
Submitted:
26 May 2023
Posted:
30 May 2023
You are already at the latest version
Abstract
The methyltransferase, KMT5A has been proposed as an oncogene in prostate cancer and therefore represents a putative therapeutic target. To confirm this hypothesis we have performed a microarray study in a prostate cancer cell line model of androgen independence following KMT5A knockdown in the presence of transcriptionally active androgen receptor (AR) to understand which genes and cellular processes are regulated by KMT5A in the presence of an active AR. We observed that 301 genes were down-regulated whilst 408 were up-regulated when KMT5A expression was reduced. KEGG pathway and Gene Ontology analysis revealed apoptosis and DNA damage signaling are up-regulated in response to KMT5A knockdown whilst protein folding and RNA splicing were down-regulated. Under these conditions, the top non-AR regulated gene was found to be CDC20, a key regulator of the spindle assembly checkpoint with an oncogenic role in several cancer types. Further investigation revealed that KMT5A regulates CDC20 in a methyltransferase dependent manner to modulate both histone H4K20 methylation within its promoter region and indirectly via the p53 signalling pathway. A positive correlation between KMT5A and CDC20 expression was also observed in clinical prostate cancer samples further supporting this association. Therefore, we conclude that KMT5A is a valid therapeutic target for the treatment of prostate cancer and CDC20 could potentially be utilized as a biomarker for effective therapeutic targeting.
Keywords:
CDC20
; BIOMARKER
; KMT5A
; p53
; prostate cancer
Introduction
Prostate cancer is the most common cancer in men in the UK. Whilst androgen receptor (AR) targeting therapies have yielded significant patient benefit, relapse to treatment is a significant clinical problem. Hence, there is an urgent need to develop alternative therapeutics to treat advanced disease. The lysine methyltransferase, KMT5A, plays an oncogenic role in a number of cancers [1-3]. Indeed, KMT5A siRNA-mediated knockdown inhibits prostate cancer cell proliferation and KMT5A has been identified as an AR interacting protein that is required for the transcription of the AR regulated gene, prostate specific antigen (PSA) via promotion of H4K20Me1 at the PSA promoter [4]. Furthermore, KMT5A plays a role in epithelial-mesenchymal transition (EMT) and enhances the invasiveness of prostate cancer cell line models, independent of the AR through its interplay with ZEB1 [5]. Initially identified as the sole methyltransferase responsible for the mono-methylation of histone H4 at lysine 20 (H4K20Me1), KMT5A was subsequently shown to methylate numerous other non-histone proteins, including p53 [6]. Greater understanding of KMT5A in the context of prostate cancer is required to determine whether it is a bona fide therapeutic target.
KMT5A activity is regulated by post-translational mechanisms during specific phases of the cell cycle. During late S phase and at the G2/M transition, the levels of KMT5A are at their peak and found localized to mitotic chromosomes. As the cell moves through prophase to anaphase, KMT5A is phosphorylated at serine 29 by cdk1/cyclin B. This results in KMT5A dissociation from chromatin and stabilisation by inhibiting KMT5A association with the APCcdh1 E3 ubiquitin ligase [7]. During anaphase, KMT5A is dephosphorylated by cdc14a/b, which in turn permits protein turnover to reduce KMT5A protein levels at G1. During G1 KMT5A levels are sustained, however during the G1/S transition, SCFskp2 ubiquitin ligase targets KMT5A for protein turnover resulting in undetectable KMT5A protein. Interestingly, KMT5A interacts with proliferating cell nuclear antigen (PCNA) at DNA replication foci and is essential for correct DNA replication [8] suggesting a high turnover rate of chromatin bound KMT5A by CRL4cdt2 [7]. The alterations in the levels of KMT5A throughout the cell cycle are mirrored by H4K20Me1 levels suggesting that methyltransferase activity is predominantly regulated by cellular KMT5A levels.
Cell cycle division 20 homologue (CDC20) is a cell cycle regulatory protein implicated in the spindle assembly checkpoint (SAC) and is required for cells to progress through mitosis. Specifically, CDC20 functions as a substrate recognition molecule and activator of APC to result in ubiquitin mediated turnover of its substrates. In particular, APCCDC20 functions during metaphase to anaphase to result in the destruction of cyclin B and securin thereby allowing sister chromatids to segregate. CDC20 activity is inhibited by the Mitotic Checkpoint Complex (MCC) and is only released to target its substrates once microtubule binding to kinetochore and appropriate tension is achieved thereby preventing genomic instability. Interestingly, there are suggestions that CDC20 may play a role in the DNA damage repair pathway via RAP80 [9] and REV1 [10] down-regulation. Furthermore, DNA damage induced p53 can directly inhibit the expression of CDC20 by associating with the CDC20 promoter region and causing chromatin remodelling [11]. In addition, p21 can inhibit CDC20 mRNA by associating with CDE-CHR elements in the CDC20 promoter [12]. Depletion of PHF8, an H4K20Me1 demethylase, results in prolonged G2 and defective mitosis and is itself a substrate of APCCDC20 [13] further suggesting that chromatin remodeling can be influenced by CDC20 levels.
CDC20 has been proposed to exhibit an oncogenic role in a number of cancers including prostate cancer [14]. Indeed, biochemical recurrence free survival is lower in patients with high levels of CDC20 compared to patients with low CDC20 expression [15]. CDC20 itself is a target for ubiquitination by the E3 ligase SPOP, which is commonly mutated and non-functional in prostate cancers providing an explanation for elevated CDC20 levels [16]. Furthermore, CDC20 expression is associated with resistance to docetaxel [16,17] and is implicated in the wnt/Beta-catenin pathway which is oncogenic in advanced prostate cancer [17,18].
The aim of this study was two-fold; first to use pathway analysis to provide further evidence that KMT5A regulates oncogenic pathways and is a valid therapeutic target in prostate cancer and secondly, to identify individual genes that are regulated by KMT5A in a model of castration resistant prostate cancer as potential biomarkers for KMT5A activity. Indeed, we show that a number of oncogenic pathways are downregulated upon KMT5A knockdown and we identified and validated CDC20 as a KMT5A regulated gene.
Materials and Methods
Antibodies
Antibodies used in this study included, KMT5A (Cell signaling), CDC20 (Ab190711, AbCam), PARP1/2 (clone H250, sc-7150, Santa Cruz Biotechnology), MDM2 (Clone N-20, sc-813, Santa Cruz Biotechnology), p21 (ab-4, Calbiochem), p53 (pAb-421#OP03, Calbiochem), p53-S15-P (Cell signaling), p53-K382-Ac (ab75754, AbCam), H4K20Me1 (Ab9051, AbCam), H4 (07-108, Merck), anti-phospho-histone H2AX (Ser139) (clone JBW301, Millipore Corp) α-tubulin (clone DM1A, T9026, Sigma), GAPDH (clone 1E6D9, Proteintech).
Compounds
Dihydrotestosterone (DHT) (Sigma) was prepared in ethanol at a final concentration of 10 mM and stored at -80°C. KMT5A inhibitors UNC0379 (S7570, Selleckchem) and Ryuvidine (2609, R&D) were purchased in powder form and resuspended in DMSO to a final concentration of 50 mM and 20 mM, respectively. Solutions were stored at -80°C for no longer than 1 month. Nutlin 3 was provided by Prof. John Lunec (Newcastle Cancer Centre).
Cell Culture
LNCaP cells, a model of androgen dependence, and AR negative PC3 cells were purchased from the American Type Culture Collection (Manassas, VA, USA), LNCaP-AI cells, a model of androgen independence, were generated in house as described previously [19]. Cells were maintained as previously described [20].
Cell line authentication was performed by short-tandem repeat profiling (NewGene, Newcastle upon Tyne, UK) and mycoplasma testing was performed routinely using MycoAlert (Lonza, UK).
siRNA
Cell lines were reverse transfected with siRNA sequences (25 nM) using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Either qPCR or western blotting confirmed successful knockdown. Non-silencing (N/S): UUCUCCGAACGUGUCACGU[dT][dT]; siKMT5A_1: CCAUGAAGUCCGAGGAACA[dT][dT]; siKMT5A_2: GATGCAACTAGAGAGACA[dT][dT]; siCDC20_1 CGGAAGACCUGCCGUUACA[dT][dT]; siCDC20_2: GGGCCGAACUCCUGGCAAA[dT][dT].
Western Blotting and Quantitative Polymerase Chain Reaction
Western [21] and qPCR analysis [20] were performed as described previously. Primer sequences are detailed in Supplementary Table S1.
Microarray
RNA was isolated using Trizol® (Invitrogen) according to the manufacturer’s instructions. RNA quality was determined using Agilent bioanalyzer 2100 prior to analysis using the Illumina HT-12 v4.0 Expression BeadChip (Oxford Genomics Centre, The Wellcome Trust Centre for Human Genetics, University of Oxford).
Array processing, background correction, normalisation and quality control checks were performed using the R package ‘Lumi’. Probe intensity values were converted to VSD (variance stabilized data) using variance stabilising transformation. The robust spline normalization (RSN) was used as an array normalisation method. Outlier samples, poor quality probes (detection threshold < 0.01) and probes that were not detected were removed from downstream analysis. Differential expression analysis was then performed using the R package ‘Limma’ and p-values adjusted to control for false discovery rate (FDR) using the Benjamini-Hochberg method [22]. Analysis was performed by the Bioinformatics Support Unit (Newcastle University).
Data can be found at GSE233350
RNA-seq analysis
Fastq files were downloaded from NCBI GEO (GSE211638, [23]) and RNA-STAR [24] analysis was performed to align raw reads to genome build GRCh37/hg19, QC checks were performed with FastQC. Gene counts were generated using ht-seq count [25] and Gencode v19. Differential expression analysis was carried out using the DESeq2 [26] package (R/Bioconductor) to compare vehicle versus 10nM DHT treated samples
Chromatin Immunoprecipitation Assays
Chromatin immunoprecipitation assays were performed in LNCaP and LNCaP-AI cells reverse transfected with either 25 nM N/S or a pool of 2 KMT5A targeting siRNAs for 72 h in steroid depleted media according to the protocol described by Schmidt et al. [27].
For immunoprecipitations, 2 μg of H4K20Me1 (Ab9051, AbCam) or 2 μg of a non-specific isotype control (DAKO) were used. QPCR analysis of immunoprecipitated DNA was performed using primers specific to the CDC20 promoter (Fwd: 5’-CCGCTAGACTCTCGTGATAGC-3’; Rev: 5’-TGGCTCCTTCAAAATCCAAC-3’) as previously described [28]. Data was calculated as % input and presented as the average fold difference of % input between experimental arms for at least three independent experiments.
Sulforhodamine B Growth Analysis
Growth assays were performed as previously described [21].
Gamma H2AX assay
LNCaP-AI and LNCaP cells were reverse transfected with either N/S or KMT5A targeting siRNA and incubated for 72 hours. Cells were collected and stained for phospho-histone H2AX (Ser139) as previously described [21].
Results
Identification of KMT5A regulated genes in androgen-independent prostate cancer
KMT5A has been proposed as a therapeutic target in prostate cancer however, in this context, KMT5A is still largely understudied. Indeed, no study has identified which genes KMT5A can regulate in castration resistant prostate cancer. To this end, KMT5A mRNA was knocked down using 2 independent siRNA sequences in the LNCaP-AI cell line model of androgen independence. After 72 hours of knockdown under steroid depleted conditions the androgen, DHT (10 nM), was applied for 24 hours prior to RNA isolation and analysis using Illumina Human HT-12 microarray.
