
Submitted:
31 May 2023
Posted:
01 June 2023
You are already at the latest version
Abstract
Chaphamaparvovirus (ChPV) is an ancient virus that has been detected in a variety of hosts. In this study, based on the phylogenetic analysis and the adaptability of ChPV to multiple hosts, we evaluated the potential of feline (FeChPV) and canine ChPV (CaChPV) for cross-species transmis-sion. Phylogenetic analysis showed that FeChPV and CaChPV were closely related. Notably, two strains of ChPVs isolated from domestic cats and 2 from dogs clustered together with CaChPVs and FeChPVs, respectively, suggesting the stringent boundaries between canine and feline ChPV may be broken. Further analysis revealed that CaChPV and FeChPV were more adapted to dogs than to cats, strongly suggesting the possibility of unidirectional or bidirectional transmission be-tween dogs and cats. Mutation analysis identified several shared mutations in cross-species-transmissible strains that were not located within immune epitopes. Furthermore, the VP struc-tures of FeChPV and CaChPV exhibited a high degree of similarity across both cross-species-transmissible and non-cross-species-transmissible strains. However, definitive experimental evi-dence is lacking, and its capacity for cross-species transmission should be approached with cau-tion and elucidated in further studies.
Keywords:
Canine
; feline
; chaphamaparvovirus
; codon usage
; phylogenetic
; cross-species transmission
1. Introduction
The Parvoviridae family is divided into three subfamilies as follows: Densovirinae, Hamaparvovirinae, and Parvovirinae [1]. The unique Hamaparvovirinae subfamily can infect a wide range of hosts, including vertebrates and invertebrates [2]. Chaphamaparvovirus (ChPV) is an important genus of the Hamaparvovirinae and can infect various animals, such as bats [3], rodents [4], birds/poultry [5,6,7,8], pigs [9], dogs [10], domestic cats [11], tasmanian devils [12], wild mammals [13,14], and human [15]. Canine ChPV (CaChPV) and feline ChPV (FeChPV) are classified as Carnivore chaphamaparvovirus 1 and Carnivore chaphamaparvovirus 2, respectively [11,16]. The complete genome of FeChPV/CaChPV is approximately 3,400 bp in length and contains two major open reading frames: nonstructural protein (NS) and virion protein (VP).
In 2017, a novel parvovirus, termed Cachavirus, was detected in the feces of dogs during an outbreak of diarrhea of an unexplained origin [10]. It was subsequently detected in Canada, China, and Italy [16,17,18]. Similarly, FeChPV was first detected in Canada during an outbreak of an unknown origin in an animal shelter [19]. Both CaChPV and FeChPV are supposedly associated with gastrointestinal symptoms and are more frequently detected in animals with diarrhea than that in healthy animals. Further, FeChPV has been detected in cats with respiratory diseases and coinfections with other common viruses [20].
Previous studies have reported on the cross-host transmission of parvoviruses. Canine parvovirus (CPV) 2a/2b/2c infects cats and causes symptoms similar to those caused by feline parvovirus (FPV) infection in domestic cats [21,22]. Mink parvovirus is a variant resulting from the adaptation of the FPV to mink. Interestingly, in China, CaChPV has been detected in cats, while FeChPV has been found in dogs, strongly suggesting a range of host diversity [17,23].
In the present study, we focused on the potential of FeChPV and CaChPV for cross-species transmission. A phylogenetic analysis and codon usage analysis revealed the potential host range diversity of the carnivore ChPV and provided additional information to support the understanding of ChPV.
2. Materials and Methods
2.1. Clinical Samples and Viral Genome Sequence Collection
From July 2018 to October 2020, 58 fecal samples from domestic cats were collected using anal swabs from different animal hospitals in the Anhui province. Thirty-two swabs were obtained from cats with diarrhea, whereas the remaining samples were obtained from healthy cats without diarrhea symptoms. Virus extraction, polymerase chain reaction, and sequencing were performed as previously described [24]. This study has been approved by the Institutional Animal Care and Use Committee of Anhui Agricultural University (SYDW-P20200311059)
We collected reference sequences of FeChPV, CaChPV, porcine parvovirus 7, poultry/birds ChPV, mouse kidney parvovirus (Mus musculus), bat ChPV, hedgehog (Erinaceus amurensis) ChPV, pangolin ChPV, red fox (Vulpes vulpes) ChPV, American black bear (Ursus americanus) ChPV, and tasmanian devil (Sarcophilus harrisii) ChPV, and human (Homo sapiens) ChPV and primate (Cebus capucinus imitator and Macaca fascicularis) ChPV from the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/,accessed on 13 December 2022).
2.2. Phylogenetic Analysis
The sequences were aligned using Multiple Alignment using Fast Fourier Transform [25] and trimmed using the TrimAl software [26]. The best substitution model was selected using ModelFinder [27]. We inferred maximum likelihood phylogenies using IQ-TREE with ultrafast bootstrap analysis [28]. Phylogenetic trees were visualized using FigTree version 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree, accessed on 25 December 2022) and Interactive Tree Of Life [29].
2.3. Codon Usage Analysis
We analyzed the nucleotide compositions of NS and VP in FeChPV and CaChPV. Basic nucleotide composition (A%, T%, C%, and G%), nucleotides at the third position of synonymous codons (A3s%, T3s%, C3s%, and G3s%), and G/C content at the third synonymous codon position (GC3s) were calculated using CAIcal SERVER [30].
The effective number of codons (ENC) was calculated using the CAIcal server. ENC values range from 20 to 61, and ENC <35 indicates codon usage bias (CUB) (the smaller the value, the stronger the CUB). The ENC formulae were based on previous studies [31].
2.4. Relative Synonymous Codon Usage Analysis
We compared the codon usage patterns of FeChPV and CaChPv with those of the host. We analyzed the Relative Synonymous Codon Usage (RSCU) of the FeChPV and CaChPV genome. The RSCU value indicates the ratio of observed codon occurrence to random occurrence. RSCU helps to understand the preferential use of synonymous codons. A synonymous codon with a higher frequency of occurrence had an RSUC of >1, whereas that with a lower frequency had an RSCU of <1. An RSCU of >1.6 denotes an overrepresented synonymous codon, whereas one of <0.6 denotes an underrepresented synonymous codon [32]. The preferred codon was defined as one most used for an amino acid (one with the highest RSCU value). The RSCU of the VP and NS were calculated using CAIcal SERVER [30]. The RSCU of the host was calculated using the following equation:
where RSCUij is the value of the i-th synonymous codon of the j-th amino acid, Xij is the observed number of the i-th codon of the j-th amino acid, and "ni" denotes the number of concatenated codons encoding the j-th amino acid.
We defined the dissimilarity of in synonymous codon usage preferences between viruses and hosts using Euclidean distances (Dp):
where ni is the number of synonymous codons of amino acid I; yij is the fraction of codon j among synonymous codons of amino acid I in the viral gene; and xij is the supply of tRNA represented by the fraction of codon j among the synonymous codons in the host transcriptome. Dp was defined as the weighted set average of the equivalent weights of the 18 codons. Higher Dp values suggested greater dissimilarity, while lower values indicated less dissimilarity [33].
2.5. Codon Usage Pattern Difference Analysis
Relative codon deoptimization index (RCDI) analysis was performed using the RCDI server [34]. An RCDI equal to 1 indicated that the virus displayed a pattern of codon usage adapted to the host. Conversely, a value of >1 indicated low adaptation [35]. A higher RCDI value indicates a greater variance from the codon usage pattern of the host. The host codon collected from the Codon-and-codon pair usage tables (CoCoPUTs) [36].
We used the similarity index [SiD or D (A, B)] to estimate the influence of the host codon usage patterns on virus formation. SiD values ranged from 0 to 1, with higher values indicating a stronger host influence on viral codon usage. We used the following formula:
where ai denotes the RSCU value of the 59 synonymous codons of the virus coding sequence and bi denotes the RSCU value of the identical codon in the host [35].
2.6. Host Adaptability Analysis
The codon adaptation index (CAI) was estimated using the CAI calculation of the CAIcal server [30]. The host codon tables of Canis, Felis catus, Sarcophilus harrisii, Chiroptera, Manis, Mus musculus, Ursus, Macaca fascicularis, Macaca mulatta, and Homo sapiens were collected from the Codon and Codon pair usage tables (CoCoPUTs) [36]. For Canis and Felis catus, we used normalised CAI (nCAI) to further correct the CAI values [37]. Normalised CAI was defined as the quotient between the CAI and its expected CAI (eCAI). eCAI was estimated using the CAI calculation of the CAIcal server [30]. nCAI less than 1 are considered to be differences in CAI values due to nucleotide composition, while values of nCAI closer to 1 or greater than 1 must be interpreted as evidence of adaptation to the host codon usage pattern [37].
