
Submitted:
29 May 2023
Posted:
30 May 2023
You are already at the latest version
Abstract
Chromatin structure plays a fundamental role in regulating gene expression, with histone modifiers shaping the structure of chromatin by adding or removing chemical changes to his-tone proteins. The p53 transcription factor controls gene expression, binds target genes, and regulates their activity. While p53 has been extensively investigated in the context of cancer research, its association with histone modifiers has received limited attention. This review ex-plores the interplay between histone modifiers and p53 in regulating gene expression. We discussed how histone modifications can influence how p53 binds to target genes and how this interplay can be disrupted in cancer cells. This study provides insights into the complex mechanisms underlying gene regulation and their implications for potential cancer therapy.
Keywords:
Histone modifications
; p53
; cancer
; gene regulation
; chromatin structure
1. Introduction
Chromatin is the complex of DNA, histone proteins, and other associated proteins that make up the structure of chromosomes within the nucleus of eukaryotic cells. It is a well organized and dynamic structure that plays a fundamental role in the packaging and regulating of DNA. The chromatin structure can either promote or inhibit gene expression, depending on the specific modifications present on the histones [1,2]. Enzymes known as histone modifiers are responsible for adding or removing chemical changes in the histone proteins, thereby shaping the chromatin structure. These modifications, such as acetylation, methylation, phosphorylation, and ubiquitination, exhibit diverse effects on chromatin structure and gene expression.
p53, as a transcription factor, plays a crucial role in controlling the expression of various genes involved in cell cycle regulation, DNA repair, apoptosis, and other cellular processes. It acts as a tumor suppressor by promoting cell cycle arrest or inducing apoptosis in response to DNA damage or cellular stress. Thus, p53 is one of the most crucial tumor suppressor genes in tumorigenesis. The activity of p53 is regulated by various mechanisms, including post-translational modifications of both p53 and the histones sur- rounding it.
The interplay between histone modifiers and p53 is critical for modulating gene expression, genomic stability, chromatin remodeling and DNA Repair. The collaboration of the two ensures proper cellular responses to DNA damage, stress signals, and other regulatory cues. Since p53 is the guardian of the genome whose activity is implicated in most types of cancers, its post-transcriptional modifications have been extensively studied [3]. However, the connection between histone modifiers and p53 gained less attention despite the growing body of research on the involvement of histone modifiers in cancer. Therefore, this review primarily centers on exploring the impact of histone modifiers on p53 and their significance in regulating gene expression.
Two major mechanisms underlie the interaction between histone modifiers and p53. First, histone modifiers can impact p53 function indirectly by modifying the chromatin structure and accessibility of p53 target genes. They can establish an open or closed chromatin state, influencing the ability of p53 to bind to its target DNA sequences and regulate gene expression. Alternatively, p53 can bind to specific histone modifiers or their associated proteins, facilitating the recruitment of these modifiers to target genes. This recruitment can lead to modifications of histones in the vicinity of p53-binding sites, either promoting or repressing gene expression.
Images are recreated from [4]. (B) Protein methylation occurs on lysine and arginine residues in histone and non-histone proteins by protein methyltransferases. The specific methyltransferases and demethylase reversibly regulated lysine methylation and de- methylation from mono to trimethylation. Arginine methylations are induced in three types, including monomethylation, asymmetric dimethylation, and symmetric dimethylation. (C) Acetyltransferases (KATs) transfer the acetyl group from acetyl–CoA to specific lysine residues in proteins, while acetylation can be reversed by lysine deacetylases (KDACs). (D) Protein phosphorylation occurs on serine, threonine and tyrosine residues. Images are recreated from [5] [6].
2. Histone Modifiers on p53
Packaging DNA into chromatin affects gene expression by making specific genes accessible to transcription factors, which are proteins that bind to DNA and control gene expression. Histone modifications affect chromatin structure by altering the interactions between histones and DNA. Each of these modifications can affect chromatin structure and gene expression differently. Modifications that are associated with active transcription, such as acetylation of histone three and histone four (H3 and H4) or di- or trimethylation (me) of H3K4, are commonly referred to as euchromatin modifications [7](Figure 1A). Conversely, modifications localized to inactive genes or regions, such as H3K9me and H3K27me, are often termed heterochromatin modifications.
The enzymes responsible for regulating post-translational epigenetic modifications on histones have been categorized into four groups based on their roles [8]. Writers add changes to histones and include DNA methyltransferases (DNMTs), histone lysine methyltransferases (KMTs), and histone acetyltransferases (HATs). Conversely, erasers re- move post-translational modifications, including histone lysine demethylases (KDMs) and histone deacetylases (HDACs). The term readers describe bromodomain and chromodomain proteins that can "read" acetylated or methylated residues. Movers can re- model chromatin by moving nucleosomes, thus facilitating gene transcription.
Furthermore, histone modifications can be classified into major groups based on the type of modification and the amino acid residue being modified, including acetylation, methylation, phosphorylation, ubiquitination, sumoylation and ADP-ribosylation. Among these modifications, methylation, acetylation, and phosphorylation are the primary ones observed in the interplay between histones and p53 (Figure 1). Building upon this understanding, this review primarily centers on exploring the impact of histone modifiers on p53 and their significance in regulating gene expression.
2.1. Methylation
The primary amino acids susceptible to methylation are arginine and lysine (Figure 1B). Arginine methylation involves the monomethylation, asymmetric dimethylation or symmetric dimethylation of arginine residues, which is mediated by three types of protein arginine methyltransferases (PRMT type I, II, III), and this modification plays a role in modulating protein function and various cellular activities. On the other hand, lysine methylation leads to the formation of mono-, di-, or trimethylated lysine residues. In histones, lysine methylation is a crucial epigenetic marker that influences gene expression by affecting chromatin structure and attracting proteins involved in transcriptional regulation. Lysine methylation can also occur on non-histone proteins, governing cellular processes such as DNA repair, signal transduction, and protein-protein interactions.
SET7/9
Lysine methylation of p53 by the histone methyltransferase SET9 enzyme leads to the activation of p53 [9]. Methyltransferase Set9 methylates p53 at K372 within the carboxyl- terminus regulatory region. Set9-mediated methylation of p53-K372 resulted in stabilizing a chromatin-bound fraction of p53. Correspondingly, the methylated p53 is restricted to the nucleus and positively affects its stability, enhancing the p53 target gene p21 expression and increasing levels of p53-mediated apoptosis (Figure 2)
A similar interplay between p53 and lysine methyltransferase has already been de- scribed [10]. Pre-methylation at K372-p53 enhances subsequent acetylation of p53 by p300 upon DNA damage. However, pre-acetylation of the p53 inhibits subsequent methylation of K372 by Set7/9 (Set7 and Set9), suggesting that methylation must precede acetylation for a positive interplay between methylation and acetylation. Of note, several lysine residues near the site where Set7/9 methylates p53 can also be acetylated by CBP/p300 [10]. These lysine residues are numbered K370, K373, K381, and K382 in vitro and K373 and K382 in vivo.
Ivanov et al. used chromatin immunoprecipitation (ChIP) assays to analyze the levels of p53 binding and histone H4 acetylation at the promoter of the p21 gene [10]. Methylation of p53 by Set7/9 increased the binding of p53 to the promoter region of the p21 gene, resulting in increased acetylation of histone H4 and subsequent transcriptional activation and cell cycle arrest (Figure 2). These results suggest an interplay between histone modifiers and p53 is required for subsequent transcriptional activation. However, the exact mechanism of methylation-dependent acetylation of p53 remains to be explored.
G9a
G9a, a Set domain-containing protein, serves as the major histone lysine methyltransferase. It methylates histone H3 at both H3K9me1 and H3K9me2 modifications, earning its name G9a [11]. By adding methyl groups to histones, the G9a protein establishes and maintains epigenetic marks that regulate gene activity. It engages in various cellular processes, including embryonic development, cell differentiation, and the maintenance of cell identity.
Human G9a (hG9a) can regulate the expression of p21 in a manner that is independent of p53 and its methylation activity [12]. G9a positively regulates p21 expression independent of p53 and its histone methyltransferase activity. Oh et al. demonstrated that hG9a upregulates p21 via interaction with PCAF, and this activating complex is recruited to the p21 promoter upon DNA damage-inducing agent etoposide treatment. Ultimately, p21 induction by G9a inhibits cellular proliferation and leads to apoptosis in p53-null cells. This regulatory mechanism does not rely on the histone lysine methyltransferase activity of G9a and functions through a pathway separate from p53 (Figure 3).
Similarly, hG9a stimulates p53's activity independently of methylation by interacting with histone acetyltransferase CBP/p300, resulting in increased histone acetylation at the promoter of pro-apoptotic genes, including PUMA, thus inducing p53 transcriptional activity [13].
On the contrary, the mouse one (mG9a) blunted P53-dependent transcription in a methylation-specific manner (Figure 3). The differences in the regulation of P53 by hG9a and mG9a may be due to splicing variants. The human G9a (EHMT2) gene is present in cells as two splice variants (hG9a long and hG9a short), while mG9a is the product of the NG36-G9a transcript, which is similar to hG9a based on amino acid sequences. The findings from two independent studies have identified that human G9a (hG9a) functions in a manner independent of methylation [12,13]. However, the specific mechanisms by which hG9a targets p53 and p21, either separately or in a coordinated manner, remain unclear. Further research is needed to elucidate the precise mechanisms by which hG9a modulates the expression of p53 and p21 and determine whether these regulations occur independently or through interconnected pathways.
