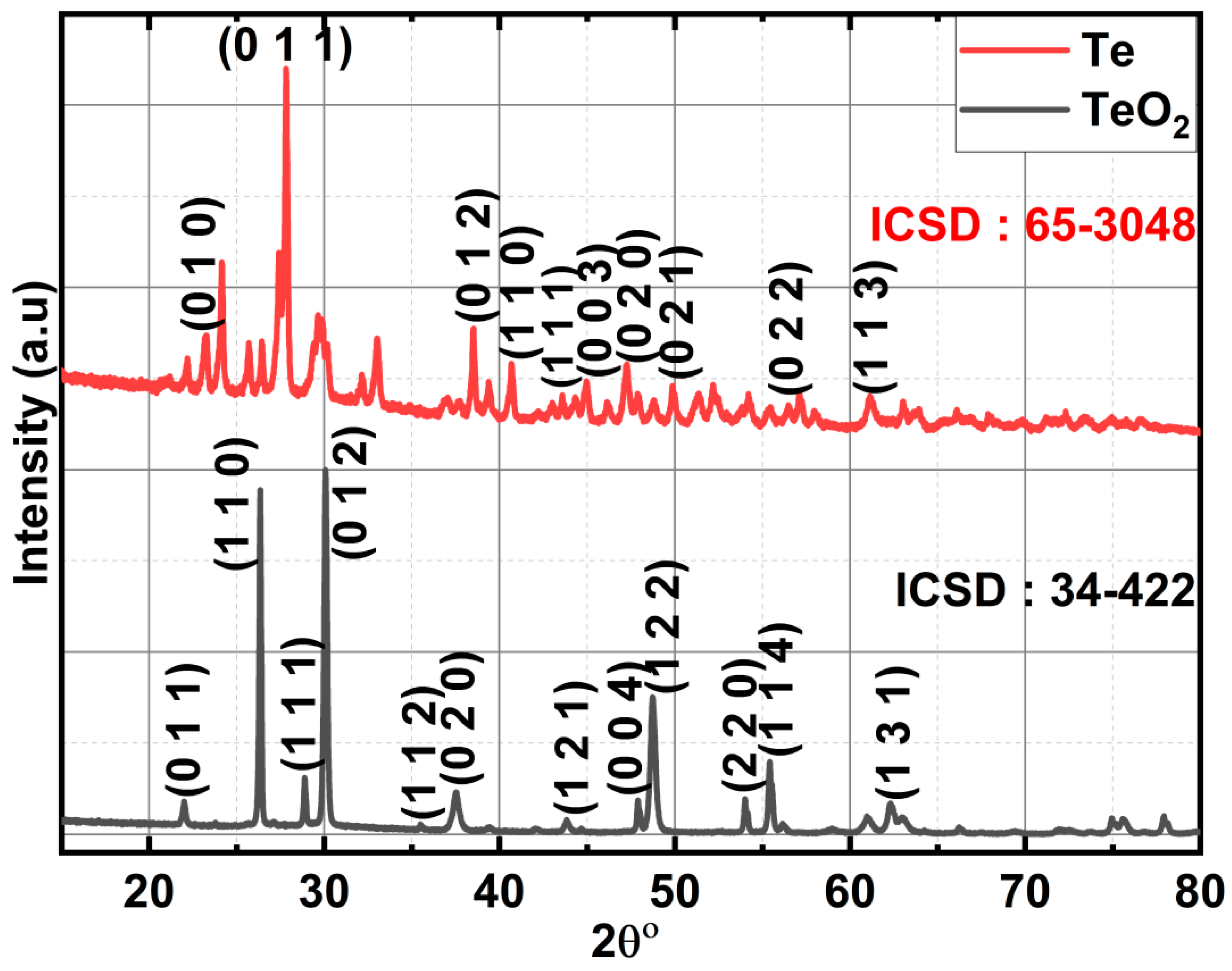
Figure 3.
Raman spectroscopy of LCO, Te, 2.5% Te-LCO & 5% Te-LCO.
