
Submitted:
30 May 2023
Posted:
31 May 2023
You are already at the latest version
Abstract
Shea tree (Vitellaria paradoxa) is an important fruit tree crop because of its oil used for cooking and industrial manufacture of cosmetics. Despite its many benefits, quantitative trait loci linked to the economic traits have not yet been studied. In this study, we performed association mapping on a panel of 374 shea tree accessions using 7,530 single-nucleotide polymorphisms (SNPs) markers for oil yield and seed related traits. Twenty three markers that were significantly (–log10 (p) = 4.87) associated to kernel oil content, kernel length; width and weight were identified. The kernel dry matter oil content and kernel width had the most significant Marker Trait Association (MTA) on chromosomes 1 and 8 respectively. Sixteen candidate genes that condition early induction of flower buds and somatic embryos, seed growth and development, substrate binding, transport, lipid biosynthesis, metabolic processes during seed germination and disease resistance and abiotic stress adaptation were identified. The presence of these genes suggest their role in promoting shea bioactive functions that condition high oil synthesis. This study provides insights into the important marker-linked seed traits with genes controlling them, useful for molecular breeding for improving oil yield in the species.
Keywords:
Linked
; marker association
; annotation
; Genes
1. Introduction
Shea tree (Vitellaria paradoxa C. F. Gaertn.) is an important economic tree crop known for its oil, used for the production of valuable products in the food and cosmetic industries [1]. The tree is endemic to the Sudano-Sahelian Africa, covering 21 countries [2], where it greatly sustains the socio-cultural and economic well-being of the communities there. Shea tree is recognized as the second highest oil-producing plant after the oil palm [3]. The global market for shea products was reported at US$30 billion in 2020 [4]. The high demand is owed to its use as substitute for confectionary and cosmetic industry. The demand for these natural and organic cosmetics in European market reached EUR 3.90 billion in 2019 [5]. The cosmetic sector alone exceeded US$530 in 2020 and is expected to rise up to US$1025 million in 2027. Out of this, USA market is projected to rise from US$240 million in 2020 to US$390 in 2027 and expected to grow at a Compound Annual Growth Rate (CAGR) of 7% arising from the increasing demand for facial make up products [6]. The total export of oils from different plants to Europe in 2020 was estimated at 300,000 tonnes, with Netherlands and France being the leading importers. However, both processed and unprocessed products are sold in national and international markets, contributing to national income through foreign exchange in the shea producing countries. Shea nuts are mostly traded on with some small quantities of unprocessed shea butter/oil [6]. The leading producers of shea are Nigeria (361,017 tonns/year), Mali (49,640 tonns/year), Burkina Faso (45,183 tonns/year), and Ghana (33,878 tonns/year) [6]. There is a huge and untenable supply deficit due to heightened international demand, necessitating breeding interventions to boost production across its range.
Recent advances in shea tree genomic studies Hale et al [3] and [7] gave insights to the broad opportunities in genome-assisted breeding. Despite these advancements, the genomic resources remain underused for boosting production and improving oil yield and quality. Genome-wide association studies (GWAS) provide opportunities to identify genomic regions of an organism which are putatively associated with the traits of interest to plant breeders [3]. With the availability of affordable and economic modifications of genome sequencing approaches like genotyping by sequencing (GBS), discovering and using SNP markers has become a preferred way of genotyping. One of these technologies is the Diversity Arrays Technology Sequencing (DArTseq), where a genome is partially sequenced using a specific combination of restriction enzymes and the restriction tags are used for assembling and discovering the SNP markers [8]. The discovered SNPs are generally spread all over the genome and can be used in GWAS for the study of a wide range of tree crop traits of economic importance [9]. This study was carried out to identify genomic loci associated with seed oil content, seed weight, seed length and width of the shea tree in Uganda. Determining the marker trait association shall enhance shea tree breeding by reducing on the time required to complete the breeding cycle.
2. Materials and methods
2.1. Plant materials and leaf sampling for DNA extraction
A total of 374 shea genotypes from the germplasm collection (Breeding Seedling Orchard) Uganda were used in this study. A total of 3600 shea fruits/seeds were collected from 180 trees (families) in the districts of Amuru, Arua, Katakwi, Moyo and Otuke (Figure 1). The seeds were then divided into two portions; for sowing and for oil extraction. A minimum of 10 seeds were randomly picked from each family for sowing to generate seedlings used in DNA extraction. Fifteen seeds from the remaining lot were processed and used for oil extraction
The shea seeds were sown in a tree nursery at Ngetta Zonal Agricultural Research Development Institute (NgeZARDI), Lira - Uganda in the month of June 2018. The seedlings were managed in the tree nursery for 12 months until they developed between 4 – 6 leaves before sampling the leaf tissues for DNA analysis. Leaf samples of 374 seedlings were randomly picked for DNA extraction and analysis at Biosciences Eastern and Central Africa- International Livestock Research Institute (BeCA-ILRI). Only healthy and recently flushed leaves from the previous season were sampled and place in DNA extraction kit and dried using Silica gel before shipping to BeCA-ILRI.
After leaf tissue sampling for DNA analysis, the genotypes were further managed in the nursery for another 6 months to allow them to heal and later planted in a multi-locational trial (breeding seed orchard) located in Lira (NgettaZARDI) and Serere (National Semi Arid Resources Research Institute (NASARRI) districts, using Random Complete Block Design (RCBD) in the month of October 2019. The trials are being maintained as germplasm collection for future breeding programme in Uganda.
2.1.1. Shea Oil extraction procedure
Oil content was determined using Soxhlet extraction, the American Official Agricultural Chemists' method for determination of oil content in plant materials in the months of September and October 2020. Oil was extracted with continuous reflux of petroleum ether over crushed dried Shea nut powder in a Soxhlet extractor. The oil contents of each seed lot were extracted in triplicates and presented in percentage of its dry matter content.
2.1.2. DNA extraction and SNP discovery by DArTseq™ technology
DNA samples were processed in digestion/ligation reactions as described by [8]. Total genomic DNA from silica dried leaf samples were extracted at BeCA-ILRI following the CetylTrimethylAmmonium Bromide (CTAB) /chloroform/ isoamyl alcohol method [10]. The DNA was quality checked using standard processes involving 0.8% agarose gel electrophoresis, optical measurements for 260 and 280 nm using a NanoDrop 2000 spectrophotometer (ND-2000 V3.5, NanoDrop Technologies, Inc.) and quantification using a Qubit™ 3.0 Fluorometer (Thermo Fisher scientific, Grand Island, NY). The libraries were prepared for 752 individuals using the PstI-SphI complexity reduction method [11] and partial-genome sequenced using proprietary DArTseq (1.0) methodology [8] on a HiSeq2500 Sequencer (Illumina Inc. San Diego, CA, USA) with 72 bases read length [12,13].
Sequences generated from each lane were processed using proprietary DArT analytical pipelines. DArT-Seq™ technology relies on a complexity reduction method using restriction enzymes that are sensitive to DNA methylated sites and repetitive DNA [13]. In the primary pipeline, the FASTQ files were first processed to filter poor-quality sequences, applying more selection criteria to the barcode region compared to the rest of the sequence. Approximately 2,500,000 (±7%) sequences per barcode/sample were used in marker calling. Finally, identical sequences were collapsed into “fastqcall files.” These files were used in the secondary pipeline for DArT P/L's proprietary SNP and SilicoDArT (Presence/Absence Markers in genomic representations) (present = 1 vs. absent = 0) calling algorithms (DArTsoft14). The analytical pipeline processed the sequence data. The reads were then aligned to the shea_V1 reference genome publicly available from the ORCAE database (https://bioinformatics.psb.ugent.be/orcae) (accessed on 30 December 2021).
2.2. Data analysis
2.2.1. Seed trait data analysis
The seed trait data got from 20 fruits per mother tree (180 mother trees in total) were analysed using RCBD package in R software “agricolae” for DAU test function) [14]. Analysis of variance (ANOVA) was then performed to determine the variations within and among the genotypes. The phenotypic variance and error variance of all the traits in this study were obtained sing multi-locus random-SNP-effect Mixed Linear Model (MLM), following Wang et al [15] and [16] to estimate the genetic variance. The genomic inflation factor (lambda) was also set for each studied trait with their significant level specified.
2.2.2. Genome-wide association analysis and gene annotation identification
A multi-locus random-SNP-effect mixed linear model (mrMLM) [15] was implemented in R statistical software using the mixed model equation for GWAS presented in equation 1, in accordance to Yu, et al. [17], using additive, general; dominant alternative and dominant reference gene action models for trait association study [18]. This current study selected mrMLM method to avoid bottlenecks in stringent correction using other control measures (false discovery rate (FDR) and Bonferroni correction) against false positive rate [19]. The mrMLM uses a less stringent significance threshold considering a critical probability value or log of odds (LOD) making it possible to identify any possible loci of importance.
where:
Y = Xb+Zu+e
- Y
- = the vector of the phenotypic observations estimated for the traits studied,
- X
- = the SNP markers (fixed effect) matrix,
- Z
- = the random kinship (co-ancestry) matrix,
- b
- = a vector representing the estimated SNP effects,
- u
- = a vector representing random additive genetic effects, and
- e
- = the vector for random residual errors.
The phenotypic variation explained by the model for a trait and a particular SNP was determined using stepwise regression implemented in the “lme4” R package. The SNP loci in significant association with traits were determined by adjusted p-value using Bonferroni correction [20]. Quantile–quantile (QQ) plots were generated by plotting the negative logarithms (-log10) of the p-values against their expected p-values to test the appropriateness of the GWAS model with the null hypothesis of no association and to determine how well the models accounted for the population structure.
