
Submitted:
24 May 2023
Posted:
30 May 2023
You are already at the latest version
Abstract
Endocrine and nervous systems reciprocally interact to manage physiological individual functions and homeostasis. The nervous system modulates hormone release through the hypothalamus, the main cerebrally specialized structure of the neuroendocrine system. Indeed, it is involved in various metabolic processes, administering hormone and neuropeptide release at different levels. This complex activity is affected by neurons of various cerebral areas, environmental factors, peripheral organs, and mediators through feedback mechanisms. Therefore, neuroendocrine pathways have a key role in metabolic homeostasis control, and their abnormalities have been associated with the development of Metabolic Syndrome (MetS) in children. The impaired functioning of genes, hormones, and neuropeptides of various neuroendocrine pathways involved in several metabolic processes has been related to an increased risk of dyslipidaemia, visceral obesity, insulin resistance, type 2 diabetes mellitus, and hypertension. This review examines the neuroendocrine effects on the risk of MetS in children, identifying and underlying the several conditions associated with neuroendocrine pathway disruption. Neuroendocrine systems should be considered in the complex pathophysiology of MetS, and, when genetic or epigenetic mutations in "hot" pathways occur, they could be studied for new potential target therapies in severe and drug-resistant paediatric forms of MetS.
Keywords:
Metabolic syndrome
; obesity
; neuroendocrine systems
; stress response
; genetic polymorphisms
1. Introduction
The Metabolic Syndrome (MetS) is a complex multifactorial disease with a not entirely recognized definition in childhood (Christian Flemming et al., 2020). The International Diabetes Federation (IDF) published a consensus definition in 2006, according to which MetS is diagnosed in children between 10 and 15 years if at least three of the following risk factors occur: waist circumference > 90th percentile, systolic blood pressure > 130 mmHg or diastolic blood pressure > 85 mmHg, triglycerides > 150 mg/dL, and high-density lipoprotein (HDL) cholesterol < 40 mg/dL (Alberti et al., 2006). In adolescents from the age of 15 years, the adult criteria for MetS are used, while in children younger than 10 years, the MetS diagnosis is not validated (Christian Flemming et al., 2020). In the paediatric population, excess fat storage is considered one of the most important risk factors for the development of both metabolic and cardiovascular diseases, including high blood pressure, atherosclerosis, dyslipidemia, and type 2 diabetes mellitus (T2D) (Kumar and Kelly, 2017). Specifically, pediatric obesity is caused by both environmental factors and genetic susceptibility. In 2020, 39 million children under the age of five were overweight or obese worldwide, with some variations across countries, and up to 6.3% of children is currently affected by severe forms of obesity (Hinney et al., 2022; Kaur et al., 2017; Mahmoud et al., 2022). The most frequent form of pediatric obesity certainly results from a link between a susceptible genetic background and the acquired environmental risk factors, which play a critical role in the pathophysiology (Hinney et al., 2022; Kleiser et al., 2009; Sohn, 2022). Sedentary lifestyles represent an important acquired risk factor for overweight, obesity and MetS in children. Indeed, high-calorie diets, low sleep quality, a sedentary lifestyle, and a low socio-economic status are common conditions in obese children (Hinney et al., 2022; Kleiser et al., 2009; Sohn, 2022). Notably, perinatal risk factors such as lack of breastfeeding and high birth weight may also contribute to excessive fat storage accumulation (Geserick et al., 2018; Kleiser et al., 2009; Qiao et al., 2015). Nevertheless, a considerable number of patients have a genetic form of obesity due to the abnormal functioning of different single genes and peptide hormones involved in the modulation of both hunger and satiety (Hinney et al., 2022; Sohn, 2022). Some of these are part of the neuroendocrine system, including neuropeptides and peptide hormones that interact with various types of neurons and neuroendocrine signalling pathways (Lemche et al., 2016a). These molecules are encoded by different genes, and their mutations can be responsible for genetically dependent excessive fat storage, according to neuroendocrine system abnormalities (Hinney et al., 2022; Lemche et al., 2016a). Furthermore, in several studies, some conditions have been identified and related to disruption of the neuroendocrine system, with a subsequent risk to develop metabolic and cardiovascular disorders (Lemche et al., 2016b). They include all circumstances where the "stress system" is chronically and/or repeatedly activated, such as low birth weight, accidents, surgery, trauma exposure, physical or sexual abuse, behaviour disorders, anxiety, and depression (Charmandari et al., 2003). Indeed, stress states have been associated with the hyperactivation and disruption of neuroendocrine pathways involved in the stress response and in metabolic homeostasis control (Charmandari et al., 2003). Even genetic polymorphisms and epigenetic modification, such as DNA methylation of genes and neuropeptides involved in the stress response, have been related to increased risk of MetS development (Womersley et al., 2022). These mechanisms are able to directly affect the neuroendocrine pathways of the stress system and have been associated with long-term biological consequences such as dyslipidaemia, hypertension, insulin resistance, and glucose intolerance (Lemche et al., 2016a). Nevertheless, the metabolic risk in stressed paediatric patients is strictly related to personal history, environmental and developmental influences, the kind of response to stressors, and the genetic background (Womersley et al., 2022). This review aims to describe the main neuroendocrine systems involved in the development of MetS in children, analysing the pathophysiology and the metabolic consequences of their disruption.
2. Neuroendocrine pathways of the stress response and the risk of metabolic syndrome
“Stress” is defined as a threatening condition where individual homeostasis is potentially endangered. In these circumstances, the organism activates neurobiological processes, known as “the stress response”, to face the threat and maintain homeostasis (Chrousos, 2009). The stress system is mostly represented by the hypothalamic-pituitary-adrenal axis (HPA axis) and the arousal/sympathetic system at the central level, while the periphery effectors are constituted by glucocorticoids and catecholamines (Pervanidou and Chrousos, 2011). Thus, when activated, these systems modulate the endocrine, metabolic, immune, neuropsychiatric, and cardiovascular functions of many organs and tissues (Figure 1). An appropriate response to stressors and/or a limited exposure to stress induces physiological processes that allow humans to face stressful conditions successfully (McEwen, 2007). Contrary, aberrant responses and long-term or very intense stress exposure are related to neuroendocrine responses that lead to crucial metabolic consequences (Charmandari et al., 2003). Specifically, several animal models and clinical studies, have shown that inappropriate stress response and chronic or intense exposure to stress lead to hyperactivity and consequent disruption of the stress system. These effects result in an increased risk to develop hypertension, insulin resistance, elevated blood glucose levels with decreased glucose uptake, visceral obesity and dyslipidaemia, T2D, an increased heart rate, higher blood pressure, and MetS (Pervanidou and Chrousos, 2012). Intense or chronic stress states include low birth weight, accidents, surgery, trauma exposure, physical or sexual abuse, behaviour disorders, anxiety, and depression. Interestingly, the crucial metabolic consequences of stress system dysregulation, could be irreversible if they occur when brain plasticity is massive and growth is unrestrained, such as during the foetal period, childhood and adolescence (Charmandari et al., 2003). Indeed, in these crucial periods, HPA axis and autonomic nervous system disruption are characterized by delayed myelination, increased neuronal loss, and altered synaptogenesis in the central nervous system, with neuropsychiatric and endocrine consequences that persist even in adulthood (Chrousos, 2007).
2.1. Hypothalamic-pituitary-adrenal axis
HPA axis plays a crucial role in the stress response. During the stress response, the corticotrophin-releasing hormone (CRH), is secreted by the parvocellular neurons of the hypothalamus and stimulated by neurons of the amygdala, hippocampus, mesocorticolymbic system, serotonin and acetylcholine systems, noradrenergic neurons of the sympathetic system, leptin, neuropeptide Y (NPY), arginine vasopressin (AVP) neurons in the hypothalamus, and inflammatory cytokines (Charmandari et al., 2003). Recently, also the Pituitary Adenylate Cyclase-Activating Peptide (PACAP) has been associated with CRH release after the binding with its receptor, expressed in the paraventricular nucleus of the hypothalamus (Womersley et al., 2022). CRH regulates the pituitary-adrenal axis and leads secretion of the pituitary adrenocorticotropin hormone (ACTH), with the synergic role of AVP neurons of the paraventricular nuclei (PVN), that are slightly permissive for ACTH secretion. In physiological circumstances, CRH and AVP present a pulsatile circadian release, higher in the early morning (Calogero et al., 1992; Holmes et al., 1986). Contrary, during stress, an increased secretion of CRH and AVP occurs and is mediated by inflammatory cytokines, vasoconstrictor factors, and lipidic molecules that stimulate HPA axis activity. In the periphery, the main effectors of the HPA axis are glucocorticoids, secreted by the zona fasciculata of the adrenal cortex, after the ACTH stimulus. During stress, glucocorticoid blood concentrations increase up to ten times the baseline values. Indeed, studies in paediatric patients who were chronically stressed have shown higher plasma cortisol concentrations and increased sensitivity to glucocorticoids in the evening, compared to healthy controls (Pervanidou et al., 2007). The primary mechanism of action of glucocorticoids is the regulation of gene transcription, which controls development, metabolism, and immune response when they bind their receptor. These hormones increase cardiac output, catecholamine sensitivity, hepatic and renal gluconeogenesis, glycogen synthesis, decrease glucose uptake, and lead to increased central adipose tissue (Ivović et al., 2020). Physiologically, glucocorticoids carry out negative feedback on the HPA axis, leading to limited tissue exposure to these hormones without relevant effects. Contrary, in conditions of chronic or intense acute stress, hyperactivation of the HPA axis occurs, with disrupted glucocorticoid secretion associated with long-term accumulation of fat in visceral adipose tissue, loss of muscle, arterial hypertension, osteoporosis, insulin resistance, dyslipidemia, and the potential development of the MetS (Pervanidou and Chrousos, 2011). Indeed, repeated HPA axis activation causes the glucocorticoid receptor complex to translocate, followed by glucose-induced gene transcription. It leads, in turn, to increased expression of FK506 binding protein 51 (FKBP5) with a subsequent decrease in FKBP5-glucocorticoid receptor complex sensitivity. Indeed, FKBP5 is a co-chaperone of the glucocorticoid receptor, and when bound to the receptor complex, it decreases the affinity of glucocorticoids for their receptors, causing an ultra-short feedback loop (Sidibeh et al., 2018). The overexpression of FKBP5, after HPA axis hyperactivation, reduces the physiological glucocorticoid negative feedback to the HPA axis, enhancing hypercortisolism and the related metabolic consequences (Womersley et al., 2022). Previous studies have shown that patients with diabetes with poor blood glucose control, emotional stress, inflammation, or comorbidities presented HPA axis hyperactivation and hypercortisolism. Chronic activation of the HPA axis has also been described in patients with type 1 diabetes with poor blood glucose control and in patients with T2D and diabetic neuropathy (Roy et al., 1993; Tsigos et al., 1993). Therefore, there are several conditions associated with increased HPA axis activity that are potentially related to the risk of MetS. They include depression, eating-disorders, obsessive-compulsive and panic disorders, chronic active alcoholism, attachment disorder of infancy, hyperthyroidism, and Cushing’s syndrome (Charmandari et al., 2003). However, epigenetic and genetic factors, personal history, age, ethnicity, developmental and environmental influences, psychopathology, and type of stressor could influence the HPA axis response with metabolic effects, and they should be considered consequently (Pervanidou and Chrousos, 2011).