PRMTs
PRMTs have crucial roles in various cellular processes, including transcriptional regulation, chromatin regulation, signal transduction, and DNA damage repair. They catalyze the transfer of a methyl group from S-adenosylmethionine to the guanidine nitrogen of arginine residues in proteins. [14,15]. PRMT5 specifically methylates histone H4 at argi- nine 3 (H4R3), indirectly influencing p53 activity by affecting the transcriptional regulation of p53 target genes. Following DNA damage, PRMT5 methylates p53 at arginine residues R333, R335, and R337 [16]. Bypassing p53 through arginine methylation leads to apoptosis evasion and facilitates tumor growth [17](Figure 4). Consistently, depletion of PRMT5 triggers p53-mediated apoptosis, indicating that arginine methylation plays a role in controlling p53 activity. Also, R337H mutation, prevalent in pediatric adrenocortical tumors in southern Brazil, underscores the significance of arginine methylation in regulating p53-mediated events and oncogenesis [18,19].
In addition to methylation of p53, PRMT5 has been shown to regulate chromatin structure and gene expression through its interaction with histones. By controlling the alternative splicing of crucial histone-modifying enzymes such as TIP60 and KMT5C, PRMT5 can affect chromatin structure and ultimately impact DNA repair [20](Figure 4). Therefore, PRMT5-mediated changes in histone methylation may indirectly affect p53 function by altering gene expression patterns.
JMJD2
While p53 is a target for epigenetic modulators, it can also target histone modifiers. JMJD2 (Jumonji C domain containing histone demethylase 2) family of proteins selectively de- methylate H3K9me3 and H3K36me3. JMJD2B/KDM4B is a p53-inducible gene in response to DNA damage (Figure 5). p53 regulates JMJD2B gene expression by binding to a p53-consensus motif in the JMJD2B promoter. JMJD2B induction attenuates the transcription of key p53 transcriptional targets, including p21, PIG3 and PUMA, while silencing enhances the induction of the two [21]. Also, JMJD2B-mediated histone demethylation is critical for p53-mediated autophagy and survival in Nutlin-treated cancer cells [22].
EZH2
EZH2, also known as Enhancer of Zeste Homolog 2, is a vital protein involved in epigenetic regulation. It belongs to the Polycomb group protein family and serves as the catalytic subunit of the Polycomb Repressive Complex 2 (PRC2)[23]. Functioning as a methyl- transferase, EZH2 adds methyl groups specifically to lysine 27 of histone H3 (H3K27) through its histone methyltransferase activity. This enzymatic function enables EZH2 to modify chromatin structure by depositing the repressive histone mark H3K27me3. The addition of methyl groups by EZH2 plays a pivotal role in gene silencing and epigenetic regulation.
Several studies provided valuable insights into the dynamic relationship between p53 and EZH2. Tang et al. demonstrated that activated p53 downregulates EZH2 gene expression by repressing the EZH2 gene promoter [24]. Additionally, their findings revealed that reducing EZH2 expression leads to impaired cell proliferation and G2/M arrest. These observations suggest that p53 controls the G2/M checkpoint by suppressing EZH2 expression. Also, Yuan et al. uncovered an intriguing interplay between Ezh2 and p53 in regulating inflammasome activation (Figure 6)[25]. Ezh2 competes with p53 for binding to the promoter of the lncRNA Neat1 gene. This competition allows Ezh2 to maintain the enrichment of H3K27 acetylation (H3K27ac) and chromatin accessibility, facilitating the transcription of Neat1 by p65. Consequently, inflammasome activation is promoted.
2.2. Phosphorylation
Phosphorylation is a common post-translational modification that involves adding a phosphate group (PO43-) to specific amino acid residues in proteins, typically serine, threonine, or tyrosine (Figure 1). This modification is catalyzed by protein kinases, which transfer the phosphate group from ATP to the target residue. Phosphorylation of p53 can occur at multiple sites in response to various stress signals. Phosphorylation of p53 at Ser15 in response to ionizing radiation enhances the transcriptional activity of p53 by increasing its affinity for DNA to recruit coactivators such as CBP/p300 (Figure 7)[26].
Similarly, other studies reported that Ser20 undergoes phosphorylation following ex- posure to ionizing radiation, which could potentially weaken the binding of p53 to Mdm2 to save p53 from ubiquitin-mediated degradation [27,28,29]. Also, Phosphorylation of human p53 Ser-392 in the C-terminal regulatory domain occurs following UV but not γ-irradiation [30,31] and results in enhancement of sequence-specific binding activity in vitro [32], possibly by promoting stable tetramer form of p53 (Figure 7)[33]. These observa- tions explain how the activation of p53-regulated genes following DNA damage.
MAP Kinase cascade
MAP kinase cascade is one of the major UV response pathways [34]. This pathway has three distinct components in mammalian cells: extracellular signal-regulated protein kinases (ERKs), p38 kinases, and stress-activated c-Jun N-terminal kinases (JNKs). These kinases participate in the regulation of cell proliferation, differentiation, stress responses, and apoptosis.
p38 MAP kinase
p38 can directly phosphorylate and activate p53. Upon activation, p38 phosphorylates specific serine residues on p53, such as Ser15 and Ser392 [34,35,36], leading to increased p53 stability, transcriptional activity, and subsequent induction of downstream target genes involved in cell cycle arrest, DNA repair, and apoptosis (Figure 8). Also, Ser15 phosphorylation stabilizes p53 by reducing its interaction with MDM2, a negative regulatory partner [37]. Hence, phosphorylation of p53 is likely to play an essential role in regulating its activity.
Also, p38 can indirectly influence histone modifications through various mechanisms. p38 phosphorylates and activates downstream targets, including kinases and transcription factors, which can, in turn, modulate histone modifications. For example, MSK1, a downstream target of the MAPK pathway, can be activated by p38 MAPK. Upon activation, MSK1/2 can phosphorylate specific residues on histone H3, leading to the modification of chromatin structure and the regulation of gene expression [38](Figure 8). Specifically, it has been demonstrated to phosphorylate histone H3 at serine 10 (H3S10) and serine 28 (H3S28) residues [39,40,41].
When activated by the p38 MAPK pathway, MSK1 interacts with p53 and is recruited to the p21 promoter, where it phosphorylates histone H3 in a p53-dependent manner. Therefore, MSK1 plays a role in activating the expression of the p21 gene [41]. This enhances the transcriptional activation of p21, as evidenced by in vitro chromatin transcription and cell-based analyses. Overall, p38 MAPK activates p53 and indirectly influences histone modifications, while histone modifications can modulate p53 function. These interconnected relationships contribute to the intricate regulatory networks involved in cellular stress responses, DNA damage repair, and gene expression control.
RSK2
RSK2 is a p90 ribosomal S6 kinase family member that is activated by growth factors, peptide hormones, and neurotransmitters via MAPK/ERK signaling (ERK1 and ERK2). It is critical in regulating gene transcription by phosphorylating CBP at Ser133 [42]. Additionally, RSK2 has been reported to phosphorylate both histone H3 and p53. When cells are stimulated with UV or EGF, RSK2 is activated through the MAPK cascade and phosphorylates p53 protein at Ser15[43](Figure 9). Authors further proposed that The RSK2/p53 complex then translocated to the nucleus where RSK2 phosphorylates histone H3 at Ser10 and induces expression of target genes. These findings suggest that the interplay of RSK2-p53-histone H3 may contribute to transcriptional regulation, chromatin remodeling and cell cycle regulation.
2.3. Acetylation
Acetylation plays a significant role in modulating the transcriptional activity of p53, serving as a substantial modification (Figure 1). acetylation is a post-transla- tional modification that adds an acetyl group (-COCH3) to specific amino acid residues in proteins, predominantly ly-sine. This modification is catalyzed by enzymes known as histone acetyltransferases (HATs) or lysine acetyltransferases (KATs).
CBP/p300
CREB-binding protein (CBP/p300) in mediating p53 acetylation and its consequential effect on p53 activity has been illuminated in previous studies [44]. Extensive research has delved into unraveling the impact of acetylation on the regulation of p53's functionality. The interaction between CBP/p300 and p53 leads to the acetylation of specific lysine residues within the regulatory part of p53, resulting in a conformational change that enhances its DNA binding activity [45].
The ability of p53 to be acetylated was subsequently confirmed using acetylation- specific antibodies [46]. Sakaguchi et al. show that CBP/p300 acetylates K382 of p53 using a polyclonal antiserum specific for p53 that is phosphorylated or acetylated at specific residues [46] while ATM phosphorylates S33 and S37 in response to UV irradiation. The acetylated p53 leads to increased binding to DNA. After DNA damage from irradiation, acetylation occurs at specific lysine residues, K382 and K320 of the p53, resulting in the recruitment of coactivators such as CBP/p300 and TRRAP to the p21 promoter and increasing histone acetylation. This suggests that a cascade of acetylation, in which p53- dependent recruitment of coactivators/HATs occurs, is essential for p53 to function correctly (Figure 10)[47].
SIRT1
On the other hand, another study proposes that SIRT1 regulates the cellular response to DNA damage by modifying p53 activity through the deacetylation of lysine residues. Specifically, deacetylation of K382 by SIRT1 inhibits p53's ability to activate transcription. It promotes its degradation, leading to decreased apoptosis and increased cell survival in response to DNA damage (Figure 10)[48].