To account for the putative genes linked to traits, a window range of 5 kb (upstream and downstream) was defined [21]; and genes were searched from the V. paradoxa Whole Genome v2.0Assembly & Annotation v2.1 [22] in the ORCAE database (https://bioinformatics.psb.ugent.be/orcae) [3], with a search for candidate genes associated with oil yield traits. The genome-wide significant threshold was determined using a modified Bonferroni multiple testing corrections. The gene name, description, and AGPv4 coordinates with their protein, were then retrieved from the Vitellaria paradoxa reference genome database (https://bioinformatics.psb.ugent.be/orcae). The putative functional candidate genes linked to the associated SNPs were then annotated in line with any initially annotated genes from other species.
3. Results
3.1. Phenotypic variation for the Shea tree traits
The traits mean values ± standard deviations and the phenotypic data range of a collection of 374 open pollinated seeds from 180 shea trees from Uganda’s parklands are presented in Table 1 and the results of ANOVA for the study traits also presented (Table 2). The mean seed oil content of 180 shea genotypes was 53.53% with a range of 39.05 – 69.77%. A relatively heavy kernels (18.81) and very low weight genotypes were also observed (Table 1).
Analysis of variance showed that genotype, environment, and their interaction (genotype-environment) were all important sources of variation in seed oil content (Table 2). Variation in kernel weight and its axial dimensions were significantly influenced by genotype and the environment. However, the interaction of genotype and environment had no significant effect on kernel weight and its axial dimension (Table 2). The ANOVA results indicates that only replication was significant at 0.05. All other factors recorded significance at 0.01 or 0.001 (Table 2).
Seed oil content showed a significant positive correlation with kernel width (r = 0.1, P ≤ 0.001). However, it negatively correlated with kernel weight (-0.01) and kernel length (-0.09) (Table 3). The result further revealed a moderate (0.44) correlations between kernel width is and kernel weight and oil content (0.1), whereas oil content is negatively correlated with kernel weight (-0.1) and kernel length (-0.9) (Figure 2).
3.2. Population structure and quality analysis of SNP data
The SNP calling pipeline generated 30,733 highly polymorphic SNP markers, of which 27,063 (88.1%) were mapped across the 12 Vitellaria paradoxa chromosomes, with the remaining clustered into undefined scalfolds. Only 7,530 SNP markers (27.8%) of the mapped SNP markers were realized after filtering with >20% of missing data, <0.05 minor allele frequency (MAF) and utilized as input for the GWAS analysis.
The filtered SNPs were not uniformly distributed across the genome. Chromosome two had the highest number of markers (960 SNPs; Chr size = 74.5 Mb) followed by chromosomes one (805 SNPs; Chr size = 82 Mb), chromosome ten (780 SNPs; Chr size = 50 Mb), five and eight (650 SNPs; Chr size = 56.5 Mb and 645 SNPs; Chr size = 58 Mb respectively). Meanwhile, chromosomes four (425 Chr size = 37 Mb) and chromosome three (430 SNPs; Chr size = 38.6 Mb) had the lowest number of markers (Figure 3 and Table 3). This indicates a non-random distribution of SNPs with varying SNP frequencies on the 12 chromosomes of shea tree genome in Uganda.
Minor allele frequency (MAF) among the 7530 SNP markers varied from 0.03 to 0.50 and average of 0.13. The study further revealed a high level of heterozygosity within individuals (0.26) and markers (0.32) indicating a high non-random association of alleles at different loci that offer opportunity for association studies and allele transfer through marker-assisted selection of the population. The filtered markers varied in their Polymorphic Information Content (PIC), ranging from 0.258 (chromosome 4) to 0.269 (chromosome12) with a mean PIC of 0.26 across the chromosomes (Table 3).
There was a general high gene diversity (0.32) across the chromosomes with chromosome 12 being the highest (0.33) and chromosomes 4, 1and 6 being the lowest (0.31 respectively). Structure analysis revealed that shea tree populations in Uganda are genetically grouped into two clusters of Eastern group and West Nile/Northern Uganda group. The Eastern cluster contributed the highest (57%) proportion of individuals and West Nile/Northern Uganda cluster (43%).
Out of the 12 chromosomes in the shea genome, only two (Chromosome 1 and 8) revealed significant loci. The result of Linkage disequilibrium (LD) indicated that 187,487 loci pairs in a physical distance of 605,450 bp. Of the total loci, 3.62% (6,795) of them were in significant (p < 0.01) LD. The results further revealed that 87 (1.28%) loci pairs had R2 = 1 (were in complete LD).
3.3. Marker association for the studied traits
The association analysis was performed on shea seed-related traits and 16 significant association markers were found on chromosomes 1, 2, 3, 5, 6, 7, 8, 9, 10, 11 and 12 (Table 4 and Figure 4). The oil yield trait studied was the nut dry matter percent oil content which revealed 07 significant markers located in chromosomes 1, 3, 4, 5, 8, 9 and 11 in a panel of 374 Vitellaria paradoxa genotypes from Uganda. Quantile-Quantile plots produced by displaying −log10 p-values against individual p-values revealed suitability of GWAS for the trait's connection in the shea tree genotypes. The association analysis was performed for percent oil content of each shea tree line in a location using the V. paradoxa reference genome (https://bioinformatics.psb.ugent.be/orcae) (accessed on 8 March 2022). There were differences between the observed and expected values of the target traits, indicating a link between the phenotypic and SNP markers as indicated in Quantile-Quantile plots.
The seven SNP markers linked with shea nut oil yield (S1_60237300, S3_14843482, S4_32032310, S5_6275145, S8_41696703, S9_32689981 and S11_43126044) were located on chromosomes 1, 3, 4, 5, 8, 9 and 11 (Table 4; Figure 4) and were associated with high nut percent oil content estimated on dry matter basis. These seven loci explained an overall phenotypic variance of 4.03, however, makers S8_41696703 and S9_32689981 had negative effects on seed oil content, although they explained the most (13.31% and 11.52% respectively) of phenotypic variation. Having them will lead to a reduction in oil yield and so we have to select against them to improve oil content in the shea population.
This current study revealed six significant SNP markers linked with shea kernel length (S3_11153087, S5_15524578, S6_46530240, S8_11121701, S11_8320549 and S12_32853547) located on chromosomes 3, 5, 6, 8, 11 and 12 (Table 4; Figure 4). The proportion of phenotypic variance explained by significant QTNs ranged from 6.5% in marker S5_15524578 to 14.6% in S6_46530240. The total phenotypic variance expressed by the trait was 0.095.
The GWAS revealed 8 genomic regions that were significant associated with kernel width. The 8 significant SNP markers linked to shea kernel width (S1_32402910, S2_47786838, S2_64059706, S7_3025298, S9_43700743, S10_50604452, S12_32853547 and S12_7613999) were located on chromosomes 1, 2, 7, 9, 10 and 12 (Table 4; Figure 4). Marker S12_32853547 contributed most (13.14%) of the phenotypic variation compared to the rest (ranging from 4.5%to 9.75%) (Table 4). The total phenotypic variation explained by the trait was 0.17.
The association analysis of the 374 open pollinated shea trees and the 7530 quality SNP markers resulted in two significant SNPs (S1_30720144 and S8_43605016) located on chromosomes 1 and 8. Marker S8_43605016 contributed most (15.79%) of the phenotypic variation compared to S1_30720144 (9.21%) (Table 4). The total phenotypic variance in this trait was 0.061 Table 4; Figure 4).
Variations in the seed traits explained by the individual SNP markers (r2) varied from 4.47% in kernel width to 15.79% in kernel weight for the significant SNPs, indicating that they represent major QTLs associated with oil yield and kernel physical parameters Alleles ‘A’ of marker S1_60237300; ‘T’ of marker S11_43126044; ‘A’ of markers S4_32032310 and S5_6275145 in oil yield , had the highest positive QTN effect (0.8255, 0.8098, 0.737 and 0.683 respectively) revealing higher association with increasing oil yield. Although most of the seed related traits indicated negative QTN effects, the allele which had the highest (0.2942) positive QTN effect was allele ‘C’ in marker S12_32853547.
3.4. Potential candidate genes
A total of 23 candidate genes were identified on linking the significant SNP regions with the V. paradoxa genome (Table. 5). The annotation result revealed six putative genes associated with seed length traits. Among these were: Protein metabolism and gluconeogenesis on chromosome 12 and Protein translocation on chromosome 11. The proteins are well known to play important role in mediating plant seed oil biosynthesis [23] and early seedling morphogenesis and development.
From the kernel width, eight putative genes were discovered, of which three (Zinc Finger located in chromosome 3, protein binding located on chromosome 9 and Protein metabolism and gluconeogenesis located on chromosome 12) had linkage with shea seed oil biosynthesis pathways. Zinc Finger has been associated with playing a key role in plant seed oil biosynthesis and accumulation [24]. All the two identified genes (ATP hydrolase located on chromosome 1 and Protein Kinase on chromosome 8) in kernel weight trait are important in the biochemical pathways of plant seed oil synthesis (Table 5). The hydrolysis process is performed by the FATB acyl-ACP thioesterase or by 3-ketoacyl-ACP synthase II (KASII).
This study further identified seven gene/protein families associated with the percent dry matter oil content in shea nuts: Acyl-ACP Thioesterase Fat B (FATB); Acyl-CoA-binding protein (ACBP); Long Chain Acyl-CoA Synthetase (LACS); Fatty acid exporter (FAX2); (3-ketoacyl-ACP synthase II (KASII) and Fatty acid desaturases (FADs) on chromosomes 1, 3, 8, 9, and 11 (Table 6).
Acyl-CoA-binding protein (ACBP) was identified on chromosomes 3 at loci S3_14843482 and chromosome 5 at loci S5_6275145 that govern plant seed oil accumulation (Table 6). The genes are 1Mbs from their respective SNPs. Candidate Gene (CG) selection for shea nut oil accumulation is presented (Supplementary Table 2). The genes were annotated with protein-coding genes, using GO.OBO v2.1. The functions of these genes in enhancing shea oil content are explained in Table 6.
