
Submitted:
29 May 2023
Posted:
30 May 2023
You are already at the latest version
Abstract
Non-alcoholic fatty liver disease (NAFLD) affects about 20–40% of the adult population in high-income countries and is now a leading indication for liver transplantation and can drive to hepatocellular carcinoma. The link between gut microbiota dysbiosis and NAFLD is now clearly established. Through analyses of the gut microbiota with shotgun metagenomics, we observed that compared to healthy controls, Adlercreutzia equolifaciens is depleted in patients with liver diseases such as NAFLD. Its abundance also decreases as the disease progresses and eventually disappears in the last stages indicating a strong association with disease severity. Moreover, we show that A. equolifaciens possesses anti-inflammatory properties, both in vitro and in vivo in a humanized mouse model of NAFLD. Therefore, our results demonstrate a link between NAFLD and the severity of liver disease and the presence of A. equolifaciens and its anti-inflammatory actions. Counterbalancing dysbiosis with this bacterium may be a promising live biotherapeutic strategy of liver diseases..
Keywords:
NAFLD
; Gut Microbiota
; Inflammation
; Metagenomics
; Live Biotherapeutic Product
1. Introduction
Non-alcoholic fatty liver disease (NAFLD), defines a spectrum of liver injury ranging from the early phases of steatosis or non-alcoholic fatty liver (NAFL), to the progressive form of non-alcoholic steatohepatitis (NASH) where hepatic inflammation can drive fibrosis progression to cirrhosis and hepatocellular carcinoma. This condition is strongly linked to unhealthy diets and sedentary lifestyle, all very prevalent in industrial societies. NAFLD which is the hepatic manifestation of the metabolic syndrome, is the leading cause of chronic liver diseases in Western countries as well as in other regions, with a prevalence as high as 30% in the general population [1] and an increasing incidence [2]. Although few preventive treatments are currently assessed, no approved pharmacological treatments to stop or limit the progression of NAFLD are currently available, so the management of the disease is mainly based on measures enforcing healthy diet and lifestyle [3].
Over the last decade, several reports linked gut microbiota modifications to NAFL or NASH [4,5,6,7]. However, results of studies on dysbiosis in NAFLD patients are divergent, and therefore a clear gut microbiota signature in NAFLD patients is still a matter of debate. A meta-analysis of 13 published studies using 16S rRNA gene sequencing reported decreased abundance of Bacteroidota and Ruminococcaceae associated with increased abundance of Veillonellaceae, Lactobacillaceae, and Dorea [8]. However, another study, in an attempt to correlate the severity of the disease with gut microbiota shift, observed increased levels of Bacteroides that have been correlated with NASH, while increased abundance of Ruminococcus was reported linked with fibrosis [9]. Similarly, using shotgun metagenomics, Loomba and colleagues characterized the gut microbiome composition of NAFLD patients and provided evidence for a gut microbiota signature specific of advanced fibrosis [10]. Moreover, our lab recently published data suggesting that nonvirulent endotoxin-producing strains of opportunistic pathogens, which are more abundant in the gut microbiota of obese patients (Enterobacter cloacae, Escherichia coli, and Klebsiella pneumoniae), may act as causative agents for induction of NAFLD [11]. These results suggest that gut microbiota dysbiosis may exacerbate hepatic steatosis and control the rate of NAFLD progression. The underlying mechanisms are complex and not fully understood but several pathways have been highlighted. Notably, it has been shown that the microbiota promotes storage of triglycerides in adipocytes through suppression of intestinal expression of a circulating lipoprotein lipase (LPL) inhibitor Angiopoietin-like 4 (ANGPTL4) [12,13]. Increased ethanol production [14,15] and induction of choline deficit by the microbiota could also promote triglyceride accumulation in the liver [16,17,18]. Among the potential mediators of this association, lipopolysaccharide (LPS) exerts relevant metabolic and proinflammatory effects [19]. Inflammation is considered a major contributor to NAFLD and is linked to increased NF-κB signaling pathway in both the intestine and liver [20].
Moreover, the farnesoid X receptor (FXR) pathway has been proposed as an important mediator of gut microbiota effect on lipid and glucose metabolism through secondary bile acids production [21]. Recent studies have also shown that bacterial metabolites of branched-chain and aromatic amino acids especially 3-(4-hydroxyphenyl) lactate and phenylacetic acid were associated with steatosis and development of fibrosis [22,23]. These and other studies from the literature clearly establish the major importance to better understand relationship between gut microbiota and NAFLD onset. These and other studies from the literature clearly establish the major importance to better understand relationships between the gut microbiota and NAFLD onset, which could also lead to new therapeutic approaches.
Fecal microbiota transfer (FMT) can be a way to demonstrate causality but also to treat patients suffering from liver diseases. As an example, FMT was reported to diminish hospital stay, improve cognition and impact on gut microbiota dysbiosis in cirrhotic patients with recurrent hepatic encephalopathy [24]. Furthermore, through manipulation of the gut microbiota in animal models, the direct role of the microbiome in the development of NAFLD has been highlighted [25,26]. Our laboratory revealed that the gut microbiota contributed to the development of steatosis induced by high-fat diet in a murine model. Moreover, by using FMT from mouse to mouse, we also showed that the severity of the steatosis depends on the microbiota composition [27]. Transferring the microbiome of NAFLD-resistant mice to wild type mice renders the latter resistant to NAFLD. Conversely, the transfer of the microbiome from mice susceptible to high-fat diet-induced NAFLD renders recipients sensitive to the diet. More recently, we confirmed that the gut microbiota contributes to steatosis development using human-to-mice FMT [28]. Thus, these data suggest that modulating the gut microbiota may be a new strategy to prevent or treat NAFLD and associated metabolic disorders.
In the present study, we show that the abundance of Adlercreutzia equolifaciens, an Actinobacteriota (recently renamed Actinomycetota) from the Eggerthellaceae family, is linked to a healthy status of the human microbiome and is reduced in patients with NAFLD. This depletion is also strongly linked to the severity of the liver disease, its abundance being very low in cirrhosis with almost disappearance in decompensated patients and in acute on chronic liver failure (ACLF). We also show that this bacterium possesses anti-inflammatory properties, both in vitro and in vivo in a humanized mouse model of NAFLD. Based on these results, we propose that counterbalancing dysbiosis using A. equolifaciens as a live biotherapeutic product may be a promising strategy for the prevention and/or treatment of liver diseases..
2. Results
2.1. Decreased Abundance of A. equolifaciens in Microbiome of Liver Disease Patients NASH1 & NASH2
Shotgun metagenomic was performed on fecal DNA of all patients in NASH1 cohort, our discovery study. A non-significant reduction in global microbial diversity was observed with increasing histological severity. However, twenty-one MGS discriminating NASH (steatohepatitis with or without fibrosis/cirrhosis) from normal/fatty liver (simple steatosis) population were identified with a combined approach of Wilcoxon Rank Sum test and Kolmogorov-Smirnov tests.
In the subsequent study NASH2 conducted in 2012, three out of the 21 MGS previously identified in NASH1 study were validated and yielding significant results (Wilcoxon Rank Sum tests). We were able to establish a phylogenetic association between two of these MGS and known bacterial species. The first MGS was Escherichia coli, while the second one, which is the focus of the current study, was found to be A. equolifaciens with 71.2 % of the genes in MGS 9828_3 being annotated to Adlercreutzia equolifaciens. This particular species belongs to the Actinobacteriota phylum, now renamed Actinomycetota, and was initially isolated from fecal samples in Japan [29]. A. equolifaciens was detected in most of the healthy subjects, with its relative abundance decreasing as the severity of the disease increased (Figure 1).
Then, we downloaded publicly available shotgun metagenomic data from liver disease patients and we performed taxonomic profiling to detect and estimate the abundance of A. equolifaciens. First, we considered the study from Loomba and colleagues comparing the gut microbiome of 85 patients with biopsy-proven NAFLD (one sample with too few reads was filtered out) among which 71 had mild to moderate disease (fibrosis stage between 0 and 2), and 14 presented advanced fibrosis (stage 3 or 4) [10]. Remarkably, the prevalence and abundance of A. equolifaciens strongly decreased with the level of fibrosis. Unfortunately, the data published with this study did not include any healthy controls.
Mining a Chinese cohort of 112 liver cirrhosis patients and 102 healthy controls [30], we observed similar features, but with a more pronounced loss of A. equolifaciens in cirrhotic patients (Figure 3). The prevalence of this MGS was 57 % in healthy controls, whereas it was only found in 16% of liver cirrhosis patients.
Finally, we looked at fecal microbiome data from 166 Spanish patients with liver cirrhosis [31] and compared them to 38 Spanish healthy controls of the MetaHIT project. Again, A. equolifaciens was strongly associated with the health status of donors and was significantly depleted in cirrhotic patients. The depletion of the bacterium was all the more pronounced as the disease progressed and reached higher stages of severity.
2.2. A. equolifaciens Encompasses Two Distinct Genomospecies with Strong Associations with Geography
Exploring several public databases, we retrieved 154 high quality Metagenome-Assembled Genomes (MAGs) of A. equolifaciens and 11 genomes derived from isolates. All genomes came from fecal material, essentially from humans but also from mice and rat (respectively 154, 10 and 1). We computed Average Nucleotide Identity (ANI) between all pairs of genomes and we performed an average-linkage hierarchical clustering (UPGMA). Finally, we extracted clusters with a 95% ANI cut-off widely used to delineate microbial species [32].
Interestingly, genomes were clustered into two phylogroups, defining two distinct genomospecies (Figure 5). The first phylogroup consisted, among others, of the A. equolifaciens type strain (DSM 19450) as well as the type strain of the subspecies celata (DSM 18785). The second phylogroup was made up of the type strains of the two recently validated species, Adercreutzia rubneri [33] and Adercreutzia hattorii [34]. Notably, genomes of these two type strains were closely related (ANI = 97.2%) suggesting that Adercreutzia rubneri and Adercreutzia hattorii actually represent the same species.
Remarkably, we found a strong association between phylogroups and geographical origin of the host (chi-squared test p < 2.2 × 10-16, Figure 6). Indeed, 82% of the genomes assigned to the first phylogroup were obtained from fecal material of Asian individuals. In contrast, the second phylogroup mostly represents genomes recovered from feces of European and North American individuals (92% of genomes).
Finally, we noticed that genes responsible for equol synthesis were frequently detected in genomes assigned to the first phylogroup but were rare in the second (60% vs 8.8% of genomes respectively, chi-squared test p < 3.7 × 10-12, Supplementary Figure 3). In others words, this result highlights that equol-producing strains are much more prevalent in Asian individuals. Interestingly, in the second phylogroup, strains predicted as equol-producers were found almost exclusively in mouse feces (9/10). Thus, the equol pathway could be vanishing in strains of the second phylogroup, but only in human and not in mice.
2.3. A. equolifaciens Displays Anti-Inflammatory Properties In Vitro
We then used the strain DSM 19450 to look for its impact on the NF-B inflammatory pathway using stably transfected reporter cell lines. As illustrated on Figure 7a, HT-29 NF-κB cells used in the test responded strongly to TNF-α by a >11-fold activation of the NF-κB pro-inflammatory pathway. M104 (bacterial culture medium) in the absence of TNF-α was used as negative control. When incubated with TNF-α in the presence of the supernatant of A. equolifaciens, the fold activation was reduced to 8 indicating a mild but significative inhibition. In contrast, the supernatant of Parvibacter caecicola, a closely related member of the Eggerthellaceae family, had no effect on NF-κB activation following TNF-α stimulation. The culture supernatant of both bacterial strains did not affect HepG2 NF-κB activity stimulated with TNF-α. Supplementary figure 1 shows that the culture medium and the supernatant of both bacterial strains, A. equolifaciens and P. caecicola had no effect on cell viability as assessed by measuring MTS activity.
