
Submitted:
31 May 2023
Posted:
01 June 2023
You are already at the latest version
Abstract
Prostate cancer contributes to cancer-related deaths globally, and the etiology of this disease is not yet fully understood. While Human Papillomavirus (HPV) has been associated with several types of cancer, including cervical, anal, and oropharyngeal cancer, studies investigating the relationship between HPV and prostate cancer have shown mixed results. This systematic review aimed to evaluate the causative association between HPV and prostate cancer using Bradford Hill's criteria. A comprehensive search of PubMed was conducted, and 60 out of 482 studies were included in the review. The included studies were evaluated based on nine Bradford Hill criteria, and information on the identification and transmission of the virus and potential oncogenic mechanisms was also extracted. The strength of association criterion was not met, and other criteria, such as consistency and coherence, were not fulfilled. However, biological plausibility was supported, and potential oncogenic mechanisms were identified. While some studies have reported the presence of HPV in prostate cancer tissues, the overall quality of evidence remains low, and the association between HPV and prostate cancer is weak. Nevertheless, the prostate is a potential reservoir for the transmission of HPV, and the HPV E6 and E7 oncoproteins and inflammation are likely to be involved in any oncogenic mechanisms. Further studies with a higher level of evidence are needed to establish a definitive link between HPV and prostate cancer.
Keywords:
Prostate cancer
; human papillomavirus
; HPV
1. Introduction
Prostate cancer (PCa) is the second most common form of cancer among men, with an estimated 1.4 million new cases and 375,000 deaths worldwide in 2020 (Sung et al., 2021). PCa incidence increases with age and the disease is most commonly diagnosed in men over 50 (Rawla, 2019). In addition, age, race, genetics, and a positive family history of PCa are nonmodifiable risk factors strongly associated with PCa development (Leitzmann and Rohrmann, 2012; Pernar et al., 2018; Gandaglia et al., 2021) . Modifiable risk factors such as metabolic syndrome, smoking, diet, obesity, physical activity, and exposure to ultraviolet rays may impact the risk of developing PCa and PCa mortality (Bostwick et al., 2004; Dagnelie et al., 2004; Leitzmann and Rohrmann, 2012; Gandaglia et al., 2021). Benign prostatic hyperplasia (BPH) is a noncancerous condition characterized by an enlargement of the prostate gland. While BPH and PCa often occur together, BPH is not recognized as a confirmed risk factor for PCa (Chang, Kirby and Challacombe, 2012; Chughtai et al., 2016). Instead, BPH may increase the likelihood of detecting incidental cancer, despite not being directly responsible for increasing the risk of developing PCa (Chang, Kirby and Challacombe, 2012; Chughtai et al., 2016). Additionally, several studies have found the presence of pathogens in PCa tissues, including human papillomaviruses (HPV), human cytomegalovirus, and Epstein-Barr virus, and Propionibacterium acnes (Zambrano et al., 2002; Sfanos et al., 2008; Chen and Wei, 2015).
HPV is one of the most common sexually transmitted infections globally, and at least 12 so-called high-risk HPV subtypes have been identified as human carcinogens (Heidegger, Borena and Pichler, 2015). HPV 16 and HPV 18 are the best understood high-risk HPV types. The high-risk HPV types encode oncoproteins that can transform infected cells. The E6 and E7 oncoproteins from these viruses are well characterized in terms of their ability to inactivate the p53 and pRB tumour-suppressor proteins respectively, although they also have a variety of other cellular targets. The HPV E5 protein can also act as an oncoprotein; for example, through the stimulation of Epidermal Growth Factor activity (Leechanachai et al., 1992; Williams et al., 2005). Although it is not part of the normal HPV life-cycle, HPV-transformed cancer cells often contain viral DNA integrated into the host genome with retained expression of E6 and E7 (Jeon, Allen-hoffmann and Lambert, 1995; Mcbride and Warburton, 2017).
The relationship between HPV and PCa has been the subject of much debate and research in recent years, with results from various studies being contradictory. To clarify the connection between these two factors, statistical meta-analysis has been employed (Yang et al., 2015; Zhou et al., 2017; Moghoofei et al., 2019). However, the limitations of this method, including difficulties in evaluating study methodologies, accounting for differences in study populations, and publication bias, have left many questions unanswered. To address these limitations, this systematic review aims to evaluate the causal relationship between HPV and PCa using Bradford Hill criteria postulates (Hill, 1965), a set of well-established criteria for evaluating causality. The review will analyze the available literature on the topic by applying the expanded version of Bradford Hill's criteria for causality. These criteria, originally consisting of the strength of association, consistency, specificity, temporality, biological gradient, plausibility, coherence, experiment, and analogy, have been expanded to include the latest advances in the study of oncoviruses such as virus identification, transmission mechanisms, and oncogenic processes.
Our investigation is premised on the idea that high-risk HPV types could play a causative role in PCa. As HPV infections can be prevented through vaccination, investigating their potential role in PCa is of critical importance.
2. Methodology
2.1. Literature Search
A comprehensive examination of available research studies was carried out following the PRISMA guidelines for Preferred Reporting Items in Systematic Reviews and Meta-Analyses (Moher et al 2009). A search was conducted, without any time restriction, using the PubMed database with the specified search terms: (Prostate Cancer OR Prostate cancers OR Prostate Carcinoma OR Prostate Carcinomas OR prostate tumour OR prostate tumours OR prostate adenocarcinoma OR prostate adenocarcinomas OR intraepithelial prostatic neoplasia OR intraepithelial prostatic neoplasias OR benign prostatic hyperplasia[MeSH Terms]) AND (HPV OR humanpapillomavirus OR human papillomavirus OR human papilloma virus OR alpha papillomavirus[MeSH Terms])”. We only included studies which produced primary data, so all reviews and meta-analyses were discounted. This search was conducted on 16th May 2023.
2.2. Paper Selection
The initial search resulted in 482 papers. We screened the abstracts of the 482 papers in terms of the inclusion and exclusion criteria and relevance to the study (Table 1). All abstracts were double-checked for relevance. As seen in Figure 1, this method ended with 68 papers deemed relevant and sufficient for this systematic review. Thereafter, a full-text review was performed. At this stage, 8 papers were excluded. Reasons for exclusion were a lack of quality in the study design; a main focus on other infectious pathogens or the prostate microenvironment as a whole and lack of HPV focus; and the paper being in a language other than English.
2.3. Quality Criteria using Newcastle-Ottawa Quality Assessment Scale
The relevant 60 papers were screened for quality using the Newcastle-Ottawa quality assessment scale (Wells et al., 2021). These criteria include a set of questions that differ depending on whether a case-control study or a cohort study is being assessed. Each study was awarded a maximum of one star for each question that it met. The categories of questions, including Selection, Comparability, and Exposure/Outcome, were assessed to evaluate the risk of bias and to assess the overall strength of the evidence. The exposure category has three questions, with the last being irrelevant to this review as it is aimed at questionnaires or interviews. The quality of case-control studies was rated out of a maximum of seven (Table 2), while the quality of cohort studies was rated out of a maximum of eight (Table 3).
2.4. Relevant Data Extraction
To extract data to assess the causal relationship between HPV and PCa incidence, each paper that scored above 3 on the Newcastle-Ottawa scale was selected and scrutinized using the nine (9) central postulates of the Bradford Hill criteria. These nine (9) criteria are Strength, Consistency, Temporality, Biological Gradient, Specificity (now defunct), Biological Plausibility, Coherence, Experiment and Analogy. To aid this review, we added specific criteria for means of transmission, identification of the virus, and oncogenic mechanisms to the Bradford Hill criteria.
3. Result and Discussion
A total of 60 original papers were reviewed to investigate the causative role of HPV in PCa. Among the 58 studies, 11 (18%) showed a positive association between HPV presence and the development of PCa. Another 10 studies (17%) were uncertain and suggested further research was necessary, while the remaining 39 studies found no association.
3.1. Analogy
The analogy criterion is one of the nine criteria proposed by Bradford Hill to assess the causal relationship between an exposure and an outcome. It involves comparing the relationship between two phenomena to a previously established causal relationship. This criterion assumes that if two phenomena are similar in relevant ways, they may have similar causal relationships (Hill, 1965). In this way, the analogy criterion provides a framework for exploring potential causal relationships in situations where direct evidence may be limited.
In the case of the relationship HPV and PCa, the analogy criterion may be useful in exploring potential causal links. HPV is a well-established causal factor in several types of cancer, including cervical cancer, penile cancer, anal cancer, vaginal cancer, vulvar cancer, and cancer of the head and neck; for recent reviews, see Graham, 2017 and Araldi et al., 2018. Therefore, it is an acceptable hypothesis that it may also cause PCa.
However, while there are similarities between the effects of HPV in cervical cancer and PCa, there are also important differences. For example, PCa is glandular in origin and not squamous cell in origin like cervical cancer. Therefore, how HPV affects cervical and prostate cells may not be directly comparable, and the analogy may be weaker. In addition, while cervical dysplasia and cervical carcinoma are often regarded as different stages of the same progressive disease, PCa and BPH are regarded as two separate diseases. However, endocervical adenocarcinomas have been classified into those related to high-risk HPV and those unrelated to high-risk HPV (Stolnicu et al., 2018; Hodgson et al., 2019). Cervical adenocarcinomas may therefore be more comparable to prostate adenocarcinoma.
Despite these differences, some studies have found similarities in the association between different types of HPV and the development of benign versus malignant lesions. For example, in one study, high-risk HPV types (16/18) were found in 92% of HPV-positive PCa cases, while LR types (6/11) were found in only 8% (Singh et al., 2015). This is similar to cervical cancer, where HPV16 is the most carcinogenic type. In contrast, LR types were found in 64% of HPV-positive BPH controls (Singh et al., 2015), indicating that there may be a differential effect of different types of HPV on benign versus malignant tissue.
The analogy criterion can also be useful in exploring potential differences in the nature of the HPV DNA in malignant and pre-malignant lesions. The viral genome often persists in genital neoplasms in an integrated state and this is a key event in the development of cervical cancer. Integration is not detected in benign lesions and clinically healthy tissues that test positive for HPV (Rodriguez et al., 2016). Similarly, it has been suggested that Prostatic intraepithelial neoplasia (PIN) and prostatic carcinoma may have a causal relationship with HPV infection, based on the finding of HPV 16 in three out of 23 carcinoma samples, as well as the similarities observed between PIN and Cervical intraepithelial neoplasia (CIN) and penile intraepithelial neoplasia (Sarkar et al., 1993; Dillner et al., 1998)
However, there are limitations to the use of the analogy criterion in exploring potential causal relationships. For example, it is important to ensure that the similarities between different diseases are not superficial and that the mechanisms by which exposure may cause an outcome are comparable. It is also important to consider the possibility of confounding factors and other potential explanations for the observed association. The similarities between PCa and cervical cancer in the way that different types of HPV act suggest that further investigation into the potential role of HPV in PCa is warranted.
3.2. Biological Plausibility and Coherence
Plausibility refers to the idea that there must be a theoretical basis to support a potential association between a virus and a disease (Hill, 1965). This review indicates that it is plausible for HPV to have an oncogenic role in the development of PCa, based on the idea of plausibility in epidemiology and biology. This is supported by evidence of the oncogenic role of HPV in other types of cancer, and by many studies that have evaluated the oncogenic properties of the HPV oncoproteins (Pim and Banks, 2010; Roman and Munger, 2013; Vande Pol and Klingelhutz, 2013). The plausibility of this association is further supported by examining the hypothesized oncogenic mechanisms and means of transmission. Therefore, it is plausible that HPV could be a causative agent in PCa.
Coherence is similar in that the cause-and-effect story should make sense with all knowledge available to the researcher (Fedak et al., 2015). However, incoherence can be found when the literature is conflicting, and many of the studies around HPV and PCa are contradictory. Therefore, coherence is a criterion that is not met for this topic, based on the studies included in this review.
