Submitted:
26 May 2023
Posted:
30 May 2023
You are already at the latest version
Abstract
Inorganic chemistry has contributed to the progress of human civilization in several different ways. Its activities at the interface with other scientific disciplines has led to emergence of several new fields like organometallic chemistry, bio-inorganic chemistry, solid-state chemistry, environmental chemistry, etc. In the past few decades, it has made inroads in yet another dimension of immense relevance, i.e. nano-science and –technology. In this assay an attempt has been made to present an overview of utility of inorganic and organometallic compounds as precursors for the synthesis of nano-materials with reference to catalysis and materials science. Applications of metal nanoparticles (NPs) and metal chalcogenide NPs, generated through molecular precursor route, in catalysis have been discussed. Synthesis of ternary and quaternary metal chalcogenide nano-materials by molecular precursor route has been reviewed.

Keywords:
Nanoparticles
; catalysis
; metal chalcogenide
; ternary metal chalcogenide
; quaternary metal chalcogenide
1. Introduction
Inorganic chemistry, one of the oldest disciplines of science, has contributed to the progress of human civilization in several ways. This branch of chemistry developed empirically since the Bronze Age (3000 to 1200 BC). Following the Iron Age (1200 to 550 BC), the ancient era began and were named after different cultures and civilizations prevalent at that time, like Roman, Viking, Indus Valley, Samkhya, Vaiseshika, etc. [1,2]. During this period alchemy evolved primarily to meet two objectives, viz., (i) transmutation process to convert lead in to gold, and (ii) preparation of an elixir of life. Although alchemists were never able to accomplish these objectives, could make several important contributions including the discovery of strong acids (H2SO4, HNO3, HCl) and strong bases (NaOH) and made several significant improvements in metallurgical processes. The beginning of inorganic chemistry in the present scientific form can be traced from 18th century [1]. In the last decade of the 19th century, the coordination theory proposed by Alfred Werner in 1893 led to meteoric growth of the area and emerged as one of the major branches of inorganic chemistry, Several theories (like CFT, LFT, etc.) and processes (e.g. Haber-Bosch process for ammonia synthesis, Ziegler Natta process, etc.) were developed after the World War-II.
The scope and horizon of inorganic chemistry appear practically limitless [3]. The utility of inorganic and organometallic compounds can be found in different forms in our daily lives encompassing from food, health, energy, environment, security, etc. Activities at the interface of inorganic chemistry and other areas have led to emergence of several new fields like organometallic chemistry, solid-sate chemistry, bio-inorganic chemistry, catalysis, materials science, environmental chemistry, etc. In the past few decades, it has made inroads in yet another dimension of immense relevance, i.e. nano-science and –technology. In this assay, we intend to present an overview of inorganic and organometallic compounds as precursors for the preparation of nano-materials with reference to catalysis and materials science. Among several synthetic approaches for the preparation of nano-materials, molecular precursor route has emerged as a successful strategy. It has an edge over other preparative methods due to ease in processability and better control over size of nano-structures, morphologies and composition of the resulting material [4,5,6,7]. Being a softer chemical synthetic route, it is also advantageous in terms of isolating metastable phases, particularly for metal chalcogenides [8].
2. Nano-materials-based catalysis
Numerous catalysis processes, both homogeneous and heterogeneous, were known since the second half of the eighteenth century even before the term ‘catalysis’ was coined by Berzelius in 1835. Since then, catalysts have played a very important role in industry for production of a myriad of chemicals [9,10]. Homogeneous and heterogeneous catalysts have their own advantages and limitations and can readily be distinguished from each other according to whether the catalyst and reactants are present in the same phase. Homogeneous catalysis reactions proceed at moderately low temperatures and are fairly well understood at molecular level permitting to design and develop new catalysts. Homogeneous catalysts are the most active and selective in the catalytic process. The major drawback of homogeneous catalysis is the difficulty of separation and isolation of catalysts from the final product, although use of supported-catalysts and biphasic catalysis have circumvented the catalyst’s separation problem [9]. Heterogeneous catalysis is often carried out at high temperatures and pressure. Heterogeneous catalysts are solids with a large surface area. Active sites are generated and on the external surface of the catalyst during the reaction. As the active sites are not always accessible, the overall efficiency of the catalytic process is often compromised. To overcome these problems associated with homogeneous and heterogeneous catalysts, another approach relying on nanoparticles has emerged and is growing rapidly [11], although this approach has been known since the early twentieth century when finely divided nickel particles were used as catalysts for hydrogenation [12]. Besides the large surface area of nanoparticles (NPs), metal NPs usually exhibit enhanced reactivity under mild conditions. However, reaction selectivity in these nanoparticle-based catalytic processes is rather elusive, but can be eluded by employing facet-controlled nanoparticles [13].
2.1. Metal nanoparticle-based catalysis
Transition metal NPs are now widely used as catalysts in a wide-ranging organic transformations like hydrogenation, hydro-dehydrogenation, various C-C coupling reactions, oxidation reactions, etc. [11,14]. Among metal NPs, palladium occupies a prominent place mainly due to its versatility in catalytic reactions [15].
Although a number of preparative routes have been endowed for the synthesis of metal NPs, the use of metal NPs generated via organometallic route in catalysis is briefly covered in this section. Organometallic precursors under reducing atmosphere (e. g., H2) or photo/ thermal conditions has emerged as an effective and reproducible method for the preparation of size, shape and composition-controlled metal nanoparticles with better dispersibility. The method has been implemented for producing various platinum group metal nanoparticles. Decomposition of [PdCl2(COD)] in the presence of N-substituted 1,3,5-triaza-7-phosphaadamantane ionic ligands under H2 atmosphere in glycerol at 60°C yields uniformly dispersed palladium nanoparticles passivated by the ligand. The latter has been employed in the benchmark Suzuki–Miyaura reaction with 90% yield [16]. Thus, an effective catalyst immobilised in glycerol has been produced which can run up to ten catalytic cycles without loss of activity. Palladium nanoparticles have been deposited onto terpyridine-modified magnetic support by decomposition of Pd2(dba)3 in the presence of H2 [17]. This nano-catalyst system is active for hydrogenation of cyclohexene under mild conditions with TOFs up to ca. 58000 h−1 or 129000 h−1. Moreover, this nano-catalyst is highly selective for the formation of monohydrogenated compounds in hydrogenation of myrcene [17].
Besides hydrogen for the preparation of nano-catalysts, thermal routes have been employed routinely for the synthesis of platinum group metals nano-particles. For Instance, face-centred cubic (fcc) phase of platinum NPs have been isolated by solvothermal decomposition of [Pt2(μ-OR)2(C8H12OMe)2] in hexadecylamine (HDA) at 210°C. The methoxy-bridged complex (OR = OMe) gave spherical Pt NPs with the size varying in the range 9-12 nm, whereas the acetato-bridged (OR = OAc) complex produced rod-like particles of 18-21 nm [18]. Several palladium complexes, such as palladium phosphine complexes, palladacyclic compounds, pincer derivatives, [Pd(OAc)2]3, etc. are routinely used as catalysts. In fact these compounds act as pre-catalysts as most of the catalytic reactions are performed at above 100°C. At such temperatures, catalysts tend to produce palladium nano-particles/ colloids [19,20]. It has been shown that palladium atoms are leached out in solution from these particles which catalyse the reactions [20].
Monodispersed spherical face-centred cubic (fcc) palladium NPs of size ranging between 1.7 and 3.5 nm have been prepared by thermolysis of a toluene solution of [Pd(OAc)2]3 and n-dodecylsulfide [21]. The size of the particles can be controlled by thermolysis temperature and the duration of thermolysis. Smaller size particles (1.7 ± 0.2) are formed at lower temperature (95 °C) and shorter reaction time (1 Hr). Shorter chain thio ethers (R2S; R = Et, Prn, Bun, Hexn, Octn) at 95 °C also afforded thio ether passivated Pd NPs but the NPs are polydispersed with the size ranging from 1.5 to 7.3 nm. As synthesized Pd NPs showed excellent catalytic activity in hydrogenation of styrene to ethyl benzene [21].
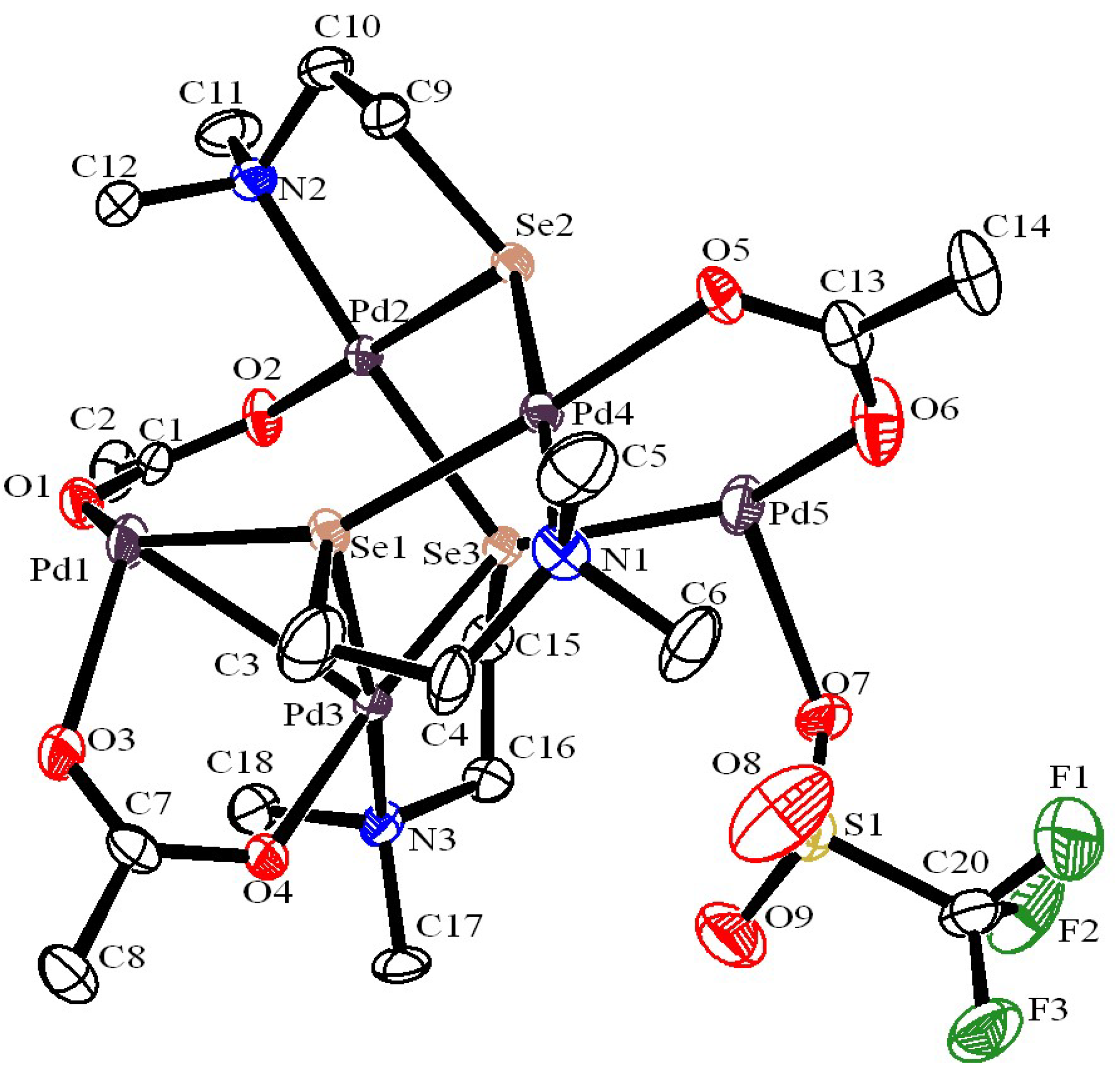
Palladium NPs generated through palladium chalcogenolate complexes route, usually form in situ, have been employed mainly in Suzuki-Miyaura C-C coupling and C-O coupling reactions. Depending on the nature of chalcogen ligand and the reaction conditions, these complexes yield either palladium NPs protected by organic moiety from the ligand or palladium chalcogenides (see later) [22,23,24]. Interestingly, the formation of palladium(0) stabilized by chalcogenolate ligand has been established by X-ray structural analysis of a pentanuclear complex, [Pd5(OAc)3(SeCH2CH2NMe2)3][OTf]2 (Figure 1) [25]. The molecular structural analysis revealed that there are four palladium centres which acquire four coordinate configuration - three of them are in a distorted square-planar configuration while geometry around the fourth one is so distorted making it nearly tetrahedral. The fifth palladium, in zero oxidation state, adopts a distorted linear configuration (<O6-Pd5-Se3 ≈ 150°). This configuration resembles with several Pd(0) phosphine complexes, [Pd(PR3)2] employed in cross-coupling reactions [25].