2.7. Parity Rule 2, ENC-Plot, and Neutrality Analysis
We performed a parity rule 2 (PR2) analysis to evaluate whether the influence of selection pressure on codon usage during evolution was consistent with mutations. A3/(A3 + T3) was the abscissa, G3/(G3+C3) was the ordinate, and the coordinate axis at 0.5 was the origin. The effects of selection pressure and mutations were considered inconsistent in cases with dots clustered around the origin and consistent in other cases [38].
ENC-plots use ENC as the ordinate and GC3s as the abscissa. Cases with dots clustered on/above and under the curve indicated that the CUB was greatly affected by mutations and other factors (e.g., selection pressure) [39].
The equation for calculating the expected ENC value was as follows:
where x is the frequency of GC at the third position of synonymous codons.
2.8. Comparative Analysis of Mutations, Immune Epitopes, and Structures
The VP protein sequences of FeChPV and CaChPV were created separately as separate datasets. Protein sequence comparisons were performed by MEGA X [40]. Mutation analysis was performed using the ESPript 3.0 (https://espript.ibcp.fr) [41]. B-cell immune epitopes were predicted using SVPPriT, and scores greater than 0.7 were adopted [42]. The structures of the Vp genes of CaChPV/Cat/MN928790.1, CaChPV/Cat/MN928791.1, CaChPV/Dog/MT123284.1, FeChPV/Dog/OQ162042.1, FeChPV/Dog/OQ162043.1, and FeChPV/Cat/MN396757.1 were predicted using AlphaFold2 [43]. We used PyMOL Molecular Graphics System (Version 2.0 Schrödinger, LLC.) for visualization and comparative analysis of the protein structures. Root Mean Square Deviation (RMCD) were used to measure the size of protein structural differences, when RMSD 1 Å was considered a cut-off for different structures [44].
2.9. Statistical Analysis
The Shapiro–Wilk test was conducted as the normality test. We performed the Mann–Whitney U test and one-way analysis of variance to analyze the significance of the Gaussian distribution data and non-conforming data, respectively. Graphpad Prism v 9.3.0 was used for the statistical analysis and data visualization.
3. Results
3.1. Clinical Samples
A total of 58 stool samples were examined, and four were positive for FeChPV. The positive rate is 6.9% (4/58). All four samples were collected from cats diagnosed with gastroenteritis, with a positive rate of 12.5% (4/32). FeChPV was not detected in asymptomatic cats. The complete genomes were uploaded to GenBank (Accession numbers: MT708230.1/HF1, MT708231.1/HF2, MZ031965/AH-03, and MZ031966.1/04).
3.2. Phylogenetic Analysis
Phylogenetic trees were established based on the complete genomes,NS and VP (Supplementary Table S1). In the genome-wide tree (Figure 1), FeChPV and CaChPV clustered on a same branch and in the same lineage as ChPV isolated from rodents (Mus musculus), Sarcophilus harrisii, Ursus americanus, primate (Cebus capucinus imitator and Macaca fascicularis), hedgehog (Erinaceus amurensis), pangolin, and bat, thus indicating a close relationship between CaChPV, FeChPV, and ChPV isolated from these hosts. Further analysis of the phylogenetic trees of NS and VP revealed that FeChPV and CaChPV belonged to two different branches. However, two strains of ChPV isolated from domestic cats clustered together with CaChPVs and 2 stains isolated from dogs clustered together with FeChPVs, thereby suggesting a close relationship between FeChPV and CaChPV and the possibility of cross-species transmission events of ChPV between dogs and cats (Figure 2).
3.4. Codon Usage Pattern Difference Analysis
To better quantify this codon usage pattern difference, we analyzed the RCDI and SiD. According to the RCDI (Figure 3), the codon usage patterns of FeChPV and CaChPV were more similar to those of dogs, thus suggesting FeChPV and CaChPV were better adapted to dogs than to cats. The codon usage patterns of CaChPV were more similar to those of dogs and cats, but not significantly different from those of FeChPV (p>0.05), suggesting that CaChPV is more adapted to the hosts. The SiD results were similar to those of RCID (Figure 3).
3.4. Host Adaptability Analysis
Variations in CUB are associated with adaptations to different hosts. We performed an adaptive analysis of dogs and cats (Supplementary Table S2). Results of the CAI (Table 1) indicated that FeChPV and CaChPV were more adaptable to dogs (Canis) than that to cats (Felis catus), thus indicating the ability of viral genes to be highly expressed in both canine and feline host cells and the potential ability for unidirectional or bidirectional cross-species transmission of both viruses between dogs and cats. The results for eCAI were also identical. Further analysis of the nCAI revealed that although the nCAI values for both FeChPV and CaChPV were less than 1 in dogs and cats, the values for FeChPV were notably closer to 1. This finding also strongly suggests that FeChPV demonstrates a higher degree of adaptability to dogs and cats. These results are consistent with the results for RCDI and SiD. However, contrary to the RCDI and SiD, the CAI showed that FeChPV is more adapted to hosts than CaChPV, which may be due to the small amount of data or statistical errors. Combination of CAI/nCAI, RCDI, and SiD results suggested the possibility of a “host spillover” event in ChPV.
3.3. CUB and RSCU Analysis
We observed deviations in codon usage for an identical virus from different hosts. Therefore, we analyzed codon usage preferences and their relationships with hosts. In the NS, neither FeChPV nor CaChPV displayed a strong CUB, with a mean ENC average value of 43.123±0.461 and 45.047±0.613, respectively. However, a stronger CUB was identified in the VP of the FeChPV group (35.081±0.417) (Table 2 and Supplementary Table S3).
CUB leads to a difference in usage of synonymous codons. We therefore analyzed the difference between the RSCU of FeChPV and CaChPV and the RSCU of the host to assess differences in synonymous codon preference. The RSCU values of CaChPV were much less different from those of dogs, indicating that the codon use preference of CaChPV is more similar to that of dogs. Moreover, CaChPV had more similar synonymous codon usage preferences with canine and cat than FeChPV in dogs and cats, strongly suggesting that CaChPV is more adaptable to the host (Figure 4 and Supplementary Table S4).
3.5. Parity Rule 2, ENC-Plot, and Neutrality Analysis
The PR2 analysis demonstrated that neither NS nor VP of FeChPV and CaChPV was clustered at the origin, thereby indicating both selection and mutation affected the CUB of these viruses in a manner that was consistent throughout the evolution (Figure 5a). The ENC-Plot depicted that all dots were distributed below the expected curve, which indicated that the selection pressure played a dominant role in these genes (Figure 5b). Consistent with these findings, the slopes of the regression lines were -0.1755, 0.08417, 0.01133, and 0.05754, respectively, in the neutrality analysis (Figure 5c), all of which were distant from 1. This aspect demonstrated the primary role of selection pressure in the formation of codon usage preferences for the FeChPV and CaChPV genes. The contributions of natural selection were 82.45% (NS of FeChPV), 91.58% (VP of FeChPV), 98.87% (NS of CaChPV), and 94.25% (VP of aChPV).
3.6. Comparative Analysis of Mutations, Immune Epitopes, and Structures
The obtained results demonstrate the influence of mutation and selection pressure on the evolution of both FeChPV and CaChPV. Therefore, we focused on the mutation characteristics of FeChPV and CaChPV, particularly in strains experiencing cross-species transmission. Among the two FeChPV strains undergoing cross-species transmission, four shared mutations were identified: G112R, L174P, S197T, and S325R. The S197 mutation in both FeChPV strains coincided with the T observed in CaChPV. Furthermore, S206 and S445 in FeChPV-OQ162042.1 mutated to the same A and N as CaChPV, while D402 in CaChPV-MN928790.1 mutated to the same N as in FeChPV. Additionally, the F131S mutation in CaChPV-MN928790.1 was also detected in FeChPV-OQ162042.1 (F151S).
In order to examine whether mutations contribute to changes in B-cell immune epitopes, the immune epitopes of both FeChPV and CaChPV were analyzed. The findings revealed no substantial differences in immune epitopes between cross-host transmitted and non-cross-host transmitted strains in either FeChPV or CaChPV. A comparison between FeChPV and CaChPV identified a conserved amino acid sequence (SIAYKEGMFK) present in the immune epitopes of both viruses.
We analyzed the structural similarity of VPs between cross-species transmitted and non-cross-species transmitted strains. Our findings demonstrated that both cross-species transmitted and non-cross-species transmitted strains displayed highly similar VP protein structures (RMSD<1) for FeChPV and CaChPV, suggesting their analogous receptor-binding configurations. Nonetheless, the presence of mutations may result in variations in binding affinity. Owing to the absence of receptor information for ChPV, we could not determine the consistency between individual virus receptor binding ability and binding site.

Table 4.