2.2. Arousal and sympathetic nervous systems
Several studies have shown a key role for the autonomic nervous system in the stress response. Specifically, during the stress response, noradrenergic neurons of the Medulla and Pons secrete norepinephrine at the Locus Coeruleus level, where the hormone is then transported to peripheral targets. Noradrenergic neurons receive stimulatory innervation from neurons of the Amygdala, Hippocampus, Mesocorticolymbic System, Serotonin and Acetylcholine Systems, and CRH neurons, and they are also stimulated by leptin, inflammatory cytokines, and glucocorticoids (Charmandari et al., 2003). The PACAP’s binding with its receptor in the hypothalamus also leads to CRH and norepinephrine release, sustaining the stress response (Womersley et al., 2022). At peripheral level, effectors of the sympathetic system are, therefore, norepinephrine and epinephrine. They reach the target organs (stomach, liver, pancreas, intestines, and adrenal gland), where they act through β-adrenoceptors, involved in metabolic effects, and α-adrenoceptors, mainly expressed on smooth muscle vessels (Lemche et al., 2016b). Specifically, catecholamines stimulate hepatic glycogenolysis and gluconeogenesis and lead to lipolysis and fatty acid release from adipose tissue. Furthermore, the adrenal medulla secretes norepinephrine and epinephrine in response to stressors that lead to an accelerated heart rate, increased blood pressure and coronary flow, bronchial dilatation, inhibition of intestinal functions, and stimulation of an inflammatory response (Lemche et al., 2016b). Indeed, both the HPA axis and noradrenergic neurons at the Locus Coeruleus have been shown to affect the release of proinflammatory cytokines with a sustained latent low grade of inflammation that sustain the metabolic disruptions during stress states. The synergic interaction between CRH and noradrenergic neurons during the central stress response, is sustained through α1-adrenoceptors and CRH type 1 receptors (Charmandari et al., 2003; Kiss and Aguilera, 1992). During the stress response, the sympathetic system is therefore hyperactive, and high levels of catecholamines are detected in blood and urine (Pervanidou and Chrousos, 2012). Interestingly, the hyperactivity of the sympathetic system occurring in stress states is related to metabolic effects, such as decreased glucose uptake, increased risk of insulin resistance, and visceral obesity with a β-adrenoceptor sensitization (Straznicky et al., 2009). Moreover, catecholamines may contribute to insulin resistance by stimulating overexpression of glucose transporters at the renal level, resulting in increased insulin secretion (Lemche et al., 2016b). On the other hand, increased blood pressure and cardiac output are directly related to a high level of norepinephrine and a higher risk of developing cardiovascular diseases (Grassi et al., 2007). In several studies, it has been observed that chronically stressed paediatric patients present increased levels of plasma and urine catecholamines compared to healthy controls (Pervanidou, 2008). The relevant metabolic consequences of the sympathetic system hyperactivation are therefore associated with increased risk of hypertension, insulin resistance, T2D, visceral obesity, and MetS (Lemche et al., 2016b).
2.3. Genetic variants and epigenetic modifications involved in the stress response
Genetic and epigenetic background presents a remarkable relevance in the stress response, with individual variable consequences. Indeed, numerous genetic polymorphisms and epigenetic modifications are documented and considered responsible for the individual genetic vulnerability (Rubens et al., 2023; Womersley et al., 2022). This suggests that genetics, developmental influences and environment modulate the stress response with different endocrine and metabolic involvements (Bartoli et al., 2013; Kesebir, 2014). It has been observed that genetic and epigenetic modifications affect genes of the sympathetic nervous system, the HPA axis, and their mediators (Lemche et al., 2016b). Specifically, the catechol-o-methyltransferase (COMT) gene regulates the expression and function of norepinephrine that represents the main hormone of the sympathetic nervous system. The COMT variant Val158Met has been related to a phenotype with higher blood pressure and heart rate and visceral obesity (Lemche et al., 2016b). Moreover, the gene haplo-insufficiency has been associated with increased glycolysis and higher plasma cholesterol and triglyceride concentrations (Napoli et al., 2015). The β1-2-3-adrenoceptors (ADRB1, ADRB2 and ADRB3) genes encode the β-adrenoceptors, and their variants and/or epigenetic modifications are also associated with a high risk of MetS, leading to a genetic vulnerability during the stress response. Interestingly, ADRB1 polymorphisms have been related to obesity, heart rate impairment, increased blood pressure, and heart failure (Gao et al., 2014). On the other hand, ADRB2 variants have been linked to insulin resistance, hyperleptinemia, obesity, and T2D, while ADRB3 variants have been associated with dyslipidaemia, obesity, and MetS (Guay et al., 2014). Moreover, the solute carrier family 6 member 2 (SLC6A2) gene encodes the norepinephrine transporter, and its variants usually lead to decreased norepinephrine reuptake. Of note, associated symptoms include headache, palpitation, and tachycardia, with an increased cardiovascular risk. SLC6A2 activity is also related to metabolic effects such as obesity and weight control, even though it has not yet been extensively studied (Lemche et al., 2016b). Genetic polymorphisms in genes encoding HPA axis components are also reported in the literature. Specifically, the CRH receptors-1 and -2 (CRHR-1-2) genes are mainly expressed in the hippocampus, liver, and adipose tissue, and their variants have been associated with depression, stress vulnerability, and cardiovascular impairment (Lemche et al., 2016b; Wolf et al., 2013). Genetic variants of genes involved in cortisol biosynthesis and catabolism have also been described and related to MetS (Lemche et al., 2016b). Interestingly, gene methylation has been reported as the main epigenetic mechanism involved in the risk of MetS during the stress response (Womersley et al., 2022). Notably, FKBP5 plays a key role in the stress response, and its methylation has been studied in depth (Womersley et al., 2022). FKBP5 is expressed in the brain, lymphocytes, muscle, and adipose tissue and modulates the nuclear transcription glucocorticoid-mediated, after translocation of the glucocorticoid receptor complex in nucleus (Häusl et al., 2019). FKBP5 is related to decreased glucose uptake in omental tissue, insulin resistance, decreased levels of high-density lipoproteins (HDL), and stimulation of the inflammatory response (Pereira et al., 2014; Womersley et al., 2022). It has been observed that chronic hypercortisolism induced methylation of CpG sites on intron 2 of FKBP5 and patients with T2D, obesity, and insulin resistance show the same methylation pathway (Womersley et al., 2022). Furthermore, the nuclear receptor subfamily 3, group C, member 1 (NR3C1) gene encodes the glucocorticoid receptor and acts both as a transcription factor that binds glucocorticoid response mediators in the promoters of glucocorticoid target genes and as a regulator of other transcriptional factors. It has been found that methylation mechanisms in this gene are associated with the development of MetS in stressed paediatric patients (Lemche et al., 2016b). Prospective studies have shown different methylation profiles of interested genes in patients with insulin resistance and variable insulin and glucose blood concentrations (Lemche et al., 2016b; Womersley et al., 2022). Even though genetic and epigenetic modifications are influenced by several factors such as individual development, environment, and social factors, they could be related to the risk of MetS, especially when they involve target genes.
3. The regulation of satiety and hunger in central neuroendocrine pathways
Central neuroendocrine system plays a key role in the balance of satiety and hunger, both decreasing and increasing food intake and fat storage deposition. The leptin-melanocortin pathway is the most important pathway promoting satiety and fat accumulation, stimulating the production and cleavage of pro-opiomelanocortin (POMC) (Hinney et al., 2022; Kleinendorst et al., 2020a). Meanwhile, the NPY/agouti-related protein (AgRP) neuroendocrine system is critical in encouraging food intake and hunger (Hinney et al., 2022; Lemche et al., 2016a; Sohn, 2022). The Hypothalamus is a central brain region for the correct regulation of satiety and hunger, especially the arcuate nucleus, with both POMC and NPY-AgRP neurons, and the paraventricular nucleus, with melanocortin neurons expressing melanocortin receptors (MCRs), such as melanocortin 4 receptors (MC4Rs) (Hinney et al., 2022; Sohn, 2022). Below, a detailed description of these important neuroendocrine pathways and their relation to metabolic consequences.