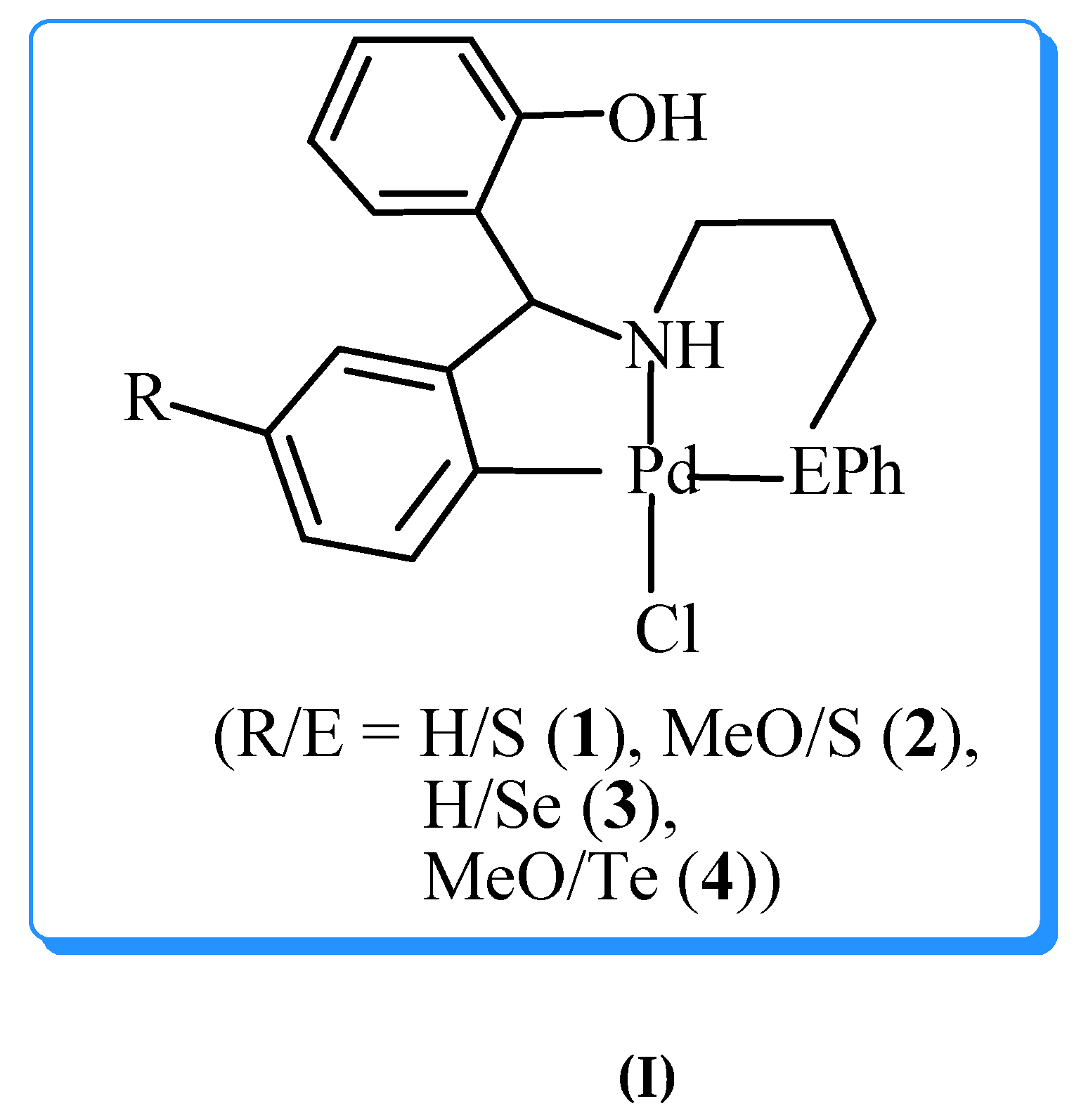
Palladium complexes derived from selenium containing Schiff bases when employed in Suzuki-Miyaura C-C coupling reactions generate Pd NPs (~ 3nm) in situ [26]. Size, dispersion and catalytic activity of the Pd NPs are controlled by the alkyl cahin length of the alkoxy group present in the Schiff base (Scheme 1) [26]. For instance, the Pd NPs formed by the complex containing OC18H37 group shows much greater activity (yield of C-C coupled product ~90%) than that formed from the complex containing OMe group (yield of C-C coupled product ~25%) [26]. Similarly, palladium(II) complexes, [PdCl2(L)] (L = 1-benzyl-4-phenylthiomethyl or 1-benzyl-4- phenylselenomethyl-1H-1,2,3-triazole), where ligand is chelated to palladium through a nitrogen atom of 1,2,3-triazole moiety and sulfur/selenium atom, generated chalcogen ligand passivated Pd NPs (3-11 nm) during the course of Heck and Suzuki-Miyaura C-C coupling reaction [27]. Palladium complex derived from telluro ether ligand generated Pd NPs stabilized by organic fragments containing tellurium, during Suzuki Miyaura coupling reactions (Scheme 2) [28].
Among platinum group metal NPs, ruthenium [29] and rhodium [30] NPs as catalysts are also gaining attention. Ruthenium nanoparticles are readily prepared from organometallic precursors like [Ru(cod)(cot)] (cod = 1,5-cyclooctadiene, cot = 1,3,5-cyclooctatriene) and [Ru(2-methylallyl)2(cod)] under H2 atmosphere in the presence of a polymer or a ligand as surface stabilizer [31]. The Ru NPs produced by thermal decomposition of [Ru(cod)(cot)] immobilized in polymer, such as polyvinylpyrrolidone (RuNP@PVP), have been employed as a catalyst for enantiospecific C-H activation of amines, amino acids, peptides [32].The same precursor when decomposed in the presence of SIDPhNp ((4S,5S)-1,3-di(naphthalen-1-yl)-4,5-diphenylimidazolidine) and SIPhOH ((S)-3-((1S,2R)-2-hydroxy-1,2-diphenylethyl)-1-((R)-2-hydroxy-1,2-diphenylethyl)-4,5-dihydro-3H-imidazoline) as stabilizers under H2 atmosphere at room temperature, gave ruthenium nanoparticles. The latter showed catalytic activity in various hydrogenation reactions [33]. Although interesting selectivity differences has been detected, enantio-selectivity has not been observed. Recently, Leitner and co-workers [34] immobilized Ru NPs prepared by H2 reduction (50 bar and 100°C) of [Ru(2-methylallyl)2(cod)] in imidazolium-based supported ionic liquid phases. The resulting Ru@SILP with diameters in the range of 0.8-2.9 nm is an efficient catalyst for hydrogenation of CO2 to formate in a mixture of water and triethylamine. Furthermore, ionic liquid modifiers with SO3− groups enhanced turnover number (TON) and turnover frequency (TOF) at lower metal loadings. For example, with the Ru@SILP(SO3H-OAc) catalyst, TONs up to 16100 at an initial TOF of 1430 h−1 has been realized [34].
A mixture of fullerene-C60/Rh(acac)3 in water under an inert atmosphere at elevated temperatures produced Rh NPs /Fullerene-C60 nano-catalyst [35]. Rh-nanoparticles (av. size of 4.3 nm) were uniformly dispersed on the surface of fullerene. Rh NPs /Fullerene-C60 nano-catalyst showed excellent activity in the reduction of 4-nitrophenol with NaBH4 in water as well as in Suzuki cross-coupling reaction.
2.2. Metal chalcogenide nanoparticles-based catalysis
Platinum group metal chalcogenides, in particular palladium chalcogenides, exhibit catalytic activity in a variety of reactions [22,23,36]. Metal sulfides, PdS, PtS, Rh2S3, Ir2S3, RuS2, have been employed as catalysts in hydrodesulfurization (HDS) reactions [36]. Platinum group chalcogenides (e.g., PdS2, Ir2S3, Ru2Se3) are also used as catalysts in hydrogenation reaction, e.g., reduction of nitrobenzene to aniline. Interestingly, these catalysts are insensitive to sulfur poisoning [36].
