
Submitted:
31 May 2023
Posted:
02 June 2023
You are already at the latest version
Abstract
Mycobacterium tuberculosis (Mtb) is the etiological agent of tuberculosis (TB), a disease that alt-hough preventable and curable, remains a global epidemic due to the emergence of resistance and a latent form responsible for a long period of treatment. Drug discovery in TB is a challenging task due to the heterogeneity of the disease, the emergence of resistance and an uncomplete knowledge of the pathophysiology of the disease. The limited permeability of the cell wall and the presence of multiple efflux pumps remain a major barrier to achieve effective intracellular drug accumulation. While the complete genome sequence of Mtb has been determined and several potential protein targets have been validated, the lack of adequate models for in vitro and in vivo studies is a limit-ing factor in TB drug discovery programs. In current therapeutic regimens, less than 0.5% of bac-terial proteins are targeted being the biosynthesis of the cell wall and the energetic metabolism two of the most important processes exploited for TB chemotherapeutics. This review provides an overview on the current challenges in TB drug discovery and emerging Mtb druggable proteins, and how chemical probes for protein profiling enabled the identification of new targets and bi-omarkers, paving the way to disruptive therapeutic regimens and diagnostic tools.
Keywords:
Mycobacterium tuberculosis
; target identification
; activity-based probes
; affinity-based probes
1. Introduction
Mycobacterium tuberculosis (Mtb), the etiological agent of tuberculosis (TB), is an obligate human pathogen spread by aerial transmission. One of the key mechanisms associated to Mtb virulence it is the ability to subvert the host immune response, and to effectively avoid complete elimination by the host immune system [1,2,3]. In addition, the bacilli can co-exist within the host in a latent, non-replicative form, metabolically and physiologically different from the replicative state. Patients infected with latent Mtb bacilli are asymptomatic, and in low TB prevalence settings, most of new active tuberculosis cases result from reactivation of these pathogen forms [2,3,4].
Although TB is curable and preventable, with a treatment success of around 85%, the disease remains a global epidemic, estimated to be the second leading caused by a single infectious agent in 2021, only after COVID-19. Additionally, recent World Health Organization (WHO) data showed that COVID-19 pandemic resulted into a rise on TB incidence, with a predicted observed maximum in 2022. Furthermore, around 25% of the world’s population is estimated to be infected, with 5 to 10% of those expected to develop active TB during their lifetime [5].
Most TB patients can be treated with the currently approved drug regimens with reasonable efficiency, and in recent years some novel drugs have been approved for the treatment of the clinically more challenging drug-resistant-TB (DR-TB), namely bedaquiline, delamanid and linezolid. Nevertheless, there are still issues in anti-TB therapy that are yet to be fulfilled, which must be urgently addressed in order to achieve TB control [5,6,7]. Most anti-TB drugs have limited efficiency against the latent bacilli, and therefore current therapeutic regimens require long durations to eliminate all forms of bacilli, usually leading to high adverse effects [8,9,10], poor compliance, and emergence of drug resistance, that consequently hampers TB control. Thus, there is an urgent, unmet need for the development of anti-TB drugs that target latent bacilli and resistant strains [11].
Lack of knowledge about the Mtb biology, still limits the development of new diagnosis techniques and conversion of new hit compounds into clinical candidates. Thus, there is a need to better understand the Mtb pathophysiology and find suitable biomarkers that allow the prediction of the response to treatment, relapse risk, assure cure and accelerate drug development [6,7]. Currently, a diverse array of strategies is used for the development of new anti-TB therapies. The most common include genetic approaches for the identification of new molecular targets, large-scale cell-based screening trials using Mtb, virtual screening, structural biology approaches, and optimizing existent drugs through molecular modifications. Combinations between approaches based on validated targets and cell-based screening trials have been gaining attention in recent years and seem a promising strategy to discover new active drugs [12,13,14].
After several decades without any novel anti-TB drugs being approved, major breakthroughs have been achieved in the last decade in the search for new therapeutic tools and regimens. Herein we review the current challenges in TB drug discovery, discuss the emerging molecular targets that can leverage the discovery of new drugs, and the development of chemical probes as a strategy to identify and validate novel targets in Mtb.
2. Challenges in TB drug discovery
TB drug discovery remains a challenging task due to the nature of its etiological agent, the heterogeneity of the disease, the emergence of resistance, and a lack of knowledge regarding the disease’s pathophysiology. One of the crucial requirements to achieve efficient treatment is the ability of a drug to enter into the target cell [15]. Compared to other bacteria, the Mtb cell wall is significantly less permeable to chemotherapeutical agents. The movement of small hydrophobic molecules occurs quickly through the mycobacterial cell wall, while the movement of hydrophilic molecules is mediated by water-filled channels [16]. Moreover, when the bacteria are found intracellularly, a second permeability barrier exists, which further reduces the movement of drugs into the bacilli [17]. Additionally, the Mtb cell envelope also include an array of efflux pumps, that have an essential role in the physiology, metabolism, and cell-signalling processes. These efflux pumps assist the expulsion of drugs from the mycobacteria and cause a natural high innate resistance to many anti-TB drugs [18].
Progression from latent infection to active TB constitute a major source of active disease in developed nations, and is becoming clear that tools to effectively address latency are needed to control the TB epidemy [4]. The Mtb latent state is characterized by a distinctive, reduced metabolism, where ribosomal functions and aerobic respiration decrease, and lipid metabolism increases; and by a thickened cell wall, with decreased permeability for hydrophilic molecules [19]. Thus, latent Mtb bacteria have an antibiotic tolerance, achieved by a combination of reduced antibiotic uptake and the lack of druggable targets resultant of the metabolic reconfiguration. Since anti-TB drugs specific and effective to latent bacteria are in short supply, current TB treatment is based on prolonged administration of the traditional anti-TB drugs. With the emergence of resistance and the pressure to shorten TB treatments, the ability to directly address latent subpopulations has become a priority in TB drug discovery, and the desire for agents capable of targeting all Mtb subpopulations has been emphasized [17]. Furthermore, the emergence of resistance has already led to the development of disease forms not treatable by any currently available therapeutical tool, such as total drug resistant-TB (TDR-TB), which remain programmatically incurable [7,20]. Consequently, there is a pressing need to develop drugs with activity in unexplored targets and pathways, that are not predisposed to resistance.
While historically effective, high-throughput screenings encounter several challenges in TB drug discovery. Despite they have general high hit rates, many compounds have undesirable physicochemical attributes, low selectivity, or mammalian cytotoxicity [17,21]. With the evolution of genomic tools, target-based screenings on validated drug targets presumed to be indispensable for the survival of Mtb and pathogenicity have gained some attention. Combination of ligand-based and structure-based chemogenomic approaches followed by biophysical and biochemical validation have also been used to identify targets for Mtb phenotypic hits [22]. However, this technique has several challenges as hits identified through target-based screenings may not translate to a whole cell system due to metabolic, permeability, and drug efflux issues. Since the publication of the entire Mtb genome sequence, and although several potential TB drug targets have been validated for use in target-based screening, no single clinically effective anti-TB agent has been discovered by this strategy [23,24].
Furthermore, the lack of predictive models for the heterogeneous bacterial subpopulations is a limiting factor in TB drug discovery. To reproduce the environmental conditions of Mtb subpopulations, several in vitro models have been developed, such as hypoxia [25,26], nutrient starvation [27], low pH [28], multi-stress [29,30], and biofilm models [31], all with some limitations. Also, while very useful in early drug discovery stages, in vitro models cannot reproduce all the host-pathogen interactions. Currently, the challenge of an adequate in vivo TB model remains, since existing animal TB models do not replicate important features of human disease [17,32]. An increased understanding of the microenvironments relevant to infection is difficult to achieve, but urgently required to identify and validate new pharmacological targets and suitable biomarkers, and consequently develop diagnostic techniques and improve therapy, through several mechanisms such as the development of new pharmacological agents, optimization of treatment durations, and triage of high-risk patients to preventive treatment [33,34].
3. Emerging Mtb drug targets
The success of TB drug discovery requires the identification of compounds targeting proteins that are essential for the growth and survival of Mtb. Ideally, these molecular targets should also display low probability to undergo mutations and to prevent or delay the emergence of drug resistance [35]. While the whole genome sequencing expanded our knowledge on the Mtb cellular machinery, less than 0.5% of bacterial proteins are targeted in current therapeutic regimens [36]. Due to the development and spread of resistance to current drugs and the high toxicity associated to therapeutic regimens used in drug resistant TB, there is an urgent need to discover new and safer drugs with novel mechanisms of action. The biosynthesis of the cell wall and the energetic metabolism of Mtb are critical cellular processes that are being exploited for TB chemotherapeutics.
3.1. Cell Wall
The cell wall of Mtb is the primary host-pathogen interaction spot, and a major determinant of the bacillus durability and robustness. The complex and dynamic structure of the cell wall (Figure 1) is essential for maintaining cellular integrity, enabling adaptation of the bacilli to the host conditions, and plays a crucial role in long-term infection and virulence. It comprises three essential substructures: a peptidoglycan (PG) inner layer, a mycolic acid (MA) outer layer, and an arabinogalactan polysaccharide (AGP) middle layer. The inhibition of key enzymes responsible for the biosynthesis of these substructures are excellent targets for novel drug development due to the absence of homologous characteristics in the host [37].
Peptidoglycan layer. The peptidoglycan is composed of N-acetylglucosamine, GlcNAc, and N-acetylmuramic acid, MurNAc, that are cross-linked with short peptides. The biosynthesis of peptidoglycan is a complex sequence of reactions, starting with the synthesis of Lipid II, in which a hydrophobic polyisoprene tail embedded in the membrane is connected to a monomer of cell-wall peptidoglycan through a pyrophosphate linker. This step is followed by translocation of Lipid II bound to the membrane formation, Lipid II polymerization and cross-linking by penicillin-binding proteins, PBPs, including L,D-transpeptidases [38]. Lipid II is targeted by the antibiotics ramoplanin and teixobactin, inhibiting the transglycosylation process and affecting peptidoglycan formation.
Figure 1.
Composition of the cell wall of Mycobacterium tuberculosis.