3.1. Leptin and LEPR, a key neuroendocrine tandem with metabolic implications
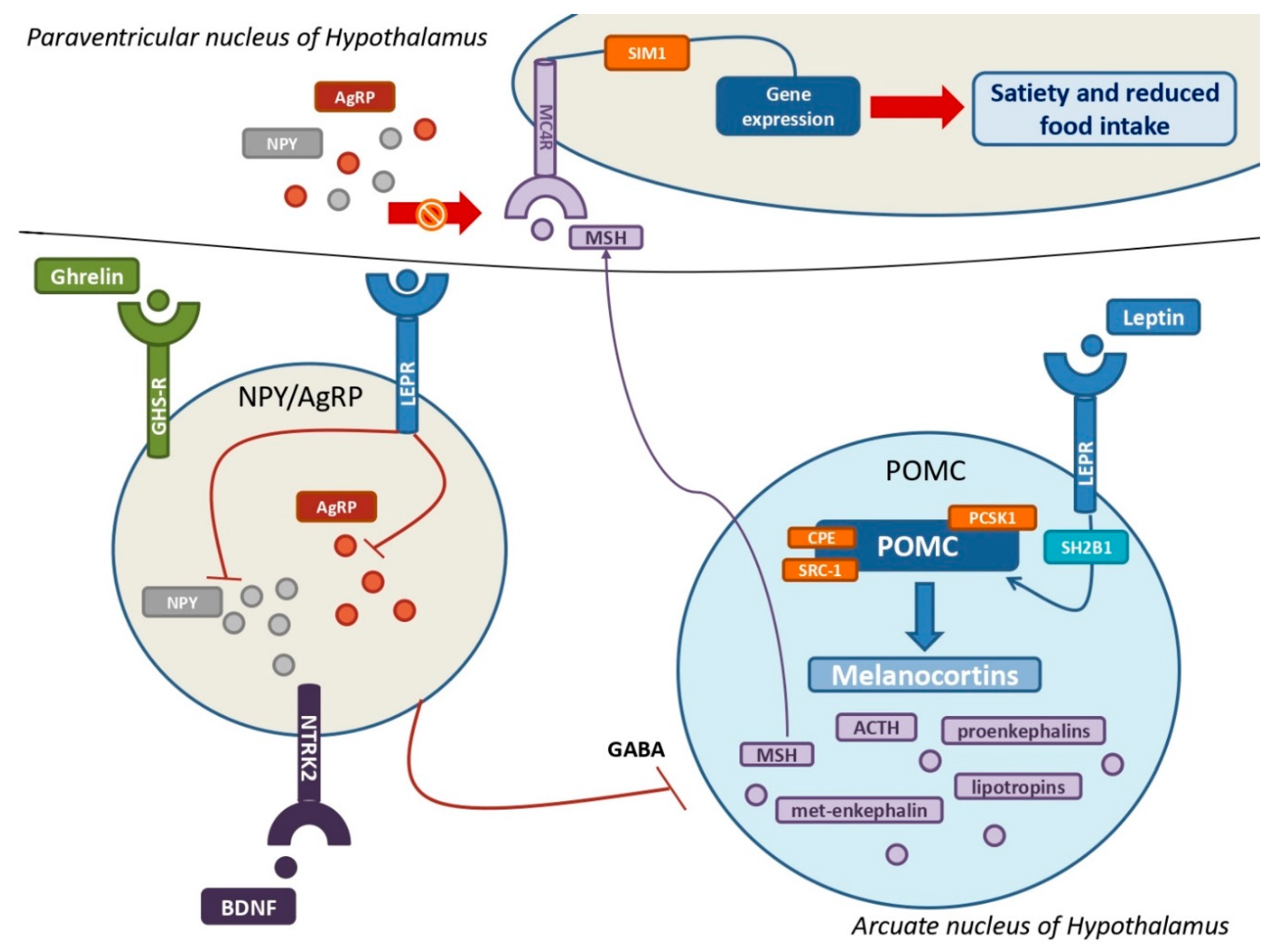
Leptin is a protein encoded by the gene Lep (7q31.3), which is regarded as a crucial molecule in the stimulation of satiety and promotion of energy expenditure (Hinney et al., 2022; Nordang et al., 2017; Obradovic et al., 2021; Sohn, 2022). Leptin is produced primarily by white adipose cells in proportion to fat mass, but it is also secreted by neurons in the hypothalamus and pituitary (Lemche et al., 2016a; Morash et al., 1999; Obradovic et al., 2021). Low leptin levels correlate with a poor energy state for the organism, eliciting responses to protect and enhance the energy reserve, such as altered behavioral and metabolic adaptation mechanisms (Pan and Myers, 2018). Instead, obese children and adults have higher leptin levels than healthy individuals (Mahmoud et al., 2022; Obradovic et al., 2021; Singh et al., 2017). Leptin is an integral part of the neuroendocrine system due to its ability to cross the blood-brain barrier and interact with its receptors LEPRs, which are highly expressed by GABAergic neurons in the arcuate nucleus of the hypothalamus (Friedman and Halaas, 1998; Lemche et al., 2016a; Mahmoud et al., 2022; Obradovic et al., 2021; Sohn, 2022). The binding of leptin and LEPRs in the arcuate nucleus stimulates the expression of POMC by a large population of POMC neurons, resulting in its cleavage into melanocortins, reducing hunger (Figure 2) (Dayton and Miller, 2018; Gregoric et al., 2021; Sohn, 2022). Notably, the SH2B adaptor 1 (SH2B1) protein has been identified as a key adapter in leptin-induced signal transduction of POMC, with also clinical implications (Doche et al., 2012; Li et al., 2007; Rui, 2014). Furthermore, the binding of LEPR and leptin inhibits NPY-AgRP neurons via a distinct neuroendocrine pathway, reducing food intake and fat accumulation (Friedman and Halaas, 1998; Lemche et al., 2016a; Mahmoud et al., 2022; Obradovic et al., 2021; Sohn, 2022). Due to its ability to stimulate the sympathetic nervous system and promote adrenoceptor activity, chronic leptin infusion also causes systemic inflammation and increases blood pressure (Lemche et al., 2016a; Reyes et al., 2015). This reveals the multiple interconnections between leptin and the cardiovascular system and explains why children and adults with leptin deficiency have numerous metabolic implications (Lemche et al., 2016a; Wasim et al., 2016). Insulin resistance, dyslipidemia, and visceral obesity are observed in obese patients with leptin impairment (Hinney et al., 2022; Lemche et al., 2016a; Sohn, 2022). Indeed, some forms of monogenic obesity brought on by loss-of-function LEP mutations are associated with severe childhood obesity, hyperphagia, and frequently T2DM (Antunes et al., 2009; Nordang et al., 2017; Wabitsch et al., 2015). In recessive forms, it is also possible to observe hypothyroidism, central hypogonadism, and delayed puberty (Nordang et al., 2017; Wabitsch et al., 2015; Wasim et al., 2016). LEPRs also play a crucial role in the satiety neuroendocrine system (Hinney et al., 2022; Kleinendorst et al., 2020a; Lemche et al., 2016a; Sohn, 2022). These neuronal receptors are encoded by the gene LEPR (1p31.3) and widely expressed in the arcuate nucleus of the hypothalamus with different implications, as discussed above, including promoting POMC expression and reducing NPY-AgRP activity (Hinney et al., 2022; Lemche et al., 2016a). However, LEPRs have also been detected in leptinergic neurons of the rostroventral lateral medulla, where they regulate blood pressure and the sympathetic system via interactions with the kidneys (Barnes and McDougal, 2014; Lemche et al., 2016a). Extremely rare, affecting a few more than 80 patients worldwide, pathogenic LEPR mutations are linked to severe childhood obesity, hyperphagia, hyperinsulinemia, hyperlipidemia, and, less frequently, multiple pituitary hormone deficiencies (Hinney et al., 2022; Kleinendorst et al., 2020a, 2020b; Poitou et al., 2020).
