Submitted:
28 May 2023
Posted:
31 May 2023
You are already at the latest version
Abstract
Diabetes mellitus is one of the most common metabolic diseases worldwide, and its long-term complications include neuropathy, referring both to the peripheral and to the central nervous system. Detrimental effects of dysglycemia, especially hyperglycemia, on the structure and function of blood-brain barrier (BBB) seem to be a significant backgrounds of diabetic neuropathy pertaining to the central nervous system (CNS). Effects of hyperglycemia, including excessive glucose influx to insulin independent cells, may induce oxidative stress and secondary innate immunity dependent inflammatory response which can damage cells within the CNS, thus promoting neurodegeneration and dementia. Advanced glycation end products (AGE) may exert similar, pro-inflammatory effects through activating receptors for advanced glycation end products (RAGE), as well as some pattern-recognition receptors (PRR). Moreover, long-term hyperglycemia can promote brain insulin resistance, which may in turn promote Aβ aggregate accumulation and tau hyperphosphorylation. This review is focused on a detailed analysis of the effects mentioned above towards the CNS, with special regard to mechanisms taking part in the pathogenesis of central long-term complications of diabetes mellitus initiated by the loss of BBB integrity.
Keywords:
diabetes mellitus
; blood-brain barrier disruption
; dysglycemia
; hyperglycemia
; insulin resistance
; neurodegenerative disease
; diabetic complications
; dementia
; diabetic encephalopathy
1. Diabetes mellitus - a short resume
Diabetes, also known as diabetes mellitus (DM), is a group of the most common endocrine/metabolic diseases worldwide, characterized by sustained high blood glucose levels [1]. It results from deficiency of insulin actions, either because of insulin deficiency or because of insulin resistance. [2] It impairs mainly glucose metabolism, but it results in a disease affecting almost each tissue in the organism. Complications of diabetes mellitus may include myocardial infarction, blindness, chronic renal failure, neuropathy, and loss of cognitive functions [3]. Impaired transport of glucose to insulin-dependent cells results in glucose excess in extracellular fluids and in insulin-independent cells, which is accompanied by severe deficiency of glucose within insulin-dependent cells. Thus, some manifestations of diabetes mellitus are related to glucose depletion inside insulin-dependent cells, others are related to glucose excess toxicity in relation to other cells and tissues, yet others - to oxidative stress or using alternative substances as energy sources. Many long-term complications of diabetes mellitus depend on disorders of blood vessels, which can be divided into those referring to large vessels (macroangiopathies) and those referring to small vessels (microangiopathies). Diabetic macroangiopathies increase the risk of atherosclerosis and its related cardiovascular diseases, while diabetic microangiopathies can lead to chronic renal failure, neuropathy, and blindness, among others. While diabetic retinopathy is regarded as the most common cause of blindness in western countries, effects of diabetes mellitus towards blood-brain barrier (BBB) are much less studied. It is quite astonishing, since retina is a specialized part of the central nervous system (CNS), and thus blood-retina barrier is regarded as a specialized region of blood-brain barrier [4]. Optic nerve is a part of CNS, and thus its capillaries carry out some barrier functions. Complications of DM in relation to BBB share many common features with those referring to diabetic retinopathy.
There are two main kinds of diabetes mellitus - type 1 DM and type 2 DM. Type 1 DM is usually an acute onset disease of young people, underlied by autoimmune destruction of beta cells of pancreatic islets (islets of Langerhans) and subsequent deficiency of insulin. Type 2 DM is usually a chronic disease with insidious onset, underlied by tissue insulin resistance and resulting deficiency od insulin actions despite quite decent concentration of insulin in the plasma. Type 2 DM is strongly associated with obesity and accounts for 90% of DM cases worldwide. The incidence of type 2 DM has been increasing for about 40 years and nowadays affects about 9% of people worldwide [5].
2. Blood-brain barrier
Blood-brain barrier has been described in detail, both morphologically and physiologically [6]. At the cellular level BBB is formed by cerebral capillary endothelial cells and the closely apposed astrocyte end-feet processes [7]. Endothelium within BBB shows a specific pattern of expression of transmembrane transport systems, regulating the transport of various substances inside and outside the cerebrospinal fluid [8,9]. BBB endothelial cells are connected to one another with tight junctions, are devoid of fenestrations, and hardly perform pinocytic transport, which promotes intracerebral environment homeostasis and is a unique trait of endothelium within cerebral microcirculation. Tight junctions connecting the adjacent endothelial cells comprise a diffusion barrier that prevents most plasma components, such as electrolytes, ionized xenobiotics, and other water-soluble substances from crossing BBB through paracellular route. On the other hand, the co-existing specialized efflux transport mechanisms, dependent on such proteins as P-glycoprotein (P-gp), breast cancer resistance protein (BRCP), and multidrug resistance protein (MRP-4), regulate trafficking of amphipathic and hydrophobic substances across BBB to protect the brain from the exposal for potentially toxic substances [10]. section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
2.1. Glucose transport across BBB
Glucose transport across BBB may depend on two independent classes of glucose transporters: facilitated diffusion transporter proteins (GLUTs) and secondary active transporter proteins, also called sodium-glucose cotransporters (SGLTs) [11,12]. GLUTs include 14 proteins, the first identified of which, and the one mainly accounting for glucose transport across BBB is GLUT-1 [13,14,15]. GLUT proteins are saturable glucose carriers, transporting glucose down its concentration gradient [16,17]. While GLUT-1 molecules present in astrocytes and endothelial cells transport glucose across BBB, GLUT-3 is thought to be the most important glucose carrier transporting glucose from the cerebrospinal fluid to the neurons, although not the only one [14]. In mature and completely differentiated endothelial cells, GLUT-1 transporters are not homogenously distributed, and more of them can be found at luminal part of their cell membrane (i.e. adjacent to the lumen of brain capillaries) than at the opposite (abluminal) part [18,19].
Increased glucose transport across BBB is correlated with increased luminal expression of GLUT-1 molecules. On the other hand, increased abluminal expression of GLUT-1 molecules is observed when there is a downregulation of GLUT-1; in such situation, luminal expression of GLUT-1 molecules is reduced, while its abluminal expression rises [20], suggesting that altered distribution of GLUT-1 molecules can modulate glucose transport from the plasma to the cerebrospinal fluid.
GLUT-4, an insulin dependent glucose transporter, together with insulin receptors located within endothelial cells of brain capillaries, can also play a significant role in glucose transporting to the CNS. In normal conditions, insulin is hardly synthesized in the brain, but insulin transport across BBB has been found to increase in the course of DM [21,22,23,24]. Insulin can get to the brain through the cerebrospinal fluid, reaching it within locations devoid of BBB, e.g. in circumventricular organ, or crossing BBB via insulin receptors acting also as insulin transporters [25,26]. A schematic diagram of BBB with participation of individual cells in glucose transport is presented in Figure 1.
Effects of insulin action towards the brain are interesting in the context of discovery suggesting that DM can promote Alzheimer’s disease (AD) due to mechanisms including attenuation of intracellular insulin receptor dependent signaling within BBB. Although complete elucidation of those mechanisms may require some additional studies, insulin may play a role in regulating tau protein phosphorylation in the course of tauopathies, through glycogen synthase kinase-3 (GSK-3) activation and stimulating the expression of soluble receptors for advanced glycation end products (s-RAGE) [29,30]. High expression of these receptors is correlated with an increased incidence of ischemic heart disease as a complication of DM, as well as with higher mortality due to all complications of DM, both type 1 and type 2 [31,32]. Higher levels of these receptors can also reflect the expression of tissue receptors for advanced glycation end products (RAGE) in the course of DM [33]. Advanced glycation end products (AGE)-RAGE system activation seems to play a crucial role in the pathogenesis of DM-related angiopathy and thrombosis [34,35]. Disruption of BBB integrity and function may have a profound effect on the CNS, thus being a prodromal manifestation of serious neurological disorders. DM-related disruption of BBB integrity and function can have an essential effect on the CNS.
2.1.1. Hyperglycemia and its effect on glucose transport across BBB
On the basis of general physiology, it is thought that when a substance occurs in excess, expression of receptors for the substance falls, undergoing an effect known as downregulation. These effects are aimed at homeostasis maintenance in terms of substrates delivery to the cells, and adjusting it to the cell demand. Therefore, it is suggested that biological response to hyperglycemia may include downregulation of proteins accounting for glucose transport from plasma to peripheral tissues. Downregulation of glucose transporters in response to hyperglycemia has been confirmed in some research studies [36,37,38] but not in others [39,40,41,42,43].
Studies on the expression of glucose transporters in rats with streptozotocin-induced DM show that chronic hyperglycemia reduces both GLUT-1 and GLUT-3 expression at the level of transcription and translation; downregulation of these transporters in vivo is observed regardless of the method of DM induction [37]. Local brain glucose utilization rises in the course of chronic, but not acute, hyperglycemia in Sprague-Dawley rats. It is accompanied by moderate, yet clinically significant, fall of GLUT-1 expression in cerebral capillaries, but not of GLUT-3 expression [44].
2.1.2. Hypoglycemia and its effect on glucose transport across BBB
Hypoglycemia can be a major threat in the course of DM, since it can result in a serious disruption of CNS functions. Frequent attempts of reducing the plasma glucose concentration in patients with DM may lead to hypoglycemia and hypoglycemic damage of the brain cells, which can induce hypoglycemia-associated autonomic failure (HAAF). General concept of HAAF background assumes that adrenalin release from adrenal medulla, which normally would inhibit insulin secretion and boost glucagon secretion, is blunted in the course of DM. This can be accompanied by being unaware of hypoglycemia, which can lead to a vicious circle of recurrent hypoglycemia and further impairment of hypoglycemia-preventing mechanisms. Although the clinical significance of HAAF is widely known, its detailed mechanisms and mediators remain grossly unknown [45,46]. Most DM patients have difficulties with maintenance of normal plasma glucose concentration because of HAAF and hypoglycemia unawareness. Diabetic rats exposal to acute hypoglycemia does not lead to increased glucose concentration in extracellular fluid of the inferior colliculus [47] thus showing that short-lasting recurrent hypoglycemia affect neither glucose transport to the brain nor brain glucose metabolism rate. However, increased expression of GLUT-1 within BBB at the level of transcription and translation in response to chronic hypoglycemia indicates the existence of a compensatory mechanism, increasing glucose transport across BBB in case of chronic hypoglycemia [48].
Increased glucose transport across BBB in response to chronic hypoglycemia may result both from increased expression of GLUT-1 and from GLUT-1 molecules redistribution within BBB [48]. Brain glucose uptake in vivo in response to chronic hypoglycemia increases regardless of hypoglycemia-inducing method [49]. Similar observations (glucose concentration in the brain increased by 48%) can be made in rats exposed to hypoglycemia for 12-14 days [49]. Furthermore, hypoglycemia results in the increase of brain glucose uptake by 25 – 45%, as well as increased GLUT-1 expression by 23% and GLUT-1 molecules redistribution from abluminal to luminal part of endothelial cells within BBB [39].
Increased GLUT-1 expression, combined with redistribution of its molecules to luminal pole of endothelial cells can additionally aggravate hypoglycemia through stimulating glucose transport across BBB. In addition, acute or mild hypoglycemia stimulates the expression of GLUT-1, GLUT-4, angiotensinogen and mitogen activated protein kinase (MAPK) phosphatase-1 [50]. Increased expression of angiotensinogen may promote vasodilation with locally increased blood flow. This may in turn locally elevate glucose concentration, resulting in overestimation of blood glucose level by the hypothalamus and subsequently inhibit hypothalamic mechanisms aimed at counteracting hypoglycemia.
