
Submitted:
30 May 2023
Posted:
31 May 2023
You are already at the latest version
Abstract
Osteoarthritis (OA) is the most common degenerative joint disease that causes chronic pain and disability. Different innate immune components, including macrophages, T cells, and neutrophils, participate in osteoarthritis pathophysiology. Neutrophils are the most abundant circulating leukocytes with multiple specialized functions contributing to innate and adaptive immune functions. Although neutrophils produce proinflammatory cytokines and chemokines, reactive oxygen species (ROS), matrix-degrading enzymes, and neutrophil extracellular traps (NET) that promote joint degradation as the first recruit cells in an inflamed joint, these cells also play an important role in joint repair by regulating the immune response, releasing anti-inflammatory factors, and activating some protective genes. In this review, various aspects of neutrophil biology, their role in inflammation and its association with osteoarthritis, and possible therapeutic approaches to target neutrophils for the treatment of osteoarthritis are described. Understanding neutrophil heterogeneity and their mechanisms of action in joint inflammation, provides a potential strategy for OA management.
Keywords:
Keywords: Osteoarthritis
; Neutrophil
; Innate Immunity
; Neutrophil Extracellular Traps (NET).
‡ These senior authors contributed equally to this work.
1. Introduction
Osteoarthritis (OA) is a painful chronic degenerative disease that causes limited movement, cartilage degradation, bone remodeling, and osteophyte formation leading to the deterioration of joint function [1]. Although the precise cause of this condition is unknown, several known risk factors, including age, obesity, gender, acute trauma, repetitive high-load activities, and irregular or repetitive overload of articular structures, all increase the likelihood of developing OA [2]. OA is a complication that affects humans and animals, particularly horses. According to some researchers, OA is the leading cause of lameness in horses, and approximately 250 million people worldwide are affected by knee OA [3]. OA is distinguished by the progressive destruction of articular cartilage and commonly affects the interphalangeal joints, spine, hips, knees, and foot [4]. Neutrophils contribute to the advancement of OA through a variety of pro-inflammatory and degenerative mechanisms [3]. This review discusses recent findings on neutrophil involvement in OA pathophysiology, focusing on secreted cytokines, chemokines, metalloproteinases, and microRNAs. Understanding the mechanisms of action of neutrophils may lead to developing novel treatments to slow the progression of OA and restore joint homeostasis.
2. Osteoarthritis
2.1. Osteoarthritis Epidemiology in Horse
One of the most commonly reported causes of lameness in horses is OA, caused by discomfort and automated annotations. Age, joint overloading, conformation, poor shoeing, prolonged joint immobility, and training intensity can all be risk factors for primary OA [5]. Furthermore, some horses might be genetically predisposed to OA because of their age or training, whereas others might never be at risk [6]. One of the critical risk factors for OA in horses is age advancement. On the other hand, OA in horses has been observed in animals as young as two years old [7]. The tremendous stresses that horses are subjected to during training and competition may cause damage to joints if they are trained at a young age. Training and racing may speed up age-related changes that occur naturally. Pathological and arthroscopic examinations of older horses have shown that OA is prevalent in their joints. Natural OA worsens with age in untrained wild horses [8]. The forces acting on the joint surfaces may vary due to improper shoeing, which can alter the limb configuration of the horse. The deterioration of articular cartilage is attributable to increased abnormal wear and loading on the joint surface due to unsuitable footwear [9]. It has been demonstrated that the prevalence of this disease in horses older than 15 years is approximately 50% and increases to 80%–90% in horses over 30 years [10]. The metacarpophalangeal (fetlock) joint is more commonly affected by OA due to the high overload received during training. A study examined the prevalence and severity of metacarpophalangeal joint OA, revealing that one-third of horses aged 2-3 years old had thickness and OA [11]. The other effective factors in the occurrence of this disease include nutrition, genetics, and management [12].
2.2. Osteoarthritis Epidemiology in Human
OA is the most prevalent joint disease, leading to disability and reduced quality of life. Epidemiologic principles can be applied to describe the prevalence of OA in the population and to investigate risk factors for its onset and progression. Most of the current information on the epidemiology of OA comes from population-based radiographic surveys [13]. A higher rate of OA has been associated with several factors, including lifestyle, gender, and jobs with high mechanical demand. In addition, the chance of getting OA can be influenced by age and the obesity epidemic. Obesity is a risk factor since the extra strain on the joints over time may result in unnecessary tension and damage [14,15]. Strength is related to physical function; increasing quadriceps strength reduces pain and improves function. Research shows that quadriceps muscle strengthening is essential for knee joint protection, and strong thigh muscles may help prevent OA worsening [16]. Based on abundant evidence, synovitis is widely accepted as a hallmark of OA. It has been possible to diagnose synovitis through histology and immunohistochemistry. Moreover, modern imaging techniques, namely magnetic resonance imaging and ultrasound, can show the features of synovitis, such as increased vascular flow, hypertrophic synovium, and effusion [17]. Mechanisms can be elucidated and prevention opportunities are identified with the help of advances in risk factor measurement via imaging, systemic and local biomarkers, and improved methods of measuring symptoms and their impact [18].
2.3. Pathogenesis
The pathogenesis of OA is characterized by continuous molecular and cellular changes in subchondral bone, articular cartilage, ligaments, synovium, and periarticular muscles [19]. Modern imaging technology identifies OA as a joint disease involving multiple tissues that confer various phenotypes. In particular, the subchondral bone is a critical part of the pathogenesis of OA. Extracellular matrix (ECM) interactions are mediated by cell surface integrins, which play a crucial role in the pathophysiology of articular cartilage. Integrins regulate cell/ECM signaling in physiological conditions. It is necessary for controlling maturation, differentiation, and cartilage homeostasis [20].
During the OA process, overuse and mechanical stress result in the induction of pro-inflammatory cytokines and regulators. One of the most critical cytokines in the inflammatory cycle of OA is IL-1, which has several roles and induces the release of some matrix metalloproteinases (MMPs) as the leading cause of enzymatic destruction of the joint capsule. In addition, IL-1 stimulates fibroblasts in chronic inflammation to increase the production of types I and III collagen, which could lead to the fibrosis of articular cartilage [3,21]. Tumor necrosis factor-α (TNF-α) stimulates the release of proteases, namely aggrecanases, collagenases, and MMPs, which degrade aggrecan and collagen [3]. Furthermore, these cytokines also stimulate the synthesis of prostaglandin E2 (PGE2) and nitric oxide (NO) [3].
Other important cytokines include insulin-like growth factor-1, transforming growth factor beta (TGF-β), and some pro-catabolic cytokines, which have been shown to trigger the enzymatic degradation of the cartilage matrix. Mediators of inflammation and oxidative stress conspire to damage chondrocyte function and viability by reprogramming them to undergo hypertrophic differentiation called early ‘senescence’ [2].
The two most notable structural changes associated with OA are the loss of cartilage and the development of osteophytes. These changes are readily noticed radiographically, and the objective measurement of disease severity is based on the degree of joint space loss (reflecting cartilage loss) and the existence of osteophytes. Moreover, subchondral osteosclerosis was present in the early phases of the OA, a procedure possibly accompanied by microfractures and recommended as a pathogenic factor in cartilage degeneration. In addition to these structural changes, OA has multiple joint and periarticular soft tissue modifications. These may include synovial hyperplasia and synovial fluid retention [22].
3. Involvement of Neutrophils in the Pathophysiology of Osteoarthritis
3.1. Immune Cells in Joint Injury and Repair
The innate immune system is essential in the host’s defense against microbial invasion and the modulation of various types of tissue injury and repair [23]. Immune responses within the joint cavity have regulatory roles in driving cartilage injury toward repair or destruction. Restoration and healing, if they occur, are accompanied by the secretion of anti-inflammatory factors from immune cells and chondrogenesis. However, once damaged, the affected cartilage cannot regenerate itself. In this pathological condition, augmented inflammatory responses develop cartilage injury to OA [24]. Articular cartilage has a limited repair capacity due to the absence of lymph tissue, blood vessels, and nerves. Macrophages, T cells, natural killer cells, dendritic cells (DCs), and neutrophils are among the immune cells primarily involved in cartilage injury and repair [24]. Under osteoarthritic conditions, neutrophils are the first immune cells to enter the synovium and affect tissue degeneration via neutrophil elastase (NE) and the release of inflammatory cytokines and chemokines [2]. This review focuses on the importance of neutrophils in OA and its therapeutic prospects.
3.2. Trafficking of Circulating Blood Neutrophils to the Synovial Cavity
Synovial fluid in a healthy joint contains no or few immune cells. The synovium is the most critical joint structure in its ability to mount an inflammatory response [25]. Neutrophils are the most common immune cell found in inflamed joint synovial fluid. Neutrophils in synovial fluid were highly activated, as evidenced by increased CD11b and CD66b [26]. Stimulation of nuclear factor κB (NF- κB) ligand (RANKL) expression in extravasated neutrophils is primarily received by toll-like receptor 4 (TLR4). In more detail, TLR4 activation and downstream signaling pathways are triggered by endogenous products, such as fibronectin, hyaluronic acid, and tenascin C released from the digested matrix. In turn, the expression of transcription factor NF-κB in neutrophils results in the accumulation of other leukocytes and the production of chemokines and pro-inflammatory cytokines in involuting joints [27].
3.3. Neutrophil-Derived Cytokines, Chemokines, and Enzymes
The role of neutrophils in bone repair is still being debated, and many studies are being conducted to manipulate neutrophils to improve healing. These investigations include targeting neutrophil maturation, interfering with neutrophil accumulation at the injury site, and reversing the detrimental changes in neutrophil function [28]. In acute and chronic inflammatory states, neutrophils are activated and release superoxide, cytokines, chemokines, and destructive tissue proteinases, such as elastase, in response to various stimuli [29]. The C-C motif chemokine ligands, including CCL2, CCL3, and CCL4, and C-X-C motif chemokine ligand CXCL8 (IL-8), are the most chemokines released by neutrophils detected in active synovial inflammation. Chemokines, such as monocyte chemoattractant protein (MCP-1) or CCL2 and macrophage inflammatory protein (MIP-1) or CCL4, have a positive association with joint pain. CCL19 and CCR7 are highly expressed in OA, leading to vascular endothelial growth factor (VEGF) secretion and further neoangiogenesis [30]. In addition, the expression of CXCL8 and CXCR2 as neutrophil chemoattractants is elevated during OA [31]. Cytokines are involved in cartilage homeostasis, degeneration, synovial activation, and inflammation. TNF-α accelerates chondrocyte apoptosis by increasing reactive oxygen species (ROS) and altering mitochondrial activity [32,33]. Chondrogenic progenitor cells (CPCs) are more famous for their anti-inflammatory function and cartilage repair during joint disturbance. These cells are attracted to the site of injury by the chemoattractant high-mobility group box I protein (HMGB1) [34]. However, neutrophilic TNF-α inhibits CPCs migration and interferes with joint healing [35]. IL-1 plays a role in tissues’ physiological and pathological processes [36]. IL-1 signaling can induce neutrophil production, recruitment, degranulation, and NET formation directly or indirectly. In turn, neutrophil proteases can activate IL-1 family members, potentially amplifying inflammation at the sites of injury or infection. IL-1 beta stimulates the expression of MMP-13, a collagenase and a critical enzyme that promotes the irreversible destruction of cartilage collagen in OA (Figure 1) [37,38]. IL-22 plays a crucial role in the development of arthritis. In the face of the large body of studies about the function of fibroblast-like synoviocytes (FLSs) in rheumatoid arthritis (RA) pathogenesis, little evidence is available about the interaction of FLSs with neutrophils in the joint microenvironment of OA. Carrion et al. showed that in addition to synovial fluid neutrophils as a source of alarmins, the expression of S100A8/A9 heterodimer is also incited by IL-22 in OA FLSs. In a feed-forward loop, S100A8 and S100A9, as the ligands of TLR4, activate the IL-22/IL-22R1 axis leading to the exacerbation of joint inflammation [39]. IL-17 and IL-22 are mainly released by Th17 and evoke immune responses by activating and recruiting neutrophils and increasing these cells’ life span in the inflamed synovium. Deligne et al. found that these cytokines are expressed at higher levels in inflamed synovium than non-inflamed synovium and promote the OA pathophysiology. In this process, TGFB1, IL-23, and IL-6 improve the production of IL-17 and IL-22 by regulating the differentiation of Th17 cells [40]. Furthermore, IFN-γ and IL-17 aggravate cartilage erosion by the up-regulation of Fc gamma receptors (FcgammaR) in macrophages and neutrophils and by raising local FcgammaR-carrying neutrophils, respectively (Supplementary Table S1) [41].