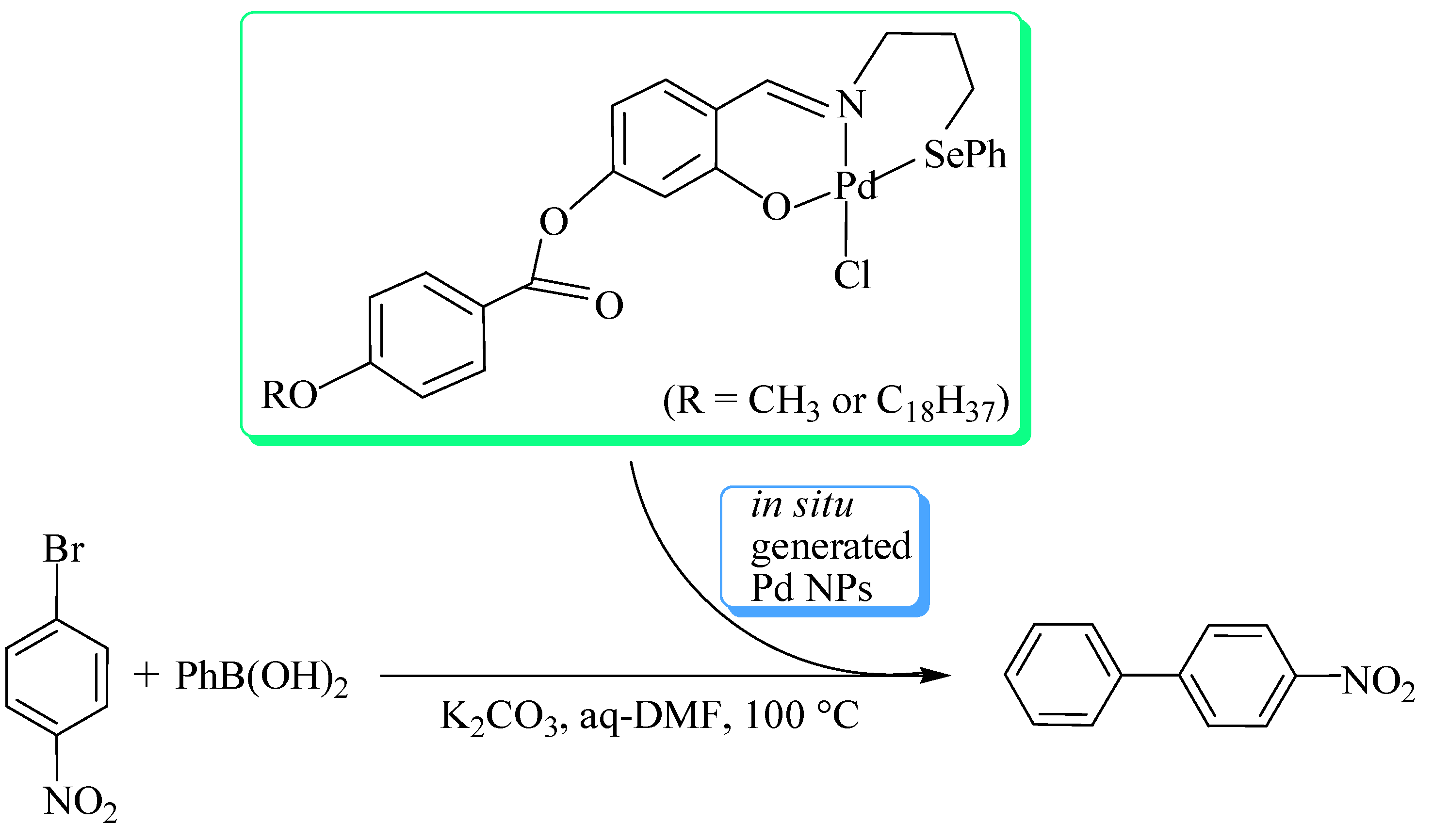
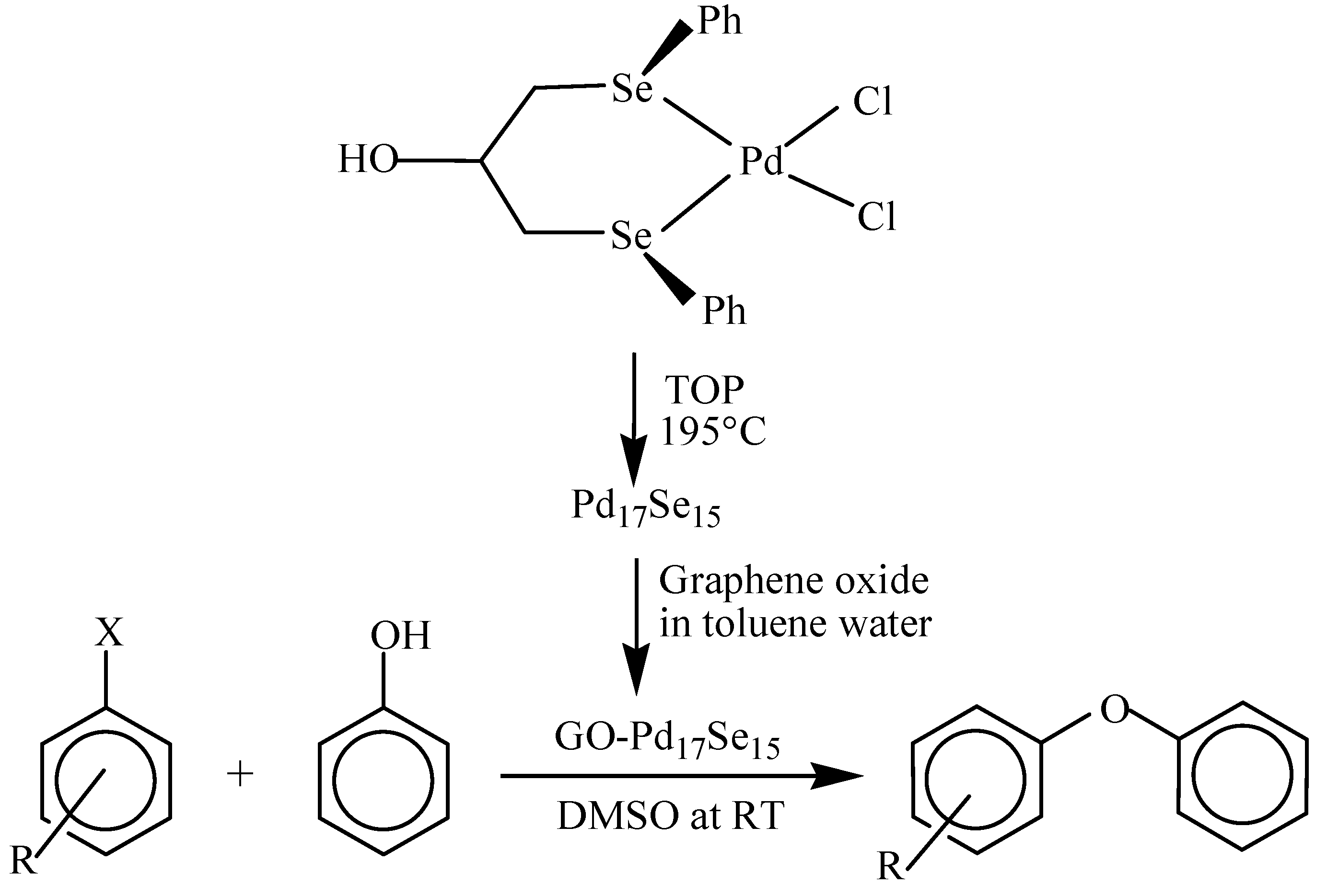
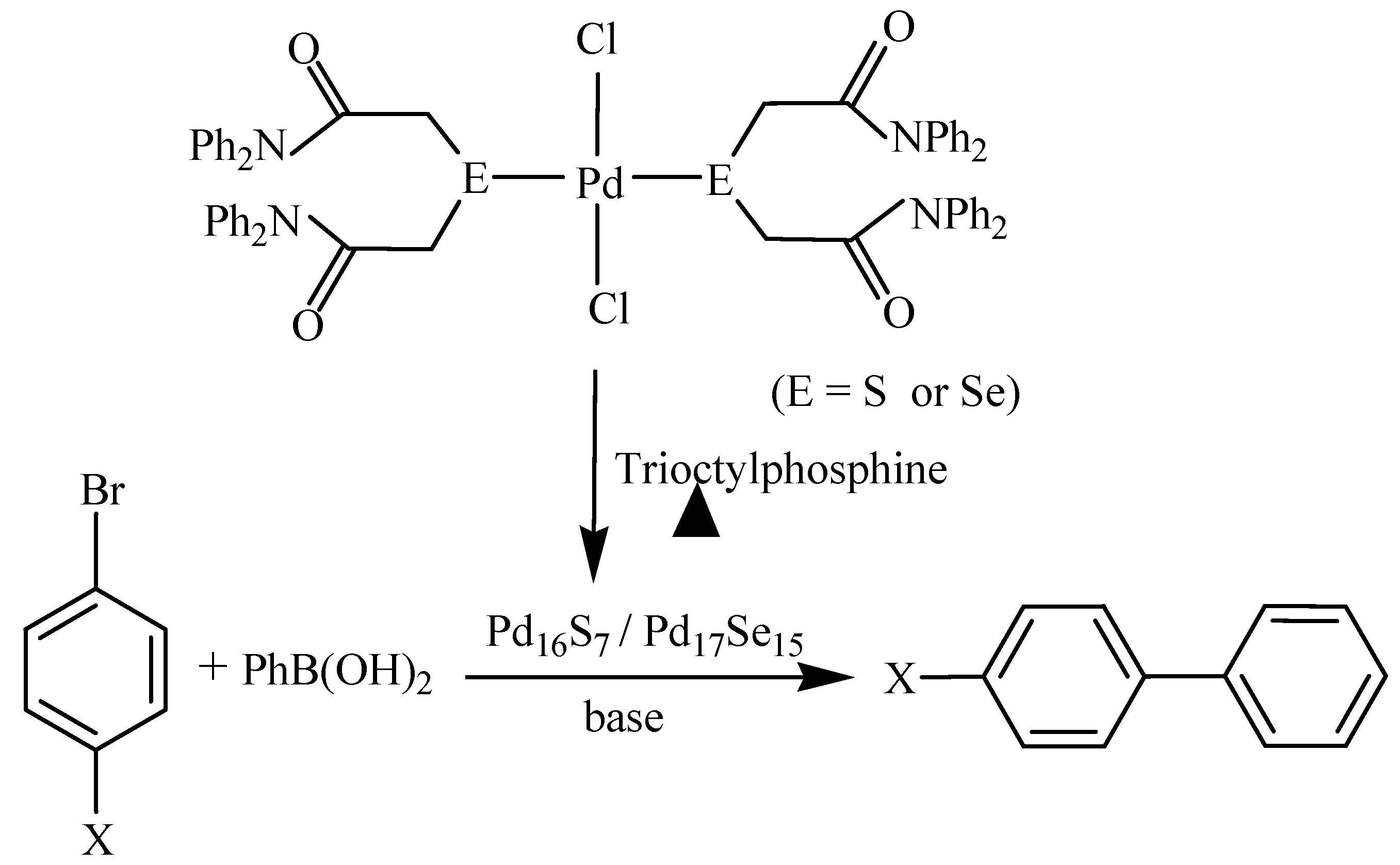
Palladium selenide nanoparticles, Pd17Se15, prepared from single source molecular precursors either by their thermolysis in trioctylphosphine [37,38] or in ionic liquid [39], catalyse Suzuki-Miyaura C-C coupling and C-O coupling reactions. The nanoparticles immobilized on to graphene oxide (GO-Pd17Se15) show better catalytic activity in C-O coupling reaction between an arylhalide and phenol at room temperature (Scheme 3) than most of the other catalysts (based on copper and palladium) which require either high temperature, long reaction time or high mol% of the catalyst [37]. The catalyst is even active in the reactions involving aryl chloride – well-known for their poor reactivity. The catalyst is recyclable up to four runs.