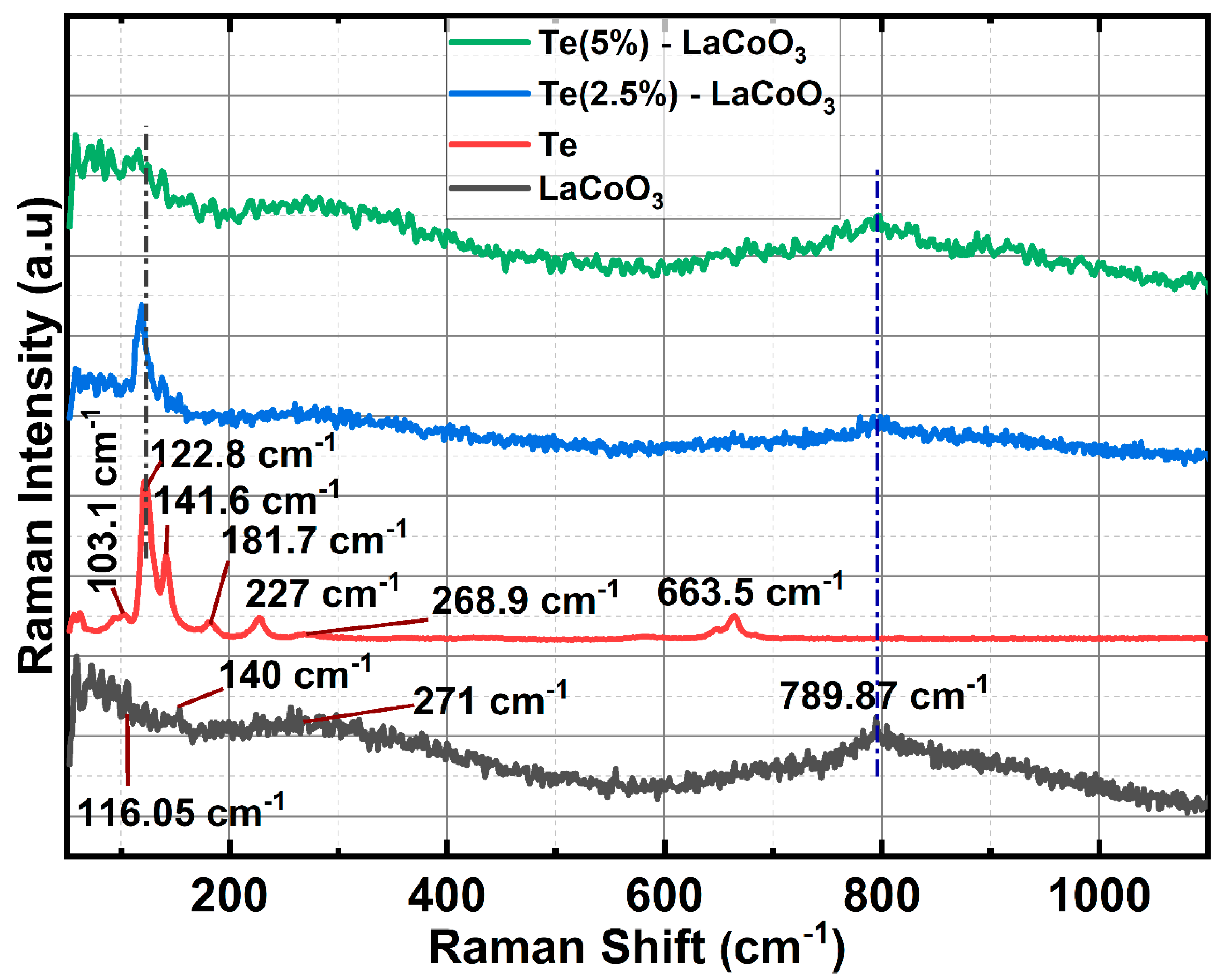
Figure 4.
SEM and EDAX image of (a) TeO2, (b) Te, (c) LaCoO3, (d) Te(2.5%) - LCO and Te(5%) - LCO.