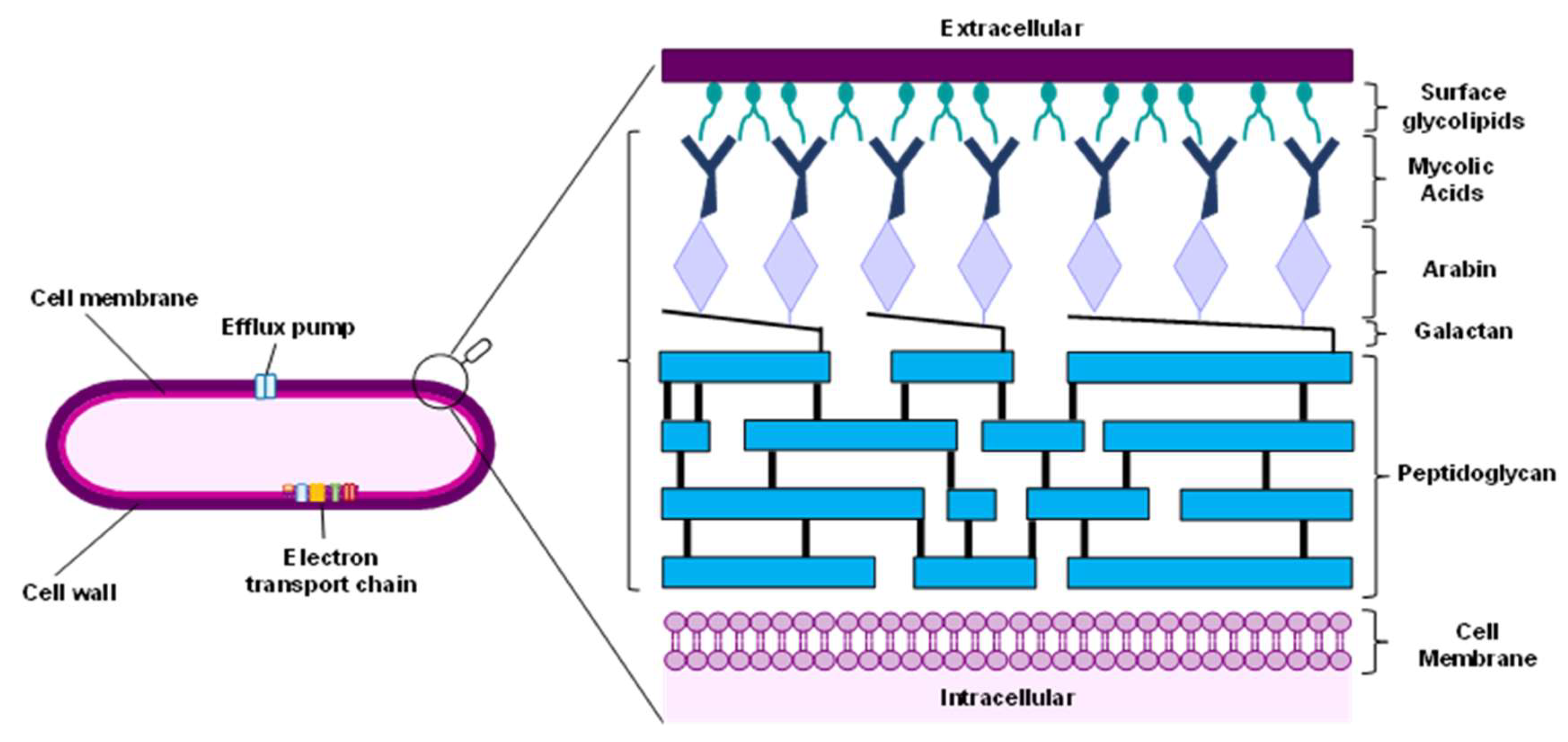
Mtb also produces β-lactamase, an enzyme that catalyses the hydrolysis of β-lactam antibiotics, a reason why the use of these antibiotics is not included in TB treatment. However, carbapenems (Figure 2A) are resistant to inactivation by β-lactamases, and thus are included in the treatment of multidrug-resistant TB, since they target the biosynthesis of peptidoglycan by inhibiting L,D-transpeptidases.
Mycolic acid layer. Mycolic acids are very long-chain (C60-90) α-alkyl β-hydroxy fatty acids that contribute to the hydrophobic, impermeable, and rigid structure of the outer membrane [39,40]. Mycolic acids are synthesised from acetyl-CoA, by at least two elongation systems, the type I and type II fatty acid synthases, FAS-I and FAS-II. The FAS-II system can only be found in bacteria, turning this system a potential selective antibacterial target. The NADH-dependent enzyme 2-trans-enoyl-acyl carrier protein reductase, InhA, is involved in the FAS-II system and is targeted by isoniazid (Figure 2B), a first-line agent used for treatment of TB. Other small molecules including ethionamide (Figure 2B), which is structurally related to isoniazid, and triclosan, have their mode of action related with InhA inhibition.
Subsequent cycles of fatty acid elongation are carried out by β-ketoacyl synthase KasA, which completes chain elongation via condensation of FAS-I-derived acyl-CoAs with malonyl-ACP (acyl carrier protein). KasA is the only essential member of three β-ketoacyl synthases encoded in the Mtb genome [41], and has been reported as a validated target for the treatment of TB [42]. A structure-based approach was used to optimize existing KasA inhibitor DG167 [43,44] to afford indazole JSF-3285 (Figure 2B) with a 30-fold increase in mouse plasma exposure. Biochemical, genetic, and X-ray studies further confirmed that JSF-3285 targets KasA.
Mycolic acids are transported to the outer membrane by bacterial membrane proteins called Mycobacterial membrane protein large (MmpL), which are part of the RND (Resistance, Nodulation and Cell Division) family. The primary role of RND proteins is to translocate a broad range of compounds across the plasma membrane to the periplasmic space, including virulence-associated envelope lipids and siderophores. Mtb genome encodes 13 MmpL proteins, of which MmpL3 has been reported in the biosynthesis of the mycobacterial outer membrane. The ethylenediamine derivative SQ109 (Figure 2B) is a MmpL3 inhibitor and has completed phase IIb-III clinical trials. SQ109 also accumulates in the lungs, the site of infection, increasing the drug efficacy [6]. Other promising MmpL3 inhibitors include indolocarboxamides and adamantylureas. As part of a drug scaffold repurposing program, the cannabinoid receptor modulator rimonabant (Figure 2B) and its diaryl pyrazole analogs, was reported to display potent anti-TB activity [45,46].
Figure 2.
Selected compounds that target the cell envelope of Mycobacterium tuberculosis. (A) carbapenems as inhibitors of L,D-transpeptidases; (B) inhibitors of the biosynthesis of mycolic acids; (C) inhibitors of the biosynthesis of arabinogalactan.
Figure 2.
Selected compounds that target the cell envelope of Mycobacterium tuberculosis. (A) carbapenems as inhibitors of L,D-transpeptidases; (B) inhibitors of the biosynthesis of mycolic acids; (C) inhibitors of the biosynthesis of arabinogalactan.
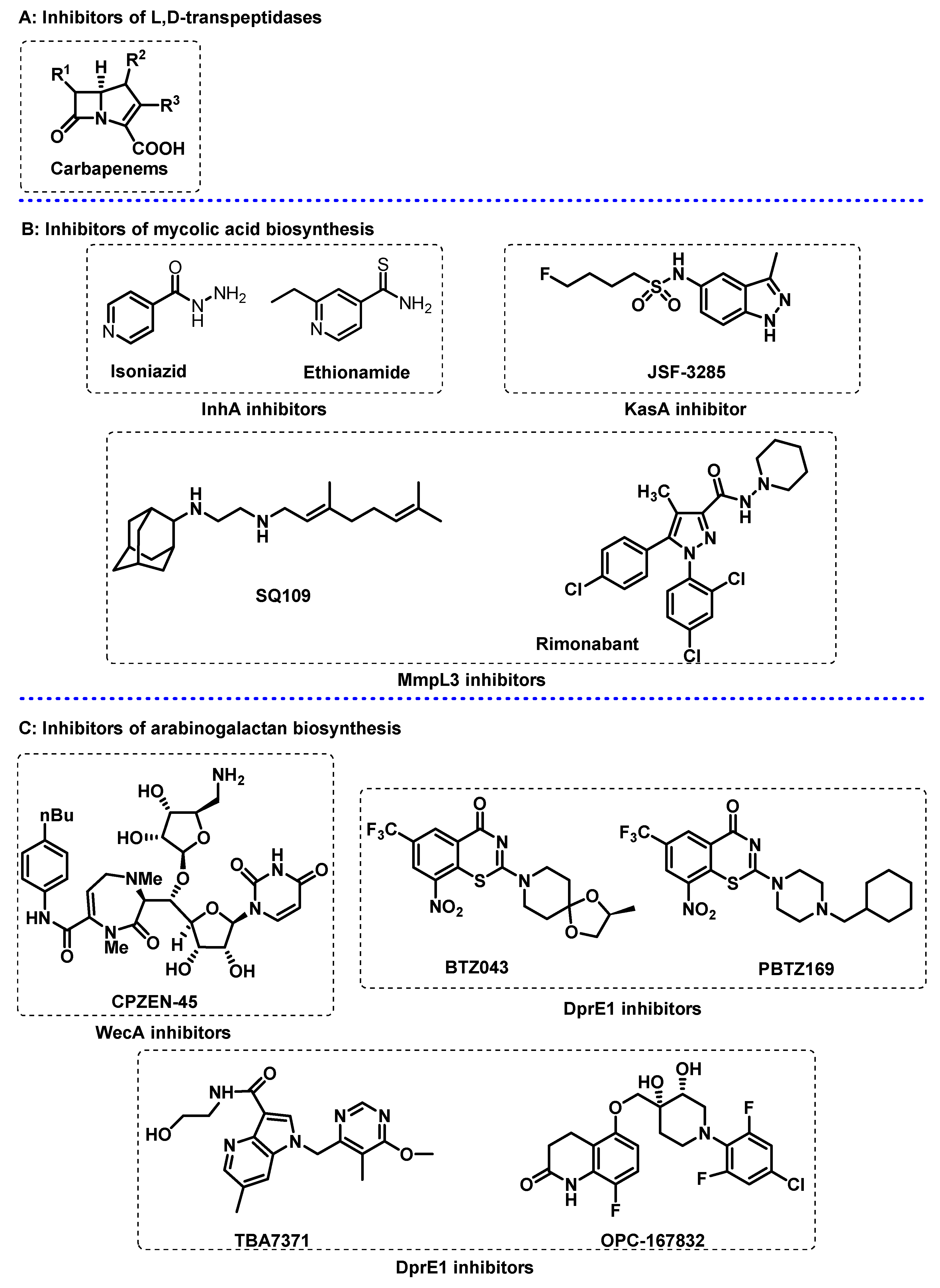
Arabinogalactan polysaccharide layer. The branched-chain arabinogalactan (AG) is the major cell wall polysaccharide, representing ca 35% of the cell wall, being composed by arabinose and galactose residues, both in the furanose configuration. This middle layer is covalently attached to peptidoglycan and mycolic acid layers requiring several enzymes that are potential targets for the design of novel inhibitors to block the formation of arabinogalactan polysaccharide [47,48,49,50]. An example are the enzymes arabinosyltransferases (EmbA, EmbB, and EmbC), which are a known targets for the drug ethambutol [51]. Another target to block the arabinogalactan polysaccharide formation is the enzyme arabinofuranosyltransferase (Aft), responsible for the polymerization of arabinofuranyl residues in decaprenylphosphoryl-D-arabinose (DPA), the lipid donor of D-arabinofuranosyl residues of AG. The DPA synthetic pathway is a potential drug target, and several arabinosyltransferases are essential in the growth of Mtb, as AftA, AftB, AftC and AftD. While AftA and AftB are responsible for the transference of the arabinofuranosyl residue, AftC and AftD introduce the α-1,3-branching in the segments of α-1,5-linked D-Araf residues [47,52].