3.3. Identification of Human Papillomaviruses in Prostate Tissues
Establishing the identity of an exposure is crucial in determining a causal relationship between an exposure and outcome. Different methods are used to identify HPV in different studies, each with its strengths and limitations. Polymerase chain reaction (PCR) is a widely used method for detecting the HPV genome, wherein specific primers are used to amplify and detect a DNA fragment of interest. 50 studies used PCR as a detection method, a summary and the primer sets are presented in Table 4. However, the choice of primers varies between studies, and this variability can affect the detection rate of HPV. For instance, one study found that primers targeting a 126 bp fragment of the E6 gene of HPV16 had a higher detection rate than primers targeting a 99 bp fragment of E6 gene, or a consensus primer pair targeting a 450 bp fragment of the L1 gene (Terris and Peehl, 1997). The lower detection rate of the L1 gene could be due to it not always integrating into the host cell genome, and longer DNA segments like L1 can be damaged during formalin-fixation and paraffin-embedding, which are common procedures for tissue storage. Indeed, formalin-fixed paraffin-embedded DNA may not always provide results if the PCR product is over 200 bp (Lenze, Müller and Hummel, 2012). Fresh frozen samples are therefore preferred over formalin-fixed samples as they tend to give more consistent results. Furthermore, different primers may have varying degrees of success in detecting the HPV genome. One study found that while 23/29 cases were positive when testing for the L1 region, additional testing with E6/E7 primers revealed another 6 HPV-positive cases (Carozzi et al., 2004). Despite its sensitivity, a limitation of PCR as a method for HPV detection is that it only shows current exposure. Therefore, previously cleared HPV infection and its possible role in cancer development cannot be assessed. Furthermore, not all PCR primers can identify HPV in PCa. It is possible that the negative HPV identification results in some studies could be due to the use of PCR primers that are not sensitive enough to detect HPV in PCa samples. It is also important to note that the viral load of HPV in PCa is generally lower than that in cervical cancer (Lawson and Glenn, 2020), making it more challenging to detect HPV in PCa samples using PCR.
To confirm the findings of PCR, gel electrophoresis, or hybridisation techniques such as in situ hybridisation (ISH) or Southern blots are often used, although these require a significant amount of viral DNA and can be time-consuming (Michopoulou et al., 2014). Furthermore, Sanger sequencing has been utilised to validate the PCR findings (Whitaker et al., 2013; Ahmed et al., 2023)
Alternative methods of identifying HPV in PCa have been explored due to the limitations of PCR and Southern blotting. In situ PCR has been used to detect high-risk HPVs in the nuclei of PCa cells, and is less susceptible to contamination compared to standard PCR (Whitaker et al., 2013). Hybridization methods have also been used to identify HPV16/18 in PCa (Ibrahim et al., 1992; Sarkar et al., 1993), Additionally, using next generation gene sequencing, high-risk HPV types 16 and 18 were identified in 12 of 502 invasive PCa from The Cancer Genome Atlas (Glenn et al., 2017). These findings suggest that alternative methods are valuable in identifying HPV in PCa cases.
Serology is another method used to identify HPV infection, by detecting antibodies to HPV capsid proteins in blood samples. One advantage of serology is that it can detect any prior exposure to HPV since antibodies can persist for a lifetime, even after the clearance of HPV DNA (Dillner et al., 1998). However, serology lacks anatomical specificity, and a positive serology result only suggests a past HPV infection from any part of the body, not exclusively from the prostate (Zhao et al., 2017).
To supplement the information obtained from HPV detection, Immunohistochemistry (IHC) can be used to detect specific proteins in the samples, such as the E6/E7 oncoproteins and tumor markers. This additional method can provide a wider picture of the oncogenic processes, and how HPV may interact with other cellular components. For example, oncogenic HPV E7 proteins were identified through IHC in 112 (75%) of 150 PCa (Pascale et al., 2013). Moreover, another study reported that HPV infection was present in 10% (3/30) of cases with adenocarcinoma and 1.1% (1/90) of cases with BPH using IHC (Mokhtari, Taghizadeh and Hani, 2013). However, this study's limitations included the absence of a specific antibody for IHC to detect various HPV subtypes and imprecise data on the exact HPV prevalence in the cancer patients vs normal controls (Mokhtari, Taghizadeh and Hani, 2013)
Understanding the possible reasons for discrepancies in results between studies is crucial for accurately assessing the role of HPV in PCa development. This review highlights the importance of considering differences in laboratory techniques when interpreting the results of HPV detection in PCa studies.
4. Strength of Association
Strength of association typically refers to the degree to which a larger association indicates a more likely causal relationship. In this review, the strength of association is evaluated based on whether a statistically significant result was found. However, not all studies performed statistical tests, so this information is not available for all of them.
4.1. Serology
All accessible published studies based on serology have been included, indicating no selection bias in this review. 13 studies in this systematic review used serology as a detection method (Table 5). Only three studies found statistically significant results in seropositivity rates for high-risk HPV types and an increased risk of PCa (Dillner et al., 1998; Adami et al., 2003; Zhao et al., 2017). In contrast, opposing results reported that HPV18 was more prevalent in controls rather than PCa cases (Hrbacek et al., 2011). Just three of the 13 serology studies included performed PCR on PCa tissues. Interestingly, in two of these cases, the PCR results did not align with the serology findings. (Chen et al., 2011; Tachezy et al., 2012). However, one of these studies showed consistent results with negative PCR findings (Strickler et al., 1998). To identify HPV antibodies in serum samples, Zhao et al., used a cutting-edge technique termed seroscreening by microarray (Zhao et al., 2017). Serum samples from males with PCa were tested, and HPV16 antibodies were found in 48 of 75 samples (64%), compared to 14 of 80 controls (17.5%) (p = 0.001).
Sero-epidemiological studies do not provide a clear case for HPV as a cause of PCa. In contrast, HPV-related cervical cancer shows a significant increase in the prevalence of HPV serum antibodies among patients compared to healthy individuals (Combes et al., 2014).
4.2. Polymerase Chain Reaction
All accessible published studies based on PCR have been included, indicating no selection bias in this review. Of these, only 22 of concluded whether their results were significant or not regarding HPV prevalence in malignant versus benign tissue. Among these, 7 studies report significant associations between HPV and PCa, with p values < 0.05 (Serth et al., 1999; Leiros et al., 2005; Martinez-Fierro et al., 2010; Mokhtari, Taghizadeh and Hani, 2013; Singh et al., 2015; Glenn et al., 2017; Medel-Flores et al., 2018). The odds ratios (ORs) reported by the studies that used PCR and concluded significant results varied greatly, ranging from 2.3 to 9.88 (Martinez-Fierro et al., 2010; Mokhtari, Taghizadeh and Hani, 2013; Medel-Flores et al., 2018) indicating that the strength of association between the HPV and PCa varied greatly between the studies. 15 studies reported no significant results, but not all of them reported ORs or P values, with some just stating "no significant findings" or "P > 0.05" (Table 6).
From the available evidence, it can be concluded that there is no significant difference in the presence of HPV and the risk of developing PCa. Hence, it appears that the strength of association between HPV and PCa is not strong.
4.3. Specificity
The original Bradford Hill criteria state that exposure must only cause one disease to support an epidemiological relationship. This is not the case for many exposure-disease relationships, so this criterion is now defunct and was not considered in this review.
4.4. Transmission of Virus
The mode of transmission of a pathogen is a critical factor in developing effective preventive strategies. HPV, a sexually transmitted infection (STI), is often linked to sexual behaviour and to an increased risk of anogenital tumors. Interestingly, sexual behaviours including early age at first intercourse and multiple sexual partners may also increase the risk of PCa (Cirakoglu, Benli and Yuce, 2018; Jian et al., 2018). A significant portion 216/1272 (17.4%) of the sexually active men population in Britain has detectable HPV DNA in their urine (Johnson et al., 2012). Both men and women who engage in sexual activity are at an increased risk of contracting high-risk HPV types. Sexual transmission of HPV occurs through cell-to-cell contact during sexual activity, but recent evidence suggests that HPV can also spread throughout the body via circulating extracellular vesicles (Guenat et al., 2017; De Carolis et al., 2019). Exosomes and extracellular vesicles have also been implicated in HPV transmission and carcinogenesis (Guenat et al., 2017).
Interestingly, Saudi Arabia’s population has low incidence rates of PCa which may be associated with genetic and environmental factors, including circumcision, religion, and dietary habits (Gazzaz and Mosli, 2009). This may support the role of HPV and sexual activity in PCa promotion, as HPV rates are also lower in this population, and there is lower STI prevalence (Madani, 2006; Gazzaz and Mosli, 2009). However, PCa incidence is higher in Iran despite similar cultural and religious practices, although the prevalence of HPV in Iranian populations is lower than in the Western World and Africa (Eghbali et al., 2012). Six studies in this review are from Iran, and five of them do not support the association between HPV and PCa with a P-value >0.05 (Aghakhani et al., 2010; Ghasemian et al., 2013; Mokhtari, Taghizadeh and Hani, 2013; Atashafrooz and Rokhbakhsh-Zamin, 2016). A serological study with PCa samples from the US found no association between HPV16 or 18 infection status and sexual history (Rosenblatt et al., 2003). The demographic information collected in the study by Rosenblatt et al was self-reported, which may affect the validity of the results.
Overall, while a possible link between HPV and PCa through sexual activity is suggested, the evidence is not conclusive, and there may be other factors at play.
4.5. Temporality
Temporality is an essential causal criterion and states that exposure must precede the onset of disease for a relationship to be causal. Many of the studies in this review are case-control based, and in these instances, the temporality criterion is not met. Due to the design of a case-control study, it is not possible to look back in time before the cancer was diagnosed and, therefore, not possible to prove that HPV infection preceded cancer onset. Only six studies in this review meet the temporality criteria and they report conflicting evidence for a link between HPV and PCa; they are a mix of serology and PCR-based studies.
The first study to meet this criterion is a nested case-control study using a Finnish serum bank. The study reported a relative risk of PCa when seropositive for HPV18 as 2.59 (CI 95% = 1.17-5.75), and found the association between HPV18 and PCa to be highly significant (p<0.05) (Dillner et al., 1998). The risk also tended to be higher for samples taken more than 10 years before diagnosis. However, a similar study of a nested case-control study, using Nordic biobanks and serum of 200,000 men, did not find an association between serologic markers of HPV16, 18, and 33 infections and risk of PCa (Korodi et al., 2005). In fact, there was a tendency for an inverse association between PCa risk and HPV18 antibody levels (OR 0.49, CI 0.22-1.09). Furthermore, no association was found between HPV16, 18, and 33 and PCa (Sutcliffe et al., 2007). Consistent results in a different study were produced, and once again found no association (Sutcliffe et al., 2010).
For these four studies, the conflicting results cannot always be explained. The Finnish and Nordic studies are very similar in terms of methodology and use the same threshold levels for the ELISA assays. The Nordic study includes three different cohorts from three different countries: Norway, Finland, and Sweden, and tests for associations between and within countries. This indicates that geographical region is also not a cause of differing results, as both use Finnish patients. The main difference between the two studies is the sample number, and the Nordic study likely had a higher statistical power that was able to exclude results due to chance. The inverse relationship between HPV18 seropositive and PCa found in the Nordic study needs further analysis. The negative findings of the US studies could be down to the difference in timings between serum collection and diagnosis. In the 2010 study, blood was collected closer to diagnosis at the second visit. The 2007 study took blood between 1993-1995, and the follow-up period was a diagnosis in 2000. In contrast, the two Scandinavian studies collected serum several years before diagnosis and so an early HPV oncogenic mechanism may not be detected.
Two studies that also meet the temporality criterion used archival formalin-fixed paraffin-embedded (FFPE) tissue samples, rather than serum. The first study was conducted in an Australian population of men with benign prostate biopsies who, 1 to 10 years later, developed PCa (Glenn et al., 2017). 52 sets of benign and PCa specimens from the same patients were collected, and PCR and IHC were used to analyse the samples. The study reported that there were no statistically significant differences in the prevalence of HPV L1 and E7 DNA (as assessed by PCR). However, differences in HPV E7 oncoprotein expression (as assessed by IHC) were highly significant, with much higher expression in the benign as compared to the later PCa in the same patient (p<0.001). In contrast, a Swedish study conducted by Bergh et al. (2007) used prostate tissue samples from 201 men with BPH who had undergone transurethral resection of the prostate (TURP) and subsequently developed PCa, along with 201 matched controls who did not develop PCa. A transurethral resection of the prostate (TURP) is a type of surgical procedure that removes a portion of the prostate gland. Using PCR, no HPV DNA was detected in any of the samples. Dodd and colleagues also provided evidence that transcripts of the HPV E6/E7 viral genes may be detected in both benign and malignant tissues of the prostate (Dodd, Paraskevas and McNicol, 1993).