3.4. Neutrophil Elastase
As polymorphonuclear leukocytes, neutrophils are usually the first to be recruited to the inflammatory site [42]. During inflammation, neutrophils release NE, a granule serine protease, which is a functional biomarker for the progression of OA. NE is a neutrophil serine protease found in neutrophil cytoplasmic blue granules. The degeneration of articular cartilage, menisci, ligaments, and capsules may be caused by NE [43,44]. Elastase, a novel pro-MMP-13 activator, impacts cartilage collagen destruction in OA patients with synovitis (Figure 1)[38].
3.5. MicroRNAs Expression by Neutrophils
MicroRNAs (miRNAs) are small non-coding RNAs essential in regulating gene expression. They bind to their cognate mRNAs, degrade the target mRNA, or block the translation process. Several miRNAs are released from neutrophils and other cells and modulate different molecular mechanisms, such as cytokine and chemokine secretion, apoptosis, and chondrogenesis [45,46]. These miRNAs’ high stability and specificity provide a foundation for their use as biomarkers for diagnostic purposes. The feasibility of profiling circulating cell-free miRNAs as biomarkers for various diseases has been demonstrated in cancer, myeloproliferative disorders, cardiac hypertrophy, failure, viral infections, nervous system disorders, and OA [47]. Sequence analyses on many mRNAs and miRNAs revealed the differential expression between lesioned and preserved OA cartilage [48]. Neutrophils secrete miRNAs, which can be upregulated or downregulated in OA environments. Specific miRNAs, such as miRNA-141, have been proposed to be essential in bone resorption inhibition [2]. MiR-223 is found in granulocytes, namely macrophages, and neutrophils, which secrete matrix proteases and inflammatory cytokines. MiR-223 expression was significantly related to keratan sulfate derived from articular cartilage degradation and was more abundant in OA synovium than in healthy synovium. Moreover, it is thought to be an essential factor in osteoclastogenesis and to likely participate in cartilage destruction, particularly in the early stages of OA [49]. While the expression miR-223 increases during the early stages of OA, the high amount of miR-155 is more related to late-stage OA [48]. Also, miR-146a-5p and miR-155-5p overexpression is reported to reduce the secretion of pro-inflammatory cytokines from neutrophils. MiRNAs play a critical role in the pathogenesis of OA. [2].
3.6. Neutrophils in Cartilage Degradation
Multiple inflammatory mediators released into joint synovial fluid are key in synovial inflammation and cartilage degradation. Metalloproteinases’ degradation of type II collagen is the primary cause of progressive joint damage [50]. Neutrophils are recruited to the injury site as inflammatory cells to initiate tissue repair. Serine proteases, such as NE, released by immune cells in inflammatory areas damage articular cartilage and subchondral bone [51]. When NE and ROS are combined, they aid in the degradation of phagocytic microorganisms in phagocytic lysosomes, which controls inflammation and the immune response. Furthermore, NE was found in significantly higher concentrations in the synovial fluid of OA patients, indicating that NE may play a significant role in the pathophysiology of OA [42].
3.7. Synovium, Cartilage, Subchondral Bone, and Innate Immunity
All the pathological stages of OA are affected by innate immunity. The high expression of TLR4-mediated inflammatory pathway proteins, including TLR4, MyD88, and NF-κB, positively correlates with more severe tissue damage[52]. The synovium is the primary site of articular inflammation in OA and is frequently characterized by synovial lining cell hyperplasia and fibrosis, combined with the infiltration of inflammatory cells. Synovitis may cause progressive ossification and articular cartilage deterioration in the early stages of OA. Therefore, synovial membrane lesions can be predictive markers for progression of articular cartilage damage. However, it is not completely clear how these changes are related. The activation of pattern-recognition receptors (PRRs) within tissues, such as the joint, causes rapid-onset inflammatory responses, followed by the initiation of adaptive immune responses, and, finally, healing reactions in the case of tissue injuries [23,53]. Interactions between immune cells localized in the synovium initiate enzymatic activity and aberrant inflammatory reactions in the afflicted areas. Well-documented evidence implicates those macrophages and neutrophils are drivers of the cellular and molecular mechanisms of sterile and inflammatory arthritis pathology in a significant number of patients. A high neutrophil-to-lymphocyte ratio (NLR) in the synovial fluid of patients with severe knee OA corresponds to OA perpetuation [54,55,56,57]. Macrophages are the most common type of immune cells in the synovium of OA patients. The response of synovial macrophages to cartilage fragments and intracellular proteins released by necrotic cells induces the production of cytokines, including IL-1β and TNF-α, both of which contribute to the degradation of cartilage and alterations in bone[17,58]. However, neutrophil activity heavily influences the progression of OA by releasing different cytokines and chemokines within the synovial fluid, subchondral bone remodeling, and by triggering cartilage destruction and chondrocyte apoptosis [54].
Subchondral bone differs from cartilage because it is highly vascularized, allowing significant tissue turnover and remodeling to adapt to mechanical loads. Subchondral bone inflammation can cause the production of angiogenic factors and local MMPs, which are thought to promote cartilage degeneration and osteophyte formation [59]. The subchondral bone is a foundation for a joint’s articular cartilage. OA was thought to result from articular cartilage wear and tear, but new evidence suggests that subchondral bone disturbance and synovial inflammation can initiate and exacerbate the disease [60]. CXCL10 may be used as a biomarker for disease activity in arthritis. Neutrophil-derived CXCL10 is up-regulated in inflamed synovial fluids and chondrocytes and plays an essential role in inflammation enhancement and disease establishment [61,62].
The CXCR3/CXCL10 axis controls Neutrophil-NK cell reciprocal interaction. More specifically, the localization of CXCR3-expressing cells, including NK cells, T cells, and B cells, in the synovial fluid of OA patients is regulated by neutrophils. To exert their pathogenic role, neutrophils activate the CXCR3/CXCL10 axis, leading to cartilage breakdown and bone remodeling [61].
The neutrophil-osteoclast cross-talk and subsequent osteoclastogenic processes are initiated by the secretion of neutrophil CXCL2, which activates the NF-kB pathway within osteoclast precursor cells. Osteoclasts are also important cells in subchondral bone remodeling. Osteoclasts are recruited to the subchondral bone plate, creating calcified chondral microcracks and causing focal subchondral bone loss. Microfissures and invading blood vessels facilitate the diffusion of osteoclast mediators into the cartilage interface and enhance cartilage matrix degradation [63].
4. Neutrophil Biology, Recruitment, and Function in Inflammation
4.1. Neutrophil Life Cycle (Neutrophil Mobilization, Clearance, and Circulation)
Neutrophils are produced and stored in the bone marrow in a large “bone marrow reserve storage pool.” During inflammation or in response to infection, these mature neutrophils could be quickly mobilized, resulting in a drastic increase in circulating neutrophils [64]. This rapid egress of mature neutrophils from the bone marrow can lead to a 10-fold rise in circulating neutrophils within hours. The process of neutrophil mobilization represents an essential step in the trafficking of these cells to inflammation sites [65,66]. For several years, it has been demonstrated that most inflammatory responses are associated with the selective and rapid mobilization of mature neutrophils from the bone marrow; however, the exact factors that mediate these reactions have remained unknown.
Nevertheless, a wide variety of chemotactic factors, such as the chemokines interleukin IL-8, C5a, and B4, can induce a rapid increase in blood neutrophils when injected intravenously into mice and rabbits, indicating these factors create chemotactic gradients that in turn lead to a neutrophil exit from the bone marrow [65,66]. Chemokines generated at the sites of inflammation generally organize the recruitment of neutrophils from the blood into tissues [67]. Following the recruitment of neutrophils to the sites of inflammation, there is an urgent need for the clearance of infiltrating cells to prevent tissue damage caused by the excessive release of cytotoxic mediators from dying cells. Neutrophils die by a process called apoptosis, and tissue macrophages, which are present at the site of inflammation, recognize and phagocytize apoptotic neutrophils by the specific molecules expressed on their surface [68,69]. The spleen and liver have been considered the primary sites destructing mature neutrophils. However, it has recently been demonstrated that radioactivity can be detected in the bone marrow up to 24 h after intravenous injection of radiolabeled neutrophils [70,71]. The distribution of neutrophils was 29% in the liver, 32% in the bone marrow, and 31% in the spleen, which shows that these three sites equally contribute to neutrophil clearance [72].
4.2. Neutrophils in Acute and Chronic Inflammation
Under normal conditions, neutrophils are the first responders to acute inflammation. These cells contribute to tissue repair and resolution by phagocytizing dead and necrotic cells to prevent them from absorbing more immune cells. They also express some proteases essential for tissue repair, such as serum MMP-9, a critical extracellular matrix-digesting enzyme. Serum MMP-9 degrades various intracellular matrix components: annexin, actin, tubulin, and HMGB1 [73]. Metalloproteinase also promotes revascularization by activating VEGF at an injured site [74]. In addition, neutrophils directly release VEGFA to promote angiogenesis. Neutrophils also actively synthesize and secrete inflammatory mediators, such as chemokines, cytokines, prostaglandins, and leukotrienes [75]. In OA and synovial inflammation, neutrophils are recruited into the injury site and release proinflammatory mediators that promote OA progression. Serine proteases, including protease 3 (PR3), neutrophil elastase (NE), trypsin, and cathepsin G (CG), can cause damage to joint cartilage [44]. Furthermore, neutrophilic proinflammatory cytokines such as TNF-α, IL-6, IL-7, and TGF-β found in the synovial fluid of OA joints [76].
Neutrophils have also been shown to play an important role in chronic inflammation in recent years. These white blood cells continuously migrate to the sites of chronic inflammation and help drive the process by releasing some enzymes, such as serine proteases, forming neutrophil extracellular traps (NETs), and activating different immune cells [77].
4.3. Neutrophil Crosstalk/Interaction with other Circulation Cells (Platelets, Adaptative Immune Cells, Monocytes/Macrophages)
In chronic inflammation, the mutual activation of platelets and neutrophils leads to a prothrombotic state. Several kinds of neutrophil-platelet complexes are detected in humans with diverse inflammatory diseases. This interaction between neutrophils and platelets triggers critical neutrophil processes, such as forming NETs and adhesion to the endothelium. For example, it has been shown that in type 2 diabetes mellitus, the number of platelets increases due to NET secretion. NETs affect thrombopoietin and platelet production [73,74]. Neutrophils can interact with adaptive immune cells, including B cells, by secreting contact-independent signals, such as B-cell activating factor. The level of this factor, expressed on the surface of neutrophils and in intracellular stores, increases chronic inflammation and is involved in the differentiation, maturation, and survival of B cells. Some populations of neutrophils, such as myeloid-derived suppressor cells (MDSCs), also interact with T cells in inflammatory conditions [78]. At the site of inflammation, neutrophils release some factors, such as cathepsin G, proteinase 3, LL-37, and human neutrophil peptides 1–3, leading to monocyte recruitment. Neutrophils can help macrophage polarization and monocyte differentiation [74,75,79]. Macrophages can recruit neutrophils to the site of inflammation by releasing various chemotactic factors, such as the chemokine C-X-C motif ligand 1 and the C-C motif chemokine ligand CCL2. In addition, macrophages can extend the neutrophil lifespan by secreting TNF-α, granulocyte colony-stimulating factor (G-CSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF) [74,75,80].