Figure 7b shows that HT-29 NF-κB cells used in the test responded strongly to TNF-α by a >15-fold activation of the NF-κB pro-inflammatory pathway. PBS used to resuspend bacterial pellet in the absence of TNF-α was used as negative control. When incubated with TNF-α in the presence of the pellet of A. equolifaciens, the fold activation was reduced to 2 indicating a very strong inhibition. In contrast, the pellet of P. caecicola exerted a mild and non-statistically significant effect on NF-κB activation following TNF-α stimulation. Again, cell viability was not affected by both strains as assessed by measuring MTS activity (supplementary Figure S1).
On Figure 7c, it is shown that HepG2 NF-κB cells responded strongly to TNF-α by a 5-fold activation of the NF-κB pro-inflammatory pathway. PBS used to resuspend bacterial pellet in the absence of TNF-α was used as negative control. When incubated with TNF-α in the presence of the pellet of A. equolifaciens, the fold activation was strongly reduced to a level closed to non-activated cells. In contrast, the pellet of P. caecicola had no effect on NF-κB activation following TNF-α stimulation. Cell viability was not affected by the pellet of A. equolifaciens and P. caecicola.
2.4. A. equolifaciens Displays Anti-Inflammatory Effect In Vivo and Impacts Mice Metabolism
The effect of DSM 19450 A. equolifaciens strain given by oral gavage and used as a biotherapeutic agent was evaluated in a mouse model of hepatic steatosis, i.e. C57Bl/6J SPF male mice treated with antibiotics and then inoculated with the gut microbiota of patients with NASH and further exposed to a high fat high fructose diet. Mice were force-fed daily with A. equolifaciens resuspended in PBS or only PBS as control. Food consumptions during the 10 weeks of follow up were similar in the two groups (not shown). Interestingly, compared to controls, mice that were gavaged with A. equolifaciens had lower weight gain (Figure 8 left and middle) which persisted during the 10 weeks of the experiment. We also observed that mice that received A. equolifaciens displayed a reduced hyperglycemia at 1 month as compared to control mice (figure 8 right). However, when an OGTT was performed on day 70, after one and a half month of induction and gavage, there was no longer any difference in fasting blood glucose or glucose tolerance between the two groups (not shown). A. equolifaciens did not influence liver steatosis induced by the high fat high sugar diet. Plasma cholesterol or ferritin were not modified by A. equolifaciens.
Upon sacrifice, fecal content was collected and weighted. The gavage with A. equolifaciens resulted in a reduction of the full caecum weight, associated with an increase but statistically non-significant increase in total SCFA. Moreover, an important increase in the amount of butyrate was observed and a reduction in the proportion of the two branched-chain fatty acids, iso-butyrate and iso-valerate (Figure 9).
Finally, both caecum wall and liver were collected to measure the level of expression of interleukin-6, a pro-inflammatory cytokine by RT-qPCR. In both tissues, the gavage with A. equolifaciens reduced the expression of IL-6 although the effect was stronger on the caecum than on the liver (Figure 10).
3. Discussion
3.1. Decreased Abundance of A. equolifaciens in the Microbiome of NAFLS/NASH Patients
Based on decreased abundance in patients with liver diseases, we can hypothesize that this bacterium is playing a key protective role in the healthy state, as shown for Faecalibacterium prauznitzii in patient with inflammatory bowel diseases or for Akkermancia muciniphila in patients with metabolic diseases such as obesity and diabetes. Although the latter are dominant bacteria in human microbiome, other such as Christensenella minuta or A. equolifaciens, are sub-dominant but essential micro-organisms for the gut ecosystem [35,36,37]. Interestingly, although we focused our work on liver diseases, A. equolifaciens is also depleted in other pathologies including ulcerative colitis [38,39,40].
3.2. A. equolifaciens Presence in Human Gut
A the time of writing, A. equolifaciens comprises two species, the original one represented by the DSM 19450 strain, and a new one named A. rubneri , defined by the strain ResAG-91 (DSM 111416) also called A. hattorii defined by the stain 8CFCBH1T (DSM 112284) [33,34]. In addition, the original species was divided in two subspecies [41], A. equolifaciens subsp. equolifaciens and A. equolifaciens subsp. celata represented respectively by the strains DSM19450 and DSM18785 [29,42]. Yet, our genomic analysis revealed that genomes of these strains are closely related (ANI = 97.2%) and the existence of such subspecies is not supported after considering dozens of other genomes. Interestingly, although A. equolifaciens is named due to its capacity to transform isoflavonol into equol, metabolic potential predictions based on the presence of genes coding for key enzymes showed that the equol production pathway is absent in genomes of many strains. In fact, most strains of the Asian phylogroup are equol producers while this metabolic capacity is rare in the phylogroup associated with North Americans and Europeans (Figure 5 and supplementary Figure S3). It is noteworthy that most bacteria from the Coriobacteriia class have metabolic capacities regarding polyphenols, independently of equol production, thus this functionality may be of major interest for therapeutic purposes. However, considering the link between A. equolifaciens and NAFLD, we may consider that the equol-producing function is probably not essential as we found that this bacterium was not only enriched in Asian healthy controls, but in Europeans as well. Moreover, in our mouse experiments, no isoflavonol is added in the diet, thus equol could not be the essential anti-inflammatory metabolite. In contrast, in multiple sclerosis, the protective effect provided by the association of isoflavonol and A. equolifaciens was due to equol production [43].
3.3. A. equolifaciens Presents Anti-Inflammatory Properties
Our in vitro data show that A. equolifaciens pellet and, to a lower extent its supernatant, possesses interesting anti-inflammatory properties through the inhibition of the NF-κB pathway, both in human intestinal epithelial cells and in hepatocytes. These properties are not present for the closely related species, P. caecicola used as a negative control. Here, we showed the results obtained with the strain DSM 19450, however we also tested about 12 other strains of A. equolifaciens belonging to the 2 different phylotypes, and found that all strains have the same properties (data not shown). Similar in vitro anti-inflammatory properties have been observed with F. prauznitzii on the same reporter cell line [35]. However, it was mostly caused by secreted compounds, including butyrate [44]. For A. equolifaciens, the effect of the supernatant is not due to butyrate (or propionate) production since they are not produced by this microbial strain.
However, we cannot rule out the role of SCFA in the in vivo model, since we observed an important increase of butyrate and a decrease of the branched SCFA production, iso-butyrate and isovalerate. Interestingly, an increased production of iso-butyrate and isovalerate has been found in the caecum of mice prone to NAFLD development compared to mice resistant to NAFLD [27]
Considering the in vivo mouse model, we observed interesting effect on weight gain, and glycemia at 1 month. Our PCR analysis of the fecal microbiota of our mice force-fed with A. equolifaciens showed different abundance from animal to animal at day 55 (suppl figure S2), but almost complete absence at the time of sacrifice. Remarkably, our data showed lower glycemia in these mice at month 1 and no significant difference at sacrifice. The absence of detectable bacteria by PCR at sacrifice may explain why no major effect was observed at that period, besides the reduction in IL-6 cytokine expression.
Considering the in vivo mouse model, we observed a clear effect on weight gain and glycemia at 1 month but not at the end of the experiment. Interestingly, PCR analysis of the fecal microbiota of mice force-fed with A. equolifaciens performed at day 55 showed variable abundance of the added between different animals (suppl figure S2), while at the time of sacrifice there was almost complete absence of A. equolifaciens. This mirrored the return to normal of weight and serum glucose at the time of sacrifice. The disappearance of this bacterial strain may explain why the effect was not sustained at this later time point.
Thus, as a conclusion, A. equolifaciens is a particularly interesting commensal bacterium, especially in view of its anti-inflammatory effects on intestinal and hepatic cells, and its action on the metabolism. Even if further studies are necessary, it could represent, alone or in combination, a Live Biotherapeutic Product candidate for the treatment of steatotic diseases whose epidemiology is worrying and therapeutic options are scarce. This also warrants further investigations in other diseases including IBD.
4. Materials and Methods
4.1. Metagenome-Wide Association Study
4.1.1. Study Population NASH1 Cohort
In a discovery study, 96 patients (56 % men; mean BMI 29.6 kg/m²) with suspected NAFLD were recruited from November 2011 to January 2012 in the department of Hepatology of the Hospital Pitié-Salpêtrière. Patients younger than 70 years old, with a liver biopsy performed within the last 3 years or having a scheduled liver biopsy during the period study, with a consent form signed were included. Patients with incomplete or non-exploitable anamnestic and/or histological data, with bariatric surgery or recent colonoscopy, with antibiotic or probiotic intake or daily consumption of alcohol were excluded. Patients were stratified in three groups of increasing histological severity: normal/fatty liver alone (FL, n=29), steatohepatitis with no/minimal fibrosis (n = 34) and steatohepatitis with bridging fibrosis/cirrhosis (n = 33) based on a liver biopsy performed prior (median 33 months) to the stool collection.
4.1.2. Study Study Population NASH2 Cohort
In a validation study, 260 patients (40.4 % of men; mean BMI 34.9 kg/m2) with suspected NAFLD were recruited from December 2012 to July 2013. Patients younger than 70 years old, with a liver biopsy performed within the last year or having a scheduled liver biopsy during the period study, with a consent form signed were included. Patients with incomplete or non-exploitable anamnestic and/or histological data, with bariatric surgery or recent colonoscopy, other causes of liver disease such as viral, auto-immune, genetic, drug-induced steatohepatitis (methotrexate, tamoxifen, corticotherapy, amiodarone and diltiazem), with situation at risk of change in intestinal microbiota, chronic enteropathy with immunogical or infectious components, treatment with antibiotic that has been discontinued less than 8 weeks prior to samples collection, daily probiotics intake in the last 3 weeks prior to samples collection, long term treatment with immunosuppressant drugs, colonoscopy within the last 3 months were excluded. Patients were stratified in four groups of increasing histological severity: normal liver (< 5% of steatosis, n = 25), simple steatosis / fatty liver (> 5% of steatosis, n = 52), steatohepatitis with no/minimal fibrosis (n = 134) and steatohepatitis with bridging fibrosis/cirrhosis (n = 49) based on a liver biopsy performed prior to the stool collection.
4.1.3. Reference Healthy Volunteer Populations Spanish MetaHit
As a reference healthy population, 91 metagenomics samples were used from the MetaHit cohort [45] (NCBI = PRJEB1220). The healthy volunteers were adults and from Spain.
4.1.4. Analysis of MGS in NASH1, NASH2 and Healthy Cohort (MetaHit)
Patient samples have been procured, collected, stored and disseminated in accordance with the highest ethical standards and in the strictest compliance with all applicable rules and regulations. This includes ensuring that consents are fully informative to donors and that the donor's wishes in relation to the use of his/her samples are strictly complied with. Feces microbiota analysis was performed according to previously used methodology [46,47]. DNA was extracted from the stool samples, sequenced using SOLiD™ technology, and mapped to the MetaHIT reference catalog. Quantitative metagenomics analysis consisted in microbial gene count and identification of 900 MetaGenomic Species (MGS = clusters of > 500 genes that co-vary at the same level of abundance from one sample to another) [45].
4.1.5. Data Availability
Shotgun metagenomic data of public cohorts were downloaded from the European Nucleotide Archive. Full list of samples used in this study is available in Supplementary Table S1.
4.1.6. Sequencing Data Preprocessing
Sequencing data quality control was performed with fastp [Chen et al., 2018] (parameters: --cut_front --cut_tail --n_base_limit 0 --length_required 60) to remove low quality reads and sequencing adapters. Then, reads mapped to the human genome (T2T CHM13v2.0 GCA_009914755.4) with bowtie2 [49] were removed. Finally, read subsampling was performed with fastq-sample (github.com/fplaza/fastq-sample) to take into account variable sequencing depth across samples. The number of reads subsampled in each sample is indicated in Supplementary Table 1.