The positive findings from studies that meet the temporality criteria are consistent with a ‘hit and run’ hypothesis for the role of HPV in PCa. This suggests that HPV acts early in the oncogenic process, supported by its presence in benign prostate tissue years before cancer diagnosis (Glenn et al., 2017), and is reflected in positive serology results (Dillner et al., 1998). This hypothesis proposes that HPV infects cells transiently, and this begins the process of malignant transformation perhaps as a consequence of immune responses or HPV-induced DNA damage. However, the presence of HPV is not needed to maintain cancer and hence cells lose the viral genome, as shown by its lowered presence at later stages.
In conclusion temporality is a key Bradford Hill criterion; however, it is hard to test for. As results between these studies are conflicting, further studies need to be done that meet this criterion to prove that exposure precedes disease.
4.6. Oncogenic Mechanism
To become cancerous, cells need to acquire specific characteristics, known as the "Hallmarks of cancer" (Hanahan and Weinberg, 2011). These include sustaining growth signals, evading natural growth blockers, avoiding cell death (Apoptosis), becoming immortal, invading other tissues, inducing blood vessel growth (angiogenesis), changing cellular metabolism, and avoiding the immune system (Hanahan and Weinberg, 2011). Additionally, two factors that contribute to the development of these hallmarks are genome instability and inflammation (Hanahan and Weinberg, 2011). HPV may play a role in the development of some of these hallmarks in prostate cells, as for its established role in causing cervical cancer and other malignancies. Understanding the hallmarks of human cancer and the factors that contribute to their development, such as genome instability and inflammation, is crucial in identifying potential mechanisms by which HPV may contribute to prostate oncogenesis, particularly through the action of the HPV oncoproteins. The most commonly proposed mechanism by which HPV could contribute to PCa involves the E6 and E7 oncoproteins. Many studies test the presence of the E6 and E7 regions of the HPV genome using specific PCR primers.
The HPV E6 and E7 oncoproteins play a crucial role in cancer development by inactivating host tumor suppressor proteins, p53 and pRb respectively, and subsequently inducing genome instability and leading to cell proliferation, immortalisation, and malignant transformation (reviewed by McLaughlin-Drubin and Münger, 2009; Moody and Laimins, 2010). Another potential way in which the HPV E6 and E7 oncoproteins may contribute to the development of PCa is by creating a chronic inflammatory environment. Chronic inflammation has been identified as an enabling characteristic of cancer, and previous studies suggest that it plays a role in the development and metastasis of human cancers (Zhao et al., 2021). The E6/E7 proteins disrupt the interferon signaling pathway, which is the body's first line of defense against viral infection. As a result, they suppress the action of the immune system, making it more difficult for the body to eliminate cancerous cells (Ghasemian et al., 2013). Additionally, chronic inflammation can damage DNA by creating oxygen-reactive species (Gazzaz and Mosli, 2009).
Many of the studies discussed in this review use benign prostatic hyperplasia (BPH) tissue as a control, but BPH is also associated with inflammation. This presents a limitation because using BPH patients as controls may make it difficult to draw meaningful conclusions if both conditions have a shared cause (Hrbacek et al., 2011). If inflammation is a common factor between the two conditions, it becomes challenging to distinguish the role of HPV in PCa. However, it is rare to find healthy prostates in the age group of interest, and BPH is the most common control used in studies. Consequently, there may be a significant difference in HPV DNA prevalence in healthy prostate tissue compared to cancer and BPH, but it is challenging to test for this.
In addition, to expand the scope of the investigation, microRNAs (miRNAs) and cell regulator proteins, such as survivin, Bcl-2, and c-Myc, can be tested. miRNAs are a family of small, endogenous non-coding RNAs that regulate a wide variety of biological processes and have been found to be dysregulated in a range of cancers (Peng and Croce, 2016). They can act as either tumor suppressors or oncogenes. This miRNA has been shown to bind directly to human Toll-like receptor 8, activating a Toll-like receptor-mediated inflammatory response that can result in tumor growth and metastasis (Fabbri et al., 2012). The study by Khatami et al., (2022) reported no significant association between the presence of HPV infection with PCa (P = 0.102). Although, the study did observe different miRNA expression in the HPV-positive PCa group compared to the normal prostate tissues of age-homogeneous healthy individuals (control): miRNA-21, -150-5p, and -155 levels were significantly upregulated. However, there was no statistically significant difference in the expression level of any selected cellular miRNAs between HPV-positive PCa samples and HPV-negative normal prostate tissues (P > 0.05). Therefore, based on this study alone, it cannot be concluded that HPV infection confers oncogenic potential associated with differing miRNA expression in PCa. The expression of miRNA-150-5p has been implicated in promoting cancer by encouraging cell migration and invasion, this miRNA is upregulated in recurrent ovarian cancer (Tung et al., 2020). Another miRNA, miRNA-155, was also expressed in hematopoietic stem cells and several solid tumours, and it plays a crucial role in regulating the immune response (Gironella et al., 2007; Faraoni et al., 2009; Mahesh and Biswas, 2019). In addition, miRNA-155 has been implicated in cervical cancer, where it promotes cell proliferation, migration, and invasion, ultimately allowing a cell to evade apoptosis (Zhang et al., 2018). Thus, the increased expression of these miRNAs in PCa with oncogenic potential may contribute to the development of hallmarks of cancer in HPV-positive PCa cells.
Specific cell regulator proteins were also found to be differentially expressed HPV-positive PCa group compared to the HPV-negative PCa group (Khatami et al., 2022). A statistically significant increase of approximately 1.8-fold to 4-fold in the mean levels of matrix metalloproteinase (MMP) -9, c-Myc, survivin, Bcl-2, and MMP-2 were observed. p53 was expressed at lower levels in the HPV-positive tissue samples compared to HPV-negative samples (fold-change: 0.22, P-value < 0.0001), which supports the role of HPV in the degradation of this crucial tumor suppressor (Khatami et al., 2022). Survivin and Bcl-2 are both anti-apoptotic mediators, and so promote cancer progression by allowing cells to survive, even when carrying mutations. c-Myc is an oncogene that is a ‘master regulator’ of cell growth and metabolism (Miller et al., 2012). Overexpression of the c-Myc protein is present in over 70% of human cancers (Madden et al., 2021), highlighting its role in malignant transformation. The upregulation of c-Myc in HPV-positive tissue and significant association of c-Myc expression level with E7 and E6 expression level in this study presents the possibility of HPV indirectly affecting PCa development in this way. MMPs are the main enzymes involved in the extracellular matrix (ECM) breakdown. The ECM holds cells together and plays a role in cell growth and survival (Itoh and Nagase, 2002). Cancer cells have to be able to degrade the ECM to invade nearby cells and metastasis, and increased MMP-2 and -9 levels in HPV-positive tissue may highlight an indirect role that HPV plays in this process.
According to Anwar et al., (1992), HPV18 infection was observed in 12 out of 15 HPV-positive cases with bone metastasis, which accounts for 80% of the cases. Also, in a recent study by Fatemipour et al., (2021) it was suggested that HPV may play a role in promoting metastasis in PCa through various molecular mechanisms; as the expression levels of certain genes associated with metastasis, including N-cadherin, SLUG, and TWIST, were found to be higher in HPV-positive specimens, while the expression levels of other genes associated with tumor suppression, such as PTPN-13 and E-cadherin, were lower in HPV-positive specimens. This finding supports the notion that HPV promotes metastasis. In cervical cancer, HPV18 infection has been linked to cervical small-cell neuroendocrine carcinomas, which exhibit aggressive behavior, rapid metastasis, and high mortality rates (Stoler et al., 1991). However, it is not clear whether HPV18 infection is associated with metastasis to specific organs, and more research is needed to establish the role of HPV in this regard. On the one hand, Tu et al., (1994) found that HPV18 was only detected in one out of 17 pelvic lymph nodes in metastatic PCa (PCa) cases, which suggests that HPV may not play a role in metastasis. Conversely, Ghasemian et al., (2013) reported that three out of five PCa patients infected with HPV developed distant metastasis, which suggests a possible role of HPV in promoting metastasis. These conflicting results underscore the need for further research in this specific area.
Several studies included in this review suggest that HPV may be linked to later stages of the disease by associating virus presence with clinical data such as the Gleason score (GS). The GS is a system used to grade PCa, with higher grades indicating more abnormal cells and a greater likelihood of cancer growth and spread. Positive associations between HPV and high GS suggest that the virus may play a later role in carcinogenesis by increasing inflammation in prostate tissue, which may lead to tumour development and metastasis. However, the results are conflicting. The first study to support this hypothesis reports a significant increase in HPV infection levels with increasing GS (P < 0.005) (Anwar et al., 1992). This study used a small sample size, and the higher GS observed in PCa samples may be due to chance. More recent studies reporting a larger sample size have produced consistent results, with significant associations between HPV infection and high GS with P values of 0.014, 0.0008, and 0.003 respectively. However, other studies have found no association between HPV infection and tumour aggressiveness represented by GS (Rosenblatt et al., 2003; Carozzi et al., 2004; Martinez-Fierro et al., 2010), and one study even found a low GS in its one HPV-positive sample out of sixty PCa samples investigated for HPV presence (Aydin et al., 2017). These studies that oppose this hypothesis that HPV may be involved in the later stages of PCa development and tumor maintenance, rather than being the primary cause of cancer initiation have larger sample sizes but use heterogeneous study designs. Therefore, further research focusing on the clinical characteristics of the cancer is needed to confirm the role of HPV in later carcinogenic processes reflected by higher GS, and to determine whether HPV is needed for tumour maintenance. Ideally, future studies should use the same HPV detection methodology. Several studies suggest that the prostate can act as a reservoir for HPVs with carcinogenic potential, but the presence of HPV does not necessarily imply a direct causal relationship with PCa.
Recent investigations have focused on the emergence of apolipoprotein B mRNA editing enzyme, catalytic-polypeptide-like (APOBEC) enzymes as a protective mechanism against viral infections (Ohba et al., 2014; Vieira et al., 2014; Gansmo et al., 2018). Studies have suggested that the oncogenic effect of HPV may not be a direct result of viral infection, but rather an indirect consequence of the ability of HPV to inhibit the protective function of APOBEC enzymes (Ohba et al., 2014; Vieira et al., 2014). APOBEC mutations have been linked to certain human cancer genomes, and the APOBEC3A/B deletion polymorphism has been associated with a higher risk of some cancers, including PCa (Burns, Temiz and Harris, 2013; Caval et al., 2014; Nik-Zainal et al., 2014; Gansmo et al., 2018). Specifically, changes in the protective effects of APOBEC3B against oncogenic viruses can occur, leading to host genome instability and cancer progression after HPV viral DNA integration (Ohba et al., 2014; Vieira et al., 2014; Cheng et al., 2019). The intricate relationship between viruses and APOBEC in cancer development is further complicated by the presence of two viruses, HPV and Epstein Barr virus (EBV), which have both been identified in PCa (Whitaker et al., 2013). The negative impact of EBV on the integrity of APOBEC adds an additional layer to this complexity (Cheng et al., 2019). Therefore, it appears that the main mechanism by which HPV influences PCa is indirect, involving the inhibition of enzymes such as APOBEC3B that would otherwise help protect against the harmful effects of viruses.
Despite these findings, the role of HPV in prostate carcinogenesis remains unclear, as there are conflicting reports between studies. The "hit and run" hypothesis opposes the later-acting mechanism, which is observed by correlation with a higher GS and metastasis. The use of immunohistochemistry (IHC) proposes interesting mechanisms for how HPV may interact with cellular components, but more research is needed to confirm these findings (Khatami et al., 2022). Moreover, HPV infection may contribute to inflammation in the gland, but the use of BPH as a control makes it challenging to distinguish this role due to inflammation possibly being part of the etiology of both conditions.
5. Consistency
Various studies examining this topic have yielded inconsistent results, which may be due in part to differences in methodology. While there have been previous discussions about different techniques for detecting HPV, this section will specifically examine discrepancies in the collection and storage of tissue samples (Table 4).
5.1. Consistency in Tissue Collection
The initial investigation on HPV DNA in prostate tissue relied primarily on TURP for sample collection, with suprapubic prostatectomy (SPP) used for two samples (McNicol and Dodd, 1990). The study found high HPV detection rates: 4 out of 4 (100%) cases of PCa and 14 out of 15 (93%) BPH controls tested positive. In a subsequent larger study by the same researchers, HPV detection rates were still relatively high, but lower than the first study: 56% of PCa cases, 61% of BPH controls, and 20% of healthy tissue controls tested positive (McNicol and Dodd, 1991). TURP and SPP were again the primary collection methods, but there were concerns that TURP might introduce HPV from the male urethra into the prostate tissue, leading to an overestimation of HPV presence. The male urethra is susceptible to HPV infection, and HPV DNA may be present in this area (Murphy et al., 1983). This issue prompted other studies to explore other tissue collection methods to minimize the potential for contamination.