3.5. Linkage disequilibrium (LD) and Marker association
The association analysis of the 374 highly heterozygous shea trees and the 7530 quality SNPs resulted in two most significant SNPs. Variations in the seed traits explained by the individual SNP markers (r2) varied from 4.47 to 15% for the significant SNPs. Allele ‘A’ of S1_60237300 marker had the highest allele effect (0.83) revealing higher association with increasing oil yield in shea tree, followed (0.81) by allele C in marker S11_43126044 and allele A (0.68) in S5_6275145 marker. Furthermore, for kernel width, allele ‘C’ in S12_32853547 also had a moderate effect (0.29). None the less, allele ‘T’ in marker S6_46530240 revealed the highest (-0.25) negative effect for the studied traits.
4. Discussion
4.1. Phenotypic data
Shortening juvenile maturity period to fruiting and increasing oil yield per acre and quality aspects in shea oil are the major concerns in the shea industry worldwide. This study aimed at selecting shea parent materials for future breeding programme and bring about farmer solutions by establishing Multi-location Breeding Seed Orchards as a short term remedy to quality source of shea tree planting materials.
There was variation in the Seed trait characteristics in the shea accessions. The results for seed traits indicated a significant variation within the populations and a non-significant variation among the populations. Such a non-significant variation observed among the populations is important in breeding for varieties that can easily be adapt across all the geographical range in Uganda. Furthermore, any newly bred variety shall be acceptable by all the communities within the shea parkland in Uganda. With the reliable heritability, selecting traits with marker association for high oil yield in shea tree will result in good genetic progress of the species. Earlier studies by [25,26] reported similar oil content (52.26%) in the species with this study (53.5%). The results of this study was slightly higher due to the participatory selection which suggests a potential genetic gain from selection given a number of genotypes with known higher yield (69.77%). Such results can be important in assessing the G x E interactions for some traits.
4.2. Candidate gene scan in the oil content traits
The shea genome revealed associated SNP markers, important for identification of QTL regions controlling the variations of the quantitative traits [7]. Most important of this was the identification of the seven significant SNPs located close to genes that encode different proteins related to plant metabolic mechanisms and transport of biosynthetic products and materials.
GWAS has the ability to increase detectability of genomic association in plants [27]. In fact, GWAS has gained increasing popularity as a tool for analysing complex traits in plants [28]. It has been used to reveal the genes controlling polygenic traits including the genetic loci associated with the trait of interest in fruit trees [9]. Kumar et al [29] used Mixed Linear Model statistical model for GWAS to study six commercial fruit traits in apple seedlings suggesting the potential of the tool in shortening the breeding cycle of tree species like shea.
The advances in omics technologies have enabled researchers to identify candidate genes that promote improvement of associated traits of commercial importance in plants. Earlier very few studies were conducted in determining these functional genes in shea tree [30,31,32]. However, recent sequencing of shea reference genome [3] and identification of genes in shea tree [7], has paved new avenues of genomic studies in the species. Studies for biochemical pathways of oil synthesis in plant seeds have been advanced [33] and several gene expression and enzyme activities in plant seed oil accumulation fronted [34]. Interestingly, 45 seed oil biosynthesis genes were reported in shea tree genome [3]. This study discovered 23 such genes that are potentially associated with shea nut oil biosynthesis pathways (Table 5 and 6). Of all, acetyl-CoA carboxylase (ACC) is notably the major enzyme catalyst in shea oil biosynthesis [7]. Earlier studies revealed (9) gene copies of Ketoacyl-ACP synthase (KAS) in shea, 6 of these were also reported in Theobroma cacao, suggesting their contribution to the increased lipid content in shea than in cocoa. Other genes with higher number of copies in shea include: FAD2, FAD3, and LACS genes [34]. The biological effect of LACs genes discovered on chromosome 4 includes modification of fatty acids chain lengths along the plant oil biosynthesis path ways [35].
The validity of the interrelations among the traits of study was assessed using correlation matrix. It was observed that oil content in V. paradoxa was only moderately correlated with kernel width. As observed in the biochemical functions of the genes conditioning the seed related traits that condition seed development and seedling germination. In concordance with this study, [36] reported that plant seed oils in angiosperms act as an important reserve of carbon and energy soon after seedling germination until it starts photosynthesis. The presence of proteins suggest their role in promoting shea bioactive functions that condition high oil yield in the species. Previous studies in shea [7] revealed similar proteins that play a major role in oil biosynthesis pathways in oil plant seeds. In fact, Wei et al. [7] predicted presence of more genes associated with oil metabolism in the shea tree genome. Another study Hale, et al. [3] predicted expansion of gene families involved in stearic acid biosynthesis in shea tree which is in agreement with this current study.
The second significant candidate gene for oil content in this study, Acyl-CoA-binding protein (ACBP) was located on chromosome 3 and 5 associated to markers S3_14843482 and S5_6275145with annotated transcriptional regulation of oil biosynthesis in seed plants. The enzyme plays a role during early fruit formation and also play multiple functions such as: tissue growth, cellular trafficking, and physiological processes [37]. The enzymes are usually in the nucleus, are expressed predominantly in developing seeds during maturation. Similar findings were also reported in Arabidopsis thaliana seeds (Yang et al., 2022). Moreover, the strong association with annotated function and Acyl-CoA-binding protein (ACBP) genes could be taken advantage of to breed for high oil yield shea tree varieties in Uganda. The biological effect of ACBP includes lipid metabolism, cellular signalling for stress management and disease resistance in plants [38]. This gene encodes metal ion binding enzyme, mostly carbonic anhydrase and alcohol dehydrogenase enzymes that contain zinc as part of their molecule. This zinc finger gene family has been reported to play a major role in oil biosynthesis pathways in the oil palm [37].
The third significant candidate genes was Ketoacyl-ACP synthase (KAS) genes. The gene plays a major role in lipid biosynthesis path ways in shea nuts, thereby increasing oil content in the species [3]. Similar findings on Chinese seed oil shrub, Paeonia lactiflora have been advanced [39]. KAS II for example is key in the biosynthesis pathways of fatty acids in plant seeds [40] and early nudling. The Fatty acid exporter (FAX2) candidate genes play a major role in biosynthesis transportation and significantly increases oil content in shea tree. In another study, Janik et al. [41] reported the involvement of FAX in Chlamydomonas reinhardtii oil synthesis, similar to this current study. On the other hand, Acyl-ACP Thioesterase Fat B (FATB) was also discovered in other plants like Koelreuteria paniculata known to be involved in the synthesis of saturated fatty acids in the species [42], which is in line with this current study. Further still, FADS genes reported in this study, is responsible for the synthesis of unsaturated fatty acids and important for plant development and response to biotic and abiotic stresses [43]. The report therefore confirms the findings in this current study for the role played by the genes in significantly controlling high oil yield in V. paradoxa Subsp. nilotica.
4.3. Candidate gene scan within the seed related traits
The seed related traits with significant SNPs under this study were considered to be having linkage with oil yield in shea nuts. The proteins responsible for oil biosynthesis identified in kernel length trait was associated to marker S8_11121701 in chromosome 8. In kernel width trait, S1_32402910 marker discovered on chromosomes 1 had proteins which are linked with processes involved in plant seed oil biosynthesis pathways [24]. For kernel weight trait, S1_30720144 and S8_43605016 markers in chromosomes 1 and 8 were associated with the proteins responsible for oil biosynthesis. In fact, Hale et al. [3,7] reported similar results with QTLs identified at different locations of shea tree genome. The proteins play a major role in ATP hydrolysis and prediction of protein residues as disordered, during plant seed oil biosynthesis processes. The first evidence was reported by Botha et al. [44] linking the functions of the genes to seed development and early seedling growth in Ricinus communis oil seeds. The genes reportedly play a major role during seed drying by concentrating inorganic phosphate while de-concentrating the extracellular pyrophosphate which inhibits formation of minerals [45].
4.4. Linkage disequilibrium (LD) and Marker association
The LD reveals the evolutionary and demographic events of a population and in mapping genes that are associated with quantitative traits. The implication of this association is that the marker loci contain a causal variant in LD with the identified marker by GWAS. This is further revealed by the small blocks in heat map where the causal variant(s) can be sought. Therefore, it is important in increasing our understanding of joint evolution of linked sets of genes. A wide range of LD (r2>0.2) in the shea tree population used in this study, is similar to that obtained in citrus [46]. Such a range of LD is expected in every heterozygous outcrossing species like shea tree [46]. The mean r2 (0.2) in the shea tree population indicates that the marker densities in the shea tree population is sufficient for genomic selection as LD is maintained by selection. This study describes the potential candidate genes associated with oil yield in shea tree. It further describes the locations of these significant genes in the chromosomes for any further verification. The significant association was discovered on chromosome 1 and 8 for seed related and oil yield traits, explaining 58% of the phenotypic variation.
Inbreeding creates LD owing to the recent common ancestry by increasing the covariance between alleles at different loci. This, therefore, offers opportunities to design association studies and allele transfer using marker-assisted selection [47,48]. LD therefore presents an opportunity in this study in that if an upper positive selection of preferred traits in shea tree is conducted, it will accelerate the frequency of alleles conferring the preferred trait during breeding. This is because as the linked loci strongly remain in LD with that allele.
4.5. Marker Assisted Selection in shea tree
The oil content candidate genes identified in this present study will be cross validated in the established multi-locational trials in NgetaZARDI and NASARRI to determine the ideal molecular markers for enhanced shea tree oil content breeding programs in the country. This can be made possible by stacking the novel genes into the shea tree genotypes with high oil content using marker-assisted selection. A combination of novel QTLs can further enhance oil content in the shea tree. Furthermore, determination of the allelic status at the markers with significant alleles for oil content will enable the selection of those significant markers for shea oil yield improvement in Uganda. The variations observed in the traits within the location but not across confirms that the species is highly outcrossing [31] or segregating population. The Analysis of variance (ANOVA) in Table 3 indicates a significant variation within the population and this further re-affirms the level of variation in the species. The results of this study points to potential QTNs that explain the genetic variations in the population. In this study, the putative major QTN for oil content explains up to 58% of the phenotypic variance in the species.