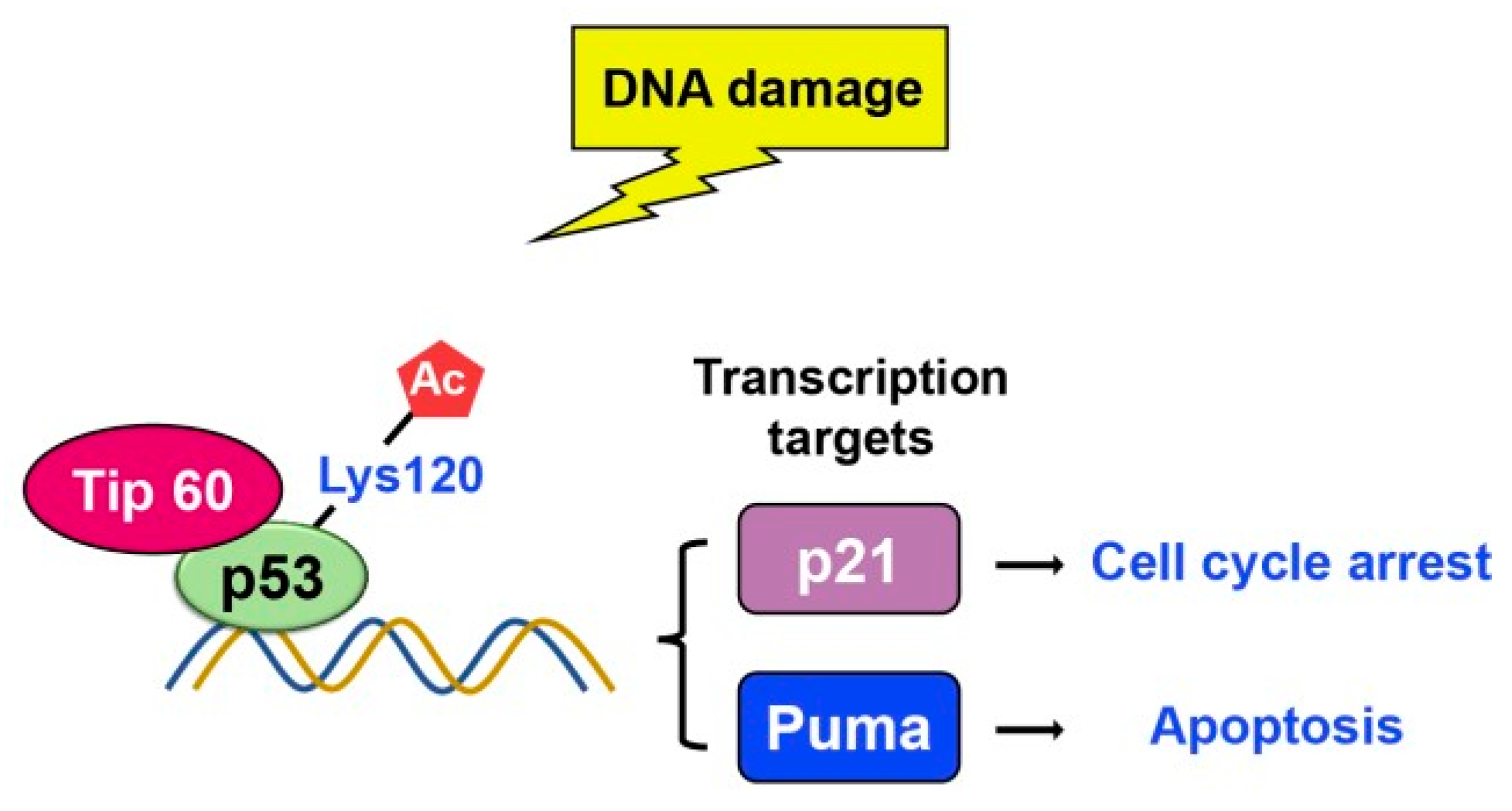
TIP60
Tip60 is another histone acetylase involved in acetylation activity, linked to DNA damage repair and apoptosis [49]. Two research groups found that Tip60 induces K120 acetylation in the DNA binding domain upon DNA damage [50,51]. Lysine 120 (K120) acetylation occurs rapidly after DNA damage and is catalyzed by the MYST histone acetyl- transferases hMOF and TIP60 (Figure 11)[51]. Mutation of K120 to arginine (K120R) debilitates K120 acetylation and blocks the transcription of pro-apoptotic target genes such as BAX and PUMA, which in turn diminishes p53-mediated apoptosis without affecting cell-cycle arrest. Also, the acetyl-K120 of p53 specifically accumulates at pro-apoptotic target genes. Additionally, studies indicate that Tip60-TRRAP complexes relocated to gamma-H2AX foci in response to DNA damage [52,53] and are crucial for the apoptotic response [54].
K120 Mutation found in human cancers further suggests that defective K120 acetylation may contribute to tumorigenesis [51]. Likewise, tumor-associated K120R mutation abrogated p53-dependent apoptosis, suggesting that p53 activity was blocked in human cancer with the same mutations [50]. As such, the relationship between Tip60-mediated p53 acetylation and the consequent induction of apoptosis is a prominent issue that war- rants further investigation.
Li et al. generated mutant mice with lysine to arginine mutations at one (K117R, K120 in humans) or three (3KR; K117R+K161R+K162R) sites in the p53. The results showed that K117R cells could still cause cell-cycle arrest and senescence but not apoptosis, while 3KR cells failed to perform any of these processes, indicating that a fine tune of acetylation modulates downstream of the DNA damage repair pathway. Consistently, while acetylation at K120 enhances apoptosis induction, acetylation at K164 promotes cell-cycle arrest suggesting that acetylation of the two lysine residues helps distinguish the cell-cycle arrest and apoptotic functions of p53 [55,56]. This highlights the importance of the interaction between histone acetyltransferases and p53 in regulating various cellular processes. Further studies are needed to understand the molecular mechanisms underlying this complex interplay fully.
Above mentioned results indicate that histone modifications are vital for regulating p53 function, with specific enzymes responsible for acetylation and lysine methylation playing a role in activating and stabilizing p53. These findings significantly impact our comprehension of p53 and its involvement in cancer, as aberrant p53 activity is frequently observed in various cancer types.
3. Perspectives
The relationship between p53 and histone modifiers is intricate and bidirectional, with both factors capable of modifying each other. Upon DNA damage or cellular stress, p53 recruits histone modifiers to specific genomic loci, influencing chromatin structure and gene expression. Conversely, histone modifiers can impact p53 activity by altering its post-translational modifications or its binding to DNA. This interplay between p53 and histone modifiers is crucial for regulating gene expression, maintaining genomic stability, facilitating chromatin remodeling, and supporting DNA repair. It ensures appropriate cellular responses to DNA damage, stress signals, and regulatory cues, although their interaction varies depending on the specific cellular environment or stimulus.
Dysregulation of histone modifiers and p53 pathways can contribute to treatment resistance in cancer cells. This involvement of histone modifiers and p53 in treatment resistance has prompted the development of multiple epigenetic anticancer drugs approved by regulatory authorities. By unraveling the molecular mechanisms underlying treatment resistance, novel combination therapies targeting both histone modifiers and p53 can be designed. These combination therapies may aim to overcome drug resistance and im- prove treatment outcomes for cancer patients [57,58].
Despite the extensive individual research conducted on histone modifiers and p53, their interplay has received relatively less attention. However, the relationship between p53 and histone modifiers plays a critical role in gene expression regulation, genomic stability, chromatin remodeling, and DNA repair. Understanding the intricate mechanisms of p53 and histone modifiers is highly relevant in cancer therapy, especially considering the frequent occurrence of p53 mutations and dysregulation. Overall, comprehending the interplay between p53 and histone modifiers is crucial for understanding treatment resistance and holds promise for developing innovative cancer therapies that can effectively target and modulate these critical factors.
Author Contributions
XYZ. literature survey, figure preparation, proofreading, and reference collection; EL. lit- erature survey and reference collection; HMK. conceptualization, literature survey, writing manuscript, review and editing, reference collection, figure editing, project administration, funding acquisition, and supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Jiangsu Kunshan Shuangchuang Grant Award (KSSC202202060), the National Natural Science Foundation of China grant (NSFC No 31972876), and the Synear and Wang-Cai grant of Duke Kunshan University to HMK.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank members of Kim laboratory for discussions and proof- reads.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writ- ing of the manuscript; or in the decision to publish the results.