Significant knockdown of KMT5A, with both siRNAs, was confirmed within the microarray data set as >80% prior to further analysis (Figure 1A). In the presence of an active AR by stimulation with DHT for 24 hours, we found 408 genes up-regulated and 310 genes down-regulated. (Supplementary Tables S2 and S3). Of these genes, 29% have previously been shown to be AR regulated in LNCaP cells (Supplementary Tables S2 and S3) [23]. In order to understand which cellular pathways and biological processes are affected under these conditions the gene lists generated were used in KEGG pathway analysis and gene ontology using DAVID [29,30]. This analysis revealed significant up-regulation of PI3K-Akt signaling, apoptosis, p53 signalling and signal transduction whilst pathways found to be significantly downregulated included splicing, protein folding, cell division and transcriptional regulation (Supplementary Tables S4 and S5). Taken together, the cellular processes and genes altered in this analysis further supports our hypothesis that KMT5A is a potential therapeutic target for prostate cancer.
In terms of individual genes which were down-regulated in response to KMT5A knockdown, CDC20 was identified as the 6th most down-regulated gene after AR regulated genes such as KRT8 and SPDEF (Figure 1B,C). Due to its role in the cell cycle and previous characterization as an oncogene [14,17], this gene was chosen for further study as a potential pharmacodynamic biomarker for KMT5A therapeutic targeting and a KMT5A effector protein.
KMT5A depletion reduces CDC20 expression
In order to validate CDC20 as a KMT5A regulated gene, further experiments were conducted in both LNCaP-AI cells and the parental, androgen sensitive LNCaP cell line. KMT5A targeting siRNAs were transfected into both cell lines in steroid depleted media for 72 hours prior to stimulation with 10 nM DHT or vehicle for a further 24 hours. QPCR confirmed a significant reduction in the expression of CDC20 in LNCaP-AI cells (Figure 2A) when KMT5A was knocked down (p<0.01) (Figure 2B), consistent with our microarray data (Figure 1C). In addition, parental LNCaP cells also exhibited a significant reduction in CDC20 expression (Figure 2C) irrespective of DHT stimulation upon significant KMT5A knockdown (p<0.001) (Figure 2D). Furthermore, a robust reduction in CDC20 protein levels was consistently observed in both cell lines (Figure 2E,F) confirming that CDC20 is regulated by KMT5A, at the level of transcription, in both cell lines irrespective of AR activation.
CDC20 depletion does not enhance KMT5A protein expression
KMT5A is phosphorylated to protect it from ubiquitin mediated degradation by APCcdh1 during late mitosis [7]. In addition, due to the similarity in recognition mechanisms between cdh1 and cdc20 for targeting proteins to the APC complex, it was suggested that CDC20 may also bind and recognise KMT5A in the absence of phosphorylation [7]. This raised the question of whether a feedback mechanism exists between these two proteins to help maintain correct cell cycle progression. To test this theory, CDC20 was knocked down in our cell line models and KMT5A levels were assessed at both the transcript and protein level. Interestingly, when KMT5A protein levels were examined subsequent to CDC20 knockdown no change was observed (Figure 3A,B) suggesting that KMT5A protein turnover is not taking place when CDC20 is present in the cell. However, a decrease in KMT5A transcripts by ~50% was observed in both cell lines (Figure 3C,D) upon robust depletion of CDC20 (Figure 3E,F) although this was not statistically significant. Therefore, it was concluded that CDC20 was not playing a significant role in KMT5A protein regulation under our experimental conditions and KMT5A sits upstream of CDC20.
KMT5A expression correlates with CDC20 expression in prostate cancer patients
To confirm whether our in vitro findings could be translated into clinical specimens of prostate cancer we interrogated publicly available datasets to confirm a positive correlation between CDC20 and KMT5A transcripts. Upon interrogation of data sets available in cBioportal [31,32] we found a significant positive correlation between KMT5A and CDC20 transcripts in a number of data sets. In a cohort of 65 treatment naïve radical prostatectomies [33] a positive correlation between CDC20 and KMT5A transcripts was observed (Spearman = 0.45; p = 0.0002) (Figure 4A). Similarly, in the MCTP dataset [34], a positive correlation was also observed (Spearman = 0.23; p = 0.024) (Figure 4B). However, this dataset contains samples from primary (blue) and metastatic prostate cancer (red). Upon correlation analysis for these individual sample types it was observed that the correlation between CDC20 and KMT5A was strongest in the primary prostate samples (Spearman = 0.28; p = 0.033; n = 59) and no statistically significant correlation was observed in the metastatic samples (Spearman = 0.034; p = 0.85; n=35), although the sample numbers are lower. However, in the metastatic cohort reported by Robinson et al [35] a statistically significant correlation was observed between CDC20 and KMT5A (Spearman = 0.22; p= 0.016; n=118) (Figure 4C). Furthermore, significant correlation were observed in bone metastases (Spearman = 0.41; p= 0.0003; n=72) (Figure 4D) and liver metastases (Spearman = 0.408; p= 0.011; n=38)(Figure 4E) in the samples from Abida et al [36]. Interestingly, the highest positive correlation between CDC20 and KMT5A expression was seen in prostate neuroendocrine carcinoma samples (Spearman = 0.68; p <0.0001; n=49) (Figure 4F) [37]. Taken together, this suggests that the positive correlation between KMT5A and CDC20 observed in our cell line models is also observed in advanced prostate cancer.
KMT5A inhibition reduces CDC20 expression and reduces prostate cancer cell proliferation.
KMT5A plays a role in the cell cycle and as such, knockdown of KMT5A has been shown to inhibit cellular proliferation [38,39]. Indeed, we observed a reduction in proliferation upon KMT5A knockdown in both the cell line models used in this study (Supplementary Figure S1A-C). In particular, proliferation was most affected under steroid depleted conditions. Furthermore, as expected, knockdown of CDC20 resulted in a robust and significant inhibition of cellular proliferation (Supplementary Figure S1D,E). Together this provides supporting evidence that both proteins play a role in prostate cancer cell proliferation.
In order to confirm that the methyltransferase activity of KMT5A is important for the regulation of CDC20 expression in prostate cancer cell lines we used two molecules that have shown inhibitory activity against KMT5A, namely UNC0379 and Ryuvidine [40,41] (Supplementary Figure S2). Firstly, we determined the GI50 values for LNCaP and LNCaP-AI cells (Figure 5A). Interestingly, we found that Ryuvidine was a more potent inhibitor than UNC0379 in LNCaP cells, however in LNCaP-AI cells there was not such a large difference in efficacy. Secondly, we used a titration of doses of both inhibitors and investigated the dose dependent effects on both KMT5A and its target histone mark, H4K20Me1. We observed that UNC0379 resulted in a robust decrease in H4K20Me1 levels in LNCaP-AI and a modest reduction in LNCaP cells when total H4 levels are taken into account. This coincided with a decrease in KMT5A protein levels in LNCaP cells whilst KMT5A levels showed minimal change in LNCaP-AI cells. Ryuvidine also demonstrated a dose dependent reduction in KMT5A activity in LNCaP cells whilst a decrease in H4K20Me1 was more difficult to achieve in LNCaP-AI cells thereby reflecting the sensitivity differences to Ryuvidine between these two cell lines (Supplementary Figures S2 and S3).
Using the GI50 concentrations for each drug in LNCaP-AI cells we investigated the levels of CDC20 by western blotting, demonstrating that both drugs result in CDC20 reduction (Figure 5B). Furthermore, when the more potent Ryuvidine was used in LNCaP cells at the GI50 dose a reduction in CDC20 levels was observed (Figure 5C). Together, this led us to conclude that KMT5A enzymatic activity is important in the regulation of CDC20 protein levels.
KMT5A knockdown reduces H4K20Me1 at the CDC20 promoter
In order to confirm that KMT5A can directly regulate the expression of CDC20 via mono-methylation of its only histone target, H4K20, chromatin immunoprecipitation assays were performed in both LNCaP-AI and LNCaP cells subsequent to KMT5A knockdown using an siRNA pool of siKMT5A_1 and siKMT5A_2. Upon KMT5A knockdown in both cell lines growing in steroid depleted media, a significant reduction in H4K20Me1 was observed at the CDC20 promoter region (Figure 6A,B). This led us to conclude that KMT5A can directly modulate the expression of CDC20 by methylation of H4K20 within the promoter region.
P53 mediates KMT5A regulation of CDC20 expression
Whilst KMT5A methyltransferase activity is important in regulating the expression of CDC20 via regulation of H4K20Me1 within the CDC20 promoter, KMT5A can also methylate non-histone proteins, including p53, to regulate functional activity. Interestingly, in our pathway analysis we uncovered some pathways within which CDC20 can be modulated. In particular, p53 directly down-regulates CDC20 expression by association with its promoter in response to DNA damage [11]. Secondly, in the absence of DNA damage, p53 can regulate CDC20 expression via a CDE-CHR element, independent of p21, when p53 is over-expressed [11,14,42]. To determine whether DNA damage in response to KMT5A knockdown was influencing this mechanism we investigated the levels of γ-H2AX in both LNCaP-AI and LNCaP cells after KMT5A knockdown. Consistent with other reports [39], KMT5A knockdown resulted in an increased level of DNA damage as denoted by a robust ~3.5 fold and ~2 fold increase in γ-H2AX levels in LNCaP-AI cells and LNCaP cells, respectively (Figure 7A) further suggesting that p53 could be a mediator of KMT5A effects on CDC20 levels. Indeed, KMT5A is well known to methylate p53 at K382 to reduce p53 activation [6] and down-regulation of KMT5A in response to DNA damage has been shown to result in conversion of this mono-methylation state to a di/tri- methylation state on K382 to increase p53 stability [6,43]. Therefore, we hypothesised that knockdown of KMT5A would shift the equilibrium from mono-methylated p53 to acetylated p53 thereby resulting in p53 activation and subsequent repression of CDC20 expression. To test this theory KMT5A was knocked down in both LNCaP-AI and LNCaP cells prior to western blotting for changes in p53 post-translational modifications and MDM2. We observed no alteration in total p53 protein levels in either cell line with siKMT5A_2 however, p53 levels were increased with siKMT5A_1. Nonetheless, a robust increase in acetylation at K382 and an increase in p53-phosphorylation at serine 15 which is associated with enhanced DNA binding was still observed with both siRNA sequences (Figure 7B,C). To confirm that p53 activation results in down-regulation of CDC20 protein levels we treated both LNCaP-AI and LNCaP cells with the MDM2 inhibitor, Nutlin 3. In both cell lines CDC20 protein levels were reduced when p53 was activated (Figure 7D). Taken together this suggests that p53 activation by KMT5A knockdown results in repression of CDC20.