Developing MAS options that use the identified molecular markers linked to traits of interest is of importance for speeding the selection process in shea tree with high oil content [49]. The use of significant SNP markers identified through GWAS analysis are important for performing MAS for shea tree breeding. In fact, the application of MAS in shea tree breeding is now made easy with the availability of genomic information on the species [3] coupled with sequencing transcriptome that now makes it possible to align them with the identified markers of interest [3,7]. The six identified markers (S1_30720144, S1_32402910, S1_60237300, S8_11121701, S8_41696703 and S8_43605016) in this study could be applied in MAS for enhanced oil content in V. paradoxa Subsp. niltica. The MAS can play a very important role in this kind of trait useful for early nursery selection of late expressing traits in the species, and therefore, by performing MAS at seedling stage (far earlier than the juvenile maturity) will greatly reduce the breeding circle.
In this current study, the application of MAS will enable the selection of S1_30720144, S1_32402910, S1_60237300, S8_11121701, S8_41696703 and S8_43605016 markers linked with high oil content genes in the shea nuts. Selection of genotypes with a combination of preferred traits accumulated in one accession would therefore augment the process of shea tree improvement. More value to the communities as an upstream selection would also require prioritizing the genotypes with significant SNPs but from sweet pulped ethnovariety to meet the community’s food and nutrition requirement [50,51]. The availability of markers linked to the identified genes will even make it possible to take the advantage of MAS in identifying heterozygous genotypes and therefore apply positive MAS selection for the alleles resulting in a very informative phenotypic traits selected for. On the other hand, MAS could also be applied in negative selection in order to introgress the target trait.
5. Conclusion
The study of marker trait association presents an important advance for identifying the genomic regions associated with the traits of interest to further marker-assisted breeding in shea tree. We performed GWAS using mrMLM 4.0 on 623 open pollinated half sib shea genotypes for future breeding programs in Uganda. This current study identified 23 putative markers associated with oil accumulation in shea nut. Candidate genes located on chromosomes 1 and 8 were the most important genes in oil biosynthesis and accumulation in V. paradoxa. Therefore, the oil yield trait hotspots were identified on chromosomes 1and 8. It’s important to note in this study that the position of the seed related traits candidate genes were in agreement with the locations of the oil yield hotspots on chromosomes1and 8. This is in support of the need for application of MAS in shea tree and presents the first ever breakthrough in identification of chromosomes 1 and 8 hotspots in the improvement and breeding of shea treein Uganda for increased oil yield. This study therefore presents the first ever genomic information on associated genes responsible for V. paradoxa Subspecies nilotica nut oil biosythesis. The data thereof establishes the foundation for explaining the molecular mechanisms of oil biosynthesis for V. paradoxa Subspecies nilotica. The markers and their linked genes provide a powerful resource for improving oil content in the species. The study therefore sets pace for genomic assisted breeding in V. paradoxa Subsp. nilotica and also broadens our understanding in the role of genomic approaches in advancing yield component traits. The findings of this study will contribute to the initiation of shea breeding for increased oil yield in Uganda. This information could also be used for future gene pyramiding, increasing genetic gain, trait introgression, marker-assisted selection and selection of parental lines for multiplication and generation of putative genotypes for shea tree breeding programs in Uganda. The study further presents gaps for future validation of the hot spot regions identified on chromosomes 1 and 8.
Author Contributions
J.B.O., and S.G. conceived and designed the study. J.B.O., E.A.A, S.G, and T.L.O. wrote the original manuscript. J.B.O. and E.A.A analysed the data. H.P, E.A.A., M.T.B and A.D helped to edit the manuscript. All authors read and approved the manuscript.
Funding
The authors would like to extend their sincere thanks to the following institutions/organizations for funding this study: World Agroforestry (ICRAF), Makerere Regional Centre for Crop Improvement (MaRCCI), the Integrated Genotyping Support and Service (IGSS) and the Intra-Africa Academic Mobility Project for Training Scientists in Crop Improvement for Food Security in Africa (SCIFSA).
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to the fact that it will still be used in another ongoing study as part of the whole PhD study.
Acknowledgments
The authors would wish to thank the management and administration of the Cocoa Research Institute of Ghana (CRIG) for their moral support and hosting the principal author of this paper during its write up and submission. The University of Ghana, West African Centre for Crop Improvement (WACCI) is here by also greatly acknowledged for coordination of the principal authors’ stay in Ghana to finalize this paper. Finally, great thanks go to the National Agricultural Research Organization (NARO), through the Director of Research (Dr Hillary Agaba), National Forestry Resources Research Institute (NaFORRI) for the immense support rendered that made data collection, analysis and write up of this paper be a success.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Wei, Y.; Ji, B.; Siewers, V.; Xu, D.; Halkier, B.A.; Nielsen, J. Identification of genes involved in shea butter biosynthesis from Vitellaria paradoxa fruits through transcriptomics and functional heterologous expression. Appl. Microbiol. Biotechnol. 2019, 103, 3727–3736. [Google Scholar] [CrossRef]
- Naughton, C.C.; Lovett, P.N.; Mihelcic, J.R. Land suitability modeling of shea (Vitellaria paradoxa) distribution across sub-Saharan Africa. Appl. Geogr. 2015, 58, 217–227. [Google Scholar] [CrossRef]
- Hale, I.; Ma, X.; Melo, A.T.O.; Padi, F.K.; Hendre, P.S.; Kingan, S.B.; Sullivan, S.T.; Chen, S.; Boffa, J.-M.; Muchugi, A.; et al. Genomic Resources to Guide Improvement of the Shea Tree. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef]
- Global Market Insight, 2021.
- Abdul-Mumeen, I.; Beauty, D. and Adam, A. Shea butter extraction technologies: Current status and future perspective. African Journal of Biochemistry Research 2019, 13, 9–22. [Google Scholar]
- Global shea Alliance, (2021). Shea production and market.
- Wei, Y.; Ji, B.; Siewers, V.; Xu, D.; Halkier, B.A.; Nielsen, J. Identification of genes involved in shea butter biosynthesis from Vitellaria paradoxa fruits through transcriptomics and functional heterologous expression. Appl. Microbiol. Biotechnol. 2019, 103, 3727–3736. [Google Scholar] [CrossRef]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C.; et al. Diversity Arrays Technology: A Generic Genome Profiling Technology on Open Platforms. In Data Production and Analysis in Population Genomics. Methods in Molecular Biology (Methods and Protocols); Humana Press, Totowa, NJ, USA, 2018; Volume 888, pp. 67–89. [CrossRef]
- Zahid, G.; Kaçar, Y.A.; Dönmez, D.; Küden, A.; Giordani, T. Perspectives and recent progress of genome-wide association studies (GWAS) in fruits. Mol. Biol. Rep. 2022, 49, 5341–5352. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J. J. and Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 1–15. [Google Scholar]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. Diversity Arrays Technology (DArT) and next-generation sequencing combined: genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc. 2011, 5, P54. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Raman, H.; Raman, R.; Kilian, A.; Detering, F.; Carling, J.; Coombes, N.; Diffey, S.; Kadkol, G.; Edwards, D.; McCully, M.; et al. Genome-Wide Delineation of Natural Variation for Pod Shatter Resistance in Brassica napus. PLOS ONE 2014, 9, e101673. [Google Scholar] [CrossRef]
- R Core Team. (2020). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. https://www.R-project.org/.
- Wang, S.-B.; Feng, J.-Y.; Ren, W.-L.; Huang, B.; Zhou, L.; Wen, Y.-J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.-M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444–19444. [Google Scholar] [CrossRef] [PubMed]
- Laporte, F.; Charcosset, A.; Mary-Huard, T. Efficient ReML inference in variance component mixed models using a Min-Max algorithm. PLOS Comput. Biol. 2022, 18, e1009659. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Rosyara, U.R.; De Jong, W.S.; Douches, D.S.; Endelman, J.B. Software for Genome-Wide Association Studies in Autopolyploids and Its Application to Potato. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Tamba, C.L.; Wen, Y.-J.; Li, P.; Ren, W.-L.; Ni, Y.-L.; Gao, J.; Zhang, Y.-M. mrMLM v4.0.2: An R Platform for Multi-locus Genome-wide Association Studies. Genom. Proteom. Bioinform. 2020, 18, 481–487. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gatarira, C.; Agre, P.; Matsumoto, R.; Edemodu, A.; Adetimirin, V.; Bhattacharjee, R.; Asiedu, R.; Asfaw, A. Genome-Wide Association Analysis for Tuber Dry Matter and Oxidative Browning in Water Yam (Dioscorea alata L.). Plants 2020, 9, 969. [Google Scholar] [CrossRef]
- Verde, I.; Jenkins, J.; Dondini, L.; Micali, S.; Pagliarani, G.; Vendramin, E.; Paris, R.; Aramini, V.; Gazza, L.; Rossini, L.; et al. The Peach v2.0 release: high-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genom. 2017, 18, 1–18. [Google Scholar] [CrossRef]
- Ma, W.; Kong, Q.; Mantyla, J.J.; Yang, Y.; Ohlrogge, J.B.; Benning, C. 14-3-3 protein mediates plant seed oil biosynthesis through interaction with AtWRI1. Plant J. 2016, 88, 228–235. [Google Scholar] [CrossRef]
- Yang, Y.; Kong, Q.; Lim, A.R.; Lu, S.; Zhao, H.; Guo, L.; Yuan, L.; Ma, W. Transcriptional regulation of oil biosynthesis in seed plants: Current understanding, applications, and perspectives. Plant Commun. 2022, 3, 100328. [Google Scholar] [CrossRef]
- Gwali, S.; Nakabonge, G.; Okullo, J.B.L.; Eilu, G.; Forestier-Chironc, N.; Piombod, G. and Davrieux, F. (2012). Fat content and fatty acid profiles of shea tree (Vitellaria paradoxa subspecies nilotica) ethno-varieties in Uganda. Forests, Trees and Livelihoods Vol. 21, No. 4, December 2012, 267–278. 20 December; 4.