References
- Berger, S.L. Histone modifications in transcriptional regulation. Curr Opin Genet Dev 2002, 12, 142–148. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Lee, H.T.; et al. The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis. J Lipid Atheroscler 2020, 9, 419–434. [Google Scholar] [CrossRef]
- Keck, F.; et al. Phosphorylation of Single Stranded RNA Virus Proteins and Potential for Novel Therapeutic Strategies. Viruses 2015, 7, 5257–5273. [Google Scholar] [CrossRef] [PubMed]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol Cell 2017, 65, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E. ; Epigenetic Therapies for Cancer. N Engl J Med 2020, 383, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Chuikov, S.; et al. Regulation of p53 activity through lysine methylation. Nature 2004, 432, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, G.S.; et al. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol Cell Biol 2007, 27, 6756–6769. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; et al. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem 2001, 276, 25309–25317. [Google Scholar] [CrossRef]
- Oh, S.T.; et al. H3K9 histone methyltransferase G9a-mediated transcriptional activation of p21. FEBS Lett 2014, 588, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Rada, M.; et al. Human EHMT2/G9a activates p53 through methylation-independent mechanism. Oncogene 2017, 36, 922–932. [Google Scholar] [CrossRef]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: who, what, and why. Mol Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Karkhanis, V.; et al. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem Sci 2011, 36, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.; et al. Arginine methylation regulates the p53 response. Nat Cell Biol 2008, 10, 1431–1439. [Google Scholar] [CrossRef]
- Li, Y.; Diehl, J.A. PRMT5-dependent p53 escape in tumorigenesis. Oncoscience 2015, 2, 700–702. [Google Scholar] [CrossRef] [PubMed]
- Custodio, G.; et al. Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J Clin Oncol 2013, 31, 2619–2626. [Google Scholar] [CrossRef]
- McBride, K.A.; et al. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat Rev Clin Oncol 2014, 11, 260–271. [Google Scholar] [CrossRef]
- Hamard, P.J.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep 2018, 24, 2643–2657. [Google Scholar] [CrossRef]
- Castellini, L.; et al. KDM4B/JMJD2B is a p53 target gene that modulates the amplitude of p53 response after DNA damage. Nucleic Acids Res 2017, 45, 3674–3692. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; et al. The histone demethylase JMJD2B is critical for p53-mediated autophagy and survival in Nutlin-treated cancer cells. J Biol Chem 2019, 294, 9186–9197. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; et al. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene 2004, 23, 5759–5769. [Google Scholar] [CrossRef]
- Yuan, J.; et al. Ezh2 competes with p53 to license lncRNA Neat1 transcription for inflammasome activation. Cell Death Differ 2022, 29, 2009–2023. [Google Scholar] [CrossRef]
- Lambert, P.F.; et al. Phosphorylation of p53 serine 15 increases interaction with CBP. J Biol Chem 1998, 273, 33048–33053. [Google Scholar] [CrossRef]
- Chehab, N.H.; et al. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci U S A 1999, 96, 13777–13782. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Taya, Y.; Prives, C. DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J 1999, 18, 1815–1823. [Google Scholar] [CrossRef]
- Unger, T.; et al. Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J 1999, 18, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Lozano, G. Functional activation of p53 via phosphorylation following DNA damage by UV but not gamma radiation. Proc Natl Acad Sci U S A 1998, 95, 2834–2837. [Google Scholar] [CrossRef]
- Lu, H.; et al. Ultraviolet radiation, but not gamma radiation or etoposide-induced DNA damage, results in the phosphorylation of the murine p53 protein at serine-389. Proc Natl Acad Sci U S A 1998, 95, 6399–6402. [Google Scholar] [CrossRef]
- Hupp, T.R.; et al. Regulation of the specific DNA binding function of p53. Cell 1992, 71, 875–886. [Google Scholar] [CrossRef]
- Sakaguchi, K.; et al. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 1997, 36, 10117–10124. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, N.; Premkumar Reddy, E. Signaling by dual specificity kinases. Oncogene 1998, 17, 1447–55. [Google Scholar] [CrossRef] [PubMed]
- She, Q.B.; Chen, N.; Dong, Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J Biol Chem 2000, 275, 20444–20449. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.L.; Meek, D.W. Phosphorylation of serine 392 in p53 is a common and integral event during p53 induction by diverse stimuli. Cell Signal 2010, 22, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; et al. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Sattarifard, H.; et al. Mitogen- and stress-activated protein kinase (MSK1/2) regulated gene expression in normal and disease states. Biochem Cell Biol 2023. [Google Scholar] [CrossRef] [PubMed]
- Crosio, C.; et al. Chromatin remodeling and neuronal response: multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J Cell Sci 2003, 116 Pt 24, 4905–4914. [Google Scholar] [CrossRef]
- Clayton, A.L.; et al. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J 2000, 19, 3714–3726. [Google Scholar] [CrossRef]
- Ahn, J.; et al. MSK1 functions as a transcriptional coactivator of p53 in the regulation of p21 gene expression. Exp Mol Med 2018, 50, 1–12. [Google Scholar] [CrossRef]
- Xing, J.; Ginty, D.D.; Greenberg, M.E. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor- regulated CREB kinase. Science 1996, 273, 959–963. [Google Scholar] [CrossRef]
- Cho, Y.Y.; et al. The p53 protein is a novel substrate of ribosomal S6 kinase 2 and a critical intermediary for ribosomal S6 kinase 2 and histone H3 interaction. Cancer Res 2005, 65, 3596–3603. [Google Scholar] [CrossRef]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Lill, N.L.; et al. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Barlev, N.A.; et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Ikura, T.; et al. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000, 102, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; et al. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 2006, 24, 827–839. [Google Scholar] [CrossRef]
- Sykes, S.M.; et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Murr, R.; et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol 2006, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Robert, F.; et al. The transcriptional histone acetyltransferase cofactor TRRAP associates with the MRN repair complex and plays a role in DNA double-strand break repair. Mol Cell Biol 2006, 26, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Tyteca, S.; et al. Tip60 and p400 are both required for UV-induced apoptosis but play antagonistic roles in cell cycle progression. EMBO J 2006, 25, 1680–1689. [Google Scholar] [CrossRef]
- Li, T.; et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; et al. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef]
- Gao, Y.; et al. Dual inhibitors of histone deacetylases and other cancer-related targets: A pharmacological perspective. Biochem Pharmacol 2020, 182, 114224. [Google Scholar] [CrossRef]
- Seo, Y.H. Dual Inhibitors Against Topoisomerases and Histone Deacetylases. J Cancer Prev 2015, 20, 85–91. [Google Scholar] [CrossRef]
Figure 1.
Histone modifications. (A) Methylation and demethylation are catalyzed by histone methyltransferase and histone demethylase, respectively. Euchromatin is characterized by specific molecular marks that indicate active gene expression, including his- tone acetylation, H3K4 trimethylation (H3K4me3), and H3K36 trimethylation (H3K36me3). In contrast, heterochromatin is marked by different modifications associated with gene repression and chromatin compaction. These marks include H3K9 trimethylation (H3K9me3) and H3K27 trimethylation (H3K27me3). Acetylation occurs at lysine residues catalyzed by histone acetyltransferase, while deacetylation is catalyzed by histone acetyltransferase. Phosphorylation: Kinases and phosphatases are enzymes involved in the addition and removal of phosphate groups, respectively, on proteins.
Figure 1.
Histone modifications. (A) Methylation and demethylation are catalyzed by histone methyltransferase and histone demethylase, respectively. Euchromatin is characterized by specific molecular marks that indicate active gene expression, including his- tone acetylation, H3K4 trimethylation (H3K4me3), and H3K36 trimethylation (H3K36me3). In contrast, heterochromatin is marked by different modifications associated with gene repression and chromatin compaction. These marks include H3K9 trimethylation (H3K9me3) and H3K27 trimethylation (H3K27me3). Acetylation occurs at lysine residues catalyzed by histone acetyltransferase, while deacetylation is catalyzed by histone acetyltransferase. Phosphorylation: Kinases and phosphatases are enzymes involved in the addition and removal of phosphate groups, respectively, on proteins.
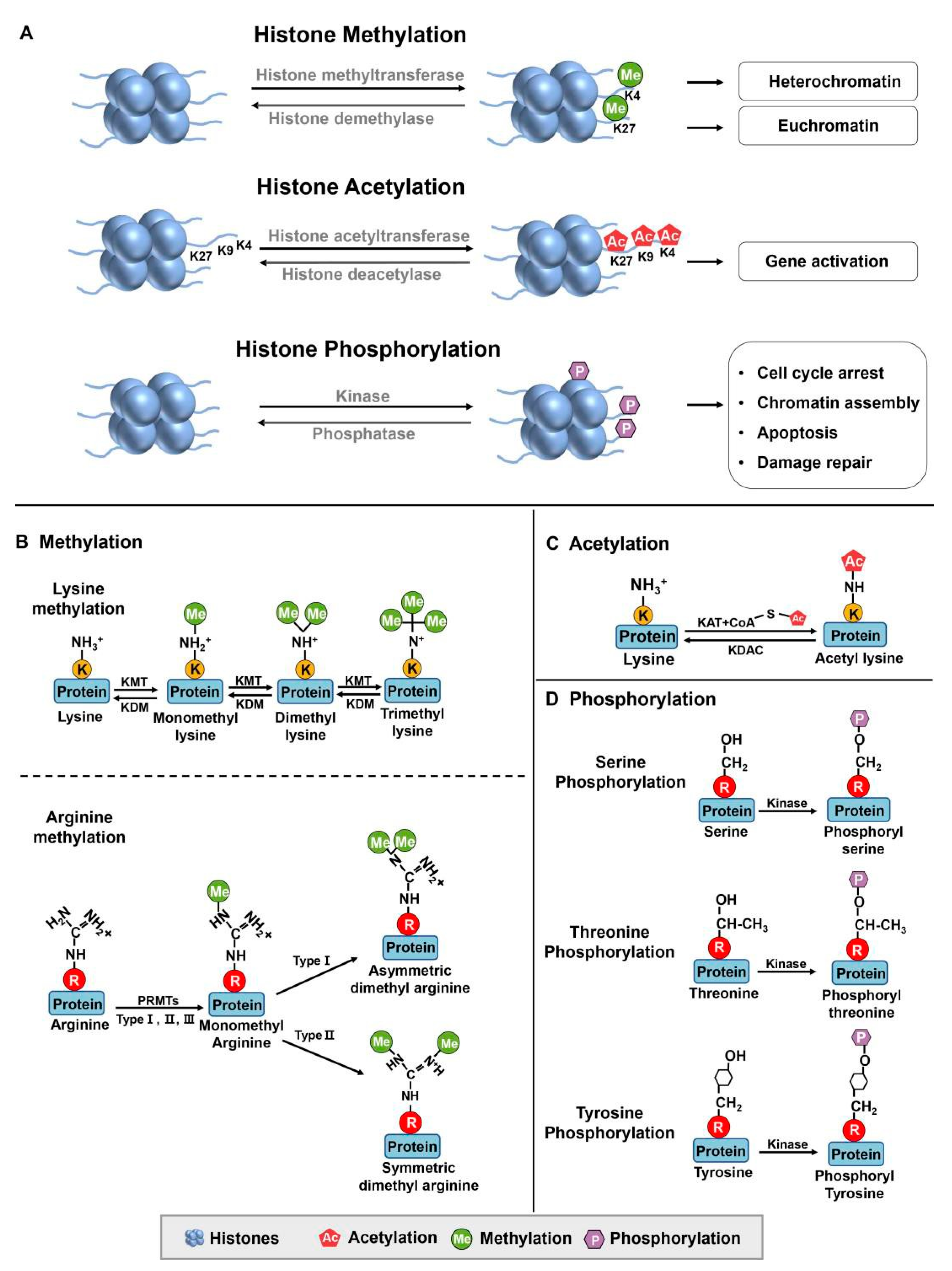
Figure 2.