2.2. Alterations in amino acid transport across BBB in the course of DM
Choline is the precursor of acetylcholine – a neurotransmitter taking part in muscle control and memory, among others. Choline is transported to the brain by a saturable transporter acting within BBB. Prolonged hyperglycemia can inhibit choline transport across BBB [51]. As to other amino acids, transport of branched chain neutral amino acids is increased in DM, while the opposite is true for basic amino acids and some essential amino acids, such as methionine, lysine, phenylalanine, and tryptophane. Interestingly, these alterations in amino acid transport across BBB result from their altered concentration in the plasma, not from the altered function of amino acid transporters across BBB [52].
2.3. Effects of DM on BBB integrity and permeability
Both in vitro and in vivo studies show that DM disrupts BBB integrity, which results in its increased permeability [53,54,55,56]. In vitro studies using co-culture of human brain microvascular endothelial cells and juxtaposed astrocytes indicate loss of BBB integrity (evaluated on the basis of transendothelial electric resistance) when hyperglycemia-mimicking conditions (glucose concentration in the medium elevated to 25 mmol/l) are maintained for at least 5 days. The integrity can be brought back to normal level after normoglycemia-mimicking conditions are restored, or when antioxidants are added [57]. In other studies [58] hyperglycemia has been found to significantly increase the expression of pro-inflammatory cytokines (TNF-α, IL-1, IL-4, IL-6) which is followed by upregulation of NF-κB and signal transducer and activator of transcription 3 (STAT-3) proteins. There are also research studies performed on brain slices obtained from diabetic rats, showing impaired communication between astrocytes and endothelial cells through gap junctions and increased production of reactive oxygen and nitrogen species, although in an unclear mechanism [59,60]. In addition, increased production of VEGF has been observed in response to the presence of advanced glycation end products [61].
High glucose concentration (30 mmol/l) in endothelial cell cultures stimulates the expression of hypoxia-inducible factor 1 alpha (HIF-1α) transcription factor and vascular endothelial growth factor (VEGF) as its downstream effector. VEGF enhances GLUT-1 molecule translocation to cell surface within BBB, as well as downregulates proteins that are responsible for proper functioning of tight junctions between endothelial cells (e.g. zonula occludens-1 (ZO-1) and occludin) [62]. In this way, hyperglycemia may increase BBB permeability. Occludin expression in brain microcapillaries in diabetic mice is significantly lower than in their healthy counterparts [63]. Inhibition of VEGF expression normalizes the expression of occludin and ZO-1, thus attenuating interendothelial leakage. Similar effect can be achieved through inhibition of HIF-1α activity, which normalizes BBB integrity and permeability [64].
Research study results in humans are a bit different. Human plasma VEGF level falls in hyperglycemia and rises in hypoglycemia, which may constitute a neuroprotective mechanism, maintaining a constant rate of glucose influx to the brain [65]. Such findings implicate a key role of VEGF as a regulator of vascular permeability in the course of BBB dysfunction due to hypo- or hyperglycemia. Regardless of VEGF, recent studies suggest a significant role of matrix metalloproteinases (MMPs) in the course of dysglycemia-induced loss of BBB integrity. In diabetic rats, BBB permeability for 14C radiolabeled sucrose rises in response to increased MMP-2 expression, which is accompanied by reduced expression of occludin and ZO-1 [53, 66–68]. Advanced glycation end products, similarly to VEGF, stimulate MMP-2 release [61].
Acute transient hyperglycemia may also induce inflammatory response and endothelial damage, which has been studied on rat model of ischemia-reperfusion damage. Increased expression of HMGB1 and intercellular adhesion molecule 1 (ICAM-1) has been found as a result of ischemia-reperfusion damage, both with co-existing mild hyperglycemia (plasma glucose concentration = 150 mg/dl) and with transient severe hyperglycemia (plasma glucose concentration = 400 mg/dl). These effects have been correlated with disrupted integrity of BBB [69]. In addition, increased expression of ICAM-1 is accompanied by increased expression of IL-1β in diabetic rats after 3 days of reperfusion [68].
Hyperglycemia-induced expression of HIF-1α and VEGF may act synergistically, enhancing detrimental response of BBB to occlusion and stopped blood flow [70], thus additionally contributing to loss of BBB integrity observed during reperfusion, although this hypothesis requires confirmation. Both brain capillary permeability and the level of pro-inflammatory cytokines rise in DM type 1 patients with ketoacidosis. Diabetic ketoacidosis, especially pronounced in the course of type 1 DM, increases BBB permeability and promotes such complications as brain edema [71]. What can be observed in a similar study is the absence of proteins responsible for proper functioning of tight junctions, such as occludin, claudin-5, ZO-1, and junctional adhesion molecule-1 (JAM-1), as well as albumin extravasation and increased expression of inflammatory response markers, such as NF-κB, C-C motif chemokine ligand 2 (CCL-2), and nitrotyrosine. This indicates that neuroinflammation combined with loss of BBB integrity plays an essential role in the pathogenesis of brain edema in the course of diabetic ketoacidosis [67]. All these findings suggest that DM significantly impairs BBB integrity and maintenance of CNS homeostasis, which may promote the risk of major neurological disorders.
Hyperglycemia can also impair vitamin C delivery, both to the retina and to the brain. Vitamin C is transported across BBB with GLUT-1 transporter, in the form of dehydroascorbic acid (DHA), subsequently transformed to ascorbic acid. Vitamin C is required for biosynthesis of collagen, catecholamines and peptide neurotransmitters. In rats with streptozotocin-induced DM, transport of DHA to the brain is reduced by 84,1 % [10]. Expression of P-glycoprotein (a glycoprotein ATP-binding cassette transporter) may also be altered in diabetic mice [72,73,74] and brought back to normal with insulin treatment [73].
Additional histologic alterations and vascular abnormalities observed within cerebral microcirculation in the course of DM may include thickening of capillary basal membrane, collagen deposition, accumulation of lipid peroxidation byproducts, and endothelial degeneration [75]. These alterations may promote aberrant neovascularization and brain remodeling, which can contribute to vascular damage, increased risk of hemorrhage, and neurodegeneration as complications of DM [76].
Glucose metabolism for energy obtaining begins from glycolysis, during which glucose is converted to pyruvate that can be further irreversibly transformed to acetyl-coenzyme A. Acetyl-coenzyme A may further react with oxalacetate to begin tricarboxylic acid cycle (TCA) – an eight step enzymatic process coupled with nicotinamide-adenine dinucleotide (NAD) conversion to the reduced form (NADH) and flavin adenine dinucleotide (FAD) conversion to the reduced FADH. NADH and FADH transfer free electrons to respiratory chain which is coupled with ATP synthesis. Oxidative glycolysis of one glucose molecule, with H2O and CO2 as final products, can yield 6 NADH molecules, 2 FADH molecules and 2 ATP molecules, which is an equivalent of 36 ATP molecules. Aerobic glycolysis also provides production of reducing equivalents, such as NADH and NADPH, which counteract oxidative stress caused by endogenous and exogenous reactive oxygen species (ROS) [77,78,79]. In normal conditions, bioenergetic demand of mechanisms transporting various substances across BBB is provided directly or indirectly by ATP. Neurons and astrocytes cooperate to fulfill their energy requirements. Glycolysis occurs mainly in astrocytes, while TCA – in neurons. Each type of cell contains both sets of enzymes, and metabolites produced in the course of glycolysis may enter other metabolic pathways, such as pentose phosphate pathway (PPP), hexosamine biosynthetic pathway (HBP), protein kinase C pathway (PKC) and AGE biosynthesis pathway. Glucose may also enter polyol biosynthetic pathway, being finally isomerized to fructose [77,78,79].
3. Correlations between hyperglycemia and oxidative stress within BBB
Hyperglycemia can result in the excessive influx of glucose to the cells, which especially refers to insulin independent cells and may result in the increased ROS production, through accelerating TCA reactions with inhibition of respiratory chain and ATP synthesis at the same time. This „hyperglycemic stress” is due to the fact that TCA reactions are coupled with transforming NAD into NADH, and in normal conditions, this NADH is used to transfer free electrons to the respiratory chain. In hyperglycemia, however, intracellular ATP level is already high, which inhibits the rate of respiratory chain reactions and ATP synthesis. Thus, there is too many NADH molecules in the mitochondria, and those NADH molecules may finally transfer the electrons into atomic oxygen, producing ROS. Increased ROS production in endothelial cells which are susceptible to this kind of stress results in microangiopathies and macroangiopathies as complications of DM [77,78,80,81]. In addition, this correlation between hyperglycemia and increased ROS production may be essential for elucidating the pathogenesis of neurodegeneration as a possible complication of DM [82,83].
On the other hand, both acute and chronic hyperglycemia may stimulate the activity of PPP in cultured astrocytes, which is a source of reduced glutathione (GSH) – a coenzyme for glutathione peroxidase, inactivating ROS. Increased activity of PPP promotes reduced glutathione regeneration, thanks to which the cells are capable of counteracting the oxidative stress [84]. In accordance with this fact, nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor has been found to translocate to the cell nucleus, together with binding immunoglobulin protein (BiP). Nrf2, taking part in cell defense against oxidative stress, is usually found in the cytoplasm, but is translocated to the cell nucleus in response to oxidative stress, thus initiating an antioxidative response dependent on stimulation of expression of some endogenous antioxidant enzymes, such as NADPH-dependent quinone oxidoreductase, heme oxygenase, and glutathione S-transferase [84].
Increased ROS production in the course of hyperglycemia inhibits GADPH activity, and thus promotes glucose entering alternative metabolic pathways (glyceraldehyde 3-phosphate processing to protein kinase C (PKC) activating metabolites and AGE-producing metabolic pathway) [75,84] (Figure 2.).
Both endothelial cells and astrocytes use insulin-independent facilitative glucose transporter 1 (GLUT1). These cells overloaded with glucose in hyperglycemic conditions show mitochondrial dysfunction in which more than usual electrons are directly transferred to O2 to generate reactive oxygen species (ROS) in the electron transport chain. Thus, hyperglycemia-driven mitochondrial tricarboxylic acid (TCA) cycle and its intermediates orchestrating mitochondrial oxidative phosphorylation, are a significant center for ROS production. Increased ROS production causes in turn inhibition of glyceraldehyde 3 phosphate dehydrogenase (GADPH) activity, and thus promotes glucose entering alternative metabolic pathways: glyceraldehyde 3-phosphate processing to protein kinase C (PKC) activating metabolites and advanced glycation end products (AGE)-producing metabolic pathway. Other glucose metabolic pathways are also overloaded, including polyol pathway, pentose phosphate pathway (PPP), and hexosamine biosynthetic pathway (HBP). The last of the mentioned metabolic pathways is used for synthesis uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), a nucleotide sugar and a coenzyme in metabolism.
* interdependent diabetic complications at the level of brain-blood barrier (BBB), including micro- and macroangiopathies as well as neurodegenerative disorders
Angiogenic edema occurring in the course of strokes accompanied by hyperglycemia has been found to result mainly from excessive activation of β isoform of PKC (PKCβ). Further activation of PKC promotes increased BBB permeability through ZO-1 phosphorylation, impaired function of tight junctions, and raised expression of VEGF [90]. Increased intracellular AGE concentrations may damage the cells through AGE-dependent modification of various proteins, affecting their interactions with surface components of cell membranes (e.g. integrins) and receptors for advanced glycation end products (RAGE). It can refer to macrophages, endothelial cells, and smooth muscle cells. RAGE activation enhances ROS production, which in turn activates NF-κB dependent metabolic pathways, promoting the expression of pro-inflammatory mediators [77,78,91,92] and enhancing innate immunity dependent inflammatory response. As a matter of fact, it has been found that in people suffering from AD, AGE accumulation promotes neuronal death and degeneration, which confirms a hypothesis that DM can increase the risk of AD and dysglycemia can be detrimental to BBB. In addition, oxidative stress activates matrix metalloproteinases, such as MMP-1, MMP-2 and MMP-9, which is accompanied by reduced activity of their tissue inhibitors (TIMP-1 and TIMP-2) and occurs in tyrosine kinase dependent manner [82].