4.1.7. Gene Coverage Table Generation
The gene abundance table was generated with the METEOR software suite (forgemia.inra.fr/metagenopolis/meteor). First, selected high quality reads from each sample were mapped with bowtie2 [49] against the updated Integrated Gene Catalogue of the human gut microbiome [50] (IGC2, 10.4 million genes). Alignments with nucleotide identity ≥ 95 % were kept to account for intra-species nucleotidic variability and the non-redundant nature of the catalogue. Then, raw gene counts were computed with a two-step procedure previously described that handles multi-mapped reads [51]. Finally, gene coverage was computed by dividing raw gene counts by gene length multiplied by average read length in sample.
4.1.8. Species-Level Taxonomic Profiling
Using MSPminer [52], the IGC2 catalogue was previously organized into 1990 MetaGenomic Species (MGS) which were clusters of co-abundant genes corresponding to the same microbial species [53]. The MGS named msp_0396 was identified as representative of A. equolifaciens. The abundance of this MGS in a sample was defined as the mean coverage of its 100 marker genes (i.e. species-specific core genes that correlate the most altogether). If less than 10% of the marker genes were seen in a sample, the abundance of the MGS was considered as null. The estimated sequencing coverage of A. equolifaciens (msp_0396) across samples is reported in Supplementary Table 1.
4.1.9. Statistical Analysis
Statistical analysis was performed using R software suite. MGS differentially abundant between controls and patients were identified using Mann–Whitney U tests.
4.2. Population Genomics
4.2.1. Data Availability
154 high quality Metagenome-Assembled Genomes (MAGs) (completeness ≥ 80% and contamination ≤ 5% assessed with CheckM2 [54]) of A. equolifaciens were downloaded from various sources including: 1) the Unified Human Gastrointestinal Genome collection (UHGG) [55], 2) the Human Reference Gut Microbiome (HRGM) [56] and 3) the Murine Intestinal Microbiota Integrated Catalog v2 (MIMIC2) [57]. In addition, 11 genome sequences derived from cultured isolates were downloaded from the NCBI GenBank (September 2022). Full list of genomes used in this study is available in Supplementary Table 2.
4.2.2. Bioinformatics and Biostatistics
Average Nucleotide Identity (ANI) between all pairs of genomes was computed with fastANI [58]. A 165x165 distance matrix was built where distance was defined as ‘1-ANI’. Then, this matrix was submitted to a Non-Metric Multidimensional Scaling process (NMDS) implemented vegan R package [59]. The NMDS output was used to map the genomes in a two-dimensional plot. Finally, genomes were clustered into phylogroups using average-linkage hierarchical clustering (UPGMA) using a 95% ANI cut-off.
Sequences of proteins involved in the equol production pathway were downloaded from the NCBI: daidzein reductase (dzr) = WP_022741749.1, dihydrodaidzein reductase (ddr) = WP_022741751.1, tetrahydrodaidzein reductase (tdr) WP_022741752.1 and WP_022741755.1 (racemase). These four proteins were searched in the genomes with tblastn [60] (amino-acid identity ≥ 90%, query coverage ≥ 90%). A strain was considered as an equol producer if at least 75% of the proteins were detected in its genome.
4.3. In vitro Experiments
4.3.1. Cell Culture and Reagents
HepG2 human hepatocarcinoma cells and HT-29 human intestinal epithelial cells were grown in DMEM (Sigma) with 2mM L-glutamine, 50 IU/mL penicillin, 50 µg/mL streptomycin and 10% heat-inactivated fetal calf serum (FCS - Lonza) in a humidified 5% CO2 atmosphere at 37°C. Absence of mycoplasma contamination was controlled using the MycoAlert kit (Lonza).
Construction and validation of the NF-κB reporter clones HepG2/κb-seap was done following the same protocol as the one used to generate HT-29 NF-κB-seap reporter cell lines and described previously [61].
4.3.2. Commensal strains and preparation of conditioned media
A. equolifaciens (DSM 19450) and Parvibacter caecicola (DSM 22242) bacteria were grown in anaerobic M104 medium completed with 0,5 % arginine at 37°C following the Hungate culture method [62]. At the end of the incubation period, bacterial cultures were centrifuged at 5000xg for 10 minutes. Bacteria conditioned media (supernatant) were then collected and filtered on 0.2µm PES filters. Non-inoculated bacteria culture medium served as control. Cell pellet were resuspended in PBS and tested in parallel.
4.3.3. Analyses of NF-κB Activation
For each experiment, reporter cells were seeded at 40 000 cells per well, into 96-wells plates and incubated 24 hours. Then cells were stimulated for 24 hours with 10 µL of tested bacteria supernatant or resuspended bacterial pellet, for a final volume of 100µl per well (i.e 10% vol/vol), in the presence or absence of TNF-α (10ng/ml final). Secreted Embryonic Alkaline Phosphatase (SEAP) in the supernatant was revealed using the Quanti-BlueTM reagent (Invivogen) according to the manufacturer’s protocol and quantified at 655nm OD. All measurements were performed using a microplate reader (Infinite 200, Tecan). Cell viability was controlled using the MTS assay (CellTiter 96 Aquous One, Promega) following manufacturer’s instruction.
4.3.3. Statistical Analysis
Results are expressed as mean ± SD. Data were analyzed using Student’s t test.
4.4. Animal Experiments
4.4.1. Clinical Cohort
A cohort of 20 NAFLD patients with moderate obesity, aged 62 on average was recruited; a liver biopsy determined a diagnosis of NAFL or NASH. All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the French CPP Ethics Committee. A donor with NASH was selected among the 20 donors.
4.4.2. Preparation and Preservation of Fecal Transplants
Stool were collected and stored at -80°c in a preservation solution in MD diluent kindly provided by MaaT Pharma (Lyon, France). Fecal transplant was prepared as previously described [63].
4.4.3. Animal Experimentation
Procedures were performed according to the European Guidelines for the Care and Use of Laboratory Animals and approved by the French Veterinary Authorities (Authorization number 78-60). The experimental protocol was agreed by the French Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche (APAFIS#18425-2018110521086756 v2). Twenty-four specific pathogen free (SPF) C57Bl/6J male mice, 7 weeks old, were purchased from Charles River Laboratories, France, from different litters. On arrival, animals were housed under controlled conditions of temperature, hygrometry and 12h light/dark cycle in a SPF animal facility (Infectiologie Expérimentale des Rongeurs et des Poissons, IERP) at INRAE, Jouy en Josas. They were individually weighted, microchipped and randomly housed in new cages, 4 mice per cage; they received a conventional gamma-irradiated 45 kGy mice control diet (CD) SAFEA03 R03-40 ad libitum (3.24 kcal/g: 14 % energy from fat, 25 % energy from proteins, 61 % energy from carbohydrates, (SAFE, Augy, France, CD).
On day 6 after arrival, feces of the mice were individually harvested (basal microbiota). Then, they received in the autoclaved drinking water for 2 weeks a mixture of broad spectrum antibiotics (vancomycin 45 µL/mL, streptomycin 1 mg/mL, colistin 1 mg/mL and ampicillin 1 mg/mL) ad libitum, to clean their endogenous microbiota as previously described [64]. On day 16, an all-bacteria-qPCR was performed to confirm complete absence of detectable bacteria in the feces (threshold of 99.99 % depletion was considered to be equivalent to germ-free mice). The stools of the selected human NASH patient were inoculated (200 µL per mouse) by two gavages at 48h intervals (day 21 and 22). Fecal transplants contained more than 70 % of viable bacteria as shown by flow cytometry. Mice were fed ad libitum for 10 weeks a high-fat, high-fructose diet (2HFD), to induce NAFLD following the protocol previously described [28]. A first group of 12 mice was gavaged five times a week from day 27 until euthanasia, i.e. 50 days, with sterile PBS. A second group of 12 mice was gavaged with a resuspension of the dry pellet of A. equolifaciens diluted in PBS. Body weight, food and liquid consumption were monitored weekly. Mice stools were collected individually at five time points: on day 6 (basal), 16 (after 2 weeks antibiotics treatment), 27 (1 week after fecal microbiota transplant), 55 (1 month after fecal microbiota transplant), 84 (1 week before the end of the experiment). On day 90, mice were euthanatized, and liver, epididymal and mesenteric adipose tissue, caecum and blood were then harvested.
4.4.4. Preparation of A. equolifaciens for Gavage
After rehydratation of the lyophilized vial with 500 μL of JCM 663 rehydration medium, 100 μL was spread in a Petri dish on Wilkins Chalgren anaerobic agarose medium containing 0.5% arginine and incubated for 3 days at 37°C in a Freter chamber. Two pre-cultures were performed in order to obtain 64 Petri dishes. For each dish, bacteria were resuspended in sterile PBS and aliquoted in 2mL tubes at an optical density of one.
4.4.5. 16 S rRNA Sequencing Analysis
DNA was extracted from feces or caecum content using Gnome DNA Isolation Kit (MP Biomedicals, Santa Ana, CA, USA). Quantifications of the total bacteria DNA were performed by real-time qPCR following the procedure previously described [65]. The V3-V4 region of the 16S rRNA genes was amplified using MolTaq (Molzym, Bremen, Germany) with the primers described previously [66]. Purified amplicons were sequenced using the MiSeq sequencing technology (Illumina, San Diego, CA, USA) at the GeT-PLaGe platform (Genotoul, Toulouse, France). Paired-end reads obtained from MiSeq sequencing were analyzed using the Galaxy supported pipeline named find, rapidly, operational taxonomic units (OTU) with Galaxy Solution (FROGS) [67]. For the preprocessing, reads with length between 380 bp and 500 bp were retained. The clustering and chimera removal tools, followed the guidelines of FROGS. Assignment was performed using the Silva 132 database updated in December 2017 with top quality pintail 100 (https://www.arb-silva.de/). OTU with abundances lower than 0.005 % were removed from the analysis [68]. 16S sequencing data were analyzed using the Phyloseq, DESeq2 and ggplot2 R packages in addition to custom scripts as described previously [69].
4.4.6. Short Chain Fatty Acids Quantification of Cecal Contents
Measurement of the short-chain fatty acids (SCFA) and branched-chain fatty acids from mice cecal contents was performed using gas chromatography as previously described [70]. The analyses were performed on an Agilent (Les Ulis, France) gas-chromatograph equipped with a split-splitless injector 7850 and ionization flame detector. Carrier gas (H2) flow rate was 10mL/min and inlet, column and detector temperatures were 200, 100 and 240°C, respectively. Data were collected and peaks integrated using OpenLabChem station software (Les Ulis, France).
4.4.7. Plasma Assays
On day 90, blood was collected from mice before euthanasia by submandibular puncture into tubes containing 5 µL EDTA 0.5 mol/L. After centrifugation (6,000 x g, 20 min, 4 °C) plasma was aliquoted and frozen at -80 °C until further analysis. Measurements of plasma AST, ALT, triglycerides (TG), high-density lipoprotein cholesterol (HDL), total cholesterol, and ferritin were performed by the biochemistry platform (CRI, UMR 1149, Paris) with an Olympus AU400 Chemistry Analyzer. LDL (low-density lipoprotein) cholesterol was calculated according to the Friedewald formula: LDL-Cholesterol = Total Cholesterol – HDL-Cholesterol – TG/2.2 (mmol/L), with all TG < 4.6 mmol/L.
Measurements of non-fasting plasma insulin and leptin were performed using mouse-specific insulin and leptin ELISA Kit (Merck Millipore).
4.4.8. Real-Time Quantitative Polymerase Chain Reaction (qPCR)
A 1/4 portion of the left liver lobe was stored in RNAlaterTM stabilization solution (Invitrogen, Carlsbad, CA, USA) at -80 °C until further analysis. Total RNA was extracted from the liver with RNAeasy Plus Mini Kit (Qiagen, Hilden, Germany). RNA integrity and concentration were checked with RNA 6000 Nano chips on an Agilent 2100 bioanalyser (Agilent Technologies, Amsterdam, Netherlands). Total RNA (10 µg per reaction) was reverse transcribed into complementary DNA using high-capacity cDNA reverse transcription kit (Applied Biosystems, Thermofisher Scientific, Foster City, California, USA) according to the manufacturer’s instructions. Real time qPCR was performed on an Applied Biosystems Step One Plus machine. Primers references of genes used for qPCR are listed in Supp. Table. 3. The relative gene expressions were normalized to two housekeeping genes: gapdh and actb (glyceraldehyde-3-phosphate dehydrogenase, actin beta) chosen on the basis of results obtained from TaqMan mouse endogenous control arrays (Applied Biosystems).