3.2. The key role of POMC, melanocortins, and related receptors in the development of MetS
POMC neurons are primarily found in the arcuate nucleus of the hypothalamus, where they represent key neuroendocrine actors in leptin-induced modulation of appetite and food intake (Gregoric et al., 2021; Sohn, 2022). POMC is encoded by the gene POMC (2p23.3) in response to leptin and LEPR binding stimulation, and represents the precursor hormone peptide of melanocortins (Hinney et al., 2022; Lemche et al., 2016a; Sohn, 2022). POMC is metabolized into multiple melanocortins via the cleavage of proprotein convertase subtilisin/kexin type 1 (PCSK1) and the cooperation of carboxypeptidase E (CPE) and steroid receptor coactivator 1 (SRC-1) (Figure 3) (Durmaz et al., 2021; Gregoric et al., 2021; Nordang et al., 2017; Sohn, 2022; Yang et al., 2019). Melanocortins are a large family of tiny neuropeptides derived from POMC, which includes the ACTH and several types of smaller melanocyte-stimulating hormones (MSH), such as α-MSH, β-MSH, and two γ-MSH isoforms (Ericson et al., 2017; Lemche et al., 2016a). ACTH is essential for the regulation of cortisol, which is produced by the adrenal gland. Other neuroendocrine molecules produced by ACTH cleavage are the endorphins, such as proenkephalins-A and -B, met-enkephalin, β-lipotropin and γ-lipotropin, which are essential for lipolysis, lipid transport, and steroidogenesis (Lemche et al., 2016a). MSH interacts with its transmembrane G-coupled melanocortin receptors (MCRs) in the hypothalamus, and, among these, MC4R and MC3R are the most important, being particularly noteworthy the MC4R's ability to bind not only melanocortins but also AgRP (Ericson et al., 2017; Hinney et al., 2022; Lemche et al., 2016a). MC4R and MC3R are widely distributed: MC4Rs are expressed in the paraventricular nucleus and amygdala and aim to regulate food intake, whereas MC4Rs elsewhere regulate energy consumption (Balthasar, 2006; Lemche et al., 2016a). MC4R may play a critical role in the pathophysiology of MetS. Indeed, the interaction between melanocortins and MC4R results in the expression of various genes with the goal of regulating body weight and energy expenditure, with the aid of the transcription factor single-minded homologue 1 (SIM1) (Farooqi et al., 2003; Hinney et al., 2022, 2013; Holder et al., 2000; Lemche et al., 2016a; Yeo et al., 2021). In addition, the melanocortins-MC4R pathway is also essential for regulating cholesterol metabolism, particularly the HDL/LDL ratio (Lemche et al., 2016a; Perez-Tilve et al., 2010). Pathogenic mutations affecting the gene MC4R (18q21.32) can be considered as the greatest risk factors for the co-occurrence of obesity and T2D, being MC4R deficiency the most common genetic cause of obesity, affecting nearly 5 percent of all obese children (Lemche et al., 2016a). Indeed, several autosomal dominant (AD) and autosomal recessive (AR) loss-of-function mutations of MC4R cause hyperphagia, severe pediatric obesity, and hyperinsulinemia due to peripheral receptorial impairment (Hinney et al., 2013; Kühnen et al., 2019; Yeo et al., 2021). Furthermore, gain-of-function mutations of MC4R have been found to improve body mass index and to decrease appetite, cardiovascular complications, and other metabolic features (Hinney et al., 2022; Lemche et al., 2016a; Lotta et al., 2019). MC3R has also its own role in the relationship between neuroendocrine systems and MetS (Lemche et al., 2016a). Unlike other melanocortin receptors, MC3R is predominantly found in limbic and hypothalamic regions of the brain, including the ventromedial hypothalamus, with the aim to regulate metabolic homeostasis and food intake (Lemche et al., 2016a). Pathogenic mutations of the gene MC3R (20q13.2) are also responsible for the accumulation of fat mass, diet-induced obesity, cycling hyperinsulinemia, and glucose intolerance (Lemche et al., 2016a). Conversely, pathogenic mutations affecting POMC can result in genetically-related severe obesity and MetS with pediatric onset (Hinney et al., 2022; Krude et al., 1998). Hence, the phenotype of POMC-associated monogenic obesity is typically characterized by pale skin and red hair, hyperphagia, severe obesity, adrenal insufficiency, and occasionally cholestasis (Graves et al., 2021; Krude et al., 1998). Interestingly, maternal diet during foetal development has a significant effect on the DNA methylation of POMC, underscoring the critical importance of environmental factors in regulating the leptin-melanocortin pathway and the neuroendocrine systems (Candler et al., 2019; Hinney et al., 2022). However, also mutations in enzymes and adaptor proteins implicated in these neuroendocrine pathways may result in abnormal phenotypes with metabolic implications. In fact, loss-of-function mutations of PCSK1 have been related to severe obesity, malabsorptive diarrhea, adrenal insufficiency, hypogonadism, hypothyroidism, and postprandial hypoglycemia (Hinney et al., 2022; Pépin et al., 2019; Sohn, 2022). Instead, clinical pictures due to loss-of-function mutations of the gene SH2B1 (16p11.2) are characterized by childhood obesity, insulin resistance, and hyperphagia (Doche et al., 2012; Rui, 2014). In addition, loss-of-function mutations of the gene CPE (4q32.3) can inhibit the cleavage of POMC into melanocortins, resulting in a triad composed of severe obesity, developmental delay, and hypogonadotropic hypogonadism (Bosch et al., 2021; Durmaz et al., 2021). The early onset of severe obesity, hyperphagia, persistent diarrhea, and metabolic abnormalities may be linked to mutations in the gene SRC-1, that inhibit the leptin-induced synthesis of POMC (Cacciottolo et al., 2022; Yang et al., 2019). Lastly, haploinsufficiency of the gene SIM1 (6q16.3) can result in a severe form of genetic obesity in children (Holder et al., 2000). Intriguingly, SIM1 plays a crucial role in the development of the Drosophila brain, and its impairment has been linked to obesity and high leptin levels in mice, although the underlying mechanisms are not entirely clear, providing further evidence for the connections between neuroendocrine signalling pathways and MetS (Holder et al., 2000; Michaud et al., 2001; Xia and Grant, 2013).
3.3. Ghrelin, the NPY-AgRP pathway, and metabolic implications
Even though the most primary feature of ghrelin is to stimulate the secretion of growth hormone (GH), it is also considered the most important hunger-stimulating hormone, counteracting leptin's effects. Ghrelin is encoded by the gene GHRL (3p25.3) and produced by neuroendocrine cells in the gastric mucosa and pancreas, as well as by neurons in the hypothalamus and anterior pituitary. Its effects are mediated by NPY-AgRP neurons in the arcuate nucleus, whose aim is to antagonize MC4Rs. Interestingly, low levels of ghrelin are correlated with MetS, while high concentrations seem to be protective against cardiovascular risk (Laurila et al., 2014; Lemche et al., 2016a). The ghrelin receptor (GHS-R), which is expressed by cells of the nervous and gastrointestinal tract, is also involved in the neuroendocrine system with effects on lipid and glucose metabolism. The primary effect of ghrelin is the stimulation of orexigenic NPY-AgRP neurons in the arcuate nucleus, promoting their neuroendocrine pathway whose purpose is to promote appetite and fat mass deposition (Hinney et al., 2022; Lemche et al., 2016a). NPY-AgRP neurons are a large population of hypothalamic neurons that secrete AgRP and NPY, which inhibit the binding of melanocortins to MC4R (Lemche et al., 2016a). NPY-AgRP are tightly regulated, as their expression is inhibited by leptin via the expression of LEPR and stimulated by ghrelin, brain-derived neurotrophic factor (BDNF), and its receptor encoded by the neurotrophic tyrosine kinase receptor type 2 gene (NTRK2) (Hinney et al., 2022). The NPY/AgRP neuroendocrine system is regarded as an important neurophysiological link between sympathetic stress reactivity, the glucocorticoid system, and the melanocortin system (Lemche et al., 2016a). In fact, NPY/AgRP neurons release γ-aminobutyric acid (GABA) to inhibit POMC neurons. AgRP is produced by a large group of neurons in the arcuate nucleus and paraventricular nucleus, which express both ghrelin and leptin receptors (Lemche et al., 2016a). AgRP enhances food intake and appetite in response to pro-inflammatory cytokines by inhibiting the interaction between MSH and MC4R (Hinney et al., 2022; Lemche et al., 2016a). NPY is widely present throughout the cerebrum, although the hypothalamus and limbic system are its primary sites of activity (Lemche et al., 2016a; Thorsell, 2010). Indeed, NPY is primarily produced in the arcuate nucleus, with its orexigenic effects manifesting in the paraventricular nucleus, thereby inducing hunger, lowering energy expenditure, and contributing to obesity (Lemche et al., 2016a). Some of the NPY receptors mediate neuroendocrine effects and, among these, NPY1R expression in fat cells is associated with the development of MetS by promoting insulin resistance and fat mass accumulation, whereas some variants of NPY2R may influence cardiometabolic traits (Lemche et al., 2016a; Sitticharoon et al., 2013; Thorsell, 2010; Wei et al., 2013). In addition, the gene NPY2R (4q31) plays a crucial role in an animal model of MetS, causing abdominal fat deposition and angiogenesis in adipose tissue in response to sympathetic nerve stimulation in response to social or environmental stress (Kuo et al., 2007; Lemche et al., 2016a). NPY's metabolic effects include vasoconstriction and a reduction in blood pressure via direct stimulation of potassium channels and intracellular calcium, as well as a promotion of fat deposition (Kuo et al., 2007; Lemche et al., 2016a). In addition, the release of NPY by paraventricular neurons may contribute to the complex pathophysiology underlying the onset of T2D (Lemche et al., 2016a). Notably, a specific leucine7 to proline7 (Leu7Pro7) polymorphism and the single nucleotide polymorphism rs16147 of the gene NPY (7p15.1) have been linked to elevated cholesterol and abdominal fat deposition, confirming the central role of this neuroendocrine system in the MetS (Jaakkola et al., 2009; Lemche et al., 2016a; Lin et al., 2015).
4. Conclusion
Several neuroendocrine pathways involved in metabolic processes have a key role in maintaining individual homeostasis, and their disruption has been associated with cardiovascular and metabolic disorders. Specifically, chronic or intense acute stress impairs the neuroendocrine systems of the stress response, leading to their hyperactivation. At the periphery, catecholamine and glucocorticoid activity have increased, with elevated risk of dyslipidaemia, hypertension, visceral obesity, insulin resistance, and glucose intolerance. Genetic and epigenetic modifications of genes that encode neuroendocrine pathways of the stress response also influence the individual's genetic vulnerability and the cardiometabolic risk related to the stress states. Also, central neuroendocrine pathways play their own role in promoting satiety or hunger states. The leptin-melanocortins pathway and the ghrelin-NPY-AgRP neuroendocrine system have different roles but are both critical for a correct metabolic balance. Neuroendocrine systems should be considered in the complex pathophysiology of MetS, and, when genetic or epigenetic mutations in "hot" pathways occur, they could be studied for new potential target therapies in severe and drug-resistant paediatric forms of MetS. However, the underlying mechanisms and genetic background behind paediatric forms of MetS related to impaired functioning of neuroendocrine systems should be further studied and better understood in order to allow a full comprehension of these critical pathways in human pathophysiology.