Thermolysis of trans-[PdCl2{E(CH2CONPh2)2}}2] (E = S or Se) in trioctylphosphine yields nanoparticles of palladium sulfide (Pd16S7 NPs, size ~31-40 nm) and palladium selenide (Pd17Se15 NPs, size ~36-45 nm) [38]. Both the palladium chalcogenide nanoparticles exhibit catalytic activity in Suzuki- Miyaura C-C coupling between aryl bromides and phenylboronic acid (Scheme 4) as well as C-O coupling reaction between aryl bromides and phenol in the presence of a base. The Pd17Se15 nanoparticles show better catalytic activity than Pd16S7 NPs. Similarly, Pd17Se15 NPs, prepared from a selenium containing carbene complex of palladium in an ionic liquid, 1-butyl-3-methylimidazolium bis(trifluoromethysulfonium)imide or propylene carbonate, catalyses Suzuki-Mayaura C-C coupling giving biaryls in 86-91% yields [39].
Unlike metal sulfides and selenides, catalytic potential of metal tellurides has been explored to some extent. Palladium forms numerous binary tellurides, but catalytic properties of only a few of them have been examined [23,36]. These tellurides are generated in situ from single source molecular precursors. Binary palladium tellurides, PdTe [40] and Pd9Te4 [40,41], generated from single source molecular precursors, catalyse Suzuki-Mayaura C-C coupling reactions and are even fairly efficient to catalyse the reactions of supposedly unreactive or poorly reactive aryl chlorides [40,41]. The Pd9Te4, being rich in palladium contents, shows better performance than PdTe [40]. These tellurides also catalyse reduction of nitrophenol to aminophenol (Scheme 5) [40]. These catalysts can be recycled up to six times.
Palladacycles (I) in Suzuki-Mayaura C-C coupling reaction serve as pre-catalyst and generate palladium chalcogenides nanoparticles (Pd16S7, Pd17Se15 and Pd3Te2) as the active catalytic species [42,43,44]. Efficiency of the catalyst is influenced by the size of the nanoparticle. For instance, efficiency of Pd16S7 NPs generated from complex 2 (size 6 nm) is significantly lower than that of NPs formed from 1 (size 2 nm) in the C-C coupling reaction [42]. The palladium rich compositions are more efficient in Suzuki-Mayaura coupling reactions even with larger particle size. For example, Pd9Te4 (size 2-4 nm) shows better activity than that of Pd3Te2 (size 1-2 nm) [41,44]. The nature of chalcogen in binary palladium chalcogenides also has a pronounced effect on the catalytic activity as is evident from the fact that nanoparticles of Pd9Te4 are better than those of palladium sulfide (Pd16S7) and palladium selenide (Pd17Se15) in terms of performance [42,43,44].
2.3. Metal chalcogenides as photo-/ electro-catalysts
Metal chalcogenides are emerging as promising photo-/ electro-catalysts. Unlike conventional oxide-based photo-catalysts like, TiO2, ZnO, etc., metal chalcogenides exhibit high absorption coefficient over a wide spectral region of the solar energy due to their narrow-band gap, consequently show enhanced photo-catalytic activity [45].The charge carriers (electron and hole, e- /h+) in these materials are generated with high separation efficiency which have been exploited in water splitting reaction, photo-degradation of various pollutants, photo-catalytic microbial activity [45].
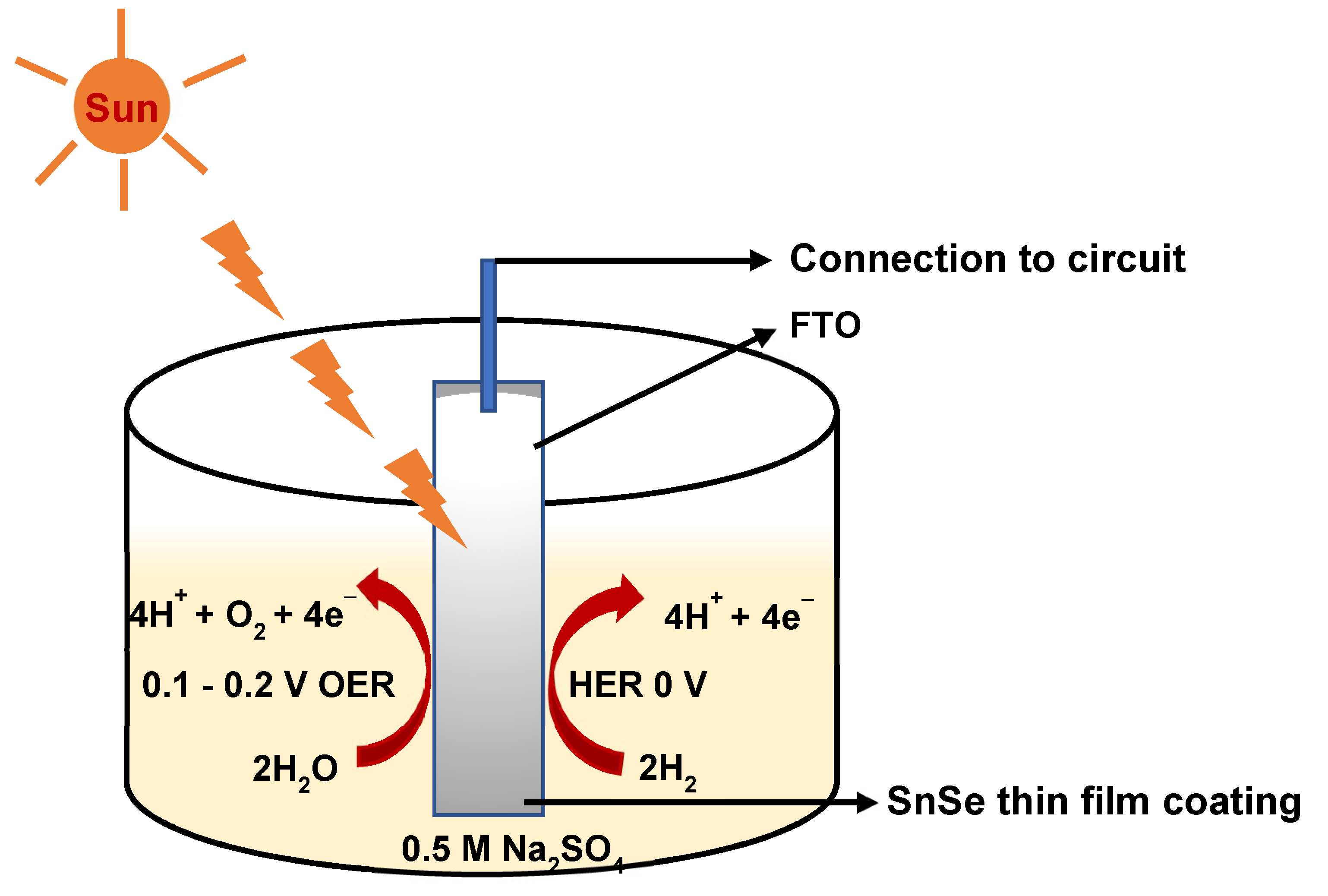
Tin selenide (SnSe, a p-type narrow-band semiconductor) thin films deposited from [Bun2Sn(SeCOPh)2] on FTO (fluorine doped tin oxide) coated glass substrate exhibit photoelectrochemical (PEC) splitting of water [46]. The films show bifunctional behaviour as they can be used for both hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) by switching from photocathode to photoanode by changing the bias voltages (Figure 2) [46].
Electrically conducting palladium selenides (tetragonal Pd4Se; orthorhombic Pd7Se4 and cubic Pd17Se15) films deposited on glass substrate by drop-cast method using different molar ratios of palladium acetate and dodecyl selenide [(C10H21)2Se] show electrocatalytic activity towards HER. For hydrogen evolution reaction, the onset potentials for Pd4Se; Pd7Se4 and Pd17Se15 are -0.03, -0.07 and -0.08 vs reversible hydrogen electrode, respectively [47]. Palladium rich selenide (Pd4Se) exhibits better electro-catalytic activity than Pd7Se4 and Pd17Se15. All the three palladium selenides require lower over potentials for HER than other known catalysts like MoS2, MoSe2. Suitability of Pd7Se4 and Pd17Se15 as counter electrode in dye-sensitized solar cells has also been evaluated. These selenides are stable under strong acid and alkaline environments and towards I-3 /I- redox couple. The photocurrent conversion efficiency of 6.88% and 7.45% is reported for Pd7Se4 and Pd17Se15, respectively [48].
3. Metal Chalcogenide Materials
The past few decades have witnessed a meteoric rise in research activities on metal chalcogenide nanomaterials due to their immense potential in wide spread technological and biomedical applications owing to their wide range of tunable band gaps across the energy spectrum and high absorption coefficient, etc. Metal chalcogenides can broadly be classified as binary (MxEn), ternary (MxMIyEn or MnExE′y) and quaternary (MxMIyMIIzEn or MnMIn′ExE′y or MnExE′yE″z) (where M, MI, MII are metals and E = S, Se, or Te) metal chalcogenides, depending on the number of elements present in these metal chalcogenides. Binary chalcogenides are the simplest class of inorganic materials. Depending on the formal oxidation state of the metal, different compositions of binary systems, like ME, ME2, M2E3 (where M = metal and E = S, Se, Te) are isolated. These materials exhibit remarkable electronic, optical, magnetic and mechanical prophesies which have been exploited in several useful ways. Modification in the crystalline structures of these materials by introducing defects, like dislocations, vacancies, edges, impurities and functional groups, further widens the horizon of their applications [49]. Although there are several synthetic approaches for the preparation of nanostructures of binary compounds, the single source precursor route offers numerous advantages [4,5,6,7]. This approach for ternary and quaternary metal chalcogenide nano-materials is evolving.
Both ternary and quaternary chalcogenide semiconductor materials find applications in photovoltaic, thermoelectric devices and photo-catalysis [50,51,52]. For instance, CuInSe2 has been investigated since 1975 as solar cell material with solar conversion efficiency in laboratory devices [53] now exceeding 21%. Various families of ternary and quaternary chalcogenide materials have been derived by a process of cross-substitution or swapping of elements in binary/ ternary systems while abiding the octet rule - a strategy pioneered by Goodman [54] and Pamplin [55]. Two types of ternary or quaternary systems can be generated from binary compounds. For instance, substitution of the cation by two or more different metal atoms while retaining the chalcogenide ligand results in ternary (e.g., Cd1-xZnXS; Mo1-xWxS2) and quaternary (e.g., Cu2ZnSnS4) metal chalcogenides of the type MxMIyEn and MxMyIMzIIEn, respectively. Alternatively replacing the chalcogenide ligand by two or three different types of chalcogen keeping cation site undisturbed can lead to ternary (e.g., MoSSe) and quaternary (e.g., CuInSSe) metal chalcogenide derivatives of type MxMIyEn and MnMIn′ExE′y/ MxMIyMzIIEn. This allows further tuning the optical and electronic properties in the resulting ternary system. Among metal chalcogenides, ternary and quaternary metal chalcogenides have relatively more distinct merits over binary counterparts as a consequence of an increase in degrees of freedom for tuning their properties. For example, their optoelectronic properties (e.g., band gap) not only can be suitably tuned over a wide range by manipulating size or shape but also changing the stoichiometry of constituent elements. In addition, these multinary metal chalcogenides possess higher flexibility in electrochemical properties owing to their hierarchical morphology. Besides, introduction of cation or anion in these metal chalcogenides boost the vacancies and also assist in realizing improved properties in the metal chalcogenide anodes in storage devices. Despite their immense technological relevance, preparation of compositionally and phase pure materials, however, remains elusive primarily due to two reasons. Firstly, formation of lower phases - existence of binary product in ternary system and binary and ternary phases in quaternary products, is often associated with their synthesis. Secondly, there are several structural configurations with more complicated electronic properties with increasing the number of elements on moving from binary to higher systems.