Although all insulin independent cells are exposed to increased glucose concentration in the course of DM, only some kinds of cells become damaged in hyperglycemia dependent manner (e.g. retinal cells, endothelial cells), probably because they cannot reduce the expression of glucose transporters. As it has been mentioned above, hyperglycemic cell damage is dependent on five mechanisms: ❶ - increased influx of metabolites to polyol pathway; ❷ - increased intracellular production of AGE; ❸ - increased expression of RAGE; ❹ - activation of protein kinase C; and ❺ - increased influx of metabolites to hexosamine biosynthetic pathway. All these mechanisms share the same downstream effector, which is increased mitochondrial ROS production [77,93].
4. Role of HMGB1 as RAGE ligand in detrimental effects of DM towards the CNS
High mobility group box 1 (HMGB1) protein is a non-histone chromosomal protein [94] regulating gene transcription through binding to DNA or chromatin, mediated by specific receptors, including RAGE and TLRs [95,96,97,98]. Because of its binding to RAGE and TLR4, HMGB1 may act as a pro-inflammatory mediator taking part in the pathogenesis of neurodegenerative diseases, such as AD [99,100]. RAGE, initially discovered as binding advanced glycation end products, can also bind other ligands, including beta-amyloid (Aβ) peptide, S100 proteins, and HMGB1 [101,102,103]. Some research studies suggest that hyperglycemia and insulin resistance underlying type 2 DM increase HMGB1 and RAGE expression both in diabetic mice and humans [104,105,106]. Furthermore, it has been found that in the course of DM complications, HMGB1 activates NF-κB signaling pathways through interactions with RAGE and TLR4 [95] (Figure 3.).
It was demonstrated that hyperglycemia and insulin resistance may increase expression of both, high mobility group box 1 protein (HMGB1) and receptors for advanced glycation end products (RAGE). HMGB1 protein is a non-histone chromosomal protein regulating gene transcription through binding to DNA or chromatin, mediated by specific receptors, including RAGE and toll-like receptors (TLRs). Therefore, activation of HMGB1-RAGE-TLR4 axis in type 2 DM may induce inflammatory response via NF-κB signaling pathway. Pro-inflammatory cytokines, activated matrix metalloproteinases, and reactive oxygen species (ROS), accumulating at the border of the brain and the capillary (vascular) compartments, are responsible for degradation of tight junction proteins such as zonula occludens-1 (ZO-1) and claudin-5. Consequently, cerebral microvessel leakage leads to increased blood-brain barrier (BBB) permeability with glial activation and the infiltration of immune cells into the brain parenchyma. Cognitive and memory impairments caused by chronic inflammation and oxidative damage are responsible for the clinical picture of diabetic encephalopathy. AGE – advanced glycation end products; Aβ – beta-amyloid; MyD88 – myeloid differentiation primary response 88 (adapter protein); JNK – c-Jun N-terminal kinase; p50/p65 – NF-κB heterodimer.
Recent studies suggest that both in the brains of AD patients and in the CSF samples collected from them, HMGB1 concentration is elevated, just like in a mouse model of AD [108,109]. Several in vitro studies have revealed that stimulation of HMGB1-RAGE-TLR4 signaling pathway promotes hippocampal neuron damage and memory loss in the course of AD [110,111,112]. In addition, RAGE interactions with HMGB1 are correlated with axonal overgrowth and neuroinflammation [103,113,114]. Another study has found that HMGB1 and TLR4 interaction promotes hippocampal neuron death in patients with DM [115]. HMGB1 activates astrocytes and promotes the release of proinflammatory cytokines, as well as stimulates inducible nitric oxide synthase (iNOS) expression in cortical astrocytes, thus stimulating TLR4 signaling [116]. Interaction between HMGB1, RAGE, and TLR4 promotes Aβ aggregate accumulation, stimulates neuroinflammation, dampens insulin dependent signaling and impairs spatial memory [117,118]. Interactions between HMGB1, RAGE, and TLR4 are related to both DM and AD associated complications, such as Aβ accumulation, neuroinflammation, insulin dependent signaling, memory deficits, and microglial cells activation.
In the course of AD, there is an increased activity of metalloproteinases which can disrupt BBB integrity, thus contributing to neuronal and cognitive dysfunctions [119,120]. DM and dementia share some common features, such as severe and chronic neuroinflammation, brain insulin resistance, overaccumulation of Aβ and disrupted BBB integrity [121].
Elevated levels of HMGB1 are correlated both with type 2 DM, and with hyperglycemia [122,123,124]. In addition, HMGB1 impairs axonal growth through its interaction with RAGE [103,125], which may impair cognitive functions [126]. Besides, HMGB1 promotes Aβ accumulation and disrupts BBB integrity [127,128]. On a mouse model of AD, HMGB1 promotes axonal degeneration through myristoylated alanine rich protein kinase C substrate (MARCKS) protein phosphorylation, dependent on TLR4 signaling [127]. TLR4, a transmembrane protein belonging to pattern-recognition receptors (PRR) family, often takes part in innate immunity dependent inflammation which has been correlated with AD associated pathology. NF-κB signaling, which is downstream to TLR4 activation, promotes biosynthesis and release of pro-inflammatory cytokines [129]. TLR4 expression is markedly increased in the brains of AD patients, which promotes amyloid peptide binding and phagocytosis by microglial cells [130,131]. In the course of Aβ-induced neuroinflammation, HMGB1 can be localized in hippocampal neurons, where it is co-responsible for AD progression through activating RAGE- and TLR4-dependent signaling pathways [100]. In AD patients, HMGB1 accumulates both in the extracellular space and intracellular space of some brain regions [132].
Studies on in vitro models of AD show that HMGB1 is activated upon Aβ injection, which is accompanied by pro-inflammatory cytokine release and NLRP3 inflammasome assembly in microglial cells [133]. Furthermore, extracellular HMGB1 can impair microglia-dependent Aβ clearance, thus promoting AD through interactions with RAGE and TLR4 [134,135]. The same interactions may take part in the impairment of memory formation in mice [136].
HMGB1, as RAGE ligand, may promote insulin resistance of the brain through activating TLR4-JNK signaling pathway [118,137,138], as well as through stimulating TNF-α dependent signaling pathway [139,140] (Figure 4.).
Downstream signaling then activates inducible transcription factors: NFAT5, AP-1, and NF-κB. Pro-inflammatory environment develops, because the target genes for all these transcription factors include cytokines such as IL-6, IL-1β, IL-18, and TNF-α. TNF-α, acting on its own receptor TNFR1, may interfere with insulin signaling through phosphorylation of some serine/threonine residues in IRS, especially IRS-1. Such an inhibitory phosphorylation of IRS-1 was also demonstrated for the components of the signaling pathways (marked in the figure with “ * ”). Interestingly, the activation of NF-κB pathway could in turn induce the expression of HMGB1 and its receptors, forming a positive feedback loop to sustain inflammatory conditions. AC – astrocyte; AP-1 – activator protein 1; BBB – blood-brain barrier; EC – endothelial cell; ERK1/2 – extracellular signal-regulated kinases 1 and 2, also known as classical mitogen-activated protein (MAP) kinases; HMGB1 – high mobility group box 1; IκB – inhibitory κB protein; IKKα, IKKβ, IKKγ – the members of the inhibitor of κB (IκB) kinase (IKK) family; INS – insulin; INSR – insulin receptor; ISF – interstitial fluid; p65/p50 – nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) heterodimer; JNK – c-Jun N-terminal kinase; MyD88 – myeloid differentiation primary response 88 (adapter protein); NFAT5 – nuclear factor of activated T cells 5; P – phosphorylation; Raf – rapidly accelerated fibrosarcoma; RAGE –– receptors for advanced glycation end-products; ROS – reactive oxygen species; Ser – serine; TLR4 – toll-like receptor 4; TNF-α – tumor necrosis factor alpha; ZO-1 – zonula occludens-1 (also known as tight junction protein-1)
5. Summary and conclusions
Insulin dependent signaling in the CNS seems to be crucial for maintenance of cognitive functions, through regulation of neurotransmitter release, synaptic transmission, and glucose uptake by neurons [143,144,145]. Disrupted brain glucose metabolism in combination with insulin resistance of the hippocampus may contribute to synaptic dysfunction, cognitive function impairment, and development of AD [146,147]. In addition, brain insulin resistance reduces cerebral blood flow [148] and cerebral cortex perfusion, which leads to cognitive deficits [149]. Moreover, insulin resistance promotes excessive Aβ aggregate accumulation and hyperphosphorylation of tau proteins, which results in cognitive impairment in patients with Alzheimer’s disease [147,150]. Normal activity of insulin dependent signaling pathways facilitates Aβ aggregate clearance and inhibits senile plaque formation [151]. Insulin resistance has been found to accelerate Aβ formation in the vicinity of presynaptic neuronal cell membranes [152,153] and has been correlated with activation of JNK dependent signaling pathway, with subsequent inhibitory phosphorylation of insulin receptor substrate 1 (IRS-1) at S616 [154]. Thus, TLR4 activation by AGE or HMGB1 can increase the risk of dementia trough promoting brain insulin resistance.
RAGE activation by AGE or by HMGB1 may exert a pro-inflammatory effect, because it activates intracellular signaling pathways stimulating NF-κB activity [100,155], while AGE themselves can induce pro-inflammatory cytokine release, thus activating innate immunity dependent inflammatory response [156].
The sum of detrimental effects of dysglycemia towards BBB consists of its pro-oxidative effect promoting ROS production [77,78], which in turn inhibits glyceraldehyde 3 phosphate dehydrogenase (GADPH) activity [77], thus redirecting glucose metabolism to AGE [77,93]. Subsequently, the excess of AGE may stimulate RAGE as well as some TLR receptors (e.g., TLR4), which induces NF-κB activity and promotes neuroinflammation [77,78,91,92]. Of note, pro-oxidative effects of dysglycemia can be neutralized with Nrf2 transcription factor induction [157].
Detrimental effects of dysglycemia towards BBB include: disrupting its integrity and increasing permeability, mainly through promoting oxidative stress [77,90], inducing inflammatory response through RAGE and TLR4 activation, redistribution of glucose transporters, such as GLUT-1, and related alteration of BBB permeability for choline and DHA [10,51]. These effects may in turn impair acetylcholine biosynthesis and antioxidative defense dependent on vitamin C.
As a result of the effects mentioned above, DM can increase the risk of neurodegeneration and dementia. Firstly, through disrupting BBB integrity. Secondly, through promoting brain insulin resistance with its detrimental effects on cognitive functions [121], Thirdly, through inducing excessive production of some substances, such as AGE and HMGB1, which may promote neuroinflammation, thus abrogating the function of microglia and Aβ clearance [158,159]. Fourthly, through increased production of AGE and HMGB1 that can directly stimulate Aβ aggregate production [127,134,160], thus accelerating their accumulation in the CNS.