4.4.9. Oral Glucose Tolerance Test (OGTT)
Fasting glycemia and insulinemia measurements as well as OGTT were performed on day 70, i.e. 7 weeks after the 2HFD regimen, and 21 days before euthanasia. After 6 h of fasting, a glucose solution (2 g glucose/kg body weight) was administered by oral gavage. Blood glucose levels at time 0 (fasting glycemia, determined before glucose gavage) and 15, 30, 60, 90 and 120 min after glucose gavage were analyzed using an Accu-Check glucometer (Roche, Meylan, France). The glucose levels were plotted against time, and the AUC (area under curve) was calculated. The plasma insulin concentrations at times 0 (fasting insulinemia) and after 30 min, were analyzed in venous blood (collected in EDTA-coated tubes), harvested from marginal tail vein, using a mouse-specific Insulin ELISA Kit (Merck Millipore, St Quentin en Yvelines, France). Insulin resistance was estimated by homeostasis model assessment (HOMA-IR index) and calculated according to the following formulas: HOMA-IR (fasting) = fasting glucose (mmol/L) x fasting insulin (mU/L)/22.5. HOMA-IR (non-fasting) = non fasting glucose (mmol/L) x non fasting insulin (mU/L)/22.5.
4.4.10. Statistical Analysis
Datasets normality was tested using the D’Agostino & Pearson normality test. Normally distributed data with equal group variances were expressed as mean ± standard errors of the mean (SEM). Non-normally distributed data, or belonging to unequal group variances, were expressed as medians (interquartile ranges). The level of significance was set at p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001). Calculations were performed with R 3.5 software and GraphPad Prism software (version 7.00, La Jolla, CA, USA).
5. Patents
Part of this work is patented under the application number PCT/EP2021/073417.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: (a). Effect of the supernatant of A. equolifaciens and P. caecicola on NF-κB activity in HepG2 reporter cell line. (b-c). Effect of supernatant of both bacterial strains on HT-29 (b) and HepG2 (c) cell viability as assessed by measuring MTS activity. (d-e). Effect of bacterial pellet on HT-29 (d) and HepG2 (e) cell viability as assessed by MTS viability test; Figure S2: Presence and abundance of A. equolifaciens in the feces of mice at different time point after the beginning of the experiment and at sacrifice Figure S3: Non-Metric Multidimensional Scaling (nMDS) plot representing genomes similarity assessed from pairwise ANI. Table S1: Data used for fig 2, 3 and 4; Table S2: Full list of genomes used in this study.
Author Contributions
F.O.P, P.G., L.C., J.D. and H.M.B. conceived and designed the experiments; V.R., J.M.P., J.B were responsible of the clinical part; C.C, S.B.D, M.M, C.P, M.B, and C.P. performed the experiments; F.O.P, C.C, S.D.B., N.L., L.C, W.F., P.G, H.M.B. analyzed the data; F.O.P, C.C, P.G and H.M.B wrote the manuscript. All authors have read, amended and agreed to the published version of the manuscript. .
Funding
This research was funded by the grant “FunAMetaGen” from the French National Research Agency ANR-15-CE14-0021 and by the grant MetaGenoPolis ANR-11-DPBS-0001.
Institutional Review Board Statement
Procedures were performed according to the European Guidelines for the Care and Use of Laboratory Animals and approved by the French Veterinary Authorities (Authorization number 78-60). The experimental protocol was agreed by the French Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche (APAFIS#18425-2018110521086756 v2). The study was conducted in accordance with the Declaration of Helsinki, and the protocol 2016-A01074-47 was approved in January 2012 by the French Ethics Committee of CPP (Comité de Protection des Personnes)-Ile de France VI.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author unless specified in the Material & Methods section.
Acknowledgments
We acknowledge the staff of the animal facility (IERP-UE907, INRAE, Jouy-en-Josas, France) in which animal experiments were performed. The IERP Facility belongs to the National Distributed Research Infrastructure for the Control of Animal and Zoonotic Emerging Infectious Diseases through In Vivo Investigation. (EMERG’IN; doi:10.15454/1.5572352821559333E12) The authors are thankful to Fabienne Béguet-Crespel for expert anaerobic bacterial culture..
Conflicts of Interest
C.C., M.B., and P.Y.M are employees of NovoBiome. L.C., W.F., and J.M.P. are employees of the Enterome. P.Y.M., J.D and H.M.B. are co-founders of Novobiome. The other authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Younossi ZM, Golabi P, Paik JM, Henry A, Van Dongen C, Henry L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology. 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Le MH, Le DM, Baez TC, Wu Y, Ito T, Lee EY, Lee K, Stave CD, Henry L, Barnett SD, et al. Global incidence of non-alcoholic fatty liver disease: a systematic review and meta-analysis of 63 studies and 1,201,807 persons. J Hepatol 2023, S0168-8278(23)00219-2. [CrossRef]
- Ratziu V, Francque S, Sanyal A. Breakthroughs in therapies for NASH and remaining challenges. J Hepatol. 2023, 76, 1263–12782022. [Google Scholar] [CrossRef]
- Schnabl B, Brenner DA. Interactions Between the Intestinal Microbiome and Liver Diseases. Gastroenterology, 2014, 46, 1513–24. [Google Scholar] [CrossRef]
- Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to Cirrhosis, new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–36. [Google Scholar] [CrossRef]
- Moschen AR, Kaser S, Tilg H. Non-alcoholic steatohepatitis : a microbiota-driven disease. Trends Endocrinol. Metab., 2013, 24, 537–45. [Google Scholar] [CrossRef]
- Safari Z, Gérard P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD). Cell Mol Life Sci., 2019, 76, 1541–58. [Google Scholar] [CrossRef] [PubMed]
- Demir M, Lang S, Martin A, Farowski F, Wisplinghoff H, Vehreschild MJGT, Krawczyk M, Nowag A, Scholz CJ, Kretzschmar A, et al. Phenotyping non-alcoholic fatty liver disease by the gut microbiota: Ready for prime time? J. Gastroenterol. Hepatol., 2020, 35, 1969–77. [Google Scholar] [CrossRef]
- Boursier J, Müller O, Barret M, Machado MV, Fizanne L, Araujo-Perez F, Guy CD, Seed PC, Rawls JF, David LA, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology, 2016, 63, 764–75. [Google Scholar] [CrossRef]
- Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, Dulai PS, Caussy C, Bettencourt R, Highlander SK, et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab., 2017, 25, 1054–62.e5. [Google Scholar] [CrossRef]
- Fei N, Bruneau A, Zhang Z, Wang R, Wang J, Rabot S, Gérard P, Zhao L. Endotoxin Producers Overgrowing in Human Gut Microbiota as the Causative Agents for Non-alcoholic Fatty Liver Disease. mBio, 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA, 2004, 101, 15718–23. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA, 2007, 104, 979–84. [Google Scholar] [CrossRef]
- Meijnikman AS, Davids M, Herrema H, Aydin O, Tremaroli V, Rios-Morales M, Levels H, Bruin S, de Brauw M, Verheij J, et al. Microbiome-derived ethanol in nonalcoholic fatty liver disease. Nat Med. 2022, 28, 2100–2106. [Google Scholar] [CrossRef]
- Yuan J, Chen C, Cui J, Lu J, Yan C, Wei X, Zhao X, Li N, Li S, Xue G, et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab., 2019, 30, 1172. [Google Scholar] [CrossRef]
- Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology; 2013, 57, 601–9. [Google Scholar] [CrossRef] [PubMed]
- Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–86. [Google Scholar] [CrossRef] [PubMed]
- Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci USA, 2006, 103, 12511–6. [Google Scholar] [CrossRef]
- Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes, 2012, 3, 279–88. [Google Scholar] [CrossRef] [PubMed]
- Wong VW, Chitturi S, Wong GL, Yu J, Chan HL, Farrell GC. Pathogenesis and novel treatment options for non-alcoholic steatohepatitis. Lancet Gastroenterol Hepatol., 2016, 1, 56–67. [Google Scholar] [CrossRef]
- Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–35. [Google Scholar] [CrossRef] [PubMed]
- Caussy C, Hsu C, Lo MT, Liu A, Bettencourt R Ajmera VH, Bassirian S, Hooker J, Sy E, Richards L, et al. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology, 2018, 68, 918–32. [Google Scholar] [CrossRef] [PubMed]
- Hoyles L, Fernández-Real JM, Federici M, Serino M, Abbot J, Charpentier J, Heymes C, Luque JL, Anthony E, et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med., 2018, 24, 1070–80. [Google Scholar] [CrossRef]
- Bajaj JS, Kassam Z, Fagan A, Gavis EA, Liu E, Cox IJ, Kheradman R, Heuman D, Wang J, Gurry T, et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology, 2017, 66, 1727–1738. [Google Scholar] [CrossRef]
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature, 2012, 482, 179–85. [Google Scholar] [CrossRef]
- De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L, Mingarelli E, Facinelli B, Magi G, Palmieri C, et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology, 2014, 59, 1738–49. [Google Scholar] [CrossRef] [PubMed]
- Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot, Bevilacqua C, Martin P, Philippe C, Walker F, Bado A, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut, 2013, 62, 1787–94. [Google Scholar] [CrossRef]
- Burz SD, Monnoye M, Philippe C, Farin W, Ratziu V, et al. Fecal Microbiota Transplant from Human to Mice Gives Insights into the Role of the Gut Microbiota in Non-Alcoholic Fatty Liver Disease (NAFLD). Microorganisms, 2021, 9, 199. [Google Scholar] [CrossRef]