Among ternary chalcogenide nano-materials, compounds from the group I-III/IV/V-VI and II-III-VI (e.g., ZnIn2S4, CdIn2S4) represent highly functional materials. Molecular precursor route for their synthesis of some of the materials is briefly described here.
3.1. I-III-VI2 (I = Cu or Ag, III = Ga or In; VI = S or Se) compounds
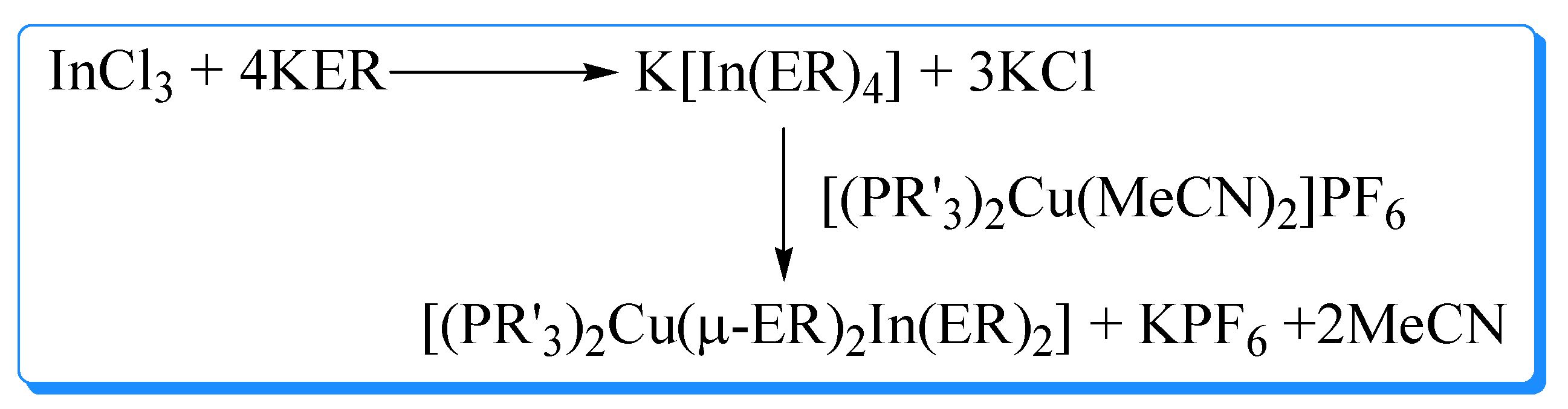
Copper based ternary materials (CuInE2) may exist in three different structural polymorphs, viz., (i) chalcopyrite (tetragonal), (ii) zinc blende (cubic closed pack (fcc) or sphalerite), and (iii) wurtzite (hexagonal). These materials have been prepared by molecular precursor routes. Kanatzidis and coworkers [56] reported the synthesis of binuclear chalcogenolate-bridged complexes of the general formula, [(PR′3)2Cu(μ-ER)2In(ER)2] (Scheme 6). Subsequently, a one-pot synthesis of these complexes has been described [57,58] which can be utilized to isolate products up to 500 g scale (Scheme 7). In an alternative method, [(PPh3)2Cu(μ-Cl)2InCl2] obtained by the reaction of [CuCl(PPh3)2] with InCl3, is treated with sodium thiolate in benzene and depending on stoichiometry of the reactants, products of the type [(PPh3)2CuIn(SEt)xCl4-x] (x = 1-4) are isolated [59]. This procedure has been extended to prepare [(PPh3)2M(μ-SEt)2M′(SEt)2] (M = Cu or Ag; M′ = Al, Ga, In) using [(PPh3)2M(μ-Cl)2M′Cl2] and NaSEt in hot benzene [60]. The SEt group in [(PPh3)2Cu(μ-SEt)2In(SEt)2] can be substituted by other thiolate / selenolate groups by treatment with REH and products of general formula [(PPh3)2Cu(μ-ER)2In(ER)2] (ER = SBu, SHex, SDodec, SPh, SePh) and [(PPh3)2Cu(μ-SEt)2In(EPh)2] ( E = S or Se) are isolated in nearly quantitative (97-99%) yields [61]. This method has been scaled up to produce [(PPh3)2Cu(μ-SPh)2In(SPh)2] on a 500 g scale in less than one hour [61].
Molecular structures of several of these complexes revealed tetrahedrally coordinated metal ions (Cu/Ag and Al/Ga/In) bridged by two chalcogenolate groups forming a four membered “MM′(μ-ER)2” ring (RE = SMe, SEt, SPh, SeEt, SePh, SeBz) (Figure 3) [56,57,60,61,62,63]. The four-membered ring, in general, adopts a planar conformation [56,60,61,62,63], but it is puckered in [(PPh3)2Ag(μ-SEt)2M′(SEt)2] (M′ = Al, Ga, In) and [(PPh3)2Cu(μ-SEt)2Ga(SEt)2] [60]. The coordination environment around each metal is completed by two tertiary phosphines on copper/silver and two terminal chalcogenolate on group-III metal ion.
The % weight loss in TG analysis of [(PR′3)2M(μ-ER)2M′(ER)2] (PR′3 = PBu3, PPh3; ER = SEt, SPrn , SBui, SPh, SeEt; M = Cu, Ag; M′ = Al, Ga, In) is consistent with the formation of MM′E2 materials [56,60,64]. Nano-materials of CuInS2 have been prepared through decomposition of [(PR′3)2Cu(μ-ER)2In(SR)2], [(PPh3)2Cu(μ-SEt)2In(SEt)2] being the most common precursor, by conventional thermolysis [65,66,67], photolysis [68] and microwave irradiation [58,69,70].
Thermolysis of a dioctylphthalate solution of [(PPh3)2Cu(μ-ER)2In(ER)2] (ER = SEt, SePh) in the presence [66] or absence [65] of hexanethiol in the temperature range 200-250°C yields nearly spherical nanoparticles of CuInE2, while in the absence of hexanethiol the particles were relatively large in size (3-30 nm) which were aggregated to form spherical clusters of ~ 500 nm [65], but in the presence of hexanethiol, thiol passivated particles of 2-4 nm size were isolated. The fluorescence in the latter particles arises from intra-band gap transitions [66]. Thermolysis of [(PPh3)2Cu(μ-ER)2In(ER)2] (ER = SEt or SePh) in THF solution in an autoclave at 140-200 °C for ER = SEt in the presence of elemental sulfur and 100-200°C for ER = SePh gives wurtzite phase of CuInE2 nanoparticles of size 2-5 nm [67]. The size of the nanoparticles can be controlled by modulation of thermolysis temperature.
Thermolysis of [(PPh3)2Cu(μ-SeBz)2In(SeBz)2] in oleylamine (OAm) or HDA at 300oC results in the formation of body centred tetragonal phase of CuInSe2 nanoparticles of irregular shape with size varying in the range of 20-30 nm. The optical direct band gap of these nanoparticles, determined from diffuse reflectance spectroscopy, is 1.86 eV which is blue shifted as compared to the bulk CuInSe2 (i.e. 1.04 eV) [63]. CuInSe2 nanowires of 33 ± 10 nm in diameter and up to several micrometers in length, have been prepared by injecting a TOP solution of [(PPh3)2Cu(μ-SePh)2In(SePh)2] to a pre-heated (270-300°C) mixture of TOP and oleic acid followed by (gold) bismuth catalysts nanoparticles. These nanowires were crystalline in nature and grew along (112) direction and had a slightly metals rich composition of CuInSe2 [71].
Thin films of CuInE2 have been deposited on various substrates like Mo, Si(100), fused silica, glass by spray CVD using toluene solutions of [(PR′3)2Cu(μ-ER)2In(ER)2] (PR′3 = PBu3, PPh3; ER = SMe, SEt or SePh) at 390-400oC/30 mm Hg. Film microstructures and orientations are influenced by deposition parameters [57,72,73,74]. However, silver complexes afforded AgInS2 and AgIn5S8 [57].
Photolytic decomposition of benzene/ toluene/ or pentane solution of [{P(octyl)3}2Cu(μ-SR)2In(ER)2] (R = Prn, But) by ultraviolet radiation (266 or 355nm) yields nanoparticles of CuInS2 of ~ 2 nm which are much smaller and more crystalline than those prepared by thermolysis of the precursors [67]. Dioctylphthalate or benzyl acetate solution of [(PPh3)2Cu(μ-SEt)2In(SEt)2] in the presence of 1, 2-ethanedithiol on microwave irradiation produced CuInS2 nanoparticles with the diameter ranging from 1.8-10.8 nm [68]. The size of the particles could be controlled by reaction time and temperature.
Thermolysis of a mixture of dual chalcogenolate precursors has also been successfully employed in one-pot synthesis of copper based ternary materials. Thus thermolysis of a 1:1 mixture of either [Me2In{SeC5H4N-4}]n and [Cu(SeC5H4N-4)(PEt3)2] in OAm [75] at 300oC or [In{SeC5H3(Me-3)N}3] and [Cu(SeC5H3(Me-3)N}]4 in OAm/ HDA/ TOPO [76] at 330oC yields tetragonal phase of CuInSe2. The former combination of precursors yields a product slightly contaminated by In2Se3. The latter precursors in OAm give nearly spherical particles of tetragonal CuInSe2 with an average diameter of 200 nm whereas in other coordinating solvents much larger particles of nearly spherical nature could be formed [76]. Similarly, xanthate complexes of copper and indium have also been employed for the synthesis of CuInS2. Thus a toluene solution of a mixture of [Cu{S2COCH(Et)But}] and [In{S2COCH(Et)But}3] in 1:1 molar ratio in the presence of OAm at RT yield NPs of chalcopyrite phase of CuInS2 with an average diameter of ~3 nm [77].