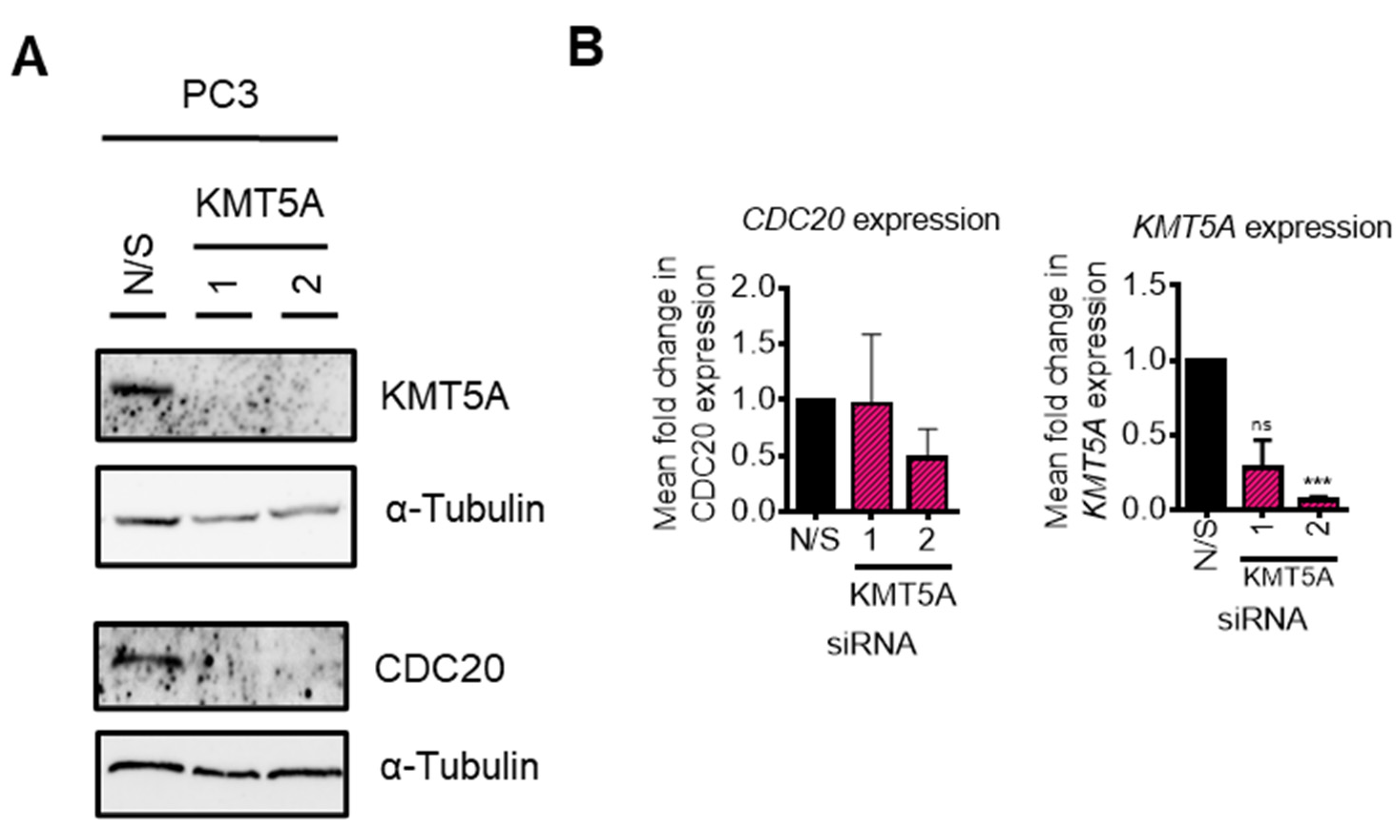
CDC20 is down-regulated by protein turnover in the absence of p53.
As p53 signalling was found to be up-regulated in our KEGG pathway analysis (Supplementary Table S4) we questioned whether KMT5A was able to regulate CDC20 if p53 was not present. As p53 loss is a common phenomenon in cancers this raised questions regarding the applicability of CDC20 as a KMT5A biomarker for those patients whose tumours lack p53 expression. To investigate further, we performed KMT5A knockdowns in p53 null, PC3 cells and performed western blotting and qPCR analysis of CDC20 levels. Surprisingly, KMT5A knockdown was still able to robustly reduce CDC20 protein levels in this cell line (Figure 8A). However, CDC20 mRNA levels were unaffected (Figure 8B) suggesting CDC20 post-translational changes occurred causing alterations in protein turnover in the absence of p53. Therefore, if a protein biomarker read-out could be used then CDC20 may remain as a valid KMT5A activity biomarker.
Discussion
Alternative therapeutic targets are urgently required for the treatment of advanced prostate cancers which have relapsed current standard of care therapies. As the androgen receptor remains a driver of disease at therapy relapse, proteins which positively modulate the transcriptional activity of the androgen receptor are proposed as putative therapeutic targets. The protein methyltransferase, KMT5A, has been shown to interact with the androgen receptor [4] and was proposed to offer therapeutic benefit to prostate cancer patients. However, the mechanisms by which KMT5A contributes to prostate cancer progression remains poorly understood.
We uncovered that KMT5A can regulate the levels of the cell cycle regulator protein CDC20 both directly at the chromatin level via modulation of histone methylation, and indirectly via methylation of the tumour suppressor protein, p53. This relationship between CDC20 and KMT5A is supported by a significant positive correlation between KMT5A and CDC20 transcripts in prostate cancer patients (Figure 4). Whilst this relationship is independent of the androgen receptor, both proteins are described as oncogenes in prostate cancer. Critically, there are no reports describing methylation specific regulation of CDC20.
KMT5A is the only known methyltransferase to monomethylate histone H4K20. As the H4K20Me1 mark is traditionally associated with a compact chromatin landscape and gene repression [44-47], it is counterintuitive that KMT5A should function to facilitate CDC20 transcription, however KMT5A mediated H4K20Me1 is now well documented to function as a transcriptional activator for some genes [48,49]. Where this has been observed there is generally a transcription factor which is implicated, for example TWIST [48]. Furthermore, H4K20Me1 is associated with actively transcribing gene bodies [50] and more recently has been found to result in chromatin accessibility at highly transcribed genes throughout the cell cycle [51]. A role for KMT5A in the pause and release of RNA pol II has also been revealed [52] further supporting the complex role of KMT5A in the positive regulation of gene transcription.
The methylation dependent regulation of p53 activity by KMT5A is key to ensuring transcriptional activation [6,43] further highlighting the ability of KMT5A to influence gene expression programmes at multiple levels. Consistently, we observe that knockdown of KMT5A results in enhanced p53 acetylation at K382 which can only occur if this residue is not methylated. Importantly, it is the subsequent phosphorylation event at S15 which facilitates the association of p53 with DNA which is enhanced upon KMT5A knockdown. This would permit the recruitment of HDAC1 and mSin3a to the CDC20 promoter to allow chromatin remodelling to occur and thereby inhibit transcription of CDC20 [11]. Importantly, we observed slightly different effects with each KMT5A targeting siRNA in this experiment with regards to the ability of KMT5A knockdown to stabilise p53 making interpretation of the p53 post-translational modifications more complex. However, densitometry confirms that both p53 phosphorylation and acetylation do increase with KMT5A knockdown with both siRNA sequences (Figure 7C). Therefore, it appears that there are two complementary mechanisms working together at the CDC20 promoter modulated by KMT5A to ensure timely expression of this gene.
Both KMT5A and CDC20 are essential cell cycle regulator proteins. KMT5A, is regulated by ubiquitin mediated protein turnover specifically at G1/S transition and between metaphase and anaphase [7] whilst CDC20 regulates the SAC to control the progression from metaphase to anaphase and ensure successful separation of sister chromatids. It is thought that the methylation of H4K20 is key to successful mitosis with the turnover of KMT5A being the major mode of H4K20 methylation regulation. Indeed, H4K20Me mediated chromosome condensation is important in this process and KMT5A knock out studies resulted in chromosome decondensation leading to cell cycle arrest at G2/M [46]. Furthermore, H4K20Me1 is required for kinetochore assembly at centromeres by recruitment of CENP-T [53]. Hence, it is logical to hypothesise that a lack of KMT5A, resulting in a decrease in H4K20Me1, will result in impaired kinetochore assembly and thereby invoke the SAC preventing CDC20 from facilitating the onset of anaphase. Therefore, due to the importance of tightly regulating CDC20 to ensure effective mitosis, modulation of CDC20 levels themselves by KMT5A would provide a failsafe to prevent mitotic catastrophe.
CDC20 is required for nuclear movement prior to anaphase where its activity, as part of the APCCDC20 complex, results in destruction of cyclin B and inactivation of CDK1. Interestingly, CDK1 mediated phosphorylation of KMT5A at serine 29 has been reported to occur during metaphase resulting in the removal of KMT5A from chromatin, holding it in a stabilised state without affecting methylase activity. It is not until anaphase that dephosphorylation by cdc14a/b permits KMT5A protein turnover via APCcdh1[7]. Furthermore, APCCDC20 targets the H4K20Me1 demethylase, PHF8, for ubiquitin mediated destruction [13] further highlighting the important relationship between CDC20 and the enzymes which modulate H4K20 methylation state.
CDC20 has been found to be overexpressed in a number of cancers, including prostate cancer [15,54] and there are a number of studies which demonstrate the relevance of CDC20 to prostate cancer development and progression. For example, CDC20 has been identified as a hub gene, alongside CDK1, in castration resistant prostate cancer [55], and contributes to cell migration, disease progression and a poorer prognosis in metastatic prostate cancer [56] with another study showing CDC20, alongside PLK1 and cyclin A, playing a critical role in prostate cancer metastasis [57]. CDC20 and PLK1 are both located at chromosomal region 9p, which is often amplified in cancer. Indeed, high expression of CDC20, PLK1 and CDK1 correlate with prostate cancer occurrence [58] and worse biochemical recurrence survival rates [59]. Furthermore, CDC20 is a target protein of Speckle-type POZ protein (SPOP), which functions to promote ubiquitin mediated protein turnover. SPOP is mutated in up to 15% of prostate cancers [60] and these mutations have been shown to result in an inability of SPOP to associate with CDC20 preventing CDC20 protein turnover and consequently resistance to CDC20 inhibitors [16]. Both SPOP mutation [61] and CDC20 overexpression are important in docetaxel resistance with inhibition or knockdown of CDC20 able to resensitise cells to docetaxel [62] highlighting the importance of CDC20 as a therapeutic target in prostate cancer at several disease stages. With inhibitors for both CDC20 and KMT5A being developed it would be important to determine whether they are able to synergise with each other both in drug resistant models of prostate cancer.
KMT5A has an important role in the DNA damage repair pathway whereby it is recruited to double strand breaks to deposit H4K20Me1 to facilitate Suv4-20 mediated H4K20Me2 which is required for 53BP1 binding and successful repair by NHEJ [63,64]. In addition, the ubiquitination of KMT5A by RNF8 increases KMT5A association with RNF168 which in turn promotes H2A ubiquitination [65]. The ubiquitination of these and other chromatin components results in the recruitment of BRCA1/BARD1/Abraxas and RAP80 to sites of γH2AX to allow the repair process to take place. Interestingly, RAP80 is a target of CDC20 and its overexpression prevents mitotic progression irrespective of DNA damage [9]. This again supports a connection between the functions of KMT5A and CDC20 in cellular processes. Additionally, the role for KMT5A in the suppression of important anti-tumourigenic processes such as positive regulation of the apoptotic process, response to gamma and ionising irradiation are also highlighted suggesting a utility of KMT5A inhibition in combination with other DNA damage inducing therapeutics such as radiotherapy or cytotoxic agents.
The cellular processes regulated by KMT5A identified in this study are consistent with those already described such as genome integrity, cell cycle progression, gene transcription and DNA damage repair. However, some novel processes were identified including RNA splicing and mRNA processing which require further investigation. This is particularly important in prostate cancer where aberrant RNA splicing, particularly of the androgen receptor, is associated with therapy resistance and poor prognosis [66].