Another study used a microdissection approach that excluded urethral mucosa and found no HPV DNA in PCa cases (Effert et al., 1992), while another found HPV DNA in BPH specimens obtained via TURP and SPP, suggesting that prostate tissue may be the source of HPV. However, other studies that used TURP did not find any HPV DNA (Masood et al., 1991; Anderson et al., 1997; Strickler et al., 1998; Bergh et al., 2007; Gazzaz and Mosli, 2009), indicating that contamination may be a significant confounding factor.
It is conceivable that high occurrence in the initial of HPV and PCa was due to contamination from urethral tissue. Nonetheless, it is more likely that the small sample size was the actual cause, as evidenced by other TURP-based studies that did not produce results. Most studies in this review have shifted away from the TURP method, particularly for PCa cases, which implies that contamination is not a significant confounding variable to consider in this review.
5.1.1. Consistency in Tissue Storage
Another difference in study methodology is the way in which tissue is stored. Most studies used FFPE blocks, fresh, or frozen tissue. In the earlier studies, fresh or frozen tissue was primarily used, but most studies in this review used FFPE blocks as the storage method (Table 4). The use of FFPE samples allows superior morphological assessment of tissue for hybridisation analysis and less chance of carry-over contamination during PCR (Ibrahim et al., 1992). However, the fixation process can influence the yield of high quality DNA, which may affect its suitability for PCR (Díaz-Cano and Brady, 1997). This is probably not an issue in this review as most studies tested for the quality of DNA extracted from samples using a reference human gene, such as beta-globin. Furthermore, when testing for this, no significant difference was seen in HPV prevalence between FFPE and fresh frozen samples (P = 1.00) (Ibrahim et al., 1992).
In conclusion, the studies reviewed here are inconsistent with each other in terms of HPV detection rates, HPV detection methodology, and tissue collection and storage methodology. Although some studies find these differences to have non-significant effects on the results, it is not clear what the true reasons are for discrepancies between studies. Perhaps a review which focuses on studies using solely the same techniques would mean the consistency criterion could be met but this would require many further studies.
6. Biological Gradient
The original Bradford Hill criteria state that a dose-response relationship supports a causal association. However, for HPV and PCa, demonstrating different exposure dosages is difficult as most studies only report whether the virus is present. A few studies in this review do consider the HPV copy number per cell (McNicol and Dodd, 1991; Rotola et al., 1992; Moyret-Lalle et al., 1995; Serth et al., 1999), but the results are inconsistent, likely due to the heterogeneity of prostate specimens (McNicol and Dodd, 1990). The L1 consensus primers GP5+/6+ can detect 1 HPV DNA copy per 100 PCa cells meaning it is sensitive enough to detect low copy numbers of HPV DNA (Strickler et al., 1998). One study that used ISH states that the probe used could detect 10 copies, and hence a weak positive ISH analysis may be due to the assay not being sensitive enough to detect low copy numbers (Chen et al., 2011). Therefore, a dose-response relationship cannot be concluded. It is not a crucial causal criterion since the "hit and run" hypothesis suggests that a high viral load may be unnecessary for prostate carcinogenesis.
7. Experimental Evidence
Bradford Hill described this criterion as evidence drawn from experimental manipulation (Fedak et al., 2015). In this case, manipulation would refer to removal of the exposure and observing if risk declines. Cessation of exposure, such as treating HPV to see if PCa rates decrease, has also not been conducted in this review. This is an example of an intervention study, whereas the ones in this review are observational: case-control and cohort studies. However, it is possible to link this criterion to vaccination programmes against HPV and cessation of exposure in this regard. Information on rates of PCa following an HPV vaccination programme are not available. However, this is an effective measure of prevention against cervical cancer (Falcaro et al., 2021; Torjesen, 2021). Hence, rates of PCa in countries following HPV vaccination could be a way forward in the future to provide experimental evidence on HPV as a cause of PCa.
To conclude, in this review, experimental manipulation of HPV as an exposure was not tested for and cannot be commented on for causal inference in PCa. Notably, expression of HPV E6 and E7 16 has been shown to immortalise prostate epithelial cells (Choo et al., 1999), which provides strong experimental evidence that HPV oncoproteins allow prostate cells to avoid normal cell control mechanisms.
8. Conclusions
Although the evidence available to date provides interesting insights into the potential role of high-risk HPV in PCa, it is not yet conclusive enough to meet key Bradford Hill criteria such as strength of association, consistency, and coherence. Additionally, the complex nature of HPV transmission and oncogenic mechanisms, coupled with the difficulty of testing criteria such as temporality, biological gradient, and experiment in this context, make it challenging to draw definitive conclusions.
While there are some similarities between PCa and cervical cancer, they also have significant differences that limit the extent to which analogies can be drawn between the two. Although the identification and transmission of HPV provide valuable information regarding the virus's plausibility as a causative agent in PCa, the conflicting nature of the literature prevents us from drawing a definitive conclusion about the causal role of HPV in this disease.
However, it is possible that the prostate serves as a reservoir for HPV transmission. If high-risk HPV does play a role in prostate carcinogenesis, it is likely through the actions of the viral oncoproteins, the promotion of inflammation, which could potentially contribute to the oncogenic process at various stages, and/or damage to host DNA. Further studies are required to gain a better understanding of this topic, potentially focusing on homogeneous study designs and result analyses. Therefore, while the evidence to date suggests a possible causal role for HPV in PCa, it is not yet strong enough to use the term "highly likely" without further research to support this claim.
Author Contributions
All authors collaboratively executed the study in this manuscript.
Funding
This research is funded by UK Medical Research Council (MR/S009086/1).
Acknowledgments
KG is grateful to the UK Medical Research Council (MR/S009086/1) for research funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Abumsimir, B. et al. (2022) ‘Molecular characterization of human papillomavirus and mouse mammary tumor virus-like infections in prostate cancer tissue and relevance with tumor characteristics.’, Molecular and clinical oncology, 16(5), p. 97. [CrossRef]
- Adami, H.-O. et al. (2003) ‘Prostate cancer risk and serologic evidence of human papilloma virus infection: a population-based case-control study.’, Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology, 12(9), pp. 872–875.
- Aghakhani, A. et al. (2010) ‘The role of human papillomavirus infection in prostate carcinoma’, Scandinavian Journal of Infectious Diseases, 43(1), pp. 64–69. [CrossRef]
- Ahmed, M.Y. et al. (2023) ‘Detection of high - risk Human Papillomavirus in prostate cancer from a UK based population’, Scientific Reports, pp. 1–9. [CrossRef]
- Anderson, M. et al. (1997) ‘Analysis of prostate tissue DNA for the presence of human papillomavirus by polymerase chain reaction, cloning, and automated sequencing.’, Journal of medical virology, 52(1), pp. 8–13. [CrossRef]
- Anwar, K. et al. (1992) ‘Presence of ras oncogene mutations and human papillomavirus DNA in human prostate carcinomas.’, Cancer research, 52(21), pp. 5991–5996.
- Araldi, R.P. et al. (2018) ‘Biomedicine & Pharmacotherapy The human papillomavirus ( HPV ) -related cancer biology : An overview’, Biomedicine & Pharmacotherapy, 106(June), pp. 1537–1556. [CrossRef]
- Atashafrooz, F. and Rokhbakhsh-Zamin, F. (2016) ‘Frequency and type distribution of human papilloma virus in patients with prostate cancer, Kerman, southeast of Iran’, Asian Pacific Journal of Cancer Prevention, 17(8), pp. 3951–3956.
- Aydin, M. et al. (2017) ‘Lack of evidence of HPV etiology of prostate cancer following radical surgery and higher frequency of the Arg/Pro genotype in Turkish men with prostate cancer.’, International braz j urol : official journal of the Brazilian Society of Urology, 43(1), pp. 36–46. [CrossRef]
- Bergh, J. et al. (2007) ‘No link between viral findings in the prostate and subsequent cancer development.’, British journal of cancer, 96(1), pp. 137–139. [CrossRef]
- Bostwick, D.G. et al. (2004) ‘Human prostate cancer risk factors’, Cancer, 101(10 SUPPL.), pp. 2371–2490. [CrossRef]
- Burns, M.B., Temiz, N.A. and Harris, R.S. (2013) ‘Evidence for APOBEC3B mutagenesis in multiple human cancers.’, Nature genetics, 45(9), pp. 977–983. [CrossRef]
- De Carolis, S. et al. (2019) ‘HPV DNA Associates With Breast Cancer Malignancy and It Is Transferred to Breast Cancer Stromal Cells by Extracellular Vesicles’, Frontiers in Oncology, 9(September), pp. 1–12. [CrossRef]
- Carozzi, F. et al. (2004) ‘Association of human papillomavirus with prostate cancer: analysis of a consecutive series of prostate biopsies.’, The International journal of biological markers, 19(4), pp. 257–261. [CrossRef]
- Caval, V. et al. (2014) ‘A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3’UTR enhances chromosomal DNA damage.’, Nature communications, 5, p. 5129. [CrossRef]
- Chang, H.-J. et al. (2023) ‘A matched case-control study in Taiwan to evaluate potential risk factors for prostate cancer’, Scientific Reports, 13(1), pp. 1–10. [CrossRef]
- Chang, R.T.M., Kirby, R. and Challacombe, B.J. (2012) ‘Is there a link between BPH and prostate cancer?’, The Practitioner, 256(1750), pp. 2,13-16.
- Chen, A.C.H. et al. (2011) ‘Human papillomavirus in benign prostatic hyperplasia and prostatic adenocarcinoma patients’, Pathology and Oncology Research, 17(3), pp. 613–617. [CrossRef]
- Chen, Y. and Wei, J. (2015) ‘Identification of pathogen signatures in prostate cancer using RNA-seq’, PLoS ONE, 10(6), pp. 1–13. [CrossRef]
- Cheng, A.Z. et al. (2019) ‘Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity.’, Nature microbiology. England, pp. 78–88. [CrossRef]
- Choo, C.K. et al. (1999) ‘Immortalization of human prostate epithelial cells by HPV 16 E6/E7 open reading frames.’, The Prostate, 40(3), pp. 150–158. [CrossRef]
- Chughtai, B. et al. (2016) ‘Benign prostatic hyperplasia.’, Nature reviews. Disease primers, 2, p. 16031. [CrossRef]
- Cirakoglu, A., Benli, E. and Yuce, A. (2018) ‘Polygamy, sexual behavior in a population under risk for prostate cancer diagnostic: an observational study from the Black Sea Region in Turkey.’, International braz j urol : official journal of the Brazilian Society of Urology, 44(4), pp. 704–708. [CrossRef]
- Combes, J.D. et al. (2014) ‘Antibodies against high-risk human papillomavirus proteins as markers for invasive cervical cancer’, International Journal of Cancer, 135(10), pp. 2453–2461. [CrossRef]
- Dagnelie, P.C. et al. (2004) ‘Diet, anthropometric measures and prostate cancer risk: a review of prospective cohort and intervention studies.’, BJU international, 93(8), pp. 1139–1150.
- Díaz-Cano, S.J. and Brady, S.P. (1997) ‘DNA extraction from formalin-fixed, paraffin-embedded tissues: protein digestion as a limiting step for retrieval of high-quality DNA.’, Diagnostic molecular pathology : the American journal of surgical pathology, part B, 6(6), pp. 342–346. [CrossRef]
- Dillner, J. et al. (1998) ‘Sero-epidemiologal association between human-papillomavirus infection and risk of prostate cancer’, International Journal of Cancer, 75(4), pp. 564–567. [CrossRef]
- Dodd, J.G., Paraskevas, M. and McNicol, P.J. (1993) ‘Detection of human papillomavirus 16 transcription in human prostate tissue.’, The Journal of urology, 149(2), pp. 400–402. [CrossRef]
- Eghbali, S.S. et al. (2012) ‘Oncogenic human papillomavirus genital infection in southern Iranian women : population-based study versus clinic-based data’, pp. 1–6.
