
Submitted:
31 May 2023
Posted:
01 June 2023
You are already at the latest version
Abstract
Compared to other 2D materials, MBenes are at an early stage in both experimental and theoretical approaches. However, the wide range of possible 2D structures leads to novel and challenging properties and their consequent applications. From all the possible stoichiometries, we have performed a theoretical study of orthorhombic and hexagonal M2B2 MBenes in the framework of the Density Functional Theory. We have found that both symmetries of Cr2B2, Fe2B2, and Zr2B2 show a metallic behavior and could be grown under certain conditions, as they are demonstrated to be dynamically stable. Moreover, the values of the magnetic moment, in specific ferromagnetic cases exceeding 2.5 μB/M2B2, make them suitable as robust 2D magnets. All our findings represent an important step in the understanding of MBenes and open several windows to future research in fields like energy conversion and storage, sensing, catalysis, biochemistry, or nanotechnology, among others.
Keywords:
Transition metal borides
; MBenes
; Energy conversion and storage
; 2D magnets
1. Introduction
Transition metal borides (MBs) can be regarded as new efficient earth-abundant materials for energy storage/conversion systems, including metal ion batteries, metal-air batteries, capacitors, oxygen evolution reaction (OER), and other electrochemical fields [1]. Single-crystalline ternary transition metal borides (MAB phases, where M are transition metals, A are p-block elements and B is boron) were first reported in 2015 by Ade and Hillebrecht and recently gained attention as promising layered materials [2]. The two-dimensional counterparts (MBenes) can be obtained from the chemical exfoliation of the MAB phases. It is noted that MBenes possess different stoichiometries and variable modes of 2D layer sandwiching compared to the corresponding MXenes [3].
The MAB phases with M and B with one-to-one stoichiometry are orthorhombic and hexagonal crystals with the chemical formula of MAB and MAB[3]. Experimentally, orthorhombic MAlB (M = Mo and W), MAlB (M = Cr, Mn, and Fe), and hexagonal TiInB[4] have already been synthesized. The MAB phases are promising candidates for obtaining new 2D MBenes. 2D MoB was reported to be obtained by partial etching of MoAlB phases through the deintercalation of Al layers from ordered stacking faults region [5,6]. Selectively HCl-etching Al layers from CrAlB yielded the bulk layered CrB nanosheets [7,8]. Removal of the indium layer through a high-temperature dealloying of TiInB yields a bulk layered TiB structure [4]. To date, however, the synthesis of individual single-layer MBenes has not been realized.
The present work concerns computational studies of the structural, energetic, electronic, and transport properties of selected MBenes compounds with M = Cr, Fe, and Zr. Some of the studied 2D structures have been proven to be stable by previous studies [9]. The studied MBenes present either an orthorhombic structure (ortho-MBenes) with (no. 51) space group symmetry or a hexagonal structure (hex-MBenes) with (no. 191) space group symmetry. In the structures, each atom is surrounded by six neighbors, and the buckled bilayers are sandwiched between transition metal (TM) layers. On the other hand, in the structures the honeycomb-type boron layer is sandwiched between two TM layers on both sides, and every TM atom is located above or below the centroid of the honeycomb structure.
The bulk counterparts of our metal monoboride nanosheets are the ferromagnetic and modifications of FeB and the nonmagnetic CrB and ZrB compounds, all widely studied both experimentally and theoretically [10,11,12]. The structure of -FeB is debatable [12], whereas -FeB and CrB are orthorhombic crystals with (no. 62) and (no. 63) space group symmetries, respectively. The ZrB solid is rock-salt structured and crystallizes in the cubic (no. 225) space group symmetry. The -FeB and CrB solids exhibit very interesting structures since both enclose boron double chain (BDC) stripes which are very common motives of all-boron nanostructures [13,14].
The purpose of this work is to understand the origin of the physical properties of the MBenes (M = Cr, Fe, and Zr) compounds, as well as to identify features that may affect the transport properties of these compounds.