- Okullo, J.B.L.; Omujai, F.; Agea, J.G.; Vuzi, P.C.; Namutebi, A.; Okello, J.B. and Nyanzi, S.A. (2010). Physico-chemical characteristics of Shea butter (Vitellaria Paradoxa C. F. Gaertu) oil from the shea districts of Uganda. AJFAND, African Journal of Food Agriculture Nutrition and Development, 10(1), 2070-2084.
- Mattia, M.R.; Du, D.; Yu, Q.; Kahn, T.; Roose, M.; Hiraoka, Y.; Wang, Y.; Munoz, P.; Gmitter, F.G. (2022). Genome-Wide Association Study of Healthful Flavonoids among Diverse Mandarin Accessions. Plants. 11, 317. 11. [CrossRef]
- Hall, D.; Tegström, C.; Ingvarsson, P.K. Using association mapping to dissect the genetic basis of complex traits in plants. Briefings Funct. Genom. 2010, 9, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chagne´, D.; Bink, M.C.A.M.; Volz, R.K.; Whitworth, C.; Carlisle, C. (2012). Genomic selection for fruit quality traits in apple (Malus 3 domestica Borkh.). PLoS One; 7:e36674.
- Abdulai, I.; Krutovsky, K.V.; Finkeldey, R. Morphological and genetic diversity of shea tree (Vitellaria paradoxa) in the savannah regions of Ghana. Genet. Resour. Crop. Evol. 2016, 64, 1253–1268. [Google Scholar] [CrossRef]
- Gwali, S.; Vaillant, A.; Nakabonge, G.; Okullo, J.B.L.; Eilu, G.; Muchugi, A.; Bouvet, J.-M. Genetic diversity in shea tree (Vitellaria paradoxasubspeciesnilotica) ethno-varieties in Uganda assessed with microsatellite markers. For. Trees Livelihoods 2014, 24, 163–175. [Google Scholar] [CrossRef]
- Fontaine, C.; Lovett, P.N.; Sanou, H.; Maley, J.; Bouvet, J.-M. Genetic diversity of the shea tree (Vitellaria paradoxa C.F. Gaertn), detected by RAPD and chloroplast microsatellite markers. Heredity 2004, 93, 639–648. [Google Scholar] [CrossRef]
- Bates, P.D.; Stymne, S.; Ohlrogge, J. Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 2013, 16, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Xianghan, L.; Tianxiang, T.; Chao, S.; Libo, S.; Hui, Z.; Chuanli, Z.; Liping, L.; Liangbin, L. (2017). Several Key Enzymes in Oil Synthesis of the Brassica napus. Journal of the Chinese Cereals and Oils Association Issue, 1: Journal of the Chinese Cereals and Oils Association Issue; 12, 100–104.
- Zhao, H.; Kosma, D.K.; Lü, S. Functional Role of Long-Chain Acyl-CoA Synthetases in Plant Development and Stress Responses. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Jasinski, S.; Chardon, F.; Nesi, N.; Lécureuil, A.; Guerche, P. Improving seed oil and protein content in Brassicaceae: some new genetic insights from Arabidopsis thaliana. OCL 2018, 25, D603. [Google Scholar] [CrossRef]
- Osorio-Guarín, J.A.; Garzón-Martínez, G.A.; Delgadillo-Duran, P.; Bastidas, S.; Moreno, L.P.; Enciso-Rodríguez, F.E.; Cornejo, O.E.; Barrero, L.S. Genome-wide association study (GWAS) for morphological and yield-related traits in an oil palm hybrid (Elaeis oleifera x Elaeis guineensis) population. BMC Plant Biol. 2019, 19, 1–11. [Google Scholar] [CrossRef]
- Raboanatahiry, N.; Wang, B.; Yu, L.; Li, M. Functional and Structural Diversity of Acyl-coA Binding Proteins in Oil Crops. Front. Genet. 2018, 9, 182. [Google Scholar] [CrossRef]
- Meng, J.-S.; Tang, Y.-H.; Sun, J.; Zhao, D.-Q.; Zhang, K.-L.; Tao, J. Identification of genes associated with the biosynthesis of unsaturated fatty acid and oil accumulation in herbaceous peony ‘Hangshao’ (Paeonia lactiflora ‘Hangshao’) seeds based on transcriptome analysis. BMC Genom. 2021, 22, 1–21. [Google Scholar] [CrossRef]
- Wu, G-Z. and Xue H-W. Arabidopsis b-Ketoacyl-[Acyl Carrier Protein] Synthase I Is Crucial for Fatty Acid Synthesis and Plays a Role in Chloroplast Division and Embryo Development. The Plant Cell, 2010, 22, 3726–3744.
- Peter, J.; Huleux, M.; Spaniol, B.; Sommer, F.; Neunzig, J.; Schroda, M.; Li-Beisson, Y.; Philippar, K. Fatty acid export (FAX) proteins contribute to oil production in the green microalga Chlamydomonas reinhardtii. Front. Mol. Biosci. 2022, 9, 939834. [Google Scholar] [CrossRef]
- Martins-Noguerol, R.; DeAndrés-Gil, C.; Garcés, R.; Salas, J.; Martínez-Force, E.; Moreno-Pérez, A. Characterization of the acyl-ACP thioesterases from Koelreuteria paniculata reveals a new type of FatB thioesterase. Heliyon 2020, 6. [Google Scholar] [CrossRef]
- Hajiahmadi, Z.; Abedi, A.; Wei, H.; Sun, W.; Ruan, H.; Zhuge, Q.; Movahedi, A. Identification, evolution, expression, and docking studies of fatty acid desaturase genes in wheat (Triticum aestivum L.). BMC Genom. 2020, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Botha, F.C.; Dennis, D.T. Phosphoglyceromutase activity and concentration in the endosperm of developing and germinating Ricinus communis seeds. Can. J. Bot. 1987, 65, 1908–1912. [Google Scholar] [CrossRef]
- Golub, E.; Boesze-Battaglia, K. The role of alkaline phosphatase in mineralization. Curr. Opin. Orthop. 2007, 18, 444–448. [Google Scholar] [CrossRef]
- Minamikawa, M.F.; Nonaka, K.; Kaminuma, E.; Kajiya-Kanegae, H.; Onogi, A.; Goto, S.; Yoshioka, T.; Imai, A.; Hamada, H.; Hayashi, T.; et al. Genome-wide association study and genomic prediction in citrus: Potential of genomics-assisted breeding for fruit quality traits. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Kim, S.; Plagnol, V.; Hu, T.T.; Toomajian, C.; Clark, R.M.; Ossowski, S.; Ecker, J.R.; Weigel, D.; Nordborg, M. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 2007, 39, 1151–1155. [Google Scholar] [CrossRef]
- Thomson, M.J.; Ismail, A.M.; McCouch, S.R.; Mackill, D.J. (2009). Abiotic Stress Adaptation in Plants, eds Pareek A, Sopory SK, Bohnert HJ (Springer Netherlands, Dordrecht, The Netherlands), pp 451–469.
- Tartarini, S. and Sansavini, S. (2003). Advances in the use of molecular markers in Pome fruit breeding. XXVIth international Horticultural conference and exhibition, Toronto, -17 2002, Canada ISHS. Acta, Hort. 11 August.
- Odoi, J.B.; Muchugi, A. .; Okia, C.A..; Gwali, S.. and Odong, T.L. (2020). Local knowledge, identification and selection of shea tree (Vitellaria paradoxa) ethnovarieties for pre-breeding in Uganda. The Journal of Agriculture and Natural Resources Sciences 2020, 7, 22–33. [Google Scholar]
- Agúndez, D.; Nouhoheflin, T.; Coulibaly, O.; Soliño, M.; Alía, R. Local Preferences for Shea Nut and Butter Production in Northern Benin: Preliminary Results. Forests 2019, 11, 13. [Google Scholar] [CrossRef]
Figure 1.
Map of Africa indicating location of Uganda and study sites. The circles in the study sites show the actual location within the districts where shea trees were selected from in Uganda.
Figure 1.
Map of Africa indicating location of Uganda and study sites. The circles in the study sites show the actual location within the districts where shea trees were selected from in Uganda.