Attachment of the arabinogalactan to the peptidoglycan structure is performed via an essential linker, the disaccharide L-rhamnose-D-N-acetylglucosamine. The enzyme N-acetylglucosamine-1-phosphate transferase, GlcNAc-1-P transferase (WecA) catalyses the first step of this linker biosynthesis. For this reason, WecA inhibitors such CPZEN-45 (Figure 2C), a caprazamycin derivative, prevents the growth of Mtb [53,54].
The enzymes decaprenylphosphoryl-β-D-ribose 2’-oxidase (DprE1) and decaprenylphosphoryl-D-2-keto erythropentose reductase (DprE2) are involved in the two-step epimerization of decaprenylphosphoryl-β-D-ribofuranose (DPR) into decaprenylphosphoryl-β-D-arabinofuranose [55]. Diverse chemical scaffolds as azaindoles, aminoquinolones, benzothiazinones, benzothiazoles, dinitrobenzamides, nitrobenzamides, pyrazolopyridines, quinoxalines, triazoles, and thiadiazoles demonstrated DprE1 inhibition. The benzothiazinone derivatives BTZ-043 and PBTZ169 (Figure 2C) are currently in phase II clinical trials and demonstrated high efficacy against M. tuberculosis. Additionally, the non-covalent inhibitors, azaindole TBA-7371 and OPC-167832, currently in phase II and phases I/II clinical trials, respectively, have shown promising results [56].
3.2. Energy Metabolism
Mtb operates its energetic metabolism in a modular and compartmentalized mode to support distinct and key cellular functions [2,19].
Electron Transport Chain. Mtb relies on oxidative phosphorylation (OxPhos) via the electron transport chain (ETC) to produce energy for growth and division. During the OxPhos process, electrons are transferred from electron donors produced in the central metabolic pathways to molecular oxygen through the ETC. The energy released in this process is conserved by proton-pumping transmembrane proteins that establish a proton gradient and thus generate an electrochemical gradient, called proton motive force (PMF). This bioenergetic pathway generates ATP from the phosphorylation of ADP [57,58].
The Mtb ETC is a highly conserved collection of membrane-bound and membrane-associated enzymes and co-factors. It is comprised by five main primary dehydrogenases, which fuel the ETC as electron donors; two main terminal oxidoreductases, which catalyse the transfer of electrons to terminal electron acceptor; and an ATPsynthase, that produces ATP through the dissipation of the PMF. A schematic representation of the Mtb ETC is presented in Figure 3 [59,60,61].
The respiratory flexibility of Mtb, that allows the bacilli to vary the ETC enzyme composition in response to environmental conditions, and the existence of human homologs to most ETC enzymes, had hampered the development of selective inhibitors. However, the discovery of bedaquiline, an ATPsynthase inhibitor, leads to an increase of research focused on targeting OxPhos. Currently, more than 30% of all new antimycobacterial drugs in clinical trials target the OxPhos, and more than 65% of Phase III trial regimens include a OxPhos inhibitor [59].
Cytochrome bc1-aa3. The Mtb Cyt bc1-aa3 supercomplex is comprised by two tightly associated protein complexes: a menaquinol-cytochrome c oxidoreductase, or cyt bc1, and an aa3 oxidase, or cyt aa3. This supercomplex acts as the primary terminal oxidase under normoxia and during exponential growth and its inhibition results in growth arrest. However, cyt bc1-aa3 is not essential for cell survival and as long as the alternate cyt bd is expressed, bc1-aa3 inhibitors do not induce bactericidal effects. The central role of the bc1-aa3 complex in the ETC and the significant differences to the mammalian counterpart make the supercomplex a good therapeutical target [58,59,60,61,62,63,64,65,66].
Imidazopyridine derivatives are examples of inhibitors that have shown to be particularly potent, the most prominent example being Q203, which is currently in phase II clinical trials and is capable of inhibiting multidrug resistant (MDR) and extensively drug resistant (XDR) Mtb strains [59,67,68,69]. Structurally similar to Q203, TB-47, is currently in pre-clinical studies and is active against drug sensitive (DS) and drug resistant (DR) Mtb strains, both active and latent bacilli [70,71]. Lansoprazole, a gastric proton-pump inhibitor, was found to be a potent hit compound in a screen of FDA-approved drugs. Lansoprazole acts as prodrug and is converted in vivo into lansoprazole sulphide, that was identified to be a cyt bc1-aa3 inhibitor on a distinct site from the one targeted by imidazopyridines (Figure 4A) [61,66,72].
Cytochrome bd. Cytochrome bd-type menaquinol (MKH2) oxidase, or cyt bd, is a non-proton pumping, less energetically efficient terminal oxidase that transfer electrons from MKH2 to molecular oxygen. Cyt bd is exclusive to prokaryotic ETC and unlike cyt bc1-aa3, the enzyme is more versatile, with multiple functions reported. The terminal oxidase is capable of detoxifying ROS and antibacterials, and protects the bacilli against hypoxia, and is capable of compensating the inactivation of cyt bc1-aa3. Cyt bd may also play a role into the Mtb’s natural drug tolerance, namely to drugs that directly target the ETC [59,60,73,74]. Thus, this cytochrome contributes to Mtb virulence, and since the enzyme is not encoded in animal genomes, it can serve as an attractive promising therapeutical target for new, selective anti-TB drugs [67,72,75].
Inhibition of cyt bd alone does not have any antimycobacterial effects. However, cyt bd inhibitors have synergetic effects with isoniazid, quicken the bactericidal activity of ATPsynthase inhibitors, and turn bc1-aa3 inhibitors bactericidal [59,60,61]. Thus, cyt bd inhibitors appear to be particularly attractive in combination therapy, namely in combination with cyt bc1-aa3 inhibitors, as the simultaneous inhibition of both terminal oxidases is highly bactericidal in a short period of time, and successful at killing both active and latent bacilli. The non-essentiality of cyt bd represents a challenge to identify its inhibitors, and thus not many cyt bd inhibitors are known. To this date, only few were identified and only one, aurachin D (Figure 4B), is characterized. Aurachins are isoprenoid quinoline alkaloids, originally extracted from myxobacteria. Further development of aurachin D is complicated by its toxicity and lack of selectivity, but optimized derivatives of aurachin D have great potential as anti-TB drugs [67,72,74,75,76].
Delamanid (DLM) and pretomanid (PTM) (Figure 4B) are two structurally related nitroimidazoles recently approved for the treatment of MDR-TB that were found to inhibit the biosynthesis of mycolic acid. However, the observance that these drugs were bactericidal against both active and latent bacilli suggested an alternative mechanism of action, as mycolic acid biosynthesis is downregulated in latency. Both DLM and PTM are pro-drugs that require activation by an F420 nitroreductase, an enzyme which produces des-nitro metabolites, with the release of NO. The putative additional mechanism of action is that the intracellular release of NO poisons the cytochrome oxidases, resulting in respiration arrest, and consequent cell death. DLM and PTM treatment results on a quick decrease in intracellular ATP levels, an increased menaquinol/menaquinone (MKH2/MK) ratio, and upregulation of cyt bd and nitrate reductase, which further support the terminal oxidases as a target of these nitroimidazoles [59,60,69].
Figure 4.
Selected compounds that target the energy metabolism of Mycobacterium tuberculosis. (A) inhibitors of cytochrome bc1-aa3; (B) inhibitors of cytochrome bd; (C) inhibitors of ATP synthase.
Figure 4.
Selected compounds that target the energy metabolism of Mycobacterium tuberculosis. (A) inhibitors of cytochrome bc1-aa3; (B) inhibitors of cytochrome bd; (C) inhibitors of ATP synthase.