2. Computational Approach
First-principles spin-polarized calculations have been performed in the framework of the Density Functional Theory (DFT) within the generalized gradient corrected approximation of Perdew–Burke–Ernzerhof (PBE) [15] for the exchange-correlation functional, using projector plane-wave (PAW) pseudopotentials [16] as implemented in the Quantum ESPRESSO (QE) suite of codes [17]. Every unit cell consists of two atoms of boron and two atoms of the TM. To avoid interactions between adjacent MBenes, we have considered an empty space of thickness of 15 Å along the normal direction. Optimized geometries were reached allowing the unit cell shape, volume, and the ions to relax until the residual forces on the atoms have been less than 0.3 meV/Å and the total energy convergence has been set to 10 Ry. We expanded the electronic wave functions and charge density in plane-wave basis sets with an energy cutoff of 70 and 700 Ry, respectively, while the -centered k-point grid in the Brillouin zone, in the Monkhorst-Pack scheme, was set to for the geometry optimization and for the DOS calculations, with Gaussian smearing of 0.02 Ry; these values ensure the accuracy of the total energy. All the structures have been considered to be initially spin-polarized and in order to determine the magnetic ground states of each, we have calculated two magnetic configurations, one ferromagnetic (FM) and one antiferromagnetic (AFM), to reach the most energetically favorable.
For the structural characterization of the systems studied, an important descriptor we use is the cohesive energy per atom (), that is, the difference in energy between the total energy of the compound and the sum of the total energies of the isolated atoms,
which means the released energy when a compound dissociates into isolated free atoms, where M represents the TM atom, is the total energy of the MBene, and are the total energies of the isolated atoms (B and TM atom), and and are the numbers of boron and TM atoms per unit cell, respectively, directly obtained from spin-polarized calculations. The phonon-dispersion curves have been obtained by means of density functional perturbation theory (DFPT), calculating the dynamical matrices in the linear response approach on a q-grid of . The transport integrals have been computed using the Boltzmann transport theory and a constant scattering rate model [18]. The charge transfer has been obtained by a Bader analysis [19] and all the visualizations have been performed using the Visualization for Electronic and STructural Analysis (VESTA) software [20].
3. Results and Discussion
3.1. Structure and Stability
Since boron is electron deficient, it is expected that a mixture with TM atoms will lead to stable structures. As mentioned above, among all the possible MBenes, we have focused on those which possess either orthorhombic or hexagonal MB structures which are shown in Figure 1. After a full structural optimization, we have found that the unit cells of ortho-MBenes become almost rectangular with when the TM is Fe or Cr ( is 1.005 and 1.013 for chromium and iron, respectively), whereas for ortho-ZrB (). All the cell parameters are described in Table 1 and compared with other literature reports.
The and phonon dispersion curves are good indicators of the bond strength and dynamical stability of the materials. The dynamical matrix gives us the frequency whose square is negative when there exist instabilities for a particular phonon mode with k (imaginary frequencies), which means that this mode does not generate the restoring force needed by the lattice vibrations and could take the structure away from the original configuration. The results of our calculations are summarized in Table 2 and in Figure 2.
All our MBenes exhibit large cohesive energies ranging from 6.222 to 8.087 eV, as shown in Table 2, which means strong internal binding and good stability. Moreover, our results are in good agreement with previous works. For instance, Zhang et al. [28] have obtained a value of 6.30 eV for ortho-CrB. For the sake of comparison, we have also computed the diamond structure of carbon using the same optimization parameters, resulting in a of 7.757 eV, which is a comparable value to that of other theoretical and experimental reports [29]. Interestingly, other studies have remarked a dependence of the structure stability with the atomic mass of the TM [22], a fact that is also reproduced in our results, being hex-ZrB the MBene with the highest (8.087 eV). From a structural point of view, CrB and FeB prefer adopting orthorhombic structures whereas ZrB accommodates better in the hexagonal one. However, the orthorhombic and hexagonal phases are close in energy (within some tens of meV), and according to recent reports [9,30], ortho-MBenes might transform into hex-MBenes at high temperatures.