B-cell immune epitopes of FeChPV and CaChPV strains.
| Strains | Location | Epitope | Score |
|---|---|---|---|
| CaChPV-MN928790.1 | 388-407 | WGPWTWKDIYGIGSNTRMYS | 1.000 |
| 475 - 494 | PEMIEMQELHHTDDEEIEVI | 0.980 | |
| 123 - 142 | WKDSSMKDSSIAYKEGMFKS | 0.908 | |
| 24 - 43 | NNTLATIVAAETGGNAINTG | 0.797 | |
| 363 - 382 | TTQGCFQVTLHLACKKRRSR | 0.758 | |
| CaChPV-MN928791.1 | 479 - 498 | EMQELPHTDDEEIEIITADE | 1.000 |
| 388-407 | WGPWTWKDIYGIGSDTRMYS | 0.858 | |
| 24 - 43 | NNTLATIVAAETGGNAINTG | 0.724 | |
| Other strains | 475 - 494 | PEMIEMQELHHTDDEEIEVI | / * |
| 388-407 | WGPWTWKDIYGIGSDTRMYS | / | |
| 24 - 43 | NNTLATIVAAETGGNAINTG | / | |
| 123 - 142 | WKDSSMKDSSIAYKEGMFKS | / | |
| 363 - 382 | TTQGCFQVTLHLACKKRRSR | / | |
| FeChPV-OQ162042.1 | 144 - 163 | VTNPLKDSSIAYKEGMFKQG | 1.000 |
| FeChPV-OQ162043.1 | 145 - 164 | TNPLKDFSIAYKEGMFKQGT | 1.000 |
| Other strains | 145 - 164 | TNPLKDFSIAYKEGMFKQGT | 1.000 |
Note: * Scores were different between strains. Identical amino acid sequences are underlined.
4. Discussion
Chaphamaparvovirus is a recently defined genus in Parvoviridae family, which comprises a broad reservoir of hosts, including both vertebrates and invertebrates. ChPV is supposedly an ancient virus existing in animal hosts for millions of years [45]. Further, transmission may have occurred between distantly related host species [45]. In this study, we detected FeChPV in fecal samples from domestic cats; all positive samples were obtained from domestic cats with diarrhea, thus suggesting FeChPV is associated with diarrhea, similar to CaChPV [39]. ChPV is often associated with feces, and, therefore, strongly suggests an association with intestinal disease [45]; however, it supposedly has the potential to cause other diseases, such as respiratory disease [11], hepatitis [46], encephalitis [13], and chronic interstitial nephropathy [47], in other species. FeChPV was identified in cats exhibiting respiratory symptoms; however, the samples gathered in this study did not include the respiratory tract. As a result, the potential association between the ChPV and respiratory diseases cannot be conclusively assessed, warranting further exploration and study. However, the exact pathogenic capacities of FeChPV and CaChPV have not been confirmed, thus warranting further research.
A previous study reported on CaChPV in domestic cats and FeChPV in dogs [17,23]. Therefore, we proposed that it displays a possibility of cross-species transmission. To examine this hypothesis, we performed phylogenetic and codon usage analyses. The phylogenetic analysis demonstrated that ChPV detected in cats displayed higher homology with CaChPV, thus breaking the restrictive host boundary easily. FeChPV and CaChPV are closely related to ChPV isolated from rodents (Mus musculus), Sarcophilus harrisii, Ursus americanus, primate (Cebus capucinus imitator and Macaca fascicularis), hedgehog (Erinaceus amurensis), pangolins, and bats. Wildlife, such as pangolins, bats, and hedgehogs, are considered natural hosts for numerous viruses, independent of frequent host spillover events [48,49]. Humans are closely associated with dogs and cats, which are companion animals. A unidirectional or bidirectional cross-host transmission of viruses can occur between companion animals and humans [50,51]. Therefore, further epidemiological investigations are required to elucidate the close relationship between FeChPV and CaChPV with human and wildlife ChPV in the future.
The CAI value has been used to assess the expression of an exogenous gene within the cell. A high CAI value represents high levels of gene expression and a closer match to the host's codon usage preferences, which may replicate more efficiently [30]. Therefore, we assessed the CAI values and the similarity of codon usage patterns between FeChPV and CaChPV across various hosts. FeChPV and CaChPV demonstrated greater similarity in the same high-frequency use codons as those in dogs, suggesting that dogs rather than cats are potential hosts for FeChPV and CaChPV. In other words, FeChPV may be able to adapt to dogs, but not cats. Usually, gene expression requires the aid of transfer RNA (tRNA), and the abundance of tRNA corresponding to the codon used in the host gene is higher within the host cell. The differential codon use pattern would limit the rate of viral gene replication because of the use of a low abundance of tRNA. Generally, for exogenous genes, the closer the pattern of use to the host codon, the more efficient the gene expression and the more harmful it is to the host because it inhibits the expression of the host gene [33,52]. The CAI, RCID, and SiD analyses yielded similar results. Altogether, this evidence suggests that both FeChPV and CaChPV may infect dogs, while FeChPV or CaChPV can be transmitted between dogs and cats. Notably, the detection of ChPV in the intestine may be a result of food residues caused by a predatory relationship, suggested by the high correlation between ChPV and feces [45]. In one study, ichthyic ChPV was detected in tilapia-fed crocodiles [53]. However, the risk of this phenomenon in cats and dogs is low, as these animals do not have a predatory relationship. Therefore, the potential for transmission of ChPV between dogs and cats is high, and the risk of transmission to other hosts should be monitored. The role of a predatory relationship in the cross-species transmission is suggested by the close relationship between FeChPV and rodents ChPV. Moreover, the CAI value is influenced by a multitude of factors, including selection pressure, host codon usage, and mutations, among others [54]. Consequently, a comprehensive analysis and examination of these pertinent factors is essential for a more accurate understanding.
The CUB analysis demonstrated a weak bias for FeChPV, however, in VP of FeChPV, CUB was stronger than others. CUB causes differences in preferences for using synonymous codons when encoding the same amino acid. Further analysis revealed much lower difference in RSCU values with dogs for both FeChPV and CaChPV. The usage of more similar synonymous codons with the host implies that the virus is able to use the higher abundance of tRNA in the host as raw material for the efficient expression of viral proteins [52]. Thus, the low variability of FeChPV and CaChPV strongly suggests that this virus is highly adapted to dogs.
The gene mutation pressure, natural selection pressure, secondary protein structure, and external environment are the chief factors contributing to codon bias [24]. Therefore, we conducted PR2, ENC-plot, and neutrality analyses to evaluate the key dynamics influencing the CUB. Although both mutation and selection pressure play a role in the evolution of codon usage, the selection pressure was the primary influence, similar to CPV, FPV, and feline bocavirus [24,55]. CPV-2 constantly generates new subtypes, such as CPV-2C, which have the potential for cross-species transmission [21]. Selection pressure in virus evolution refers to the various factors and conditions (such as host immune response, antiviral drug and vaccine use, and environmental variables, among others) that drive changes in viral populations over time. These pressures can influence the survival and replication of viral strains, with some variants becoming more successful due to their specific adaptations. The selection pressure drives the process of natural selection, which can result in the emergence of new viral strains with improved fitness in a given environment [56,57,58,59]. Therefore, the selective pressure on CaChPV and FeChPV warrants attention, and further monitoring of the virus should be intensified to assess its evolutionary status.
Mutation analysis identified co-occurring mutations in cross-species transmitted strains, such as R449K, S197T, and G112R, among others. Notably, some FeChPV mutations correspond to the same amino acids as those in CaChPV, including S197T, D402N, and S206A. While the implications of these mutations for cross-species transmission could not be further explored in this study, they warrant attention. In CPV, specific mutations result in altered host range and receptor-binding capacity [60]. These mutations do not appear in corresponding B-cell immune epitopes, thus exerting minimal impact on B-cell immunity. Partial immune epitope overlap between CaChPV and FeChPV suggests potential cross-immunity, though experimental validation is required. Consequently, these mutations in CaChPV and FeChPV merit further investigation. Concerning structural similarity, our analysis revealed a high degree of resemblance between VP of cross-species transmissible and non-cross-species transmissible strains. This similarity in VP protein structure suggests a strong potential for binding to respective canine and feline receptors, providing a foundation for infection across different species.
5. Conclusions
This study highlights the possibility of cross-species transmission of ChPV between dogs and cats. The virus has demonstrated a high level of adaptability to dogs, as evidenced by high CAI/eCAI/nCAI to dogs, similar synonymous codon usage, similar codon usage pattern, and VP structure. These findings underscore the need for increased vigilance and detection of ChPV, as well as further research to evaluate the potential harm to dogs and cats and the risk of cross-species transmission. Given the limitations of the current sequence data and potential analytical errors, additional experiments are required to corroborate these findings.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Supplementary Table S1: The information of chaphamaparvovirus sequence used in the study; Supplementary Table S2: Codon usage analysis of Feline Chaphamaparvovirus and Canine Chaphamaparvovirus using NS and VP; Supplementary Table S3: Detailed information of relative synonymous codon usage analysis (RSCU) of FeChPV and CaChPV with hosts; Supplementary Table S4: The detailed results of Codon Adaptation Index, Relative Synonymous Codon Usage, Relative Codon Deoptimization Index and Similarity index.