3.2. I-IV-VI (I = Cu, IV = Sn; VI = S or Se) compounds
Nanostructures of Cu2SnE3 (E = S or Se) have been prepared by using a mixture of dual chalcogenolate precursors [78,79]. Thus, thermolysis of a 1:2 mixture of [But2Sn(Spyz)2] and [Cu(Spyz)(PPh3)2] (Spyz = 2-pyrazinylthiolate) (Figure 4) in OAm at 300oC affords spherical nanoparticles of monoclinic Cu2SnS3 (CTS) with average diameter of ∼ 40 nm [78]. The high resolution TEM (HRTEM) image revealed crystalline nature with lattice fringe spacing of 3.1Å (Figure 5). These nanoparticles show better photoluminescence than Cu1.8S [78]. Thermolysis of [Me2Sn(SepyMe-3)2] and [Cu(SepyMe-3)4] in OAm or TOPO at 300oC yields phase pure cubic Cu2SnSe3 nano-crystals which could be isolated in different morphologies (flower-shaped, dish-like, nano-sheets) by varying the reaction conditions. These nano-structures are photo responsive in a photo electrochemical cell with power conversion efficiency of 1.7% [79]. Similarly, phase pure cubic Cu2GeS3 nanostructures of size 11 nm have been synthesized by thermolysis of [Cu(S2COPri)2TMEDA] and Ge(S2COPri)4 in OAm. HRTEM image of these nanostructures unveil lattice fringes with a d-spacing of ~ 0.31 nm corresponding to (111) lattice plane of cubic Cu2GeS3 corroborating with Bragg reflections observed in the pXRD pattern of the same [80]. The direct band gap of these nanostructures has been estimated as 2.36 eV. Photo-electrochemical cell made from these Cu2GeS3 nanostructures exhibit photovoltaic properties under light conditions.
3.3. I-V-VI (I = Cu or Ag; IV = Sb or Bi; VI = S or Se) compounds
The lattice of these compounds is formed mostly based on antimony and bismuth with complimentary Cu(I) or Ag(I) ions. Copper antimony sulfide exists in four dif different forms, viz., (i) CuSbS2 (chalcostibite), (ii) Cu3SbS3 (skinnerite), (iii) Cu3SbS4 (famatinite) and (iv) Cu12Sb4S13 (tetrahedrite). Dual source precursors have been employed for the preparation of a variety of compositions and shapes of copper antimony sulfides through a one-pot synthesis route. Xanthate and dithiocarbamate complexes have been used as precursors. Thus thermal decomposition of copper and antimony diethyldithiocarbamates at 220°C yields CuSbS2, rhombic Cu3SbS3 and pyramidal Cu12Sb4S13 depending on molar ratios of the precursors and the nature of the solvent (OAm or a mixture of OAm and 1-dodecanethiol) [81]. The same precursors have been employed for the synthesis of CuSbS2 and Cu3SbS4 (fematinite) at a low Cu:Sb precursor ratio while tetrahedrite (Cu12Sb4S13) at a high Cu:Sb precursor ratio through the solventless thermolysis and aerosol assisted chemical vapour deposition (AACVD) methods [82]. A chlorobenzene solution of a mixture of 1:1 or 3:1 ratio of [Cu{S2COCH(Et)But}] and [Sb(S2COPri)3] after spin coating followed by treatment with n-hexylamine results in the formation of CuSbS2, Cu12Sb4S13 thin films, respectively at room temperature. The band gaps of these films are 1.57 and 1.74 eV respectively which are well suited for PV applications [83].
3.4. II-III-VI (II = Zn or Cd; III = In or Ga; VI = S or Se) compounds
ZnM2E4 (where M = Ga or In and E = S or Se) belongs to a family of tetrahedral semiconductors of type II-III2-VI4. ZnIn2S4 is a layered semiconductor occurring in three crystal polymorphs viz., cubic, hexagonal and rhombohedral lattices. Hexagonal phase ZnIn2S4 (ZIS) spherical nanocrystals (NCs) of size ranging from 2.9 to 8.5 nm have been prepared by co-thermolysis of xanthate precursors, [In(S2COPri)3] and [Zn(S2COPri)2] in OAm at 280°C at different time (10, 15 and 20 mins) [84]. Moreover, the size dependent optical properties of luminescent ZIS NCs with quantum yields in the range of 7-11% have been investigated. Life time measurements of these NCs exhibit life times in the range of 1.3-1.5 ns and 5.7-7.3 ns for the fast- and slow-decaying components, respectively.
3.5. I-IIIx-III′1-x-VI2 (I = Cu; III or III′ = Ga or In; VI = S or Se) quaternary compounds
The title compound semiconductors can be produced by replacing cations or anions of ternary semiconductors with ions of the same oxidation state. Such type of semiconductors are also known as pseudo-quaternary compounds. CuInxGa1−xS2 (0 ≤ x ≤1) nanoparticles have been prepared by co-decomposition of two single source molecular precursors, viz. (Ph3P)2Cu(μ-SEt)2In(SEt)2 and (Ph3P)2Cu(μ-SEt)2Ga(SEt)2 under microwave irradiation [85]. Tunable band gaps in the range of 1.4 to 2.3 eV have been achieved by controlling the stoichiometry in the quaternary CuInxGa1−xS2 nanoparticles.
3.6. I2-II-IV-VI4 (I = Cu; II = Zn or Cd; IV = Sn; VI = S or Se) quaternary compounds
Quaternary I2–II–IV–VI4 compound semiconductors can be derived ternary I–III–VI2 compounds by replacing two trivalent cations with one divalent and one tetravalent cation. Cu2ZnSnS4 (CZTS) and Cu2ZnSnSe4 (CZTSe) are evolving as cheap alternative materials with similar physical properties such as band gaps, intrinsic p-type nature and absorption coefficients to that of CuInS2 and CuInSe2. Nanostructured CZTS thin films have been deposited by spraying a solution containing metal dithiocarbamates of Cu(S2CNEt2)2, Zn(S2CNEt2)2 and Sn(But)2(S2CNEt2)2 in 2:1:1 ratio onto the preheated glass substrates at 450°C under argon for 1 h and the formation of CZTS has been established by pXRD and Raman spectroscopy [86].
4. Conclusions
From the foregoing discussion it is evident that the molecular precursors provide a convenient synthesis route for the preparation of a wide variety of exotic nano-materials of technological importance. This approach also offers high versatility for the synthesis of rather complex ternary and quaternary chalcogenides. It is hoped that this assay would facilitate further progress in designing new molecular precursors and their use in the synthesis of nano-materials of desired compositions, phases and morphologies.
Acknowledgments
The authors are grateful to the Department of Atomic Energy for funding. We sincerely acknowledge the contributions of our collaborators, colleagues, and students whose names appear as co-authors in the publications.
References
- Banerjea, D. ‘Inorganic chemistry: A modern treatise’, Asian Books Pvt. Ltd., New Delhi, 2012.
- Ray, P.C. ‘A history of Hindu chemistry’, Volume 1, 1902.
- Yam, V.W.W. Angew. Chem. Int. Ed. 2015, 54, 8304-8305.
- Jain, V.K.; Kedarnath, G., ‘Applications of metal-selenium and /-tellurium compounds in materials science’ Chapter 11, pp 383-443, in ‘Selenium and tellurium reagents in chemistry and materials science’; Eds. Laitinen, R.; Oilunkaniemi,R., Walther de Gruyter, Germany, 2019.
- Kedarnath, G., ‘Synthesis of advanced inorganic materials through molecular precursors’, Chapter-15, in ‘Handbook on synthesis strategies for advanced materials’ Eds. Tyagi A.K.; Ningthoujam, R.S., Springer Nature Singapore Pte Ltd., 2021.
- ‘Nanomaterials via single source precursors: Synthesis, processing and applications’, Eds. Apblett, A. W.; Barron, A. R.; Hepp, A. F., Elsevier, 2022.
- Jain, V.K.; Chauhan, R.S., ‘Metal chalcogenolates: Synthesis and applications to materials science’, Chapter-3; pp 58-82; in ‘Chalcogen Chemistry: Fundamentals and Applications’, Eds. Lippolis, V.; Santi, C.; Lenardao, E.J.; Braga, A.L., RSC (UK) 2023.
- Gu, C.; Xu, H. M.; Han, S. K.; Gao, M. R.; Yu, S. H., Chem. Soc. Rev. 2021, 50, 6671-6683.
- Steinborn, D. ‘Fundamentals of organometallic catalysis’ Wiley-VCH, Weinheim, Germany, 2012.
- Colacot, T.J. (Ed), ‘New trends in cross-coupling: Theory and applications’, Royal Society of Chemistry, UK, 2015.
- Serp, P.; Philippot, K. (Eds.), ‘Nanomaterials in catalysis’, Weinheim, Germany, 2013.
- Astruc, D., Chem. Rev. 2020, 120, 2, 461–463.
- Lee, K.; Kim, M.; Kim, H. J. Mater. Chem., 2010, 20, 3791-3798.
- Thematic issue of Chemical reviews dedicated to Nanoparticles in Catalysis, Chem. Rev. 2020, 120, issue No. 2.
- Favier, I.; Pla, D.; Gómez, M., Chem. Rev. 2020, 120, 1146−1183; Biffis, A.; Centomo, P.; Zotto, A.D.; Zecca, M., Chem. Rev. 2018, 118, 2249-2295.
- Chahdoura, F.; Favier, I.; Pradel, C.; Mallet-Ladeira, S.; Gómez, M., Catal. Commun. 2015, 63, 47-51.
- Guerrero, M.; Costa, N.J.S.; Vono, L.L.R.; Rossi, L.M.; Gusevskayad, E.V.; Philippot, K., J. Mater. Chem. A, 2013, 1, 1441.
- Ghawale, N.; Dey, S.; Jain, V.K.; Tewari, R., Bull. Mater. Sci. 2009, 32, 15-18.
- De Vries, J.G., Dalton Trans. 2006, 421-429.
- Gaikwad, A.V.; Holuigue, A.; Thathagar, M.B.; ten Elshof J.E.; Rothenberg, G., Chem. Eur. J. 2007, 13, 6908-6913.
- Ganesan, M.; Freemantle, R.G.; Obare, S.O., Chem. Mater. 2007, 19, 3464-3471.
- Arora, A.; Oswal, P.; Rao, G.K.; Kumar, S.; Kumar, A., Dalton Trans., 2021, 50, 8628-8656.
- Arora, A.; Oswal, P.; Datta, A; Kumar, A., Coord. Chem. Rev., 2022, 459, 214406.
- Oswal, P.; Arora, A.; Gairola, S.; Datta, A.; Kumar, A., New J. Chem. 2021, 45, 21449-21487.
- Paluru, D.K.; Dey, S.; Wadawale, A.P.; Bhuvanesh, N.; Grupp, A.; Kaim, W.; Jain, V.K., Chem. Asian J. 2016, 11, 401-410.
- Rao, G.K.; Kumar, A.; Kumar, B.; Kumar, D.; Singh, A. K., Dalton Trans. 2012, 41, 1931-1937.
- Saleem, F.; Rao, G.K.; Kumar, A.; Kumar, S.; Singh, M.P.; Singh, A.K., RSC Adv., 2014, 4, 56102-56111 .
- Kumar, S.; Rao, G.K.; Kumar, A.; Singh, M.P.; Singh, A.K., Dalton Trans. 2013, 42, 16939-16948.
- Axet, M. R.; Philippot, K., Chem. Rev. 2020, 120, 1085−1145.
- Luo, L.; Li, H.; Peng, Y.; Feng, C.; Zeng, J., ChemNanoMat, 2018, 4, 451-466.