The crosstalks of neutrophils in chronic arthritis inflammation are limited to rheumatoid arthritis or indirectly related to OA [81,82]. The expression of serine elastase gives this ability neutrophils to transmigrate extracellular matrices and altering their endothelial-associated chemotactic activities, modulate the trafficking of different leukocyte subsets, including T cells [83].
4.4. Neutrophil Heterogeneity in Chronic Inflammation
It has been proven that, similar to other immune cells, neutrophils also can switch into various phenotypes with different functions during inflammation. These differences reflect the developmental stage of neutrophils as well as their activation status. Therefore, neutrophils differ in surface markers, nuclear morphology, NET generation, buoyancy, migratory and phagocytic capacity, and immunomodulatory function. Inflammation-related neutrophils have different characteristics than neutrophils involved in other diseases, and these cells have significant heterogeneity even in a similar environment [79]. In chronic inflammation, a heterogeneous population of immature and mature neutrophils, known as low-density neutrophils (LDNs), are present in the mononuclear cell fraction [80]. The LDNs were first described in RA and systemic lupus erythematosus [84]. The LDNs include granulocytic/polymorphonuclear MDSCs (PMN-MDSCs), which have immunosuppressive effects. In addition, low-density granulocytes (LDGs) are a group of the LDN population characterized by pro-inflammatory properties [78].
4.5. New Insights into Neutrophil Extracellular Traps in Inflammation
Neutrophils are crucial cells that play a significant role in the innate immune system’s fight against invading pathogens. During acute inflammation, they are the first responders that kill microbes through phagocytosis and degranulation [85]. These cells also release some structures known as NETs into the extracellular environment to kill pathogens. NETs mainly comprise histones, chromatin DNA, and granular proteins [86]. The granule proteins include myeloperoxidase (MPO), NE, proteinase 3, pentraxin 3, cathepsin G, peptidoglycan-binding proteins, lactoferrin, and DNA-free histones [87]. In addition to their role in killing extracellular pathogens, NETs can be a source of autoantigens in cases of autoimmune diseases [88]. During NET formation, neutrophils may undergo a cell death program. In this process, the citrullination of histone and activation of NADPH oxidase is essential. Histone citrullination helps chromatin decondensation, a vital part of NET formation. Nuclear peptidyl arginine deiminase-4, expressed by neutrophils, catalyzes histone hypercitrullination. Furthermore, by promoting nuclear membrane disappearance, ROS also helps chromatin and granule proteins to be released into the cytoplasm [86,89].
It has been shown that NETs and neutrophil elastase play a crucial pathogenic role in promoting synovial inflammation and cartilage damage. In addition, during NET formation, some potent enzymes that can promote tissue injury are secreted into the extracellular space. Khandpur et al. (2013) showed accelerated NETosis in rheumatoid arthritis pathogenesis which may promote aberrant adaptive and innate immune responses in the joint [90].
5. Neutrophils as a Target for Osteoarthritis Treatment
As one of the major immune cells, neutrophils have a dual role in the pathogenesis of arthritis. On the one hand, these cells act as a mediator with considerable potential to destroy the articular cartilage and other joint components by releasing pro-inflammatory cytokines, destructive enzymes, and chemotactic factors that stimulate the migration of other immune cells. On the other hand, neutrophils have long-term homeostatic functions, including orchestrating the resolution of inflammation and contributing to articular cartilage repair as a regulator of immune response and a player in changing their phenotypic plasticity [24,91,92]. Because of the intimate role of neutrophils in arthritis, they have emerged as therapeutic targets [2]. Several aspects of neutrophil biology may be therapeutically targeted, including production, recruitment, function, and apoptosis (Supplementary Table S2) [93,94].
Neutrophils contribute to producing and releasing many cytokines, chemokines, and enzymes within the joint that promote OA progression [2]. Non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and hyaluronic acid are the most commonly used medications for OA to alleviate joint pain, inflammation, and function [95,96]. NSAID-mediated anti-inflammatory mechanisms independent of inhibiting COX and PGE2 release have been proposed [97]. These medicines inhibit neutrophil aggregation and degranulation [98]. Moreover, NSAIDs can reduce C5a- and CXCL8-induced neutrophil migration and F-actin polymerization through integrin downregulation or PI3K/Akt pathway [97]. Despite the immunosuppressive effect glucocorticoids have on immune cells, they exert multiple and even contradictory effects on neutrophils. They can inhibit or induce apoptosis in neutrophils. Likewise, glucocorticoids may have anti-inflammatory or pro-inflammatory effects on neutrophils [99]. Glucocorticoids also influence neutrophils’ maturation, extravasation, adhesion, metabolism, and activation via signaling pathways. Hyaluronic acid exerts protective effects against arthritis through different mechanisms [99,100]. Hyaluronic acid interacts with joint cells, including synoviocytes, chondrocytes, osteocytes, and immune cells, and affects inflammatory mediators [101,102]. AKT has a pivotal role in OA, and it was found that hyaluronic acid significantly reduced the p-AKT expression level in synovial-fluid neutrophils. Furthermore, hyaluronic acid can reduce the levels of phosphorylated p38MAPK, NF-κB, p53, Bax, and Caspase-3 in synovial fluid neutrophils that indicates the modulatory effect of hyaluronic acid on pro-inflammatory responses and pro-apoptotic events [103].
Different anti-cytokine strategies, including antibodies against pro-inflammatory cytokines, such as TNF-a and IL-1β, or the use of anti-inflammatory cytokines, namely TNF-β, IL-4, and IL-10, have been investigated for arthritis treatment. TNF-α is a pro-inflammatory cytokine produced by neutrophils with a considerable role in inflammatory arthritis and cartilage and bone degeneration [67,104,105,106,107]. Different TNF-α blockers have been evaluated for treating arthritis, especially inflammatory RA [105,106]. A reduction in peripheral blood neutrophil count was reported following treatment with anti-TNF-α in arthritic patients [107]. TNF-α inhibitors may affect neutrophils in different ways. They attenuate the generation of pro-inflammatory cytokines, chemokines, and MPO by neutrophils and also contribute to the upregulation of adhesion molecules expression and priming of respiratory burst in adherent neutrophils [108,109].
Moreover, anti-TNF-α agents can reduce neutrophil ROS production and the influx of neutrophils from inflamed joints [108]. CD69 is a type II membrane protein expressed on T and B cells, platelets, eosinophils, and activated neutrophils with a crucial role in inflammatory joint diseases [109,110]. Anti-TNF-α has been capable of inhibiting CD69 expression on arthritic neutrophils and downregulating neutrophil chemoattractant IL-8. Therefore, these cells can be targets for anti-TNF-α treatment [111,112]. It was demonstrated that TNF-α inhibition did not modify neutrophil immune functions against pathogen agents and was tolerated in terms of serious adverse effects. However, it has been suggested that neutropenia caused by TNF inhibitors may be due to accelerated apoptosis, and patients at higher risk of developing neutropenia had a low baseline neutrophil [107,111,113].
IL-1 family, particularly IL-1β and interleukin-1 receptor, is the key component linked to the pathogenesis of arthritis [114,115]. Consequently, the reduced synthesis of proteoglycans by chondrocytes and increased synthesis of proteolytic enzymes, NO, and other cytokines mediated by IL-1β cause cartilage destruction in OA [116,117]. This cytokine induces neutrophil production, recruitment, degranulation, and NETosis. Moreover, it delays neutrophil apoptosis through different mechanisms, such as upregulating neutrophil chemoattractants (i.e., CXC- and CCL- chemokines), inducing adhesion molecule expression and local chemokine production (IL-8), as well as upregulating anti-apoptotic agents (i.e., Bcl-2 family and Mcl-1) [37,118]. Furthermore, releasing IL-1β by neutrophils causes positive feedback during the autoinflammatory process by binding to the type 1 IL-1 receptor [119]. IL-1Ra inhibits cytokine-induced catabolism, and IL-1 deficiency protects joints from inflammation in induced arthritis [116,120]. It has been proposed that the therapeutic function of IL-1 receptor antagonist (IL-1Ra) may be more effective by reducing neutrophil recruitment into the joint cavity rather than increasing apoptosis or inhibiting the activation of neutrophils.[118]. Evidence of the beneficial role of IL-1Ra as a disease-modifying OA drug (DMOAD) was demonstrated in experimental autoimmune arthritis and OA models [120]. Anakinra is a recombinant form of IL-1Ra indicated for improving clinical signs and slowing the progression of structural damage in OA, RA, and other immune-mediated arthritis types [113,116,121]. A recent in vitro study suggested that anakinra inhibited NET formation in neutrophils, and this process was dependent on IL-1β signaling pathways [122]. Canakinumab is a human monoclonal anti-IL-1β antibody with an indication for RA and juvenile idiopathic arthritis [113]. Different studies demonstrated that canakinumab decreased OA symptoms and had a chondroprotective effect. This agent induces neutrophil apoptosis through MAPK14, NF-κB downregulation, and GRP78 upregulation [123,124,125]. Data from an experimental model of arthritis showed that the administration of recombinant IL-37 suppressed joint inflammation associated with the decreased neutrophil influx into the joint parallel with a reduction in neutrophil chemo-attractant chemokines (C-X-C motif) ligand 1 (CXCL1), macrophage inflammatory protein 1-alpha (MIP1α/CCL3), and IL-1α. In addition, reduction in pro-inflammatory cytokines TNF-α, IL-1β, and IL-6, and the neutrophil enzyme MPO were related to the therapeutic potential of IL-37 [121]. Blockade of anti-IL-6 receptors through reducing neutrophil infiltration and NET formation can have a therapeutic role in arthritis [126].
Recent evidence identified that miRNAs play an important role in regulating cartilage and bone homeostasis, catabolism, and anabolism ad repair [127,128]. The miRNAs are a group of endogenous small noncoding RNAs, and their up- or downregulation has been suggested to be linked to the pathogenesis of arthritis. Anti-cytokine therapy can affect miRNAs due to interactions between cytokines and them [129,130,131]. It has been suggested that treatment with anti-TNF-α and anti-IL-6 receptors modulates miRNA levels in neutrophils and attenuates inflammation [132]. In vitro treatment of RA neutrophils with anti-TNF-α and anti-IL-6 receptors has been accompanied by diminished miRNA levels in neutrophils and enhanced inflammatory profile [133].
Although cytokine-modulating therapies have been considered for arthritis treatment, a question of interest is whether cytokine-modulating therapies with just one cytokine or chemokine will be sufficient to stop inflammation and improve OA. It is important to note that neutrophils and other immune cells contribute to releasing many cytokines and chemokines with a potential synergistic effect in a cascade reaction during OA initiation and progression. Therefore, blocking one member can interrupt this synergy [3,134], and different pro-inflammatory and anti-inflammatory cytokines and their interactions can be considered for new therapeutic approaches to arthritis [135].
NE, secreted by neutrophils during inflammation, can potentially be targeted for OA treatment. NE is a degenerative protease that can induce cartilage destruction and pain correlated with arthritis severity during OA [43,136]. NE participates in neutrophil migration by cleaving adhesion molecules and modifying chemokine and cytokine activity and interaction with specific cell surface receptors [137]. Sivelestat sodium hydrate is a synthetic, potent, selective inhibitor of NE, which is used for treating acute lung injury and has raised considerable interest in arthritis treatment. Sivelestat can reduce inflammation and pain by inhibiting TNF-α, IL-6, NO secretion and PAR2, p44/42 MAPK activity [43,136,138,139]. Furthermore, in the OA condition, sivelestat inhibited NF-κB and HMGB1, which is known to be contributed to the NET formation and neutrophil recruitment [138,140,141]. Besides inhibiting NE, targeting the MMPs produced by neutrophils may also be influential for arthritis therapy [142]. However, the inhibition of MMPs has so far provided only limited therapeutic benefits probably due to the bilateral function of different MMPs in pathological conditions [93,143,144]. Another promising therapeutic approach for arthritis is considering pathways with the ability for NET formation inhibition or have an effect on the component in NETs. [145]. It was found that polydatin a natural precursor of resveratrol treatment markedly inhibited NET formation mediated by neutrophils and protected the joint against arthritis [146].