Author Contributions
Conceptualization, W.Y., Y.K., and G.X.; methodology, G.X. Z.Y., and Y.K.; software, G.X. and Z.Y..; validation, G.X., Z.Y. and W.Y.; investigation, P.Y. and T.X.; data curation, W.Y.; writing—original draft preparation, G.X., P.Y., and Z.Y.; writing—review and editing, W.Y. and Y.K..; visualization, G.X. and Z.Y.; supervision, W.Y.; project administration, W.Y.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Hefei Rural Revitalization Science and Technology Expert Assistance Team.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from Supplementary files.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Penzes, J.J.; Soderlund-Venermo, M.; Canuti, M.; Eis-Hubinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. , Reorganizing the family Parvoviridae: a revised taxonomy independent of the canonical approach based on host association. Arch Virol 2020, 165, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Verhoeven, J.T.P.; Munro, H.J.; Roul, S.; Ojkic, D.; Robertson, G.J.; Whitney, H.G.; Dufour, S.C.; Lang, A.S. , Investigating the Diversity and Host Range of Novel Parvoviruses from North American Ducks Using Epidemiology, Phylogenetics, Genome Structure, and Codon Usage Analysis. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Souza, W.M.; Romeiro, M.F.; Fumagalli, M.J.; Modha, S.; de Araujo, J.; Queiroz, L.H.; Durigon, E.L.; Figueiredo, L.T.M.; Murcia, P.R.; Gifford, R.J. , Chapparvoviruses occur in at least three vertebrate classes and have a broad biogeographic distribution. J Gen Virol 2017, 98, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, Z.; Wang, Y.; Li, W.; Fu, X.; Lin, Y.; Shen, Q.; Wang, X.; Wang, H.; Zhang, W. , A novel rodent Chapparvovirus in feces of wild rats. Virol J 2016, 13, 133. [Google Scholar] [CrossRef] [PubMed]
- Hargitai, R.; Boros, A.; Pankovics, P.; Matics, R.; Altan, E.; Delwart, E.; Reuter, G. , Detection and genetic characterization of a novel parvovirus (family Parvoviridae) in barn owls (Tyto alba) in Hungary. Arch Virol 2021, 166, 231–236. [Google Scholar] [CrossRef]
- Lima, D.A.; Cibulski, S.P.; Tochetto, C.; Varela, A.P.M.; Finkler, F.; Teixeira, T.F.; Loiko, M.R.; Cerva, C.; Junqueira, D.M.; Mayer, F.Q.; Roehe, P.M. , The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics. Virus Res 2019, 261, 9–20. [Google Scholar] [CrossRef]
- Liu, X.; Wang, H.; Liu, X.; Li, Y.; Chen, J.; Zhang, J.; Wang, X.; Shen, S.; Wang, H.; Deng, F.; Wang, M.; Guan, W.; Hu, Z. , Genomic and transcriptional analyses of novel parvoviruses identified from dead peafowl. Virology 2020, 539, 80–91. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Klaassen, M.; Bhatta, T.R.; Alexandersen, S. , Metagenomic characterisation of avian parvoviruses and picornaviruses from Australian wild ducks. Sci Rep 2020, 10, 12800. [Google Scholar] [CrossRef]
- Xing, X.; Zhou, H.; Tong, L.; Chen, Y.; Sun, Y.; Wang, H.; Zhang, G. , First identification of porcine parvovirus 7 in China. Arch Virol 2018, 163, 209–213. [Google Scholar] [CrossRef]
- Fahsbender, E.; Altan, E.; Seguin, M.A.; Young, P.; Estrada, M.; Leutenegger, C.; Delwart, E. , Chapparvovirus DNA Found in 4% of Dogs with Diarrhea. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Di Profio, F.; Sarchese, V.; Palombieri, A.; Fruci, P.; Massirio, I.; Martella, V.; Fulvio, M.; Di Martino, B. , Feline chaphamaparvovirus in cats with enteritis and upper respiratory tract disease. Transbound Emerg Dis 2022, 69, 660–668. [Google Scholar] [CrossRef]
- Chong, R.; Shi, M.; Grueber, C.E.; Holmes, E.C.; Hogg, C.J.; Belov, K.; Barrs, V.R. , Fecal Viral Diversity of Captive and Wild Tasmanian Devils Characterized Using Virion-Enriched Metagenomics and Metatranscriptomics. J Virol 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Alex, C.E.; Fahsbender, E.; Altan, E.; Bildfell, R.; Wolff, P.; Jin, L.; Black, W.; Jackson, K.; Woods, L.; Munk, B.; Tse, T.; Delwart, E.; Pesavento, P.A. , Viruses in unexplained encephalitis cases in American black bears (Ursus americanus). PLoS One 2020, 15, e0244056. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Shi, M.; Que, T.C.; Cui, X.M.; Ye, R.Z.; Xia, L.Y.; Hou, X.; Zheng, J.J.; Jia, N.; Xie, X.; Wu, W.C.; He, M.H.; Wang, H.F.; Wei, Y.J.; Wu, A.Q.; Zhang, S.F.; Pan, Y.S.; Chen, P.Y.; Wang, Q.; Li, S.S.; Zhong, Y.L.; Li, Y.J.; Tan, L.H.; Zhao, L.; Jiang, J.F.; Hu, Y.L.; Cao, W.C. , Trafficked Malayan pangolins contain viral pathogens of humans. Nat Microbiol 2022, 7, 1259–1269. [Google Scholar] [CrossRef]
- Fahsbender, E.; Charlys da-Costa, A.; Elise Gill, D.; Augusto de Padua Milagres, F.; Brustulin, R.; Julio Costa Monteiro, F.; Octavio da Silva Rego, M.; Soares D'Athaide Ribeiro, E.; Cerdeira Sabino, E.; Delwart, E. , Plasma virome of 781 Brazilians with unexplained symptoms of arbovirus infection include a novel parvovirus and densovirus. PLoS One 2020, 15, e0229993. [Google Scholar] [CrossRef] [PubMed]
- Palombieri, A.; Di Profio, F.; Lanave, G.; Capozza, P.; Marsilio, F.; Martella, V.; Di Martino, B. , Molecular detection and characterization of Carnivore chaphamaparvovirus 1 in dogs. Vet Microbiol 2020, 251, 108878. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Hu, W.; Liu, Q.; Zuo, K.; Zhi, G.; Xu, X.; Kan, Y.; Yao, L.; Xie, Q. , Genetic Analysis of Cachavirus-Related Parvoviruses Detected in Pet Cats: The First Report From China. Front Vet Sci 2020, 7, 580836. [Google Scholar] [CrossRef]
- Canuti, M.; Mira, F.; Sorensen, R.G.; Rodrigues, B.; Bouchard, E.; Walzthoni, N.; Hopson, M.; Gilroy, C.; Whitney, H.G.; Lang, A.S. , Distribution and diversity of dog parvoviruses in wild, free-roaming and domestic canids of Newfoundland and Labrador, Canada. Transbound Emerg Dis 2022, 69, e2694–e2705. [Google Scholar] [CrossRef]
- Li, Y.; Gordon, E.; Idle, A.; Altan, E.; Seguin, M.A.; Estrada, M.; Deng, X.; Delwart, E. , Virome of a Feline Outbreak of Diarrhea and Vomiting Includes Bocaviruses and a Novel Chapparvovirus. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Li, Y.; Chen, B.; Wang, H.; Wang, X.; Xiao, X.; Zhou, P.; Li, S. , Detection of FeChPV in a cat shelter outbreak of upper respiratory tract disease in China. Front Microbiol 2022, 13, 1064747. [Google Scholar] [CrossRef]
- Jing, Z.; Ji, P.; Wei, Y.; Hao, F.; Wei, Y. , Isolation and identification of a novel canine parvovirus type 2c strain in domestic cats in Dalian, China. Front Vet Sci 2022, 9, 1001604. [Google Scholar] [CrossRef]
- Wu, J.; Gao, X.T.; Hou, S.H.; Guo, X.Y.; Yang, X.S.; Yuan, W.F.; Xin, T.; Zhu, H.F.; Jia, H. , Molecular epidemiological and phylogenetic analyses of canine parvovirus in domestic dogs and cats in Beijing, 2010-2013. J Vet Med Sci 2015, 77, 1305–10. [Google Scholar] [CrossRef]
- Ji, J.; Cui, H.; Xu, S.Q.; Xu, X.; Liu, Q.; Kan, Y.C.; Xie, Q.M.; Yao, L.G. , Molecular Characterization of Feline Chaphamaparvovirus (Carnivore chaphamaparvovirus 2) Firstly Detected in Dogs from China. Transboundary and Emerging Diseases 2023, 2023. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, X.; Li, W.; Cui, Y.; Zhang, D.; Xu, F.; Jiang, S.; Zhou, T. , Phylogenetic analysis and evolution of feline bocavirus in Anhui Province, eastern China. Comp Immunol Microbiol Infect Dis 2021, 77, 101676. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. , MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002, 30, 3059–66. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. , trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–3. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. , ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. , IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 2015, 32, 268–74. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. , Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Puigbo, P.; Bravo, I.G.; Garcia-Vallve, S. , CAIcal: a combined set of tools to assess codon usage adaptation. Biol Direct 2008, 3, 38. [Google Scholar] [CrossRef]