Conclusions
A number of key oncogenic signalling pathways are regulated by the methyltransferase, KMT5A. Here, we provide evidence for a role of KMT5A in both metaphase to anaphase control via regulation of CDC20 and propose that close links between mitosis and DNA damage repair processes are present via this relationship. As both CDC20 and KMT5A are up-regulated in cancer via a number of mechanisms the relationship between the two proteins may be dysregulated thereby promoting genomic instability. This presents an opportunity to identify beneficial therapeutic combinations to treat patients based on a number of criteria such as SPOP mutation status, KMT5A expression status and therapeutic resistance.
Supplementary Materials
Supplementary Figure S1. KMT5A and CDC20 knockdown inhibits proliferation of prostate cancer cells. Supplementary Figure S2. KMT5A inhibition by UNC0379 reduces KMT5A levels and H4K20Me1. Supplementary Figure S3. KMT5A inhibition by Ryuvidine reduces KMT5A levels and H4K20Me1. Table S1. QPCR primers. Table S2. Genes significantly up-regulated by KMT5A knockdown. Table S3. Genes significantly down-regulated by KMT5A knockdown. Table S4. KEGG pathways down-regulated in response to KMT5A knockdown in the presence of DHT stimulation. Table S5. KEGG pathways up-regulated in response to KMT5A knockdown in the presence of DHT stimulation. Table S6. Biological Processes down-regulated in response to KMT5A knockdown in the presence of DHT stimulation. Table S7. Biological Processes up-regulated in response to KMT5A knockdown in the presence of DHT stimulation.
Author Contributions
Conceptualization, K.C. and C.R.; Methodology, K.C. and C.R.; Validation, K.C., Z.A., O.T. and S.N.; Formal Analysis, S.N., Z.A.; Investigation, K.C., Z.A., M.A., D.A., K.J., H.C., R.G.; Resources, K.C., C.R.; Data Curation, Z.A.; Writing – Original Draft Preparation, Z.A., M.A. and K.C.; Writing – Review & Editing, K.C., C.R., E.L., E.S.; Visualization, K.C.; Supervision, K.C., C.R.; Project Administration, K.C.; Funding Acquisition, K.C., Z.A., D.A.
Funding
This work was supported by a Movember funded Prostate Cancer UK Career Development Fellowship (CDF12-006) to KC, The Higher Committee for Education Development in Iraq, as a PhD studentship to ZA, and The Public Authority of Applied Education and Training, Kuwait to DA. ES is supported by a Prostate Cancer UK Travelling Prize fellowship [TLD-PF19-002]. ELW is supported by a Prostate Cancer UK Research Innovation Award (RIA19-ST2-005). RG is supported by a William Edmond Harker Foundation Studentship. For the purpose of Open Access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript (AAM) version arising from this submission.
Data Availability Statement
The microarray data presented in this study are openly available in GEO: GSE233350. Publicly available datasets were analysed in this project. This data can be found in cBioportal.
Acknowledgements
Nutlin 3 was provided as a gift from Professor John Lunec (Newcastle Cancer Centre, Newcastle University). We thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 090532/Z/09/Z and MRC Hub grant G0900747 91070) for the generation of the Gene Expression data. We thank Mr Oswald To for contributions to chromatin generation for this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Veschi, V.; Liu, Z.; Voss, T.C.; Ozbun, L.; Gryder, B.; Yan, C.; Hu, Y.; Ma, A.; Jin, J.; Mazur, S.J.; et al. Epigenetic siRNA and Chemical Screens Identify SETD8 Inhibition as a Therapeutic Strategy for p53 Activation in High-Risk Neuroblastoma. Cancer Cell 2017, 31, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Qiao, K.; Du, Y.; Zhang, X.; Cheng, H.; Peng, L.; Guo, Z. Downregulation of histone methyltransferase SET8 inhibits progression of hepatocellular carcinoma. Sci Rep 2020, 10, 4490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hou, W.; Chai, M.; Zhao, H.; Jia, J.; Sun, X.; Zhao, B.; Wang, R. MicroRNA-127-3p inhibits proliferation and invasion by targeting SETD8 in human osteosarcoma cells. Biochem Biophys Res Commun 2016, 469, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Li, Y.; Du, F.; Han, X.; Li, X.; Niu, Y.; Ren, S.; Sun, Y. Histone H4 Lys 20 methyltransferase SET8 promotes androgen receptor-mediated transcription activation in prostate cancer. Biochem Biophys Res Commun 2014, 450, 692–696. [Google Scholar] [CrossRef]
- Hou, L.; Li, Q.; Yu, Y.; Li, M.; Zhang, D. SET8 induces epithelialmesenchymal transition and enhances prostate cancer cell metastasis by cooperating with ZEB1. Mol Med Rep 2016, 13, 1681–1688. [Google Scholar] [CrossRef]
- Shi, X.; Kachirskaia, I.; Yamaguchi, H.; West, L.E.; Wen, H.; Wang, E.W.; Dutta, S.; Appella, E.; Gozani, O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell 2007, 27, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, W.; Kong, X.; Congdon, L.M.; Yokomori, K.; Kirschner, M.W.; Rice, J.C. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev 2010, 24, 2531–2542. [Google Scholar] [CrossRef]
- Wu, S.; Rice, J.C. A new regulator of the cell cycle: the PR-Set7 histone methyltransferase. Cell Cycle 2011, 10, 68–72. [Google Scholar] [CrossRef]
- Cho, H.J.; Lee, E.H.; Han, S.H.; Chung, H.J.; Jeong, J.H.; Kwon, J.; Kim, H. Degradation of human RAP80 is cell cycle regulated by Cdc20 and Cdh1 ubiquitin ligases. Mol Cancer Res 2012, 10, 615–625. [Google Scholar] [CrossRef]
- Chun, A.C.; Kok, K.H.; Jin, D.Y. REV7 is required for anaphase-promoting complex-dependent ubiquitination and degradation of translesion DNA polymerase REV1. Cell Cycle 2013, 12, 365–378. [Google Scholar] [CrossRef]
- Banerjee, T.; Nath, S.; Roychoudhury, S. DNA damage induced p53 downregulates Cdc20 by direct binding to its promoter causing chromatin remodeling. Nucleic Acids Res 2009, 37, 2688–2698. [Google Scholar] [CrossRef]
- Kidokoro, T.; Tanikawa, C.; Furukawa, Y.; Katagiri, T.; Nakamura, Y.; Matsuda, K. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene 2008, 27, 1562–1571. [Google Scholar] [CrossRef]
- Lim, H.J.; Dimova, N.V.; Tan, M.K.; Sigoillot, F.D.; King, R.W.; Shi, Y. The G2/M regulator histone demethylase PHF8 is targeted for degradation by the anaphase-promoting complex containing CDC20. Mol Cell Biol 2013, 33, 4166–4180. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol Ther 2015, 151, 141–151. [Google Scholar] [CrossRef]
- Mao, Y.; Li, K.; Lu, L.; Si-Tu, J.; Lu, M.; Gao, X. Overexpression of Cdc20 in clinically localized prostate cancer: Relation to high Gleason score and biochemical recurrence after laparoscopic radical prostatectomy. Cancer Biomark 2016, 16, 351–358. [Google Scholar] [CrossRef]
- Wu, F.; Dai, X.; Gan, W.; Wan, L.; Li, M.; Mitsiades, N.; Wei, W.; Ding, Q.; Zhang, J. Prostate cancer-associated mutation in SPOP impairs its ability to target Cdc20 for poly-ubiquitination and degradation. Cancer Lett 2017, 385, 207–214. [Google Scholar] [CrossRef]
- Li, K.; Mao, Y.; Lu, L.; Hu, C.; Wang, D.; Si-Tu, J.; Lu, M.; Peng, S.; Qiu, J.; Gao, X. Silencing of CDC20 suppresses metastatic castration-resistant prostate cancer growth and enhances chemosensitivity to docetaxel. Int J Oncol 2016, 49, 1679–1685. [Google Scholar] [CrossRef]
- Li, J.; Karki, A.; Hodges, K.B.; Ahmad, N.; Zoubeidi, A.; Strebhardt, K.; Ratliff, T.L.; Konieczny, S.F.; Liu, X. Cotargeting Polo-Like Kinase 1 and the Wnt/beta-Catenin Signaling Pathway in Castration-Resistant Prostate Cancer. Mol Cell Biol 2015, 35, 4185–4198. [Google Scholar] [CrossRef]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O'Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res 2013, 41, 4433–4446. [Google Scholar] [CrossRef]
- Bainbridge, A.; Walker, S.; Smith, J.; Patterson, K.; Dutt, A.; Ng, Y.M.; Thomas, H.D.; Wilson, L.; McCullough, B.; Jones, D.; et al. IKBKE activity enhances AR levels in advanced prostate cancer via modulation of the Hippo pathway. Nucleic Acids Res 2020, 48, 5366–5382. [Google Scholar] [CrossRef]
- Coffey, K.; Blackburn, T.J.; Cook, S.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Hewitt, L.; Huberman, K.; McNeill, H.V.; Newell, D.R.; et al. Characterisation of a Tip60 specific inhibitor, NU9056, in prostate cancer. PLoS One 2012, 7, e45539. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple hypothesis testing. J R Stat Soc B 1995, 57. [Google Scholar] [CrossRef]
- Labaf, M.; Li, M.; Ting, L.; Karno, B.; Zhang, S.; Gao, S.; Patalano, S.; Macoska, J.A.; Zarringhalam, K.; Han, D.; et al. Increased AR expression in castration-resistant prostate cancer rapidly induces AR signaling reprogramming with the collaboration of EZH2. Front Oncol 2022, 12, 1021845. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef]
- Schmidt, D.; Wilson, M.D.; Spyrou, C.; Brown, G.D.; Hadfield, J.; Odom, D.T. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods 2009, 48, 240–248. [Google Scholar] [CrossRef]