- Fabbri, M. et al. (2012) ‘MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response’, Proceedings of the National Academy of Sciences of the United States of America, 109(31). [CrossRef]
- Falcaro, M. et al. (2021) ‘The effects of the national HPV vaccination programme in England, UK, on cervical cancer and grade 3 cervical intraepithelial neoplasia incidence: a register-based observational study’, The Lancet, 398(10316), pp. 2084–2092. [CrossRef]
- Faraoni, I. et al. (2009) ‘miR-155 gene: a typical multifunctional microRNA.’, Biochimica et biophysica acta, 1792(6), pp. 497–505. [CrossRef]
- Fatemipour, M. et al. (2021) ‘Human papillomavirus and prostate cancer: The role of viral expressed proteins in the inhibition of anoikis and induction of metastasis’, Microbial Pathogenesis, 152, p. 104576. [CrossRef]
- Fedak, K.M. et al. (2015) ‘Applying the Bradford Hill criteria in the 21st century: How data integration has changed causal inference in molecular epidemiology’, Emerging Themes in Epidemiology, 12(1), pp. 1–9. [CrossRef]
- Gandaglia, G. et al. (2021) ‘Epidemiology and Prevention of Prostate Cancer’, European urology oncology, 4(6), pp. 877–892. [CrossRef]
- Gansmo, L.B. et al. (2018) ‘APOBEC3A/B deletion polymorphism and cancer risk.’, Carcinogenesis, 39(2), pp. 118–124. [CrossRef]
- Gazzaz, F.S. and Mosli, H.A. (2009) ‘Lack of detection of human papillomavirus infection by hybridization test in prostatic biopsies.’, Saudi medical journal, 30(5), pp. 633–637.
- Ghasemian, E. et al. (2013) ‘Evaluation of human papillomavirus infections in prostatic disease: A cross-sectional study in Iran’, Asian Pacific Journal of Cancer Prevention, 14(5), pp. 3305–3308. [CrossRef]
- Gironella, M. et al. (2007) ‘Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development.’, Proceedings of the National Academy of Sciences of the United States of America, 104(41), pp. 16170–16175. [CrossRef]
- Glenn, W.K. et al. (2017) ‘High risk human papilloma viruses (HPVs) are present in benign prostate tissues before development of HPV associated prostate cancer’, Infectious Agents and Cancer, 12(1), pp. 1–10. [CrossRef]
- Graham, S. V (2017) ‘The human papillomavirus replication cycle, and its links to cancer progression : a comprehensive review’, 0(July), pp. 2201–2221.
- Guenat, D. et al. (2017) ‘Exosomes and other extracellular vesicles in HPV transmission and carcinogenesis’, Viruses, 9(8), pp. 1–17. [CrossRef]
- Hanahan, D. and Weinberg, R.A. (2011) ‘Hallmarks of cancer: the next generation.’, Cell, 144(5), pp. 646–674. [CrossRef]
- Hill, A. (1965) ‘President’ s Address The Environment and Disease: Association or causation?’, Proc R Soc Med, (58), pp. 295–300.
- Hodgson, A. et al. (2019) ‘International Endocervical Adenocarcinoma Criteria and Classification (IECC): correlation with adverse clinicopathological features and patient outcome.’, Journal of clinical pathology, 72(5), pp. 347–353. [CrossRef]
- Hrbacek, J. et al. (2011) ‘Serum antibodies against genitourinary infectious agents in prostate cancer and benign prostate hyperplasia patients: a case-control study.’, BMC cancer, 11, p. 53. [CrossRef]
- Ibrahim, G.K. et al. (1992) ‘Detection of human papillomavirus in the prostate by polymerase chain reaction and in situ hybridization’, Journal of Urology, pp. 1822–1826. [CrossRef]
- Itoh, Y. and Nagase, H. (2002) ‘Matrix metalloproteinases in cancer.’, Essays in biochemistry, 38, pp. 21–36. [CrossRef]
- Jeon, S., Allen-hoffmann, B.L. and Lambert, P.F. (1995) ‘Integration of Human Papillomavirus Type 16 into the Human Genome Correlates with a Selective Growth Advantage of Cells †’, 69(5), pp. 2989–2997.
- Jian, Z. et al. (2018) ‘Sexual Activity and Risk of Prostate Cancer: A Dose-Response Meta-Analysis.’, The journal of sexual medicine, 15(9), pp. 1300–1309. [CrossRef]
- Johnson, A.M. et al. (2012) ‘Epidemiology of, and behavioural risk factors for, sexually transmitted human papillomavirus infection in men and women in Britain’, Sexually Transmitted Infections, 88(3), pp. 212–217. [CrossRef]
- Khatami, A. et al. (2022) ‘Human papilloma virus (HPV) and prostate cancer (PCa): The potential role of HPV gene expression and selected cellular MiRNAs in PCa development’, Microbial Pathogenesis, 166(March), p. 105503. [CrossRef]
- Korodi, Z. et al. (2005) ‘Human papillomavirus 16, 18, and 33 infections and risk of prostate cancer: A Nordic nested case-control study’, Cancer Epidemiology Biomarkers and Prevention, 14(12), pp. 2952–2955. [CrossRef]
- Lawson, J.S. and Glenn, W.K. (2020) ‘Evidence for a causal role by human papillomaviruses in prostate cancer-A systematic review’, Infectious Agents and Cancer, 15(1), pp. 1–11. [CrossRef]
- Leechanachai, P. et al. (1992) ‘The E5 gene from human papillomavirus type 16 is an oncogene which enhances growth factor-mediated signal transduction to the nucleus.’, Oncogene, 7(1), pp. 19–25.
- Leiros, G.J. et al. (2005) ‘Detection of human papillomavirus DNA and p53 codon 72 polymorphism in prostate carcinomas of patients from Argentina.’, BMC urology, 5, p. 15. [CrossRef]
- Leitzmann, M.F. and Rohrmann, S. (2012) ‘Risk factors for the onset of prostatic cancer: age, location, and behavioral correlates.’, Clinical epidemiology, 4, pp. 1–11. [CrossRef]
- Lenze, D., Müller, H.H. and Hummel, M. (2012) ‘Considerations for the use of formalin-fixed and paraffin-embedded tissue specimens for clonality analysis’, Journal of Hematopathology, 5(1–2), pp. 27–34. [CrossRef]
- Madani, T.A. (2006) ‘Sexually transmitted infections in Saudi Arabia.’, BMC infectious diseases, 6, p. 3. [CrossRef]
- Madden, S.K. et al. (2021) ‘Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c-Myc.’, Molecular cancer, 20(1), p. 3. [CrossRef]
- Mahesh, G. and Biswas, R. (2019) ‘MicroRNA-155: A Master Regulator of Inflammation.’, Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research, 39(6), pp. 321–330. [CrossRef]
- Martinez-Fierro, M.L. et al. (2010) ‘Identification of viral infections in the prostate and evaluation of their association with cancer.’, BMC cancer, 10, p. 326. [CrossRef]
- Masood, S. et al. (1991) ‘Human papillomavirus in prostatic cancer: no evidence found by in situ DNA hybridization.’, Southern medical journal, 84(2), pp. 235–236. [CrossRef]
- Mcbride, A.A. and Warburton, A. (2017) ‘The role of integration in oncogenic progression of HPV-associated cancers’, pp. 1–7.
- McLaughlin-Drubin, M.E. and Münger, K. (2009) ‘Oncogenic activities of human papillomaviruses’, Virus Research, 143(2), pp. 195–208. [CrossRef]
- McNicol, P.J. and Dodd, J.G. (1990) ‘Detection of human papillomavirus DNA in prostate gland tissue by using the polymerase chain reaction amplification assay’, Journal of Clinical Microbiology, 28(3), pp. 409–412. [CrossRef]
- McNicol, P.J. and Dodd, J.G. (1991) ‘High prevalence of human papillomavirus in prostate tissues’, Journal of Urology, 145(4), pp. 850–853. [CrossRef]
- Medel-Flores, O. et al. (2018) ‘Association between HPV infection and prostate cancer in a Mexican population.’, Genetics and molecular biology, 41(4), pp. 781–789. [CrossRef]
- Michopoulou, V. et al. (2014) ‘Detection of human papillomavirus (HPV) DNA prevalence and p53 codon 72 (Arg72Pro) polymorphism in prostate cancer in a Greek group of patients’, Tumor Biology, 35(12), pp. 12765–12773. [CrossRef]
- Miller, D.M. et al. (2012) ‘c-Myc and cancer metabolism.’, Clinical cancer research : an official journal of the American Association for Cancer Research, 18(20), pp. 5546–5553. [CrossRef]
- Moghoofei, M. et al. (2019) ‘Association between human papillomavirus infection and prostate cancer: A global systematic review and meta-analysis’, Asia-Pacific Journal of Clinical Oncology, 15(5), pp. e59–e67. [CrossRef]
- Mokhtari, M., Taghizadeh, F. and Hani, M. (2013) ‘Is prostatic adenocarcinoma in a relationship with Human Papilloma Virus in Isfahan- Iran’, Journal of Research in Medical Sciences, 18(8), pp. 707–710.
- Moody, C.A. and Laimins, L.A. (2010) ‘Human papillomavirus oncoproteins : pathways to transformation’, Nature Publishing Group, 10(8), pp. 550–560. [CrossRef]
- Moyret-Lalle, C. et al. (1995) ‘ras, p53 and HPV status in benign and malignant prostate tumors.’, International journal of cancer, 64(2), pp. 124–129. [CrossRef]
- Murphy, W.M. et al. (1983) ‘Papillomavirus structural antigens in condyloma acuminatum of the male urethra.’, The Journal of urology, 130(1), pp. 84–85. [CrossRef]
- Nik-Zainal, S. et al. (2014) ‘Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer.’, Nature genetics, 46(5), pp. 487–491. [CrossRef]
- Ohba, K. et al. (2014) ‘In vivo and in vitro studies suggest a possible involvement of HPV infection in the early stage of breast carcinogenesis via APOBEC3B induction.’, PloS one, 9(5), p. e97787. [CrossRef]
- Pascale, M. et al. (2013) ‘Is human papillomavirus associated with prostate cancer survival?’, Disease Markers, 35(6), pp. 607–613. [CrossRef]
- Peng, Y. and Croce, C.M. (2016) ‘The role of MicroRNAs in human cancer’, Signal Transduction and Targeted Therapy, 1(1), p. 15004. [CrossRef]
- Pereira, N.M. et al. (2023) ‘Presence of HPV in prostate tissue from patients submitted to prostate biopsy’, Acta Cirurgica Brasileira, 37(12), pp. 1–8. [CrossRef]
- Pernar, C.H. et al. (2018) ‘The Epidemiology of Prostate Cancer.’, Cold Spring Harbor perspectives in medicine, 8(12). [CrossRef]
- Pim, D. and Banks, L. (2010) ‘Interaction of viral oncoproteins with cellular target molecules: Infection with high-risk vs low-risk human papillomaviruses’, Apmis, 118(6–7), pp. 471–493. [CrossRef]
- Vande Pol, S.B. and Klingelhutz, A.J. (2013) ‘Papillomavirus E6 oncoproteins’, Virology, 445(1–2), pp. 115–137. [CrossRef]
- Rawla, P. (2019) ‘Epidemiology of Prostate Cancer.’, World journal of oncology, 10(2), pp. 63–89. [CrossRef]
- Rodriguez, M.I.D. et al. (2016) ‘Human papilloma virus detection by INNOLiPA HPV in prostate tissue from men of Northeast Mexico’, Asian Pacific Journal of Cancer Prevention, 17(11), pp. 4863–4865. [CrossRef]
- Roman, A. and Munger, K. (2013) ‘The papillomavirus E7 proteins’, Virology, 445(1–2), pp. 138–168. [CrossRef]
- Rosenblatt, K.A. et al. (2003) ‘Serologic evidence of human papillomavirus 16 and 18 infections and risk of prostate cancer.’, Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology, 12(8), pp. 763–768.
- Rotola, A. et al. (1992) ‘Presence and physical state of HPV DNA in prostate and urinary-tract tissues’, International Journal of Cancer, 52(3), pp. 359–365. [CrossRef]
- Sarkar, F.H. et al. (1993) ‘Detection of human papillomavirus (HPV) DNA in human prostatic tissues by polymerase chain reaction (PCR).’, The Prostate, 22(2), pp. 171–180. [CrossRef]
- Serth, J. et al. (1999) ‘Increased levels of human papillomavirus type 16 DNA in a subset of prostate cancers.’, Cancer research, 59(4), pp. 823–825.