- Peng, Q.; Zhang, X.; Li, J.; He, W.; Fan, B.; Ni, Y.; Liu, M.; Li, B. , Comprehensive analysis of codon usage patterns of porcine deltacoronavirus and its host adaptability. Transbound Emerg Dis 2022, 69, e2443–e2455. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.U.; Rehman, H.U.; Rahman, I.U.; Rauf, A.; Alshammari, A.; Alharbi, M.; Haq, N.U.; Suleria, H.A.R.; Raza, S.H.A. , Analysis of codon usage bias of lumpy skin disease virus causing livestock infection. Front Vet Sci 2022, 9, 1071097. [Google Scholar] [CrossRef]
- Chen, F.; Yang, J.R. , Distinct codon usage bias evolutionary patterns between weakly and strongly virulent respiratory viruses. iScience 2022, 25, 103682. [Google Scholar] [CrossRef] [PubMed]
- Puigbo, P.; Aragones, L.; Garcia-Vallve, S. , RCDI/eRCDI: a web-server to estimate codon usage deoptimization. BMC Res Notes 2010, 3, 87. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Ding, S.; Wang, Z.; Jiang, R.; He, Z. , Host Plants Shape the Codon Usage Pattern of Turnip Mosaic Virus. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Alexaki, A.; Kames, J.; Holcomb, D.D.; Athey, J.; Santana-Quintero, L.V.; Lam, P.V.N.; Hamasaki-Katagiri, N.; Osipova, E.; Simonyan, V.; Bar, H.; Komar, A.A.; Kimchi-Sarfaty, C. , Codon and Codon-Pair Usage Tables (CoCoPUTs): Facilitating Genetic Variation Analyses and Recombinant Gene Design. J Mol Biol 2019, 431, 2434–2441. [Google Scholar] [CrossRef]
- Puigbo, P.; Bravo, I.G.; Garcia-Vallve, S. , E-CAI: a novel server to estimate an expected value of Codon Adaptation Index (eCAI). BMC Bioinformatics 2008, 9, 65. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, M.; Liu, Q.; Cao, Y.; Zhang, W.; Liang, Y.; Song, X.; Ji, K.; Shao, Y.; Qi, K.; Tu, J. , Epidemiology and Evolution of Emerging Porcine Circovirus-like Viruses in Pigs with Hemorrhagic Dysentery and Diarrhea Symptoms in Central China from 2018 to 2021. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, W.; Wang, R.; Zhang, W.; Li, G.; Lu, M.; Shao, Y.; Yang, Y.; Wang, N.; Gao, Q.; Su, S. , Analysis of the Codon Usage Pattern of HA and NA Genes of H7N9 Influenza A Virus. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. , MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. , Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 2014, 42, (Web Server issue), W320–4. [Google Scholar] [CrossRef]
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. , SVMTriP: a method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PLoS One 2012, 7, e45152. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; Bridgland, A.; Meyer, C.; Kohl, S.A.A.; Ballard, A.J.; Cowie, A.; Romera-Paredes, B.; Nikolov, S.; Jain, R.; Adler, J.; Back, T.; Petersen, S.; Reiman, D.; Clancy, E.; Zielinski, M.; Steinegger, M.; Pacholska, M.; Berghammer, T.; Bodenstein, S.; Silver, D.; Vinyals, O.; Senior, A.W.; Kavukcuoglu, K.; Kohli, P.; Hassabis, D. , Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baggio, G.; Filippini, F.; Righetto, I. , Comparative Surface Electrostatics and Normal Mode Analysis of High and Low Pathogenic H7N7 Avian Influenza Viruses. Viruses 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Penzes, J.J.; de Souza, W.M.; Agbandje-McKenna, M.; Gifford, R.J. , An Ancient Lineage of Highly Divergent Parvoviruses Infects both Vertebrate and Invertebrate Hosts. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Matos, M.; Bilic, I.; Viloux, N.; Palmieri, N.; Albaric, O.; Chatenet, X.; Tvarogova, J.; Dinhopl, N.; Heidl, S.; Liebhart, D.; Hess, M. , A novel Chaphamaparvovirus is the etiological agent of hepatitis outbreaks in pheasants (Phasianus colchicus) characterized by high mortality. Transbound Emerg Dis 2022, 69, e2093–e2104. [Google Scholar] [CrossRef]
- Edmondson, E.F.; Hsieh, W.T.; Kramer, J.A.; Breed, M.W.; Roelke-Parker, M.E.; Stephens-Devalle, J.; Pate, N.M.; Bassel, L.L.; Hollingshead, M.G.; Karim, B.O.; Butcher, D.O.; Warner, A.C.; Nagashima, K.; Gulani, J. , Naturally Acquired Mouse Kidney Parvovirus Infection Produces a Persistent Interstitial Nephritis in Immunocompetent Laboratory Mice. Vet Pathol 2020, 57, 915–925. [Google Scholar] [CrossRef]
- Han, Z.; Xiao, J.; Song, Y.; Zhao, X.; Sun, Q.; Lu, H.; Zhang, K.; Li, J.; Li, J.; Si, F.; Zhang, G.; Zhao, H.; Jia, S.; Zhou, J.; Wang, D.; Zhu, S.; Yan, D.; Xu, W.; Fu, X.; Zhang, Y. , Highly diverse ribonucleic acid viruses in the viromes of eukaryotic host species in Yunnan province, China. Front Microbiol 2022, 13, 1019444. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, X.; Si, X.; Ye, L.; Lawrence, K.; Lu, Y.; Du, C.; Xu, H.; Yang, Q.; Xia, Q.; Yu, G.; Xu, W.; Yuan, F.; Hao, J.; Jiang, J.F.; Zheng, A. , Hedgehogs as Amplifying Hosts of Severe Fever with Thrombocytopenia Syndrome Virus, China. Emerg Infect Dis 2022, 28, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Kannekens-Jager, M.M.; de Rooij, M.M.T.; de Groot, Y.; Biesbroeck, E.; de Jong, M.K.; Pijnacker, T.; Smit, L.A.M.; Schuurman, N.; Broekhuizen-Stins, M.J.; Zhao, S.; Duim, B.; Langelaar, M.F.M.; Stegeman, A.; Kooistra, H.S.; Radstake, C.; Egberink, H.F.; Wagenaar, J.A.; Broens, E.M. , SARS-CoV-2 infection in dogs and cats is associated with contact to COVID-19-positive household members. Transbound Emerg Dis 2022. [Google Scholar] [CrossRef]
- Seang, S.; Burrel, S.; Todesco, E.; Leducq, V.; Monsel, G.; Le Pluart, D.; Cordevant, C.; Pourcher, V.; Palich, R. , Evidence of human-to-dog transmission of monkeypox virus. Lancet 2022, 400, 658–659. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wu, P.; Deng, S.; Zhang, H.; Hou, Y.; Hu, Z.; Zhang, J.; Chen, X.; Yang, J.R. , Dissimilation of synonymous codon usage bias in virus-host coevolution due to translational selection. Nat Ecol Evol 2020, 4, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, W.; Chan, J.F.; Wang, G.; Huang, Y.; Yi, Y.; Zhu, Z.; Peng, R.; Hu, X.; Wu, Y.; Zeng, J.; Zheng, J.; Cui, X.; Niu, L.; Zhao, W.; Lu, G.; Yuen, K.Y.; Yin, F. , Identification of a Novel Ichthyic Parvovirus in Marine Species in Hainan Island, China. Front Microbiol 2019, 10, 2815. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.B.; Kudla, G. , Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Tucciarone, C.M.; Cecchinato, M.; Drigo, M. , Canine parvovirus type 2 (CPV-2) and Feline panleukopenia virus (FPV) codon bias analysis reveals a progressive adaptation to the new niche after the host jump. Mol Phylogenet Evol 2017, 114, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Day, T.; Kennedy, D.A.; Read, A.F.; Gandon, S. , Pathogen evolution during vaccination campaigns. PLoS Biol 2022, 20, e3001804. [Google Scholar] [CrossRef]
- Brand, M.; Kesmir, C. , Evolution of SARS-CoV-2-specific CD4(+) T cell epitopes. Immunogenetics 2023, 1–11. [Google Scholar] [CrossRef]
- Chen, Y.; Xue, Y.; Yang, J. , Gilteritinib: Repurposing of AXL-targeting kinase inhibitors against COVID-19. J Med Virol 2023. [Google Scholar] [CrossRef]
- Mandya Naganayak, M.; Kuralayanapalya Puttahonnappa, S.; Indrabalan, U.B.; Paramanandham, K.; Jacob, S.S.; Subramaniam, S.; Patil, S.S.; Seethakempanahalli Kempanna, K.; Goroshi, S. , An extensive analysis of Codon usage pattern, Evolutionary rate, and Phylogeographic reconstruction in Foot and mouth disease (FMD) serotypes (A, Asia 1, and O) of six major climatic zones of India: A comparative study. Acta Trop 2022, 236, 106674. [Google Scholar] [CrossRef]
- Chen, X.; Wang, J.; Zhou, Y.; Yue, H.; Zhou, N.; Tang, C. , Circulation of heterogeneous Carnivore protoparvovirus 1 in diarrheal cats and prevalence of an A91S feline panleukopenia virus variant in China. Transbound Emerg Dis 2022, 69, e2913–e2925. [Google Scholar] [CrossRef]
Figure 1.