References
- Alberti, K.G.M.M., Zimmet, P., Shaw, J., 2006. Metabolic syndrome-a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet. Med. 23, 469–480. [CrossRef]
- Antunes, H., Santos, C., Carvalho, S., 2009. Serum leptin levels in overweight children and adolescents. Br. J. Nutr. 101, 1262–1266. [CrossRef]
- Balthasar, N., 2006. Genetic dissection of neuronal pathways controlling energy homeostasis. Obes. Silver Spring Md 14 Suppl 5, 222S-227S. [CrossRef]
- Barnes, M.J., McDougal, D.H., 2014. Leptin into the rostral ventral lateral medulla (RVLM) augments renal sympathetic nerve activity and blood pressure. Front. Neurosci. 8, 232. [CrossRef]
- Bartoli, F., Carrà, G., Crocamo, C., Carretta, D., Clerici, M., 2013. Metabolic Syndrome in People Suffering from Posttraumatic Stress Disorder: A Systematic Review and Meta-Analysis. Metab. Syndr. Relat. Disord. 11, 301–308. [CrossRef]
- Bosch, E., Hebebrand, M., Popp, B., Penger, T., Behring, B., Cox, H., Towner, S., Kraus, C., Wilson, W.G., Khan, S., Krumbiegel, M., Ekici, A.B., Uebe, S., Trollmann, R., Woelfle, J., Reis, A., Vasileiou, G., 2021. BDV Syndrome: An Emerging Syndrome With Profound Obesity and Neurodevelopmental Delay Resembling Prader-Willi Syndrome. J. Clin. Endocrinol. Metab. 106, 3413–3427. [CrossRef]
- Cacciottolo, T.M., Henning, E., Keogh, J.M., Bel Lassen, P., Lawler, K., Bounds, R., Ahmed, R., Perdikari, A., Mendes de Oliveira, E., Smith, M., Godfrey, E.M., Johnson, E., Hodson, L., Clément, K., van der Klaauw, A.A., Farooqi, I.S., 2022. Obesity Due to Steroid Receptor Coactivator-1 Deficiency Is Associated With Endocrine and Metabolic Abnormalities. J. Clin. Endocrinol. Metab. 107, e2532–e2544. [CrossRef]
- Calogero, A.E., Norton, J.A., Sheppard, B.C., Listwak, S.J., Cromack, D.T., Wall, R., Jensen, R.T., Chrousos, G.P., 1992. Pulsatile activation of the hypothalamic-pituitary-adrenal axis during major surgery. Metabolism 41, 839–845. [CrossRef]
- Candler, T., Kühnen, P., Prentice, A.M., Silver, M., 2019. Epigenetic regulation of POMC; implications for nutritional programming, obesity and metabolic disease. Front. Neuroendocrinol. 54, 100773. [CrossRef]
- Charmandari, E., Kino, T., Souvatzoglou, E., Chrousos, G.P., 2003. Pediatric Stress: Hormonal Mediators and Human Development. Horm. Res. Paediatr. 59, 161–179. [CrossRef]
- Christian Flemming, G.M., Bussler, S., Körner, A., Kiess, W., 2020. Definition and early diagnosis of metabolic syndrome in children. J. Pediatr. Endocrinol. Metab. 33, 821–833. [CrossRef]
- Chrousos, G.P., 2009. Stress and disorders of the stress system. Nat. Rev. Endocrinol. 5, 374–381. [CrossRef]
- Chrousos, G.P., 2007. Organization and Integration of the Endocrine System: The Arousal and Sleep Perspective. Sleep Med. Clin. 2, 125–145. [CrossRef]
- Dayton, K., Miller, J., 2018. Finding treatable genetic obesity: strategies for success. Curr. Opin. Pediatr. 30, 526–531. [CrossRef]
- Doche, M.E., Bochukova, E.G., Su, H.-W., Pearce, L.R., Keogh, J.M., Henning, E., Cline, J.M., Saeed, S., Dale, A., Cheetham, T., Barroso, I., Argetsinger, L.S., O’Rahilly, S., Rui, L., Carter-Su, C., Farooqi, I.S., 2012. Human SH2B1 mutations are associated with maladaptive behaviors and obesity. J. Clin. Invest. 122, 4732–4736. [CrossRef]
- Durmaz, A., Aykut, A., Atik, T., Özen, S., Ayyıldız Emecen, D., Ata, A., Işık, E., Gökşen, D., Çoğulu, Ö., Özkınay, F., 2021. A New Cause of Obesity Syndrome Associated with a Mutation in the Carboxypeptidase Gene Detected in Three Siblings with Obesity, Intellectual Disability and Hypogonadotropic Hypogonadism. J. Clin. Res. Pediatr. Endocrinol. 13, 52–60. [CrossRef]
- Ericson, M.D., Lensing, C.J., Fleming, K.A., Schlasner, K.N., Doering, S.R., Haskell-Luevano, C., 2017. Bench-top to clinical therapies: A review of melanocortin ligands from 1954 to 2016. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 2414–2435. [CrossRef]
- Farooqi, I.S., Keogh, J.M., Yeo, G.S.H., Lank, E.J., Cheetham, T., O’Rahilly, S., 2003. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 348, 1085–1095. [CrossRef]
- Friedman, J.M., Halaas, J.L., 1998. Leptin and the regulation of body weight in mammals. Nature 395, 763–770. [CrossRef]
- Gao, Y., Lin, Y., Sun, K., Wang, Y., Chen, J., Wang, H., Zhou, X., Fan, X., Hui, R., 2014. Orthostatic Blood Pressure Dysregulation and Polymorphisms of β-Adrenergic Receptor Genes in Hypertensive Patients. J. Clin. Hypertens. 16, 207–213. [CrossRef]
- Geserick, M., Vogel, M., Gausche, R., Lipek, T., Spielau, U., Keller, E., Pfäffle, R., Kiess, W., Körner, A., 2018. Acceleration of BMI in Early Childhood and Risk of Sustained Obesity. N. Engl. J. Med. 379, 1303–1312. [CrossRef]
- Grassi, G., Quarti-Trevano, F., Seravalle, G., Dell’Oro, R., 2007. Cardiovascular risk and adrenergic overdrive in the metabolic syndrome. Nutr. Metab. Cardiovasc. Dis. 17, 473–481. [CrossRef]
- Graves, L.E., Khouri, J.M., Kristidis, P., Verge, C.F., 2021. Proopiomelanocortin deficiency diagnosed in infancy in two boys and a review of the known cases. J. Paediatr. Child Health 57, 484–490. [CrossRef]
- Gregoric, N., Groselj, U., Bratina, N., Debeljak, M., Zerjav Tansek, M., Suput Omladic, J., Kovac, J., Battelino, T., Kotnik, P., Avbelj Stefanija, M., 2021. Two Cases With an Early Presented Proopiomelanocortin Deficiency-A Long-Term Follow-Up and Systematic Literature Review. Front. Endocrinol. 12, 689387. [CrossRef]
- Guay, S.-P., Brisson, D., Lamarche, B., Biron, S., Lescelleur, O., Biertho, L., Marceau, S., Vohl, M.-C., Gaudet, D., Bouchard, L., 2014. ADRB3 gene promoter DNA methylation in blood and visceral adipose tissue is associated with metabolic disturbances in men. Epigenomics 6, 33–43. [CrossRef]
- Häusl, A.S., Balsevich, G., Gassen, N.C., Schmidt, M.V., 2019. Focus on FKBP51: A molecular link between stress and metabolic disorders. Mol. Metab. 29, 170–181. [CrossRef]
- Hinney, A., Körner, A., Fischer-Posovszky, P., 2022. The promise of new anti-obesity therapies arising from knowledge of genetic obesity traits. Nat. Rev. Endocrinol. 18, 623–637. [CrossRef]
- Hinney, A., Volckmar, A.-L., Knoll, N., 2013. Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis. Prog. Mol. Biol. Transl. Sci. 114, 147–191. [CrossRef]
- Holder, J.L., Butte, N.F., Zinn, A.R., 2000. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum. Mol. Genet. 9, 101–108. [CrossRef]
- Holmes, M.C., Antoni, F.A., Aguilera, G., Catt, K.J., 1986. Magnocellular axons in passage through the median eminence release vasopressin. Nature 319, 326–329. [CrossRef]
- Ivović, M., Marina, L.V., Šojat, A.S., Tančić-Gajić, M., Arizanović, Z., Kendereški, A., Vujović, S., 2020. Approach to the Patient with Subclinical Cushing’s Syndrome. Curr. Pharm. Des. 26, 5584–5590. [CrossRef]
- Jaakkola, U., Kallio, J., Heine, R.J., Nijpels, G., t’ Hart, L.M., Maassen, J.A., Bouter, L.M., Stehouwer, C.D.A., Dekker, J.M., 2009. Neuropeptide Y polymorphism significantly magnifies diabetes and cardiovascular disease risk in obesity: the Hoorn Study. Eur. J. Clin. Nutr. 63, 150–152. [CrossRef]
- Kaur, Y., de Souza, R.J., Gibson, W.T., Meyre, D., 2017. A systematic review of genetic syndromes with obesity. Obes. Rev. Off. J. Int. Assoc. Study Obes. 18, 603–634. [CrossRef]
- Kesebir, S., 2014. Metabolic syndrome and childhood trauma: Also comorbidity and complication in mood disorder. World J. Clin. Cases 2, 332. [CrossRef]
- Kiss, A., Aguilera, G., 1992. Participation of α1-Adrenergic Receptors in the Secretion of Hypothalamic Corticotropin- Releasing Hormone during Stress. Neuroendocrinology 56, 153–160. [CrossRef]
- Kleinendorst, L., Abawi, O., van der Kamp, H.J., Alders, M., Meijers-Heijboer, H.E.J., van Rossum, E.F.C., van den Akker, E.L.T., van Haelst, M.M., 2020a. Leptin receptor deficiency: a systematic literature review and prevalence estimation based on population genetics. Eur. J. Endocrinol. 182, 47–56. [CrossRef]