Set7/9 methylation activates p53, which leads to transcriptional activation of p21 gene expression and p53-mediated apoptosis and cell cycle arrest.
Figure 2.
Set7/9 methylation activates p53, which leads to transcriptional activation of p21 gene expression and p53-mediated apoptosis and cell cycle arrest.
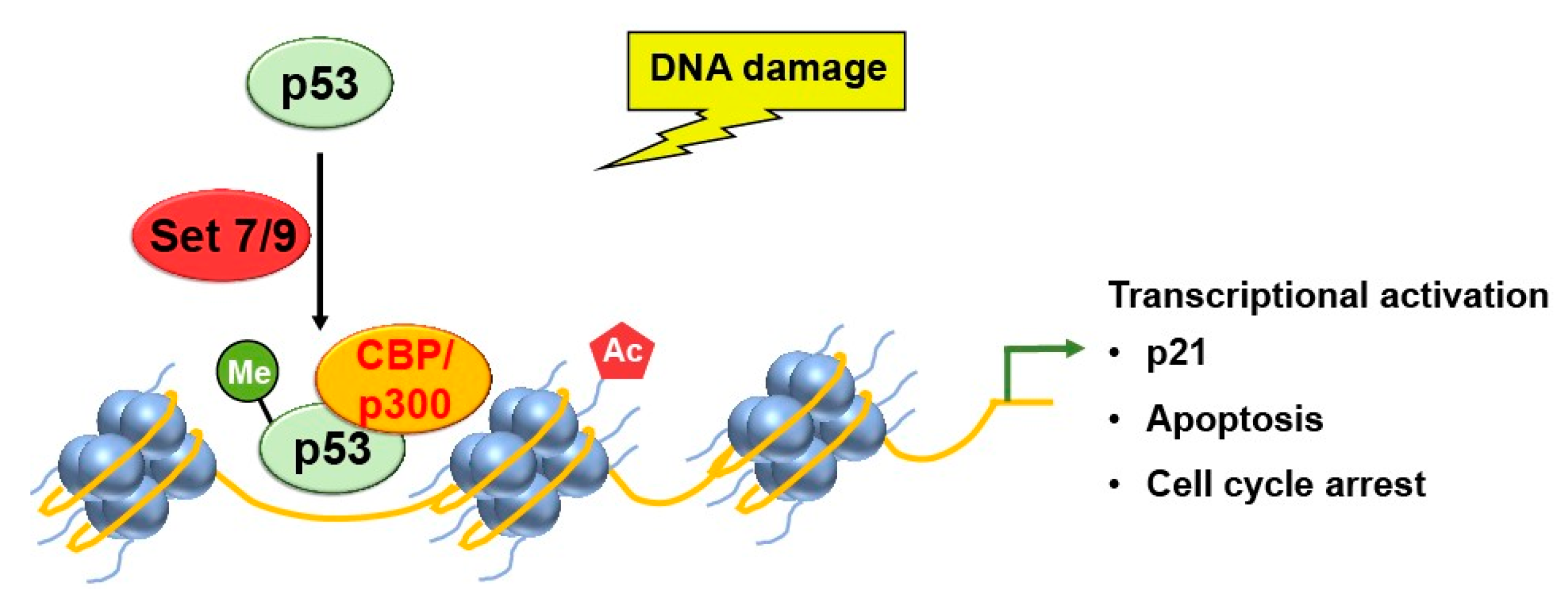
Figure 3.
G9a has the potential to modulate p53 transcriptional activity in a differential manner. (A) In humans, G9a functions as a coactivator for p53 by recruiting histone acetyltransferases (HATs) such as CBP and PCAF (B) However, mouse G9a exerts a repressive effect on p53 transcriptional activity.
Figure 3.
G9a has the potential to modulate p53 transcriptional activity in a differential manner. (A) In humans, G9a functions as a coactivator for p53 by recruiting histone acetyltransferases (HATs) such as CBP and PCAF (B) However, mouse G9a exerts a repressive effect on p53 transcriptional activity.
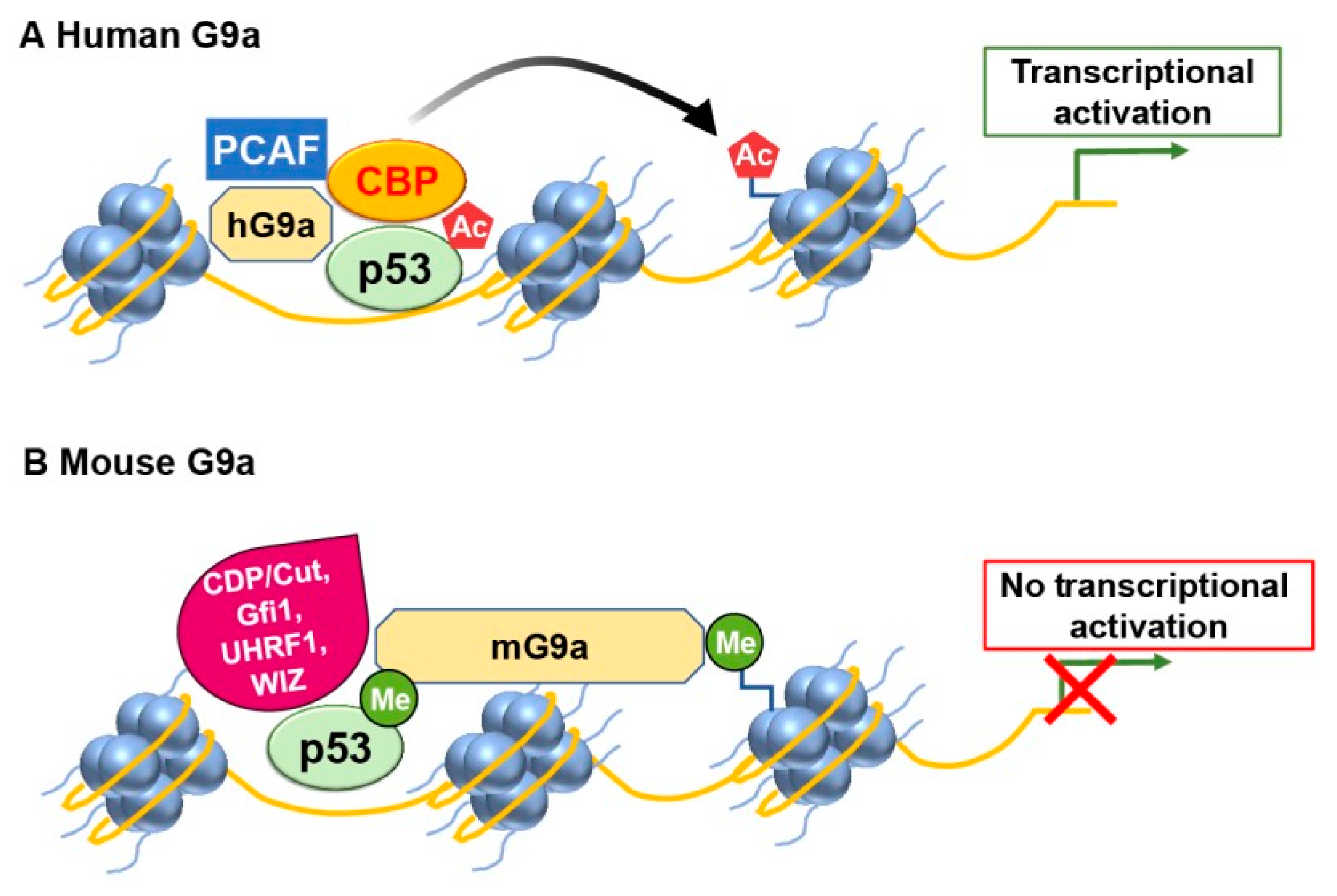
Figure 4.