Phylogenetic trees of Chaphamaparvovirus based on complete sequence (n=351). Potential cross-species transmission strains are highlighted in red. The feline chaphamaparvovirus obtained in this study uses rectangles to highlight. The best substitution model is GTR+F+R10. Canine bocavirus 2/MG025952.1 is used as an outgroup;.
Figure 1.
Phylogenetic trees of Chaphamaparvovirus based on complete sequence (n=351). Potential cross-species transmission strains are highlighted in red. The feline chaphamaparvovirus obtained in this study uses rectangles to highlight. The best substitution model is GTR+F+R10. Canine bocavirus 2/MG025952.1 is used as an outgroup;.
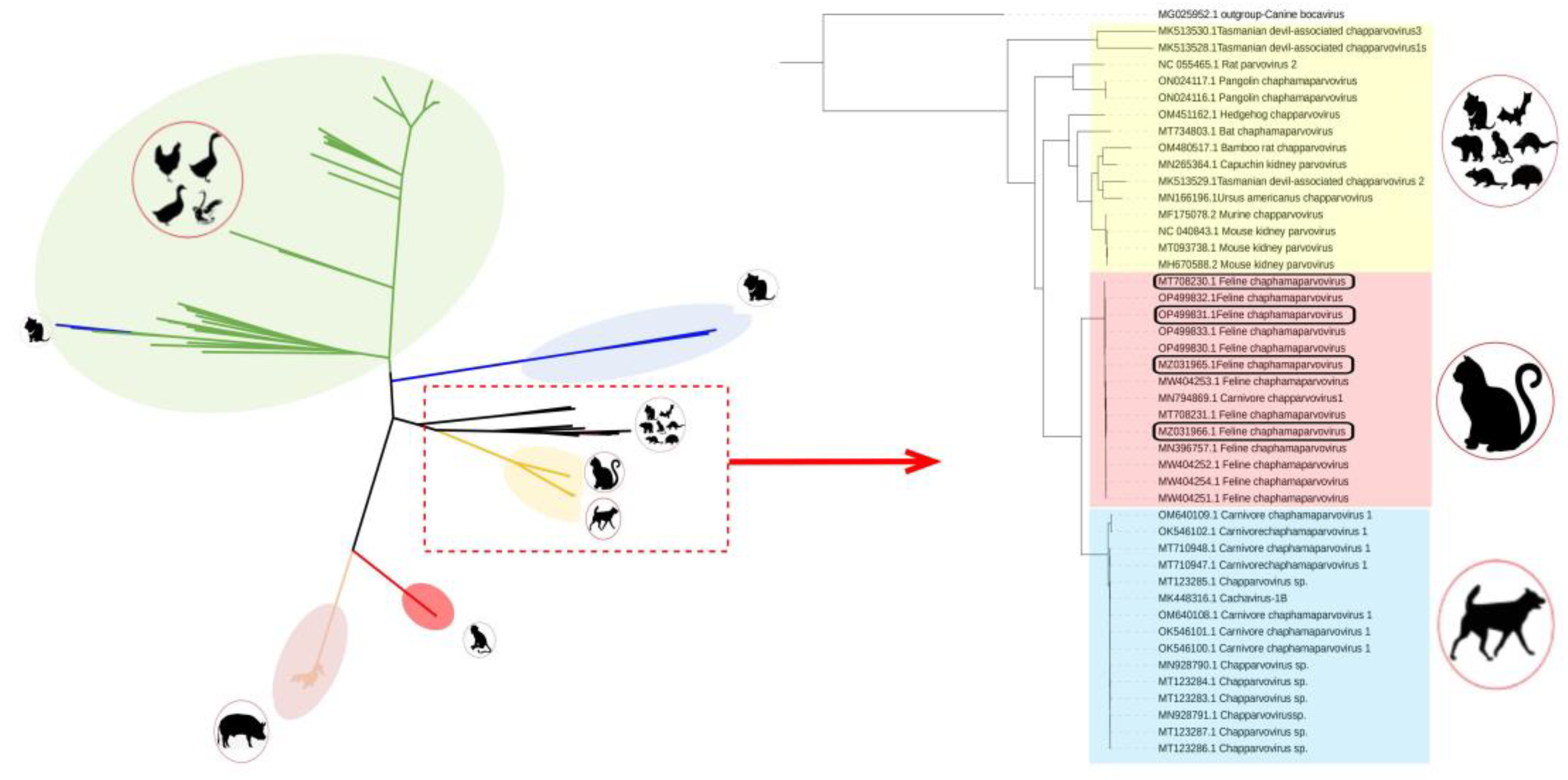
Figure 2.
Phylogenetic trees of feline chaphamaparvovirus (FeChPV) and canine chaphamaparvovirus (CaChPV) based on NS and VP. (a) NS (n=30); and (b) VP (n=33). The best substitution model is HKY+G4+F. Ursus americanus chapparvovirus/ MN166196.1 is used as an outgroup (Not shown). The red background indicates FeChPV; the blue background indicates CaChPV; Potential cross-species transmission strains are highlighted in red. FigTree and Interactive Tree Of Life are used for visualization. NS, nonstructural protein; VP, virion protein. Outgroup branch not shown.
Figure 2.
Phylogenetic trees of feline chaphamaparvovirus (FeChPV) and canine chaphamaparvovirus (CaChPV) based on NS and VP. (a) NS (n=30); and (b) VP (n=33). The best substitution model is HKY+G4+F. Ursus americanus chapparvovirus/ MN166196.1 is used as an outgroup (Not shown). The red background indicates FeChPV; the blue background indicates CaChPV; Potential cross-species transmission strains are highlighted in red. FigTree and Interactive Tree Of Life are used for visualization. NS, nonstructural protein; VP, virion protein. Outgroup branch not shown.
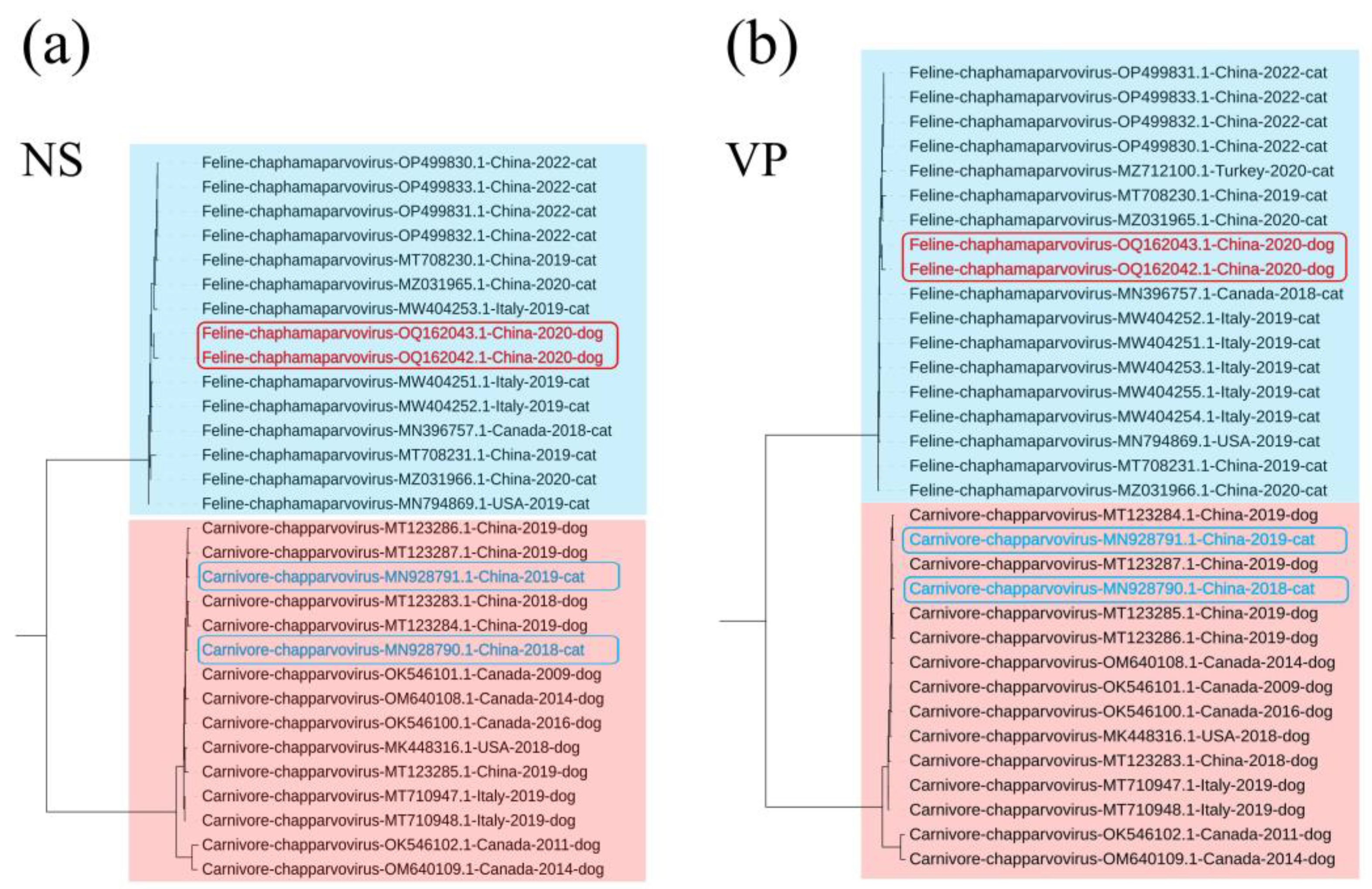
Figure 3.