- Kleinendorst, L., Abawi, O., van der Voorn, B., Jongejan, M.H.T.M., Brandsma, A.E., Visser, J.A., van Rossum, E.F.C., van der Zwaag, B., Alders, M., Boon, E.M.J., van Haelst, M.M., van den Akker, E.L.T., 2020b. Identifying underlying medical causes of pediatric obesity: Results of a systematic diagnostic approach in a pediatric obesity center. PloS One 15, e0232990. [CrossRef]
- Kleiser, C., Schaffrath Rosario, A., Mensink, G.B.M., Prinz-Langenohl, R., Kurth, B.-M., 2009. Potential determinants of obesity among children and adolescents in Germany: results from the cross-sectional KiGGS Study. BMC Public Health 9, 46. [CrossRef]
- Krude, H., Biebermann, H., Luck, W., Horn, R., Brabant, G., Grüters, A., 1998. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 19, 155–157. [CrossRef]
- Kühnen, P., Krude, H., Biebermann, H., 2019. Melanocortin-4 Receptor Signalling: Importance for Weight Regulation and Obesity Treatment. Trends Mol. Med. 25, 136–148. [CrossRef]
- Kumar, S., Kelly, A.S., 2017. Review of Childhood Obesity: From Epidemiology, Etiology, and Comorbidities to Clinical Assessment and Treatment. Mayo Clin. Proc. 92, 251–265. [CrossRef]
- Kuo, L.E., Kitlinska, J.B., Tilan, J.U., Li, L., Baker, S.B., Johnson, M.D., Lee, E.W., Burnett, M.S., Fricke, S.T., Kvetnansky, R., Herzog, H., Zukowska, Z., 2007. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat. Med. 13, 803–811. [CrossRef]
- Laurila, M., Santaniemi, M., Kesäniemi, Y.A., Ukkola, O., 2014. High plasma ghrelin protects from coronary heart disease and Leu72Leu polymorphism of ghrelin gene from cancer in healthy adults during the 19 years follow-up study. Peptides 61, 122–129. [CrossRef]
- Lemche, E., Chaban, O.S., Lemche, A.V., 2016a. Neuroendocrinological and Epigenetic Mechanisms Subserving Autonomic Imbalance and HPA Dysfunction in the Metabolic Syndrome. Front. Neurosci. 10, 142. [CrossRef]
- Li, Z., Zhou, Y., Carter-Su, C., Myers, M.G., Rui, L., 2007. SH2B1 enhances leptin signaling by both Janus kinase 2 Tyr813 phosphorylation-dependent and -independent mechanisms. Mol. Endocrinol. Baltim. Md 21, 2270–2281. [CrossRef]
- Lin, X., Qi, Q., Zheng, Y., Huang, T., Lathrop, M., Zelenika, D., Bray, G.A., Sacks, F.M., Liang, L., Qi, L., 2015. Neuropeptide Y genotype, central obesity, and abdominal fat distribution: the POUNDS LOST trial. Am. J. Clin. Nutr. 102, 514–519. [CrossRef]
- Lotta, L.A., Mokrosiński, J., Mendes de Oliveira, E., Li, C., Sharp, S.J., Luan, J., Brouwers, B., Ayinampudi, V., Bowker, N., Kerrison, N., Kaimakis, V., Hoult, D., Stewart, I.D., Wheeler, E., Day, F.R., Perry, J.R.B., Langenberg, C., Wareham, N.J., Farooqi, I.S., 2019. Human Gain-of-Function MC4R Variants Show Signaling Bias and Protect against Obesity. Cell 177, 597-607.e9. [CrossRef]
- Mahmoud, R., Kimonis, V., Butler, M.G., 2022. Genetics of Obesity in Humans: A Clinical Review. Int. J. Mol. Sci. 23, 11005. [CrossRef]
- McEwen, B.S., 2007. Physiology and Neurobiology of Stress and Adaptation: Central Role of the Brain. Physiol. Rev. 87, 873–904. [CrossRef]
- Michaud, J.L., Boucher, F., Melnyk, A., Gauthier, F., Goshu, E., Lévy, E., Mitchell, G.A., Himms-Hagen, J., Fan, C.M., 2001. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum. Mol. Genet. 10, 1465–1473. [CrossRef]
- Morash, B., Li, A., Murphy, P.R., Wilkinson, M., Ur, E., 1999. Leptin gene expression in the brain and pituitary gland. Endocrinology 140, 5995–5998. [CrossRef]
- Napoli, E., Tassone, F., Wong, S., Angkustsiri, K., Simon, T.J., Song, G., Giulivi, C., 2015. Mitochondrial Citrate Transporter-dependent Metabolic Signature in the 22q11.2 Deletion Syndrome. J. Biol. Chem. 290, 23240–23253. [CrossRef]
- Nordang, G.B.N., Busk, Ø.L., Tveten, K., Hanevik, H.I., Fell, A.K.M., Hjelmesæth, J., Holla, Ø.L., Hertel, J.K., 2017. Next-generation sequencing of the monogenic obesity genes LEP, LEPR, MC4R, PCSK1 and POMC in a Norwegian cohort of patients with morbid obesity and normal weight controls. Mol. Genet. Metab. 121, 51–56. [CrossRef]
- Obradovic, M., Sudar-Milovanovic, E., Soskic, S., Essack, M., Arya, S., Stewart, A.J., Gojobori, T., Isenovic, E.R., 2021. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 12, 585887. [CrossRef]
- Pan, W.W., Myers, M.G., 2018. Leptin and the maintenance of elevated body weight. Nat. Rev. Neurosci. 19, 95–105. [CrossRef]
- Pépin, L., Colin, E., Tessarech, M., Rouleau, S., Bouhours-Nouet, N., Bonneau, D., Coutant, R., 2019. A New Case of PCSK1 Pathogenic Variant With Congenital Proprotein Convertase 1/3 Deficiency and Literature Review. J. Clin. Endocrinol. Metab. 104, 985–993. [CrossRef]
- Pereira, M.J., Palming, J., Svensson, M.K., Rizell, M., Dalenbäck, J., Hammar, M., Fall, T., Sidibeh, C.O., Svensson, P.-A., Eriksson, J.W., 2014. FKBP5 expression in human adipose tissue increases following dexamethasone exposure and is associated with insulin resistance. Metabolism 63, 1198–1208. [CrossRef]
- Perez-Tilve, D., Hofmann, S.M., Basford, J., Nogueiras, R., Pfluger, P.T., Patterson, J.T., Grant, E., Wilson-Perez, H.E., Granholm, N.A., Arnold, M., Trevaskis, J.L., Butler, A.A., Davidson, W.S., Woods, S.C., Benoit, S.C., Sleeman, M.W., DiMarchi, R.D., Hui, D.Y., Tschöp, M.H., 2010. Melanocortin signaling in the CNS directly regulates circulating cholesterol. Nat. Neurosci. 13, 877–882. [CrossRef]
- Pervanidou, P., 2008. Biology of Post-Traumatic Stress Disorder in Childhood and Adolescence. J. Neuroendocrinol. 20, 632–638. [CrossRef]
- Pervanidou, P., Chrousos, G.P., 2012. Metabolic consequences of stress during childhood and adolescence. Metabolism 61, 611–619. [CrossRef]
- Pervanidou, P., Chrousos, G.P., 2011. Stress and obesity/metabolic syndrome in childhood and adolescence. Int. J. Pediatr. Obes. 6, 21–28. [CrossRef]
- Pervanidou, P., Kolaitis, G., Charitaki, S., Lazaropoulou, C., Papassotiriou, I., Hindmarsh, P., Bakoula, C., Tsiantis, J., Chrousos, G.P., 2007. The Natural History of Neuroendocrine Changes in Pediatric Posttraumatic Stress Disorder (PTSD) After Motor Vehicle Accidents: Progressive Divergence of Noradrenaline and Cortisol Concentrations Over Time. Biol. Psychiatry 62, 1095–1102. [CrossRef]
- Poitou, C., Mosbah, H., Clément, K., 2020. MECHANISMS IN ENDOCRINOLOGY: Update on treatments for patients with genetic obesity. Eur. J. Endocrinol. 183, R149–R166. [CrossRef]
- Qiao, Y., Ma, J., Wang, Y., Li, W., Katzmarzyk, P.T., Chaput, J.-P., Fogelholm, M., Johnson, W.D., Kuriyan, R., Kurpad, A., Lambert, E.V., Maher, C., Maia, J., Matsudo, V., Olds, T., Onywera, V., Sarmiento, O.L., Standage, M., Tremblay, M.S., Tudor-Locke, C., Church, T.S., Zhao, P., Hu, G., ISCOLE Research Group, 2015. Birth weight and childhood obesity: a 12-country study. Int. J. Obes. Suppl. 5, S74-79. [CrossRef]
- Reyes, M., Quintanilla, C., Burrows, R., Blanco, E., Cifuentes, M., Gahagan, S., 2015. Obesity is associated with acute inflammation in a sample of adolescents. Pediatr. Diabetes 16, 109–116. [CrossRef]
- Roy, M.S., Roy, A., Gallucci, W.T., Collier, B., Young, K., Kamilaris, T.C., Chrousos, G.P., 1993. The ovine corticotropin-releasing hormone-stimulation test in type I diabetic patients and controls: Suggestion of mild chronic hypercortisolism. Metabolism 42, 696–700. [CrossRef]
- Rubens, M., Bruenig, D., Adams, J.A.M., Suresh, S.M., Sathyanarayanan, A., Haslam, D., Shenk, C.E., Mathews, B., Mehta, D., 2023. Childhood maltreatment and DNA methylation: A systematic review. Neurosci. Biobehav. Rev. 147, 105079. [CrossRef]
- Rui, L., 2014. SH2B1 regulation of energy balance, body weight, and glucose metabolism. World J. Diabetes 5, 511–526. [CrossRef]