On one hand, calculations of the phonon frequencies reveal that some small imaginary frequencies appear in the surroundings of the point for orthorhombic CrB and ZrB what is shown on the left panel of Figure 2. On the other hand, none of the hexagonal structures have imaginary frequencies what is shown on the right panel of Figure 2. Similar dynamical instabilities, as for ortho-MBenes, have been reported for freestanding 2D structures in previous works [25,31]. These studies conclude that the out-of-plane acoustic mode, ZA, is responsible for such instabilities, which are against long-wavelength transversal waves that could be fixed by defects like grain boundaries or ripples [32].
The highest values of the frequencies at the -point, are collected in Table 2. All the frequencies are higher than 740 cm. Starting with the orthorhombic structures, the highest optical frequency at -point increases with the atomic number of the TM atom, that is, Cr, Fe, and Zr, in this order. These results are very close to those found in the literature [24]. Since the optical frequency is an indicator of the bond strength, ZrB is more stable than CrB and FeB. The trend is, however, the opposite for hex-mBenes, meaning that the highest frequency at the -point corresponds to the TM atom which has the smallest atomic number, in this case Cr, and decreases for Fe and Zr. We can conclude that the studied MBenes are stable and could be experimentally grown under certain conditions.
3.2. Electronic Properties
To understand the electronic behavior of the MBenes involved in our study, we have calculated the spin-polarized band structure, the density of states (DOS), and the projected density of states (PDOS) for the studied systems. The results of these calculations are shown in Figs. Figure 3 and Figure 4 for ortho-MBenes and hex-MBenes, respectively. Looking at the band structure and DOS, we may conclude that there are no band gaps between the valence band (VB) and the conduction band (CB) for any of the studied structures, which means that all the systems are metallic with partially occupied bands crossing the Fermi level (for the majority and for the minority spin channels). This metallic character of the pristine MBenes has been reported also by other works [33]. In all cases, the p orbitals of boron are deep in energy (ranging from -8 to -2 eV approximately) and, in the orthorhombic structures, partially hybridize with the d orbitals of the TM. Near the Fermi level, the PDOS for both symmetries of CrB and FeB is dominated by the d orbitals of Cr and Fe, respectively (see the right panels of (a) and (b) in Figure 3 and Figure 4). Whereas, an hybridization between p and d orbitals of Zr occurs at the Fermi level of ZrB (see the right panels of (c) in Figure 3 and Figure 4). The contribution of the majority states at the Fermi level of ortho-FeB is very small, whereas an equal contribution of the minority and majority states is observed in hex-FeB and also in both symmetries of ZrB. Finally, the contribution of the majority states is larger than that of the minority states at the Fermi level for CrB.
According to our Bader analysis, the charge transfer, , always happens from the TMs to the boron atoms. This is shown in Table 2 where we collected the values of for all the cases studied. The obtained values are in agreement with other reports (e.g., for ortho-FeB as reported in Ref. [26]). In general, considering the and structures, the largest charge transfer happens between Zr and B, whereas Fe is the TM for which the charge transfer to B is the smallest.
The conductivity results are shown in Figure 5. They reveal an anisotropy of the conductivity tensor for the ortho-MBenes, especially for the case of ortho-ZrB for which the direction perpendicular to the BDC is clearly the preferred one. Whereas for ortho-FeB the conductivity is higher along the BDC. On the other hand, the hex-MBenes are isotropic with respect to conductivity. It is also worth highlighting that the orthorhombic FeB and ZrB present the highest conductivity values among all the studied cases.