PRMT5 methylates H4R3, indirectly influencing p53 activity by affecting the transcriptional regulation of p53 target genes. (A) Arginine methylation-induced bypassing of p53 leads to the evasion of apoptosis and facilitates tumor growth, whereas depletion of PRMT5 induces p53- mediated apoptosis. (B) PRMT5 controls over the alternative splicing of key histone- modifying enzymes like TIP60 and KMT5C, thereby influencing chromatin structure and influencing DNA repair pathway.
Figure 4.
PRMT5 methylates H4R3, indirectly influencing p53 activity by affecting the transcriptional regulation of p53 target genes. (A) Arginine methylation-induced bypassing of p53 leads to the evasion of apoptosis and facilitates tumor growth, whereas depletion of PRMT5 induces p53- mediated apoptosis. (B) PRMT5 controls over the alternative splicing of key histone- modifying enzymes like TIP60 and KMT5C, thereby influencing chromatin structure and influencing DNA repair pathway.
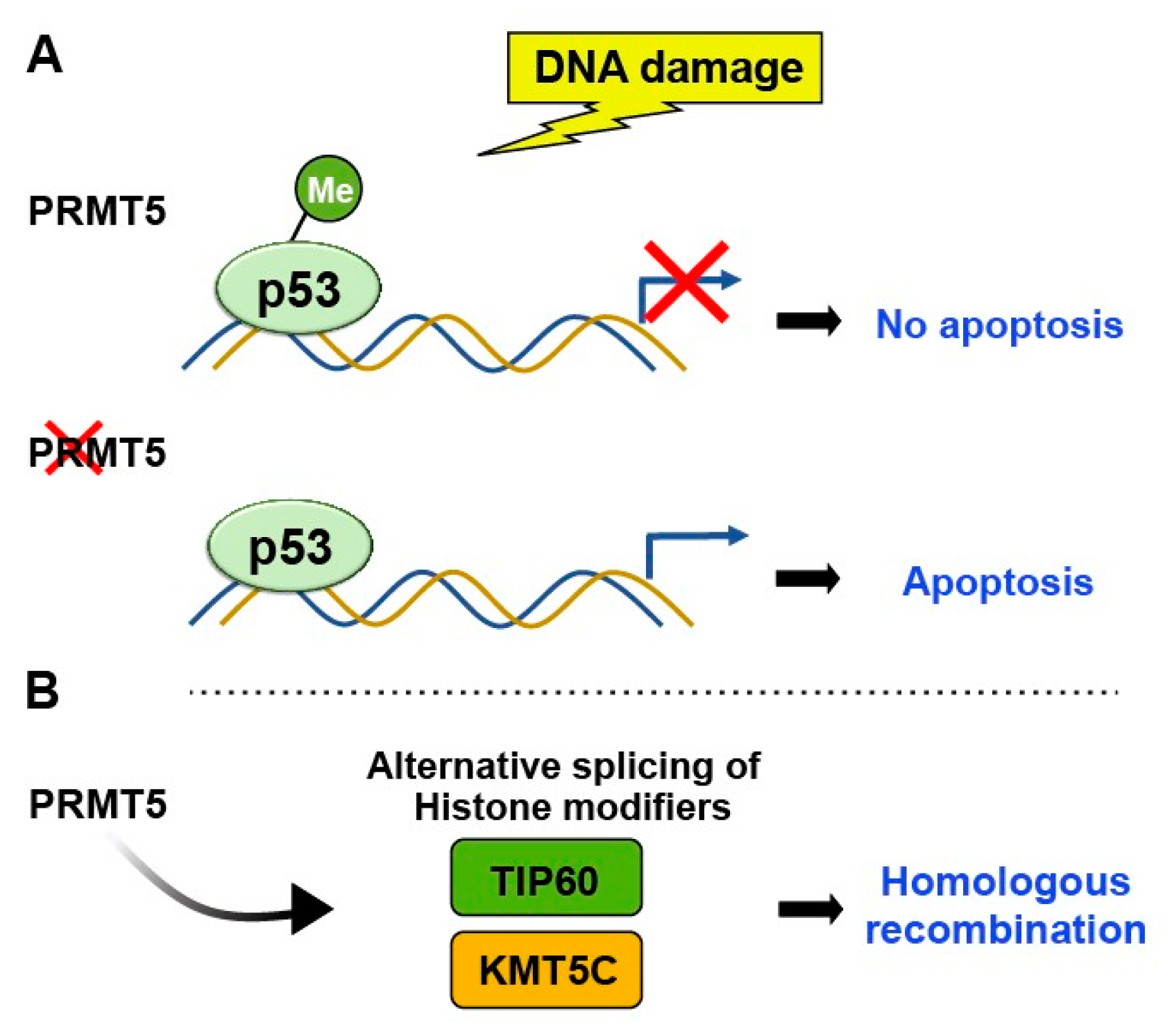
Figure 5.
In response to DNA damage, p53 binds to a p53-consensus motif in the JMJD2B promoter, hence p53 controls the expression of the JMJD2B gene. The induction of JMJD2B in turn suppresses the transcription of important p53 targets, such as p21, PIG3, and PUMA.
Figure 5.
In response to DNA damage, p53 binds to a p53-consensus motif in the JMJD2B promoter, hence p53 controls the expression of the JMJD2B gene. The induction of JMJD2B in turn suppresses the transcription of important p53 targets, such as p21, PIG3, and PUMA.
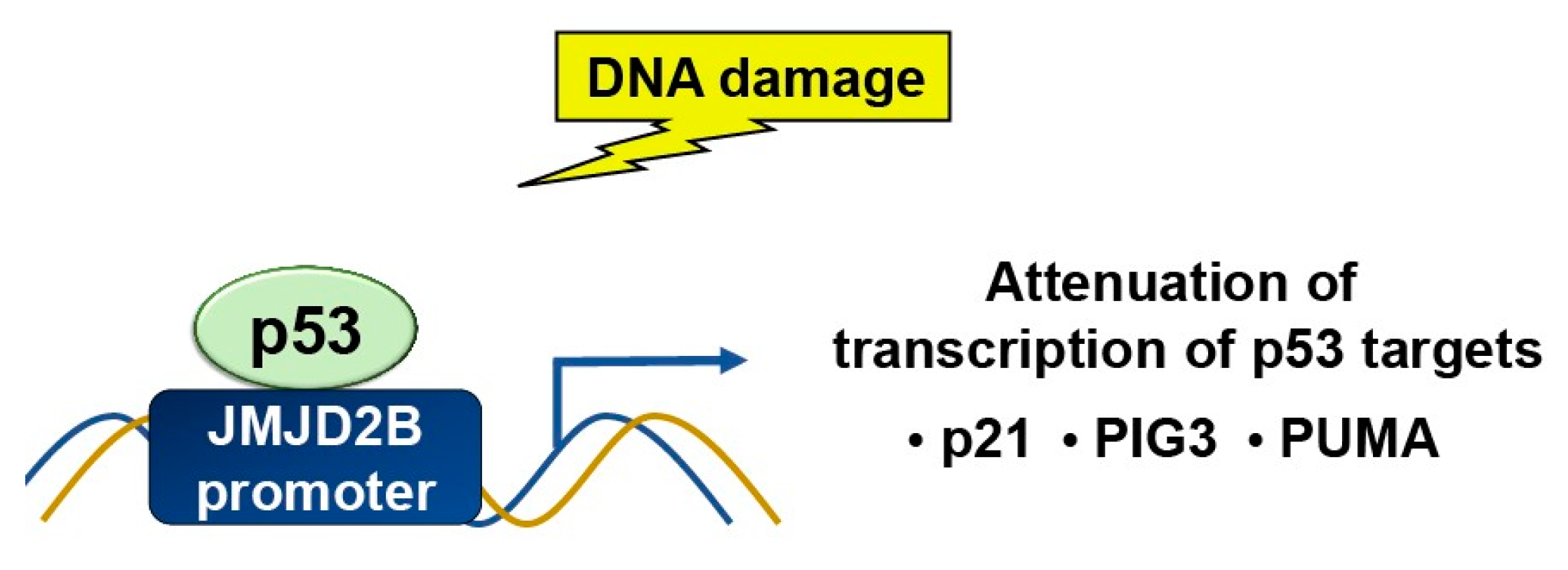
Figure 6.
The competition between Ezh2 and p53 regulates inflammasome activation. Upon exposure to inflammasome inducers, Ezh2 inhibits the binding of p53 to the promoter region of the lncRNA Neat1 gene. As a result, the recruitment of SIRT1 by p53 is also disrupted, preventing its binding to the DNA. This process leads to the enrichment of H3K27ac. Subsequently, the facilitated transcription of Neat1 by p65 promotes the activation of the inflammasome. The image is modified from [25].
Figure 6.
The competition between Ezh2 and p53 regulates inflammasome activation. Upon exposure to inflammasome inducers, Ezh2 inhibits the binding of p53 to the promoter region of the lncRNA Neat1 gene. As a result, the recruitment of SIRT1 by p53 is also disrupted, preventing its binding to the DNA. This process leads to the enrichment of H3K27ac. Subsequently, the facilitated transcription of Neat1 by p65 promotes the activation of the inflammasome. The image is modified from [25].