Other studies exist on developing therapies that target the key activators of neutrophils, such as intracellular signaling molecules (e.g., Janus kinase, spleen tyrosine kinase, and p38 MAP kinase) and adenosine receptors [109]. APPA (apocynin and paeonol) is a plant-derived compound with anti-inflammatory and chondroprotective properties that are considered a novel medication for OA treatment. It is currently being tested in clinical trials for human application [147,148,149]. An in vitro study on the effect of APPA on neutrophils showed that while APPA does not significantly impair neutrophil defense function, it modulates pathological aspects of neutrophil functions. APPA reduces neutrophil degranulation and NET formation. In addition, it dysregulates TNF-α, TNFα-mediated IL-8 expression of and ROS generation by neutrophils, as well as the inhibition of cytokine-driven signaling pathways (i.e., TNF-α-mediated activation of NF-κB and GM-CSF activation of Erk1/2) [148].
As a hemopoietic growth factor, G-CSF and its receptor are essential in neutrophil release, activation, and function [150,151]. It has been shown that G-CSF receptor blockade attenuated the progression of the inflammatory joint disease by blocking neutrophil trafficking with the suppression of chemokines (KC, MCP-1) and pro-inflammatory cytokines (IL-1β, IL-6) production as well as changes to cell adhesion receptors, including decreased CXCR2 and increased CD62L expression. This therapeutic strategy could be achieved without adverse effects on the immune response [151]. Inhibition of the pro-inflammatory pathway of the CXCR2 receptor via reducing neutrophils to the inflamed joints may be effective in arthritis treatment. Similar successful results have been reported using CXCR1/2 inhibitors on neutrophil recruitment in different inflammatory arthritic models [152].
Applying mesenchymal stem cells (MSC) for OA treatment holds immense potential and has been increasingly applied as a therapeutic method. MSCs isolated from distinct sources have strongly suggested that MSCs benefit arthritis treatment [149,153,154,155]. Although stem cells have been proposed to have unique abilities associated with tissue regeneration, the underlying mechanisms of MSCs remain largely unknown. However, other studies have concentrated on the interaction of MSCs with the immune system. [149,156,157,158]. It seems that MSCs might exert their positive effect on arthritis by modulating neutrophil migration and function. Several mechanisms, including the suppression of NO secretion, decreasing N-Formyl-L-Methionine-L-leucyl-L-phenylalanine, and induction of respiratory burst have been postulated to explain the effect of MSCs on neutrophils [159]. During the initial stage of inflammation, the first-respondent cells recruited to the inflammation site are neutrophils; their survival is the central arm for inflammation resolution and tissue repair [159,160]. It has been reported that MSCs can inhibit neutrophils apoptosis through IL-6, which is signaled by activating STAT-3 transcription factor, downregulation of Bax, and upregulation of MCL-1 [156,160]. Secretion of IL-6, IFN-β, and GM-CSF by MSCs can sustain neutrophils viability and function. MSCs also can reduce the adhesion, infiltration, and recruitment of neutrophils through TNF-stimulated gene 6, CXCL2, and CXCR2 [156,159,161]. The crosstalk between MSCs and neutrophils contributes to tissue repair and regeneration [162,163]. MSCs can polarize the pro-inflammatory N1 subset into the immune modulatory N2 subset by modulating the extracellular signal-regulated kinase pathway. Furthermore, an interplay between MSCs and neutrophils plays a role in vascular regeneration during the healing process via the effect on IL-6, PDGF, angiopoietin-1, HGF, and VEGF expression [163]. Chondrogenic progenitor cells (CPCs) are defined as stem cell-like cells in articular cartilage and are identified in different stages of OA. These cells have recently raised great interest in OA treatment because of their self-renewal, multilineage differentiation, and immunomodulatory and phagocytic properties [164,165]. Both pro- and anti-inflammatory actions of CPCs have been identified, and different treatment strategies related to CPCs are focused on their effects on the regulation of neutrophils [2,156]. Several inflammatory cytokines, chemokines, and MMPs, such as IL-6, CXCL-12, macrophage inhibitory factor, and MMP-13, are expressed by CPCs. CPCs may have an essential role in attracting neutrophils and their degranulation via IL-8 production in the early stage of cartilage injury [166]. TNF-α and IL-1β released by neutrophils inhibit the migration of CPCs. In contrast, CPCs produce an IL-1 receptor antagonist (IL-1Ra) that inhibits the binding of IL-1β [2].
Cell membrane-coated nanoparticles (NPs) are recently considered another promising strategy for arthritis treatment [167]. NPs consist of an NP core coated with membrane derived from natural cells, including white blood cells, and have an excellent biological interface and natural characteristics of source cells properties [168,169]. Neutrophil-NPs can absorb and interact with various inflammatory cytokines, including IL-1β and TNF-α. As a result, pro-inflammatory factors are neutralized, and synovial inflammation is inhibited [170,171,172]. Moreover, neutrophil-NPs mimic the natural adhesion between neutrophils and chondrocytes, which increases their cartilage penetration for targeting chondrocytes [172,173]. Anti-inflammatory activity and apoptosis inhibition of inflamed chondrocytes have been demonstrated for NPs in OA [174]. Evaluating the effect of neutrophil-NPs on the arthritis model showed their effectiveness in ameliorating joint damage and suppressing overall arthritis severity [167]. Neutrophil-based drug delivery systems were applied in treating various inflammatory conditions, including arthritis. This method has considerable biological potential to target different tissue mechanisms, overcome multiple physiological barriers, and enhance accumulation in the target tissue [175,176,177].
Several studies have identified evidence of cellular communication among different types of cells in the joints and their roles in OA pathogenesis [164,178,179]. Extracellular vesicles (EVs) are a novel form of intercellular communication, contributing to the spectrum of physiological and pathophysiological processes by transferring bioactive cargo, including lipids, proteins, nucleic acids, and metabolites, to target cells [180,181,182]. EVs are spherical lipid structures secreted by nearly all cell types. They are categorized into three main subtypes, microvesicles (MVs), exosomes, and apoptotic bodies, for their therapeutic potential [180,181,182,183,184,185,186]. Nanoenzyme-engineered neutrophil-derived exosomes with the potential to inhibit pro-inflammatory factors production have been shown to alleviate inflammatory stress in fibroblast-like synoviocytes and attenuate joint injury.
Moreover, these exosomes have relieved inflammation synovitis and ameliorated cartilage damage via inducing Th17/Treg cell balance regulation to suppress overall arthritis severity [187]. Neutrophil-derived microvesicles (NDMVs) have been demonstrated to inhibit inflammation by limiting recipient cells’ immune responses, which could encourage further research into targeting arthritis with NDMVs [186,188]. Downregulation of TNFα-induced expression of IL-5, IL-6, IL-8, MCP-1, IFNγ, and MIP-1β using NDMVs internalized by FLS isolated from OA patients, has been reported [189]. Evidence from an in vitro investigation and an in vivo study suggests that neutrophil MVs enriched in AnxA1 caused TGF-β production, matrix deposition, and chondrocyte homeostasis by inducing FPR2/ALX signaling and protecting the joint. Furthermore, neutrophil EVs reduce the loss of sulfated glycosaminoglycans. They could exert their protective effect on joints via EV mediator Annexin A1 (AnxA1) and its receptor Fpr2/3 on the chondrocyte as well as polarizing macrophages towards a more anti-inflammatory phenotype characterized by higher levels of CD206 and lower expression of MHCII and CD86. Similarly, neutrophil MVs enriched in AnxA1 interact with their receptor formyl peptide receptor 2/ALX and induce TGF-b1 production which causes ECM deposition and chondrocytes protection [92].
6. Conclusions
OA is a progressive disease in which neutrophils play an integral role in its pathogenesis and degradation of cartilage through the production of numerous cytokines and other inflammatory factors, as well as degrading enzymes. Furthermore, neutrophils contribute to the resolution of inflammation and cartilage repair via regulating immune responses. A better understanding of neutrophil functions in OA pathophysiology would allow for promising therapeutic strategies for osteoarthritis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Neutrophil-derived Products and their role in osteoarthritis; Table S2: The potential pharmacological function.
Author Contributions
Conceptualization, Y.M. and S.G..; writing—original draft preparation, Y.M., R.R., S.M., H.M. N.K., and S.G.; writing—review and editing, Y.M., N.K., and S.G; visualization, Y.M. and S.G.; funding acquisition, Y.M. and S.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received No funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Allen, K.; Thoma, L.; Golightly, Y. Epidemiology of osteoarthritis. Osteoarthr. Cartil. 2022, 30, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Chaney, S.; Vergara, R.; Qiryaqoz, Z.; Suggs, K.; Akkouch, A. The Involvement of Neutrophils in the Pathophysiology and Treatment of Osteoarthritis. Biomedicines 2022, 10, 1604. [Google Scholar] [CrossRef] [PubMed]
- Molnar, V.; Matišić, V.; Kodvanj, I.; Bjelica, R.; Jeleč, Ž.; Hudetz, D.; Rod, E.; Čukelj, F.; Vrdoljak, T.; Vidović, D.; et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9208. [Google Scholar] [CrossRef]
- Sacitharan, P.K. Ageing and osteoarthritis. Biochemistry and cell biology of ageing: part II clinical science, 2019: p. 123-159.
- Garbin, L.C.; Morris, M.J. A Comparative Review of Autologous Conditioned Serum and Autologous Protein Solution for Treatment of Osteoarthritis in Horses. Front. Veter- Sci. 2021, 8, 602978. [Google Scholar] [CrossRef]
- Ramos, S.; Pinto, A.; Cardoso, M.; Alexandre, N.; Bettencourt, E.; Monteiro, S.; Gama, L.T. Prevalence of Radiographic Signs of Osteoarthritis in Lusitano Purebred Horses. J. Equine Veter- Sci. 2020, 94, 103196. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, P.A.; Meireles, M.A.D.; Ribeiro, L.M.F.; de Lannes, S.T.; Meireles, N.F.T.; Viana, I.S.; Hokamura, H.K. Influence of Exercise, Age, Body weight, and Growth on the Development of Tarsal Osteoarthritis in Young Mangalarga Marchador Horses. J. Equine Veter- Sci. 2019, 80, 36–39. [Google Scholar] [CrossRef]
- Balamurugan, K.; Shammi, M.; George, R.S.; Kannan, T.; Siva, R.; Kar, S. A Retrospective Study on Equine Lameness and Influence of Age, Breed and Joint in Osteoarthritis. Int. J. Curr. Microbiol. Appl. Sci. 2020, 9, 3391–3393. [Google Scholar] [CrossRef]
- McIlwraith, C.W. , et al., Principles of Musculoskeletal Disease: Joint Injuries And Disease And Osteoarthritis. Adams and Stashak’s Lameness in Horses, 2020: p. 801-874.
- Ireland, J.; Clegg, P.; McGowan, C.; Platt, L.; Pinchbeck, G. Factors associated with mortality of geriatric horses in the United Kingdom. Prev. Veter- Med. 2011, 101, 204–218. [Google Scholar] [CrossRef]
- Neundorf, R.H.; Lowerison, M.B.; Cruz, A.M.; Thomason, J.J.; McEwen, B.J.; Hurtig, M.B. Determination of the prevalence and severity of metacarpophalangeal joint osteoarthritis in Thoroughbred racehorses via quantitative macroscopic evaluation. Am. J. Vet. Res. 2010, 71, 1284–1293. [Google Scholar] [CrossRef]
- McIlwraith, C.W.; Frisbie, D.D.; Kawcak, C.E. The horse as a model of naturally occurring osteoarthritis. Bone Jt. Res. 2012, 1, 297–309. [Google Scholar] [CrossRef]
- Arden, N. and M.C. Nevitt, Osteoarthritis: epidemiology. Best practice & research Clinical rheumatology, 2006. 20(1): p. 3-25.