Author Contributions
Conceptualization, M.W. and D.S.; Writing – Original Draft Preparation, M.W.; Writing – Review & Editing, D.S. and A.G.; Visualization, D.S.; Supervision, D.S. and A.G.; Software, M.W. and D.S.; Funding Acquisition, A.G.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable. This review is based on already published data listed in the references.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Aβ | beta-amyloid |
| ABC-transporter | ATP-binding cassette transporter (P-glycoprotein – P-gp) |
| AD | Alzheimer’s disease |
| AGE | advanced glycation end products |
| ATP | adenosine triphosphate |
| BBB | blood-brain barrier |
| BiP | binding immunoglobulin protein |
| BRCP | breast cancer resistance protein |
| CCL-2 | C-C motif chemokine ligand 2 (also known as MCP-1 – monocyte chemoattractant protein-1) |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DHA | dehydroascorbic acid |
| DM | diabetes mellitus |
| FAD | flavin adenine dinucleotide |
| FADH | flavin adenine dinucleotide (reduced form) |
| GADPH | glyceraldehyde 3 phosphate dehydrogenase |
| GLUT-1, GLUT-3, GLUT-4 | glucose transporter 1, 3, and 4, respectively |
| GLUTs | glucose transporters |
| GSH - | reduced L-Glutathione |
| GSK3 | glycogen synthase kinase-3 |
| HAAF | hypoglycemia-associated autonomic failure |
| HBP | hexosamine biosynthetic pathway |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| HMGB1 | high mobility group box 1 |
| ICAM-1 | intercellular adhesion molecule 1 |
| IL-1, IL-4, IL-6 | interleukin 1, 4, and 6, respectively |
| iNOS | inducible nitric oxide synthase |
| IRS-1 | insulin receptor substrate 1 |
| JAM-1 | junctional adhesion molecule-1 |
| JNK | c-Jun N-terminal kinase |
| MAPK phosphatase-1 | mitogen activated protein kinase phosphatase 1 |
| MARCKS | myristoylated alanine rich protein kinase C substrate |
| MMP-1, MMP-2, MMP-9 | matrix metalloproteinase-2 |
| MMPs | matrix metalloproteinases |
| MRP-4 | multidrug resistance protein |
| MyD88 | myeloid differentiation primary response 88 (adapter protein) |
| NAD | nicotinamide-adenine dinucleotide |
| NADH | nicotinamide adenine dinucleotide (reduced form) |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 inflammasome | leucine-rich repeat (LRR)-containing proteins (NLR) family member 3 inflammasome |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| p50/p65 | NF-κB heterodimer, member of the Rel family of transcription factors |
| P-gp | P-glycoprotein |
| PKC | protein kinase C |
| PKCβ | β isoform of protein kinase C |
| PPP | pentose phosphate pathway |
| PRR | pattern-recognition receptors |
| RAGE | receptors for advanced glycation end-products |
| ROS | reactive oxygen species |
| SGLTs | sodium-glucose co-transporters |
| s-RAGE | soluble receptor for advanced glycation end-products |
| STAT-3 | signal transducer and activator of transcription 3 |
| TCA | tricarboxylic acid |
| TIMP-1, TIMP-2 | tissue inhibitor of matrix metalloproteinase 1 and 2, respectively |
| TLR4 | toll-like receptor 4 |
| TLRs | toll-like receptors |
| TNF-α | tumor necrosis factor alpha |
| UDP-GlcNAc | uridine diphosphate N-acetylglucosamine |
| VEGF | vascular endothelial growth factor |
| ZO-1 | zonula occludens-1 (also known as tight junction protein-1) |
References
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes mellitus statistics on prevalence and mortality: facts and fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Banting, F.G.; Best, C.H. The internal secretion of the pancreas. 1922. Indian J. Med. Res. 2007, 125, 251–66. [Google Scholar] [PubMed]
- Krolewski, A.S.; Warram, J.H. Natural History of Diabetes Mellitus. In Principles and Practice of Endocrinology and Metabolism; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 1320–1327. [Google Scholar]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; A Banks, W.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. The Blood-Brain Barrier Interface in Diabetes Mellitus: Dysfunctions, Mechanisms and Approaches to Treatment. Curr. Pharm. Des. 2020, 26, 1438–1447. [Google Scholar] [CrossRef]
- Bradbury, M. The blood-brain barrier. Exp. Physiol. 1993, 78, 453–472. [Google Scholar] [CrossRef]
- Emmi, A.; Wenzel, H.J.; Schwartzkroin, P.A.; Taglialatela, M.; Castaldo, P.; Bianchi, L.; Nerbonne, J.; Robertson, G.A.; Janigro, D. Do Glia Have Heart? Expression and Functional Role forEther-A-Go-GoCurrents in Hippocampal Astrocytes. J. Neurosci. 2000, 20, 3915–3925. [Google Scholar] [CrossRef]
- Davson, H.; Segal, M.B. Physiology of the CSF and Blood-Brain Barriers; CRC Press: Boca Raton, FL, USA, 1996. [Google Scholar]
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Pooja Naik, L.C. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M.; Rogers, R.C.; McDougal, D.H.; Ritter, S.; Qualls-Creekmore, E.; Hermann, G.E.; Zhang, J.; Jiang, S.; Wei, J.; et al. Glucose transporters in the 21st Century. Am. J. Physiol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef]
- Zhao, F.-Q. Functional Properties and Genomics of Glucose Transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Augustin, R. The protein family of glucose transport facilitators: It's not only about glucose after all. IUBMB Life 2010, 62, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009, 31, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Klepper, J. GLUT1 deficiency syndrome in clinical practice. Epilepsy Res. 2012, 100, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Duelli, R.; Kuschinsky, W. Brain Glucose Transporters: Relationship to Local Energy Demand. Physiology 2001, 16, 71–76. [Google Scholar] [CrossRef]
- Vemula, S.; Roder, K.E.; Yang, T.; Bhat, G.J.; Thekkumkara, T.J.; Abbruscato, T.J. A Functional Role for Sodium-Dependent Glucose Transport across the Blood-Brain Barrier during Oxygen Glucose Deprivation. Experiment 2009, 328, 487–495. [Google Scholar] [CrossRef] [PubMed]
- McAllister, M.S.; Krizanac-Bengez, L.; Macchia, F.; Naftalin, R.J.; Pedley, K.C.; Mayberg, M.R.; Marroni, M.; Leaman, S.; A Stanness, K.; Janigro, D. Mechanisms of glucose transport at the blood–brain barrier: an in vitro study. Brain Res. 2001, 904, 20–30. [Google Scholar] [CrossRef]
- Cornford, E.M.; Hyman, S.; Swartz, B.E. The Human Brain GLUT1 Glucose Transporter: Ultrastructural Localization to the Blood—Brain Barrier Endothelia. J. Cereb. Blood Flow Metab. 1994, 14, 106–112. [Google Scholar] [CrossRef]
- Cornford, E.M.; Hyman, S. Localization of brain endothelial luminal and abluminal transporters with immunogold electron microscopy. Neurorx 2005, 2, 27–43. [Google Scholar] [CrossRef]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef]
- Bingham, E.M.; Hopkins, D.; Smith, D.; Pernet, A.; Hallett, W.; Reed, L.; Marsden, P.K.; Amiel, S.A. The Role of Insulin in Human Brain Glucose Metabolism. Diabetes 2002, 51, 3384–3390. [Google Scholar] [CrossRef]
- Frank, H.J.L.; Pardridge, W.M.; Jankovic-Vokes, T.; Vinters, H.V.; Morris, W.L. Insulin Binding to the Blood-Brain Barrier in the Streptozotocin Diabetic Rat. J. Neurochem. 1986, 47, 405–411. [Google Scholar] [CrossRef]
- Laron, Z. Insulin and the brain. Arch. Physiol. Biochem. 2009, 115, 112–116. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Eisenberg, J.; Yang, J. Human Blood?Brain Barrier Insulin Receptor. J. Neurochem. 1985, 44, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- A Banks, W.; Jaspan, J.B.; Huang, W.; Kastin, A.J. Transport of Insulin Across the Blood-Brain Barrier: Saturability at Euglycemic Doses of Insulin. Peptides 1997, 18, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Nirwane, A.; Yao, Y. Basement membrane and blood–brain barrier. Stroke Vasc. Neurol. 2018, 4, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Patching, S.G. Glucose Transporters at the Blood-Brain Barrier: Function, Regulation and Gateways for Drug Delivery. Mol. Neurobiol. 2017, 54, 1046–1077. [Google Scholar] [CrossRef]
- Li, X.-H.; Lv, B.-L.; Xie, J.-Z.; Liu, J.; Zhou, X.-W.; Wang, J.-Z. AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol. Aging 2012, 33, 1400–1410. [Google Scholar] [CrossRef]
- Lam, J.K.Y.; Wang, Y.; Shiu, S.W.M.; Wong, Y.; Betteridge, D.J.; Tan, K.C.B. Effect of insulin on the soluble receptor for advanced glycation end products (RAGE). Diabet. Med. 2013, 30, 702–709. [Google Scholar] [CrossRef]
- Thomas, M.C.; on behalf of the FinnDiane Study Group; Söderlund, J.; Lehto, M.; Mäkinen, V.-P.; Moran, J.L.; Cooper, M.E.; Forsblom, C.; Groop, P.-H. Soluble receptor for AGE (RAGE) is a novel independent predictor of all-cause and cardiovascular mortality in type 1 diabetes. Diabetologia 2011, 54, 2669–2677. [Google Scholar] [CrossRef]
- Fujisawa, K.; Katakami, N.; Kaneto, H.; Naka, T.; Takahara, M.; Sakamoto, F.; Irie, Y.; Miyashita, K.; Kubo, F.; Yasuda, T.; et al. Circulating soluble RAGE as a predictive biomarker of cardiovascular event risk in patients with type 2 diabetes. Atherosclerosis 2013, 227, 425–428. [Google Scholar] [CrossRef]