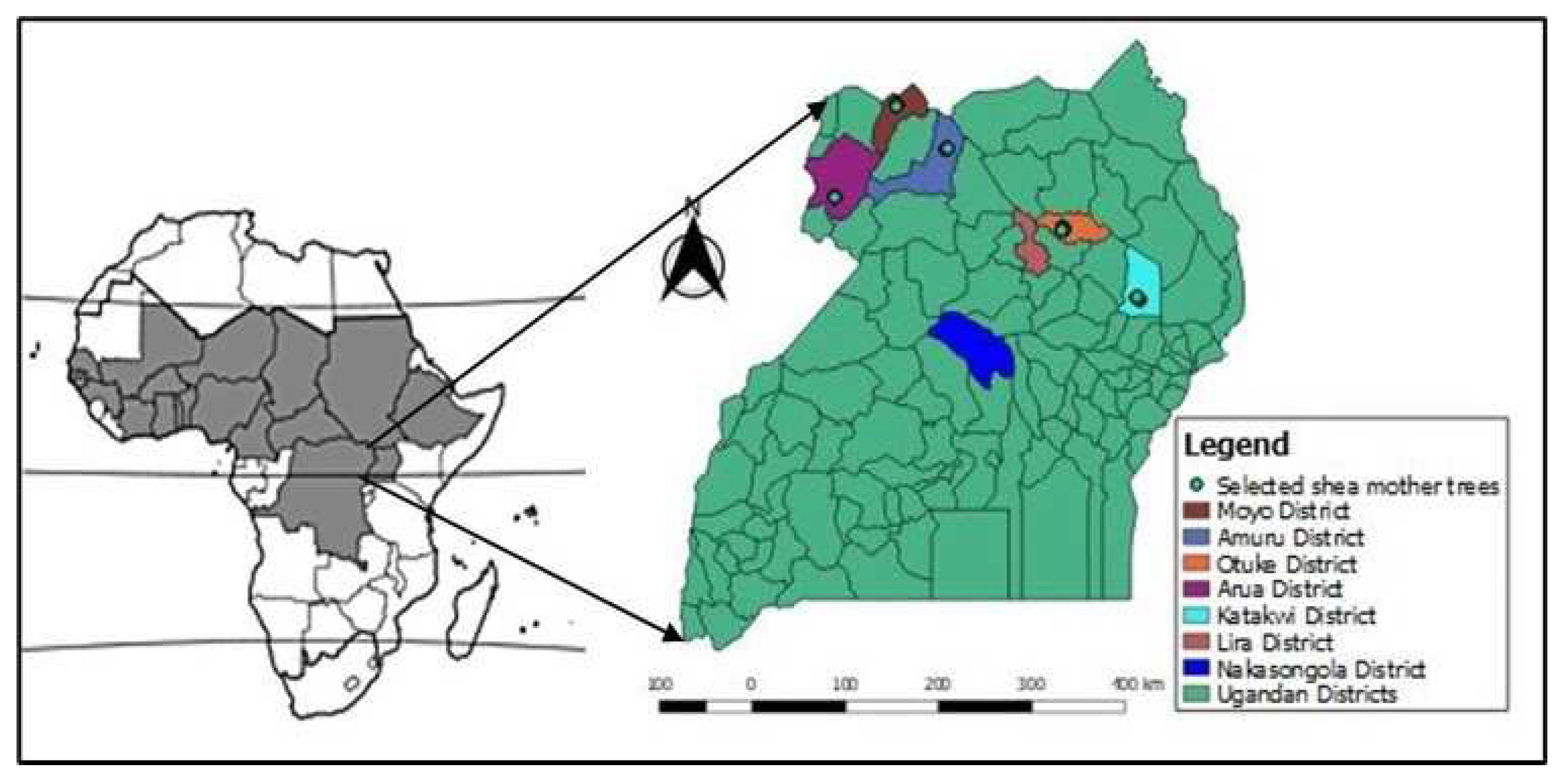
Figure 2.
Correlation (P ≤ 0.001) among four traits (width = Kernel length, Width = Kernel width and Kernel = Weight and Oil content) of the 374 Shea tree lines. Colour in the boxes indicate proportion of correlations.
Figure 2.
Correlation (P ≤ 0.001) among four traits (width = Kernel length, Width = Kernel width and Kernel = Weight and Oil content) of the 374 Shea tree lines. Colour in the boxes indicate proportion of correlations.
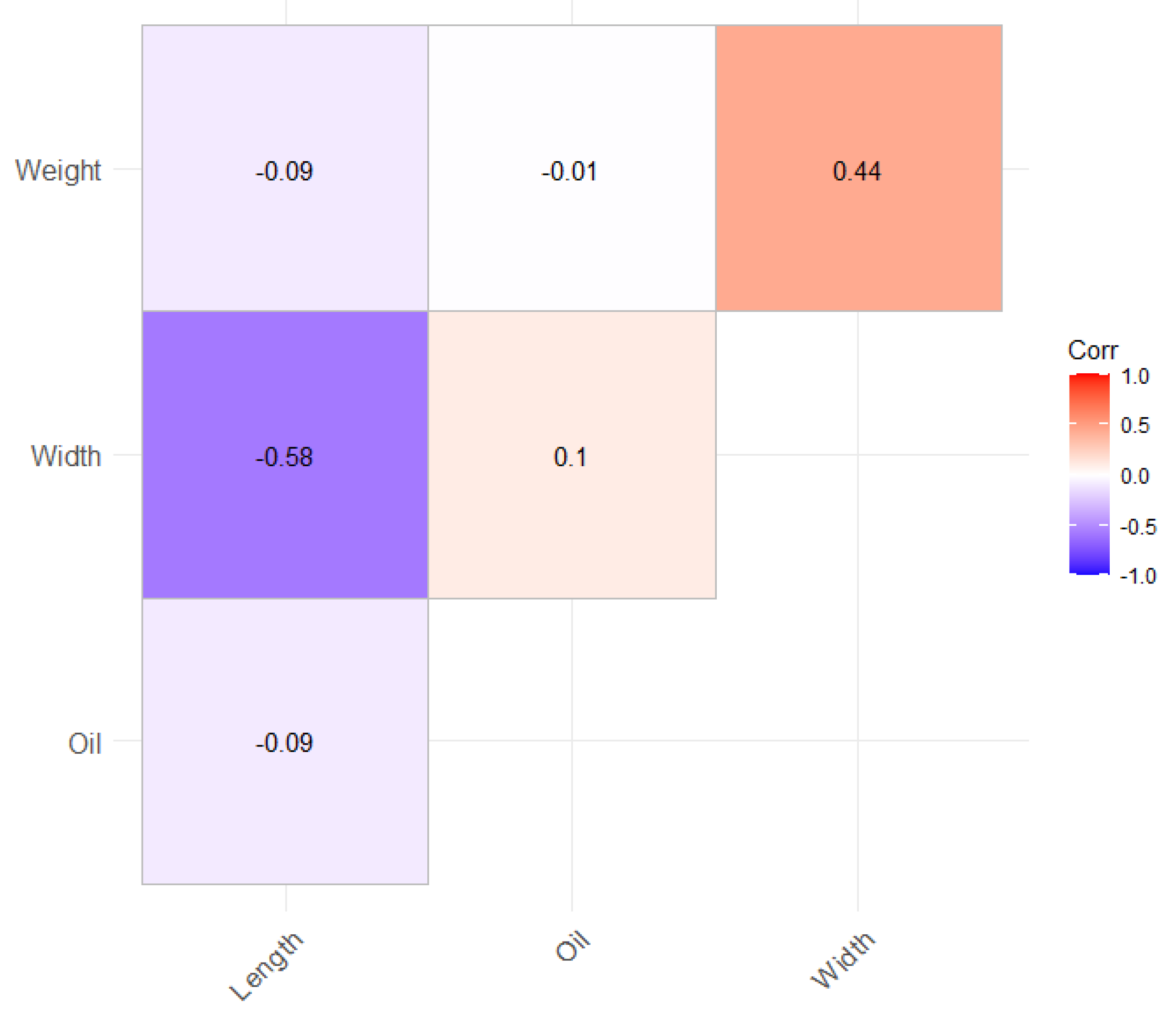
Figure 3.
The number and size of SNPs within 1Mb window size of V. paradoxa Subsp. nilotica genome.
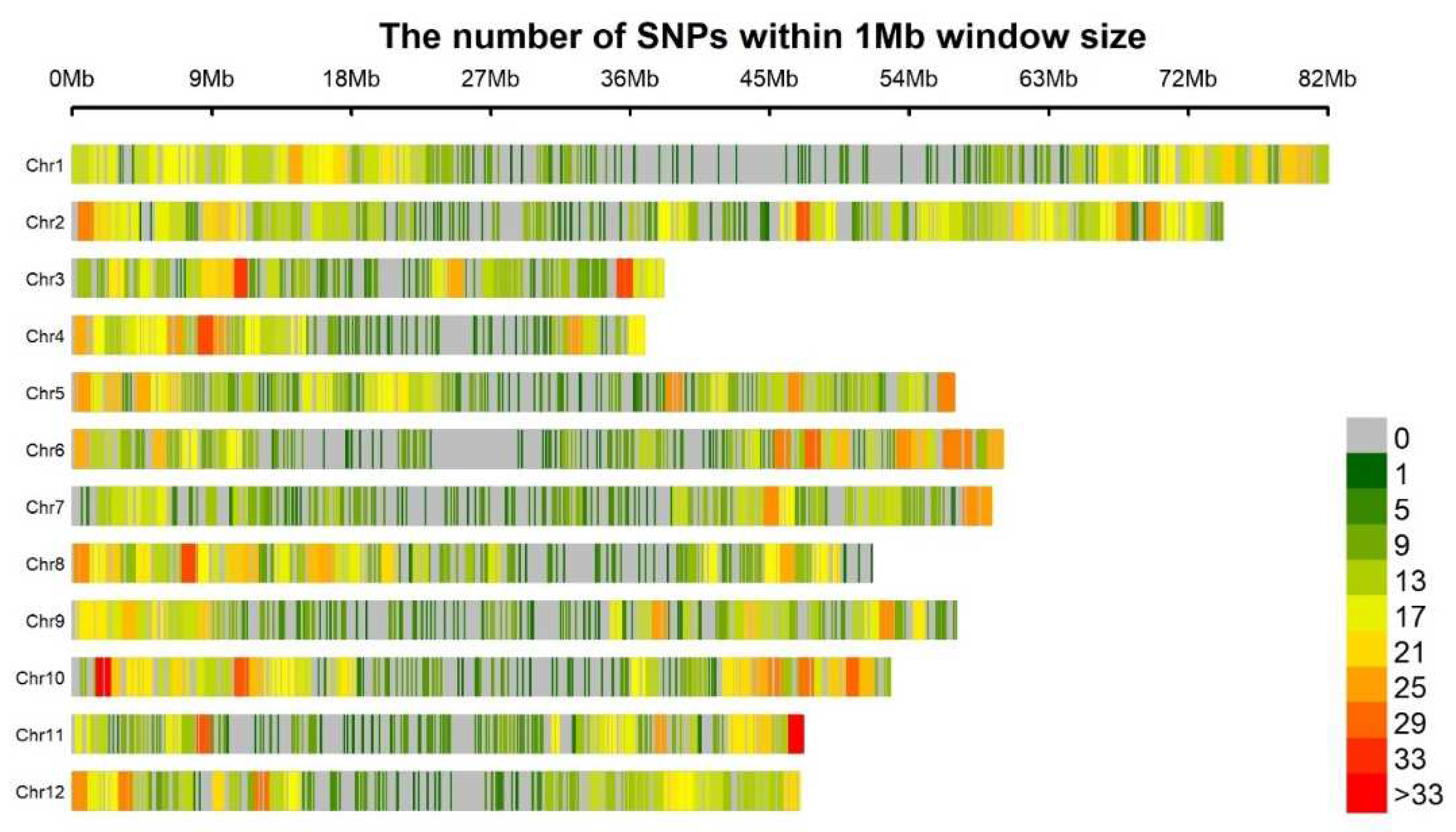
Figure 4.