Codon usage pattern difference between FechPV/CaChPV and hosts. RCDI: relative synonymous codon usage analysis; SiD: similarity index analysis. The black line represents CaChPV, and the red represents FeChPV. Parametric and non-parametric t tests are used to analyze the significance of Gaussian distribution data and non-conforming data, respectively. **** is defined as p<0.0001; p>0.05 is unlabeled.
Figure 3.
Codon usage pattern difference between FechPV/CaChPV and hosts. RCDI: relative synonymous codon usage analysis; SiD: similarity index analysis. The black line represents CaChPV, and the red represents FeChPV. Parametric and non-parametric t tests are used to analyze the significance of Gaussian distribution data and non-conforming data, respectively. **** is defined as p<0.0001; p>0.05 is unlabeled.
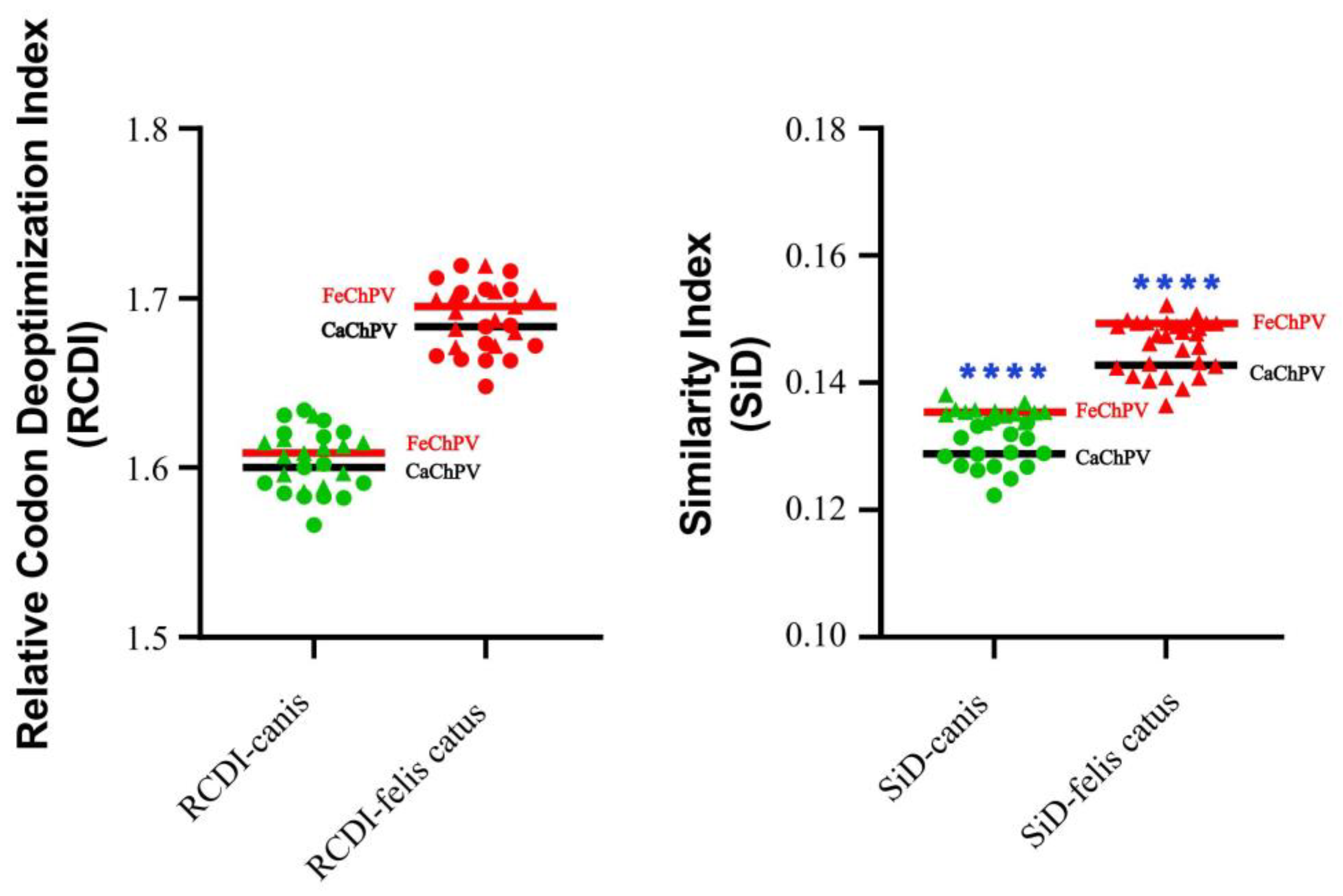
Figure 4.
Analysis of synonymous codon usage preferences between viruses and hosts. The black line represents CaChPV, and the red represents FeChPV. The Shapiro–Wilk test is per-formed for the normality test. Parametric and non-parametric t tests are used to analyze the significance of Gaussian distribution data and non-conforming data, respectively. **** is defined as p<0.0001; p>0.05 is unlabeled.
Figure 4.
Analysis of synonymous codon usage preferences between viruses and hosts. The black line represents CaChPV, and the red represents FeChPV. The Shapiro–Wilk test is per-formed for the normality test. Parametric and non-parametric t tests are used to analyze the significance of Gaussian distribution data and non-conforming data, respectively. **** is defined as p<0.0001; p>0.05 is unlabeled.

Figure 5.
Parity rule 2, ENC-plot, and neutrality analysis based on NS and VP of FeChPV and CaChPV. (a) Parity rule 2; (b) ENC-plot; and (c) neutrality analysis. Red circle and triangle represent the NS and VP of CaChPV; blue circle and triangle represent the NS and VP of FeChPV. NS, nonstructural protein; VP, virion protein; and ENC, effective number of codons.
Figure 5.
Parity rule 2, ENC-plot, and neutrality analysis based on NS and VP of FeChPV and CaChPV. (a) Parity rule 2; (b) ENC-plot; and (c) neutrality analysis. Red circle and triangle represent the NS and VP of CaChPV; blue circle and triangle represent the NS and VP of FeChPV. NS, nonstructural protein; VP, virion protein; and ENC, effective number of codons.