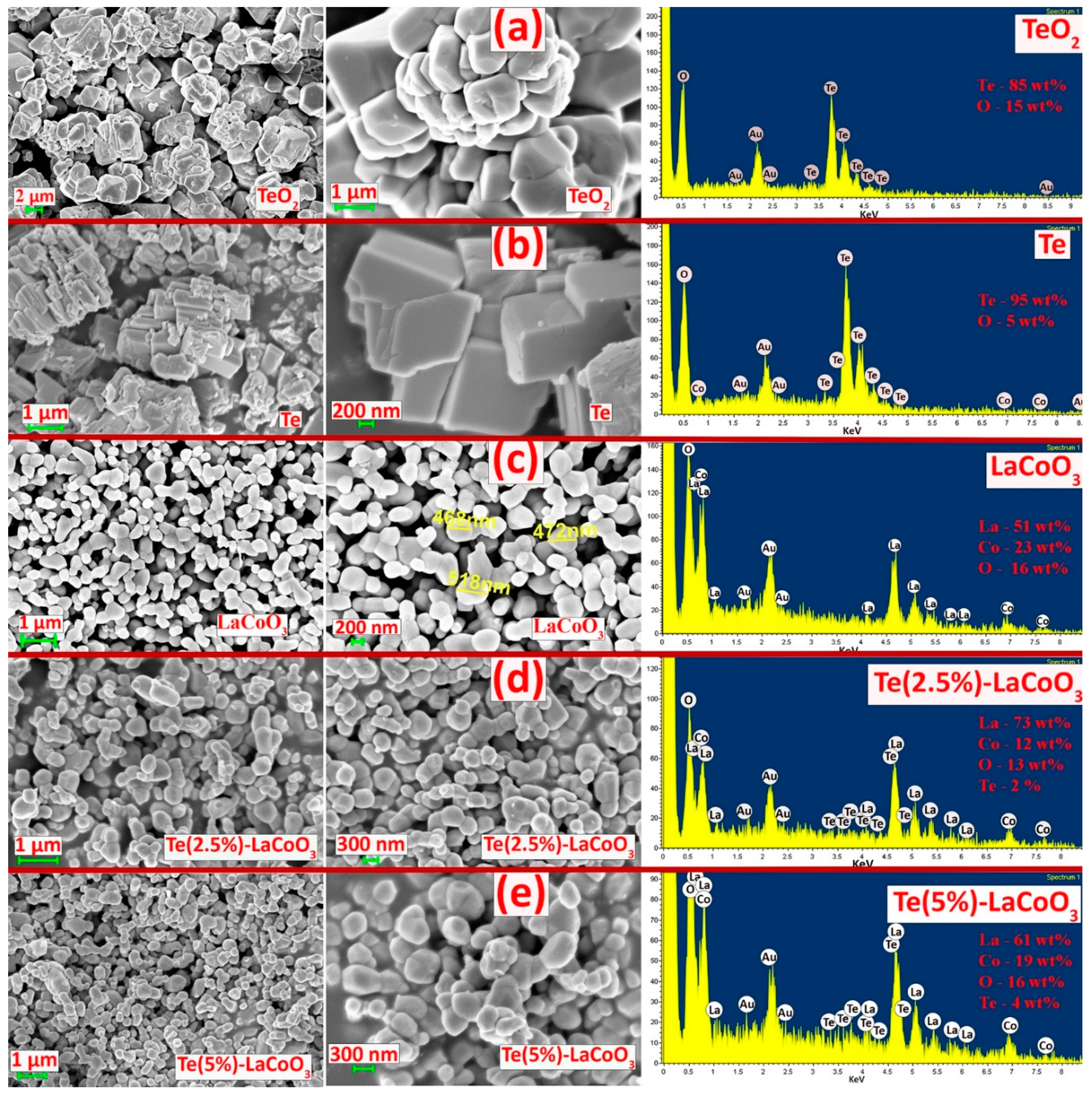
Figure 5.
XPS Survey Scan (a & b) of LCO and Te(2.5%)-LCO respectively. Detail XPS scan of La 3d (b & e), Co 2p (c & f), O 1s (g & h) of LCO and Te(2.5%)-LCO respectively and Te 3d (i) of Te(2.5%)-LCO.
Figure 5.
XPS Survey Scan (a & b) of LCO and Te(2.5%)-LCO respectively. Detail XPS scan of La 3d (b & e), Co 2p (c & f), O 1s (g & h) of LCO and Te(2.5%)-LCO respectively and Te 3d (i) of Te(2.5%)-LCO.