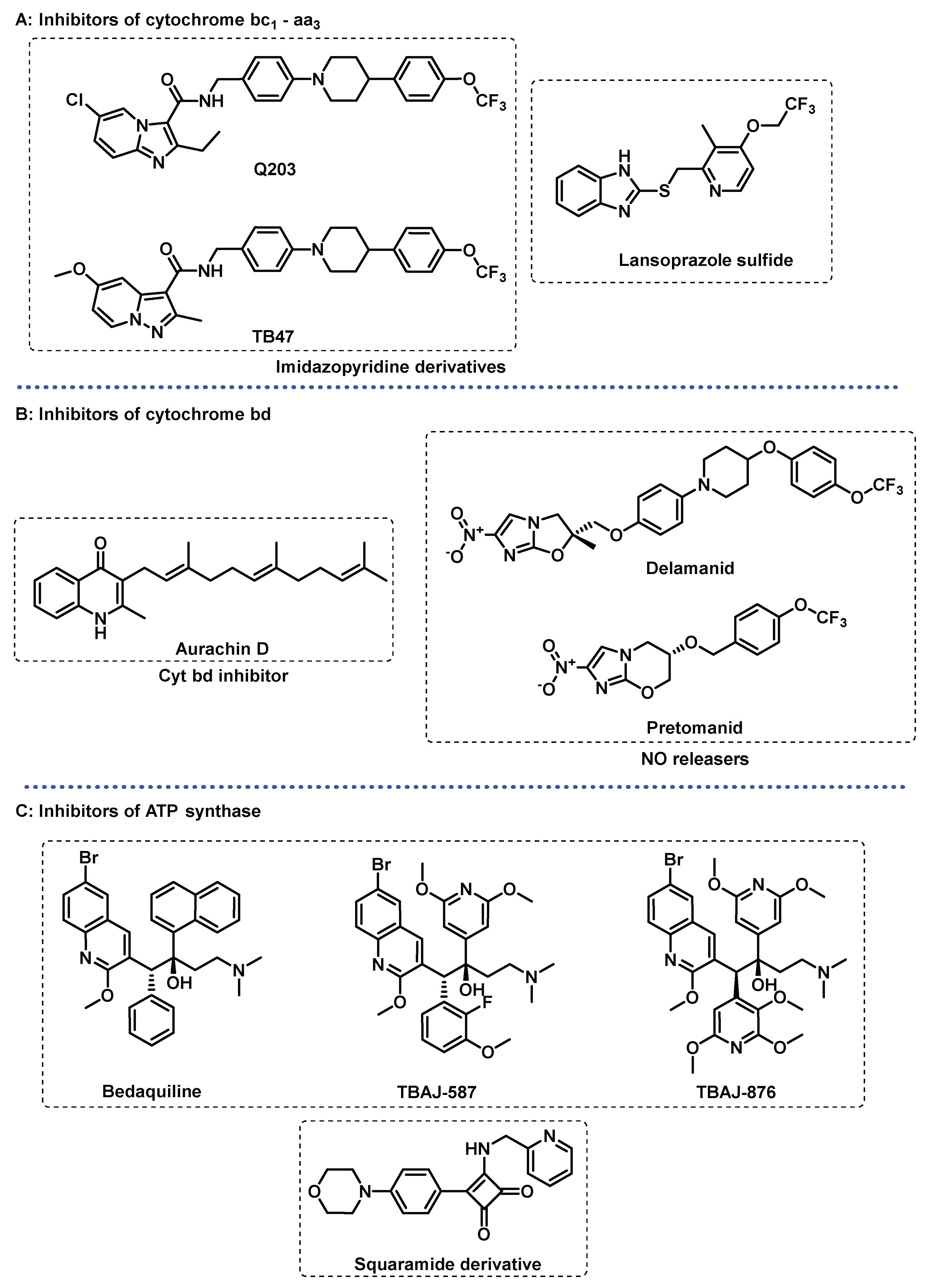
ATPsynthase. Bedaquiline (BDQ, Figure 4C), approved by the FDA in 2012, was the first approved drug specifically for TB in more than 40 years, is an inhibitor of the ATPsynthase. BDQ is a potent bactericidal, efficacious against MDR and latent bacilli and currently, is conditionally administrated for the MDR-TB treatment [59,68,77]. The activity is proposed to work through binding to two subunits on the ATPsynthase, thus inhibiting ATP synthesis and leading to a depletion of intracellular ATP levels. Additionally, bedaquiline is capable of acting as a protonophore leading to the uncoupling of the ETC via collapse of the PMF. Inhibition by BDQ depletes intracellular ATP levels, activates respiration, and induces a metabolic remodelling that upregulates ATPsynthase, NDH-2 and cyt bd. Interestingly, the bacterial activity of BDQ is delayed, i.e., does not occur immediately upon the ATP depletion, explained by the metabolic remodelling Mtb experiences upon BDQ exposure [57,59,67,77].
The toxicity associated with the drug and the emergence of bedaquiline-resistant Mtb strains restrain its use to MDR- and XDR-TB patients. Thus, to address its shortcomings, a medicinal chemistry approach was conducted to study the chemical space of diarylquinolines to find the next generation equivalents with superior safety profiles. In this context, two 3,5-diakoxy-4-pyridyl derivatives, TBAJ-587 and TBAJ-876 (Figure 4C), showed to be particularly interesting and are currently in Phase I clinical trials [58,61,68,77].
A number of recent studies have identified new ATPsynthase inhibitors with novel mechanisms of action. A family of squaramide derivatives showed to be particularly interesting, with its lead compound (Figure 4C) currently being evaluated on pre-clinical trials. These compounds target ATPsynthase through a different binding site, meaning they do not show cross-resistance to BDQ, and have shown to be active against BDQ-resistant strains [57,59,67].
Other targets on the ETC. The PMF consists of an electrical potential due to charge separation across the membrane and a chemical potential of protons. The generation and maintenance of a PMF is essential for the Mtb energy production and consequent bacterial growth and survival, in every metabolic state. PMF uncouplers generally act as protonophores and uncouple OxPhos from the ETC, thus inhibit ATP synthesis, leading to cell death [78]. Generally, this kind of compounds are not sufficiently selective to be used as antimycobacterial agents and thus, the development of specific PMF uncouplers remains an area of interest. However, there are some examples of anti-TB drugs in clinical use that act as PMF uncouplers in addition to an alternative mode of action, such as bedaquiline (Figure 4C), pyrazinamide (PZA), nitazoxanide (NTZ), (Figure 5) and SQ109 (Figure 2B) [57,78].
Although the mechanism of action of PZA is not still fully understood, current knowledge is that it acts as a multitarget drug that dissipates the PMF, inhibits ATP synthesis, inhibits membrane transport, and reduces the activity of other proteins, such as aspartate decarboxylase, a protein involved in the Coenzyme A biosynthetic pathway. Evidence of PZA’s uncoupling activity first arise with its ability to target latent bacilli. Additionally, PZA showed to synergize with other PMF uncouplers to deplete ATP depletion and enhance mycobacterial killing, implying its anti-TB activity relies substantially on its uncoupling activity [57,61].
Figure 5.
Proton motive force uncouplers currently in clinical settings.
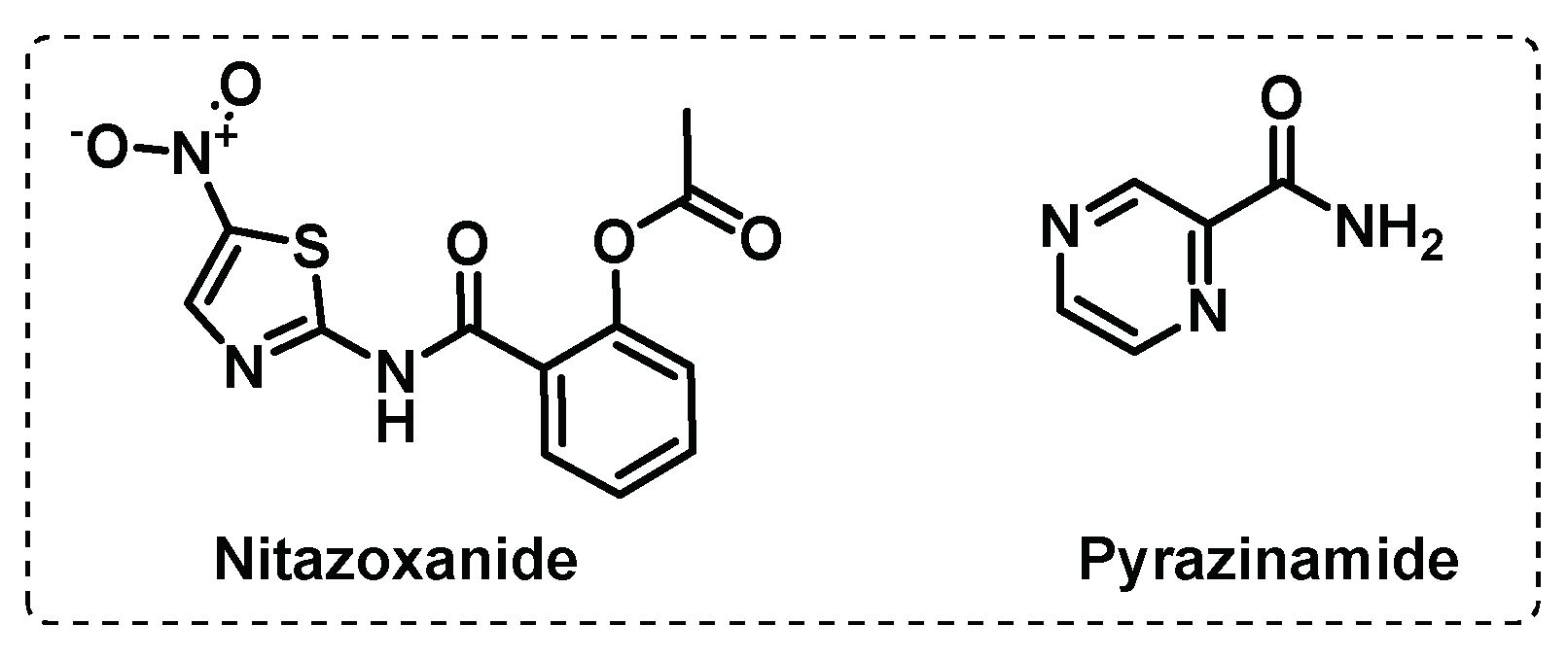
Initially, SQ109 was reported to interfere with the assembly of mycolic acids in the mycobacterial cell wall through inhibition of membrane transporter MmpL3, but recently, it was demonstrated that SQ109 interferes with respiration, through the ability to act as protonophore and dissipate the PMF [59,68].
Nitazoxanide is an FDA-approved repurposed drug with broad-spectrum antiparasitic and antiviral activity. NTZ is proposed to promote Mtb killing by enhancing autophagy through the inhibition of the human mTORC1, and additionally disrupt the PMF, through activity as a protonophore. NTZ inhibits potently both active and latent Mtb bacilli but has poor pharmacokinetic and pharmacodynamic proprieties. Thus, there is some interest in the development of NTZ derivatives with improved bioavailability [59,79,80].