3.3. Magnetic Properties
The origin of magnetism in these types of materials arises from the d orbitals of the TM atoms. Both FM and AFM configurations have been considered to determine the ground state. The results of our calculations are summarized in Table 3. Among all the considered structures, only the hex-FeB resulted in an AFM ground state since the total energy of the FM state is 46.84 meV higher in energy. On the other hand, ortho-FeB has an FM ground state in accordance with previous reports [34]. In the particular case of CrB, both structures result in an FM arrangement of the magnetic moments. In general, the total energy difference, , between the FM and AFM configurations is always higher in absolute value for the orthorhombic structures (-104.38 and -108.12 eV for CrB and FeB, respectively) than for the hexagonal ones (-0.29 and 46.84 eV for CrB and FeB, respectively), indicating that in the latter case, the magnetic ordering may not be preserved at room temperature. Both orthorhombic CrB and FeB structures show an FM ground state with a magnetic moment over 2.5 per unit cell, a suitable behavior for robust 2D magnets. Interestingly, the boron atoms are also slightly polarized for those cases for which the TM–boron distances () are the shortest (see Table 1). Finally, ZrB MBenes exhibit a non-magnetic behavior.
4. Summary and Conclusions
In summary, we have done a comparison between the structural, electronic, magnetic, and transport properties of orthorhombic and hexagonal phases for a selected group of MBenes, CrB, FeB and ZrB, that are usually studied separately in the literature. Although there are several theoretical reports that have predicted the stability of the studied MBenes, to our knowledge, hex-FeB is shown in this work to be dynamically stable for the first time. Experimentally the most studied MBenes are those composed of early TMs, whereas those composed of late TMs (like iron) remain to be synthesized.
We have observed that, from an energetic point of view, the larger the atomic weight, the higher the , which means that ZrB possess the strongest bonds. However, despite little differences between the cohesive energies of ortho- and hex-structures of each MBene, all of them can exist theoretically. This affirmation is reinforced by the calculation of the corresponding phonon dispersion plots, that predict a stable behavior of all the MBenes.
On the other hand, the metallic character of our pristine MBenes makes them efficient materials for charge transport, therefore, competing 2D materials for electronic, sensing, or electrocatalytic purposes. We have predicted that for both symmetries of CrB and for ortho-FeB the contribution of the majority spin states is larger than that of the minority spin states at the Fermi level leading to an FM ground state and opening the possibility of their use in information magnetic storage.
This comprehension of their properties together with the acknowledgment that, within the same framework, both orthorhombic and hexagonal phases are feasible and different in their properties, give us the possibility to go further in our research and consider them as potential candidates for sensing and catalytic processes. Although there exists some parallelism with MXenes, MBenes are emerging 2D materials expected to have great development in the future, due to their diverse stoichiometries and, as a consequence, structural differences and new challenging physical, chemical, and biological properties. What makes the biggest difference for MBenes is that some of them can potentially exist both in the orthorhombic and hexagonal phases.
Author Contributions
Conceptualization, I.A.-C. and N.G.S.; methodology, I.A.-C.; validation, I.A.-C.; investigation, I.A.-C. and N.G.S.; writing and original draft preparation, I.A.-C.; writing, review, and editing, N.G.S.; visualization, I.A.-C.; supervision, N.G.S.; All authors have read and agreed to the published version of the manuscript.