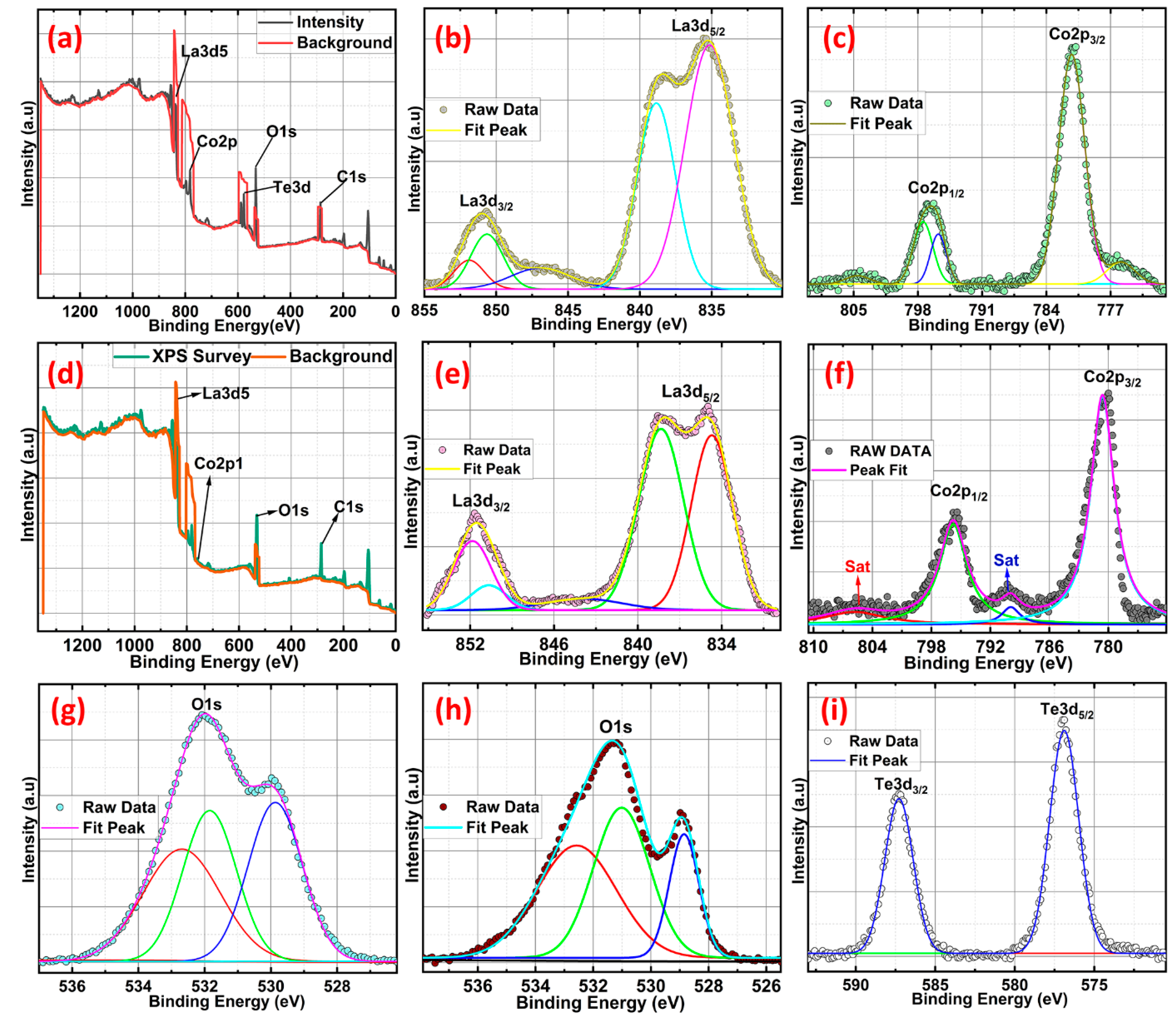
Figure 6.
Isothermal magnetization curve of (a) LCO, 2.5% Te-LCO and 5% Te-LCO at RT and (b) enlarged image of LCO, 2.5% Te-LCO and 5% Te-LCO at RT. .
Figure 6.
Isothermal magnetization curve of (a) LCO, 2.5% Te-LCO and 5% Te-LCO at RT and (b) enlarged image of LCO, 2.5% Te-LCO and 5% Te-LCO at RT. .