- Curry, Z.A.; Beling, A.; Borg-Stein, J. Knee osteoarthritis in midlife women: unique considerations and comprehensive management. Menopause 2022, 29, 748–755. [Google Scholar] [CrossRef]
- Tu, C.; He, J.; Wu, B.; Wang, W.; Li, Z. An extensive review regarding the adipokines in the pathogenesis and progression of osteoarthritis. Cytokine 2019, 113, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ashkavand, Z.; Malekinejad, H.; Vishwanath, B.S. The pathophysiology of osteoarthritis. J. Pharm. Res. 2013, 7, 132–138. [Google Scholar] [CrossRef]
- Mathiessen, A.; Conaghan, P.G. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res. Ther. 2017, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jordan, J.M. Epidemiology of Osteoarthritis. Clin. Geriatr. Med. 2010, 26, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, W.; Yong, H.; He, M.; Yang, Y.; Deng, Z.; Li, Y. Macrophages in osteoarthritis: pathophysiology and therapeutics. Am. J. Transl. Res 2020, 12, 261–268. [Google Scholar] [PubMed]
- Grässel, S.; Aszodi, A. Osteoarthritis and Cartilage Regeneration: Focus on Pathophysiology and Molecular Mechanisms. Int. J. Mol. Sci. 2019, 20, 6156. [Google Scholar] [CrossRef]
- Vincent, T.L. IL-1 in osteoarthritis: time for a critical review of the literature. F1000Research 2019, 8, 934. [Google Scholar] [CrossRef]
- Mora, J.C.; Przkora, R.; Cruz-Almeida, Y. Knee osteoarthritis: pathophysiology and current treatment modalities. J. Pain Res. 2018, 11, 2189–2196. [Google Scholar] [CrossRef]
- Liu-Bryan, R. Synovium and the Innate Inflammatory Network in Osteoarthritis Progression. Curr. Rheumatol. Rep. 2013, 15, 1–7. [Google Scholar] [CrossRef]
- Li, M.; Yin, H.; Yan, Z.; Li, H.; Wu, J.; Wang, Y.; Wei, F.; Tian, G.; Ning, C.; Li, H.; et al. The immune microenvironment in cartilage injury and repair. Acta Biomater. 2021, 140, 23–42. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.E.; Pezzanite, L.; Goodrich, L.; Santangelo, K.; Chow, L.; Dow, S.; Wheat, W. Role of Innate Immunity in Initiation and Progression of Osteoarthritis, with Emphasis on Horses. Animals 2021, 11, 3247. [Google Scholar] [CrossRef] [PubMed]
- Arve-Butler, S.; Schmidt, T.; Mossberg, A.; Berthold, E.; Gullstrand, B.; Bengtsson, A.A.; Kahn, F.; Kahn, R. Synovial fluid neutrophils in oligoarticular juvenile idiopathic arthritis have an altered phenotype and impaired effector functions. Arthritis Res. Ther. 2021, 23, 1–12. [Google Scholar] [CrossRef]
- Fattori, V.; Amaral, F.A.; Verri, W.A. Neutrophils and arthritis: Role in disease and pharmacological perspectives. Pharmacol. Res. 2016, 112, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Zdziennicka, J.; Szponder, T.; Wessely-Szponder, J. Application of Natural Neutrophil Products for Stimulation of Monocyte-Derived Macrophages Obtained before and after Osteochondral or Bone Injury. Microorganisms 2021, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Fujie, K.; Shinguh, Y.; Inamura, N.; Yasumitsu, R.; Okamoto, M.; Okuhara, M. Release of neutrophil elastase and its role in tissue injury in acute inflammation: effect of the elastase inhibitor, FR134043. Eur. J. Pharmacol. 1999, 374, 117–125. [Google Scholar] [CrossRef]
- Gong, Y.; Koh, D.-R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2009, 339, 437–448. [Google Scholar] [CrossRef]
- Scanzello, C.R. Chemokines and inflammation in osteoarthritis: Insights from patients and animal models. J. Orthop. Res. 2017, 35, 735–739. [Google Scholar] [CrossRef]
- López-Armada, M.J.; Carames, B.; Martín, M.A.; Cillero-Pastor, B.; Lires-Dean, M.; Fuentes-Boquete, I.; Arenas, J.; Blanco, F.J. Mitochondrial activity is modulated by TNFalpha and IL-1beta in normal human chondrocyte cells. Osteoarthr. Cartil. 2006, 14, 1011–1022. [Google Scholar] [CrossRef]
- Afonso, V.; Champy, R.; Mitrovic, D.; Collin, P.; Lomri, A. Reactive oxygen species and superoxide dismutases: Role in joint diseases. Jt. Bone Spine 2007, 74, 324–329. [Google Scholar] [CrossRef]
- Wagner, G.; Lehmann, C.; Bode, C.; Miosge, N.; Schubert, A. High Mobility Group Box 1 Protein in Osteoarthritic Knee Tissue and Chondrogenic Progenitor Cells: An Ex Vivo and In Vitro Study. CARTILAGE 2019, 12, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Joos, H.; Wildner, A.; Hogrefe, C.; Reichel, H.; Brenner, R.E. Interleukin-1 beta and tumor necrosis factor alpha inhibit migration activity of chondrogenic progenitor cells from non-fibrillated osteoarthritic cartilage. Arthritis Res. Ther. 2013, 15, R119–R119. [Google Scholar] [CrossRef] [PubMed]
- Blom, A.B.; van der Kraan, P.M.; Berg, W.B.v.D. Cytokine Targeting in Osteoarthritis. Curr. Drug Targets 2007, 8, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Pyrillou, K., L. C. Burzynski, and M.C. Clarke, Alternative pathways of IL-1 activation, and its role in health and disease. Frontiers in immunology, 2020. 11: p. 3288.
- Wilkinson, D.J. , et al., Matrix metalloproteinase-13 is fully activated by neutrophil elastase and inactivates its serpin inhibitor, alpha-1 antitrypsin: Implications for osteoarthritis. The FEBS journal, 2022. 289(1): p. 121-139.
- Carrión, M. , et al., IL-22/IL-22R1 axis and S100A8/A9 alarmins in human osteoarthritic and rheumatoid arthritis synovial fibroblasts. Rheumatology, 2013. 52(12): p. 2177-2186.
- Deligne, C.; Casulli, S.; Pigenet, A.; Bougault, C.; Campillo-Gimenez, L.; Nourissat, G.; Berenbaum, F.; Elbim, C.; Houard, X. Differential expression of interleukin-17 and interleukin-22 in inflamed and non-inflamed synovium from osteoarthritis patients. Osteoarthr. Cartil. 2015, 23, 1843–1852. [Google Scholar] [CrossRef]
- Grevers, L.C. , et al., Different amplifying mechanisms of interleukin-17 and interferon-γ in Fcγ receptor–mediated cartilage destruction in murine immune complex–mediated arthritis. Arthritis & Rheumatism: Official Journal of the American College of Rheumatology, 2009. 60(2): p. 396-407.
- Wang, G.; Jing, W.; Bi, Y.; Li, Y.; Ma, L.; Yang, H.; Zhang, Y. Neutrophil Elastase Induces Chondrocyte Apoptosis and Facilitates the Occurrence of Osteoarthritis via Caspase Signaling Pathway. Front. Pharmacol. 2021, 12, 666162. [Google Scholar] [CrossRef]
- Muley, M.M.; Reid, A.R.; Botz, B.; Bölcskei, K.; Helyes, Z.; McDougall, J.J. Neutrophil elastase induces inflammation and pain in mouse knee joints via activation of proteinase-activated receptor-2. Br. J. Pharmacol. 2016, 173, 766–777. [Google Scholar] [CrossRef]
- Kaneva, M.K. Neutrophil elastase and its inhibitors—overlooked players in osteoarthritis. FEBS J. 2022, 289, 113–116. [Google Scholar] [CrossRef]
- Prajzlerová, K.; Kryštůfková, O.; Hánová, P.; Horváthová, V.; Gregová, M.; Pavelka, K.; Vencovský, J.; Šenolt, L.; Filková, M. High miR-451 expression in peripheral blood mononuclear cells from subjects at risk of developing rheumatoid arthritis. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- guila, S. , et al., Micrornas as new regulators of neutrophil extracellular trap formation. International Journal of Molecular Sciences, 2021. 22(4): p. 2116.
- Gurol, T., W. Zhou, and Q. Deng, Micro RNA s in neutrophils: potential next generation therapeutics for inflammatory ailments. Immunological reviews, 2016. 273(1): p. 29-47.
- de Almeida, R.C. , et al., RNA sequencing data integration reveals an miRNA interactome of osteoarthritis cartilage. Annals of the rheumatic diseases, 2019. 78(2): p. 270-277.
- Okuhara, A. , et al., Changes in microRNA expression in peripheral mononuclear cells according to the progression of osteoarthritis. Modern rheumatology, 2012. 22(3): p. 446-457.
- McClurg, O.; Tinson, R.; Troeberg, L. Targeting Cartilage Degradation in Osteoarthritis. Pharmaceuticals 2021, 14, 126. [Google Scholar] [CrossRef]
- Majtnerová, P.; Roušar, T. An overview of apoptosis assays detecting DNA fragmentation. Mol. Biol. Rep. 2018, 45, 1469–1478. [Google Scholar] [CrossRef]
- Wang, H. , et al., Histomorphology and innate immunity during the progression of osteoarthritis: Does synovitis affect cartilage degradation? Journal of cellular physiology, 2018. 233(2): p. 1342-1358.
- Wilson, M.E.; Mccandless, E.E.; Olszewski, M.A.; Robinson, N.E. Alveolar macrophage phenotypes in severe equine asthma. Vet. J. 2020, 256, 105436. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, M.; Zhang, X.; Wellman, S.S.; Bolognesi, M.; Kraus, V.B. Synergistic Roles of Macrophages and Neutrophils in Osteoarthritis Progression. Arthritis Rheumatol. 2020, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Haraden, C.A.; Huebner, J.L.; Hsueh, M.-F.; Li, Y.-J.; Kraus, V.B. Synovial fluid biomarkers associated with osteoarthritis severity reflect macrophage and neutrophil related inflammation. Arthritis Res. Ther. 2019, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kraus, V.; McDaniel, G.; Huebner, J.; Stabler, T.; Pieper, C.; Shipes, S.; Petry, N.; Low, P.; Shen, J.; McNearney, T.; et al. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthr. Cartil. 2016, 24, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hunter, D.; Jin, X.; Ding, C. The importance of synovial inflammation in osteoarthritis: current evidence from imaging assessments and clinical trials. Osteoarthr. Cartil. 2017, 26, 165–174. [Google Scholar] [CrossRef]
- Thomson, A. and C.M. Hilkens, Synovial Macrophages in Osteoarthritis: The Key to Understanding Pathogenesis? Frontiers in Immunology, 2021: p. 1831.
- McIlwraith, C.W. , Traumatic arthritis and posttraumatic osteoarthritis in the horse, in Joint disease in the horse. 2016, Elsevier. p. 33-48.
- Donell, S. Subchondral bone remodelling in osteoarthritis. EFORT Open Rev. 2019, 4, 221–229. [Google Scholar] [CrossRef]
- Benigni, G. , et al., CXCR3/CXCL10 axis regulates neutrophil–NK cell cross-talk determining the severity of experimental osteoarthritis. The Journal of Immunology, 2017. 198(5): p. 2115-2124.