- Saeed, S., Arslan, M., Manzoor, J., Din, S.M., Janjua, Q.M., Ayesha, H., Ain, Q.-T., Inam, L., Lobbens, S., Vaillant, E., Durand, E., Derhourhi, M., Amanzougarene, S., Badreddine, A., Berberian, L., Gaget, S., Khan, W.I., Butt, T.A., Bonnefond, A., Froguel, P., 2020. Genetic Causes of Severe Childhood Obesity: A Remarkably High Prevalence in an Inbred Population of Pakistan. Diabetes 69, 1424–1438. [CrossRef]
- Sidibeh, C.O., Pereira, M.J., Abalo, X.M., J. Boersma, G., Skrtic, S., Lundkvist, P., Katsogiannos, P., Hausch, F., Castillejo-López, C., Eriksson, J.W., 2018. FKBP5 expression in human adipose tissue: potential role in glucose and lipid metabolism, adipogenesis and type 2 diabetes. Endocrine 62, 116–128. [CrossRef]
- Singh, R.K., Kumar, P., Mahalingam, K., 2017. Molecular genetics of human obesity: A comprehensive review. C. R. Biol. 340, 87–108. [CrossRef]
- Sitticharoon, C., Chatree, S., Churintaraphan, M., 2013. Expressions of neuropeptide Y and Y1 receptor in subcutaneous and visceral fat tissues in normal weight and obese humans and their correlations with clinical parameters and peripheral metabolic factors. Regul. Pept. 185, 65–72. [CrossRef]
- Sohn, Y.B., 2022. Genetic obesity: an update with emerging therapeutic approaches. Ann. Pediatr. Endocrinol. Metab. 27, 169–175. [CrossRef]
- Straznicky, N.E., Lambert, G.W., Masuo, K., Dawood, T., Eikelis, N., Nestel, P.J., McGrane, M.T., Mariani, J.A., Socratous, F., Chopra, R., Esler, M.D., Schlaich, M.P., Lambert, E.A., 2009. Blunted sympathetic neural response to oral glucose in obese subjects with the insulin-resistant metabolic syndrome. Am. J. Clin. Nutr. 89, 27–36. [CrossRef]
- Styne, D.M., Arslanian, S.A., Connor, E.L., Farooqi, I.S., Murad, M.H., Silverstein, J.H., Yanovski, J.A., 2017. Pediatric Obesity-Assessment, Treatment, and Prevention: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 102, 709–757. [CrossRef]
- Thorsell, A., 2010. Brain neuropeptide Y and corticotropin-releasing hormone in mediating stress and anxiety. Exp. Biol. Med. Maywood NJ 235, 1163–1167. [CrossRef]
- Tsigos, C., Young, R.J., White, A., 1993. Diabetic neuropathy is associated with increased activity of the hypothalamic-pituitary-adrenal axis. J. Clin. Endocrinol. Metab. 76, 554–558. [CrossRef]
- Wabitsch, M., Funcke, J.-B., von Schnurbein, J., Denzer, F., Lahr, G., Mazen, I., El-Gammal, M., Denzer, C., Moss, A., Debatin, K.-M., Gierschik, P., Mistry, V., Keogh, J.M., Farooqi, I.S., Moepps, B., Fischer-Posovszky, P., 2015. Severe Early-Onset Obesity Due to Bioinactive Leptin Caused by a p.N103K Mutation in the Leptin Gene. J. Clin. Endocrinol. Metab. 100, 3227–3230. [CrossRef]
- Wasim, M., Awan, F.R., Najam, S.S., Khan, A.R., Khan, H.N., 2016. Role of Leptin Deficiency, Inefficiency, and Leptin Receptors in Obesity. Biochem. Genet. 54, 565–572. [CrossRef]
- Wei, Z., Zhang, K., Wen, G., Balasubramanian, K., Shih, P.B., Rao, F., Friese, R.S., Miramontes-Gonzalez, J.P., Schmid-Schoenbein, G.W., Kim, H.-S., Mahata, S.K., O’Connor, D.T., 2013. Heredity and cardiometabolic risk: naturally occurring polymorphisms in the human neuropeptide Y(2) receptor promoter disrupt multiple transcriptional response motifs. J. Hypertens. 31, 123–133. [CrossRef]
- Wolf, E.J., Mitchell, K.S., Logue, M.W., Baldwin, C.T., Reardon, A.F., Humphries, D.E., Miller, M.W., 2013. CORTICOTROPIN RELEASING HORMONE RECEPTOR 2 ( CRHR-2 ) GENE IS ASSOCIATED WITH DECREASED RISK AND SEVERITY OF POSTTRAUMATIC STRESS DISORDER IN WOMEN: CRHR-2 and PTSD. Depress. Anxiety 30, 1161–1169. [CrossRef]
- Womersley, J.S., Nothling, J., Toikumo, S., Malan-Müller, S., van den Heuvel, L.L., McGregor, N.W., Seedat, S., Hemmings, S.M.J., 2022. Childhood trauma, the stress response and metabolic syndrome: A focus on DNA methylation. Eur. J. Neurosci. 55, 2253–2296. [CrossRef]
- Xia, Q., Grant, S.F.A., 2013. The genetics of human obesity. Ann. N. Y. Acad. Sci. 1281, 178–190. [CrossRef]
- Yang, Y., van der Klaauw, A.A., Zhu, L., Cacciottolo, T.M., He, Y., Stadler, L.K.J., Wang, C., Xu, P., Saito, K., Hinton, A., Yan, X., Keogh, J.M., Henning, E., Banton, M.C., Hendricks, A.E., Bochukova, E.G., Mistry, V., Lawler, K.L., Liao, L., Xu, J., O’Rahilly, S., Tong, Q., UK10K Consortium, Inês Barroso, null, O’Malley, B.W., Farooqi, I.S., Xu, Y., 2019. Steroid receptor coactivator-1 modulates the function of Pomc neurons and energy homeostasis. Nat. Commun. 10, 1718. [CrossRef]
- Yeo, G.S.H., Chao, D.H.M., Siegert, A.-M., Koerperich, Z.M., Ericson, M.D., Simonds, S.E., Larson, C.M., Luquet, S., Clarke, I., Sharma, S., Clément, K., Cowley, M.A., Haskell-Luevano, C., Van Der Ploeg, L., Adan, R.A.H., 2021. The melanocortin pathway and energy homeostasis: From discovery to obesity therapy. Mol. Metab. 48, 101206. [CrossRef]
Figure 1.
Neuroendocrine pathways of the stress response. The stress system consists of the hypothalamic-pituitary-adrenal axis (HPA axis) and the arousal/sympathetic system at the central level, while the peripheral effectors are represented by glucocorticoids and catecholamines. The two systems interact with each other at different levels. CRH and NE are the main central mediators and receive stimulatory afferents from the amygdala/hippocampus complex, the mesocorticolimbic system, serotonin, and acetylcholine systems. CRH is secreted by the parvocellular neurons of the hypothalamus, while NE is mainly synthesized in the medulla and pons at locus coeruleus level. They, in turn, stimulate the anterior pituitary gland to secrete ACTH and the adrenal medulla to secrete epinephrine and norepinephrine, respectively. The ACTH release leads to glucocorticoid synthesis by the cortex of the adrenal gland. Catecholamines and glucocorticoids then act in synergy on target organs and tissues. The main metabolic consequences are gluconeogenesis, glycogen synthesis, decreased glucose uptake, insulin resistance, visceral obesity, a low grade of chronic inflammation, and a higher risk of glucose intolerance, type 2 diabetes, and MetS. Moreover, catecholamines lead to increased cardiac output, heart rate, and blood pressure, with a higher risk of hypertension and cardiovascular diseases.CRH= corticotrophin-releasing hormone; NE= norepinephrine; ACTH= adrenocorticotropin hormone; MetS= metabolic Syndrome.
Figure 1.
Neuroendocrine pathways of the stress response. The stress system consists of the hypothalamic-pituitary-adrenal axis (HPA axis) and the arousal/sympathetic system at the central level, while the peripheral effectors are represented by glucocorticoids and catecholamines. The two systems interact with each other at different levels. CRH and NE are the main central mediators and receive stimulatory afferents from the amygdala/hippocampus complex, the mesocorticolimbic system, serotonin, and acetylcholine systems. CRH is secreted by the parvocellular neurons of the hypothalamus, while NE is mainly synthesized in the medulla and pons at locus coeruleus level. They, in turn, stimulate the anterior pituitary gland to secrete ACTH and the adrenal medulla to secrete epinephrine and norepinephrine, respectively. The ACTH release leads to glucocorticoid synthesis by the cortex of the adrenal gland. Catecholamines and glucocorticoids then act in synergy on target organs and tissues. The main metabolic consequences are gluconeogenesis, glycogen synthesis, decreased glucose uptake, insulin resistance, visceral obesity, a low grade of chronic inflammation, and a higher risk of glucose intolerance, type 2 diabetes, and MetS. Moreover, catecholamines lead to increased cardiac output, heart rate, and blood pressure, with a higher risk of hypertension and cardiovascular diseases.CRH= corticotrophin-releasing hormone; NE= norepinephrine; ACTH= adrenocorticotropin hormone; MetS= metabolic Syndrome.
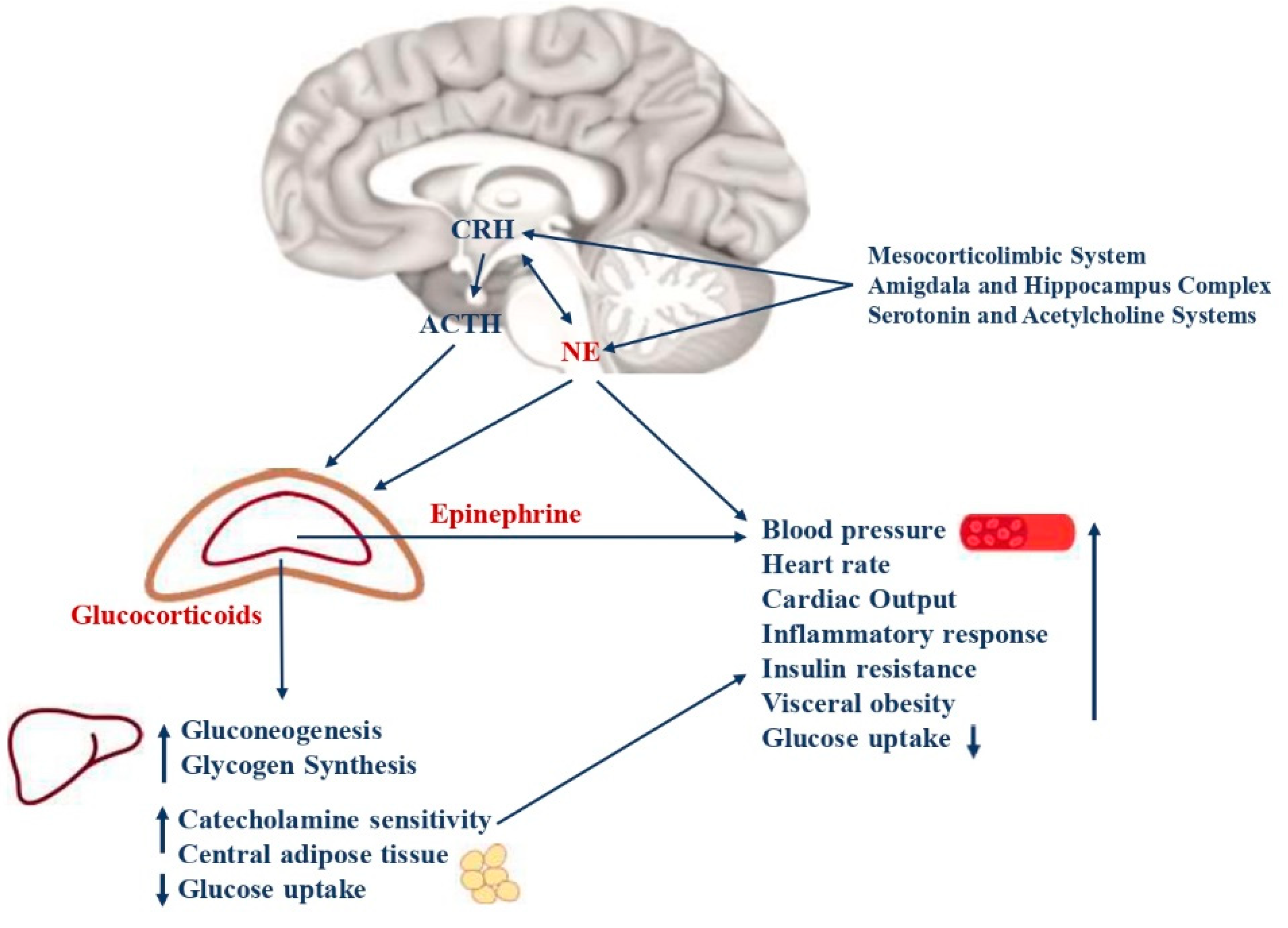
Figure 2.