- Yamagishi, S.-I.; Imaizumi, T. Serum Levels of Soluble Form of Receptor for Advanced Glycation End Products (sRAGE) May Reflect Tissue RAGE Expression In Diabetes. Arter. Thromb. Vasc. Biol. 2007, 27, e32–e32. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.-I. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp. Gerontol. 2011, 46, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Yamagishi, S.-I.; Matsui, T.; Nakamura, K.; Imaizumi, T. Role of Advanced Glycation End Products (AGEs) in Thrombogenic Abnormalities in Diabetes. Curr. Neurovascular Res. 2006, 3, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Duelli, R.; Maurer, M.; Staudt, R.; Heiland, S.; Duembgen, L.; Kuschinsky, W. Increased cerebral glucose utilization and decreased glucose transporter Glut1 during chronic hyperglycemia in rat brain. Brain Res. 2000, 858, 338–347. [Google Scholar] [CrossRef]
- Hou, W.-K.; Xian, Y.-X.; Zhang, L.; Lai, H.; Hou, X.-G.; Xu, Y.-X.; Yu, T.; Xu, F.-Y.; Song, J.; Fu, C.-L.; et al. Influence of blood glucose on the expression of glucose trans-porter proteins 1 and 3 in the brain of diabetic rats. Chin. Med J. 2007, 120, 1704–9. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Triguero, D.; Farrell, C.R. Downregulation of Blood-Brain Barrier Glucose Transporter in Experimental Diabetes. Diabetes 1990, 39, 1040–1044. [Google Scholar] [CrossRef]
- Simpson, I.A.; Appel, N.M.; Hokari, M.; Oki, J.; Holman, G.D.; Maher, F.; Koehler-Stec, E.M.; Vannucci, S.J.; Smith, Q.R. Blood-Brain Barrier Glucose Transporter. J. Neurochem. 1999, 72, 238–247. [Google Scholar] [CrossRef]
- Gruetter, R.; Novotny, E.J.; Boulware, S.D.; Rothman, D.L.; Shulman, R.G. 1H NMR Studies of Glucose Transport in the Human Brain. J. Cereb. Blood Flow Metab. 1996, 16, 427–438. [Google Scholar] [CrossRef]
- Hasselbalch, S.G.; Knudsen, G.M.; Capaldo, B.; Postiglione, A.; Paulson, O.B. Blood-Brain Barrier Transport and Brain Metabolism of Glucose during Acute Hyperglycemia in Humans1. J. Clin. Endocrinol. Metab. 2001, 86, 1986–1990. [Google Scholar] [CrossRef]
- Nielsen, J.K.; Djurhuus, C.B.; Gravholt, C.H.; Carus, A.C.; Granild-Jensen, J.; Ørskov, H.; Christiansen, J.S. Continuous Glucose Monitoring in Interstitial Subcutaneous Adipose Tissue and Skeletal Muscle Reflects Excursions in Cerebral Cortex. Diabetes 2005, 54, 1635–1639. [Google Scholar] [CrossRef]
- Seaquist, E.R.; Tkac, I.; Damberg, G.; Thomas, W.; Gruetter, R. Brain glucose concentrations in poorly controlled diabetes mellitus as measured by high-field magnetic resonance spectroscopy. Metabolism 2005, 54, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Duelli, R.; Maurer, M.; Staudt, R.; Heiland, S.; Duembgen, L.; Kuschinsky, W. Increased cerebral glucose utilization and decreased glucose transporter Glut1 during chronic hyperglycemia in rat brain. Brain Res. 2000, 858, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Cryer, P.E. Mechanisms of Hypoglycemia-Associated Autonomic Failure in Diabetes. New Engl. J. Med. 2013, 369, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Bakatselos, S.O. Hypoglycemia unawareness. Diabetes Res. Clin. Pr. 2011, 93, S92–S96. [Google Scholar] [CrossRef] [PubMed]
- McCrimmon, R.J.; Jacob, R.J.; Fan, X.; McNay, E.C.; Sherwin, R.S. Effects of recurrent antecedent hypoglycaemia and chronic hyperglycaemia on brainstem extra-cellular glucose concentrations during acute hypoglycaemia in conscious diabetic BB rats. Diabetologia 2003, 46, 1658–1661. [Google Scholar] [CrossRef]
- Kumagai, A.K.; Kang, Y.-S.; Boado, R.J.; Pardridge, W.M. Upregulation of Blood-Brain Barrier GLUT1 Glucose Transporter Protein and mRNA in Experimental Chronic Hypoglycemia. Diabetes 1995, 44, 1399–1404. [Google Scholar] [CrossRef]
- McCall, A.L.; Fixman, L.B.; Fleming, N.; Tornheim, K.; Chick, W.; Ruderman, N.B. Chronic hypoglycemia increases brain glucose transport. Am. J. Physiol. Metab. 1986, 251, E442–E447. [Google Scholar] [CrossRef]
- Mastaitis, J.W.; Wurmbach, E.; Cheng, H.; Sealfon, S.C.; Mobbs, C.V. Acute Induction of Gene Expression in Brain and Liver by Insulin-Induced Hypoglycemia. Diabetes 2005, 54, 952–958. [Google Scholar] [CrossRef]
- Mooradian, A.D. Blood-Brain Barrier Choline Transport Is Reduced in Diabetic Rats. Diabetes 1987, 36, 1094–1097. [Google Scholar] [CrossRef]
- Mans, A.M.; DeJoseph, M.R.; Davis, D.W.; Hawkins, R.A. Regional amino acid transport into brain during diabetes: effect of plasma amino acids. Am. J. Physiol. Metab. 1987, 253, E575–E583. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Lundeen, T.F.; Norwood, K.M.; Brooks, H.L.; Egleton, R.D. Increased blood–brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia 2007, 50, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.K.; Levin, E.C.; Clifford, P.M.; Han, M.; Tourtellotte, R.; Chamberlain, D.; Pollaro, M.; Coretti, N.J.; Kosciuk, M.C.; Nagele, E.P.; et al. Diabetes and Hypercholesterolemia Increase Blood-Brain Barrier Permeability and Brain Amyloid Deposition: Beneficial Effects of the LpPLA2 Inhibitor Darapladib. J. Alzheimer's Dis. 2013, 35, 179–198. [Google Scholar] [CrossRef]
- Starr, J.M.; Wardlaw, J.; Ferguson, K.; MacLullich, A.; Deary, I.J.; Marshall, I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J. Neurol. Neurosurg. Psychiatry 2003, 74, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.D.; VanGilder, R.L.; Houser, K.A. Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. Am. J. Physiol. Circ. Physiol. 2006, 291, H2660–H2668. [Google Scholar] [CrossRef]
- Allen, C.L.; Bayraktutan, U. Antioxidants attenuate hyperglycaemia-mediated brain endothelial cell dysfunction and blood-brain barrier hyperpermeability. Diabetes, Obes. Metab. 2009, 11, 480–490. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Wang, Z.; Zhang, X.; Yao, L.; Wang, F.; Liu, S.; Yin, J.; Ling, E.-A.; Wang, L.; et al. High glucose-induced expression of inflammatory cytokines and reactive oxygen species in cultured astrocytes. Neuroscience 2012, 202, 58–68. [Google Scholar] [CrossRef]
- Ball, K.K.; Harik, L.; Gandhi, G.K.; Cruz, N.F.; Dienel, G.A. Reduced gap junctional communication among astrocytes in experimental diabetes: Contributions of altered connexin protein levels and oxidative-nitrosative modifications. J. Neurosci. Res. 2011, 89, 2052–2067. [Google Scholar] [CrossRef]
- Gandhi, G.K.; Ball, K.K.; Cruz, N.F.; A Dienel, G. Hyperglycaemia and Diabetes Impair Gap Junctional Communication among Astrocytes. ASN Neuro 2010, 2, AN20090048–73. [Google Scholar] [CrossRef]
- Shimizu, F.; Sano, Y.; Tominaga, O.; Maeda, T.; Abe, M.-A.; Kanda, T. Advanced glycation end-products disrupt the blood–brain barrier by stimulating the release of transforming growth factor–β by pericytes and vascular endothelial growth factor and matrix metalloproteinase–2 by endothelial cells in vitro. Neurobiol. Aging 2013, 34, 1902–1912. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. 2009, 106, 1977–1982. [Google Scholar] [CrossRef]
- Vorbrodt, A.W.; Dobrogowska, D.H.; Tarnawski, M.; Meeker, H.C.; Carp, R.I. Immunogold study of altered expression of some interendothelial junctional molecules in the brain blood microvessels of diabetic scrapie-infected mice. Histochem. J. 2006, 37, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, Z.; Shi, H. HIF-1 is involved in high glucose-induced paracellular permeability of brain endothelial cells. Cell. Mol. Life Sci. 2012, 69, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Oltmanns, K.M.; Melchert, U.H.; Scholand-Engler, H.G.; Schultes, B.; Schweiger, U.; Peters, A. Divergent effects of hyper- and hypoglycemia on circulating vascular endothelial growth factor in humans. Metabolism 2008, 57, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Chehade, J.M.; Haas, M.J.; Mooradian, A.D. Diabetes-Related Changes in Rat Cerebral Occludin and Zonula Occludens-1 (ZO-1) Expression. Neurochem. Res. 2002, 27, 249–252. [Google Scholar] [CrossRef]
- Hoffman, W.H.; Stamatovic, S.M.; Andjelkovic, A.V. Inflammatory mediators and blood brain barrier disruption in fatal brain edema of diabetic ketoacidosis. Brain Res. 2009, 1254, 138–148. [Google Scholar] [CrossRef]
- Ding, C.; He, Q.; Li, P.-A. Diabetes increases expression of ICAM after a brief period of cerebral ischemia. J. Neuroimmunol. 2005, 161, 61–67. [Google Scholar] [CrossRef]
- Ennis, S.R.; Keep, R.F. Effect of sustained-mild and transient-severe hyperglycemia on ischemia-induced blood-brain barrier opening. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2007, 27, 1573–1582. [Google Scholar] [CrossRef]
- Ergul, A.; Elgebaly, M.M.; Middlemore, M.-L.; Li, W.; Elewa, H.; A Switzer, J.; Hall, C.; Kozak, A.; Fagan, S.C. Increased hemorrhagic transformation and altered infarct size and localization after experimental stroke in a rat model type 2 diabetes. BMC Neurol. 2007, 7, 33–33. [Google Scholar] [CrossRef]
- Vavilala, M.S.; Richards, T.L.; Roberts, J.S.; Chiu, H.; Pihoker, C.; Bradford, H.; Deeter, K.; Marro, K.I.; Shaw, D. Change in blood–brain barrier permeability during pediatric diabetic ketoacidosis treatment*. Pediatr. Crit. Care Med. 2010, 11, 332–338. [Google Scholar] [CrossRef]
- Liu, H.; Xu, X.; Yang, Z.; Deng, Y.; Liu, X.; Xie, L. Impaired function and expression of P-glycoprotein in blood–brain barrier of streptozotocin-induced diabetic rats. Brain Res. 2006, 1123, 245–252. [Google Scholar] [CrossRef]
- Liu, H.; Liu, X.; Jia, L.; Liu, Y.; Yang, H.; Wang, G.; Xie, L. Insulin therapy restores impaired function and expression of P-glycoprotein in blood–brain barrier of experimental diabetes. Biochem. Pharmacol. 2008, 75, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Maeng, H.-J.; Kim, M.-H.; Jin, H.-E.; Shin, S.M.; Tsuruo, T.; Kim, S.G.; Kim, D.-D.; Shim, C.-K.; Chung, S.-J. Functional Induction of P-glycoprotein in the Blood-Brain Barrier of Streptozotocin-Induced Diabetic Rats: Evidence for the Involvement of Nuclear Factor-κB, a Nitrosative Stress-Sensitive Transcription Factor, in the Regulation. Drug Metab. Dispos. 2007, 35, 1996–2005. [Google Scholar] [CrossRef] [PubMed]