Acknowledgments
The use of supercomputers at the Interdisciplinary Centre for Mathematical and Computational Modelling (ICM) at the University of Warsaw is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Akopov, G.; Yeung, M.T.; Kaner, R.B. Rediscovering the Crystal Chemistry of Borides. Adv. Mater. 2017, 29, 1604506. [CrossRef]
- Ade, M.; Hillebrecht, H. Ternary Borides Cr2AlB2, Cr3AlB4, and Cr4AlB6: The First Members of the Series (CrB2)nCrAl with n = 1,2,3 and a Unifying Concept for Ternary Borides as MAB-Phases. Inorg. Chem. 2015, 54, 6122–6135. [CrossRef]
- Carlsson, A.; Rosen, J.; Dahlqvist, M. Theoretical predictions of phase stability for orthorhombic and hexagonal ternary MAB phases. Phys. Chem. Chem. Phys. 2022, 24, 11249–11258. [CrossRef]
- Wang, J.; Ye, T.N.; Gong, Y.; Wu, J.; Miao, N.; Tada, T.; Hosono, H. Discovery of hexagonal ternary phase Ti2InB2 and its evolution to layered boride TiB. Nat. Commun. 2019, 10. [CrossRef]
- Alameda, L.T.; Moradifar, P.; Metzger, Z.P.; Alem, N.; Schaak, R.E. Topochemical Deintercalation of Al from MoAlB: Stepwise Etching Pathway, Layered Intergrowth Structures, and Two-Dimensional MBene. J. Am. Chem. Soc. 2018, 140, 8833–8840. [CrossRef]
- Alameda, L.T.; Lord, R.W.; Barr, J.A.; Moradifar, P.; Metzger, Z.P.; Steimle, B.C.; Holder, C.F.; Alem, N.; Sinnott, S.B.; Schaak, R.E. Multi-Step Topochemical Pathway to Metastable Mo2AlB2 and Related Two-Dimensional Nanosheet Heterostructures. J. Am. Chem. Soc. 2019, 141, 10852–10861. [CrossRef]
- Zhang, H.; Xiang, H.; zhi Dai, F.; Zhang, Z.; Zhou, Y. First demonstration of possible two-dimensional MBene CrB derived from MAB phase Cr2AlB2. J. Mater. Sci. Technol. 2018, 34, 2022–2026. [CrossRef]
- Zhang, H.; Dai, F.Z.; Xiang, H.; Wang, X.; Zhang, Z.; Zhou, Y. Phase pure and well crystalline Cr2AlB2: A key precursor for two-dimensional CrB. J. Mater. Sci. Technol. 2019, 35, 1593–1600. [CrossRef]
- Khazaei, M.; Wang, J.; Estili, M.; Ranjbar, A.; Suehara, S.; Arai, M.; Esfarjani, K.; Yunoki, S. Novel MAB phases and insights into their exfoliation into 2D MB. Nanoscale 2019, 11, 11305–11314. [CrossRef]
- Barinov, V.A.; Dorofeev, G.A.; Ovechkin, L.V.; Elsukov, E.P.; Ermakov, A.E. Structure and magnetic properties of the α-FeB phase obtained by mechanical working. Phys. Status Solidi (a) 1991, 123, 527–534. [CrossRef]
- Zhao, X.; Li, L.; Bao, K.; Zhu, P.; Tao, Q.; Ma, S.; Cui, T. Insight the effect of rigid boron chain substructure on mechanical, magnetic and electrical properties of β-FeB. J. Alloy. Compd. 2022, 896, 162767. [CrossRef]
- Saldaña, F.I.; Defoy, E.; Janisch, D.; Rousse, G.; Autran, P.O.; Ghoridi, A.; Séné, A.; Baron, M.; Suescun, L.; Godec, Y.L.; Portehault, D. Revealing the Elusive Structure and Reactivity of Iron Boride α-FeB. Inorg. Chem. 2023, 62, 2073–2082. [CrossRef]
- Gonzalez Szwacki, N.; Sadrzadeh, A.; Yakobson, B.I. B80 Fullerene: An Ab Initio Prediction of Geometry, Stability, and Electronic Structure. Phys. Rev. Lett. 2007, 98, 166804. [CrossRef]
- Szwacki, N.G. Boron Fullerenes: A First-Principles Study. Nanoscale Res. Lett. 2007, 3. [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Physics: Condens. Matter 2009, 21, 395502. [CrossRef]