- Furman, B.D.; Kent, C.L.; Huebner, J.L.; Kraus, V.B.; McNulty, A.L.; Guilak, F.; Olson, S.A. CXCL10 is upregulated in synovium and cartilage following articular fracture. J. Orthop. Res. 2018, 36, 1220–1227. [Google Scholar] [CrossRef]
- Jung, Y.-K.; Han, M.-S.; Park, H.-R.; Lee, E.-J.; Jang, J.-A.; Kim, G.-W.; Lee, S.-Y.; Moon, D.; Han, S. Calcium-phosphate complex increased during subchondral bone remodeling affects earlystage osteoarthritis. Sci. Rep. 2018, 8, 487. [Google Scholar] [CrossRef]
- Hidalgo, A.; Chilvers, E.R.; Summers, C.; Koenderman, L. The Neutrophil Life Cycle. Trends Immunol. 2019, 40, 584–597. [Google Scholar] [CrossRef]
- Capucetti, A.; Albano, F.; Bonecchi, R. Multiple Roles for Chemokines in Neutrophil Biology. Front. Immunol. 2020, 11, 1259. [Google Scholar] [CrossRef]
- V Lerman, Y. and M. Kim, Neutrophil migration under normal and sepsis conditions. Cardiovascular & Haematological Disorders-Drug Targets (Formerly Current Drug Targets-Cardiovascular & Hematological Disorders), 2015. 15(1): p. 19-28.
- Tecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-Derived Cytokines: Facts Beyond Expression. Front. Immunol. 2014, 5, 508. [Google Scholar] [CrossRef] [PubMed]
- Mócsai, A.; Walzog, B.; Lowell, C.A. Intracellular signalling during neutrophil recruitment. Cardiovasc. Res. 2015, 107, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, C.; Cassatella, M.A. Neutrophil-derived chemokines on the road to immunity. Semin. Immunol. 2016, 28, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Burn, G.L. , et al., The neutrophil. Immunity, 2021. 54(7): p. 1377-1391.
- Christoffersson, G. and M. Phillipson, The neutrophil: one cell on many missions or many cells with different agendas? Cell and tissue research, 2018. 371(3): p. 415-423.
- Furze, R.C.; Rankin, S.M. The role of the bone marrow in neutrophil clearance under homeostatic conditions in the mouse. FASEB J. 2008, 22, 3111–3119. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The role of neutrophils in inflammation resolution. Semin. Immunol. 2016, 28, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Peiseler, M.; Kubes, P. More friend than foe: the emerging role of neutrophils in tissue repair. J. Clin. Investig. 2019, 129, 2629–2639. [Google Scholar] [CrossRef]
- Tamassia, N. , et al., Cytokine production by human neutrophils: Revisiting the “dark side of the moon”. European journal of clinical investigation, 2018. 48: p. e12952.
- Soehnlein, O.; Steffens, S.; Hidalgo, A.; Weber, C. Neutrophils as protagonists and targets in chronic inflammation. Nat. Rev. Immunol. 2017, 17, 248–261. [Google Scholar] [CrossRef]
- Costa, S.; Bevilacqua, D.; Cassatella, M.A.; Scapini, P. Recent advances on the crosstalk between neutrophils and B or T lymphocytes. Immunology 2019, 156, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood 2016, 127, 2173–2181. [Google Scholar] [CrossRef]
- Scapini, P.; Marini, O.; Tecchio, C.; Cassatella, M.A. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol. Rev. 2016, 273, 48–60. [Google Scholar] [CrossRef]
- Wright, H.L.; Moots, R.J.; Edwards, S.W. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601. [Google Scholar] [CrossRef]
- Gupta, K.; Shukla, M.; Cowland, J.B.; Malemud, C.J.; Haqqi, T.M. Neutrophil gelatinase–associated lipocalin is expressed in osteoarthritis and forms a complex with matrix metalloproteinase 9. Arthritis Rheum. 2007, 56, 3326–3335. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.M.; Betz, T.V.; Lamont, D.J.; Kim, M.B.; Shaw, S.K.; Froio, R.M.; Baleux, F.; Arenzana-Seisdedos, F.; Alon, R.; Luscinskas, F.W. Elastase Release by Transmigrating Neutrophils Deactivates Endothelial-bound SDF-1α and Attenuates Subsequent T Lymphocyte Transendothelial Migration. J. Exp. Med. 2004, 200, 713–724. [Google Scholar] [CrossRef]
- Hacbarth, E. and A. Kajdacsy-Balla, Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis & Rheumatism: Official Journal of the American College of Rheumatology, 1986. 29(11): p. 1334-1342.
- Gierlikowska, B. , et al., Phagocytosis, Degranulation and Extracellular Traps Release by Neutrophils—The Current Knowledge, Pharmacological Modulation and Future Prospects. Frontiers in Pharmacology, 2021. 12: p. 666732.
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Liberale, L.; Carbone, F.; Vecchié, A.; Diaz-Cañestro, C.; Camici, G.G.; Montecucco, F.; Dallegri, F. The Pathophysiological Role of Neutrophil Extracellular Traps in Inflammatory Diseases. Arthritis Res. Ther. 2018, 118, 006–027. [Google Scholar] [CrossRef] [PubMed]
- Fousert, E.; Toes, R.; Desai, J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells 2020, 9, 915. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2017, 18, 134–147. [Google Scholar] [CrossRef]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs Are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci. Transl. Med. 2013, 5, 178ra40–178ra40. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef]
- Headland, S.E.; Jones, H.R.; Norling, L.V.; Kim, A.; Souza, P.R.; Corsiero, E.; Gil, C.D.; Nerviani, A.; Dell’accio, F.; Pitzalis, C.; et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci. Transl. Med. 2015, 7, 315ra190–315ra190. [Google Scholar] [CrossRef]
- Németh, T.; Sperandio, M.; Mócsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef]
- Filep, J.G. Targeting Neutrophils for Promoting the Resolution of Inflammation. Front. Immunol. 2022, 13, 866747. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.-P.; Martel-Pelletier, J.; Rannou, F.; Cooper, C. Efficacy and safety of oral NSAIDs and analgesics in the management of osteoarthritis: Evidence from real-life setting trials and surveys. Semin. Arthritis Rheum. 2015, 45, S22–S27. [Google Scholar] [CrossRef]
- Paglia, M.D.G.; Silva, M.T.; Lopes, L.C.; Barberato-Filho, S.; Mazzei, L.G.; Abe, F.C.; Bergamaschi, C.d.C. Use of corticoids and non-steroidal anti-inflammatories in the treatment of rheumatoid arthritis: Systematic review and network meta-analysis. PLOS ONE 2021, 16, e0248866. [Google Scholar] [CrossRef]
- Bertolotto, M.; Contini, P.; Ottonello, L.; Pende, A.; Dallegri, F.; Montecucco, F. Neutrophil migration towards C5a and CXCL8 is prevented by non-steroidal anti-inflammatory drugs via inhibition of different pathways. Br. J. Pharmacol. 2014, 171, 3376–3393. [Google Scholar] [CrossRef] [PubMed]
- Marsolais, D.; Côté, C.H.; Frenette, J. Nonsteroidal Anti-Inflammatory Drug Reduces Neutrophil and Macrophage Accumulation but Does Not Improve Tendon Regeneration. Lab. Investig. 2003, 83, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.; Ricci, E.; Migliorati, G.; Gentili, M.; Riccardi, C. How Glucocorticoids Affect the Neutrophil Life. Int. J. Mol. Sci. 2018, 19, 4090. [Google Scholar] [CrossRef]
- Cruz-Topete, D.; Cidlowski, J.A. One Hormone, Two Actions: Anti- and Pro-Inflammatory Effects of Glucocorticoids. Neuroimmunomodulation 2014, 22, 20–32. [Google Scholar] [CrossRef]
- Jin, L.; Xu, K.; Liang, Y.; Du, P.; Wan, S.; Jiang, C. Effect of hyaluronic acid on cytokines and immune cells change in patients of knee osteoarthritis. BMC Musculoskelet. Disord. 2022, 23, 1–9. [Google Scholar] [CrossRef]
- A Nicholls, M.; Fierlinger, A.; Niazi, F.; Bhandari, M. The Disease-Modifying Effects of Hyaluronan in the Osteoarthritic Disease State. Clin. Med. Insights: Arthritis Musculoskelet. Disord. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-C. , et al., Hyaluronic acid injection reduces inflammatory and apoptotic markers through modulation of AKT by repressing the oxidative status of neutrophils from osteoarthritic synovial fluid. International Journal of Biological Macromolecules, 2020. 165: p. 2765-2772.
- Chisari, E., K. Yaghmour, and W. Khan, The effects of TNF-alpha inhibition on cartilage: A systematic review of preclinical studies. Osteoarthritis and Cartilage, 2020. 28(5): p. 708-718.
- Kim, J.-R.; Yoo, J.J.; Kim, H.A. Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis. Int. J. Mol. Sci. 2018, 19, 674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shi, N.; Diao, Z.; Chen, Y.; Zhang, Y. Therapeutic potential of TNFα inhibitors in chronic inflammatory disorders: Past and future. Genes Dis. 2020, 8, 38–47. [Google Scholar] [CrossRef]
- Hastings, R.; Ding, T.; Butt, S.; Gadsby, K.; Zhang, W.; Moots, R.J.; Deighton, C. Neutropenia in patients receiving anti–tumor necrosis factor therapy. Arthritis Care Res. 2010, 62, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shu, W.; Zhou, G.; Lin, J.; Chu, F.; Wu, H.; Liu, Z. Anti-TNF-α Therapy Suppresses Proinflammatory Activities of Mucosal Neutrophils in Inflammatory Bowel Disease. Mediat. Inflamm. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.L.; Moots, R.J.; Bucknall, R.C.; Edwards, S.W. Neutrophil function in inflammation and inflammatory diseases. Rheumatology 2010, 49, 1618–1631. [Google Scholar] [CrossRef]
- Potera, R.M.; Jensen, M.J.; Hilkin, B.M.; South, G.K.; Hook, J.S.; A Gross, E.; Moreland, J.G. Neutrophil azurophilic granule exocytosis is primed by TNF-α and partially regulated by NADPH oxidase. J. Endotoxin Res. 2016, 22, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Capsoni, F.; Sarzi-Puttini, P.; Atzeni, F.; Minonzio, F.; Bonara, P.; Doria, A.; Carrabba, M. Effect of adalimumab on neutrophil function in patients with rheumatoid arthritis. Thromb. Haemost. 2005, 7, R250–R255. [Google Scholar] [CrossRef]
- Taylor, P.C.; Peters, A.M.; Paleolog, E.; Chapman, P.T.; Elliott, M.J.; McCloskey, R.; Feldmann, M.; Maini, R.N. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor α blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000, 43, 38–47. [Google Scholar] [CrossRef]
- Chevalier, X. and F. Eymard, Anti-IL-1 for the treatment of OA: dead or alive? Nature Reviews Rheumatology, 2019. 15(4): p. 191-192.
- Guma, M. , et al., Caspase 1–independent activation of interleukin-1β in neutrophil-predominant inflammation. Arthritis & Rheumatism: Official Journal of the American College of Rheumatology, 2009. 60(12): p. 3642-3650.
- Kay, J.; Calabrese, L. The role of interleukin-1 in the pathogenesis of rheumatoid arthritis. Rheumatology 2004, 43 (Suppl. S3), iii2–iii9. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef]
- Chevalier, X.; Goupille, P.; Beaulieu, A.D.; Burch, F.X.; Bensen, W.G.; Conrozier, T.; Loeuille, D.; Kivitz, A.J.; Silver, D.; Appleton, B.E. Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2009, 61, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Prince, L.R.; Allen, L.; Jones, E.C.; Hellewell, P.G.; Dower, S.K.; Whyte, M.K.; Sabroe, I. The Role of Interleukin-1β in Direct and Toll-Like Receptor 4-Mediated Neutrophil Activation and Survival. Am. J. Pathol. 2004, 165, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- zcan, A. and O. Boyman, Mechanisms regulating neutrophil responses in immunity, allergy, and autoimmunity. Allergy, 2022.