Genome-wide association of Kernel dry matter oil content in a panel of 374 Vitellaria paradoxa genotypes with 7,530 SNP markers using a Multi-Locus Random Mixed Linear Model (mrMLM). (The y-axis representing the p-value of the marker-trait association on a -log10 scale and the x-axis relates to the 12 shea tree chromosomes. The purple/pink dots above the horizontal 5% Bonferroni threshold light dotted line indicates SNPs associated with QTL that condition kernel length.
Figure 4.
Genome-wide association of Kernel dry matter oil content in a panel of 374 Vitellaria paradoxa genotypes with 7,530 SNP markers using a Multi-Locus Random Mixed Linear Model (mrMLM). (The y-axis representing the p-value of the marker-trait association on a -log10 scale and the x-axis relates to the 12 shea tree chromosomes. The purple/pink dots above the horizontal 5% Bonferroni threshold light dotted line indicates SNPs associated with QTL that condition kernel length.
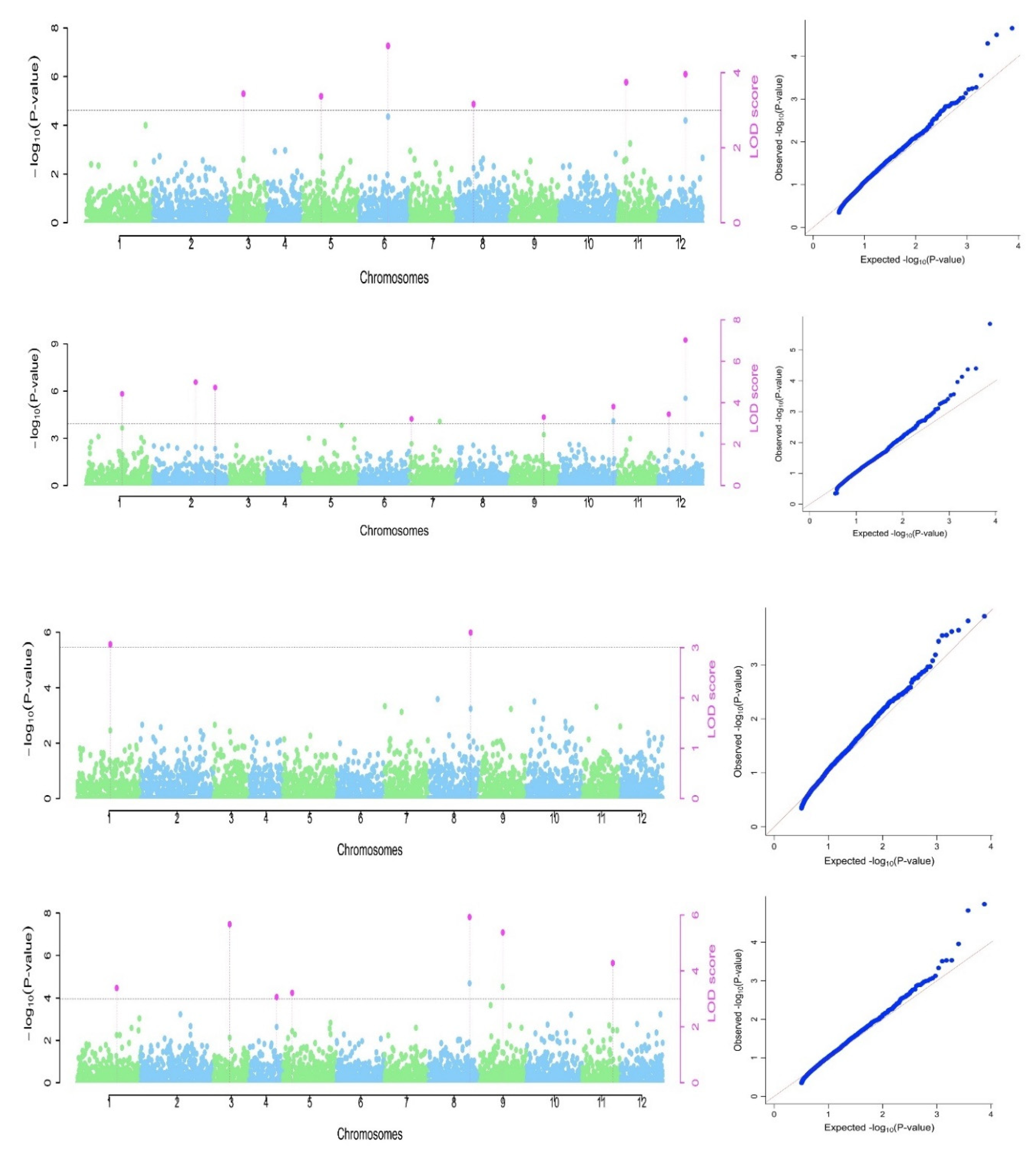
Figure 6.
Pairwise LD in r2 for a) chromosome 1 and b) chromosome 8.
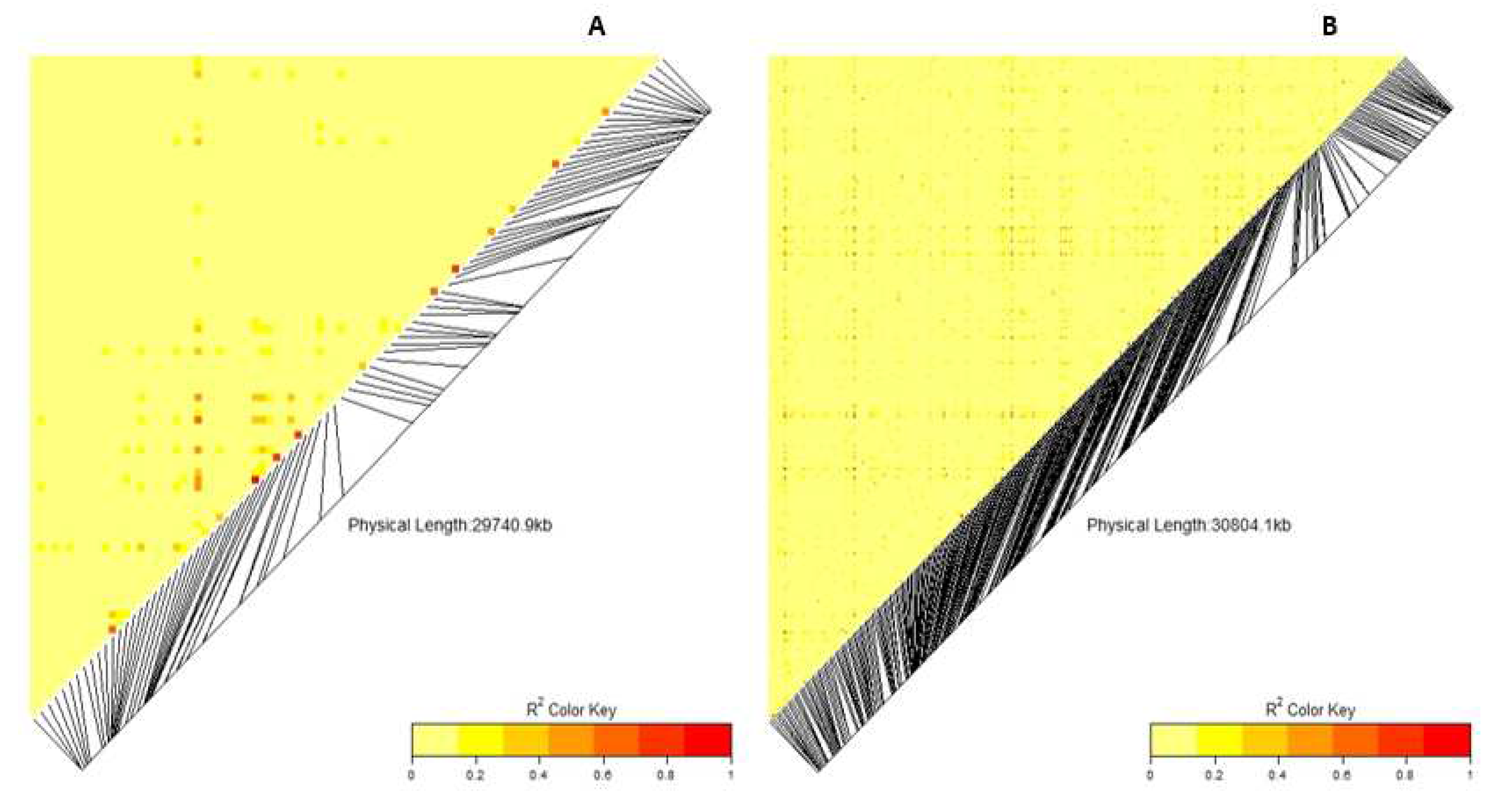

Table 1.
Summary statistics for the studied traits.
| Traits | Mean±(SD) | Minimum | Maximum |
|---|---|---|---|
| Kernel dry matter oil content (%) | 53.53±2.28 | 39.05 | 69.77 |
| Kernel length (cm) | 3.19±0.34 | 1.90 | 8.43 |
| Kernel width (cm) | 3.61±0.43 | 2.23 | 4.97 |
| Kernel weight (mg) | 10.30±0.30 | 2.00 | 18.8 |
SD = Standard Deviation.
Table 2.