- Himmetoglu, B.; Janotti, A. Transport properties of KTaO3 from first-principles. J. Physics: Condens. Matter 2016, 28, 065502. [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Apllied Crystallogr. 2011, 44, 1272–1276. [CrossRef]
- Bo, T.; Liu, P.F.; Xu, J.; Zhang, J.; Chen, Y.; Eriksson, O.; Wang, F.; Wang, B.T. Hexagonal Ti2B2 monolayer: A promising anode material offering high rate capability for Li-ion and Na-ion batteries. Phys. Chem. Chem. Phys. 2018, 20, 22168–22178. [CrossRef]
- He, Q.; Li, Z.; Xiao, W.; Zhang, C.; Zhao, Y. Computational investigation of 2D 3d/4d hexagonal transition metal borides for metal-ion batteries. Electrochim. Acta 2021, 384, 138404. [CrossRef]
- Yuan, G.; Bo, T.; Qi, X.; Liu, P.F.; Huang, Z.; Wang, B.T. Monolayer Zr 2 B 2 : A promising two-dimensional anode material for Li-ion batteries. Appl. Surf. Sci. 2019, 480, 448–453. [CrossRef]
- Mir, S.H.; Yadav, V.K.; Singh, J.K. Efficient CO2Capture and Activation on Novel Two-Dimensional Transition Metal Borides. ACS Appl. Mater. Interfaces 2022, 14, 29703–29710. [CrossRef]
- Dou, M.; Li, H.; Yao, Q.; Wang, J.; Liu, Y.; Wu, F. Room-temperature ferromagnetism in two-dimensional transition metal borides: A first-principles investigation. Phys. Chem. Chem. Phys. 2021, 23, 10615–10620. [CrossRef]
- Ozdemir, I.; Kadioglu, Y.; Yüksel, Y.; Ümit Aklncl.; Üzengi Aktürk, O.; Aktürk, E.; Ciraci, S. Columnar antiferromagnetic order of a MBene monolayer. Phys. Rev. B 2021, 103. [CrossRef]
- Qi, S.; Fan, Y.; Zhao, L.; Li, W.; Zhao, M. Two-dimensional transition metal borides as highly efficient N2 fixation catalysts. Appl. Surf. Sci. 2021, 536. [CrossRef]
- Zhang, B.; Zhou, J.; Guo, Z.; Peng, Q.; Sun, Z. Two-dimensional chromium boride MBenes with high HER catalytic activity. Appl. Surf. Sci. 2020, 500. [CrossRef]
- Shin, H.; Kang, S.; Koo, J.; Lee, H.; Kim, J.; Kwon, Y. Cohesion energetics of carbon allotropes: Quantum Monte Carlo study. J. Chem. Phys. 2014, 140, 114702. [CrossRef]
- Xu, T.; Wang, Y.; Xiong, Z.; Wang, Y.; Zhou, Y.; Li, X. A Rising 2D Star: Novel MBenes with Excellent Performance in Energy Conversion and storage. Nano-Micro Lett. 2022, 15:6. [CrossRef]
- Şahin, H.; Cahangirov, S.; Topsakal, M.; Bekaroglu, E.; Akturk, E.; Senger, R.T.; Ciraci, S. Monolayer honeycomb structures of group-IV elements and III-V binary compounds: First-principles calculations. Phys. Rev. B 2009, 80, 155453. [CrossRef]
- Fasolino, A.; Los, J.H.; Katsnelson, M.I. Intrinsic ripples in graphene. Nat. Mater. 2007, 6, 858–861. [CrossRef]
- Zhang, B.; Zhou, J.; Sun, Z. MBenes: Progress, challenges and future. J. Mater. Chem. A 2022, 10, 15865–15880. [CrossRef]
- Guo, Z.; Zhou, J.; Sun, Z. New two-dimensional transition metal borides for Li ion batteries and electrocatalysis. J. Mater. Chem. A 2017, 5, 23530–23535. [CrossRef]
Figure 1.
Side (left) and top (right) views of the (a) ortho-MBene and (b) hex-MBene structures of MB corresponding to and symmetries, respectively. The unit cells used in the calculations are shown in red.
Figure 1.
Side (left) and top (right) views of the (a) ortho-MBene and (b) hex-MBene structures of MB corresponding to and symmetries, respectively. The unit cells used in the calculations are shown in red.