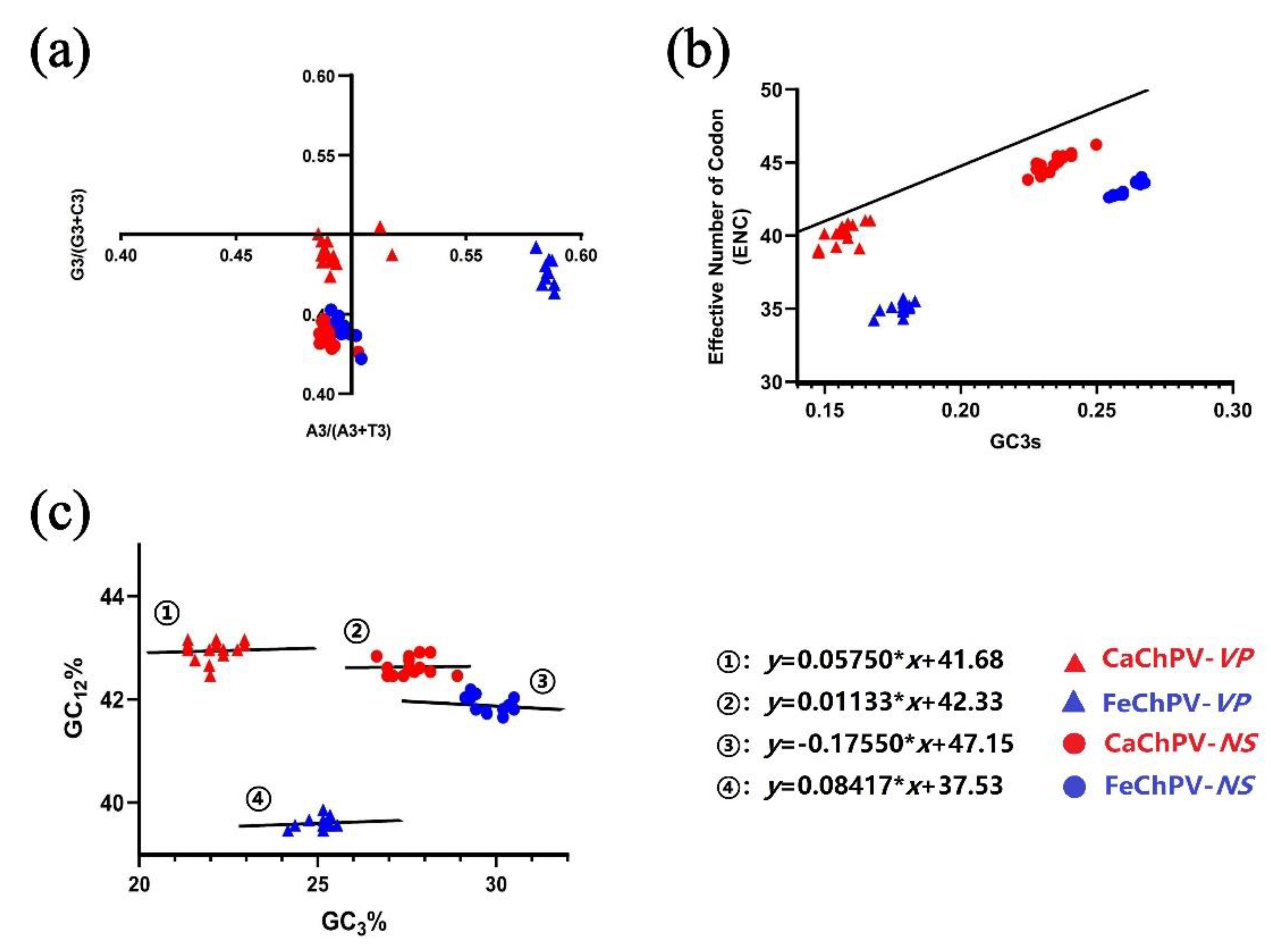
Figure 6.
Heat map of Root Mean Square Deviation of FeChPV and CaChPV. MT123283.1, MY123284.1, MN928791.1, and MN928790.1 are CaChPV, MW396757.1,MW404251.1, OQ162043.1, and OQ162.42.1 are FeChPV. Among them, MN928791.1, MN928790.1, OQ162043.1, and OQ162042.1 are cross-species transmitted strains.
Figure 6.
Heat map of Root Mean Square Deviation of FeChPV and CaChPV. MT123283.1, MY123284.1, MN928791.1, and MN928790.1 are CaChPV, MW396757.1,MW404251.1, OQ162043.1, and OQ162.42.1 are FeChPV. Among them, MN928791.1, MN928790.1, OQ162043.1, and OQ162042.1 are cross-species transmitted strains.
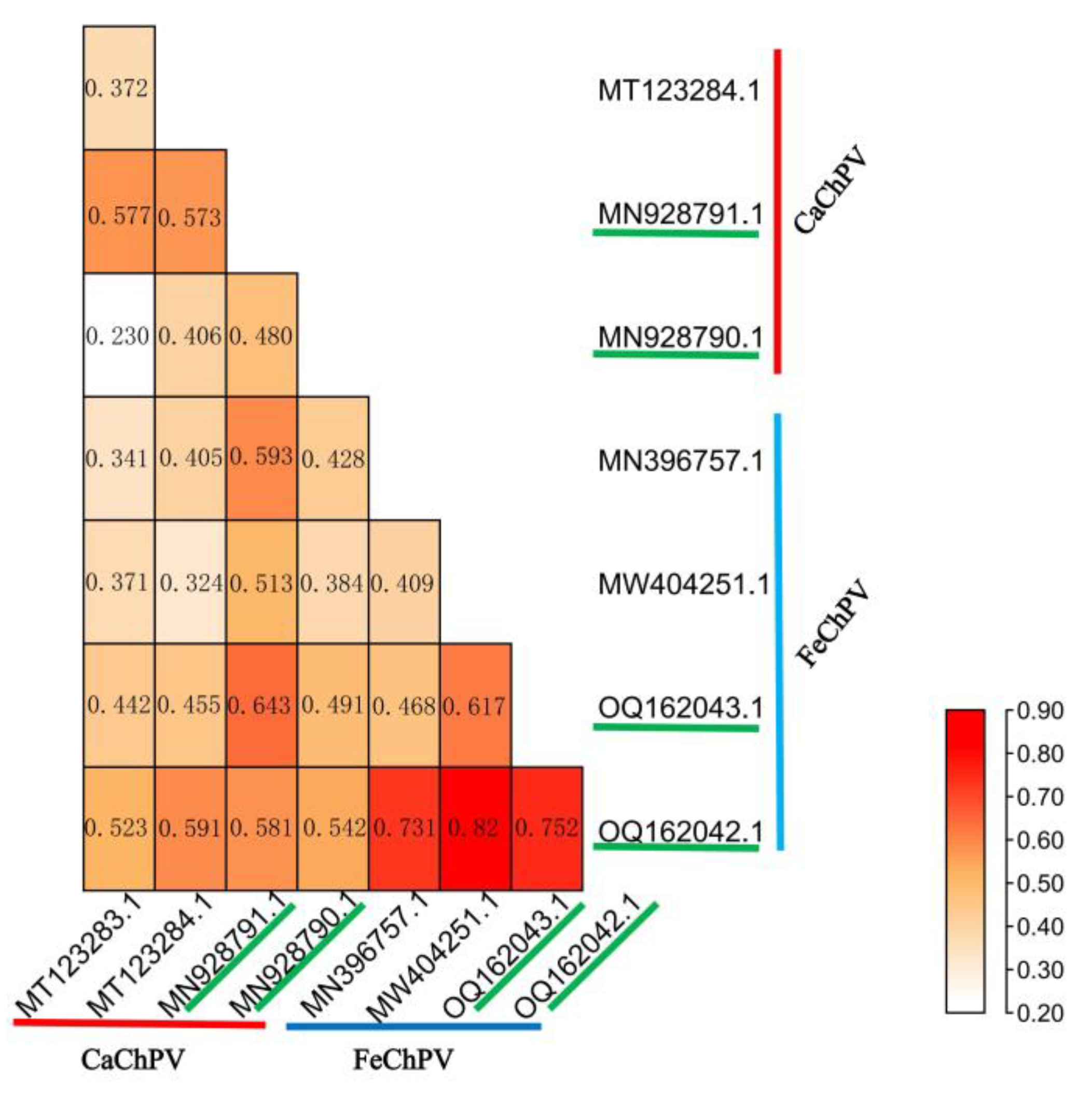
Table 1.
The codon adaptation index of feline chaphamaparvovirus (FeChPV) and canine chaphamaparvovirus (CaChPV).
Table 1.
The codon adaptation index of feline chaphamaparvovirus (FeChPV) and canine chaphamaparvovirus (CaChPV).
| Viruses | Dogs (Canis) (Mean±SD) | Cats (Felis catus) (Mean±SD) | |
| CAI | FeChPV | 0.7624±0.0008 | 0.7089±0.0009 |
| CaChPV | 0.7485±0.0020 | 0.6950±0.0020 | |
| eCAI | FeChPV | 0.7694±0.0017 | 0.7157±0.0016 |
| CaChPV | 0.7657±0.0029 | 0.7113±0.0030 | |
| Normalised CAI (CAI/eCAI) | FeChPV | 0.9909±0.0024 | 0.9906±0.0033 |
| CaChPV | 0.9775±0.0038 | 0.9771±0.0042 |
Note: The larger values between FeChPV and CaChPV are highlighted in bold.
Table 2.
The effective number of codons of feline chaphamaparvovirus (FeChPV) and canine chaphamaparvovirus (CaChPV).
Table 2.
The effective number of codons of feline chaphamaparvovirus (FeChPV) and canine chaphamaparvovirus (CaChPV).
| Virus | Gene | Range of ENC | The average of ENC (?X±S ) |
|---|---|---|---|
| FeChPV | NS | 42.6-44.0 | 43.123±0.461 |
| VP | 34.2-35.7 | 35.081±0.417 | |
| CaChPV | NS | 43.9-46.3 | 45.047±0.613 |
| VP | 39.1-41.2 | 40.16±0.761 |
Table 3.
Mutation analysis of cross-species transmission FeChPV and CaChPV strains.
| Strains | Mutation Sites |
| CaChPV-MN928790.1 | I56T, Y68C, F131S, D402N, R449K |
| CaChPV-MN928791.1 | Y22H, Q365R, R449K, H484P, |
| FeChPV-OQ162042.1 | M19T, G112R, F151S, L174P,S197T, S206A, S325R, E352G, S445N, |
| FeChPV-OQ162043.1 | G112R, L174P, S197T, S325R, |
Note: Mutations that occurred in both strains are bolded. Mutations to the same site as the other host are underlined.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