The main hypothalamic neuroendocrine pathways involved in hunger and satiety. Ghrelin is mostly produced by neuroendocrine cells of the gastrointestinal tract. White adipose cells are the primary source of leptin production, and obese patients have higher levels of leptin compared to normal-weight individuals. Ghrelin and BDNF stimulate NPY/AgRP neurons to express GABA, thereby inhibiting POMC neurons in the arcuate nucleus of the Hypothalamus. In addition, NPY and AgRP antagonize the MSH-MC4R binding to a large family of neurons in the paraventricular nucleus of the Hypothalamus, resulting in hunger and fat deposition. Also, leptin stimulates the cleavage of POMC into melanocortins, promoting satiety and energy expenditure. BDNF= brain-derived neurotrophic factor; NPY= neuropeptide Y; AgRP= agouti-related protein; GABA= γ-aminobutyric acid; POMC = pro-opiomelanocortin; MSH = melanocyte-stimulating hormones; MC4R = melanocortin receptor type 4.
Figure 2.
The main hypothalamic neuroendocrine pathways involved in hunger and satiety. Ghrelin is mostly produced by neuroendocrine cells of the gastrointestinal tract. White adipose cells are the primary source of leptin production, and obese patients have higher levels of leptin compared to normal-weight individuals. Ghrelin and BDNF stimulate NPY/AgRP neurons to express GABA, thereby inhibiting POMC neurons in the arcuate nucleus of the Hypothalamus. In addition, NPY and AgRP antagonize the MSH-MC4R binding to a large family of neurons in the paraventricular nucleus of the Hypothalamus, resulting in hunger and fat deposition. Also, leptin stimulates the cleavage of POMC into melanocortins, promoting satiety and energy expenditure. BDNF= brain-derived neurotrophic factor; NPY= neuropeptide Y; AgRP= agouti-related protein; GABA= γ-aminobutyric acid; POMC = pro-opiomelanocortin; MSH = melanocyte-stimulating hormones; MC4R = melanocortin receptor type 4.
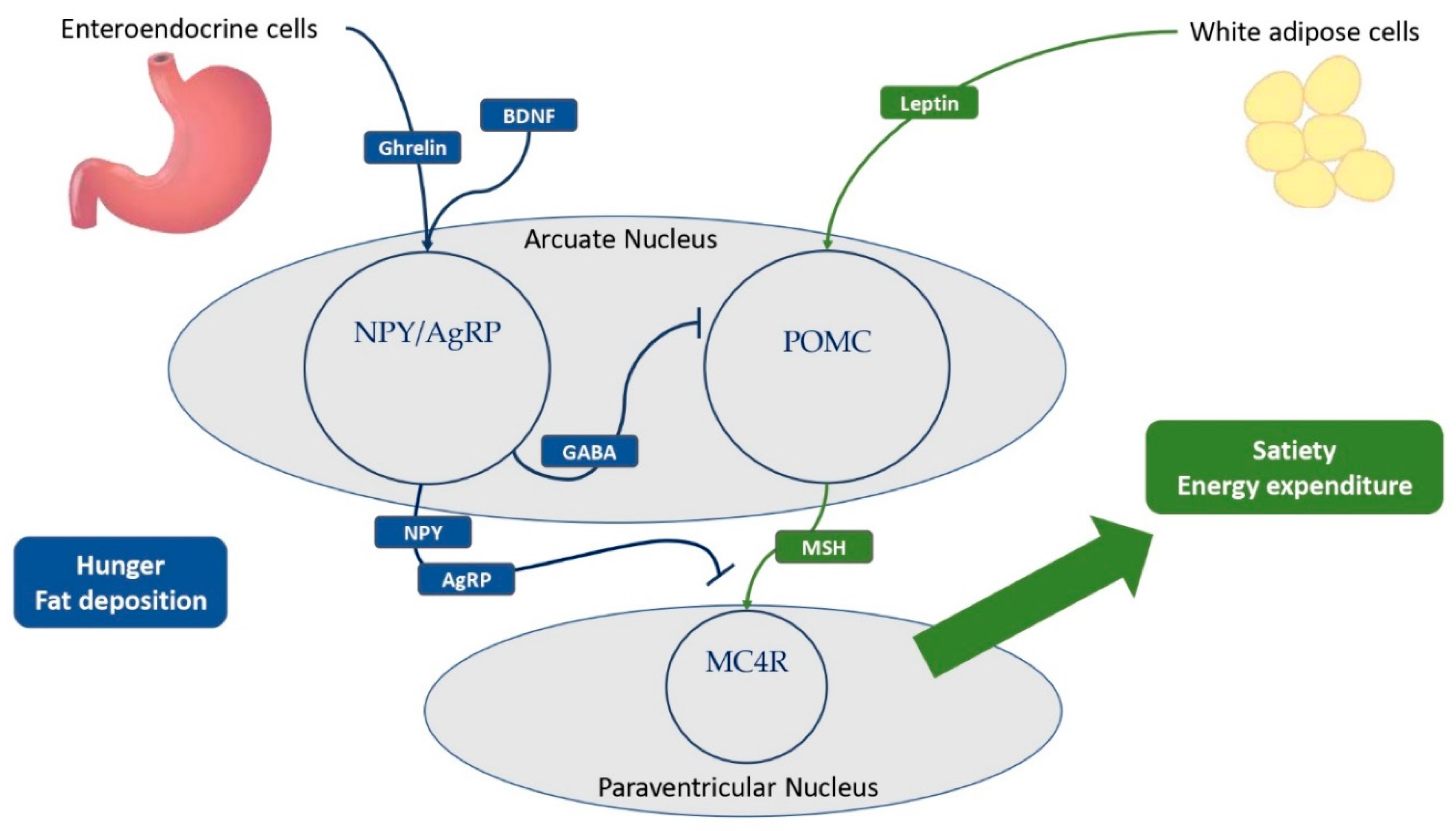
Figure 3.
The leptin-melanocortin pathway and its interactions with the NPY-AgRP neuroendocrine system A huge population of neurons in the Arcuate nucleus produce pro-opiomelanocortin (POMC), which is then cleaved into melanocortins by PCSK1, CPE, and SRC-1 upon leptin and its receptor (LEPR) binding. SH2B1 is a crucial adaptor protein involved in the leptin-LEPR pathway. The various MSH isoforms bind MC4Rs, which are highly expressed by MC4 neurons in the Paraventricular nucleus of the hypothalamus, resulting in multiple gene expression that promotes satiety and decreased food intake. The stimulation of NYP/AgRP neurons by ghrelin, BDNF, and their own receptors results in a massive production of both NPY and AgRP. Their main goal is to antagonize MSH from binding with MC4Rs to increase hunger and improve food intake. Leptin also inhibits NPY/AgRP neurons and their suppression of POMC neurons via GABAergic systems. POMC = pro-opiomelanocortin; PCSK1= proprotein convertase subtilisin/kexin type 1; CPE= carboxypeptidase E; SRC-1= steroid receptor coactivator 1; LEPR = leptin receptor; SH2B1 = SH2B adapter protein 1; MSH= melanocyte-stimulating hormones; MC4R = melanocortin receptor type 4; NPY = neuropeptide Y; AgRP = agouti-related protein; GABA = γ-aminobutyric acid.
Figure 3.
The leptin-melanocortin pathway and its interactions with the NPY-AgRP neuroendocrine system A huge population of neurons in the Arcuate nucleus produce pro-opiomelanocortin (POMC), which is then cleaved into melanocortins by PCSK1, CPE, and SRC-1 upon leptin and its receptor (LEPR) binding. SH2B1 is a crucial adaptor protein involved in the leptin-LEPR pathway. The various MSH isoforms bind MC4Rs, which are highly expressed by MC4 neurons in the Paraventricular nucleus of the hypothalamus, resulting in multiple gene expression that promotes satiety and decreased food intake. The stimulation of NYP/AgRP neurons by ghrelin, BDNF, and their own receptors results in a massive production of both NPY and AgRP. Their main goal is to antagonize MSH from binding with MC4Rs to increase hunger and improve food intake. Leptin also inhibits NPY/AgRP neurons and their suppression of POMC neurons via GABAergic systems. POMC = pro-opiomelanocortin; PCSK1= proprotein convertase subtilisin/kexin type 1; CPE= carboxypeptidase E; SRC-1= steroid receptor coactivator 1; LEPR = leptin receptor; SH2B1 = SH2B adapter protein 1; MSH= melanocyte-stimulating hormones; MC4R = melanocortin receptor type 4; NPY = neuropeptide Y; AgRP = agouti-related protein; GABA = γ-aminobutyric acid.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