- Lara, P.; Philippot, K.; Chaudret, B., ChemCatChem 2013, 5, 28-45.
- Taglang, C.; Martínez-Prieto, L.M.; del Rosal, I.; Maron, L.; Poteau, R.; Philippot, K.; Chaudret, B.; Perato, S.; Lone, A.S.; Puente, C.; Dugave, C.; Rousseau, B.; Pieters, G., Angew. Chem. Int. Ed. 2015, 54, 10474-10477.
- Martínez-Prieto, L.M.; Ferry, A.; Lara, P.; Richter, C.; Philippot, K.; Glorius, F.; Chaudret, B., Chem. Eur. J. 2015, 21, 17495-17502.
- Anandaraj, S. J. L.; Kang, L.; DeBeer, S.; Bordet, A.; Leitner, W., Small 2023, 2206806.
- Gopiraman,M.; Saravanamoorthy, S.; Ullah,S.; Ilangovan, A.; Kim, I.S.; Chung, I. M., RSC Adv., 2020, 10, 2545-2559.
- Dey, S.; Jain, V.K., Platinum Metals Rev., 2004, 48, 16-29.
- Joshi, H.; Sharma, K. N.; Sharma, A. K.; Prakash, O.; Singh, A.K., Chem. Commun., 2013, 49, 7483-7485.
- Singh, P.; Singh, A. K., Dalton Trans., 2017, 46, 10037-10049.
- Klauke, K.; Gruber, I.; Knedel, T.O.; Schmolke, L.; Barthel, J.; Breitzke, H.; Janiak, C., Organometallics, 2018, 37, 298-308.
- Arora, A.; Oswal, P., Rao, G. K.; Kumar, S.; Singh, A. K.; Kumar, A., RSC Adv., 2021, 11, 7214-7224.
- Arora, A.; Oswal, P., Rao, G. K.; Kumar, S.; Singh, A. K.; Kumar, A., Catal. Lett., 2022, 152, 1999-2011.
- Rao, G. K., Kumar, A.; Kumar, S.; Dupare, U. B.; Singh, A. K., Organometallics, 2013, 32, 2452-2458.
- Rao, G. K.; Kumar, A.; Ahmed, J.; Singh, A. K., Chem. Commun., 2010, 46, 5954-5956.
- Rao, G. K.; Kumar, A.; Singh, M. P.; Singh, A. K., J. Organomet. Chem., 2014, 749, 1-6.
- Rahman, A.; Khan, M.M., New J. Chem. 2021, 45, 19622-19635.
- Khan, M.D.; Amir, M.; Sohail, M.; Sher, M.; Baig, N.; Akhtar, J.; Malik, M.A.; Revaprasadu, N., Dalton Trans. 2018, 47, 5465-5473.
- Kukunuri, S.; Austeria, P.M.; Sampath, S., Chem. Commun. 2016, 52, 206-209.
- Kukunuri, S.; Karthick, S.N..; Sampath, S., J. Mater. Chem. A 2015, 3, 17144-17153.
- Zheng, Y.; Slade, T. J.; Hu, L.; Tan, X. Y.; Luo, Y.; Luo, Z. Z.; Xu, J.; Yan, Q.; Kanatzidis, M. G., Chem. Soc. Rev. 2021, 50, 9022-9054.
- Fan, F. J.; Wu, L.; Yu, S. H., Energy Environ. Sci., 2014, 7, 190-208.
- Coughlan, C.; Ibanez, M.; Dobrozhan, O.; Singh, A.; Cobat, A.; Ryan,K., Chem. Rev., 2017, 17, 5865-6109.
- Zheng, X.; Song, Y.; Liu, Y.; Yang, Y.; Wu, D.; Yang, Y.; Feng, S.; Li, J.; Liu, W.; Shen, Y.; Tian, X., Coord. Chem. Rev. 2023, 475, 214898.
- Stanbery, B. J., Critical Rev. Solid State Mater. Sci., 2002, 27, 73-117.
- Goodman, C. H. L. J. Phys. Chem. Solids, 1958, 6, 305-314.
- Pamplin, B. R. Nature, 1960, 188, 136-137.
- Hirpo, W.; Dhingra, S.; Sutorik, A. C.; Kanatzidis, M. G., J. Am. Chem. Soc., 1993, 115, 1597-1599.
- Banger, K. K.; Jin, M. H. C.; Harris, J. D.; Fanwick, P. E.; Hepp, A.F., Inorg. Chem., 2003, 42, 7713-7715.
- Gardner, J. S.; Shrudha, E.; Wang, C.; Lau, L. D.; Rodriguez, R. G.; Pak, J. J., J. Nanopart. Res., 2008, 10, 633-641.
- Margulieux, K. R.; Sun, C.; Zakharov, L. N.; Holland, A. W.; Pak, J. J., Inorg. Chem., 2010, 49, 3959-3961.
- Margulieux, K. R.; Sun, C.; Kihara, M. T.; Colson, A. C.; Zakharov, L. N.; Whitmire, K. H.; Holland, A. W.; Pak, J. J., Eur. J. Inorg. Chem., 2017, 2068-2077.
- Sun, C.; Westover, R. D.; Margulieux, K. R.; Zakharov, L. N.; Holland, A. W.; Pak, J. J. Inorg. Chem., 2010, 49, 4756-4758.
- Masnovi, J.; Banger, K. K.; Fanwick, P. P.; Hepp, A. F., Polyhedron, 2015, 102, 246-252.
- Pal, M. K.; Dey, S.; Neogy, S.; Kumar, M., RSC Adv., 2019, 9, 18302-18307.
- Banger, K. K.; Cowen, J.; Hepp, A.F., Chem. Mater., 2001, 13, 3827-3829.
- Castro, S. L.; Bailey, S. G.; Raffaelle, R. P.; Banger, K. K.; Hepp, A. F., Chem. Mater., 2003, 15, 3142-3147.
- Castro, S. L.; Bailey, S. G.; Raffaelle, R. P.; Banger, K. K.; Hepp, A.F., J. Phys. Chem. B, 2004, 108, 12429-12435.
- Zhao, X.; Huang, Y.; Corrigan, J.F., Inorg. Chem. 2016, 55, 10810-10817.
- Nairn, J. J.; Shapiro, P. J.; Twamley, B.; Pounds, T.; Wandruszka, R. V.; Fletcher, T. R.; Williams, M.; Wang, C.; Norton, M.G., Nano Letters, 2006, 6, 1218-1223.
- Sun, C.; Gardner, J. S.; Shrudha, E.; Margulieux, K. R.; Westover, R. D.; Lau, L.; Long, G.; Bajracharya, C.; Wang, C.; Thurber, A.; Punnoose, A.; Rodriguez, R. G.; Pak, J. J., J. Nanomaterials, 2009, 748567.
- Sun, C.; Gardner, J. S.; Long, L.; Bajracharya, C.; Thurber, A.; Punnoose, A.; Rodriguez, R. G.; Pak, J.J., Chem. Mater., 2010, 22, 2699-2701.
- Wooten, A. J.; Werder, D. J.; Williams, D. J.; Casson, J. L.; Hollingsworth, J. A., J. Am. Chem. Soc., 2009, 131, 16177-16188.
- Hollingsworth, J. A.; Hepp, A. F.; Buhro, W. E., Chem. Vap. Deposition, 1999, 5, 105-108.
- Banger, K. K.; Hollingsworth, J. A.; Harris, J. D.; Cowen, J. E.; Buhro, W. E.; Hepp, A. F., Appl. Organomet. Chem., 2002, 16, 617-627.
- Banger, K. K.; Harris, J. D.; Cowen, J. E.; Hepp, A. F., Thin Solid Films, 2002, 403-404, 390-395.
- Pal, M. K.; Dey, S.; Wadawale, A. P.; Kushwah, N.; Kumar , M.; Jain, V. K., Chem. Select, 2018, 3, 8575-8580.
- Sharma, R. K.; Kedarnath, G.; Kushwah, N.; Pal, M. K.; Wadawale, A.; Vishwanath, B.; Paul, B.; Jain, V. K., J. Organomet. Chem., 2013, 747, 113-118.
- Buchmaier, C.; Rath, T.; Pirolt, F.; Knall, A.C.; Kaschnitz, P.; Glatter, O.; Weweska, K.; Hofer, F.; Kunret, B.; Krenn, K.; Trimmer, G., RSC Adv. 2016, 6, 106120-106129.
- Tygai, A.; Kole, G. K.; Shah, A. Y.; Wadawale, A.; Srivastava, A. P.; Kumar, M.; Kedarnath, G.; Jain, V. K., J. Organomet. Chem., 2019, 887, 24-31.
- Tyagi, A.; Shah, A. Y.; Kedarnath, G.; Wadawale, A.; Singh, V.; Tyagi, D.; Betty, C. A.; Lal, C.; Jain, V. K., J. Mater. Sci. Mater. Electronics, 2018, 29, 8937-8946.
- Shah, A.Y.; Karmakar,K.; Tyagi, A; Kedarnath,G. New J. Chem., 2022, 46, 19817-19823.
- Xu, D.; Shen, S.; Zhang, Y.; Gu, H.; Wang, Q., Inorg Chem. 2013, 52, 12958-12962.
- Makin,F.; Alam, F.; Buckingham, M.A.; Lewis, D. J. Scientific Reports, 2022, 12, 5627.
- Rath, T.; MacLachlan, A.J.; Brown, M.D.; Haque, S. A., J. Mater. Chem. A 2015, 3, 24155-24162.
- Kushwah, N.; Kedarnath, G.; Sudarsan, V.; Srivastava, A. P. New J. Chem., 2023, 47, 307-314.
- Sun, C.; Gardner, J.S.; Long, G.; Bajracharya, C.; Thurber, A.; Punnoose, A.; Rodriguez, R. G.; Pak, J. J. Chem. Mater. 2010, 22, 2699–2701.
- Murtaza, G.; Alderhami, S.; Alharbi, Y. T.; Zulfiqar, U.; Hossin, M.; Alanazi, A. M.; Almanqur, L.; Onche, E. U.; Venkateswaran, S. P.; Lewis, D. J. ACS Appl. Energy Mater. 2020, 3, 1952−1961.
Figure 1.
Molecular structure of [Pd5(OAc)3(SeCH2CH2NMe2)3][OTf)]2 ; hydrogen atoms and triflate ions are omitted for clarity (Redrwan using data from Reference 25).
Figure 1.
Molecular structure of [Pd5(OAc)3(SeCH2CH2NMe2)3][OTf)]2 ; hydrogen atoms and triflate ions are omitted for clarity (Redrwan using data from Reference 25).
Scheme 1.
Scheme 2.
Scheme 3.
Scheme 4.
Scheme 5.