3.3. Other Targets
3.3.1. Iron uptake
Iron is fundamental in Mtb survival and for this reason all the systems involved in iron uptake are promising drug targets [81]. When in unfavourable iron-deficient environment, Mtb increases the uptake of iron through the synthesis of high-affinity iron chelators called siderophores. Targeting the biosynthesis of mycobactin siderophores from mycobacteria, has been exploited as an approach to inhibit the growth of Mtb. The Mg2+-dependent salicylate synthase (MtbI) enzyme is a validated target since it is responsible for salicylate synthesis from chorismate, in the first step of the mycobactin biosynthesis pathway. Furthermore, MtbI offers the potential to enable the discovery of highly selective inhibitors, as it is absent in the host. Using a receptor-based virtual screening procedure, several furan-based compounds (Figure 6) were identified as potent MtbI inhibitors [82,83].
Another approach to block iron uptake in Mtb is to inhibit the iron-dependent transcription factor, IdeR, that controls siderophore synthesis. This regulator is a DNA-binding protein of the DtxR family that is responsible for activation or deactivation of storage proteins, according to excess or lack of iron, respectively. A screening against IdeR revealed benzothiazole benzene sulfonic (Figure 6) as a promising scaffold to develop IdeR inhibitors [6,84,85].
3.3.2. DNA related enzymes
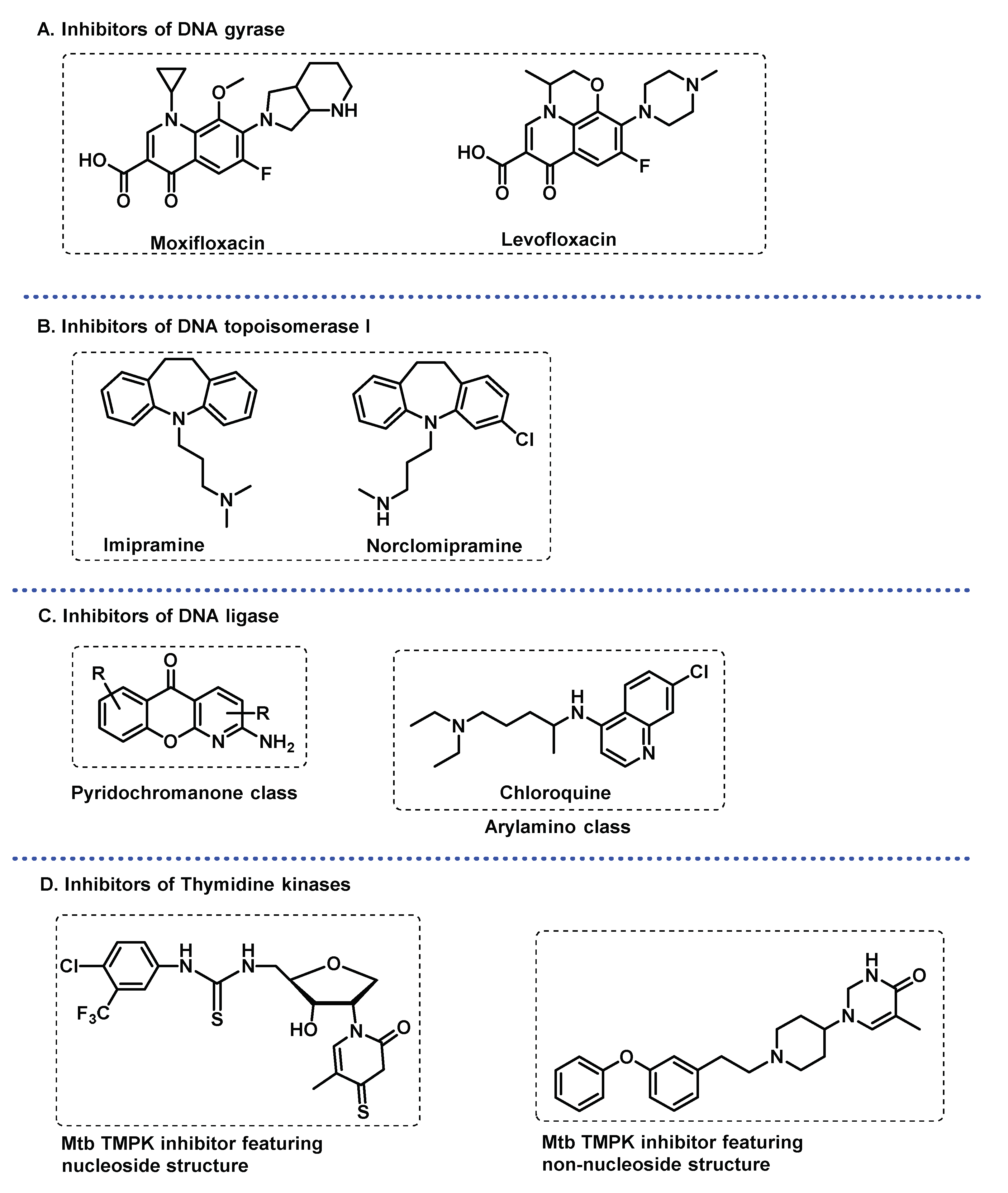
DNA gyrase. This enzyme is a validated target for anti-tubercular drug discovery. It is an ATP-dependent enzyme that is essential for efficient DNA replication, transcription, and recombination in bacteria [86]. Moreover, its absence in the mammalian organism makes this enzyme a suitable target for the development of antibacterial drugs with selective toxicity. Fluoroquinolones are effective inhibitors of this enzyme (Figure 7A). As with other antitubercular drugs, side-effects and emerging of bacterial resistance has fuelled intensive research for new chemical entities, from natural or synthetic origin, possessing DNA gyrase inhibiting properties that would be effective against MDR-TB, and it could also be effective against fluoroquinolone-resistant Mtb [87].
DNA Topoisomerase I. Imipramine and norclomipramine (Figure 7B) showed growth inhibition of both Mycobacterium smegmatis and Mtb cells. They target DNA topoisomerase I, an essential mycobacterial enzyme in the maintenance of topological homeostasis within the cell during a variety of DNA transaction processes such as replication, transcription, and chromosome segregation. It was suggested that they bind near the metal binding site of the enzyme, so targeting metal coordination in topoisomerases might be a general strategy to develop new lead molecules [88].
DNA ligases. Vital enzymes in replication and repair, DNA ligase catalyses the formation of a phosphodiester linkage between adjacent termini in double-stranded DNA through similar mechanisms. The DNA ligases utilize either ATP or NAD+ as cofactors. Those utilizing NAD+ are attractive drug targets because of the unique cofactor requirement for ligase activity and are exclusively found in eubacteria and some viruses. Gene knockout and other studies have shown NAD+-dependent DNA ligases to be indispensable in several bacteria including Mtb. Compounds belonging to arylamino and pyridochromanone classes (Figure 7C) have been identified as specific inhibitors of NAD+-dependent DNA ligases and can potentially be used to develop novel antibacterial therapies [89,90].
Thymidine kinase. Thymidine monophosphate kinase (TMPK) catalyses the γ-phosphate transfer from ATP to thymidine monophosphate (dTMP) in the presence of Mg2+, yielding thymidine diphosphate (dTDP) and ADP. Because TMPK is essential for thymidine triphosphate (dTTP) synthesis and in view of its low sequence identity (22%) with the human isozyme (TMPKh) and has a unique catalytic mechanism, it represents an attractive target for selectively inhibiting mycobacterial DNA synthesis [91]. Both industrial and academic efforts have afforded several potent Mtb TMPK inhibitors in the last two decades, including thymidine-like and non-nucleoside inhibitors (Figure 7D) [91].
Figure 7.
DNA related enzymes: (A) Second line anti-TB fluoroquinolones as DNA gyrase inhibitors; (B) DNA topoisomerase I inhibitors of Mtb; (C) Examples of inhibitors of Mtb DNA ligase; (D) Thymidine monophosphate kinase inhibitors.
Figure 7.
DNA related enzymes: (A) Second line anti-TB fluoroquinolones as DNA gyrase inhibitors; (B) DNA topoisomerase I inhibitors of Mtb; (C) Examples of inhibitors of Mtb DNA ligase; (D) Thymidine monophosphate kinase inhibitors.
4. Chemical Probes for Target Identification in Mycobacteria
The search for new biomarkers and potential drug targets in Mtb led to the development of chemical probes as tools for protein profiling proteomic methodologies. Activity-based protein profiling (ABPP) is a proteomic technique that enables quantification and functional analysis of enzymes using activity-based probes (ABP). Typically, ABPs react covalently with the active form of an enzyme or mechanistically related classes of enzymes. ABPs include (i) a reactive group (or warhead) that react with the catalytic amino acid residue of the enzyme, (ii) a reporter tag (e.g., biotin for protein pulldown, or a fluorophore for cell imaging), and (iii) a linker bridging the warhead and the tag.
In contrast to ABPP, profiling non-catalytic proteins rely on the use of photoaffinity-based probes (AfABP) that incorporate a photo-activable moiety to enable the covalent crosslinking between the probe and the target protein upon irradiation [92]. Concerning the reporter tag, a biorthogonal handle can be used instead of the biotin or the fluorophore. In this strategy, after the linkage of the probe to the target, a copper alkyne-azide cyclization is performed in the living cell or in cell lysates, to incorporate the tag for analysis (Figure 8).
4.1. Activity-based protein profiling (ABPP)
ABPP studies have been used to understand biological pathways in Mycobacterium tuberculosis, find new therapeutic targets, or identify new biomarkers.
4.1.1. Cytosolic serine hydrolases
Several serine hydrolases have been reported as putative target in bacterial infections [93,94], and, in case of Mtb, it represents 1,2% of all proteome [95]. Ortega et al. reported an extensive ABPP study in replicating and non-replicating Mtb in order to identify serine hydrolases that remain active in non-replicating persistent states. Using a pan-serine hydrolase fluorophosphonate probe, FP-ABP (Figure 9), 78 hydrolases of a total of 208 proteins were identified. The activity of these 78 hydrolases was analysed in normoxia and hypoxia and only 3 were active in hypoxic conditions, while 41 were active in aerated cultures and 34 were active in both conditions. Overall, these data provided experimental validation for previously annotated Mtb enzymes and identified 37 FP-labeled proteins that were found to be active in non-replicating Mtb that could be used as new drug targets for persistent Mtb. Specifically, mycobacterial acid resistance protease (MarP) activity was shown to remain unchanged between both phenotypes, suggesting a role in maintaining persistence. The ClpP2 subunit, included in the Mtb ClpP protease complex, was shown to be the only one detected in non-replicating Mtb [96,97].