- Mehta, S.; Akhtar, S.; Porter, R.M.; Önnerfjord, P.; Bajpayee, A.G. Interleukin-1 receptor antagonist (IL-1Ra) is more effective in suppressing cytokine-induced catabolism in cartilage-synovium co-culture than in cartilage monoculture. Arthritis Res. Ther. 2019, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Koenders, M.; Kalabokis, V.; Kim, J.; Tan, A.C.; Garlanda, C.; Mantovani, A.; Dagna, L.; Joosten, L.A.B.; Dinarello, C.A. Treating experimental arthritis with the innate immune inhibitor interleukin-37 reduces joint and systemic inflammation. Rheumatology 2016, 55, 2220–2229. [Google Scholar] [CrossRef]
- Mistry, P.; Carmona-Rivera, C.; Ombrello, A.K.; Hoffmann, P.; Seto, N.L.; Jones, A.; Stone, D.L.; Naz, F.; Carlucci, P.; Dell’orso, S.; et al. Dysregulated neutrophil responses and neutrophil extracellular trap formation and degradation in PAPA syndrome. Rheumatology 2018, 77, 1825–1833. [Google Scholar] [CrossRef]
- Cai, X.; Yuan, S.; Zeng, Y.; Wang, C.; Yu, N.; Ding, C. New Trends in Pharmacological Treatments for Osteoarthritis. Front. Pharmacol. 2021, 12, 645842. [Google Scholar] [CrossRef]
- Cheleschi, S.; Cantarini, L.; Pascarelli, N.A.; Collodel, G.; Lucherini, O.M.; Galeazzi, M.; Fioravanti, A. Possible chondroprotective effect of canakinumab: An in vitro study on human osteoarthritic chondrocytes. Cytokine 2014, 71, 165–172. [Google Scholar] [CrossRef]
- Ghouri, A.; Conaghan, P.G. Prospects for Therapies in Osteoarthritis. Calcif. Tissue Int. 2020, 109, 339–350. [Google Scholar] [CrossRef]
- Ohyama, A.; Osada, A.; Kawaguchi, H.; Kurata, I.; Nishiyama, T.; Iwai, T.; Ishigami, A.; Kondo, Y.; Tsuboi, H.; Sumida, T.; et al. Specific Increase in Joint Neutrophil Extracellular Traps and Its Relation to Interleukin 6 in Autoimmune Arthritis. Int. J. Mol. Sci. 2021, 22, 7633. [Google Scholar] [CrossRef]
- Yu, X.-M.; Meng, H.-Y.; Yuan, X.-L.; Wang, Y.; Guo, Q.-Y.; Peng, J.; Wang, A.-Y.; Lu, S.-B. MicroRNAs’ Involvement in Osteoarthritis and the Prospects for Treatments. Evidence-Based Complement. Altern. Med. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Malemud, C.J. MicroRNAs and Osteoarthritis. Cells 2018, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Lu, X. , et al., miR-335-5P contributes to human osteoarthritis by targeting HBP1. Experimental and therapeutic medicine, 2021. 21(2): p. 1-1.
- Kmiołek, T.; Paradowska-Gorycka, A. miRNAs as Biomarkers and Possible Therapeutic Strategies in Rheumatoid Arthritis. Cells 2022, 11, 452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, C.; Zhang, C.; Luo, C.; Zhong, B.; Yu, X. MiRNA-132 regulates the development of osteoarthritis in correlation with the modulation of PTEN/PI3K/AKT signaling. BMC Geriatr. 2021, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Castro-Villegas, C.; Pérez-Sánchez, C.; Escudero, A.; Filipescu, I.; Verdu, M.; Ruiz-Limón, P.; Aguirre, M.A.; Jiménez-Gomez, Y.; Font, P.; Rodriguez-Ariza, A.; et al. Circulating miRNAs as potential biomarkers of therapy effectiveness in rheumatoid arthritis patients treated with anti-TNFα. Arthritis Res. Ther. 2015, 17, 1–15. [Google Scholar] [CrossRef]
- De la Rosa, I.A. , et al., Impaired microRNA processing in neutrophils from rheumatoid arthritis patients confers their pathogenic profile. Modulation by biological therapies. Haematologica, 2020. 105(9): p. 2250.
- Dinarello, C.A. Anti-inflammatory Agents: Present and Future. Cell 2010, 140, 935–950. [Google Scholar] [CrossRef]
- Liu, S.; Deng, Z.; Chen, K.; Jian, S.; Zhou, F.; Yang, Y.; Fu, Z.; Xie, H.; Xiong, J.; Zhu, W. Cartilage tissue engineering: From proinflammatory and anti-inflammatory cytokines to osteoarthritis treatments (Review). Mol. Med. Rep. 2022, 25, 1–15. [Google Scholar] [CrossRef]
- Muley, M.M.; Krustev, E.; Reid, A.R.; McDougall, J.J. Prophylactic inhibition of neutrophil elastase prevents the development of chronic neuropathic pain in osteoarthritic mice. J. Neuroinflammation 2017, 14, 1–12. [Google Scholar] [CrossRef]
- Mannelli, L.D.C.; Micheli, L.; Cinci, L.; Maresca, M.; Vergelli, C.; Pacini, A.; Quinn, M.T.; Giovannoni, M.P.; Ghelardini, C. Effects of the neutrophil elastase inhibitor EL-17 in rat adjuvant-induced arthritis. Rheumatology 2016, 55, 1285–1294. [Google Scholar] [CrossRef]
- Yu, X.; Zhao, L.; Yu, Z.; Yu, C.; Bi, J.; Sun, B.; Cong, H. Sivelestat sodium hydrate improves post-traumatic knee osteoarthritis through nuclear factor-κB in a rat model. Exp. Ther. Med. 2017, 14, 1531–1537. [Google Scholar] [CrossRef]
- Crocetti, L.; Giovannoni, M.P.; Cantini, N.; Guerrini, G.; Vergelli, C.; Schepetkin, I.A.; Khlebnikov, A.I.; Quinn, M.T. Novel Sulfonamide Analogs of Sivelestat as Potent Human Neutrophil Elastase Inhibitors. Front. Chem. 2020, 8, 795. [Google Scholar] [CrossRef] [PubMed]
- Tadie, J.-M.; Bae, H.-B.; Jiang, S.; Park, D.W.; Bell, C.P.; Yang, H.; Pittet, J.-F.; Tracey, K.; Thannickal, V.J.; Abraham, E.; et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L342–L349. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Ming, B.; Dong, L. The Role of HMGB1 in Rheumatic Diseases. Front. Immunol. 2022, 13, 815257. [Google Scholar] [CrossRef] [PubMed]
- Li, R.-L.; Duan, H.-X.; Liang, Q.; Huang, Y.-L.; Wang, L.-Y.; Zhang, Q.; Wu, C.-J.; Liu, S.-Q.; Peng, W. Targeting matrix metalloproteases: A promising strategy for herbal medicines to treat rheumatoid arthritis. Front. Immunol. 2022, 13, 1046810. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E. and C. Libert, Is there new hope for therapeutic matrix metalloproteinase inhibition? Nature reviews Drug discovery, 2014. 13(12): p. 904-927.
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors — Novel strategies bring new prospects. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef]
- Clarke, J. NETs directly injure cartilage in RA. Nat. Rev. Rheumatol. 2020, 16, 410–410. [Google Scholar] [CrossRef]
- Yang, F.; Luo, X.; Luo, G.; Zhai, Z.; Zhuang, J.; He, J.; Han, J.; Zhang, Y.; Zhuang, L.; Sun, E.; et al. Inhibition of NET formation by polydatin protects against collagen-induced arthritis. Int. Immunopharmacol. 2019, 77, 105919. [Google Scholar] [CrossRef]
- Larkins, N.; King, C.M. Effectiveness of apocynin-paeonol (APPA) for the management of osteoarthritis in dogs: comparisons with placebo and meloxicam in client-owned dogs. 2017, 3. 3. [CrossRef]
- Cross, A.L.; Hawkes, J.; Wright, H.L.; Moots, R.J.; Edwards, S.W. APPA (apocynin and paeonol) modulates pathological aspects of human neutrophil function, without supressing antimicrobial ability, and inhibits TNFα expression and signalling. Inflammopharmacology 2020, 28, 1223–1235. [Google Scholar] [CrossRef]
- Han, S. Osteoarthritis year in review 2022: biology. Osteoarthr. Cartil. 2022, 30, 1575–1582. [Google Scholar] [CrossRef]
- Semerad, C.L.; Liu, F.; Gregory, A.D.; Stumpf, K.; Link, D.C. G-CSF Is an Essential Regulator of Neutrophil Trafficking from the Bone Marrow to the Blood. Immunity 2002, 17, 413–423. [Google Scholar] [CrossRef]
- Campbell, I.K.; Leong, D.; Edwards, K.M.; Rayzman, V.; Ng, M.; Goldberg, G.L.; Wilson, N.J.; Scalzo-Inguanti, K.; Mackenzie-Kludas, C.; Lawlor, K.E.; et al. Therapeutic Targeting of the G-CSF Receptor Reduces Neutrophil Trafficking and Joint Inflammation in Antibody-Mediated Inflammatory Arthritis. J. Immunol. 2016, 197, 4392–4402. [Google Scholar] [CrossRef]
- Alam, M.J. , et al. Therapeutic blockade of CXCR2 rapidly clears inflammation in arthritis and atopic dermatitis models: demonstration with surrogate and humanized antibodies. in MAbs. 2020. Taylor & Francis.
- Freitag, J.; Bates, D.; Boyd, R.; Shah, K.; Barnard, A.; Huguenin, L.; Tenen, A. Mesenchymal stem cell therapy in the treatment of osteoarthritis: reparative pathways, safety and efficacy – a review. BMC Musculoskelet. Disord. 2016, 17, 1–13. [Google Scholar] [CrossRef]
- Maumus, M.; Guérit, D.; Toupet, K.; Jorgensen, C.; Noël, D. Mesenchymal stem cell-based therapies in regenerative medicine: applications in rheumatology. Stem Cell Res. Ther. 2011, 2, 14–14. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C., W. Wu, and X. Qu, Mesenchymal stem cells in osteoarthritis therapy: A review. American journal of translational research, 2021. 13(2): p. 448.
- Le Blanc, K.; Davies, L.C. Mesenchymal stromal cells and the innate immune response. Immunol. Lett. 2015, 168, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M. , et al., The immune properties of mesenchymal stem cells. International journal of biomedical science: IJBS, 2007. 3(2): p. 76.
- Jiang, W.; Xu, J. Immune modulation by mesenchymal stem cells. Cell Prolif. 2019, 53, e12712. [Google Scholar] [CrossRef] [PubMed]
- Joel, M.D.M.; Yuan, J.; Wang, J.; Yan, Y.; Qian, H.; Zhang, X.; Xu, W.; Mao, F. MSC: immunoregulatory effects, roles on neutrophils and evolving clinical potentials. 2019, 11, 3890–3904.
- Rosales, C. , Neutrophil: a cell with many roles in inflammation or several cell types? Frontiers in physiology, 2018. 9: p. 113.
- Molnar, V.; Pavelić, E.; Vrdoljak, K.; Čemerin, M.; Klarić, E.; Matišić, V.; Bjelica, R.; Brlek, P.; Kovačić, I.; Tremolada, C.; et al. Mesenchymal Stem Cell Mechanisms of Action and Clinical Effects in Osteoarthritis: A Narrative Review. Genes 2022, 13, 949. [Google Scholar] [CrossRef]
- Planat-Benard, V.; Varin, A.; Casteilla, L. MSCs and Inflammatory Cells Crosstalk in Regenerative Medicine: Concerted Actions for Optimized Resolution Driven by Energy Metabolism. Front. Immunol. 2021, 12, 626755. [Google Scholar] [CrossRef]
- Harrell, C.R.; Djonov, V.; Volarevic, V. The Cross-Talk between Mesenchymal Stem Cells and Immune Cells in Tissue Repair and Regeneration. Int. J. Mol. Sci. 2021, 22, 2472. [Google Scholar] [CrossRef] [PubMed]
- Li, Z., Z. Huang, and L. Bai, Cell interplay in osteoarthritis. Frontiers in Cell and Developmental Biology, 2021: p. 2132.