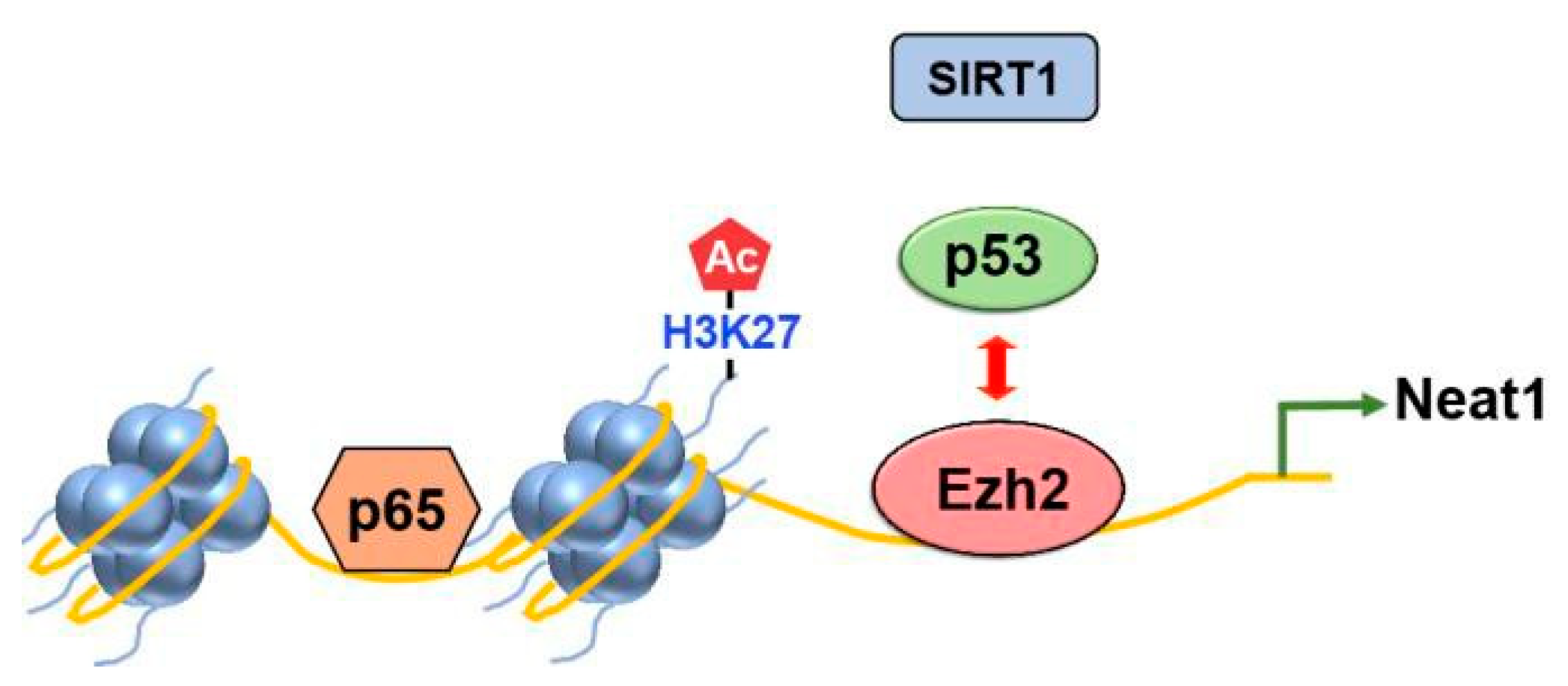
Figure 7.
Phosphorylation of p53 at multiple sites in response to DNA damage regulates its transcriptional activity, DNA binding affinity, and protection against degradation. Phosphorylation at Ser15 and Ser20 enhances transcriptional activity and prevents ubiquitin-mediated degradation respectively. Phosphorylation at Ser-392 enhances sequence-specific DNA binding and stabilizes tetramer formation following UV irradiation.
Figure 7.
Phosphorylation of p53 at multiple sites in response to DNA damage regulates its transcriptional activity, DNA binding affinity, and protection against degradation. Phosphorylation at Ser15 and Ser20 enhances transcriptional activity and prevents ubiquitin-mediated degradation respectively. Phosphorylation at Ser-392 enhances sequence-specific DNA binding and stabilizes tetramer formation following UV irradiation.
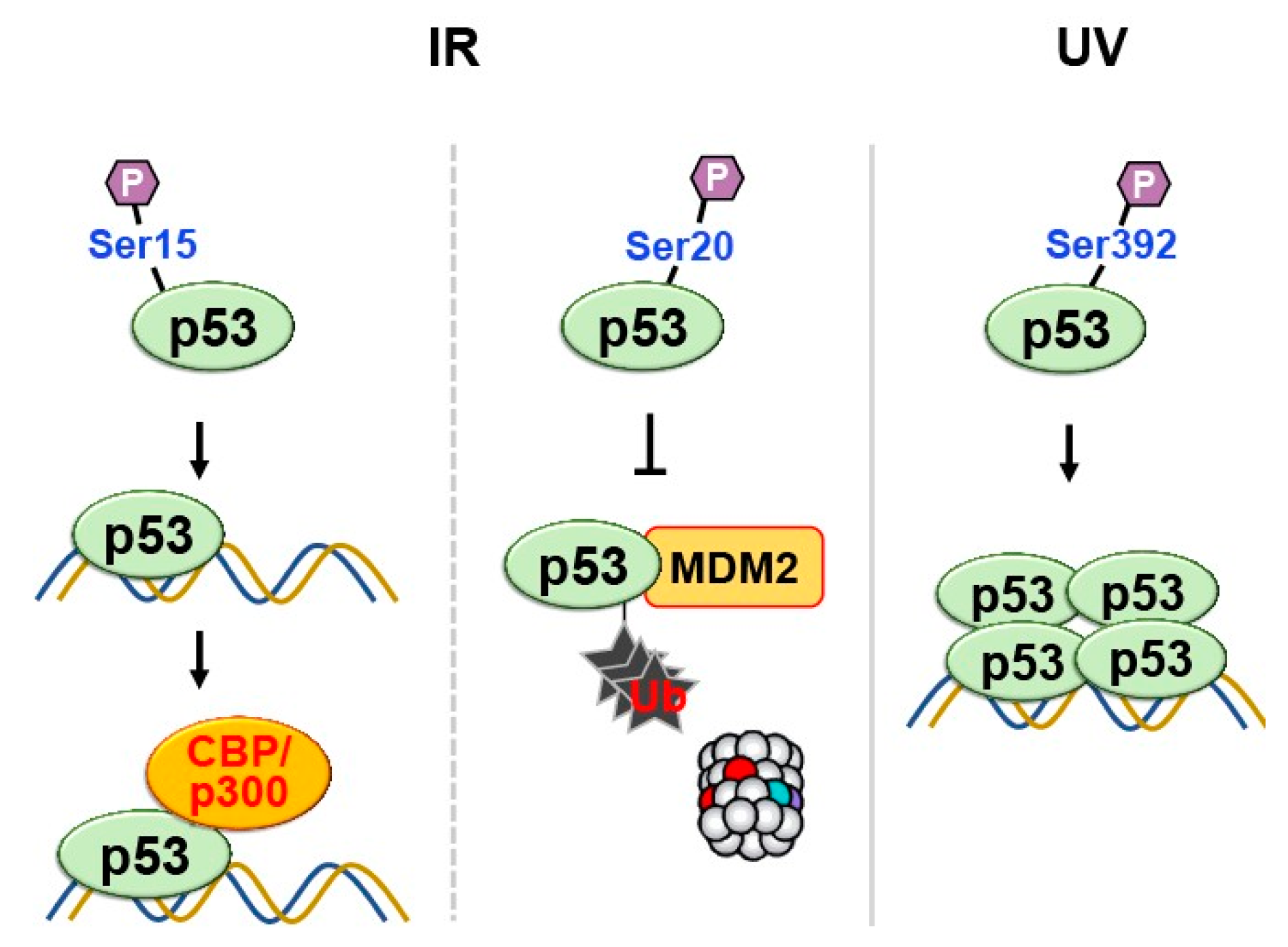
Figure 8.
p38 (A) p38 phosphorylates p53 at Ser15 and Ser392, activating p53 and increasing its stability, transcriptional activity, and induction of target genes involved in cell cycle arrest, DNA repair, and apoptosis. Ser15 phosphorylation also stabilizes p53 by reducing its interaction with MDM2. (B) Upon activation by p38, MSK1/2 phosphorylates histone H3 at Ser10 and Ser28, modulating chromatin structure and gene expression. (C) When activated by the p38 MAPK pathway, MSK1 interacts with p53 and is recruited to the p21 promoter, where it phosphorylates histone H3 in a p53- dependent manner.
Figure 8.
p38 (A) p38 phosphorylates p53 at Ser15 and Ser392, activating p53 and increasing its stability, transcriptional activity, and induction of target genes involved in cell cycle arrest, DNA repair, and apoptosis. Ser15 phosphorylation also stabilizes p53 by reducing its interaction with MDM2. (B) Upon activation by p38, MSK1/2 phosphorylates histone H3 at Ser10 and Ser28, modulating chromatin structure and gene expression. (C) When activated by the p38 MAPK pathway, MSK1 interacts with p53 and is recruited to the p21 promoter, where it phosphorylates histone H3 in a p53- dependent manner.

Figure 9.