Lentz et al., designed activity-based probes (ABPs) to selectively target Mtb “Hydrolase important to pathogenesis” (Hip 1). This is a cell-envelope-associated serine protease whose proteolytic activity is required for the immunomodulation of host inflammatory responses and has weak homology to other host proteases. From a library of serine-reactive electrophiles, a series of 7-amino-4-chloro-3-(2-bromoethoxy)isocoumarins were identified as potent time-dependent inhibitors of Hip1, and used to synthesise the fluorescence probe (Figure 10A). While this ABP displayed high potency but low selectivity, optimization of the isocoumarin scaffold led to the isocoumarin (Figure 10B), an inhibitor with nanomolar activity against Hip1 and improved selectivity [98].
Isocoumarins were also identified as inhibitors of Mtb growth in a screening of a library containing electrophiles that react with serine hydrolases. In particular, the 7-urea chloroisocoumarin JCP276, was active against Mtb and Mycobacterium kansasii, but against other non-tuberculous mycobacteria had no effect. A competitive gel-based ABPP assay with fluorophosphonate-tetramethylrhodamine (FP-TMR) and JCP276, showed that the hit compound was able to interact with several proteins. The proteomic ABPP study using the BMB034 probe allowed the identification of 7 major targets, mostly between lipases and esterases (Figure 11). However, the inhibition of these targets individually does not affect cell growth, which suggests that the potency of JCP276 may arise from the inhibition of multiple targets [99].
In another study, combining the usual tools of ABPP with a competitive approach, Li et al. screened a 1,2,3-triazole urea library of ca 200 molecules with the aim of identifying the serine hydrolase that are implicated in the Mtb growth. First, using a fluorophosphonate-biotin probe, FP-biotin (Figure 12), several serine hydrolase targets were revealed, and the selected targets were then tested for the two most active 1,2,3-triazole ureas. The results showed that the antimycobacterial activity displayed by these compounds is related with the inhibition of several key serine hydrolases essential in lipid metabolism and cell wall biosynthesis. The competitive ABPP study with FP-TMR showed the multiple target inhibition that led to disruption of the cell wall and lipid metabolism [100].
4.1.2. Membrane serine and cysteine hydrolases
ABPP also proved to be instrumental in revealing the key role of serine hydrolases in mycobacterial cell wall biosynthesis, leading to the identification of potential inhibitors of these enzymes. β-Lactam antibiotics interferes with the final phase of the biosynthesis of peptidoglycan inhibiting irreversibly D,D-transpeptidases serine hydrolases known as penicillin binding-proteins (PBP’s) and so preventing the formation of bonds between peptide chains of peptidoglycan (“cross-linking”). However, in Mtb the peptidoglycan layer contains different peptide crosslinks, which mostly requires catalysis by L,D-transpeptidases, which are cysteine hydrolases [101,102].
The presence of β-lactamase in TB bacilli, raises the question of whether this group of antibiotics can be used against Mtb. However, the combination of certain β-lactams, as carbapenem and meropenem, with a β-lactamase inhibitor as clavulanic acid revealed an antitubercular activity improvement, fact that reiterates the importance of β-lactams in TB treatment.
Quezada et al. screened a library of β-lactams against Mtb under replicating and non-replicating conditions and found two cephalosporins, exclusively active against non-replicating Mtb. To explore the possibility of an alternative, less known, pathway beyond the action of transpeptidases, chemical probes, were designed to perform ABPP studies (Figure 13) [103].
Focused on the L,D-transpeptidases relevance for the cell wall biosynthesis, Munnick et al. developed an assay based on the use of cysteine-selective fluorogenic probes for testing the reactivity with L,D-transpeptidases, which appears to be of special importance for Mtb virulence. In this assay two fluorogenic probes based on benzoxadiazole and fluorescein, were tested in the presence of competitive inhibitors for the L,D-transpeptidases, as well as several β-lactams antibiotics (Figure 14). This study revelead penems and carbapenems as potent inhibitors of L,D-transpeptidases [104].
Similarly to other bacterial infections, penicillin and cephalosporin β-lactam antibiotics fail in TB therapeutics due to inactivation by β-lactamases. However, based on the observation that carbapenems can reduce the activity of this enzyme, led Levine et al. to developed activity-based probes based in a carbapenem derivative meropenem, Mero-Cy5 (Figure 15) with the aim of finding the class of enzymes involved and the mechanism of action of meropenem. This probe inhibited L,D-transpeptidases, through binding to an active-site cysteine residue, but also binds to other transpeptidases, carboxypeptidases and to β-lactamase. The probe designed by Levine et al. proved to be a powerful tool for target identification and highlights the potential of carbapenem β-lactam antibiotics to treat TB [105].
Serine-hydrolases play a crucial role in catalysing essential transacylation reactions, namely in the binding of mycolates as β–keto or hydroxyesters. The similarity between the β-lactone pattern and the mycolates, enable the covalent acylation of catalytic serine residues by β-lactones. Based on that, Lehmann et al., developed β-lactones that could covalently inhibit these enzymes preventing the formation of the mycobacterium membrane. A β-lactone developed by this group exhibited good activity and selectivity for mycobacteria and they synthesised an alkyne probe, EZ120P, to identify the molecular targets (Figure 16). Through ABPP standard procedures, the serine proteases Pks13 and Ag85, essential proteins in the biosynthesis of the mycobacterial cell wall, were identified as possible targets [106].
Tetrahydrolipstatin (THL) is an inhibitor of fatty acid synthetase, an enzyme that plays an important role in latent tuberculosis since fatty acids are crucial for the survival of mycobacterium in this phase. This inhibitor contains a β-lactone group which forms covalent adducts with serine residues of target enzymes. Ravidran et al. synthesised a THL-ABP (Figure 17) and using click chemistry, the fluorescent tagged THL-proteins allowed the determination of their targets in mycobacteria. From 14 possible targets (α/β-hydrolases, including many lipid esterases), two of them were validated through several experimental techniques, the lipH and tesA, that are fundamental lipolytic enzymes in the dormant state of mycobacteria [107].
Tallman et al. developed probes to identify Mtb esterases in active and dormant conditions, leading to the discovery of several esterases involved in the different states of the Mtb. With a red-fluorescent TAMRA-FP probe, serine hydrolases were detected in replicating, dormant and reactivation conditions but the enzymatic activity is reduced in dormancy. However, using ABPP (desthiobiotin-fluorophosphonate) and fluorogenic (DCF-AME) probe-based profiling, was possible to identify esterases presents in dormant conditions (Culp1, LipH, LipM, LipN and Rv3036c) or in both states (CaeA, Rv0183, and Rv1683) (Figure 18) [108,109,110].
4.1.3. Other membrane targets
Acyltrehaloses biosynthesis. Acyltrehaloses are components of the outer membrane of Mtb and have been identified as antigens, with interest as diagnostic markers with the potential to distinguish between tuberculous and nontuberculous mycobacteria [111].
Polyacyltrehalose (PAT) is the predominant acyltrehalose in Mtb, and together with diacyltrehalose (DAT) have a structural function in the cell envelope and a role in the ability of Mtb to replicate and persist in the host, by allowing the Mtb intracellular survival and modulating host immune responses [112]. The enzymes involved in PAT biosynthesis are not identified yet. The PAT biosynthetic gene locus is identical to that of sulfolipid 1, a trehalose glycolipid structurally analogous to PAT, which is also unique to virulent Mtb. Chp1, a cutinase-like hydrolase protein, was already described as the terminal acyltransferase in sulfolipid 1 biosynthesis [113]. Chp2 may have a role in the biosynthesis of PAT, which is reinforced by the coordinate upregulation of chp2 gene, a homologue of chp1 (rv3822). However, the specific role of Chp2 is still not confirmed.
Touchette et al. hypothesized that Chp2 is the acyltransferase responsible for the transformation of DAT in PAT, once the PAT biosynthetic gene cluster includes chp2 (rv1184c). To confirm the enzymatic activity of Chp2, the authors have resorted to an activity-based probe FP-TMR, (Figure 11B), consisting in a fluorescent labelling reagent that specifically modifies the active-site residue of serine hydrolases. Thus, it was verified that Chp2 contains a C-terminal serine hydrolase domain that is inhibited by the lipase inhibitor tetrahydrolipstatin (THL). Results also showed that THL inhibits Chp2, leading to decreased levels of PAT and the accumulation of the DAT, suggesting that Chp2 is responsible for the synthesis of PAT from DAT, having an analogous role to the Chp1 in sulfolypid 1 byosynthesis [111].
Fatty acid biosynthesis. Knowing that fatty acids play an important role in mycobacteria, Ishikawa et al. developed probes with the aim of identify dehydratase (DH) enzymes in fatty acid synthases (FASs). These probes contain a specific reactive sulfonyl-3-alkyne warhead to prevent hydrolysis or non-enzymatic inactivation. The designed probes in Figure 19 were based in 3-decynoyl-N-acetylcysteamine [3-decynoyl-NAC] structure, a known inhibitor of dehydratase FabA, an important enzyme in fatty acid biosynthesis mechanism. The different experiments performed led to the following conclusions: (i) these fluorescent probes are selective inhibitors for dehydratase enzymes in FASs, (ii) the sulfonyl alkyne scaffold is required for stability, (iii) the probes exhibit antibiotic activity, and (iv) identification and selective isolation of DH-containing enzymes [114].
Sulfomucins degradation. Mucins, where sulfomucins are included, are a mucosal fluid with antimicrobial properties that plays a crucial role in protecting the host from the invasion of pathogens, forming a physical barrier with direct antimicrobial activity. Mucin degradation by bacteria is often regarded as an initial stage in pathogenesis since it would disturb the protection of the host mucosal surfaces [115,116]. In mycobacteria, sulfatases are responsible for sulfomucins degradation and thus play a role in the pathogenicity of mycobacteria and in the hydrolysis of the N-sulfate group, present in sulfated glycosaminoglycans and thereby modulating bacterial adhesion [117,118]. To detect the sulfatase activity in mycobacteria, Yoon et al., developed an activity-based probe, that forms a N-methyl isoindole compound, after intramolecular cyclization by the action of sulfatase enzyme, responsible for a coloured precipitate (Figure 20). It was verified in cultures of Mycobacterium avium and Mycobacterium smegmatis that the probe gave rise to a coloured precipitate after cleavage, indicating that this probe can be very useful in the detection of sulfatase activity in Mtb, and presenting the advantage of being detected in naked eye [119].