- Sfanos, K.S. et al. (2008) ‘A molecular analysis of prokaryotic and viral DNA sequences in prostate tissue from patients with prostate cancer indicates the presence of multiple and diverse microorganisms.’, The Prostate, 68(3), pp. 306–320. [CrossRef]
- Singh, N. et al. (2015) ‘Implication of high risk Human papillomavirus HR-HPV infection in prostate cancer in Indian population-A pioneering case-control analysis’, Scientific Reports, 5, pp. 7–10. [CrossRef]
- Stoler, M.H. et al. (1991) ‘Small-cell neuroendocrine carcinoma of the cervix. A human papillomavirus type 18-associated cancer.’, The American journal of surgical pathology, 15(1), pp. 28–32. [CrossRef]
- Stolnicu, S. et al. (2018) ‘International Endocervical Adenocarcinoma Criteria and Classification (IECC): A New Pathogenetic Classification for Invasive Adenocarcinomas of the Endocervix.’, The American journal of surgical pathology, 42(2), pp. 214–226. [CrossRef]
- Strickler, H.D. et al. (1998) ‘A multifaceted study of human papillomavirus and prostate carcinoma.’, Cancer, 82(6), pp. 1118–1125.
- Sung, H. et al. (2021) ‘Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries’, CA: A Cancer Journal for Clinicians, 71(3), pp. 209–249. [CrossRef]
- Sutcliffe, S. et al. (2007) ‘Plasma antibodies against Chlamydia trachomatis, human papillomavirus, and human herpesvirus type 8 in relation to prostate cancer: a prospective study.’, Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology, 16(8), pp. 1573–1580. [CrossRef]
- Sutcliffe, S. et al. (2010) ‘Human papillomavirus types 16, 18, and 31 serostatus and prostate cancer risk in the Prostate Cancer Prevention Trial.’, Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology, 19(2), pp. 614–618. [CrossRef]
- Tachezy, R. et al. (2012) ‘HPV persistence and its oncogenic role in prostate tumors.’, Journal of medical virology, 84(10), pp. 1636–1645. [CrossRef]
- Terris, M.K. and Peehl, D.M. (1997) ‘Human papillomavirus detection by polymerase chain reaction in benign and malignant prostate tissue is dependent on the primer set utilized.’, Urology, 50(1), pp. 150–156. [CrossRef]
- Torjesen, I. (2021) ‘HPV vaccine cut cervical cancer rates in England by 87\%’, BMJ, 375. [CrossRef]
- Tu, H. et al. (1994) ‘Rare incidence of human papillomavirus types 16 and 18 in primary and metastatic human prostate cancer.’, Urology, 44(5), pp. 726–731. [CrossRef]
- Tung, C.-H. et al. (2020) ‘MicroRNA-150-5p promotes cell motility by inhibiting c-Myb-mediated Slug suppression and is a prognostic biomarker for recurrent ovarian cancer.’, Oncogene, 39(4), pp. 862–876. [CrossRef]
- Vieira, V.C. et al. (2014) ‘Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B.’, mBio, 5(6). [CrossRef]
- Whitaker, N.J. et al. (2013) ‘Human papillomavirus and Epstein Barr virus in prostate cancer: Koilocytes indicate potential oncogenic influences of human papillomavirus in prostate cancer’, Prostate, 73(3), pp. 236–241. [CrossRef]
- Williams, S.M.G. et al. (2005) ‘Requirement of Epidermal Growth Factor Receptor for Hyperplasia Induced by E5, a High-Risk Human Papillomavirus Oncogene’, (15), pp. 6534–6542.
- Yang, L. et al. (2015) ‘Worldwide Prevalence of Human Papillomavirus and Relative Risk of Prostate Cancer: A Meta-analysis’, Scientific Reports, 5, pp. 1–10. [CrossRef]
- Yow, M.A. et al. (2014) ‘Detection of infectious organisms in archival prostate cancer tissues’, BMC Cancer, 14(1), pp. 1–5. [CrossRef]
- Zambrano, A. et al. (2002) ‘Detection of human polyomaviruses and papillomaviruses in prostatictissue reveals the prostate as a habitat for multipleviral infections’, Prostate, 53(4), pp. 263–276. [CrossRef]
- Zhang, Z. et al. (2018) ‘MicroRNA-150 promotes cell proliferation, migration, and invasion of cervical cancer through targeting PDCD4.’, Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 97, pp. 511–517. [CrossRef]
- Zhao, H. et al. (2021) ‘Inflammation and tumor progression: signaling pathways and targeted intervention’, Signal Transduction and Targeted Therapy, 6(1), p. 263. [CrossRef]
- Zhao, X. et al. (2017) ‘Role of antibodies to human papillomavirus 16 in prostate cancer: A seroscreening by peptide microarray.’, Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine, 39(6), p. 1010428317698371. [CrossRef]
- Zhou, Y. et al. (2017) ‘Diagnosis of cancer as an emergency: A critical review of current evidence’, Nature Reviews Clinical Oncology, 14(1), pp. 45–56. [CrossRef]
Figure 1.
PRISMA flowchart for selecting and screening the resultant papers of the initial literature search.
Figure 1.
PRISMA flowchart for selecting and screening the resultant papers of the initial literature search.
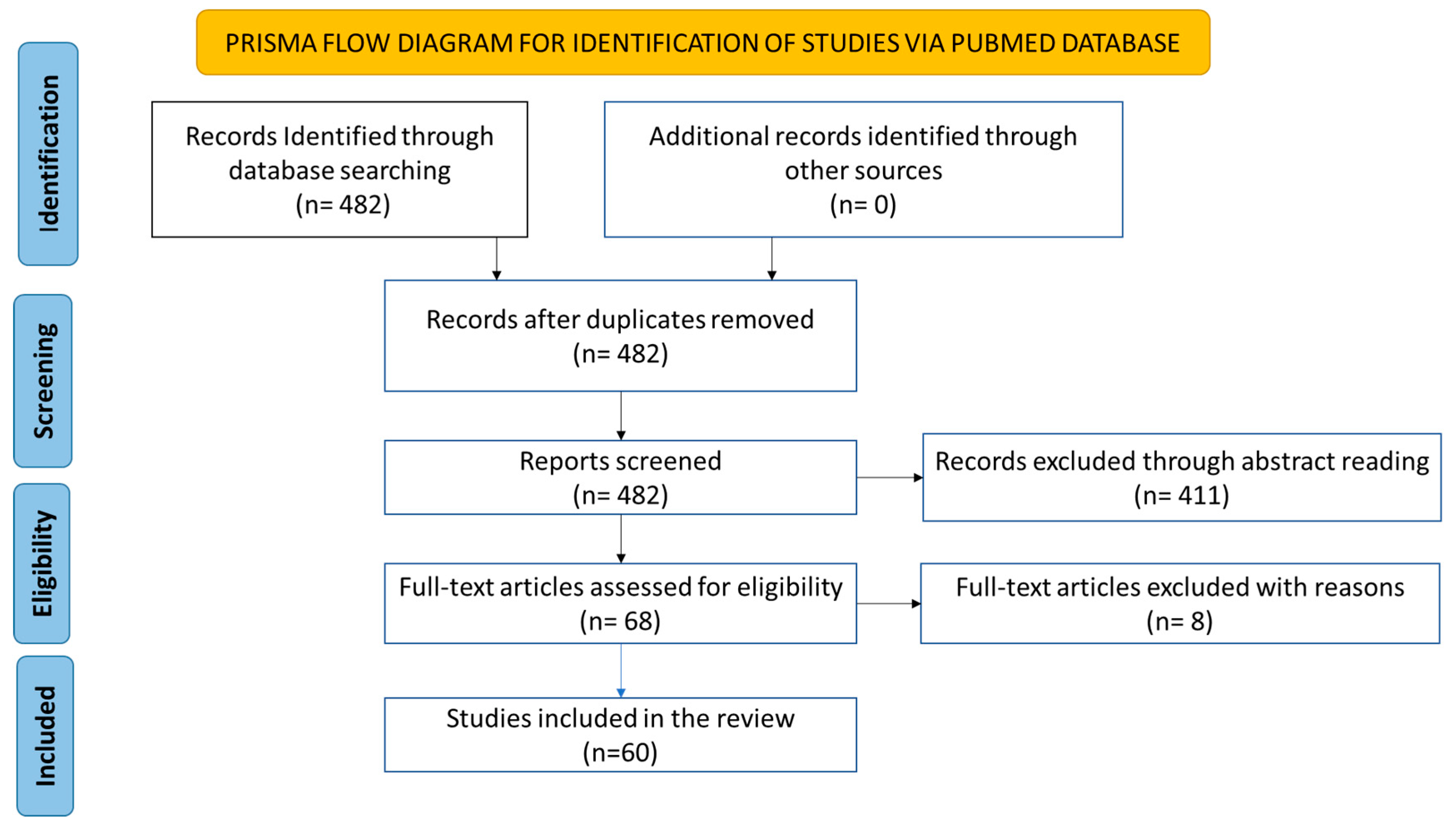

Table 1.
The inclusion and exclusion criteria used during abstract screening.
| Inclusion Criteria | Exclusion Criteria |
|---|---|
| Primary studies | Secondary data (e.g., Meta-Analysis and Systematic reviews) |
| Tissues only infected with HPV | Coinfections |
| Male participants over the age of 18 | Male participants with age < 18 |
| Paper in English | Studies published languages other than English |
| Duplicated papers |
Table 2.
Assessment of case-control study quality using the Newcastle-Ottawa Quality Assessment Scale. 1–Is the case definition accurate? 2–Representativeness of the cases. 3-Selection of controls. 4-Definition of controls. 5-Comparability of cases and controls of the basis of the design or analysis (star awarded when study tested the quality of its samples). 6-Ascertainment of exposure. 7-Same method of ascertainment for cases and controls.
Table 2.
Assessment of case-control study quality using the Newcastle-Ottawa Quality Assessment Scale. 1–Is the case definition accurate? 2–Representativeness of the cases. 3-Selection of controls. 4-Definition of controls. 5-Comparability of cases and controls of the basis of the design or analysis (star awarded when study tested the quality of its samples). 6-Ascertainment of exposure. 7-Same method of ascertainment for cases and controls.