- Maruo T, Sakamoto M, Ito C, Toda T, Benno Y. Adlercreutzia equolifaciens gen. nov., sp. nov., an equol-producing bacterium isolated from human faeces, and emended description of the genus Eggerthella. Int J Syst Evol Microbiol., 2008, 58, 1221–7. [CrossRef]
- Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature, 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Solé C, Guilly S, Da Silva K, Llopis M, Le-Chatelier E, Huelin P, Carol M, Moreira R, Fabrellas N, De Prada G, et al. Alterations in Gut Microbiome in Cirrhosis as Assessed by Quantitative Metagenomics: Relationship With Acute-on-Chronic Liver Failure and Prognosis. Gastroenterology, 2021, 160, 206–18.e13. [Google Scholar] [CrossRef] [PubMed]
- Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Stoll DA, Danylec N, Soukup ST, Hetzer B, Kulling SE, Huch M. Adlercreutzia rubneri sp. nov., a resveratrol-metabolizing bacterium isolated from human faeces and emended description of the genus Adlercreutzia. Int J Syst Evol Microbiol., 2021, 71, 004987. [CrossRef]
- Sakamoto M, Ikeyama N, Yuki M, Murakami T, Mori H, Lino T and Ohkuma M. Adlercreutzia hattorii sp. nov., an equol non-producing bacterium isolated from human faeces. Int J Syst Evol Microbiol. 2021, 71, 005121. [CrossRef]
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA, 2008, 105, 16731–6. [CrossRef]
- Cani PD, Depommier C, Derrien M, Everard A, de Vos WM. Akkermansia muciniphila: paradigm for next-generation beneficial microorganisms. Nat Rev Gastroenterol Hepatol. 2022, 19, 625–637. [CrossRef]
- Mazier W, Le Corf K, Martinez C, Tudela H, Kissi D, Kropp C, Coubard C, Soto M, Elustondo F, Rawadi G, al. A New Strain of Christensenella minuta as a Potential Biotherapy for Obesity and Associated Metabolic Diseases. Cells, 2021, 10, 823. [Google Scholar] [CrossRef]
- Galipeau HJ, Caminero A, Turpin W, Bermudez-Brito M, Santiago A, Libertucci J, Constante M, Raygoza Garay JA, Rueda G, et al. Novel Fecal Biomarkers That Precede Clinical Diagnosis of Ulcerative Colitis. Gastroenterology, 2021, 160, 1532–45. [Google Scholar] [CrossRef]
- van de Guchte, M. , Mondot S., Doré J. Dynamic Properties of the Intestinal Ecosystem Call for Combination Therapies, Targeting Inflammation and Microbiota, in Ulcerative Colitis. Gastroenterology. 2021, 161, 1969–1981e12. [Google Scholar] [CrossRef]
- Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Nouioui I, Carro L, García-López M, Meier-Kolthoff JP, Woyke T, Kyrpides NC, Pukall R, Klenk HP, Goodfellow M, Göker M. Genome-Based Taxonomic Classification of the Phylum Actinobacteria. Front Microbiol. 2018, 9, 2007. [Google Scholar] [CrossRef]
- Minamida K, Ota K, Nishimukai M, Tanaka M, Abe A, Sone T, Tomita F, Hara H, Asano K. Asaccharobacter celatus gen. nov., sp. nov., isolated from rat caecum. Int J Syst Evol Microbiol. 2008, 58, 1238–40. [CrossRef]
- Jensen SN, Cady NM, Shahi SK, Peterson SR, Gupta A, Gibson-Corley KN, Mangalam AK. Isoflavone diet ameliorates experimental autoimmune encephalomyelitis through modulation of gut bacteria depleted in patients with multiple sclerosis. Sci Adv. 2021, 7, eabd4595. [Google Scholar] [CrossRef]
- Lenoir M, Martín R, Torres-Maravilla E, Chadi S, González-Dávila P, Sokol H, Langella P, Chain F, Bermúdez-Humarán LG. Butyrate mediates anti-inflammatory effects of Faecalibacterium prausnitzii in intestinal epithelial cells through Dact3. Gut Microbes, 2020, 12, 1–16. [Google Scholar] [CrossRef]
- Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, Plichta DR, Gautier L, Pedersen AG, Le Chatelier E, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–8. [Google Scholar] [CrossRef]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [CrossRef] [PubMed]
- Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–6. [CrossRef]
- Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods, 2012, 9, 357–9. [Google Scholar] [CrossRef]
- Wen C, Zheng Z, Shao T, Liu L, Xie Z, Le Chatelier E, He Z, Zhong W, Fan Y, Zhang L, et al. Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis. Genome Biol, 2017, 18, 214. [Google Scholar] [CrossRef]
- Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 2014, 32, 834-41. [CrossRef]
- Plaza Oñate F, Le Chatelier E, Almeida M, Cervino ACL, Gauthier F, Magoulès F, et al. MSPminer: Abundance-based reconstitution of microbial pan-genomes from shotgun metagenomic data. Bioinformatics. 2019, 35, 1544–52. [Google Scholar] [CrossRef]
- Plaza Onate F, Pons N, Gauthier F, Almeida M, Ehrlich SD, Le Chatelier E. Updated Metagenomic Species Pan-genomes (MSPs) of the human gastrointestinal microbiota. Recherche Data Gouv, V5. 2021. [CrossRef]
- Chklovski A, Parks DH, Woodcroft B, Tyson G. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. bioRxiv 2022. [CrossRef]
- Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, Pollard KS, Sakharova E, Parks DH, Hugenholtz P, et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol., 2021, 39, 105–114. [Google Scholar] [CrossRef]
- Kim CY, Lee M, Yang S, Kim K, Yong D, Kim HR, Lee I. Human reference gut microbiome catalog including newly assembled genomes from under-represented Asian metagenomes. Genome Med. 2021, 13, 134. [Google Scholar] [CrossRef]
- Plaza Oñate F, Gitton-Quent O, Almeida M; Le Chatelier. MIMIC2: Murine Intestinal Microbiota Integrated Catalog v2. Recherche Data Gouv, V4. 2021. [CrossRef]
- Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927-30. [Google Scholar] [CrossRef]
- Gertz EM, Yu YK, Agarwala R, Schäffer AA, Altschul SF. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 2006, 4, 41. [Google Scholar] [CrossRef]
- Lakhdari O, Cultrone A, Tap J, Gloux K, Bernard F, et al. Functional metagenomics: a high throughput screening method to decipher microbiota-driven NF-kappaB modulation in the human gut. PLoS One 2010, 5, e13092. [Google Scholar] [CrossRef]
- Hungate, RE. The anaerobic mesophilic cellulolytic bacteria. Bacteriol Rev 1950, 14, 1–49. [Google Scholar] [CrossRef]
- Burz SD, Abraham AL, Fonseca F, David O, Chapron A, et al. A Guide for Ex Vivo Handling and Storage of Stool Samples Intended for Fecal Microbiota Transplantation. Sci. Rep., 2019, 9, 8897. [Google Scholar] [CrossRef]
- Zhang X, Grosfeld A, Williams E, Vasiliauskas D, Barretto S, et al. Fructose malabsorption induces cholecystokinin expression in the ileum and cecum by changing microbiota composition and metabolism. FASEB J. 2019, 33, 7126–42. [Google Scholar] [CrossRef] [PubMed]
- Furet JP, Firmesse O, Gourmelon M, Bridonneau C, Tap J, Mondot S, Doré J, Corthier G. Comparative assessment of human and farm animal faecal microbiota using real-time quantitative PCR. FEMS Microbiol. Ecol., 2009, 68, 351–62. [Google Scholar] [CrossRef] [PubMed]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, and Schloss PD. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol., 2013, 79, 5112–20. [Google Scholar] [CrossRef] [PubMed]
- Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, Maman S, Hernandez-Raquet G, Combes S, Pascal G. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics, 2018, 34, 1287–94. [Google Scholar] [CrossRef] [PubMed]
- Bokulich NA Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, J Caporaso G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods, 2013, 10, 57–9. [Google Scholar] [CrossRef]
- Safari Z, Bruneau A, Monnoye M, Mariadassou M, Philippe C, Zatloukal K, Gérard P. Murine Genetic Background Overcomes Gut Microbiota Changes to Explain Metabolic Response to High-Fat Diet. Nutrients, 2020, 12, 2872020. [Google Scholar] [CrossRef]
- Lécuyer E, Le Roy T, Gestin A, Lacombe A, Philippe C, Ponnaiah M, Huré JB, Fradet M, Farid Ichou F, Boudebbouze S, et al. Tolerogenic Dendritic Cells Shape a Transmissible Gut Microbiota That Protects From Metabolic Diseases. Diabetes, 2021, 70, 2067–80. [Google Scholar] [CrossRef]
Figure 1.
Relative abundance of A. equolifaciens (MGS 9828_3) in groups of patients determined according to their clinical status based on biopsy reading in NASH1 (a) and NASH2 (b) cohorts. Healthy: healthy controls; NAFL: patients with steatohepatitis with no/minimal fibrosis; NASH: patients with steatohepatitis with and without fibrosis or cirrhosis. Relative abundancies are log10-transformed and “M10” is an artificial value introduced when the species is not detected. A. equolifaciens was significantly (Wilcoxon Rank Sum tests) more abundant in healthy volunteers compared to NASH/NAFL populations (NASH1: p < 0.0001; NASH2: p < 0.0001). Moreover, A. equolifaciens was more abundant in patient with NAFL patients compared to NASH population (NASH1: p = 0.018; NASH2: p = 0.0043).
Figure 1.
Relative abundance of A. equolifaciens (MGS 9828_3) in groups of patients determined according to their clinical status based on biopsy reading in NASH1 (a) and NASH2 (b) cohorts. Healthy: healthy controls; NAFL: patients with steatohepatitis with no/minimal fibrosis; NASH: patients with steatohepatitis with and without fibrosis or cirrhosis. Relative abundancies are log10-transformed and “M10” is an artificial value introduced when the species is not detected. A. equolifaciens was significantly (Wilcoxon Rank Sum tests) more abundant in healthy volunteers compared to NASH/NAFL populations (NASH1: p < 0.0001; NASH2: p < 0.0001). Moreover, A. equolifaciens was more abundant in patient with NAFL patients compared to NASH population (NASH1: p = 0.018; NASH2: p = 0.0043).
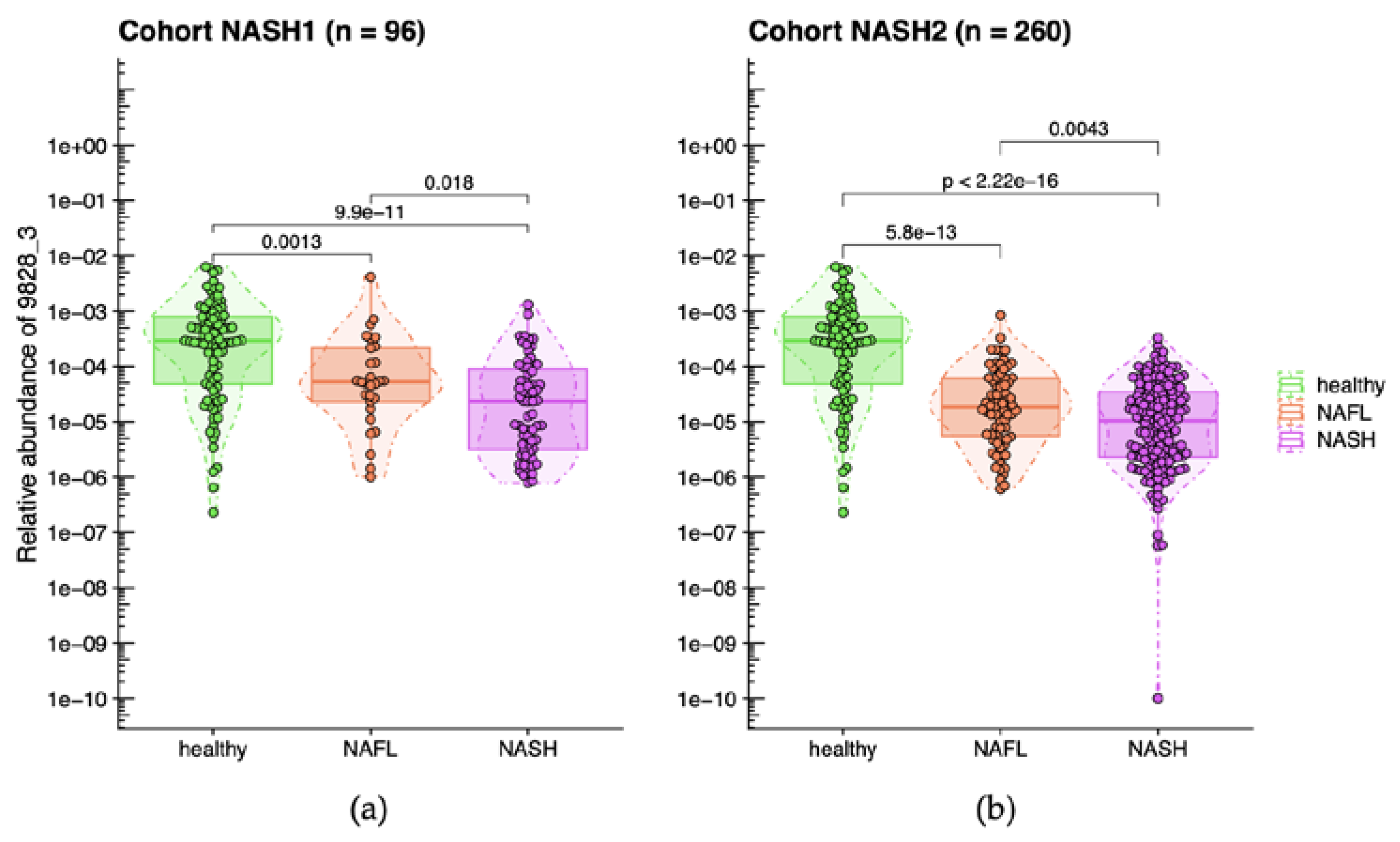
Figure 2.