- Dai, H.; Chen, R.; Gui, C.; Tao, T.; Ge, Y.; Zhao, X.; Qin, R.; Yao, W.; Gu, S.; Jiang, Y.; et al. Eliminating senescent chondrogenic progenitor cells enhances chondrogenesis under intermittent hydrostatic pressure for the treatment of OA. Stem Cell Res. Ther. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Ding, L.; Zhou, C.; Zheng, H.; Wang, Q.; Song, H.; Buckwalter, J.A.; Martin, J.A. Migrating Progenitor Cells Derived From Injured Cartilage Surface Respond to Damage-Associated Molecular Patterns. CARTILAGE 2021, 13, 755S–765S. [Google Scholar] [CrossRef]
- Zhang, Q.; Dehaini, D.; Zhang, Y.; Zhou, J.; Chen, X.; Zhang, L.; Fang, R.H.; Gao, W.; Zhang, L. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol. 2018, 13, 1182–1190. [Google Scholar] [CrossRef]
- Liu, L.; Pan, D.; Chen, S.; Martikainen, M.-V.; Kårlund, A.; Ke, J.; Pulkkinen, H.; Ruhanen, H.; Roponen, M.; Käkelä, R.; et al. Systematic design of cell membrane coating to improve tumor targeting of nanoparticles. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Narain, A.; Asawa, S.; Chhabria, V.; Patil-Sen, Y.; Cottam, E.; Pierini, R.; Roberts, R.; Wileman, T.; Kim, S.-W.; Kim, H.; et al. Cell membrane coated nanoparticles: next-generation therapeutics. Nanomedicine 2017, 12, 2677–2692. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dai, B.; Guo, J.; Zheng, L.; Guo, Q.; Peng, J.; Xu, J.; Qin, L. Nanoparticle–Cartilage Interaction: Pathology-Based Intra-articular Drug Delivery for Osteoarthritis Therapy. Nano-Micro Lett. 2021, 13, 1–48. [Google Scholar] [CrossRef]
- Collison, J. Nanoparticles in neutrophil clothing. Nat. Rev. Rheumatol. 2018, 14, 622–622. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Z.; Pang, Y.; Zhou, H. The interaction between nanoparticles and immune system: application in the treatment of inflammatory diseases. J. Nanobiotechnology 2022, 20, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gong, H.; Gao, W.; Zhang, L. Recent Progress in Capturing and Neutralizing Inflammatory Cytokines. CCS Chem. 2020, 2, 376–389. [Google Scholar] [CrossRef]
- Yu, Q.; Huang, Y.; Chen, X.; Chen, Y.; Zhu, X.; Liu, Y.; Liu, J. A neutrophil cell membrane-biomimetic nanoplatform based on l-arginine nanoparticles for early osteoarthritis diagnosis and nitric oxide therapy. Nanoscale 2022, 14, 11619–11634. [Google Scholar] [CrossRef]
- Wang, H.; Zang, J.; Zhao, Z.; Zhang, Q.; Chen, S. The Advances of Neutrophil-Derived Effective Drug Delivery Systems: A Key Review of Managing Tumors and Inflammation. Int. J. Nanomed. 2021, ume 16, 7663–7681. [Google Scholar] [CrossRef]
- Su, Y.; Gao, J.; Kaur, P.; Wang, Z. Neutrophils and Macrophages as Targets for Development of Nanotherapeutics in Inflammatory Diseases. Pharmaceutics 2020, 12, 1222. [Google Scholar] [CrossRef]
- Zhao, Z.; Ukidve, A.; Kim, J.; Mitragotri, S. Targeting Strategies for Tissue-Specific Drug Delivery. Cell 2020, 181, 151–167. [Google Scholar] [CrossRef] [PubMed]
- A Marino, A.; Waddell, D.D.; Kolomytkin, O.V.; Meek, W.D.; Wolf, R.; Sadasivan, K.K.; A Albright, J. Increased Intercellular Communication through Gap Junctions May Contribute to Progression of Osteoarthritis. Clin. Orthop. Relat. Res. 2004, 422, 224–232. [Google Scholar] [CrossRef]
- Carpintero-Fernandez, P.; Gago-Fuentes, R.; Wang, H.Z.; Fonseca, E.; Caeiro, J.R.; Valiunas, V.; Brink, P.R.; Mayan, M.D. Intercellular communication via gap junction channels between chondrocytes and bone cells. Biochim. et Biophys. Acta (BBA) - Biomembr. 2018, 1860, 2499–2505. [Google Scholar] [CrossRef]
- Doyle, L.; Wang, M. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Sánchez, G.B.; Bunn, K.E.; Pua, H.H.; Rafat, M. Extracellular vesicles: mediators of intercellular communication in tissue injury and disease. Cell Commun. Signal. 2021, 19, 1–18. [Google Scholar] [CrossRef]
- Nederveen, J.P.; Warnier, G.; Di Carlo, A.; Nilsson, M.I.; Tarnopolsky, M.A. Extracellular Vesicles and Exosomes: Insights From Exercise Science. Front. Physiol. 2021, 11, 604274. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, X.; Zhao, J.; Yang, Y.; Cai, X.; Xu, J.; Cao, P. Exosomes: A Novel Strategy for Treatment and Prevention of Diseases. Front. Pharmacol. 2017, 8, 300–300. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bréchard, S. Neutrophil Extracellular Vesicles: A Delicate Balance between Pro-Inflammatory Responses and Anti-Inflammatory Therapies. Cells 2022, 11, 3318. [Google Scholar] [CrossRef]
- Rondelli, V.; Helmy, S.; Passignani, G.; Parisse, P.; Di Silvestre, D. Integrated Strategies for a Holistic View of Extracellular Vesicles. ACS Omega 2022, 7, 19058–19069. [Google Scholar] [CrossRef]
- Johnson, L.B.; Kuethe, J.W.; Caldwell, C.C. Neutrophil derived microvesicles: emerging role of a key mediator to the immune response. In Endocrine, Metabolic & Immune Disorders-Drug Targets (Formerly Current Drug Targets - Immune, Endocrine & Metabolic Disorders); 2014; Volume 14, pp. 210–217.
- Zhang, L.; Qin, Z.; Sun, H.; Chen, X.; Dong, J.; Shen, S.; Zheng, L.; Gu, N.; Jiang, Q. Nanoenzyme engineered neutrophil-derived exosomes attenuate joint injury in advanced rheumatoid arthritis via regulating inflammatory environment. Bioact. Mater. 2022, 18, 1–14. [Google Scholar] [CrossRef]
- Hong, C.-W. Extracellular Vesicles of Neutrophils. Immune Netw. 2018, 18, e43. [Google Scholar] [CrossRef] [PubMed]
- Zhan, D.; Cross, A.; Wright, H.L.; Moots, R.J.; Edwards, S.W.; Honsawek, S. Internalization of Neutrophil-Derived Microvesicles Modulates TNFα-Stimulated Proinflammatory Cytokine Production in Human Fibroblast-Like Synoviocytes. Int. J. Mol. Sci. 2021, 22, 7409. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Miller, R.J.; Malfait, A.-M. Osteoarthritis joint pain: the cytokine connection. Cytokine 2014, 70, 185–193. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of Neutrophil elastase (NE) production is linked to an increase in cartilage degeneration by activation of MMP-13. Synovial fluid contains numerous cytokines and chemokines that promote inflammation.
Figure 1.
Schematic representation of Neutrophil elastase (NE) production is linked to an increase in cartilage degeneration by activation of MMP-13. Synovial fluid contains numerous cytokines and chemokines that promote inflammation.
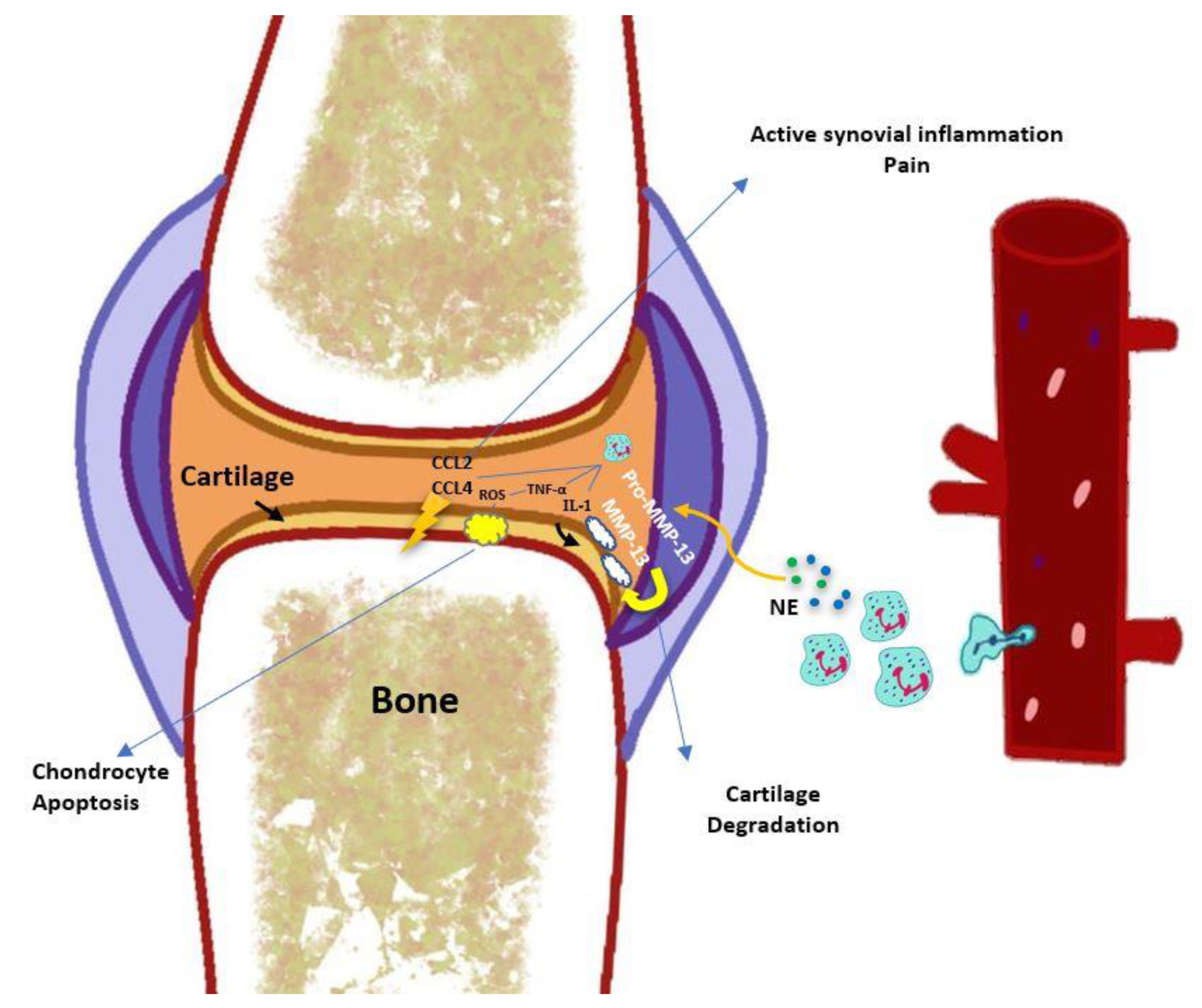
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