| S/N | Study | Selection | Comparability | Exposure | Total | ||||
|---|---|---|---|---|---|---|---|---|---|
| 1` | 2 | 3 | 4 | 5 | 6 | 7 | |||
| 1 | Medel Flores et al. (2018) | * | * | - | * | * | * | * | 6 |
| 2 | Chen et al. (2011) | * | * | - | * | * | * | * | 6 |
| 3 | Aghakhani et al. (2011) | * | * | - | * | * | * | * | 6 |
| 4 | Zhao et al. (2017) | * | * | - | * | - | * | * | 5 |
| 5 | Tachezy et al. (2012) | * | * | - | * | * | * | * | 6 |
| 6 | Ghasemian et al. (2013) | * | * | - | * | * | * | * | 6 |
| 7 | Rodriguez et al. (2016) | * | * | - | * | * | * | * | 6 |
| 8 | Khatami et al. (2022) | * | * | - | * | * | * | * | 6 |
| 9 | Rotola et al. (1992) | * | * | - | * | * | * | * | 6 |
| 10 | Moyret-Lalle et al. (1995) | * | * | - | * | * | * | * | 6 |
| 11 | Atashafrooz et al. (2016) | * | * | - | * | * | * | * | 6 |
| 12 | Singh et al. (2015) | * | * | - | * | * | * | * | 6 |
| 13 | Sarkar et al. (1993) | * | * | - | - | * | * | * | 5 |
| 14 | Noda et al. (1998) | * | * | - | * | * | * | * | 6 |
| 15 | Korodi et al. (2005) | * | * | * | * | - | * | * | 6 |
| 16 | Carozzi et al. (2004) | * | * | - | * | * | * | * | 6 |
| 17 | Adami et al. (2003) | * | * | * | * | - | * | * | 6 |
| 18 | Leiros et al. (2005) | * | * | - | * | * | * | * | 6 |
| 19 | Wideroff et al. (1996) | * | * | - | * | * | * | * | 6 |
| 20 | Martinez-Fierro et al. (2010) | * | * | * | * | * | * | * | 7 |
| 21 | Sutcliffe et al. (2010) | * | * | * | * | - | * | * | 6 |
| 22 | Silvestre et al. (2009) | * | * | - | * | * | * | * | 6 |
| 23 | Michopoulo et al. (2014) | * | * | - | * | * | * | * | 6 |
| 24 | McNicol and Dodd (1990) | * | * | - | * | * | * | * | 6 |
| 25 | Rosenblatt et al. (2003) | * | * | * | * | - | * | * | 6 |
| 26 | Aydin et al. (2017) | * | * | - | * | * | * | * | 6 |
| 27 | McNicol and Dodd (1991) | * | * | - | * | * | * | * | 6 |
| 28 | Masood et al. (1991) | * | * | - | * | - | * | * | 5 |
| 29 | Anwar et al. (1992) | * | * | - | * | - | * | * | 5 |
| 30 | Terris and Peehl (1997) | * | * | - | - | * | * | * | 5 |
| 31 | Suzuki et al. (1996) | * | * | - | * | - | * | * | 5 |
| 32 | Ibrahim et al. (1992) | * | * | - | * | * | * | * | 6 |
| 33 | Serth et al. (1999) | * | * | - | * | * | * | * | 6 |
| 34 | Dodd et al. (1993) | * | * | - | * | * | * | * | 6 |
| 35 | Salehi and Hadavi (2012) | * | * | - | * | * | * | * | 6 |
| 36 | Strickler et al. (1998) | * | * | - | * | * | * | * | 6 |
| 37 | Hrbacek et al. (2011) | * | * | - | * | - | * | * | 5 |
| 38 | Sutcliffe et al. (2007) | * | * | * | * | - | * | * | 6 |
| 39 | Bergh et al. (2006) | * | * | - | * | * | * | * | 6 |
| 40 | Groom et al. 2012) | * | * | - | * | * | * | * | 6 |
| 41 | Tu et al. (1994) | * | * | - | * | * | * | * | 6 |
| 42 | Sitas et al. (2007) | * | * | - | * | - | * | * | 6 |
| 43 | Anderson et al., (1997) | * | * | - | * | * | * | * | 6 |
| 44 | Effert et al., (1992) | * | * | - | - | - | * | 3 | |
| 45 | Noda et al., (1998) | * | * | - | - | * | * | * | 5 |
| 46 | Araujo-Neto et al., (2016) | * | * | - | - | - | * | * | 4 |
| 47 | Balis et al., (2007) | * | * | - | - | - | * | 3 | |
| 48 | Mokhtari et al., (2013) | - | * | - | * | * | - | * | 4 |
| 49 | Pascale et al., (2013) | * | * | - | - | - | * | - | 3 |
| 50 | Abumsimir et al (2022) | * | * | - | - | - | * | - | 3 |
| 50 | Nahand et al., (2020) | * | * | - | * | * | * | 5 | |
| 51 | Pereira et al., (2023) | * | * | - | - | - | * | - | 3 |
| 52 | Yow et al., (2014) | * | * | - | - | - | * | 3 | |
| 53 | Whitaker et al., (2012) | * | * | - | * | - | * | * | 5 |
| 54 | (Ahmed et al., 2023) | * | * | * | * | * | * | * | 7 |
| 55 | (Chang et al., 2023) | * | * | * | * | * | * | * | 7 |
Table 3.
Assessment of cohort study quality using the Newcastle-Ottawa Quality Assessment Scale. 1-Representativeness of the exposed cohort. 2- Selection of the non-exposed cohort. 3-Ascertainment of exposure. 4-Demonstration that outcome of interest was not present at start of study. 5-Comparability of cohorts based on the design or analysis (star awarded when study tested the quality of its samples). 6-Assessment of outcome. 7-Was follow-up long enough for outcomes to occur (3 years). 8-Adequacy of follow up of cohorts.
Table 3.
Assessment of cohort study quality using the Newcastle-Ottawa Quality Assessment Scale. 1-Representativeness of the exposed cohort. 2- Selection of the non-exposed cohort. 3-Ascertainment of exposure. 4-Demonstration that outcome of interest was not present at start of study. 5-Comparability of cohorts based on the design or analysis (star awarded when study tested the quality of its samples). 6-Assessment of outcome. 7-Was follow-up long enough for outcomes to occur (3 years). 8-Adequacy of follow up of cohorts.
| Study | Selection | Comparability | Outcome | Total score | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| Dillner et al. (1998) | * | * | * | - | - | * | * | - | 6 |
| Gazzaz and Mosli (2009) | * | * | * | * | - | * | - | - | 5 |
| Strickler et al (1998) | * | - | * | * | * | * | 5 | ||
| Glenn et al (2017) | * | - | * | - | - | * | * | 4 | |
| Dennis et al., (2009) | * | * | * | * | * | * | - | - | 6 |
Table 4.
PCR studies included in this review. Formalin-fixed paraffin-embedded, FFPE; paraffin-embedded, PE; radical prostatectomy, RP; simple prostatectomy, SP; total prostatectomy, TP; suprapubic prostatectomy, SPP; subcapsular prostatectomy, SCP; transurethral resection of the prostate, TURP; radical retropubic prostatectomy, RRP; open prostatectomy, OP; transrectal biopsy, TRB; transperineal biopsy, TPB; transvesical prostatectomy, TVP; Transrectal ultrasound, TRUS.
Table 4.
PCR studies included in this review. Formalin-fixed paraffin-embedded, FFPE; paraffin-embedded, PE; radical prostatectomy, RP; simple prostatectomy, SP; total prostatectomy, TP; suprapubic prostatectomy, SPP; subcapsular prostatectomy, SCP; transurethral resection of the prostate, TURP; radical retropubic prostatectomy, RRP; open prostatectomy, OP; transrectal biopsy, TRB; transperineal biopsy, TPB; transvesical prostatectomy, TVP; Transrectal ultrasound, TRUS.
| Subjects | HPV types | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Author | HPV detection | Collection (storage) | No. | Type | HPV | 6 | 16 | 18 | Other |
| Medel Flores et al. (2018) | L1 PCR consensus primers E6/E7 PCR for HPV 16, 18, 31, 33, 52b, 58 |
RP (FFPE) | 189 | PCa cases | 37 (20%) | 6 | 7 | 8 | 52 (17), 58 (12) |
| RP (FFPE) | 167 | BPH controls | 16 (10%) | 1 | 3 | 52 (16), 58 (5) | |||
|
Pascale et al. (2013) |
E7 IHC staining L1 PCR consensus primer |
Surgery, not fine needle biopsies (FFPE) | 150 | PCa cases | 19 (13%) | 9 | |||
|
Chen et al. (2011) |
L1 PCR consensus primer Type specific PCR primer for HPV 18 ISH for HPV 18 |
Collection not specified (snap frozen) | 51 | PCa cases | 7 (14%) | 7 | |||
| Collection not specified (snap frozen) | 11 | BPH controls | 3 (27%) | 3 | |||||
|
Aghakhani et al. (2011) |
L1 PCR consensus primers | RP, TURP (FFPE) | 104 | PCa cases | 13 (13%) | 1 | 7 | 3 | 11 (2) |
| RP, TURP (FFPE) | 104 | BPH controls | 8 (8%) | 1 | 3 | 2 | 11 (2) | ||
|
Tachezy et al. (2012) |
L1 PCR consensus primers | RRP (FFPE) | 51 | PCa cases | 1 (2%) | 42 (1) | |||
| 11 SP, 84 TURP (FFPE) | 95 | BPH controls | 2 (2%) | 1 | 1 unknown | ||||
|
Mokhtari et al. (2013) |
IHC staining | Collection not specified (PE) | 30 | PCa cases | 3 (10%) | ||||
| Collection not specified (PE) | 90 | BPH controls | 1 (1%) | ||||||
|
Balis et al. (2007) |
L1 PCR consensus primer Type specific PCR primers for HPV 11, 16, 18, 33 |
Collection not specified (22 FFPE, 20 fresh frozen) | 42 | PCa cases | 2 (5%) | Unknown | |||
|
Ghasemian et al. (2013) |
L1 PCR consensus primers | Collection not specified (FFPE) | 29 | PCa cases | 5 (17%) | ||||
| Collection not specified (FFPE) | 167 | BPH controls | 8 (5%) | ||||||
|
Rodriguez et al. (2016) |
INNO-LiPA HPV kit–L1 and 28 HPV genotypes | OP (FFPE) | 62 | PCa cases | 12 (20%) | 11 (46.1%), 51, 52, and 66 (15.4%) | |||
| TURP (FFPE) | 25 | BPH controls | 1 (4%) | 1 | |||||
|
Khatami et al. (2022) |
L1 PCR consensus primer | Collection not specified (snap frozen) | 73 | PCa cases | 21 (29%) | 1 | 9 | 7 | 11 (1), 33 (3) |
| Collection not specified (snap frozen) | 39 | Healthy controls | 7 (8%) | 3 | 3 | 11 (1) | |||
|
Rotola et al. (1992) |
E6 PCR for HPV 6/11, 16 | Collection not specified (snap frozen) | 8 | PCa cases | N/A | 4 | 6 | 11 (4) | |
| Collection not specified (snap frozen) | 17 | BPH controls | N/A | 11 | 14 | 11 (11) | |||
| Moyret-Lalle et al. (1995) | E6 PCR for HPV 16 and 18 | Collection not specified (snap frozen) | 17 | PCa cases | 9 (53%) | 9 | |||
| Collection not specified (snap frozen) | 22 | BPH controls | 7 (32%) | 7 | |||||
|
Atashafrooz et al. (2016) |
Real Time PCR HPV detection/genotyping assay kit–13 genotypes | Collection not specified (PE) | 100 | PCa cases | 20 (20%) | 1 | 16/18 (8), 31/33 (6), 54 (2), 6/11 (3) | ||
| Collection not specified (PE) | 100 | BPH Controls | 8 (8%) | 2 | 16/18 (1), 31/33 (1), 6/11 (4) | ||||
|
Araujo-Neto et al. (2016) |
L1 PCR consensus primers E6/E6 PCR for HPV 16 |
RP (fresh frozen) | 104 | PCa cases | 0 (0%) | ||||
|
Singh et al. (2015) |
L1 PCR consensus primers Type specific PCR primers for HPV 6, 11, 16, 18 |
Collection not specified (storage not specified) | 95 | PCa cases | 39 (41%) | 2 | 30 | 6 | 11 (1) |
| Collection not specified (storage not specified) |
55 | BPH controls | 11 (20%) | 6 | 3 | 1 | 11 (1) | ||
|
Sarkar et al. (1993) |
E6/E7 PCR for HPV 6, 11, 16, and 18 Southern blot hybridisation |
Surgical resection, not TURP (PE) | 23 | PCa cases | 3 (13%) | 3 | |||
| Surgical resection, not TURP (PE) | 23 | PIN controls | 0 (0%) | ||||||
|
Noda et al. (1998) |