A) Prevalence of A. equolifaciens in patients with NAFLD depending on the level of fibrosis. Analysis from publicly available metagenomic data from Loomba et al [10]. Fisher exact test: p = 0.014. B) Relative abundance of A. equolifaciens in patients with NAFLD depending on the level of fibrosis. Mann–Whitney U test: AUC=0.67 and p=0.023.
Figure 2.
A) Prevalence of A. equolifaciens in patients with NAFLD depending on the level of fibrosis. Analysis from publicly available metagenomic data from Loomba et al [10]. Fisher exact test: p = 0.014. B) Relative abundance of A. equolifaciens in patients with NAFLD depending on the level of fibrosis. Mann–Whitney U test: AUC=0.67 and p=0.023.
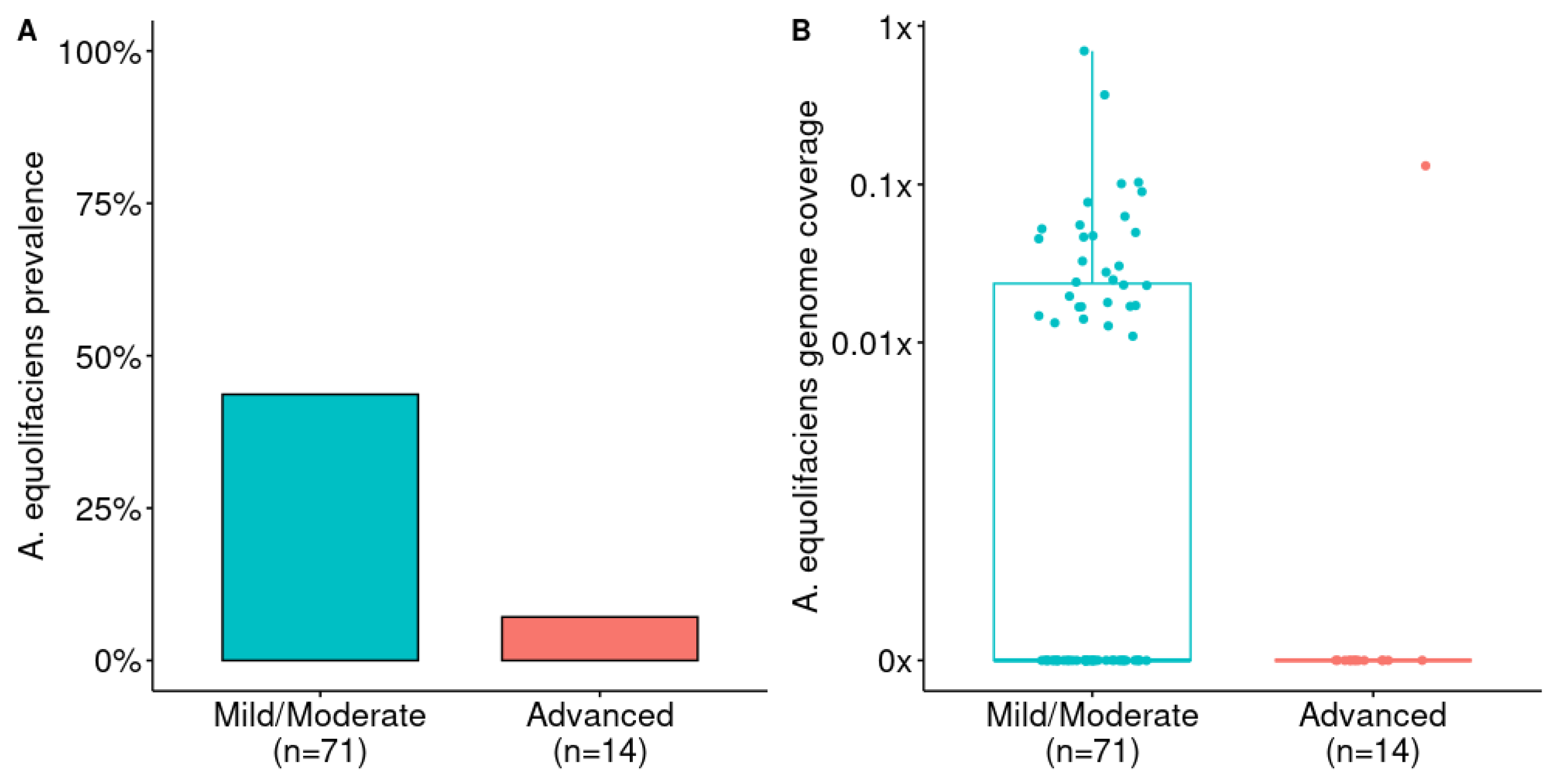
Figure 3.
A) Prevalence of A. equolifaciens in patients with liver cirrhosis compared to healthy controls. Analysis from publicly available metagenomic data from Qin et al [30]. Fisher exact test: p = 3.91 × 10-10. B) Relative abundance of A. equolifaciens in patients with liver cirrhosis compared to healthy controls. Mann–Whitney U test: AUC=0.72 and p=9.73× 10-11).
Figure 3.
A) Prevalence of A. equolifaciens in patients with liver cirrhosis compared to healthy controls. Analysis from publicly available metagenomic data from Qin et al [30]. Fisher exact test: p = 3.91 × 10-10. B) Relative abundance of A. equolifaciens in patients with liver cirrhosis compared to healthy controls. Mann–Whitney U test: AUC=0.72 and p=9.73× 10-11).
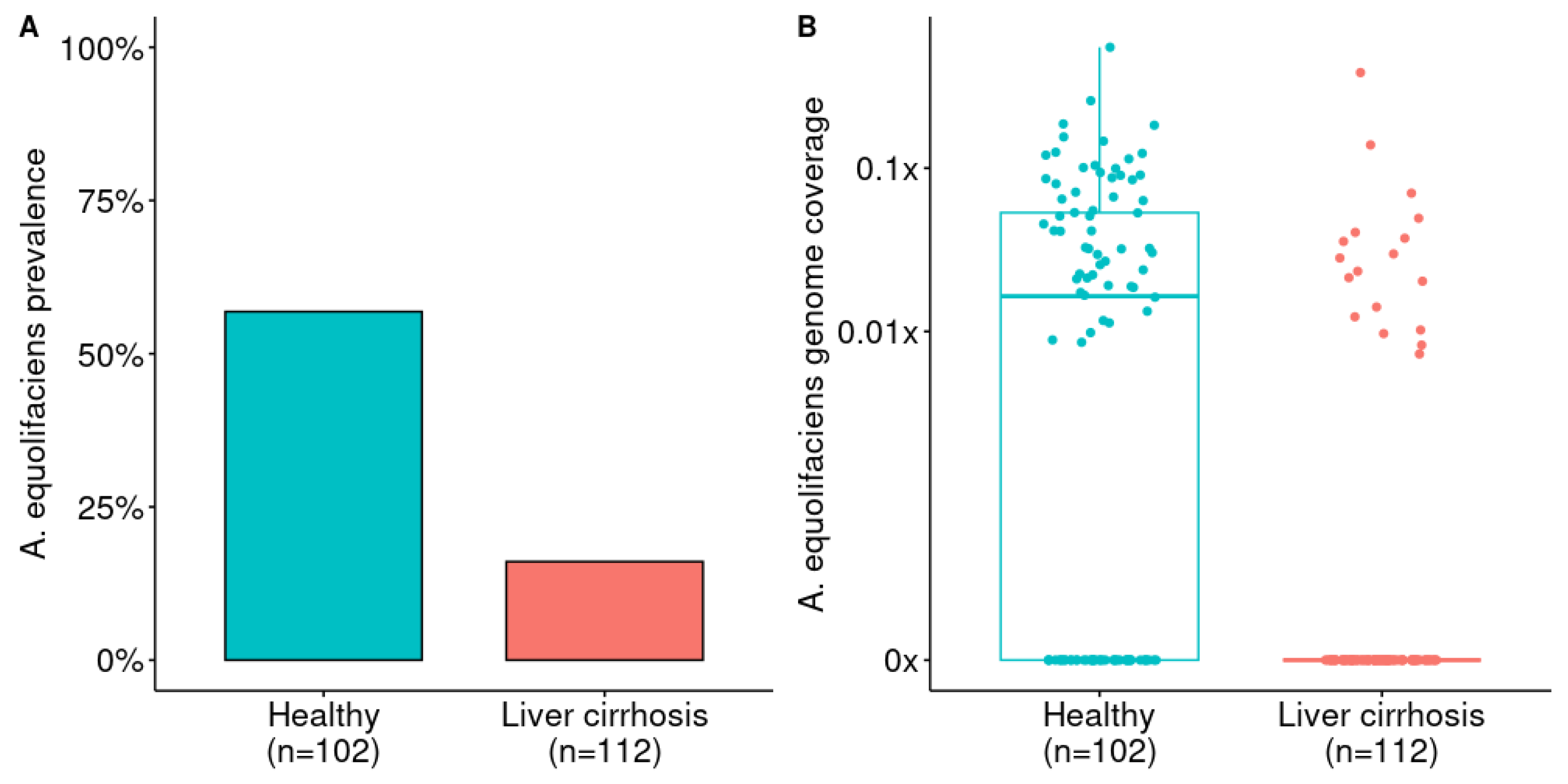
Figure 4.
A) Prevalence of A. equolifaciens in patients with liver cirrhosis according to disease severity (Compensated, Decompensated, Acute Decompensation and Acute-on-Chronic Liver Failure) and compared to healthy controls. Analysis from publicly available metagenomic data from Sole et al., 2021 [31]. Healthy controls vs all liver cirrhosis patients, Fisher exact test: p = 2.39 × 10-13. Liver cirrhosis patients only according to disease severity, chi-squared test: p = 1.37 × 10-11. B) Abundance of A. equolifaciens in patients with liver cirrhosis according to disease severity and compared to healthy controls. Healthy controls vs all liver cirrhosis patients, Mann–Whitney U test: AUC = 0.83 and p < 2.2 × 10-16. Liver cirrhosis patients only according to disease severity, Kruskal-Wallis test: p = 1.66 × 10-11.
Figure 4.
A) Prevalence of A. equolifaciens in patients with liver cirrhosis according to disease severity (Compensated, Decompensated, Acute Decompensation and Acute-on-Chronic Liver Failure) and compared to healthy controls. Analysis from publicly available metagenomic data from Sole et al., 2021 [31]. Healthy controls vs all liver cirrhosis patients, Fisher exact test: p = 2.39 × 10-13. Liver cirrhosis patients only according to disease severity, chi-squared test: p = 1.37 × 10-11. B) Abundance of A. equolifaciens in patients with liver cirrhosis according to disease severity and compared to healthy controls. Healthy controls vs all liver cirrhosis patients, Mann–Whitney U test: AUC = 0.83 and p < 2.2 × 10-16. Liver cirrhosis patients only according to disease severity, Kruskal-Wallis test: p = 1.66 × 10-11.