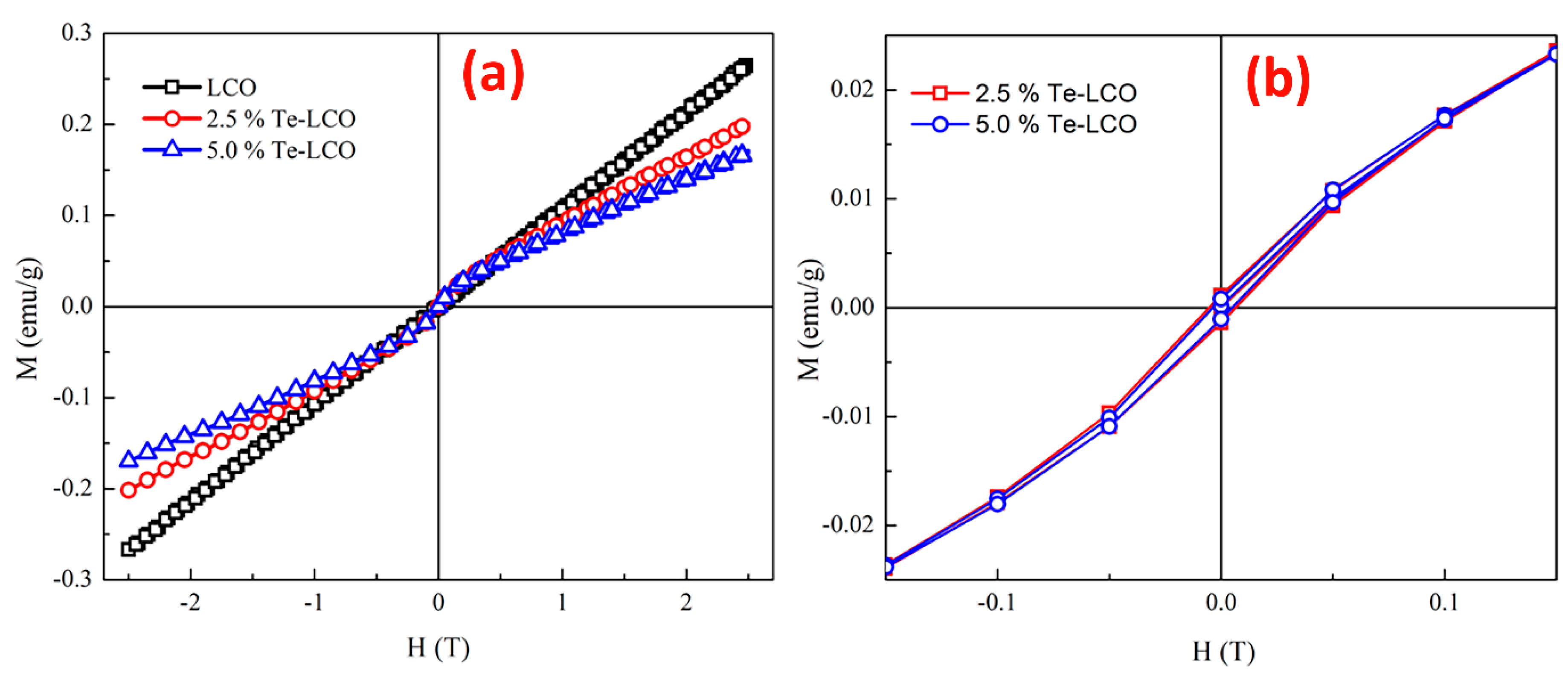
Figure 7.
TEM image of (a & b) LaCoO3, (d & e) Te(2.5%) – LCO and (g & h) Te(5%) – LCO. SAED of (c) LaCoO3, and (i) Te(5%) – LCO and EDS image of (f) Te(5%) – LCO.
Figure 7.
TEM image of (a & b) LaCoO3, (d & e) Te(2.5%) – LCO and (g & h) Te(5%) – LCO. SAED of (c) LaCoO3, and (i) Te(5%) – LCO and EDS image of (f) Te(5%) – LCO.