- Xie, Q.; Wu, Q.; Mack, S.C.; Yang, K.; Kim, L.; Hubert, C.G.; Flavahan, W.A.; Chu, C.; Bao, S.; Rich, J.N. CDC20 maintains tumor initiating cells. Oncotarget 2015, 6, 13241–13254. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 2007, 8, R183. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Wei, G.H.; Liu, D.; Wang, L.; Hou, Y.; Zhu, S.; Peng, L.; Zhang, Q.; Cheng, Y.; Su, H.; et al. Whole-genome and Transcriptome Sequencing of Prostate Cancer Identify New Genetic Alterations Driving Disease Progression. Eur Urol 2018, 73, 322–339. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Jorgensen, S.; Elvers, I.; Trelle, M.B.; Menzel, T.; Eskildsen, M.; Jensen, O.N.; Helleday, T.; Helin, K.; Sorensen, C.S. The histone methyltransferase SET8 is required for S-phase progression. J Cell Biol 2007, 179, 1337–1345. [Google Scholar] [CrossRef]
- Tardat, M.; Murr, R.; Herceg, Z.; Sardet, C.; Julien, E. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J Cell Biol 2007, 179, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Blum, G.; Ibanez, G.; Rao, X.; Shum, D.; Radu, C.; Djaballah, H.; Rice, J.C.; Luo, M. Small-molecule inhibitors of SETD8 with cellular activity. ACS Chem Biol 2014, 9, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.; Yu, W.; Xiong, Y.; Butler, K.V.; Brown, P.J.; Jin, J. Structure-activity relationship studies of SETD8 inhibitors. Medchemcomm 2014, 5, 1892–1898. [Google Scholar] [CrossRef]
- Nath, S.; Chowdhury, A.; Dey, S.; Roychoudhury, A.; Ganguly, A.; Bhattacharyya, D.; Roychoudhury, S. Deregulation of Rb-E2F1 axis causes chromosomal instability by engaging the transactivation function of Cdc20-anaphase-promoting complex/cyclosome. Mol Cell Biol 2015, 35, 356–369. [Google Scholar] [CrossRef]
- Williams, K.; Christensen, J.; Rappsilber, J.; Nielsen, A.L.; Johansen, J.V.; Helin, K. The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS One 2014, 9, e96545. [Google Scholar] [CrossRef]
- Trojer, P.; Li, G.; Sims, R.J., 3rd; Vaquero, A.; Kalakonda, N.; Boccuni, P.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Nimer, S.D.; et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 2007, 129, 915–928. [Google Scholar] [CrossRef]
- Shoaib, M.; Walter, D.; Gillespie, P.J.; Izard, F.; Fahrenkrog, B.; Lleres, D.; Lerdrup, M.; Johansen, J.V.; Hansen, K.; Julien, E.; et al. Histone H4K20 methylation mediated chromatin compaction threshold ensures genome integrity by limiting DNA replication licensing. Nat Commun 2018, 9, 3704. [Google Scholar] [CrossRef]
- Oda, H.; Hubner, M.R.; Beck, D.B.; Vermeulen, M.; Hurwitz, J.; Spector, D.L.; Reinberg, D. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell 2010, 40, 364–376. [Google Scholar] [CrossRef]
- Nishioka, K.; Rice, J.C.; Sarma, K.; Erdjument-Bromage, H.; Werner, J.; Wang, Y.; Chuikov, S.; Valenzuela, P.; Tempst, P.; Steward, R.; et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell 2002, 9, 1201–1213. [Google Scholar] [CrossRef]
- Yang, F.; Sun, L.; Li, Q.; Han, X.; Lei, L.; Zhang, H.; Shang, Y. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J 2012, 31, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Nie, F.; Wang, S.; Li, L. Histone H4 Lys 20 monomethylation by histone methylase SET8 mediates Wnt target gene activation. Proc Natl Acad Sci U S A 2011, 108, 3116–3123. [Google Scholar] [CrossRef] [PubMed]
- Vakoc, C.R.; Sachdeva, M.M.; Wang, H.; Blobel, G.A. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol Cell Biol 2006, 26, 9185–9195. [Google Scholar] [CrossRef] [PubMed]
- Shoaib, M.; Chen, Q.; Shi, X.; Nair, N.; Prasanna, C.; Yang, R.; Walter, D.; Frederiksen, K.S.; Einarsson, H.; Svensson, J.P.; et al. Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes. Nat Commun 2021, 12, 4800. [Google Scholar] [CrossRef]
- Kapoor-Vazirani, P.; Vertino, P.M. A dual role for the histone methyltransferase PR-SET7/SETD8 and histone H4 lysine 20 monomethylation in the local regulation of RNA polymerase II pausing. J Biol Chem 2014, 289, 7425–7437. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Shang, W.H.; Toyoda, A.; Misu, S.; Monma, N.; Ikeo, K.; Molina, O.; Vargiu, G.; Fujiyama, A.; Kimura, H.; et al. Histone H4 Lys 20 monomethylation of the CENP-A nucleosome is essential for kinetochore assembly. Dev Cell 2014, 29, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Sun, Y.; Chen, J.; Li, H.; Yao, K.; Liu, Y.; Liu, Q.; Lu, J. The Oncogenic Role of APC/C Activator Protein Cdc20 by an Integrated Pan-Cancer Analysis in Human Tumors. Front Oncol 2021, 11, 721797. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Hu, K.; Li, D.; Wang, Y.; Liu, M.; Wang, X.; Zhu, W.; Wang, X.; Yang, Z.; Lu, J. Identification of Core Genes and Potential Drugs for Castration-Resistant Prostate Cancer Based on Bioinformatics Analysis. DNA Cell Biol 2020, 39, 836–847. [Google Scholar] [CrossRef]
- Dai, L.; Song, Z.X.; Wei, D.P.; Zhang, J.D.; Liang, J.Q.; Wang, B.B.; Ma, W.T.; Li, L.Y.; Dang, Y.L.; Zhao, L.; et al. CDC20 and PTTG1 are Important Biomarkers and Potential Therapeutic Targets for Metastatic Prostate Cancer. Adv Ther 2021, 38, 2973–2989. [Google Scholar] [CrossRef]
- Gu, P.; Yang, D.; Zhu, J.; Zhang, M.; He, X. Bioinformatics analysis identified hub genes in prostate cancer tumorigenesis and metastasis. Math Biosci Eng 2021, 18, 3180–3196. [Google Scholar] [CrossRef]
- Wei, J.; Yin, Y.; Deng, Q.; Zhou, J.; Wang, Y.; Yin, G.; Yang, J.; Tang, Y. Integrative Analysis of MicroRNA and Gene Interactions for Revealing Candidate Signatures in Prostate Cancer. Front Genet 2020, 11, 176. [Google Scholar] [CrossRef]
- Luo, C.; Chen, J.; Chen, L. Exploration of gene expression profiles and immune microenvironment between high and low tumor mutation burden groups in prostate cancer. Int Immunopharmacol 2020, 86, 106709. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Shi, Q.; Zhu, Y.; Ma, J.; Chang, K.; Ding, D.; Bai, Y.; Gao, K.; Zhang, P.; Mo, R.; Feng, K.; et al. Prostate Cancer-associated SPOP mutations enhance cancer cell survival and docetaxel resistance by upregulating Caprin1-dependent stress granule assembly. Mol Cancer 2019, 18, 170. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Lin, Y.; Cui, P.; Li, H.; Zhang, L.; Sun, Z.; Huang, S.; Li, S.; Huang, S.; Zhao, Q.; et al. Cdc20/p55 mediates the resistance to docetaxel in castration-resistant prostate cancer in a Bim-dependent manner. Cancer Chemother Pharmacol 2018, 81, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Tuzon, C.T.; Spektor, T.; Kong, X.; Congdon, L.M.; Wu, S.; Schotta, G.; Yokomori, K.; Rice, J.C. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep 2014, 8, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Dulev, S.; Tkach, J.; Lin, S.; Batada, N.N. SET8 methyltransferase activity during the DNA double-strand break response is required for recruitment of 53BP1. EMBO Rep 2014, 15, 1163–1174. [Google Scholar] [CrossRef]
- Lu, X.; Xu, M.; Zhu, Q.; Zhang, J.; Liu, G.; Bao, Y.; Gu, L.; Tian, Y.; Wen, H.; Zhu, W.G. RNF8-ubiquitinated KMT5A is required for RNF168-induced H2A ubiquitination in response to DNA damage. FASEB J 2021, 35, e21326. [Google Scholar] [CrossRef]
- Jimenez-Vacas, J.M.; Herrero-Aguayo, V.; Montero-Hidalgo, A.J.; Gomez-Gomez, E.; Fuentes-Fayos, A.C.; Leon-Gonzalez, A.J.; Saez-Martinez, P.; Alors-Perez, E.; Pedraza-Arevalo, S.; Gonzalez-Serrano, T.; et al. Dysregulation of the splicing machinery is directly associated to aggressiveness of prostate cancer. EBioMedicine 2020, 51, 102547. [Google Scholar] [CrossRef]
Figure 1.
KMT5A regulated genes in LNCaP-AI cells (A) LNCaP-AI cells were reverse transfected with 25 nM siRNA targeting KMT5A or a non-silencing control (N/S) in steroid depleted media. After 72 hours 10 nM DHT was added to the cells for a further 24 hours. RNA was isolated, quality checked, gene expression profiles determined using Illumina HT-12 v4.0 Expression BeadChip Microarray. 3 independent experimental repeats were performed. Data analysis confirmed successful KMT5A knockdown. (B) Table of genes ranked for their downregulation in response to KMT5A knockdown in the presence of DHT stimulation. Genes which were down-regulated more the KMT5A (SETD8) are shown (full gene lists can be found in supplementary information. *GSE211638 [23] (C) CDC20 expression levels in response to KMT5A knockdown as determined by microarray analysis. One-way ANOVA with Dunnett’s multiple comparisons test *p<0.05; **p<0.01.
Figure 1.
KMT5A regulated genes in LNCaP-AI cells (A) LNCaP-AI cells were reverse transfected with 25 nM siRNA targeting KMT5A or a non-silencing control (N/S) in steroid depleted media. After 72 hours 10 nM DHT was added to the cells for a further 24 hours. RNA was isolated, quality checked, gene expression profiles determined using Illumina HT-12 v4.0 Expression BeadChip Microarray. 3 independent experimental repeats were performed. Data analysis confirmed successful KMT5A knockdown. (B) Table of genes ranked for their downregulation in response to KMT5A knockdown in the presence of DHT stimulation. Genes which were down-regulated more the KMT5A (SETD8) are shown (full gene lists can be found in supplementary information. *GSE211638 [23] (C) CDC20 expression levels in response to KMT5A knockdown as determined by microarray analysis. One-way ANOVA with Dunnett’s multiple comparisons test *p<0.05; **p<0.01.
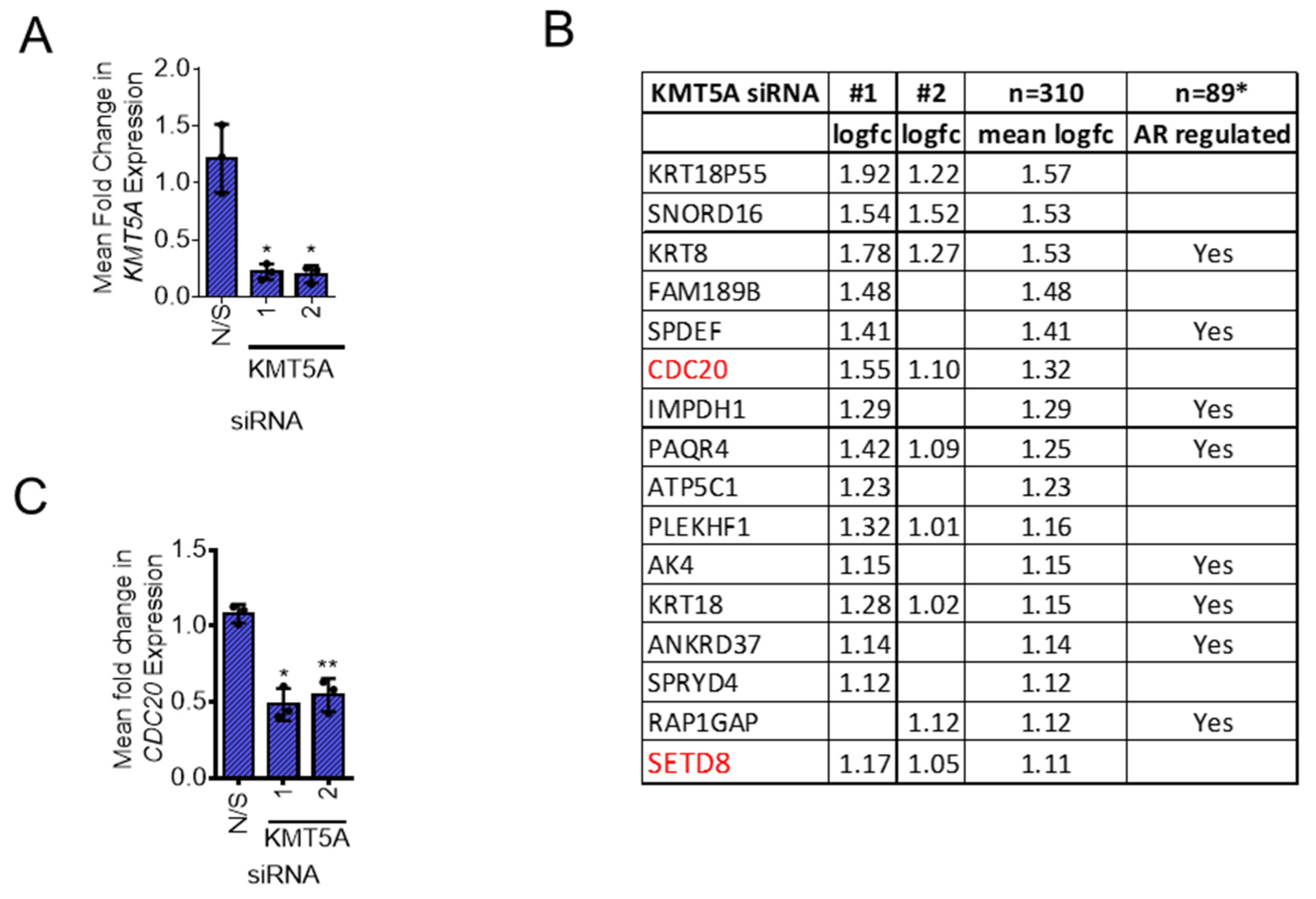
Figure 2.