PCR primers for LCR and E7 for HPV 16, 18, 31, 33, 52, 58 | TP (FFPE) | 38 | PCa cases | 0 (0%) | ||||
| 10 SCP, 61 TURP (FFPE) | 71 | BPH controls | 3 (4%) | 3 | |||||
|
Carozzi et al. (2004) |
L1 PCR consensus primer E6/E7 PCR for HPV types 16, 18, 31, 33, 35, 45, 52, 58 Hybridisation |
TPB (formalin) | 26 | PCa cases | 17 (65%) | 1 | 3 | 3 | 58 (4) |
| TPB (formalin) | 25 | BPH controls | 12 (48%) | 1 | 2 | 53 (4) | |||
|
Leiros et al. (2005) |
Type specific PCR primers for HPV 6, 11, 16, 18 L1 PCR consensus primer Southern blot hybridisation |
TRB (FFPE) | 41 | PCa cases | 17 (42%) | 5 | 11 (2) | ||
| TRB (FFPE) | 30 | BPH controls | 0 (0%) | ||||||
| Wideroff et al. (1996) | L1 PCR consensus primers E6 PCR for HPV 6, 11, 16, 18, 31, 33, 45 Hybridisation |
TURP, RP, excision biopsy (FFPE) | 56 | PCa cases | 7 (13%) | ||||
| TURP (FFPE) | 42 | BPH controls | 4 (10%) | ||||||
|
Martinez-Fierro et al. (2010) |
L1 PCR consensus primers Linear Array HPV Genotyping Test |
TRB, TURP (storage not specified) | 55 | PCa cases | 11 (20%) | ||||
| TRB, TURP (storage not specified) | 75 | Non-PCa controls | 4 (5%) | ||||||
|
Silvestre et al. (2009) |
L1 PCR consensus primer Linear Array HPV Genotyping Test |
Collection and storage not specified | 65 | PCa cases | 2 (3%) | 2 | 84 (coinfection) | ||
| Collection and storage not specified | 6 | BPH controls | 0 (0%) | ||||||
|
Michopoulo et al. (2014) |
L1 PCR consensus primer | Collection not specified (FFPE) | 50 | PCa cases | 8 (16%) | 2 | 4 | 31 (1), unknown (1) | |
| Collection not specified (FFPE) | 30 | Healthy controls | 1 (3%) | Unknown (1) | |||||
|
McNiol and Dodd (1990) |
E6 PCR for HPV 16 and 18 | 2 SPP, 17 TURP (fresh frozen) | 4 | PCa cases | 4 (100%) | 4 | |||
| 15 | BPH controls | 14 (93%) | 11 | 16 + 18 (3) | |||||
| Autopsy (fresh frozen) | 5 | Healthy controls | 1 (20%) | 1 | |||||
|
Aydin et al. (2017) |
L1 PCR consensus primer HPV sign® Q24 for genotyping |
RP (FFPE) | 60 | PCa cases | 1 (2%) | 57 | |||
| TVP (FFPE) | 36 | BPH controls | 0 (0%) | ||||||
|
McNiol and Dodd (1991) |
E6 PCR for HPV 16 and 18 Hybridisation |
TURP, SPP (fresh frozen) | 27 | PCa cases | 14 (52%) | 14 | 1 | ||
| TURP, SPP (fresh frozen) | 56 | BPH controls | 34 (63%) | 34 | 3 | ||||
| Autopsy (fresh frozen) | 5 | Healthy controls | 1 (20%) | 1 | |||||
|
Masood et al. (1991) |
In situ hybridisation for HPV 6, 11, 16, 18, 31, 33, 35 | Core needle biopsy, TURP (FFPE) | 20 | PCa cases | 0 (0%) | ||||
| Core needle biopsy, TURP (FFPE) | 20 | BPH controls | 0 (0%) | ||||||
|
Anwar et al. (1992) |
E6 PCR for HPV 16, 18, 33 | TURP, SPP (FFPE) | 68 | PCa cases | 28 (41%) | 11 | 7 | 33 (5) | |
| TURP, SPP (FFPE) | 10 | BPH controls | 0 (0%) | ||||||
| Autopsy (FFPE) | 10 | Healthy controls | 0 (0%) | ||||||
|
Rodriguez et al. (2015) |
L1 PCR consensus primer Type specific PCR for 19 HPV genotypes |
Collection not specified (FFPE) | 69 | PCa cases | 0 (0%) | ||||
| Terris and Peehl (1997) | L1 PCR consensus primer E6 PCR for HPV 16 |
RRP (FFPE) | 53 | PCa cases | 12 (23%) | 12 | |||
| 41 | Peripheral benign tissue | 15 (37%) | 15 | ||||||
| 37 | Healthy controls | 6 (16%) | 6 | ||||||
|
Suzuki et al. (1996) |
L1 PCR consensus primer | 29 TP, 22 autopsy (storage not specified) | 51 | PCa cases | 8 (16%) | 8 | |||
|
Ibrahim et al. (1992) |
L1 PCR consensus primer ISH |
RP, TURP, TRB (FFPE and fresh frozen) | 40 | PCa cases | 6 (15%) | 6 | |||
| RP, TURP, TRB (FFPE) | 12 | BPH controls | 0 (0%) | ||||||
| RP, TURP, TRB (FFPE) | 17 | Healthy controls | 2 (12%) | 2 | |||||
|
Serth et al. (1999) |
E6 PCR for HPV 16 | RP (snap frozen) | 47 | PCa cases | 10 (21%) | 10 | |||
| TVP (snap frozen) | 37 | BPH controls | 1 (3%) | 1 | |||||
|
Dodd et al. (1993) |
Reverse transcription PCR for E6/E7 mRNA of HPV16 | Collection not specified (fresh frozen) |
7 | PCa cases | 3 (43%) | 3 | |||
| Collection not specified (fresh frozen) |
10 | BPH controls | 5 (50%) | 5 | |||||
|
Salehi and Hadavi (2012) |
L1 PCR consensus primer | Collection not specified (snap frozen) |
68 | PCa cases | 3 (4%) | ||||
| Collection not specified (snap frozen) |
85 | BPH controls | 0 (0%) | ||||||
|
Abumsimir et al. (2022) |
L1 PCR consensus primers | Biopsies (fresh) | 50 | PCa cases | 8 (16%) | 8 | |||
|
Strickler et al. (1998) |
L1 PCR consensus primers E6 PCR for HPV 11, 16, 18, 51, 61 Southern blot hybridisation |
Surgery, TURP (fresh frozen) | 63 | PCa cases | 0 (0%) | ||||
| Surgery, TURP (fresh frozen) | 61 | BPH controls | 0 (0%) | ||||||
|
Glenn et al. (2017) |
L1 PCR consensus primer E7 PCR for HPV 16 and 18 IHC for E7 oncoprotein |
Collection not specified (FFPE) | 28 | PCa cases | L1 8 (29%) E7 19 (68%) E7P 8 (29%) |
||||
| 28 | BPH controls–years before | L1 13 (46%) E7 23 (82%) E7P 23 (82%) |
|||||||
|
Bergh et al. (2006) |
L1 PCR consensus primer | TURP (FFPE) | 201 | PCa cases | 0 (0%) | ||||
| TURP (FFPE) | 201 | BPH controls | 0 (0%) | ||||||
|
Groom et al. (2012) |
INNO-LiPA HPV Genotyping kit Hybridisation to HPV 16 |
Collection and storage not specified | 100 | PCa cases | 1 (1%) | 1 | |||
| Collection and storage not specified | 62 | Healthy controls | 0 (0%) | ||||||
|
Tu et al. (1994) |
L1 PCR consensus primer Hybridisation |
RP (FFPE) | 43 | PCa cases | 1 (2%) | 1 | |||
| Collection not specified (snap frozen) | 17 | Metastatic pelvic lymph nodes | 1 (6%) | 1 | |||||
| RRP (not specified) | 1 | Normal prostate | 0 (0%) | ||||||
| Effert et al. (1992) | E6 PCR for HPV 16 and 18 | RP (FFPE) | 30 | PCa cases | 0 (0%) | ||||
|
Gazzaz and Mosli (2009) |
Hybridisation using Hybrid Capture 2 kit | TURP, TRB (fresh) | 6 | PCa cases | 0 (0%) | ||||
| TURP, TRB (fresh) | 50 | 21 BPH, 29 nodular hyperplasia | 0 (0%) | ||||||
|
Anderson et al. (1997) |
L1 PCR consensus primer E6 and E2 ORF PCR for HPV 16 |
TURP (fresh frozen) | 14 | PCa cases | 0 (0%) | ||||
| TURP (fresh frozen) | 10 | Benign controls | 0 (0%) | ||||||
| Pereira et al., (2023) | L1 PCR consensus primer for HPV 16, 18, 56, 59 and 66. | TRUS Biopsies | 162 | PCa cases, | 10(6.2%) | ||||
| Nahand et al., (2020) | L1 and E7 PCR consensus primer and INNO-LiPA HPV Genotyping Kit | Surgery | 58 | PCa cases | 19(32.7%) | 9 | 6 | 33(3) | |
| Yow et al., (2014) | PapType High-Risk (HR) HPV Detection and Genotyping kit | TRUP (FFPE) RP (FFPE) |
221 | PCa cases | 0 (0%) | ||||
| Whitaker et al (2012) | L1 PCR consensus primer, and In Situ PCR | Collection not specified (FFPE; Fresh frozen) | 10 | PCa cases | 7 (70%) | 1 | |||
| Ahmed et al (2023) | HPV-HCR Genotype-Eph kit | Biopses (Fresh) | 49 | PCa cases, Benign controls | 16 (32.7%) | 4 | 33, 35, 45, 52, 56, 58 | ||
| Chang et al (2023) | Cobas 4800 HPV Test and DR HPV Genotyping IVD Kit | FFPE | 178 | PCa cases, Benign controls | 12 (6.7%) | 2 | 52 (1), 53(3), 62(1), others (5) | ||
Table 5.
Serology studies included in this review.
| Author | Year | Pathogen studied | PCa present | % Seropositive | No. of controls without PCa | % Seropositive | RR/OR | 95% CI | Evidence of association? | Method |
|---|---|---|---|---|---|---|---|---|---|---|
| Chen et al. | 2011 | HPV 16, 18,31,33, 52, 58 | 26/53 | 49 | 35/104 | 33.7 | OR = 0.71-4.16 | 0.18–48.0 | No | Fluorescent assay |
| Zhao et al. | 2017 | HPV 16 | 48/75 | 64 | 14/80 | 17.5 | N/A | N/A | Yes (p < 0.001) | Peptide microarray |
| Tachezy et al. | 2012 | HPV 16,18,31,33 | 14/51 | 27 | 172 | 21.5 | 1.44 | 0.69–2.97 | P =0.329 | ELISA |
| Dillner et al. | 1998 | HPV 16,18,33 | 31/165 | 18.8 | 35/290 | 12.1 | RR = 2.59, 2.38, 0.66 | 1.17–5.75 0.75–7.58 0.26–1.66 | Yes for HPV 16 and 18 (P< 0.001) | ELISA |
| Korodi et al. | 2005 | HPV 16,18,33 | 107/799 | 13.4 | 363/2596 | 14.0 | OR 0.94 | 0.74-1.19 | No | ELISA |
| Adami et al. | 2003 | HPV 16, 18, 33 | 129/238 | 54 | 105/210 | 50 | OR= 0.7, 0.9, 1.6 | 0.4-1.3, 0.5-1.9, 1.0-2.7 | Yes for HPV 33 | ELISA |
| Sutcliffe et al. | 2010 | HPV 16, 18,33 | 180/616 | 29.2 | 179/616 | 29 | OR 1.07, 0.87, 1.15 | 0.77-1.48 0.47-1.63 | No | ELISA |
| Rosenblatt et al. | 2003 | HPV 16, 18 | 81/642 | 12.6 | 64/570 | 11.3 | OR = 1.06, 1.36 | 0.71-1.57 0.69-2.69 | No | ELISA |
| Strickler et al. | 1998 | HPV 16 | 1/63 | 1.6 | 7/144 | 4.9 | N/A | N/A | No (p = 0.44) | ELISA |
| Hrbacek et al. | 2011 | HPV 16, 18, 31, 33 | 50/329 | 15.2 | 33/105 | 31 | OR = 0.48, 023, 073, 0.43 | 0.21-1.13 0.09-0.61 0.32-1.83 0.13-1.48 |
No | ELISA |
| Sutcliffe et al. | 2007 | HPV 16 | 144/ 691 | 20.8 | 145/691 | 21 | OR = 0.83,1.04,1.14 | 0.57-1.23 0.66-1.64 0.76-1.72 | No | ELISA |
| Sitas et al. | 2007 | HPV 16 | 59/205 | 28.78 | 173/673 | 25.71 | OR 1.33 | 0.86–2.07 | No | ELISA |
| Dennis et al | 2009 | HPV 16 and 18 | 50/267 | 19 | 45/267 | 17 | OR 1.33 | 0.73-1.75 | No | ELISA |
Table 6.
Included PCR studies with no significant differences in the presence of HPV and the risk of developing PCa.
Table 6.
Included PCR studies with no significant differences in the presence of HPV and the risk of developing PCa.
| S/N | Author | OR (95% Cl) | P value |
|---|---|---|---|
| 1 | Chen et al (2011) | P>0.05 | |
| 2 | Aghakhani et al., (2011) |
- | P>0.05 |
| 3 | Khatami et al., (2022) | 2.01 (0.8-5.68) | P= 0.102 |
| 4 | Atashafrooz et al., (2016) |
- | P= 0.413 |
| 5 | Noda et al., (1998) | - | P=0.19 |
| 6 | Wideroff et al., (1996) | 1.36(0.37, 4.98) | p>0.05 |
| 7 | Michopoulou et al., (2014) | 5.52(0.66-45.6) | P=0.086 |
| 8 | Rotola et al., (1992) | - | P>0.05 |
| 9 | McNicol and Dodd (1991) | P>0.05 | |
| 10 | Ibrahim et al., (1992) | - | P= 0.343 |
| 11 | Salehi and M. Hadavi (2012) | P=0.71 | |
| 12 | Nahand et al (2020) | P=0.078 | |
| 13 | Periera et al (2023) | P=0.487 | |
| 14 | Ahmed et al (2023) | P>0.05 | |
| 15 | Chang et al (2023) | P>0.005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