- A McCuskey, P.; McCuskey, R.S. In vivo and electron microscopic study of the development of cerebral diabetic microangiography. Microcirc. Endothel. Lymphat. 1984, 1. [Google Scholar]
- Prakash, R.; Johnson, M.; Fagan, S.C.; Ergul, A. Cerebral Neovascularization and Remodeling Patterns in Two Different Models of Type 2 Diabetes. PLOS ONE 2013, 8, e56264. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Naik, P.; Prasad, S.; Cucullo, L. Role and Function of Dehydrogenases in CNS and Blood-Brain Barrier Pathophysiology: In Dehydrogenases; Intech: London, UK, 2013. [Google Scholar]
- Takahashi, S.; Abe, T.; Izawa, Y.; Suzuki, N. Effects of fluctuating glucose concentrations on oxidative metabolism of glucose in cultured neurons and astroglia. J. Diabetes Mellit. 2012, 02, 19–26. [Google Scholar] [CrossRef]
- Okouchi, M.; Okayama, N.; Alexander, J.S.; Aw, T.Y. NRF2-Dependent Glutamate-L-Cysteine Ligase Catalytic Subunit Expression Mediates Insulin Protection Against Hyperglycemia-Induced Brain Endothelial Cell Apoptosis. Curr. Neurovascular Res. 2006, 3, 249–261. [Google Scholar] [CrossRef]
- Haorah, J.; Ramirez, S.H.; Schall, K.; Smith, D.; Pandya, R.; Persidsky, Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood?brain barrier dysfunction. J. Neurochem. 2007, 101, 566–576. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Takahashi, S.; Izawa, Y.; Suzuki, N. Astroglial Pentose Phosphate Pathway Rates in Response to High-Glucose Environments. ASN Neuro 2012, 4, AN20120002. [Google Scholar] [CrossRef] [PubMed]
- Devraj, K.; Klinger, M.E.; Myers, R.L.; Mokashi, A.; Hawkins, R.A.; Simpson, I.A. GLUT-1 glucose transporters in the blood-brain barrier: Differential phosphorylation. J. Neurosci. Res. 2011, 89, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K.; Shah, A.M.; Fan, L.M.; Teng, L.; Li, J.-M.; Datla, S.R.; et al. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Stefano, G.B.; Challenger, S.; Kream, R. Hyperglycemia-associated alterations in cellular signaling and dysregulated mitochondrial bioenergetics in human metabolic disorders. Eur. J. Nutr. 2016, 55, 2339–2345. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, M.J.; Huang, Q.; Sweet, J.G. Inhibition of Protein Kinase Cβ Reverses Increased Blood–Brain Barrier Permeability During Hyperglycemic Stroke and Prevents Edema Formation In Vivo. Stroke 2011, 42, 3252–3257. [Google Scholar] [CrossRef]
- Byun, K.; Bayarsaikhan, E.; Kim, D.; Son, M.; Hong, J.; Jeong, G.-B.; Paek, S.H.; Won, M.-H.; Lee, B. Activated microglial cells synthesize and secrete AGE-albumin. Anat. Cell Biol. 2012, 45, 47–52. [Google Scholar] [CrossRef]
- Guerin-Dubourg, A.; Catan, A.; Bourdon, E.; Rondeau, P. Structural modifications of human albumin in diabetes. Diabetes Metab. 2012, 38, 171–178. [Google Scholar] [CrossRef]
- Lyons, T.J.; Basu, A. Biomarkers in diabetes: hemoglobin A1c, vascular and tissue markers. Transl. Res. 2012, 159, 303–312. [Google Scholar] [CrossRef]
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A New Group of Chromatin-Associated Proteins with a High Content of Acidic and Basic Amino Acids. JBIC J. Biol. Inorg. Chem. 1973, 38, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous Danger Signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.-C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High Mobility Group 1 Protein (Hmg-1) Stimulates Proinflammatory Cytokine Synthesis in Human Monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Min, H.J.; Ko, E.A.; Wu, J.; Kim, E.S.; Kwon, M.K.; Kwak, M.S.; Choi, J.E.; Lee, J.E.; Shin, J.-S. Chaperone-like Activity of High-Mobility Group Box 1 Protein and Its Role in Reducing the Formation of Polyglutamine Aggregates. J. Immunol. 2013, 190, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Štros, M. HMGB proteins: Interactions with DNA and chromatin. Biochim. et Biophys. Acta (BBA) - Gene Regul. Mech. 2010, 1799, 101–113. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Balasubramaniam, V.R.; Othman, I.; Shaikh, M.F. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur. J. Pharmacol. 2019, 858, 172487. [Google Scholar] [CrossRef]
- Nan, K.; Han, Y.; Fang, Q.; Huang, C.; Yu, L.; Ge, W.; Xiang, F.; Tao, Y.-X.; Cao, H.; Li, J. HMGB1 gene silencing inhibits neuroinflammation via down-regulation of NF-κB signaling in primary hippocampal neurons induced by Aβ25–35. Int. Immunopharmacol. 2019, 67, 294–301. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Huttunen, H.; Rauvala, H. Amphoterin as an extracellular regulator of cell motility: from discovery to disease. J. Intern. Med. 2004, 255, 351–366. [Google Scholar] [CrossRef]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The Receptor for Advanced Glycation End Products (RAGE) Is a Cellular Binding Site for Amphoterin. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef]
- Yao, D.; Brownlee, M. Hyperglycemia-Induced Reactive Oxygen Species Increase Expression of the Receptor for Advanced Glycation End Products (RAGE) and RAGE Ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhong, J.; Zhang, X.; Liu, Z.; Yang, Y.; Gong, Q.; Ren, B. The Role of HMGB1 in the Pathogenesis of Type 2 Diabetes. J. Diabetes Res. 2016, 2016, 2543268. [Google Scholar] [CrossRef] [PubMed]
- Gharaati, M.E.; Nahavandi, A.; Mojarad, T.B.; Roghani, M. iabetic Encephalopathy Affecting Mitochondria and Axonal Transport Proteins. Basic Clin. Neurosci. J. 2020, 11, 781–794. [Google Scholar] [CrossRef]
- Gaikwad, S.; Puangmalai, N.; Bittar, A.; Montalbano, M.; Garcia, S.; McAllen, S.; Bhatt, N.; Sonawane, M.; Sengupta, U.; Kayed, R. Tau oligomer induced HMGB1 release contributes to cellular senescence and neuropathology linked to Alzheimer’s disease and frontotemporal dementia. Cell Rep. 2021, 36, 109419–109419. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Shaikh, M.F.; Chakraborti, A.; Kumari, Y.; Aledo-Serrano, A.; Aleksovska, K.; Alvim, M.K.M.; Othman, I. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front. Neurosci. 2018, 12, 628. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.L.; Fernandes, A.; Aguilar-Pimentel, J.A.; de Angelis, M.H.; Guedes, J.R.; Brito, M.A.; Ortolano, S.; Pani, G.; Athanasopoulou, S.; Gonos, E.S.; et al. Towards frailty biomarkers: Candidates from genes and pathways regulated in aging and age-related diseases. Ageing Res. Rev. 2018, 47, 214–277. [Google Scholar] [CrossRef]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.-J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef]
- Urbonaviciute, V.; Fürnrohr, B.G.; Meister, S.; Munoz, L.; Heyder, P.; De Marchis, F.; Bianchi, M.E.; Kirschning, C.; Wagner, H.; Manfredi, A.A.; et al. Induction of inflammatory and immune responses by HMGB1–nucleosome complexes: implications for the pathogenesis of SLE. J. Exp. Med. 2008, 205, 3007–3018. [Google Scholar] [CrossRef]
- Tian, J.; Avalos, A.M.; Mao, S.-Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9–dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Shi, Y.; Du, P.; Wang, J.; Han, Y.; Sun, B.; Feng, J. HMGB1/TLR4 promotes apoptosis and reduces autophagy of hippocampal neurons in diabetes combined with OSA. Life Sci. 2019, 239, 117020. [Google Scholar] [CrossRef] [PubMed]
- Famakin, B.M.; Tsymbalyuk, O.; Tsymbalyuk, N.; Ivanova, S.; Woo, S.K.; Kwon, M.S.; Gerzanich, V.; Simard, J.M. HMGB1 is a Potential Mediator of Astrocytic TLR4 Signaling Activation following Acute and Chronic Focal Cerebral Ischemia. Neurol. Res. Int. 2020, 2020, 3929438. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Steinle, J.J. HMGB1 inhibits insulin signalling through TLR4 and RAGE in human retinal endothelial cells. Growth Factors 2018, 36, 164–171. [Google Scholar] [CrossRef]
- Wang, X.-X.; Tan, M.-S.; Yu, J.-T.; Tan, L. Matrix Metalloproteinases and Their Multiple Roles in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 908636. [Google Scholar] [CrossRef] [PubMed]
- Nagele, R.G.; Clifford, P.M.; Siu, G.; Levin, E.C.; Acharya, N.K.; Han, M.; Kosciuk, M.C.; Venkataraman, V.; Zavareh, S.; Zarrabi, S.; et al. Brain-Reactive Autoantibodies Prevalent in Human Sera Increase Intraneuronal Amyloid-β1-42 Deposition. J. Alzheimer's Dis. 2011, 25, 605–622. [Google Scholar] [CrossRef]
- Jeong, J.-H.; Lee, D.H.; Song, J. HMGB1 signaling pathway in diabetes-related dementia: Blood-brain barrier breakdown, brain insulin resistance, and Aβ accumulation. Biomed. Pharmacother. 2022, 150. [Google Scholar] [CrossRef]
- Jr, J. .; Kalousová, M.; Švarcová, J.; Muravská, A.; Kvasnička, J.; Landová, L.; Zima, T.; Škrha, J. Relationship of Soluble RAGE and RAGE Ligands HMGB1 and EN-RAGE to Endothelial Dysfunction in Type 1 and Type 2 Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2012, 120, 277–281. [Google Scholar] [CrossRef]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased Toll-Like Receptor (TLR) Activation and TLR Ligands in Recently Diagnosed Type 2 Diabetic Subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef]