Figure 2.
Phonon dispersion relations for the (a) and (b) structures of CrB, FeB, and ZrB.
Figure 3.
Electronic band structure (left) and DOS (center) for the majority (blue) and minority (red) spin states, and orbital-resolved PDOS (right) for the structures of (a) CrB, (b) FeB, and (c) ZrB.
Figure 3.
Electronic band structure (left) and DOS (center) for the majority (blue) and minority (red) spin states, and orbital-resolved PDOS (right) for the structures of (a) CrB, (b) FeB, and (c) ZrB.
Figure 4.
Electronic band structure and DOS for the majority (blue) and minority (red) spins, and orbital-resolved projected density of states (PDOS) for the structures of (a) CrB, (b) FeB, and (c) ZrB.
Figure 4.
Electronic band structure and DOS for the majority (blue) and minority (red) spins, and orbital-resolved projected density of states (PDOS) for the structures of (a) CrB, (b) FeB, and (c) ZrB.
Figure 5.
Components of the conductivity tensor for the (left) and (right) structures of CrB, FeB, and ZrB.
Figure 5.
Components of the conductivity tensor for the (left) and (right) structures of CrB, FeB, and ZrB.
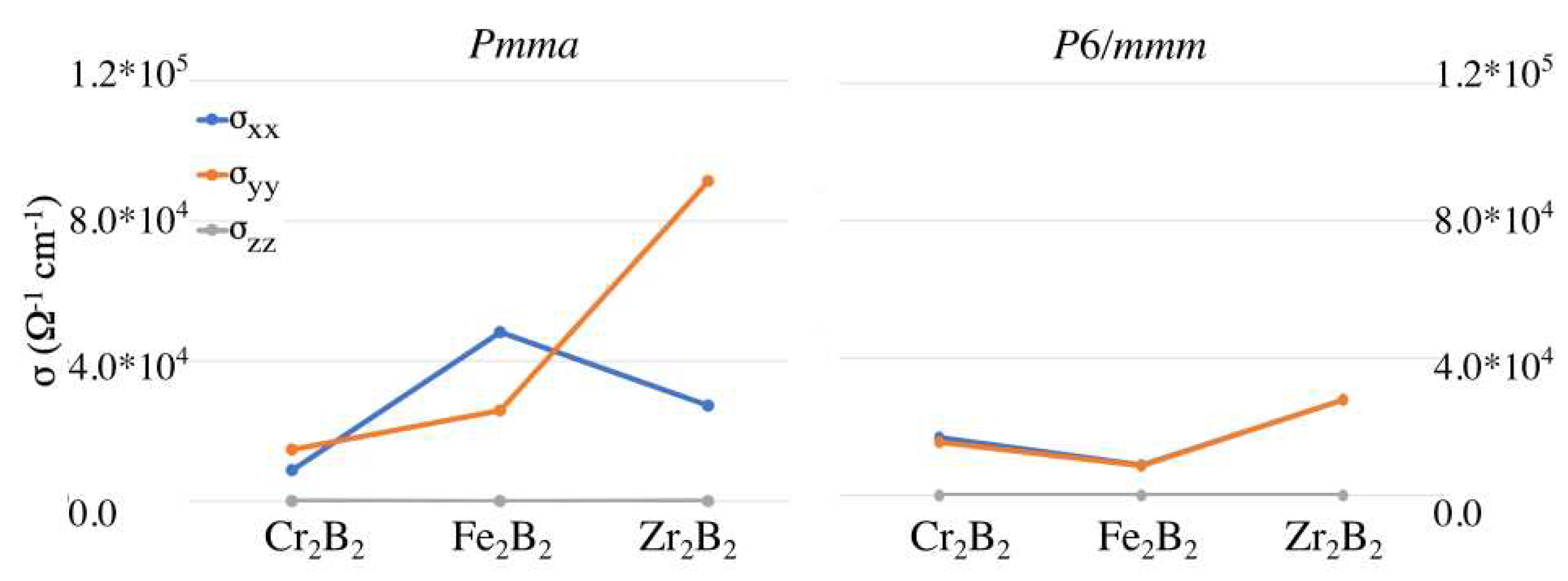

Table 1.