CDC20 is a KMT5A regulated gene. (A) LNCaP-AI cells were reverse transfected with 25 nM siRNA targeting KMT5A or a non-silencing control (N/S) in steroid depleted media. After 72 hours 10 nM DHT or vehicle control was added to the cells for a further 24 hours. RNA was isolated and CDC20 mRNA and (B) KMT5A mRNA quantified by QPCR. The same experiment was performed in (C) LNCaP cells and CDC20 levels and (D) KMT5A knockdown confirmed by QPCR. Data is expressed as mean fold change over 3 independent experiments +/- SEM. (E) Under the same experimental conditions protein levels of CDC20 and KMT5A were determined by western blotting in LNCaP-AI and (F) LNCaP cells. Alpha-tubulin was used as a loading control. Data shown is representative of 3 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test; **p<0.01; ***p<0.001; ****p<0.001. .
Figure 2.
CDC20 is a KMT5A regulated gene. (A) LNCaP-AI cells were reverse transfected with 25 nM siRNA targeting KMT5A or a non-silencing control (N/S) in steroid depleted media. After 72 hours 10 nM DHT or vehicle control was added to the cells for a further 24 hours. RNA was isolated and CDC20 mRNA and (B) KMT5A mRNA quantified by QPCR. The same experiment was performed in (C) LNCaP cells and CDC20 levels and (D) KMT5A knockdown confirmed by QPCR. Data is expressed as mean fold change over 3 independent experiments +/- SEM. (E) Under the same experimental conditions protein levels of CDC20 and KMT5A were determined by western blotting in LNCaP-AI and (F) LNCaP cells. Alpha-tubulin was used as a loading control. Data shown is representative of 3 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test; **p<0.01; ***p<0.001; ****p<0.001. .
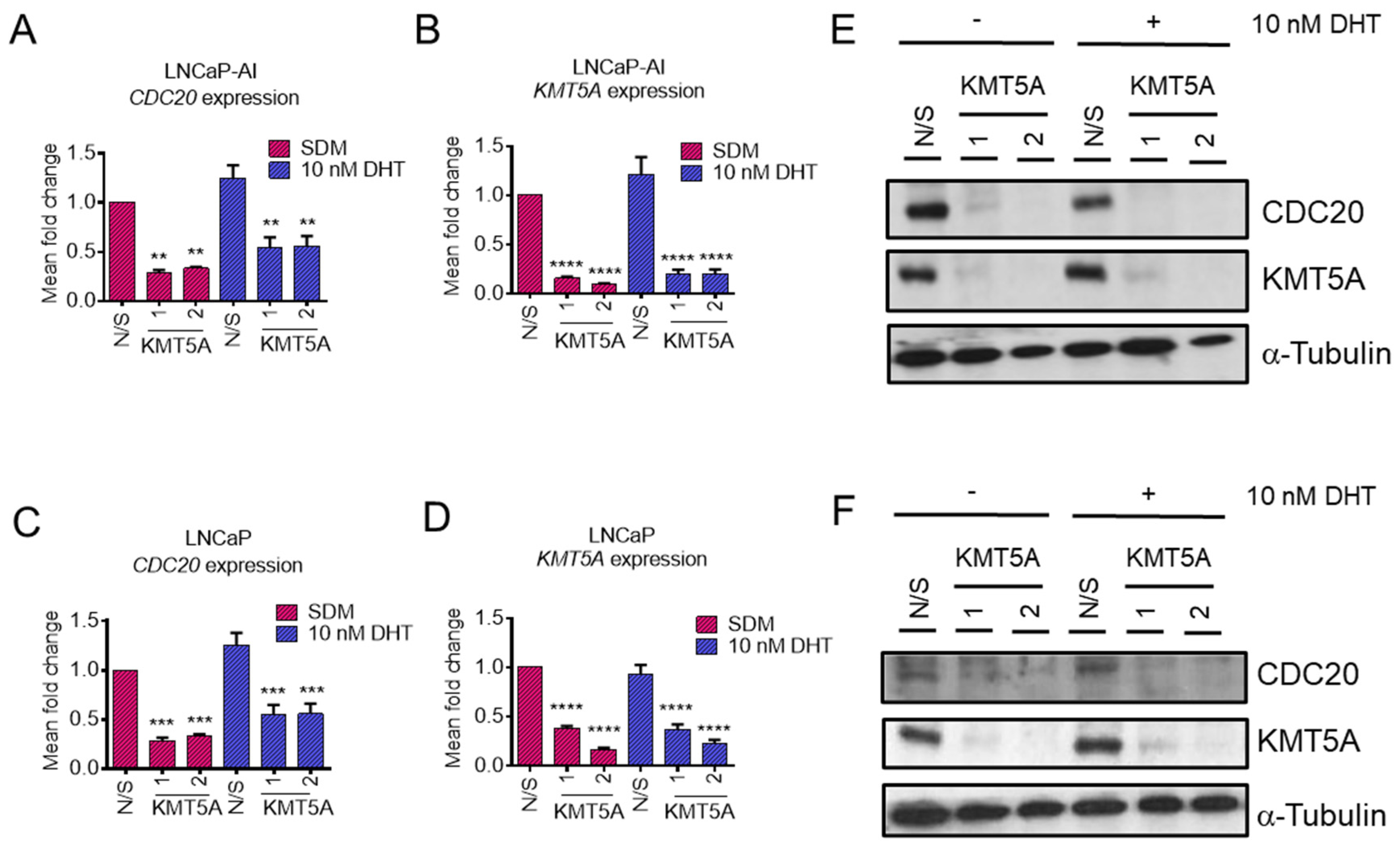
Figure 3.
CDC20 knockdown does not affect KMT5A protein levels. (A) LNCaP-AI cells and (B) LNCaP cells were reverse transfected with 25 nM siRNA targeting CDC20 or a non-silencing control (N/S) for 72 hours prior to analysis by western blot analysis. Alpha-tubulin was used as a loading control. (C) LNCaP-AI and (D) LNCaP cells were reverse transfected with 25 nM siRNA targeting CDC20 or a non-silencing control (N/S) for 72 hours prior to RNA isolation and qPCR analysis for KMT5A and (E,F) CDC20 mRNA levels. HPRT1 was used as a housekeeping gene. Data is expressed as mean fold change +/- SEM. One-way ANOVA was used to determine statistical significance. *p<0.05; ** p<0.01; ****p<0.0001.
Figure 3.
CDC20 knockdown does not affect KMT5A protein levels. (A) LNCaP-AI cells and (B) LNCaP cells were reverse transfected with 25 nM siRNA targeting CDC20 or a non-silencing control (N/S) for 72 hours prior to analysis by western blot analysis. Alpha-tubulin was used as a loading control. (C) LNCaP-AI and (D) LNCaP cells were reverse transfected with 25 nM siRNA targeting CDC20 or a non-silencing control (N/S) for 72 hours prior to RNA isolation and qPCR analysis for KMT5A and (E,F) CDC20 mRNA levels. HPRT1 was used as a housekeeping gene. Data is expressed as mean fold change +/- SEM. One-way ANOVA was used to determine statistical significance. *p<0.05; ** p<0.01; ****p<0.0001.
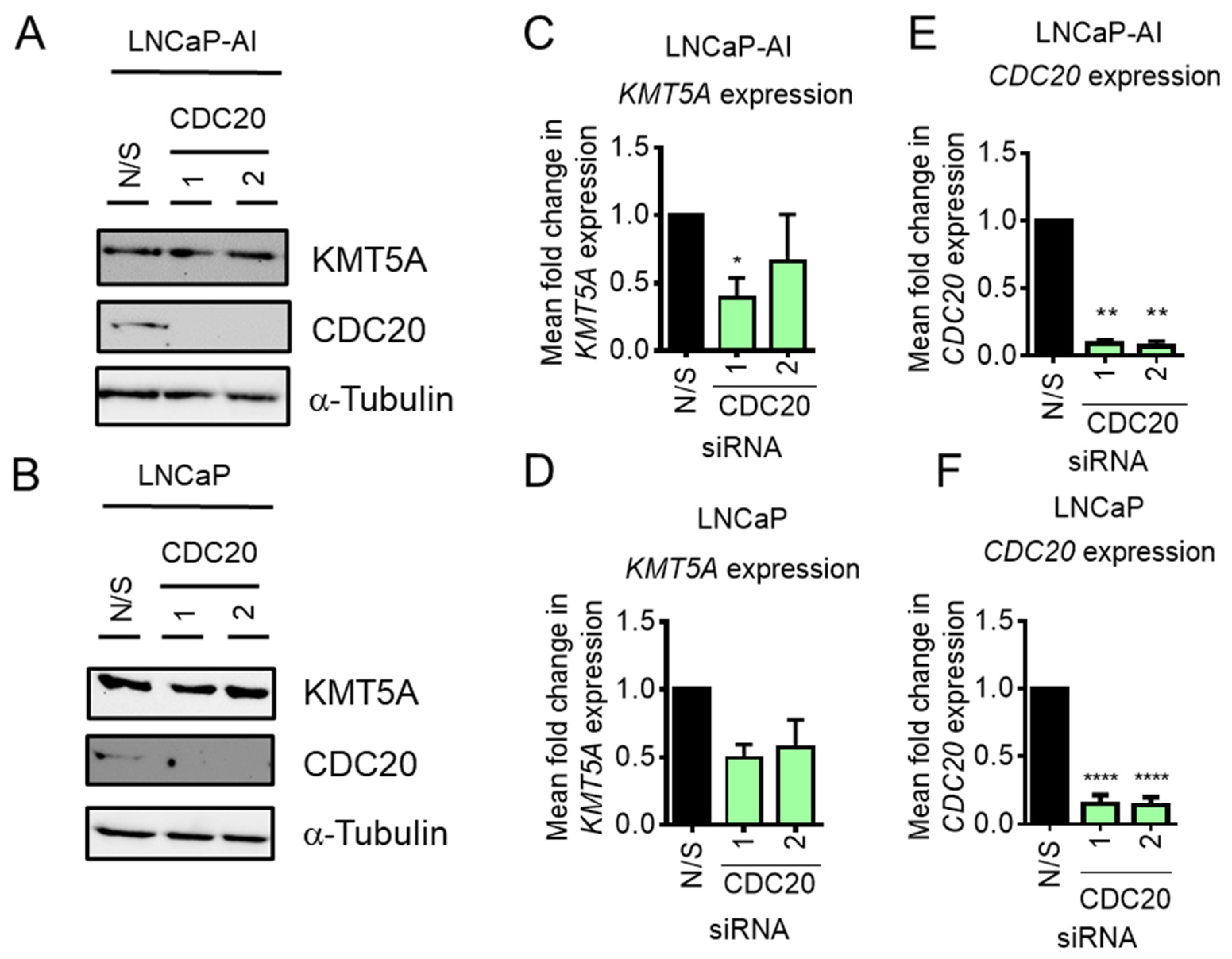
Figure 4.