- Hagiwara, S.; Iwasaka, H.; Hasegawa, A.; Koga, H.; Noguchi, T. Effects of hyperglycemia and insulin therapy on high mobility group box 1 in endotoxin-induced acute lung injury in a rat model*. Crit. Care Med. 2008, 36, 2407–2413. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, H.J.; Kuja-Panula, J.; Sorci, G.; Agneletti, A.L.; Donato, R.; Rauvala, H. Coregulation of Neurite Outgrowth and Cell Survival by Amphoterin and S100 Proteins through Receptor for Advanced Glycation End Products (RAGE) Activation. J. Biol. Chem. 2000, 275, 40096–40105. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.; Westra, J.; Limburg, P.; Bijl, M.; Kallenberg, C. HMGB1 in vascular diseases: Its role in vascular inflammation and atherosclerosis. Autoimmun. Rev. 2012, 11, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Motoki, K.; Tagawa, K.; Chen, X.; Hama, H.; Nakajima, K.; Homma, H.; Tamura, T.; Watanabe, H.; Katsuno, M.; et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 2016, 6, 31895. [Google Scholar] [CrossRef] [PubMed]
- Festoff, B.W.; Sajja, R.K.; van Dreden, P.; Cucullo, L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J. Neuroinflammation 2016, 13, 1–12. [Google Scholar] [CrossRef]
- Vaure, C.; Liu, Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [PubMed]
- Le Page, A.; Dupuis, G.; Frost, E.H.; Larbi, A.; Pawelec, G.; Witkowski, J.M.; Fulop, T. Role of the peripheral innate immune system in the development of Alzheimer's disease. Exp. Gerontol. 2018, 107, 59–66. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and Toll-Like Receptors 2 and 4 Are Required for Fibrillar Aβ-Stimulated Microglial Activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef]
- Takata, K.; Kitamura, Y.; Kakimura, J.-I.; Shibagaki, K.; Tsuchiya, D.; Taniguchi, T.; A Smith, M.; Perry, G.; Shimohama, S. Role of high mobility group protein-1 (HMG1) in amyloid-β homeostasis. Biochem. Biophys. Res. Commun. 2003, 301, 699–703. [Google Scholar] [CrossRef]
- Falcão, A.S.; Carvalho, L.A.R.; Lidónio, G.; Vaz, A.R.; Lucas, S.D.; Moreira, R.; Brites, D. Dipeptidyl Vinyl Sulfone as a Novel Chemical Tool to Inhibit HMGB1/NLRP3-Inflammasome and Inflamma-miRs in Aβ-Mediated Microglial Inflammation. ACS Chem. Neurosci. 2017, 8, 89–99. [Google Scholar] [CrossRef]
- Takata, K.; Takada, T.; Ito, A.; Asai, M.; Tawa, M.; Saito, Y.; Ashihara, E.; Tomimoto, H.; Kitamura, Y.; Shimohama, S. Microglial Amyloid-β1-40 Phagocytosis Dysfunction Is Caused by High-Mobility Group Box Protein-1: Implications for the Pathological Progression of Alzheimer’s Disease. Int. J. Alzheimer's Dis. 2012, 2012, 685739. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.-F.; Walker, D.G.; Brachova, L.; Beach, T.G.; Rogersa, J.; Schmidt, A.M.; Stern, D.M.; Du Yan, S. Involvement of Microglial Receptor for Advanced Glycation Endproducts (RAGE) in Alzheimer's Disease: Identification of a Cellular Activation Mechanism. Exp. Neurol. 2001, 171, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Mazarati, A.; Maroso, M.; Iori, V.; Vezzani, A.; Carli, M. High-mobility group box-1 impairs memory in mice through both toll-like receptor 4 and Receptor for Advanced Glycation End Products. Exp. Neurol. 2011, 232, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, Z.; Xie, J.; Kang, L.-N.; Wang, L.; Xu, B. High Mobility Group Box-1: A Missing Link between Diabetes and Its Complications. Mediat. Inflamm. 2016, 2016, 3896147. [Google Scholar] [CrossRef]
- Montes, V.N.; Subramanian, S.; Goodspeed, L.; A Wang, S.; Omer, M.; Bobik, A.; Teshigawara, K.; Nishibori, M.; Chait, A. Anti-HMGB1 antibody reduces weight gain in mice fed a high-fat diet. Nutr. Diabetes 2015, 5, e161–e161. [Google Scholar] [CrossRef]
- Guzmán-Ruiz, R.; Ortega, F.; Rodríguez, A.; Vázquez-Martínez, R.; Díaz-Ruiz, A.; Garcia-Navarro, S.; Giralt, M.; Garcia-Rios, A.; Cobo-Padilla, D.; Tinahones, F.J.; et al. Alarmin high-mobility group B1 (HMGB1) is regulated in human adipocytes in insulin resistance and influences insulin secretion in β-cells. Int. J. Obes. 2014, 38, 1545–1554. [Google Scholar] [CrossRef]
- Ghosh, G.; Wang, V.Y.-F.; Huang, D.-B.; Fusco, A. NF-κB regulation: lessons from structures. Immunol. Rev. 2012, 246, 36–58. [Google Scholar] [CrossRef]
- Neuhofer, W. Role of NFAT5 in Inflammatory Disorders Associated with Osmotic Stress. Curr. Genom. 2010, 11, 584–590. [Google Scholar] [CrossRef]
- van Beijnum, J.R.; Buurman, W.A.; Griffioen, A.W. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis 2008, 11, 91–99. [Google Scholar] [CrossRef]
- Heni, M.; Kullmann, S.; Preissl, H.; Fritsche, A.; Häring, H.-U. Impaired insulin action in the human brain: causes and metabolic consequences. Nat. Rev. Endocrinol. 2015, 11, 701–711. [Google Scholar] [CrossRef]
- Benedict, C.; Hallschmid, M.; Schultes, B.; Born, J.; Kern, W. Intranasal Insulin to Improve Memory Function in Humans. Neuroendocrinology 2007, 86, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, M.; Naghdi, N.; Choopani, S. Intra CA1 insulin microinjection improves memory consolidation and retrieval. Peptides 2007, 28, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Newcomer, J.; Kanne, S.; Dagogo-Jack, S.; Cryer, P.; Sheline, Y.; Luby, J.; Dagogo-Jack, A.; Alderson, A. Memory improvement following induced hyperinsulinemia in alzheimer's disease. Neurobiol. Aging 1996, 17, 123–130. [Google Scholar] [CrossRef]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Kanjwal, Y.; Mohanty, P.; Rao, S.; Sung, B.H.; Wilson, M.; Dandona, P. Insulin-induced vasodilatation of internal carotid artery. Metabolism 1999, 48, 1470–1473. [Google Scholar] [CrossRef]
- Hoscheidt, S.M.; Kellawan, J.M.; E Berman, S.; A Rivera-Rivera, L.; A Krause, R.; Oh, J.M.; Beeri, M.S.; A Rowley, H.; Wieben, O.; Carlsson, C.M.; et al. Insulin resistance is associated with lower arterial blood flow and reduced cortical perfusion in cognitively asymptomatic middle-aged adults. J. Cereb. Blood Flow Metab. 2017, 37, 2249–2261. [Google Scholar] [CrossRef]
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Peskind, E.R.; Asthana, S.; Purganan, K.; Wait, C.; Chapman, D.; Schwartz, M.W.; Plymate, S.; Craft, S. Insulin increases CSF A 42 levels in normal older adults. Neurology 2003, 60, 1899–1903. [Google Scholar] [CrossRef]
- Yamamoto, N.; Matsubara, T.; Sobue, K.; Tanida, M.; Kasahara, R.; Naruse, K.; Taniura, H.; Sato, T.; Suzuki, K. Brain insulin resistance accelerates Aβ fibrillogenesis by inducing GM1 ganglioside clustering in the presynaptic membranes. J. Neurochem. 2012, 121, 619–628. [Google Scholar] [CrossRef]
- de la Monte, S.M. Contributions of Brain Insulin Resistance and Deficiency in Amyloid-Related Neurodegeneration in Alzheimerʼs Disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef]
- Yoon, S.O.; Park, D.J.; Ryu, J.C.; Ozer, H.G.; Tep, C.; Shin, Y.J.; Lim, T.H.; Pastorino, L.; Kunwar, A.J.; Walton, J.C.; et al. JNK3 Perpetuates Metabolic Stress Induced by Aβ Peptides. Neuron 2012, 75, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, Inflammation, and Insulin Resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Nogueira-Machado, J.A.; Volpe, C.M.d.O.; Veloso, C.A.; Chaves, M.M. HMGB1, TLR and RAGE: a functional tripod that leads to diabetic inflammation. Expert Opin. Ther. Targets 2011, 15, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, F.; Yu, Z.; Guo, S.; Liu, N.; Jiang, Y.; Lo, E.H.; Xu, Y.; Wang, X. HDAC3 inhibition prevents blood-brain barrier permeability through Nrf2 activation in type 2 diabetes male mice. J. Neuroinflammation 2019, 16, 1–15. [Google Scholar] [CrossRef]
- Weller, R.O.; Subash, M.; Preston, S.D.; Mazanti, I.; Carare, R.O. SYMPOSIUM: Clearance of Aβ from the Brain in Alzheimer's Disease: Perivascular Drainage of Amyloid-β Peptides from the Brain and Its Failure in Cerebral Amyloid Angiopathy and Alzheimer's Disease. Brain Pathol. 2008, 18, 253–266. [Google Scholar] [CrossRef]
- Tahara, K.; Kim, H.-D.; Jin, J.-J.; Maxwell, J.A.; Li, L.; Fukuchi, K.-I. Role of toll-like receptor signalling in A uptake and clearance. Brain 2006, 129, 3006–3019. [Google Scholar] [CrossRef]
- Takata, K.; Kitamura, Y.; Tsuchiya, D.; Kawasaki, T.; Taniguchi, T.; Shimohama, S. High mobility group box protein-1 inhibits microglial A? clearance and enhances A? neurotoxicity. J. Neurosci. Res. 2004, 78, 880–891. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram of blood-brain barrier (BBB) [8,9,11,12,17,27,28]. The numbers in black circles (1–8) correspond to the glucose transporters (GLUTs) 1–8, respectively; the numbers in blue circles (1–2) corresponnd to the sodium-glucose cotransporters (SGLTs) 1–2, respectively; the main glucose transporter for a given cell type under physiological conditions is marked with an enlarged symbol. BM – basement membrane; ISF – interstitial fluid; RBC – red blood cell.
Figure 1.
Schematic diagram of blood-brain barrier (BBB) [8,9,11,12,17,27,28]. The numbers in black circles (1–8) correspond to the glucose transporters (GLUTs) 1–8, respectively; the numbers in blue circles (1–2) corresponnd to the sodium-glucose cotransporters (SGLTs) 1–2, respectively; the main glucose transporter for a given cell type under physiological conditions is marked with an enlarged symbol. BM – basement membrane; ISF – interstitial fluid; RBC – red blood cell.
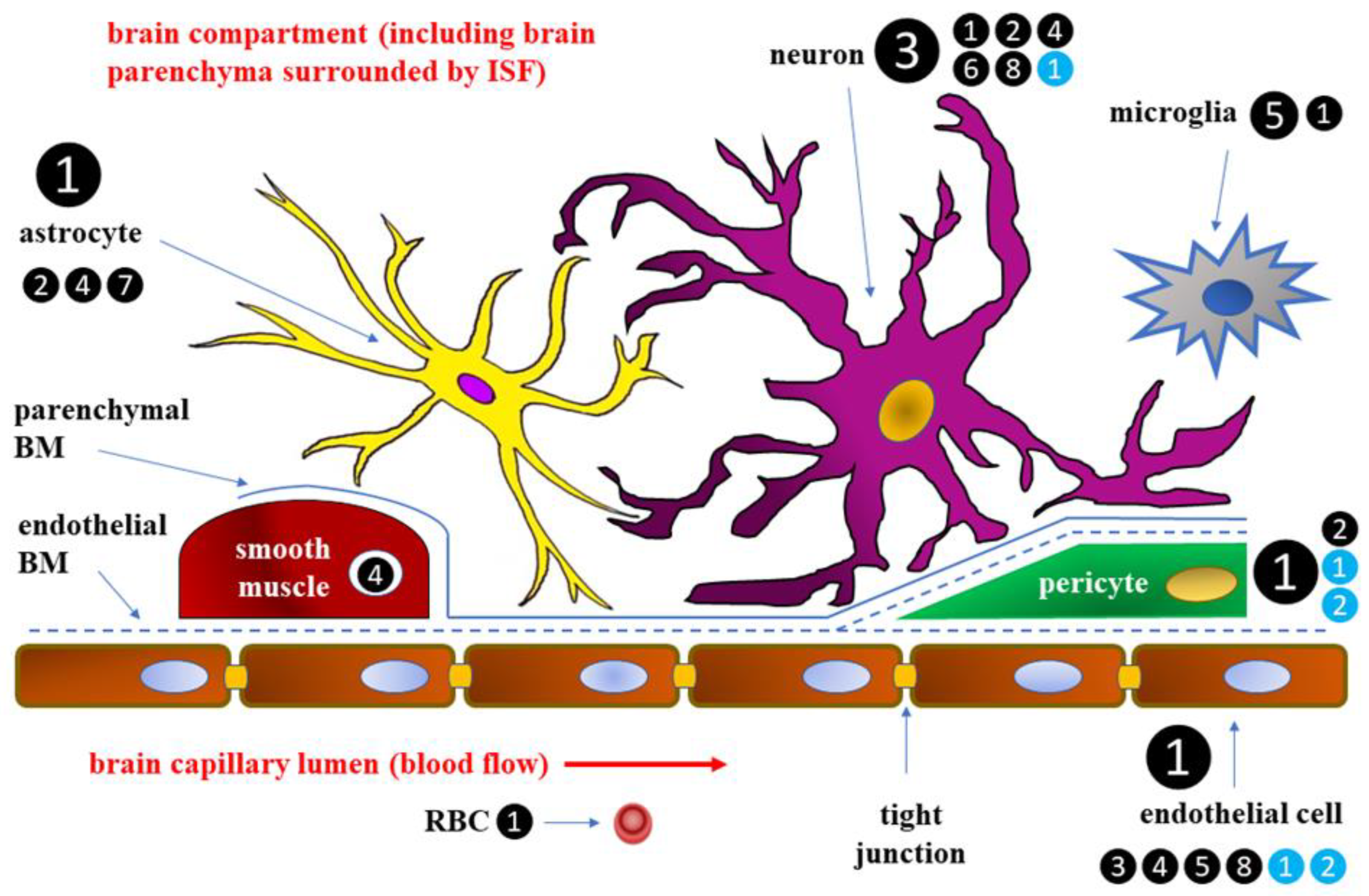
Figure 2.
Hyperglycemia and oxidative stress in endothelial cells and astrocytes resulting from increased glucose influx [84,85,86,87,88,89].