Summary analysis of variance for the studied traits.
| Source of variation | Dfa | KOCb | KLc | KWd | KWte |
|---|---|---|---|---|---|
| Replications | 2 | 4.81 | 0.01307 | 0.0249 | 0.08108* |
| Environment | 4 | 1840.82*** | 0.694*** | 0.82403*** | 0.90574*** |
| Genotypes | 373 | 60.42*** | 1.45026*** | 2.54701*** | 0.9112*** |
| Genotype x Environment | 1492 | 35.9** | 0.01524 | 20.69 | 0.01666 |
| Residuals | 3738 | 8.61 | 0.0159 | 0.01553 | 0.02156 |
a Degrees of freedom; b Kernel dry matter oil content (%); c kernel length (cm); d kernel width (cm); e Kernel weight (mg) and levels of significance ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05.
Table 3.
Number of SNPs for each chromosome before and after filtration and the average polymorphism information content for V. paradoxa Sub.Species nilotica.
Table 3.
Number of SNPs for each chromosome before and after filtration and the average polymorphism information content for V. paradoxa Sub.Species nilotica.
| Chromosomes | All SNPsa | Filtered SNPs | Chrb Size (Mbs) | PICc | Gene Divd |
|---|---|---|---|---|---|
| 1 | 2893 | 805 | 82 | 0.262 | 0.32 |
| 2 | 3450 | 960 | 74.5 | 0.260 | 0.32 |
| 3 | 1545 | 430 | 38.6 | 0.261 | 0.32 |
| 4 | 1527 | 425 | 37 | 0.258 | 0.31 |
| 5 | 2336 | 650 | 56.5 | 0.261 | 0.32 |
| 6 | 2210 | 615 | 58 | 0.259 | 0.31 |
| 7 | 2088 | 581 | 57.3 | 0.262 | 0.32 |
| 8 | 2318 | 645 | 48 | 0.260 | 0.32 |
| 9 | 2124 | 591 | 56.5 | 0.262 | 0.32 |
| 10 | 2803 | 780 | 50 | 0.265 | 0.32 |
| 11 | 1791 | 498 | 47.1 | 0.265 | 0.32 |
| 12 | 1978 | 550 | 46.9 | 0.269 | 0.33 |
| Total/Mean | 27063 | 7530 | 652.4 | 0.26 | 0.32 |
aSingle Nucluotide Polymorphism; bChromosome; cPolymorphic information content and dgene diversity.
Table 4.
List of significant markers in a panel of 374 Vitellaria paradoxa genotypes indicating the genomic regions associated with studied traits.
Table 4.
List of significant markers in a panel of 374 Vitellaria paradoxa genotypes indicating the genomic regions associated with studied traits.
| Trait | Pσa | Marker | Chrb | Position (bp) | Alleles | QTN effect | LOD score | '-log10c | r2 (%)d | MAFe |
|---|---|---|---|---|---|---|---|---|---|---|
| Oil content | S1_60237300 | 1 | 60237300 | AA | 0.83 | 3.39 | 4.11 | 6.61 | 0.12 | |
| S3_14843482 | 3 | 14843482 | AA | -1.06 | 5.67 | 6.49 | 11.80 | 0.14 | ||
| S4_32032310 | 4 | 32032310 | AA | 0.74 | 3.07 | 3.77 | 6.76 | 0.19 | ||
| 4.03 | S5_6275145 | 5 | 6275145 | AA | 0.68 | 3.21 | 3.92 | 5.11 | 0.15 | |
| S8_41696703 | 8 | 41696703 | TT | -1.06 | 5.93 | 6.76 | 13.31 | 0.17 | ||
| S9_32689981 | 9 | 32689981 | CC | -1.22 | 5.38 | 6.19 | 11.52 | 0.09 | ||
| S11_43126044 | 11 | 43126044 | CC | 0.81 | 4.28 | 5.05 | 8.18 | 0.31 | ||
| kernel length | S3_11153087 | 3 | 11153087 | TT | -0.13 | 3.44 | 4.16 | 8.19 | 0.12 | |
| S5_15524578 | 5 | 15524578 | AA | 0.10 | 3.37 | 4.09 | 6.51 | 0.32 | ||
| S6_46530240 | 6 | 46530240 | TT | -0.25 | 4.71 | 5.49 | 14.55 | 0.05 | ||
| 0.095 | S8_11121701 | 8 | 11121701 | GG | -0.14 | 3.16 | 3.87 | 9.08 | 0.10 | |
| S11_8320549 | 11 | 8320549 | CC | -0.13 | 3.74 | 4.48 | 7.28 | 0.10 | ||
| S12_32853547 | 12 | 32853547 | CC | -0.18 | 3.96 | 4.71 | 9.31 | 0.06 | ||
| kernel width | S1_32402910 | 1 | 32402910 | CC | -0.19 | 4.42 | 5.20 | 9.75 | 0.12 | |
| 0.169 | S2_47786838 | 2 | 47786838 | CC | 0.16 | 4.99 | 5.79 | 9.01 | 0.26 | |
| S2_64059706 | 2 | 64059706 | AA | 0.17 | 4.73 | 5.52 | 8.28 | 0.13 | ||
| S7_3025298 | 7 | 3025298 | CC | 0.15 | 3.22 | 3.92 | 5.29 | 0.10 | ||
| S9_43700743 | 9 | 43700743 | AA | -0.18 | 3.30 | 4.01 | 7.77 | 0.11 | ||
| S10_50604452 | 10 | 50604452 | GG | 0.19 | 3.81 | 4.55 | 8.69 | 0.10 | ||
| S12_32853547 | 12 | 32853547 | CC | 0.29 | 7.02 | 7.89 | 13.14 | 0.06 | ||
| S12_7613999 | 12 | 7613999 | TT | 0.12 | 3.44 | 4.17 | 4.47 | 0.20 | ||
| kernel weight | S1_30720144 | 1 | 30720144 | CC | -0.08 | 3.06 | 3.76 | 9.20 | 0.22 | |
| 0.061 | S8_43605016 | 8 | 43605016 | CC | -0.11 | 3.29 | 4.00 | 15.70 | 0.18 |
aPhenotypic variance bChromosome, cthe negative logarithms (-log10) of the p-values dsquared correlation coefficient eminimum allele frequency.
Table 5.
Gene annotation for the significant SNPs for shea seed related traits.
| Traits | Marker | Chra | Posb | Gene ID | GO.c | Function |
|---|---|---|---|---|---|---|
| Kernel length | S3_11153087 | 3 | 11153087 | Vitpa03g07900 | IPR006968 | UVB-sensing and in early seedling morphogenesis and development |
| S5_15524578 | 5 | 15524578 | Vitpa05g09840 | GO:0005515 | ion transportation and signal transduction | |
| S6_46530240 | 6 | 46530240 | Vitpa06g28930 | PTHR23155 | Disease resistance (R) | |
| S8_11121701 | 8 | 11121701 | Vitpa08g10570 | GO:0004017 | Predicts residues in protein biosythesis | |
| S11_8320549 | 11 | 8320549 | Vitpa11g07160 | PTHR33052 | Protein translocation | |
| S12_32853547 | 12 | 32853547 | Vitpa12g19540 | GO:0003824 | Protein metabolism and gluconeogenesis | |
| Kernel width | S1_32402910 | 1 | 32402910 | Vitpa01g21080 | GO:0005515 | Consensus disorder prediction |
| S2_47786838 | 2 | 47786838 | Vitpa02g27300 | GO:0043190 | Glutathione synthetase ATP-binding | |
| S2_64059706 | 2 | 64059706 | Vitpa02g39460 | Zinc finger | ||
| S7_3025298 | 7 | 3025298 | Vitpa07g02460 | GO:0005515 | Calcium signaling | |
| S9_43700743 | 9 | 43700743 | Vitpa09g19440 | PTHR14859 | Protein binding | |
| S10_50604452 | 10 | 50604452 | Vitpa10g25960 | GO:0003677 | Chromosome cohesion | |
| S12_32853547 | 12 | 32853547 | Vitpa12g19540 | GO:0003824 | Protein metabolism and gluconeogenesis | |
| S12_7613999 | 12 | 7613999 | Vitpa12g07520 | GO:0055114 | Catalyze the oxidation of alcohols to aldehydes and ketones | |
| Kernel weight | S1_30720144 | 1 | 30720144 | Vitpa01g20620 | GO:0003676 | Hydrolyze ATP |
| S8_43605016 | 8 | 43605016 | Vitpa08g25310 | GO:0004672 | Predict protein residues as disordered |
aChromosome, bMarker chromosome position and cGene ontology.
Table 6.
Gene annotation for the significant SNPs for oil content traits.
| Traits | Marker | Chra | Posb | Gene IDc | GO.d | Function |
|---|---|---|---|---|---|---|
| Oil content | S1_60237300 | 1 | 62536299 | Vitpa01g27780 (Acyl-ACP Thioesterase Fat B (FATB)) |
GO:0004553 | Consensus disorder prediction |
| S3_14843482 | 3 | 14843482 | Vitpa03g10720 (Acyl-CoA-binding protein (ACBP)) |
GO:0005515 | Protein binding | |
| S4_32032310 | 4 | 32032310 | Vitpa04g14070 Long Chain Acyl-CoA Synthetase (LACS)) |
G3DSA | Oxidoreductase activity | |
| S5_6275145 | 5 | 6275145 | Vitpa05g04280 (Acyl-CoA-binding protein (ACBP)) |
GO:0000160 | Transcriptional regulation of oil biosynthesis in seed plants | |
| S8_41696703 | 8 | 41696703 | Vitpa08g23790 (Fatty acid exporter (FAX2)) |
GO:0008168 | methyltransferase activity | |
| S9_32689981 | 9 | 32689981 | Vitpa09g14250 (3-ketoacyl-ACP synthase II (KASII)) |
GO:0004672 | Early noduling | |
| S11_43126044 | 11 | 43126044 | Vitpa11g24760 (Fatty acid desaturases (FADs)) | abiotic stress reduction |
aChromosome, bChromosome position, cGene identification and dGene Ontology.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