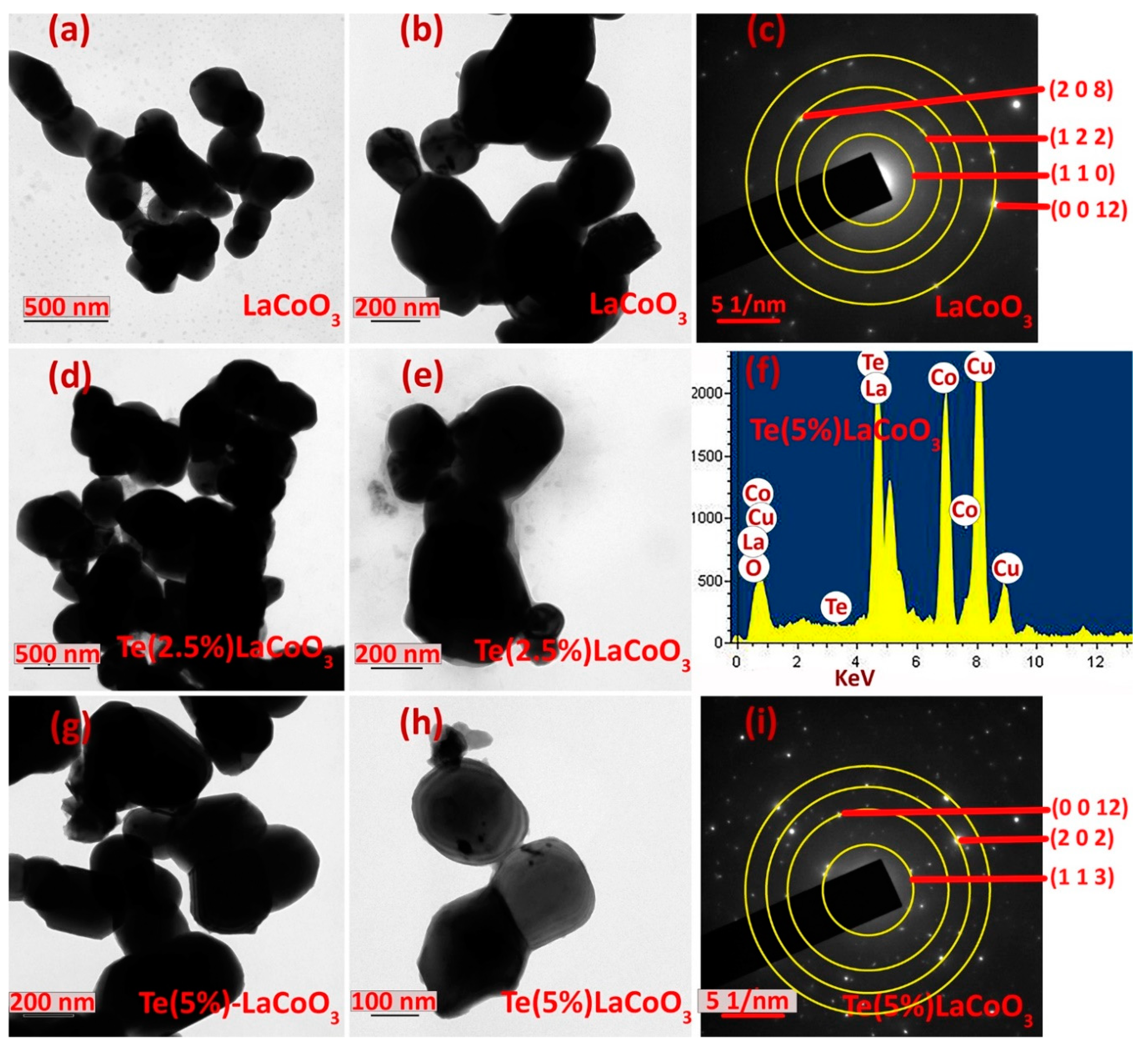

Table 1.
The crystallite size and lattice parameter of TeO2, Te, LCO, LCO/Te(2.5%) and LCO/Te(5%).
| Composition | Crystal System | Space Group | Crystallite size (nm) | Lattice Parameter (Å) | ||
| a | b | c | ||||
| TeO2 | Tetragonal | P41212 | 26.80514 | 5.40681 | 5.40865 | 13.20149 |
| Te | Hexagonal | P3121 | 28.33724 | 4.19097 | 4.19123 | 5.98099 |
| LaCoO3 | Rhombohedral | Rc | 43.42469 | 5.41768 | 5.39366 | 13.13084 |
| 2.5% Te/LCO | Rhombohedral | Rc | 31.59925 | 5.42428 | 5.39256 | 13.1579 |
| 5% Te/LCO | Rhombohedral | Rc | 31.59925 | 5.42681 | 5.39434 | 13.20149 |
Table 2.
Saturation magnetization(M), remnant magnetization (MR) and coercivity(HC) of pure and Te@LCO.
Table 2.
Saturation magnetization(M), remnant magnetization (MR) and coercivity(HC) of pure and Te@LCO.
| Composition | HC(T) | M (emu/g) @300K | MR(emu/g) |
| LCO | -- | --- | ----- |
| 2.5% Te-LCO | 0.0065 | 0.20166 | 0.00128 |
| 5% Te-LCO | 0.0049 | 0.17051 | 0.00094 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