Mycobactins biosynthesis. Iron is fundamental for Mtb survival, and to compensate the lack of iron, the bacteria developed mycobactins, a type of iron chelating molecule, also called siderophores, responsible for shuttle free extracellular iron ions into the cytoplasm of mycobacterial cells.
Since the adenylating enzyme MbtA is crucial in the biosynthesis of mycobactins, Duckworth group developed the probe Sal-AMS ABP based on a potent selective inhibitor of MbtA (Sal-AMS) (Figure 21), with benzophenone as a photoreactive group and a small alkyne as a reporter group. The assays performed with this probe demonstrated an extraordinary specificity for MbtA in crude mixtures with other enzymes and the possibility of identifying adenylating enzymes in other organisms, as E. Coli [120].
4.1.4. ATP-binding enzymes
ATP-binding proteome. Wolfe et al. developed a different approach applying activity-based chemoproteomic to selectively profile the ATP-binding proteome in normally growing and hypoxic Mtb. The study was carried out in Mtb H37Rv strain and used a desthiobiotin-conjugated ATP as a molecular probe (Figure 22), where the desired enzymes are covalently modified with biotin, and after a pull down the target proteins can be identified through chemoproteomic. This chemoproteomic technique may be used to broaden the functional annotations and physiological roles of many nucleotide-binding proteins and supports the evidence regarding the potential of antimicrobial inhibitors whose mode of action relies on competition within the ATP-binding site of select proteins. With this approach using enriched subproteome of desthiobiotin-labeled ATP-binding proteins (ATPome), originated the total identification of 176 proteins, of which 122 were labelled via the molecular probe and more than half have been reported to be essential for in vitro survival [121].
4.2. Affinity-based probes (AfBP)
The biosynthesis of mycolic acid is carried out by two fatty acid synthases, FAS-I and FAS-II, where FAS-II is responsible for the synthesis of very long acyl chains. The mycobacterial FAS-II system works through the interaction between the acyl carrier protein (AcpM), which binds the growing acyl chain, and its respective enzymes, such as, ketosynthases (KasA/KasB), reductases (MabA), dehydratases (HadAB/HadBC) and enoyl reductase (InhA), that are responsible for further processing [122,123].
Thioacetazone (TAC) is a bacteriostatic anti-TB drug whose use was restricted due to severe side effects and the frequent emergence of resistance. TAC’s mechanism of action was not confirmed, but it was presumed that it interferes with the mycolic acid biosynthesis, based on the isolation of several truncated hydroxy fatty acids leading to the suggestion that dehydratases in the FAS-II system could be possible targets. Also, there is a hypothesis that monooxygenase EthA activates TAC that then binds to the dehydratase complex HadAB through a cysteine residue (Cys61) (Figure 23). Another potential target is the dehydratase enzyme HadC in the complex HadBC, but there is no evidence of that [124].
Singh et al. developed a TAC-affinity based probe (Figure 24) with an azidonaphthalimido butanoic acid as a fluorescent template to establish the target enzymes of the drug. The results showed a formation of cross-links with the HadAB complex in presence or not of the monooxygenase EthA, indicating that HadAB complex has affinity for TAC itself. Also, it was observed that is the HadA component in HadAB and the HadC component in the HadBC are the targets of TAC or its oxidized forms. The selectivity of this probe towards the dehydratase HadAB and HadBC was also verified since the other dehydratase present in Mtb did not demonstrate to be primary targets. Additionally, this probe has the advantage to promote cross-linking with the target protein under white light exposure, contrasting to UV-activated photo-affinity probes, which can lead to protein degradation [125].
Trehalose dimycolates (TDMs) are the most abundant glycolipids in Mtb’s cell wall and play an important role in Mtb’s pathogenesis confering protection against severe environmental conditions. TDMs also have immunomodulatory functions including prevention of phagosome-lysosome fusion, allowing the bacteria to survive inside the host macrophage. Additionally, TDMs have a crucial role in granuloma formation. However, despite the importance of TDM, only one receptor is known, the macrophage inducible C-type lectin (mincle) [126].
Khan et al. synthesised an affinity-based proteome profiling (AfBPP) TDM probe (Figure 25), formed by a benzophenone group as a photoreactive trap and by an alkyne tag. The reactive carbohydrate portion is unfunctionalized and has a small and hydrophobic trap and tag system, being a good mimic of the original TDM. The authors reported that this probe was then validated to be used for proteomic studies, since can activate macrophages and appears to be a suitable TDM mimic [127].
5. Conclusions
Tuberculosis remains a global epidemic threat due to the complexity of the disease and the emergence of drug resistance. New breakthroughs and insights into the development of safer and more efficient drugs requires a more comprehensive understanding of Mtb drug targets associated to TB pathophysiology. Many of the emerging targets with potential to positively impact anti-TB drug discovery are involved in cell wall synthesis, energy metabolism, iron uptake and DNA synthesis. The development of activity-based and photoaffinity-based, combined with the most recent developments in proteomic methodologies, provided the TB scientific community with powerful tools to identify novel molecular targets and shed some light on the understanding of biological pathways in Mtb. The full integration of classical phenotypic screening and genomic approaches with the proteomic-based protein profiling will be instrumental to identify effective targets to develop new safer and efficacious drug candidates, capable to address the current challenges in TB therapy.
Funding
This study was supported by Fundação para a Ciência e Tecnologia (FCT, Portugal) through projects UIDB/04138/2020 and UIDP/04138/2020, and fellowship 2020.05735.BD (M.C.) and SFRH/BD/137459/2018 (R. F.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Nature Reviews Microbiology 2018, 16, 202-213. [CrossRef]
- Mashabela Gabriel, T.; de Wet Timothy, J.; Warner Digby, F. Mycobacterium tuberculosis Metabolism. Microbiology Spectrum 2019, 7, 1-26. [CrossRef]
- Woodman, M.; Haeusler, I.L.; Grandjean, L. Tuberculosis Genetic Epidemiology: A Latin American Perspective. Genes 2019, 10. [CrossRef]
- Kiazyk, S.; Ball, T.B. Latent tuberculosis infection: An overview. Can Commun Dis Rep 2017, 43, 62-66. [CrossRef]
- Organization, W.H. Global tuberculosis report 2022; Geneva, 2022.
- Campaniço, A.; Moreira, R.; Lopes, F. Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. European Journal of Medicinal Chemistry 2018, 150, 525-545. [CrossRef]
- Tiberi, S.; du Plessis, N.; Walzl, G.; Vjecha, M.J.; Rao, M.; Ntoumi, F.; Mfinanga, S.; Kapata, N.; Mwaba, P.; McHugh, T.D.; et al. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. The Lancet Infectious Diseases 2018, 18, 183-198. [CrossRef]
- Reid, M.J.A.; Arinaminpathy, N.; Bloom, A.; Bloom, B.R.; Boehme, C.; Chaisson, R.; Chin, D.P.; Churchyard, G.; Cox, H.; Ditiu, L.; et al. Building a tuberculosis-free world: The Lancet Commission on tuberculosis. The Lancet 2019, 393, 1331-1384. [CrossRef]
- Nuermberger Eric, L. Preclinical Efficacy Testing of New Drug Candidates. Microbiology Spectrum 2017, 5, 1-22. [CrossRef]
- Şenol, G. Recent and New Strategies for Extensively Drug-resistant Tuberculosis. Mediterranean Journal of Infection, Microbes and Antimicrobials 2018, 7. [CrossRef]
- Gupta, V.K.; Kumar, M.M.; Singh, D.; Bisht, D.; Sharma, S. Drug targets in dormant Mycobacterium tuberculosis: can the conquest against tuberculosis become a reality? Infectious Diseases 2018, 50, 81-94. [CrossRef]
- Reddy, D.S.; Sinha, A.; Kumar, A.; Saini, V.K. Drug re-engineering and repurposing: A significant and rapid approach to tuberculosis drug discovery. Archiv der Pharmazie 2022, 355, 1-26. [CrossRef]
- Macalino, S.J.Y.; Billones, J.B.; Organo, V.G.; Carrillo, M.C.O. In Silico Strategies in Tuberculosis Drug Discovery. Molecules 2020, 25. [CrossRef]
- Huszár, S.; Chibale, K.; Singh, V. The quest for the holy grail: new antitubercular chemical entities, targets and strategies. Drug Discovery Today 2020, 25, 772-780. [CrossRef]
- Velayati, A.A.; Farnia, P.; Hoffner, S. Drug-resistant Mycobacterium tuberculosis: Epidemiology and role of morphological alterations. Journal of Global Antimicrobial Resistance 2018, 12, 192-196. [CrossRef]
- Sarathy, J.; Dartois, V.; Dick, T.; Gengenbacher, M. Reduced Drug Uptake in Phenotypically Resistant Nutrient-Starved Nonreplicating Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy 2013, 57, 1648-1653. [CrossRef]
- Kumar, A.; Chettiar, S.; Parish, T. Current challenges in drug discovery for tuberculosis. Expert Opinion on Drug Discovery 2017, 12, 1-4. [CrossRef]
- da Silva, P.E.A.; Machado, D.; Ramos, D.; Couto, I.; Von Groll, A.; Viveiros, M. Efflux Pumps in Mycobacteria: Antimicrobial Resistance, Physiological Functions, and Role in Pathogenicity. In Efflux-Mediated Antimicrobial Resistance in Bacteria: Mechanisms, Regulation and Clinical Implications, Li, X.-Z., Elkins, C.A., Zgurskaya, H.I., Eds.; Springer International Publishing: Cham, 2016; pp. 527-559.