KMT5A and CDC20 are positively correlated in clinical prostate cancer samples. Correlation in expression between KMT5A and CDC20 was carried out in publicly available datasets in cBioportal. (A) Treatment naïve radical prostatectomies (n=65) from [33] (B) Both primary and metastatic prostate cancer (n=94) from [34] (C) Metastatic prostate adenocarcinoma samples (n=150) from [35] (D) Bone metastatic prostate adenocarcinoma samples (n=72/266) and (E) Liver metastatic prostate adenocarcinoma samples (n=38) from [36] (F) Prostate neuroendocrine carcinoma samples (n=49) from [37]. Spearman correlation (two-tailed) was calculated using Graphpad software.
Figure 4.
KMT5A and CDC20 are positively correlated in clinical prostate cancer samples. Correlation in expression between KMT5A and CDC20 was carried out in publicly available datasets in cBioportal. (A) Treatment naïve radical prostatectomies (n=65) from [33] (B) Both primary and metastatic prostate cancer (n=94) from [34] (C) Metastatic prostate adenocarcinoma samples (n=150) from [35] (D) Bone metastatic prostate adenocarcinoma samples (n=72/266) and (E) Liver metastatic prostate adenocarcinoma samples (n=38) from [36] (F) Prostate neuroendocrine carcinoma samples (n=49) from [37]. Spearman correlation (two-tailed) was calculated using Graphpad software.
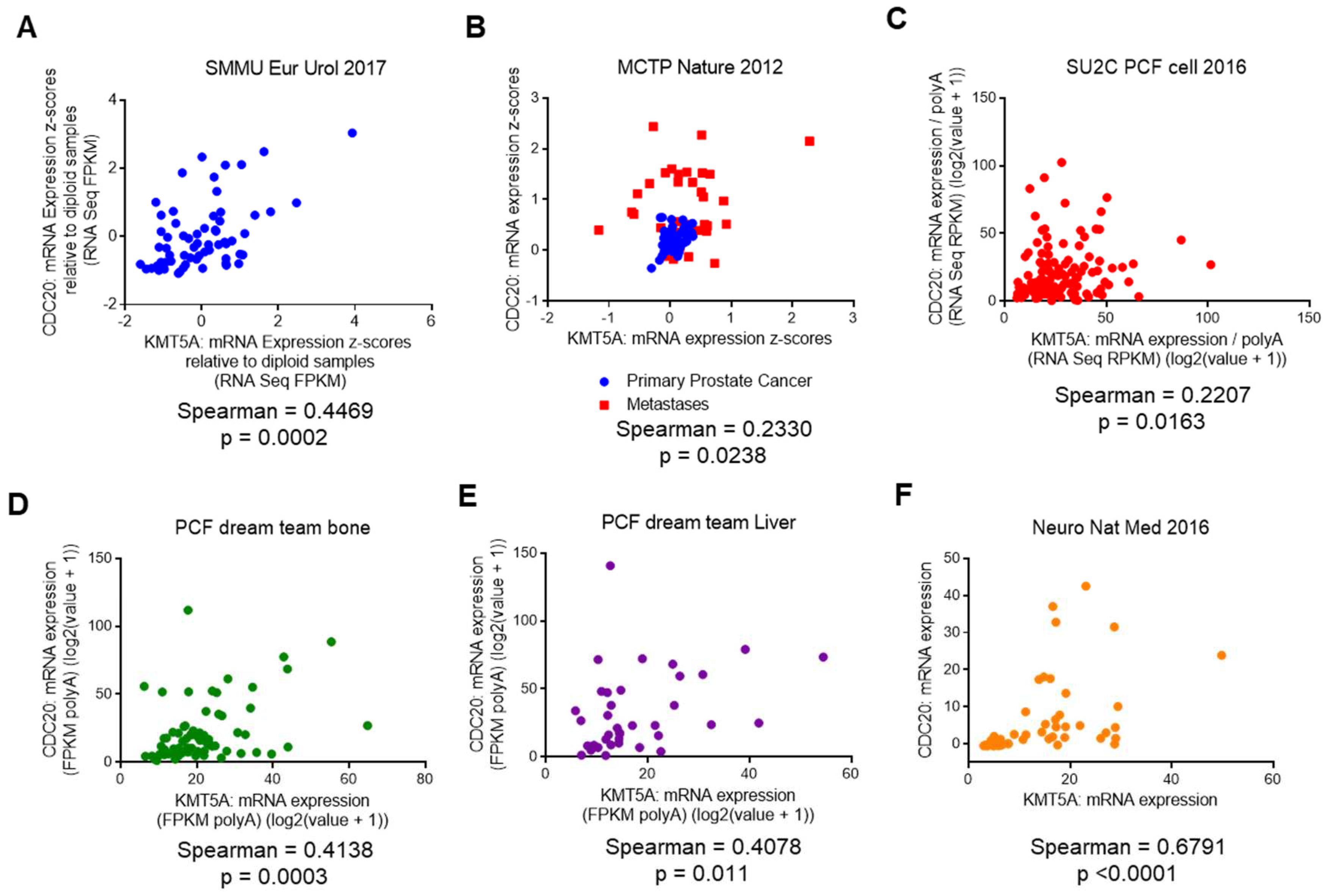
Figure 5.
KMT5A inhibitors restrict prostate cancer cell growth and down-regulate CDC20. (A) LNCaP-AI and LNCaP cells were treated with a dose range of UNC0379 or Ryuvidine and GI50 concentrations determined by SRB assay after 3 doubling times. Data is expressed as mean % growth inhibition +/- SEM from 3 independent experiments (B) LNCaP-AI cells were treated with either UNC0379 (7 μM) or Ryuvidine (1 μM or 2.77 μM) for 48 hours prior to protein analysis by western blotting. (C) LNCaP cells were treated with Ryuvidine (0.7 μM) for 48 hours prior to protein analysis by western blotting. Data shown is representative of 3 independent experiments.
Figure 5.
KMT5A inhibitors restrict prostate cancer cell growth and down-regulate CDC20. (A) LNCaP-AI and LNCaP cells were treated with a dose range of UNC0379 or Ryuvidine and GI50 concentrations determined by SRB assay after 3 doubling times. Data is expressed as mean % growth inhibition +/- SEM from 3 independent experiments (B) LNCaP-AI cells were treated with either UNC0379 (7 μM) or Ryuvidine (1 μM or 2.77 μM) for 48 hours prior to protein analysis by western blotting. (C) LNCaP cells were treated with Ryuvidine (0.7 μM) for 48 hours prior to protein analysis by western blotting. Data shown is representative of 3 independent experiments.
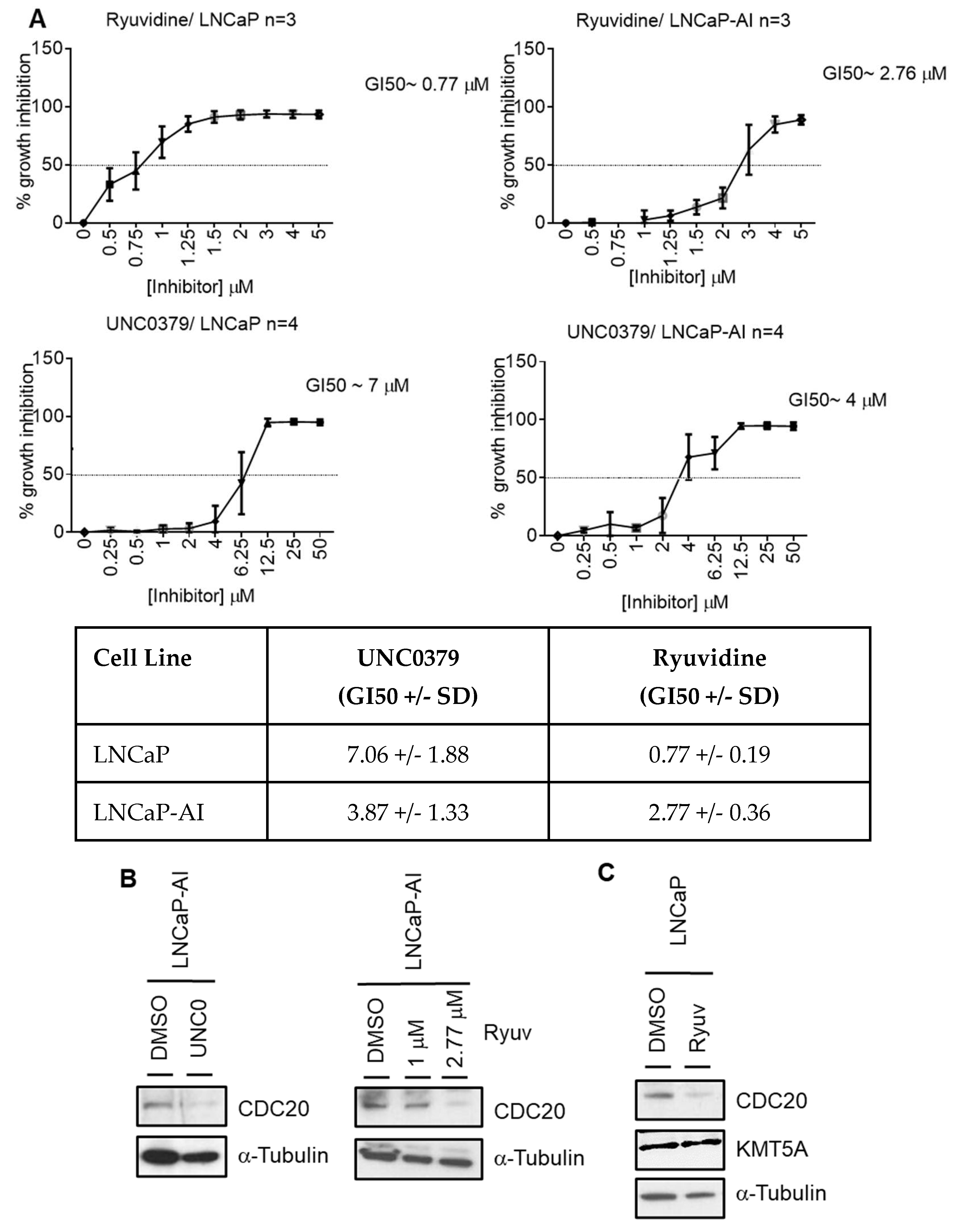
Figure 6.