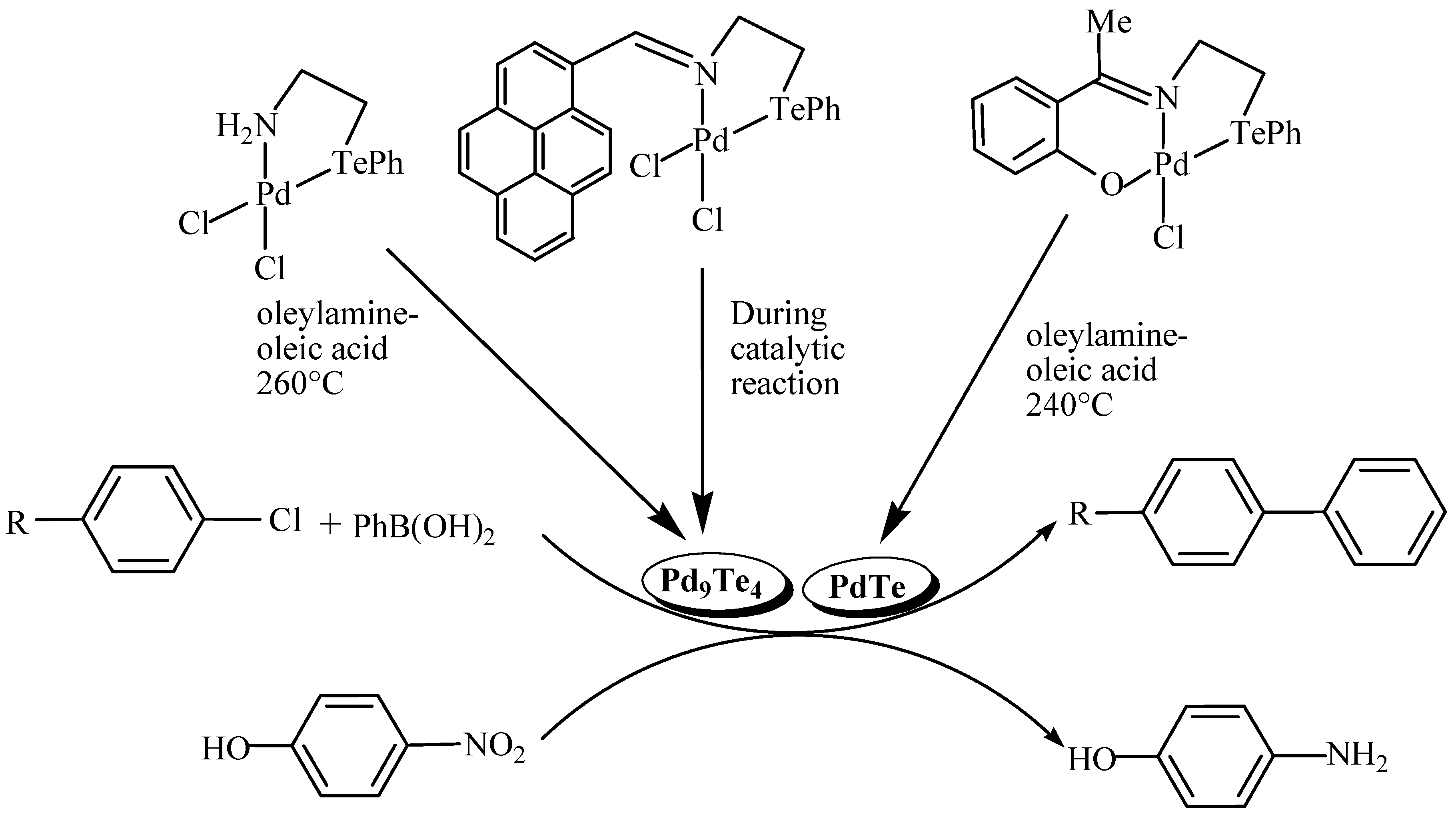
Figure 2.
Schematic diagram showing switching of photocathode to photoanode (Adapted from Reference 46).
Figure 2.
Schematic diagram showing switching of photocathode to photoanode (Adapted from Reference 46).
Scheme 6.
Scheme 7.
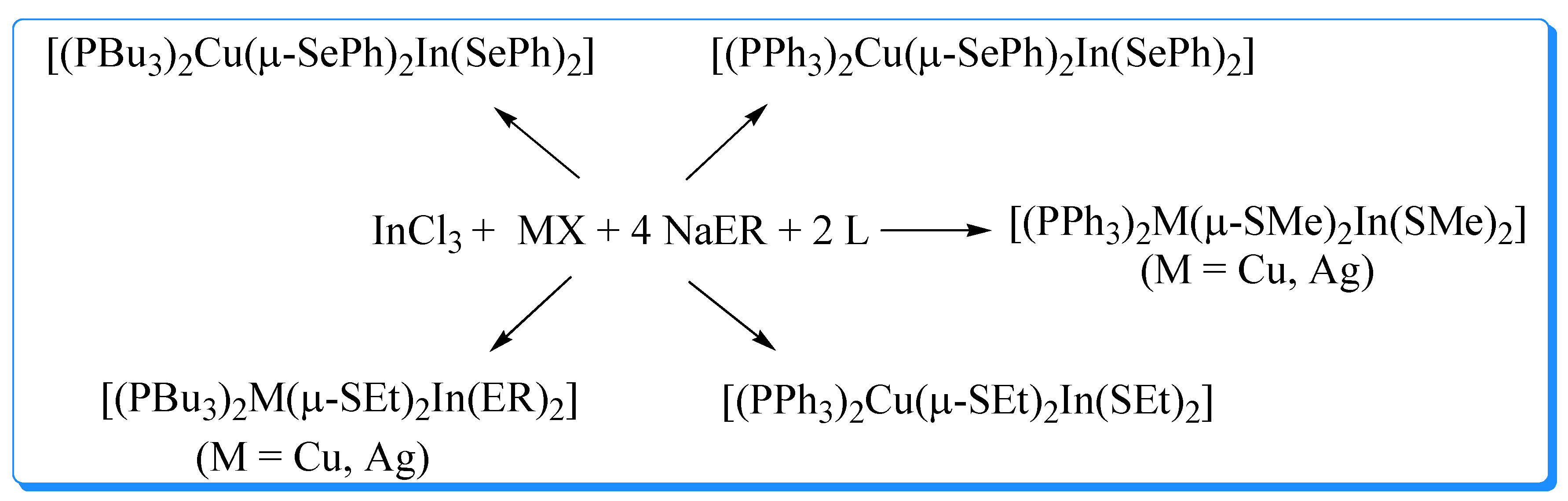
Figure 3.
Molecular structure of [(PPh3)2CuIn(SeCH2C6H5)4]. (Redrawn using data from reference 63).
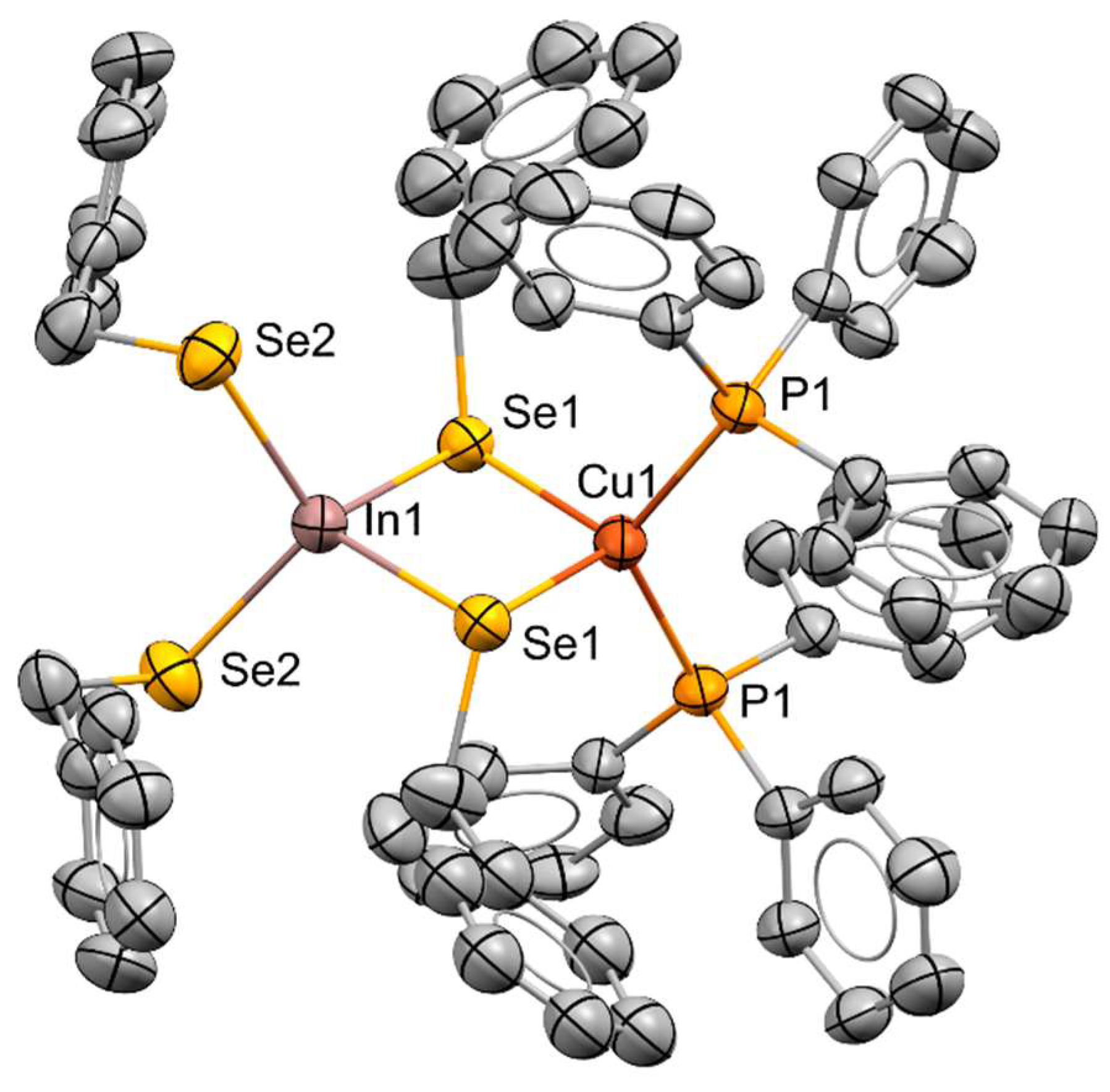
Figure 4.
Molecular structures of (a) [tBu2Sn(Spyz)2] and (b) [Cu(Spyz)(PPh3)2] (redrawn using data from reference 78).
Figure 4.
Molecular structures of (a) [tBu2Sn(Spyz)2] and (b) [Cu(Spyz)(PPh3)2] (redrawn using data from reference 78).
Figure 5.
SEM, TEM and HRTEM of a), b), c) monoclinic CTS obtained from thermolysis of a 1:1 mixture of [tBu2Sn(Spyz)2] and (b) [Cu(Spyz)(PPh3)2] in OAm (from Ref 78).
Figure 5.
SEM, TEM and HRTEM of a), b), c) monoclinic CTS obtained from thermolysis of a 1:1 mixture of [tBu2Sn(Spyz)2] and (b) [Cu(Spyz)(PPh3)2] in OAm (from Ref 78).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