Lattice parameters (a and b), boron–boron distance (), TM–boron distance (), and thickness of the 2D structure (t) for CrB, FeB, and ZrB. The , , and t parameters are defined in Figure 1. All the values are in Angstroms (Å) and for comparison purposes, the data in brackets are taken from the literature.
Table 1.
Lattice parameters (a and b), boron–boron distance (), TM–boron distance (), and thickness of the 2D structure (t) for CrB, FeB, and ZrB. The , , and t parameters are defined in Figure 1. All the values are in Angstroms (Å) and for comparison purposes, the data in brackets are taken from the literature.
| CrB | FeB | ZrB | ||||
|---|---|---|---|---|---|---|
| a | 2.885 | 2.919 | 2.823 | 2.913 | 3.084 | 3.159 |
| (2.860 4) (2.930 5) |
(2.921 1) (2.926 2) |
(2.800 4) (2.770 5) |
(3.07 7) | (3.144 1) (3.160 2) (3.134 3) |
||
| b | 2.870 | 2.919 | 2.787 | 2.913 | 3.281 | 3.159 |
| (2.850 4) (2.870 5) |
(2.921 1) (2.926 2) |
(2.680 4) (2.820 5) |
(3.27 7) | (3.144 1) (3.160 2) (3.134 3) |
||
| 1.810 | 1.685 | 1.813 | 1.682 | 1.721 | 1.824 | |
| (1.689 2) | (1.71 7) | (1.825 2) (1.891 3) |
||||
| 2.10 | 2.15 | 2.04 | 2.15 | 2.46 | 2.49 | |
| (2.11 4) | (2.05 4) | (2.43 7) | (2.49 3) | |||
| t | 2.122 | 2.662 | 2.134 | 2.680 | 2.817 | 3.382 |
| (2.651 1) (2.639 2) |
(2.13 6) | (2.84 7) | (3.391 1) (3.380 2) (3.385 3) |
Table 2.
Cohesive energy (), highest frequency at the point (), and charge transfer from TM to B () for CrB, FeB, and ZrB.
Table 2.
Cohesive energy (), highest frequency at the point (), and charge transfer from TM to B () for CrB, FeB, and ZrB.
| CrB | FeB | ZrB | ||||
|---|---|---|---|---|---|---|
| (eV) | 6.222 | 6.201 | 6.901 | 6.830 | 8.050 | 8.087 |
| (cm) | 762.45 | 1003.50 | 772.74 | 976.01 | 928.02 | 744.12 |
| (e) | -0.76 | -0.61 | -0.37 | -0.40 | -1.17 | -0.86 |
Table 3.
Total magnetic moment (), magnetic moment of the TM atoms (), magnetic moment induced on the boron atoms (), magnetic ground state (MGS) and the total energy difference between the FM and the AFM configurations () for CrB, FeB, and ZrB.
Table 3.
Total magnetic moment (), magnetic moment of the TM atoms (), magnetic moment induced on the boron atoms (), magnetic ground state (MGS) and the total energy difference between the FM and the AFM configurations () for CrB, FeB, and ZrB.
| CrB | FeB | ZrB | ||||
|---|---|---|---|---|---|---|
| (/unit cell) | 2.56 | 0.63 | 2.69 | 0.00 | 0.00 | 0.00 |
| (/ion) | 1.03 | 0.31 | 1.26 | 2.06/-2.06 | 0.00 | 0.00 |
| (/ion) | -0.05 | -0.01 | -0.05 | 0.00 | 0.00 | 0.00 |
| MGS | FM | FM | FM | AFM | Non-magnetic | Non-magnetic |
| (meV/unit cell) | -104.38 | -0.29 | -108.12 | 46.84 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