H4K20Me1 is reduced at the CDC20 promoter in response to KMT5A knockdown.KMT5A was knocked down in (A) LNCaP-AI and (B) LNCaP cells growing in steroid depleted media for 72 hours. Chromatin was collected and immunoprecipitation for H4K20Me1 carried out. Isolated DNA was purified and primers targeting the CDC20 promoter region used to determine the levels of H4K20Me1 association with this region. Experiments were performed 3 times and data is expressed as the mean fold change relative to non-silencing control +/- SEM. IgG was used as a negative control. Student’s T-test * p<0.05.
Figure 6.
H4K20Me1 is reduced at the CDC20 promoter in response to KMT5A knockdown.KMT5A was knocked down in (A) LNCaP-AI and (B) LNCaP cells growing in steroid depleted media for 72 hours. Chromatin was collected and immunoprecipitation for H4K20Me1 carried out. Isolated DNA was purified and primers targeting the CDC20 promoter region used to determine the levels of H4K20Me1 association with this region. Experiments were performed 3 times and data is expressed as the mean fold change relative to non-silencing control +/- SEM. IgG was used as a negative control. Student’s T-test * p<0.05.
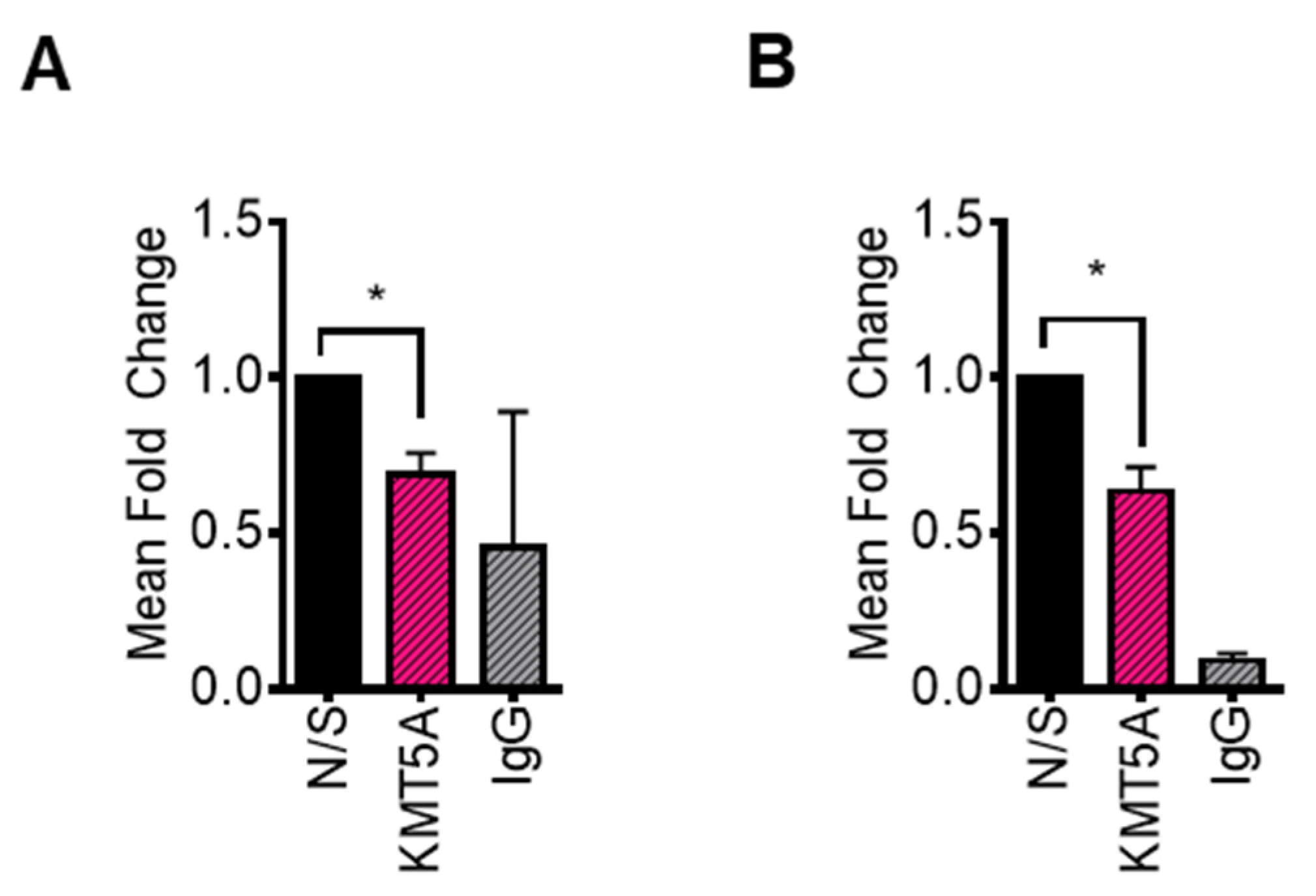
Figure 7.
KMT5A regulates CDC20 via p53 activation. (A) LNCaP-AI and LNCaP cells were reverse transfected with either a pool of 3 KMT5A targeting siRNA or non-silencing (N/S) control for 72 hours prior to assessment of γ-H2AX by flow cytometry. (B) LNCaP-AI and LNCaP cells were reverse transfected with 2 independent siRNA sequences for 72 hours prior to western blotting analysis for CDC20, p53, p-p53, p53-Ac, MDM2, KMT5A. (C) Densitometry of western blots shown in (B). Background was subtracted from intensity values prior to intensity normalisation against appropriate loading control. Intensities for post-translationally modified proteins were normalised to total protein intensity. (D) LNCaP-AI and LNCaP cells were treated with Nutlin 3 (5 μM) for 0 and 16 hours prior to western analysis. Western data shown is representative of 3 independent experiments. Student’s T-test was used to determine statistical significance for individual cell lines shown in (A).
Figure 7.
KMT5A regulates CDC20 via p53 activation. (A) LNCaP-AI and LNCaP cells were reverse transfected with either a pool of 3 KMT5A targeting siRNA or non-silencing (N/S) control for 72 hours prior to assessment of γ-H2AX by flow cytometry. (B) LNCaP-AI and LNCaP cells were reverse transfected with 2 independent siRNA sequences for 72 hours prior to western blotting analysis for CDC20, p53, p-p53, p53-Ac, MDM2, KMT5A. (C) Densitometry of western blots shown in (B). Background was subtracted from intensity values prior to intensity normalisation against appropriate loading control. Intensities for post-translationally modified proteins were normalised to total protein intensity. (D) LNCaP-AI and LNCaP cells were treated with Nutlin 3 (5 μM) for 0 and 16 hours prior to western analysis. Western data shown is representative of 3 independent experiments. Student’s T-test was used to determine statistical significance for individual cell lines shown in (A).
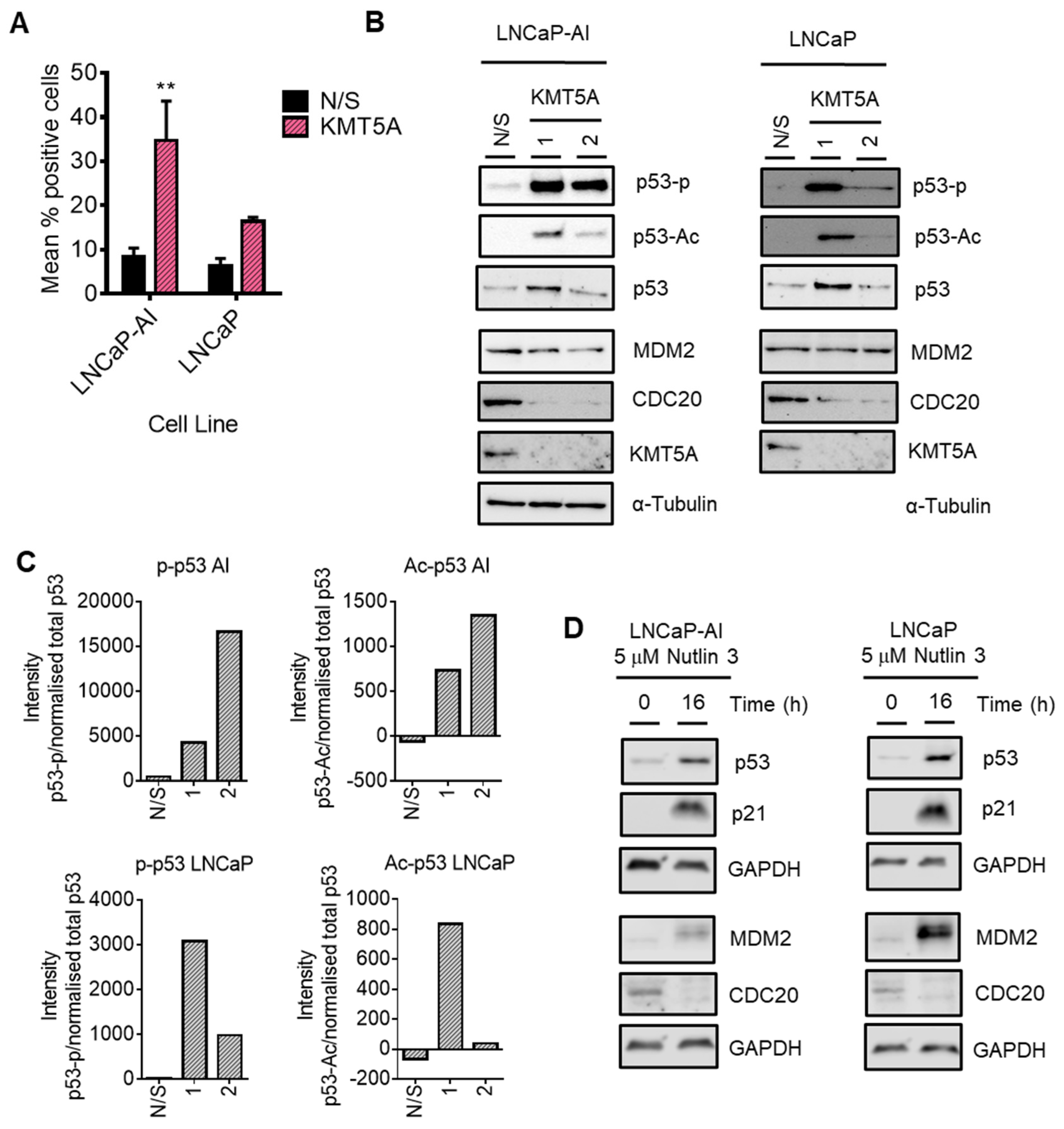
Figure 8.
KMT5A regulates protein turnover in the absence of p53. (A) KMT5A was knocked down with 2 independent siRNA for 72 hours. Protein was collected and analysed by western blotting for KMT5A, CDC20 and α-tubulin was used as a loading control. (B) In a parallel experiment RNA was collected and gene expression of CDC20 and KMT5A analysed by QPCR. Data is shown as the mean fold change relative to N/S controls over 3 experimental repeats +/- SEM. One-way ANOVA was used to determine statistical significance.
Figure 8.
KMT5A regulates protein turnover in the absence of p53. (A) KMT5A was knocked down with 2 independent siRNA for 72 hours. Protein was collected and analysed by western blotting for KMT5A, CDC20 and α-tubulin was used as a loading control. (B) In a parallel experiment RNA was collected and gene expression of CDC20 and KMT5A analysed by QPCR. Data is shown as the mean fold change relative to N/S controls over 3 experimental repeats +/- SEM. One-way ANOVA was used to determine statistical significance.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