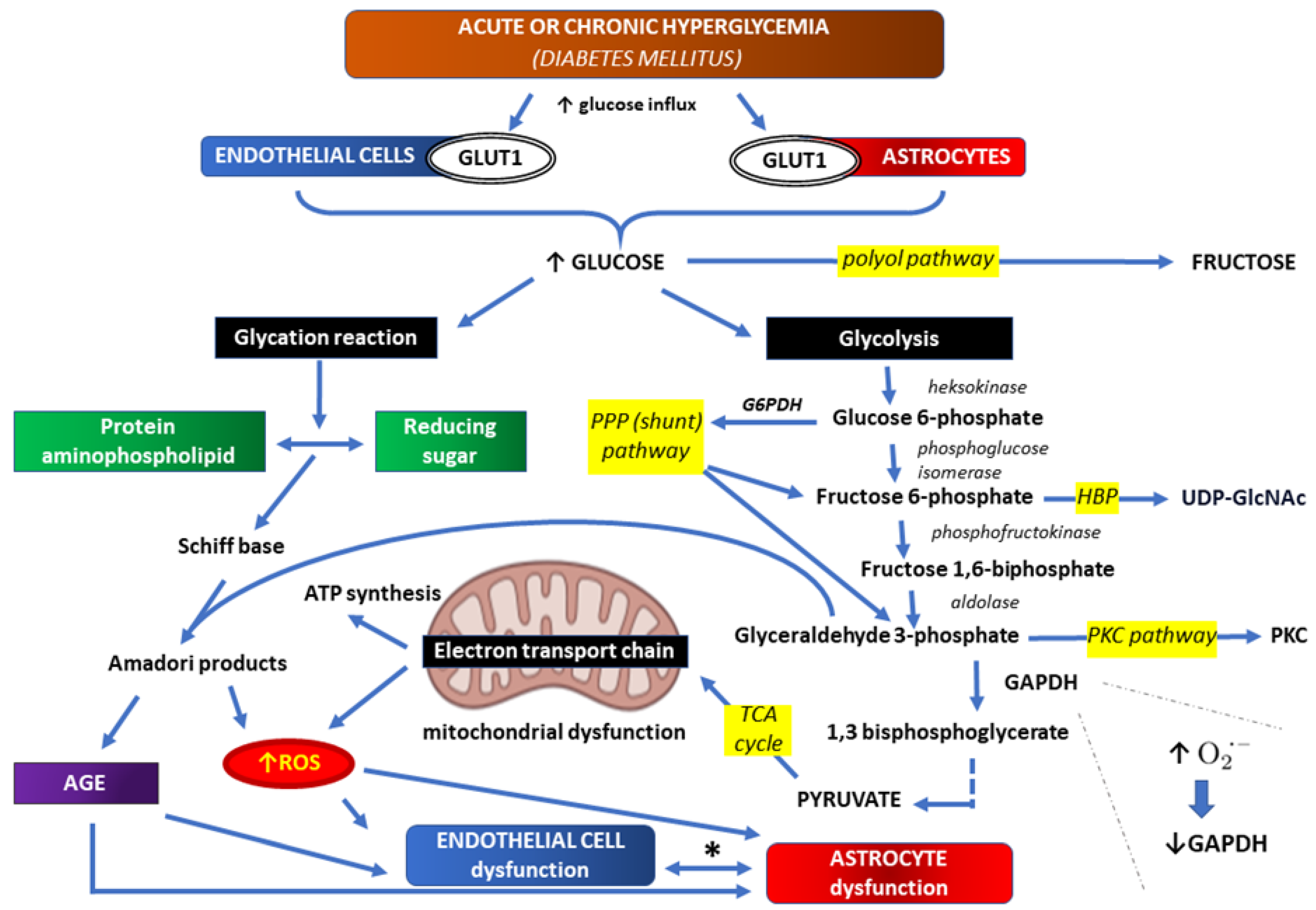
Figure 3.
HMGB1-RAGE-TLR4 signaling in type 2 diabetes mellitus (DM) may compromise the integrity of BBB and increase the risk of neurodegenerative diseases [94,95,96,97,98,99,100,101,102,103,104,105,106,107].
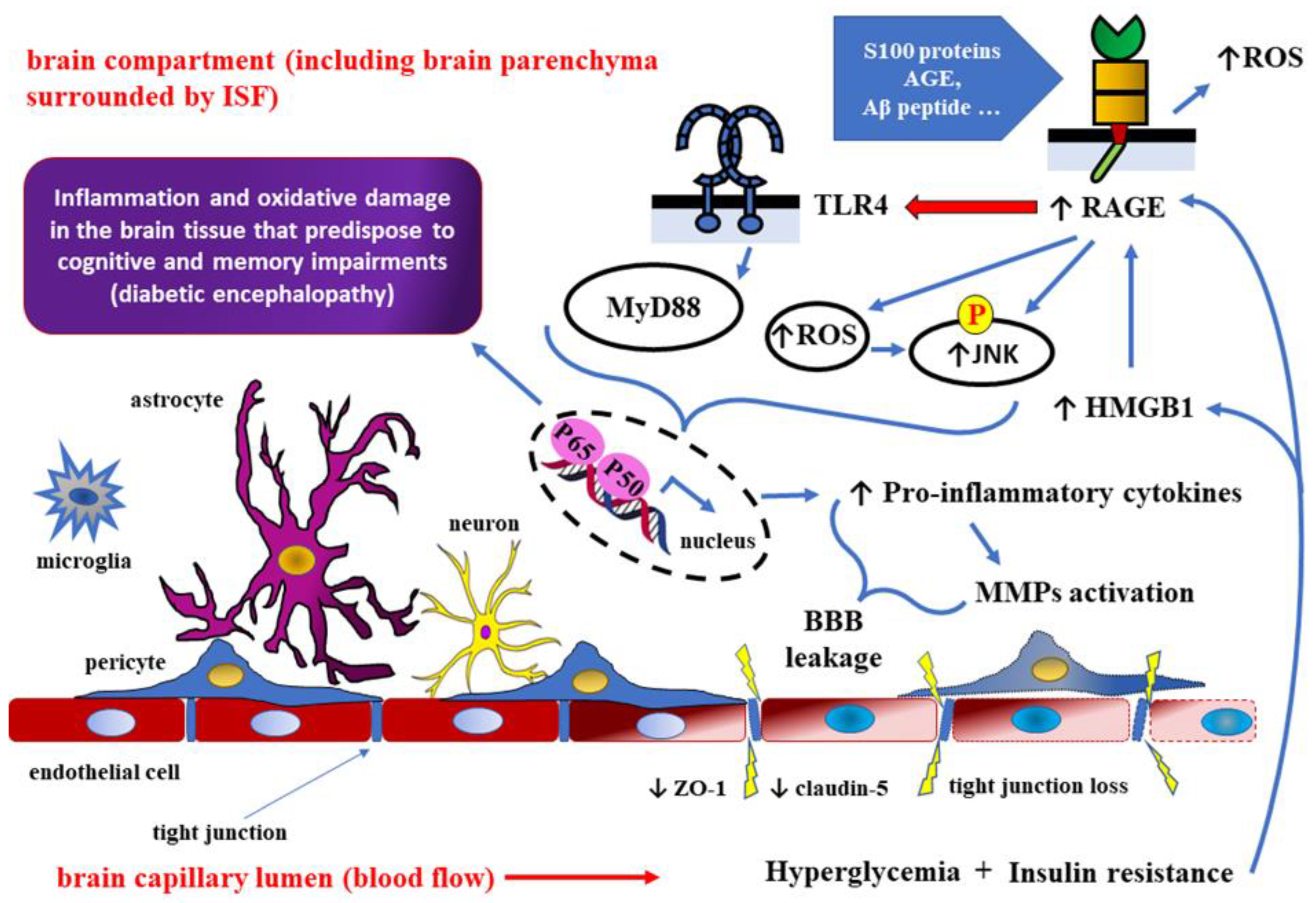
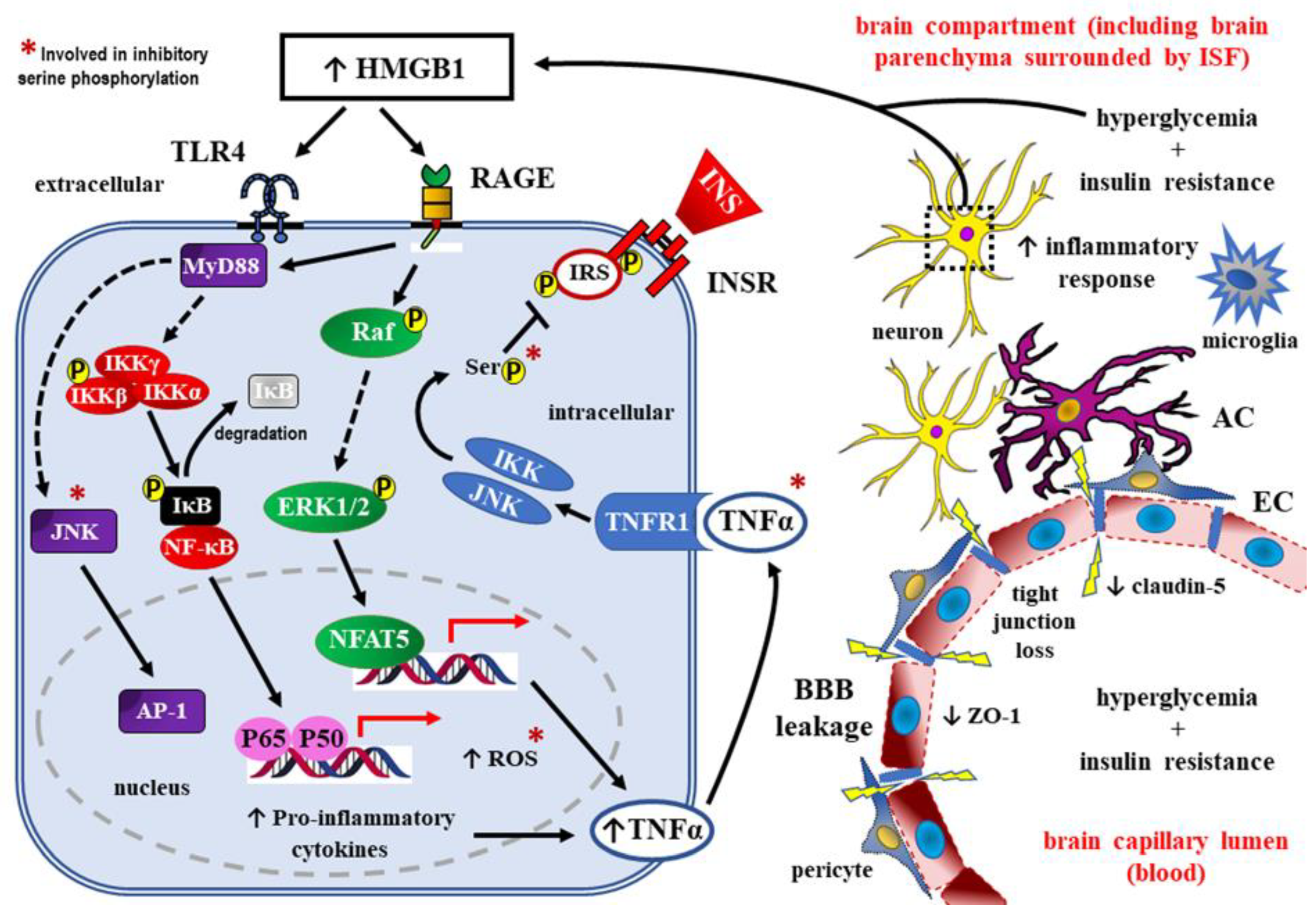
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