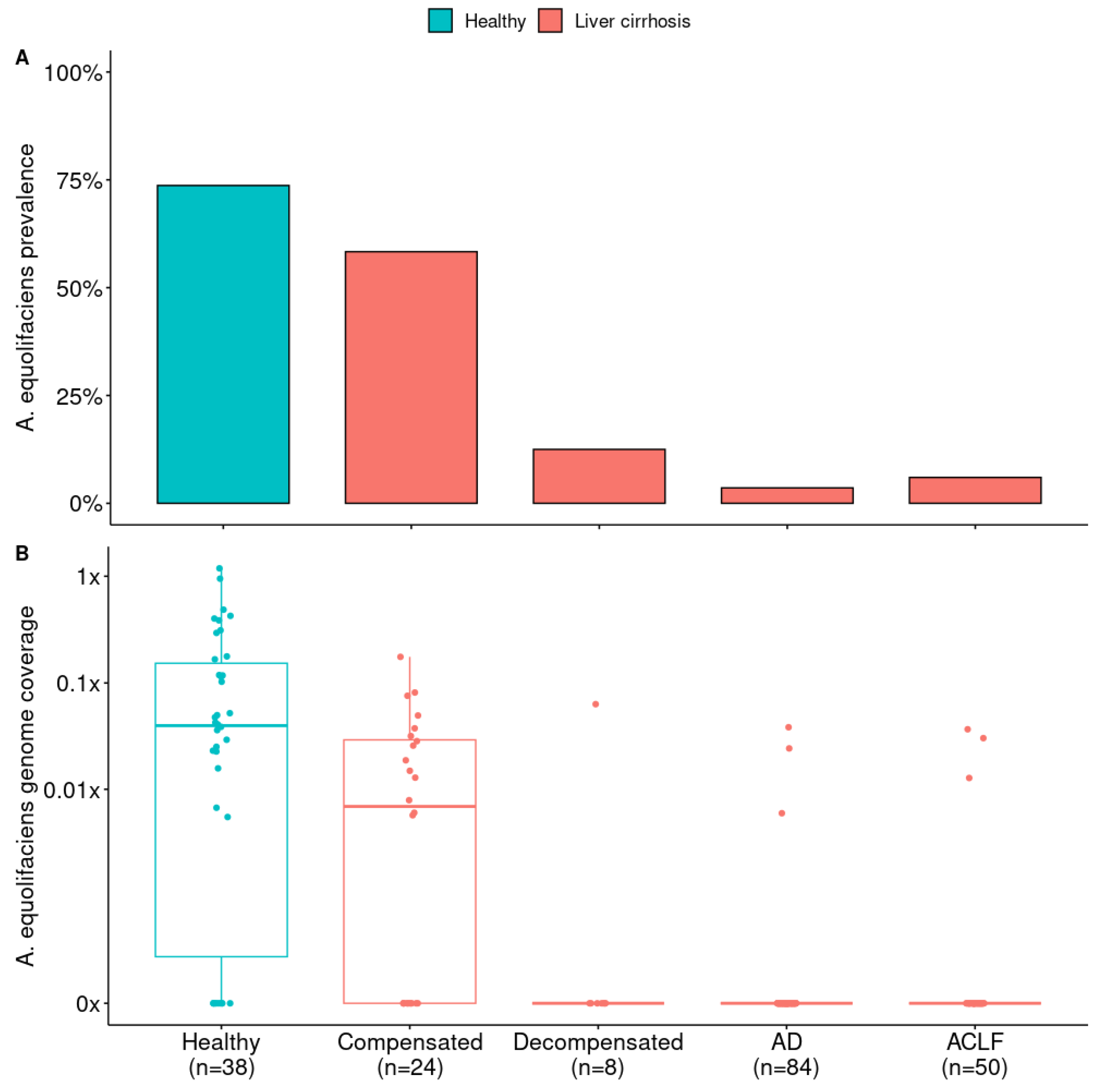
Figure 5.
Non-Metric Multidimensional Scaling (nMDS) plot representing genomes similarity assessed from pairwise ANI. Each point represents a genome and the color indicates the phylogroup (genomospecies) to which it belongs. Genomes of the four type strains are labeled.
Figure 5.
Non-Metric Multidimensional Scaling (nMDS) plot representing genomes similarity assessed from pairwise ANI. Each point represents a genome and the color indicates the phylogroup (genomospecies) to which it belongs. Genomes of the four type strains are labeled.
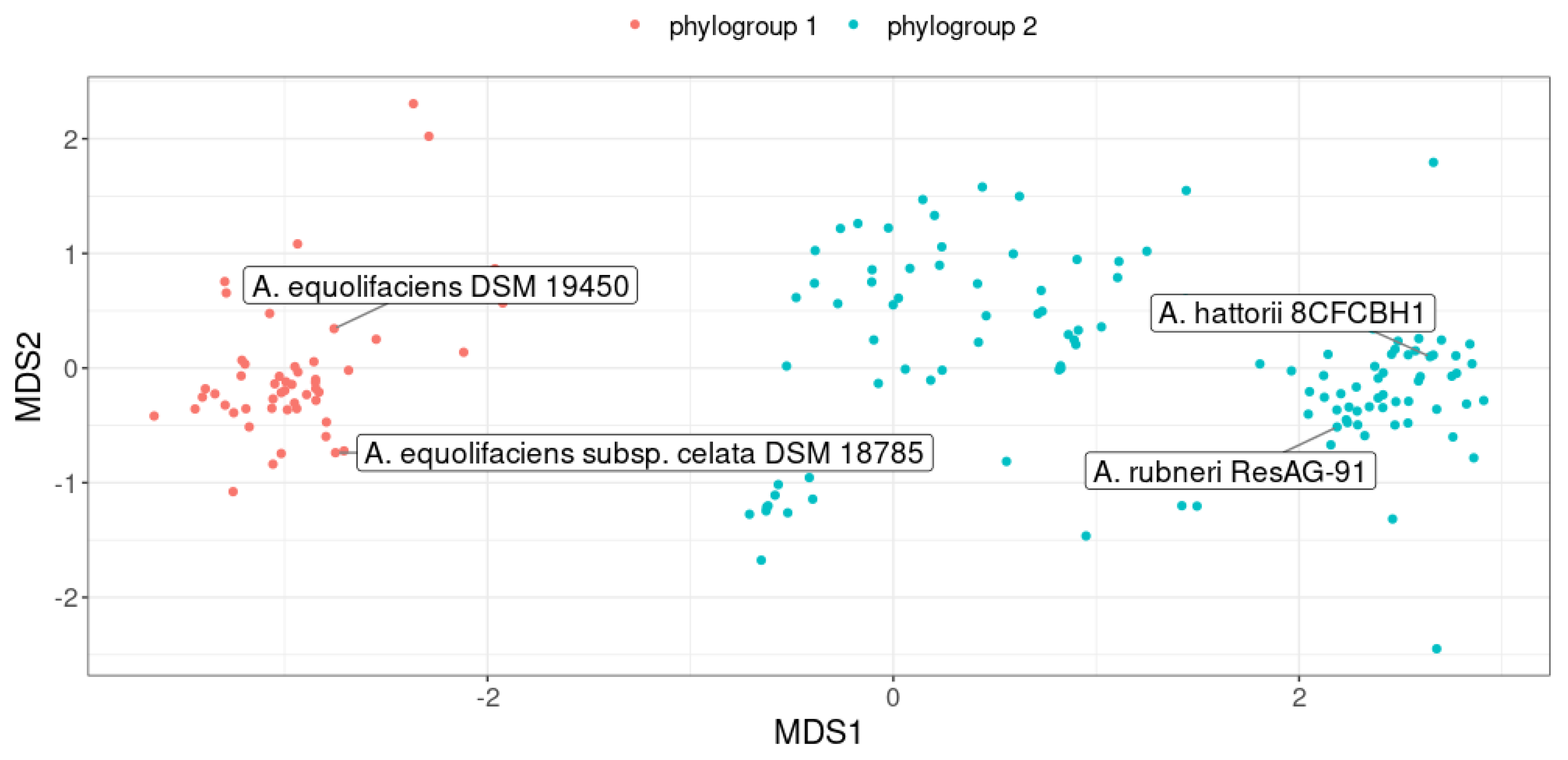
Figure 6.
Non-Metric Multidimensional Scaling (nMDS) plot representing genomes similarity assessed from pairwise ANI. Each point represents a genome and the color indicates the geographical origin of the host. The vertical dashed line separates the two phylogroups (genomospecies). Non-human related genomes are not shown.
Figure 6.
Non-Metric Multidimensional Scaling (nMDS) plot representing genomes similarity assessed from pairwise ANI. Each point represents a genome and the color indicates the geographical origin of the host. The vertical dashed line separates the two phylogroups (genomospecies). Non-human related genomes are not shown.
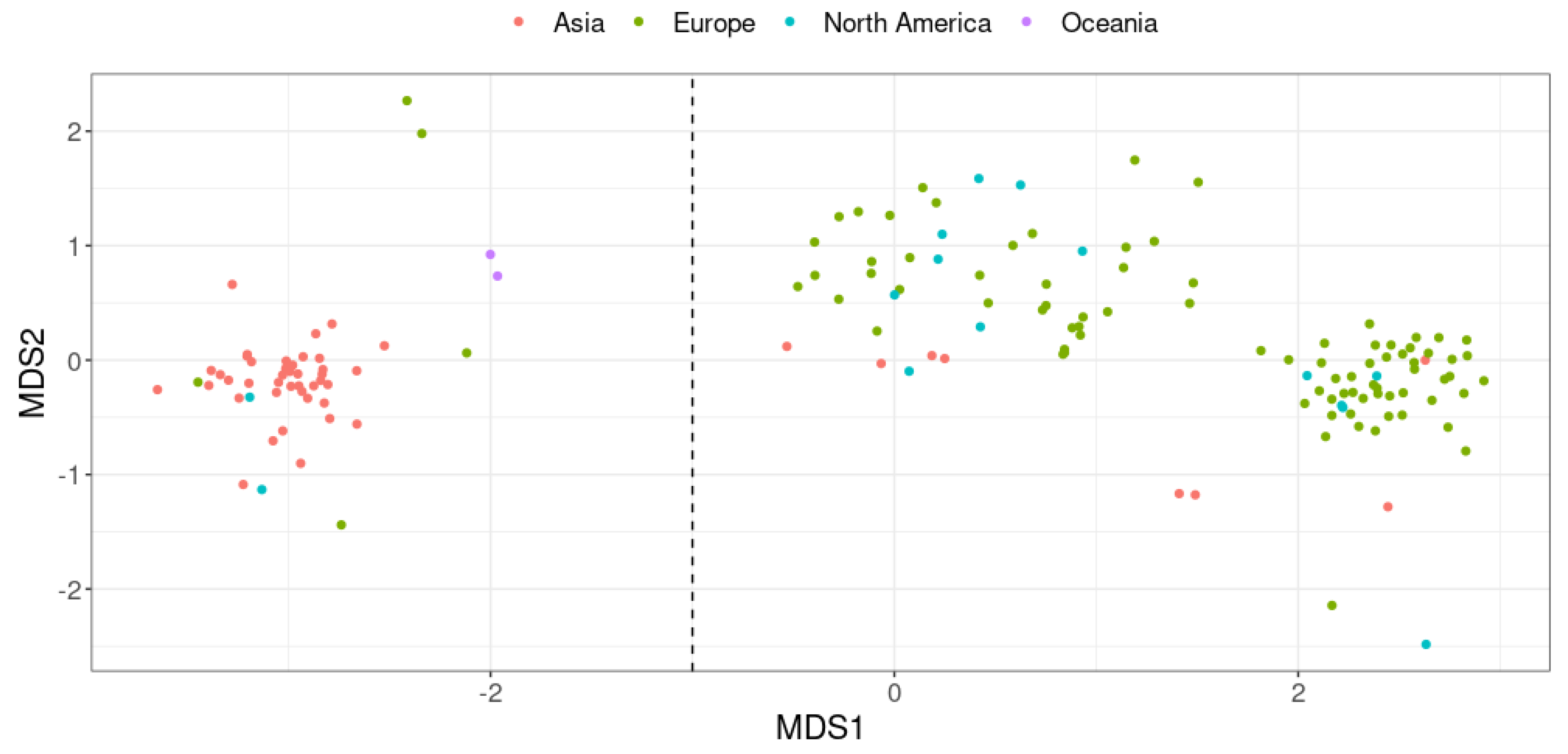
Figure 7.
Effect of supernatant and pellet of A. equolifaciens and P. caecicola on NF-κB activity on HT-29 reporter (a) and (b) and HepG2 (c) reporter cell lines. NF-κB activity is measured in the absence (first bar) or presence of TNF-α. Values represent fold increase of NF-κB activity in unstimulated cells (M104 or PBS). Results are expressed as the mean ± the SEM of at least three experiments with triplicate determinations. p-values < 0.05 were considered significant ** p < 0.0021, ***p < 0.0002, **** p < 0.0001).
Figure 7.