Upon stimulation with UV or EGF, activated RSK2 phosphorylates p53 at Ser15, and the RSK2/p53 complex trans- locates to the nucleus, where RSK2 phosphorylates histone H3 at Ser10, contributing to transcriptional regulation, chromatin remodeling, and cell cycle control.
Figure 9.
Upon stimulation with UV or EGF, activated RSK2 phosphorylates p53 at Ser15, and the RSK2/p53 complex trans- locates to the nucleus, where RSK2 phosphorylates histone H3 at Ser10, contributing to transcriptional regulation, chromatin remodeling, and cell cycle control.
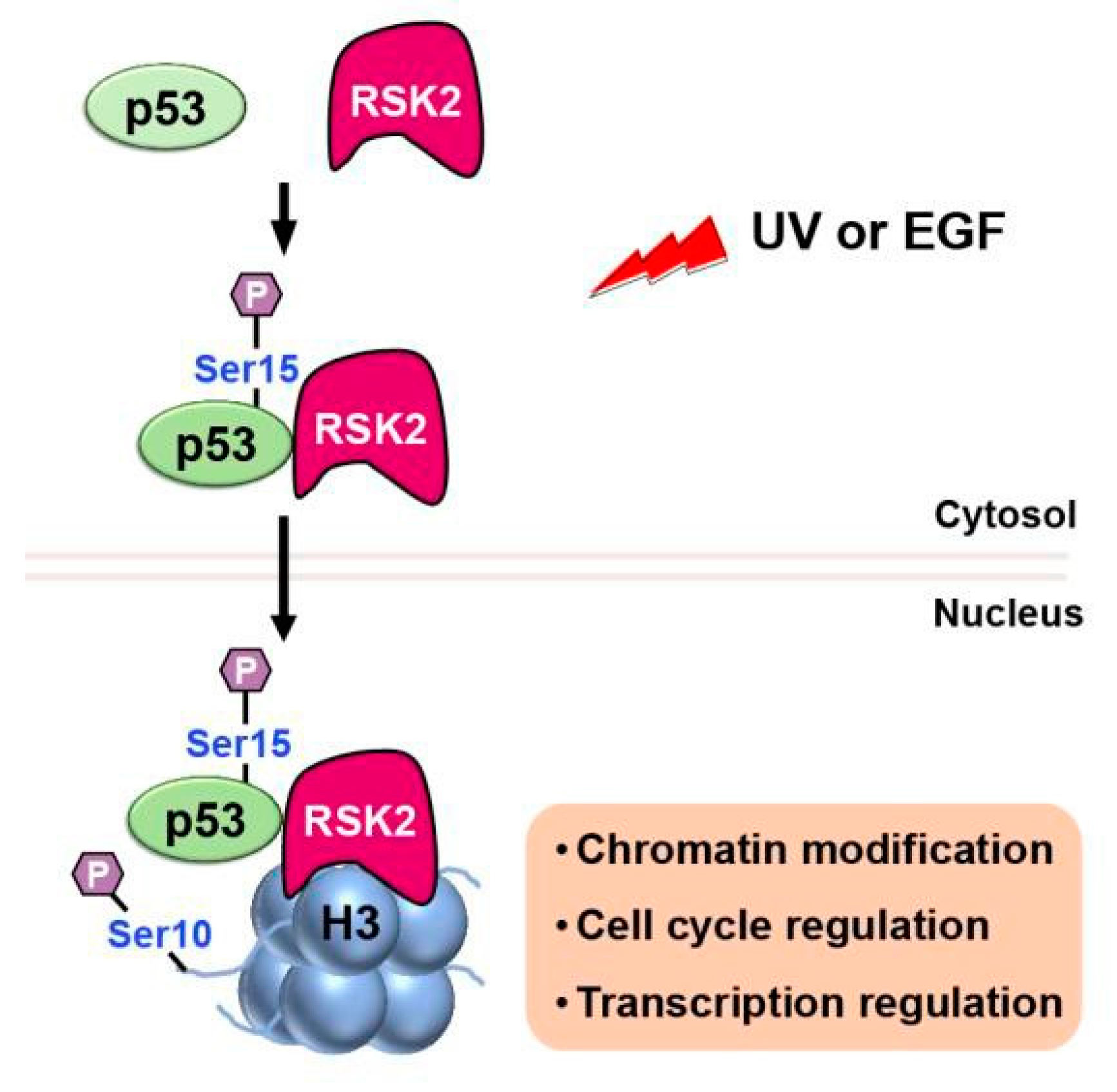
Figure 10.
Following DNA damage caused by irradiation, lysine residues of p53 undergo acetylation. This acetylation leads to the recruitment of coactivators like CBP/p300 and TRRAP to the p21 promoter, thereby increasing histone acetylation. Meanwhile, SIRT1-mediated K382 deacetylation inhibits p53's transcriptional activation, promoting its degradation.
Figure 10.
Following DNA damage caused by irradiation, lysine residues of p53 undergo acetylation. This acetylation leads to the recruitment of coactivators like CBP/p300 and TRRAP to the p21 promoter, thereby increasing histone acetylation. Meanwhile, SIRT1-mediated K382 deacetylation inhibits p53's transcriptional activation, promoting its degradation.
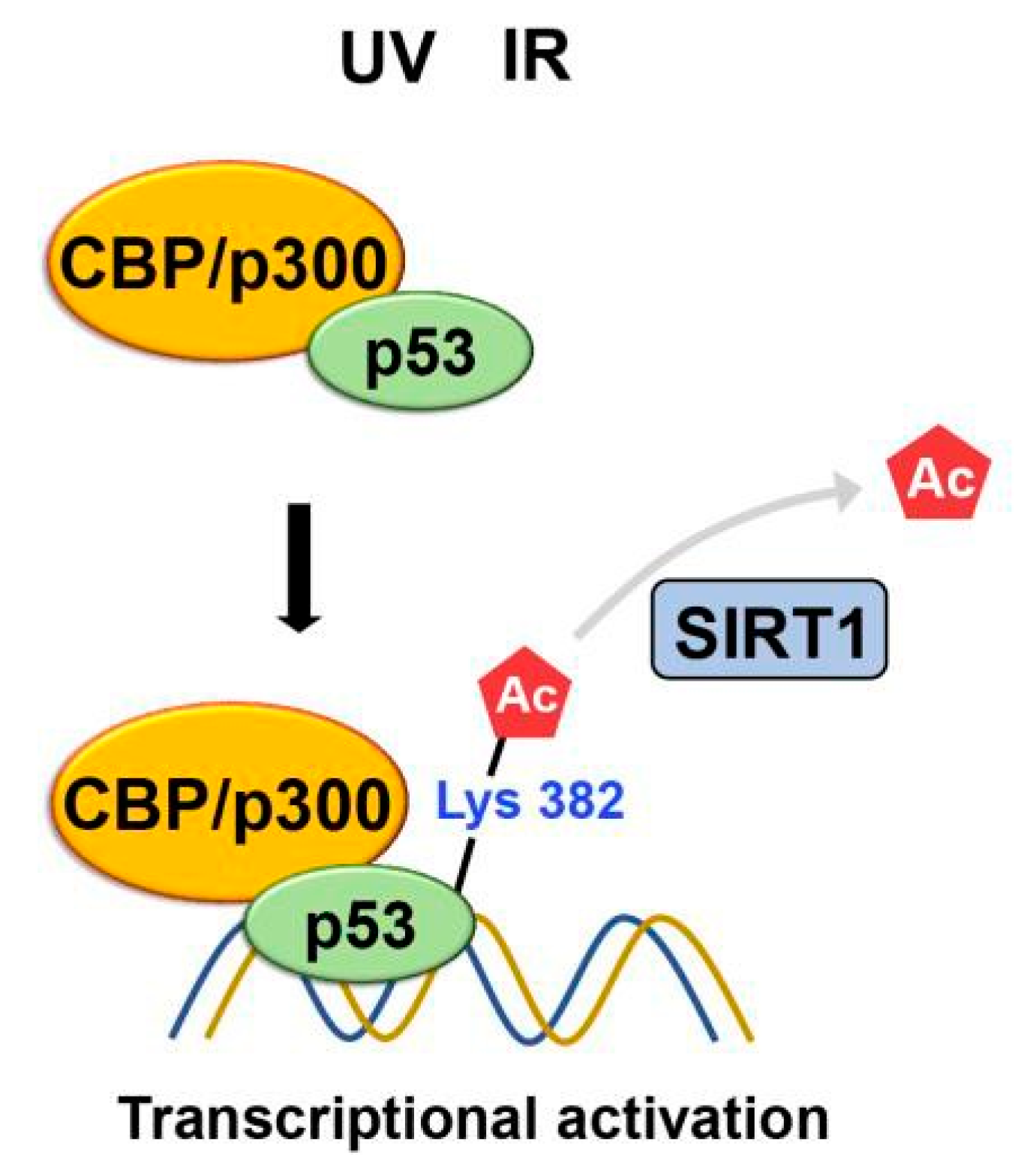
Figure 11.
Upon DNA damage, Tip60 interacts with p53 and binds to its target gene promoters, leading to p21 activation and growth arrest. Additionally, Tip60 induces K120 acetylation, resulting in activation of PUMA expression.
Figure 11.
Upon DNA damage, Tip60 interacts with p53 and binds to its target gene promoters, leading to p21 activation and growth arrest. Additionally, Tip60 induces K120 acetylation, resulting in activation of PUMA expression.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