Effect of supernatant and pellet of A. equolifaciens and P. caecicola on NF-κB activity on HT-29 reporter (a) and (b) and HepG2 (c) reporter cell lines. NF-κB activity is measured in the absence (first bar) or presence of TNF-α. Values represent fold increase of NF-κB activity in unstimulated cells (M104 or PBS). Results are expressed as the mean ± the SEM of at least three experiments with triplicate determinations. p-values < 0.05 were considered significant ** p < 0.0021, ***p < 0.0002, **** p < 0.0001).
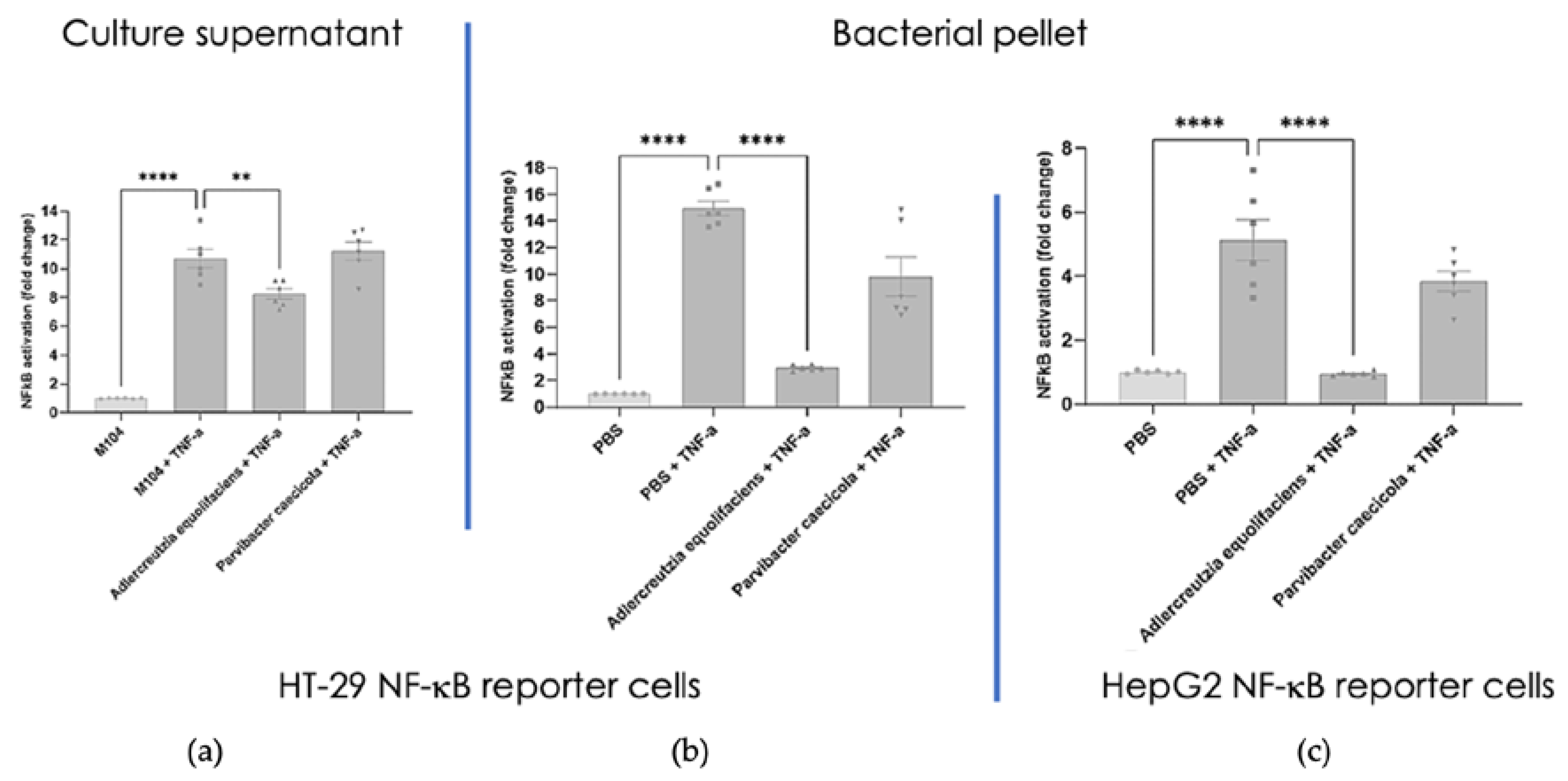
Figure 8.
(a) Average weight (+/- SEM) along time of mice receiving A. equolifaciens (MGS1) or PBS as control. (b) Weight of mice at the end of the experience. (c) Fasting glycemia of mice at the beginning of the experience (basal) and 1 month after beginning of the gavage with PBS or A. equolifaciens.
Figure 8.
(a) Average weight (+/- SEM) along time of mice receiving A. equolifaciens (MGS1) or PBS as control. (b) Weight of mice at the end of the experience. (c) Fasting glycemia of mice at the beginning of the experience (basal) and 1 month after beginning of the gavage with PBS or A. equolifaciens.
Figure 9.
A equolifaciens gavage modulates caecum weight and caecal SCFA concentration. (a) Weight of caecum/mouse weight at the end of the experience. (b) Butyrate concentration in μmol/g of caecum content. (c) D-Isovalerate concentration/g of caecum content expressed as % of total SCFA. (d) Isobutyrate concentration/g of caecum content expressed as % of total SCFA. Orange. Mice receiving PBS; blue, mice receiving A. equolifaciens.
Figure 9.
A equolifaciens gavage modulates caecum weight and caecal SCFA concentration. (a) Weight of caecum/mouse weight at the end of the experience. (b) Butyrate concentration in μmol/g of caecum content. (c) D-Isovalerate concentration/g of caecum content expressed as % of total SCFA. (d) Isobutyrate concentration/g of caecum content expressed as % of total SCFA. Orange. Mice receiving PBS; blue, mice receiving A. equolifaciens.
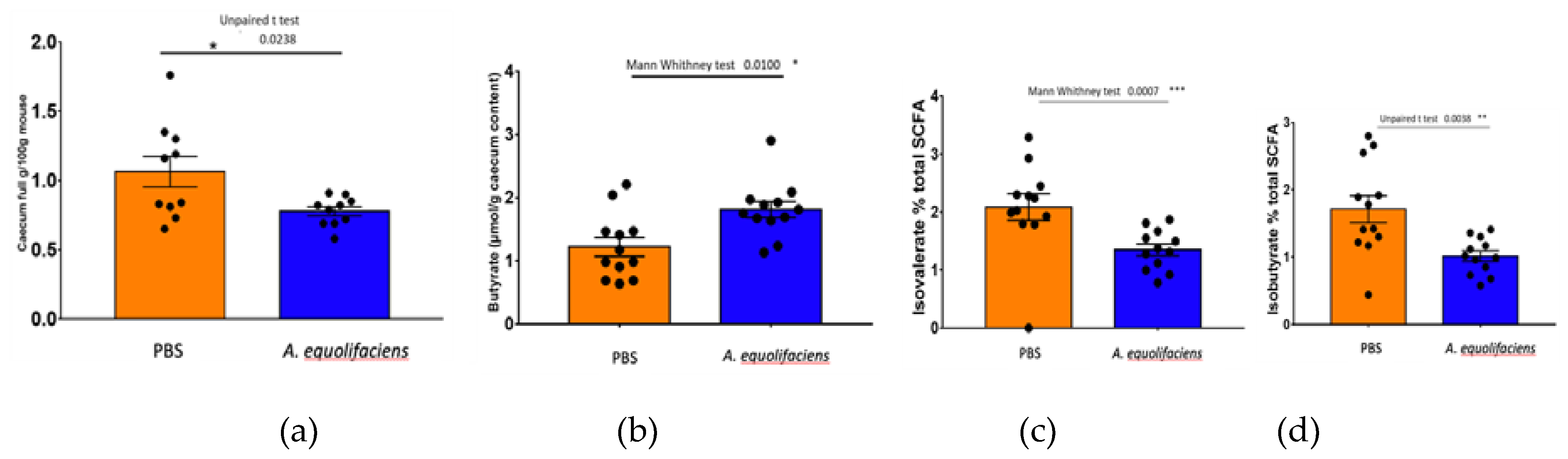
Figure 10.
A. equolifaciens gavage reduces IL-6 mRNA expression in the caecum (a) and the liver (b) of mice. GAPDH and actin- were used as housekeeping genes in the caecum and the liver, respectively.
were used as housekeeping genes in the caecum and the liver, respectively.
were used as housekeeping genes in the caecum and the liver, respectively.
Figure 10.
A. equolifaciens gavage reduces IL-6 mRNA expression in the caecum (a) and the liver (b) of mice. GAPDH and actin- were used as housekeeping genes in the caecum and the liver, respectively.
were used as housekeeping genes in the caecum and the liver, respectively.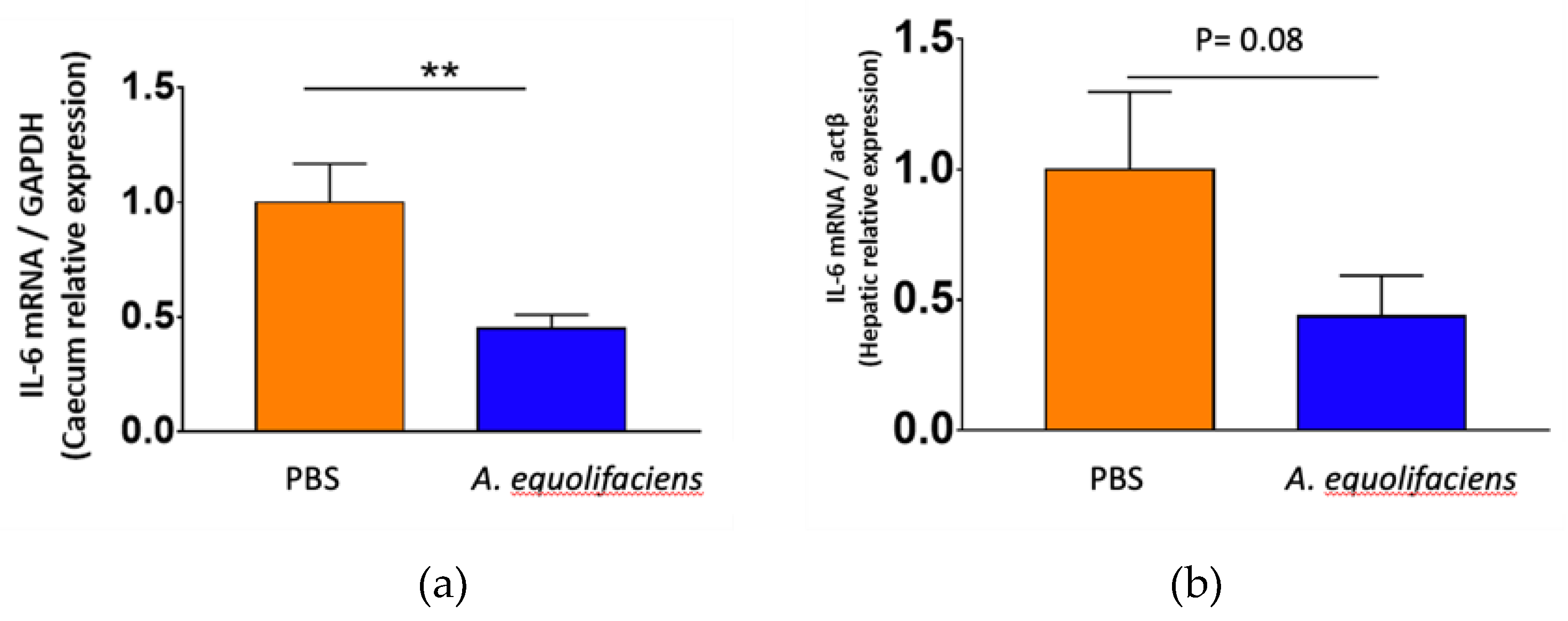
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