- Eoh, H.; Wang, Z.; Layre, E.; Rath, P.; Morris, R.; Branch Moody, D.; Rhee, K.Y. Metabolic anticipation in Mycobacterium tuberculosis. Nature Microbiology 2017, 2, 1-7. [CrossRef]
- Sharma, S.; Sharma, D.; Kalia, N.P. Editorial: Approaches to Address Resistance, Drug Discovery, and Vaccine Development in Mycobacterium tuberculosis: Challenges and Opportunities. Frontiers in Microbiology 2022, 13, 1-3. [CrossRef]
- Tomasi, F.G.; Rubin, E.J. Failing upwards: Genetics-based strategies to improve antibiotic discovery and efficacy in Mycobacterium tuberculosis. Front Cell Infect Microbiol 2022, 12, 1-8. [CrossRef]
- Mugumbate, G.; Mendes, V.; Blaszczyk, M.; Sabbah, M.; Papadatos, G.; Lelievre, J.; Ballell, L.; Barros, D.; Abell, C.; Blundell, T.L.; et al. Target Identification of Mycobacterium tuberculosis Phenotypic Hits Using a Concerted Chemogenomic, Biophysical, and Structural Approach. Frontiers in Pharmacology 2017, 8, 1-13. [CrossRef]
- Lechartier, B.; Rybniker, J.; Zumla, A.; Cole, S.T. Tuberculosis drug discovery in the post-post-genomic era. EMBO Molecular Medicine 2014, 6, 158-168. [CrossRef]
- Borsari, C.; Ferrari, S.; Venturelli, A.; Costi, M.P. Target-based approaches for the discovery of new antimycobacterial drugs. Drug Discovery Today 2017, 22, 576-584. [CrossRef]
- Cho Sang, H.; Warit, S.; Wan, B.; Hwang Chang, H.; Pauli Guido, F.; Franzblau Scott, G. Low-Oxygen-Recovery Assay for High-Throughput Screening of Compounds against Nonreplicating Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy 2007, 51, 1380-1385. [CrossRef]
- Wayne, L.G.; Hayes, L.G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infection and Immunity 1996, 64, 2062-2069. [CrossRef]
- Zhang, M.; Sala, C.; Hartkoorn Ruben, C.; Dhar, N.; Mendoza-Losana, A.; Cole Stewart, T. Streptomycin-Starved Mycobacterium tuberculosis 18b, a Drug Discovery Tool for Latent Tuberculosis. Antimicrobial Agents and Chemotherapy 2012, 56, 5782-5789. [CrossRef]
- Darby, C.M.; Ingólfsson, H.I.; Jiang, X.; Shen, C.; Sun, M.; Zhao, N.; Burns, K.; Liu, G.; Ehrt, S.; Warren, J.D.; et al. Whole Cell Screen for Inhibitors of pH Homeostasis in Mycobacterium tuberculosis. PLOS ONE 2013, 8, 1-12. [CrossRef]
- Gold, B.; Warrier, T.; Nathan, C. A Multistress Model for High Throughput Screening (HTS) Against Nonreplicating Mycobacterium tuberculosis (M. tuberculosis). In Mycobacteria Protocols, Parish, T., Kumar, A., Eds.; Springer US: New York, NY, 2021; pp. 611-635.
- Aguilar-Ayala, D.A.; Cnockaert, M.; Vandamme, P.; Palomino, J.C.; Martin, A.; Gonzalez-Y-Merchand, J. Antimicrobial activity against Mycobacterium tuberculosis under in vitro lipid-rich dormancy conditions. Journal of Medical Microbiology 2018, 67, 282-285. [CrossRef]
- Wang, F.; Sambandan, D.; Halder, R.; Wang, J.; Batt, S.M.; Weinrick, B.; Ahmad, I.; Yang, P.; Zhang, Y.; Kim, J.; et al. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proceedings of the National Academy of Sciences 2013, 110, E2510-E2517. [CrossRef]
- Egorova, A.; Salina, E.G.; Makarov, V. Targeting Non-Replicating Mycobacterium tuberculosis and Latent Infection: Alternatives and Perspectives (Mini-Review). International Journal of Molecular Sciences 2021, 22, 1-15. [CrossRef]
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nature Reviews Disease Primers 2016, 2, 1-23. [CrossRef]
- Dartois, V.A.; Rubin, E.J. Anti-tuberculosis treatment strategies and drug development: challenges and priorities. Nature Reviews Microbiology 2022, 20, 685-701. [CrossRef]
- Perveen, S.; Sharma, R. Screening approaches and therapeutic targets: The two driving wheels of tuberculosis drug discovery. Biochemical Pharmacology 2022, 197, 114906. [CrossRef]
- Abrahams, K.A.; Besra, G.S. Mycobacterial drug discovery. RSC Medicinal Chemistry 2020, 11, 1354-1365. [CrossRef]
- Xu, X.; Dong, B.; Peng, L.; Gao, C.; He, Z.; Wang, C.; Zeng, J. Anti-tuberculosis drug development via targeting the cell envelope of Mycobacterium tuberculosis. Frontiers in Microbiology 2022, 13. [CrossRef]
- Shetye, G.S.; Franzblau, S.G.; Cho, S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Translational Research 2020, 220, 68-97. [CrossRef]
- Nataraj, V.; Varela, C.; Javid, A.; Singh, A.; Besra, G.S.; Bhatt, A. Mycolic acids: deciphering and targeting the Achilles’ heel of the tubercle bacillus. Molecular Microbiology 2015, 98, 7-16. [CrossRef]
- PaweŁczyk, J.; Kremer, L. The Molecular Genetics of Mycolic Acid Biosynthesis. Microbiology Spectrum 2014, 2, 1-20. [CrossRef]
- DeJesus Michael, A.; Gerrick Elias, R.; Xu, W.; Park Sae, W.; Long Jarukit, E.; Boutte Cara, C.; Rubin Eric, J.; Schnappinger, D.; Ehrt, S.; Fortune Sarah, M.; et al. Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. mBio 2017, 8, e02133-02116. [CrossRef]
- Inoyama, D.; Awasthi, D.; Capodagli, G.C.; Tsotetsi, K.; Sukheja, P.; Zimmerman, M.; Li, S.-G.; Jadhav, R.; Russo, R.; Wang, X.; et al. A Preclinical Candidate Targeting Mycobacterium tuberculosis KasA. Cell Chemical Biology 2020, 27, 560-570. [CrossRef]
- Abrahams, K.A.; Chung, C.-w.; Ghidelli-Disse, S.; Rullas, J.; Rebollo-López, M.J.; Gurcha, S.S.; Cox, J.A.G.; Mendoza, A.; Jiménez-Navarro, E.; Martínez-Martínez, M.S.; et al. Identification of KasA as the cellular target of an anti-tubercular scaffold. Nature Communications 2016, 7, 12581. [CrossRef]
- Kumar, P.; Capodagli Glenn, C.; Awasthi, D.; Shrestha, R.; Maharaja, K.; Sukheja, P.; Li, S.-G.; Inoyama, D.; Zimmerman, M.; Ho Liang Hsin, P.; et al. Synergistic Lethality of a Binary Inhibitor of Mycobacterium tuberculosis KasA. mBio 2018, 9, e02101-02117. [CrossRef]
- Ramesh, R.; Shingare, R.D.; Kumar, V.; Anand, A.; B, S.; Veeraraghavan, S.; Viswanadha, S.; Ummanni, R.; Gokhale, R.; Srinivasa Reddy, D. Repurposing of a drug scaffold: Identification of novel sila analogues of rimonabant as potent antitubercular agents. European Journal of Medicinal Chemistry 2016, 122, 723-730. [CrossRef]
- Kwofie, S.K.; Hanson, G.; Sasu, H.; Enninful, K.S.; Mensah, F.A.; Nortey, R.T.; Yeboah, O.P.; Agoni, C.; Wilson, M.D. Molecular Modelling and Atomistic Insights into the Binding Mechanism of MmpL3 Mtb. Chemistry & Biodiversity 2022, 19, e202200160. [CrossRef]
- Abrahams, K.A.; Besra, G.S. Mycobacterial cell wall biosynthesis: a multifaceted antibiotic target. Parasitology 2018, 145, 116-133. [CrossRef]
- Zhang, L.; Zhao, Y.; Gao, Y.; Wu, L.; Gao, R.; Zhang, Q.; Wang, Y.; Wu, C.; Wu, F.; Gurcha, S.S.; et al. Structures of cell wall arabinosyltransferases with the anti-tuberculosis drug ethambutol. Science 2020, 368, 1211-1219. [CrossRef]
- Kastrinsky, D.B.; McBride, N.S.; Backus, K.M.; LeBlanc, J.J.; Barry, C.E. 1.04–Mycolic Acid/Cyclopropane Fatty Acid/Fatty Acid Biosynthesis and Health Relations. In Comprehensive Natural Products II, Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, 2010; pp. 65-145.
- Zhang, L.; Zhao, Y.; Gao, R.; Li, J.; Yang, X.; Gao, Y.; Zhao, W.; Gurcha, S.S.; Veerapen, N.; Batt, S.M.; et al. Cryo-EM snapshots of mycobacterial arabinosyltransferase complex EmbB2-AcpM2. Protein & Cell 2020, 11, 505-517. [CrossRef]
- Goude, R.; Amin, A.G.; Chatterjee, D.; Parish, T. The Arabinosyltransferase EmbC Is Inhibited by Ethambutol in Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy 2009, 53, 4138-4146. [CrossRef]
- Meniche, X.; Sousa-d, d.; apos; Auria, C.; Van-der-Rest, B.; Bhamidi, S.; Huc, E.; Huang, H.; De Paepe, D.; Tropis, M.; et al. Partial redundancy in the synthesis of the d-arabinose incorporated in the cell wall arabinan of Corynebacterineae. Microbiology 2008, 154, 2315-2326. [CrossRef]
- Xu, L.; Qian, L.; Kang, J.; Sha, S.; Xin, Y.; Lu, S.; Ma, Y. Down-regulation of N-acetylglucosamine-1-phosphate transferase (WecA) enhanced the sensitivity of Mycobacterium smegmatis against rifampin. Journal of Applied Microbiology 2016, 121, 966-972. [CrossRef]
- Young, E.F.; Durham, P.G.; Perkowski, E.F.; Malik, S.; Hickey, A.J.; Braunstein, M. Efficacy of inhaled CPZEN-45 in treating tuberculosis in the guinea pig. Tuberculosis 2022, 135, 102207. [CrossRef]
- Sammartino, J.C.; Morici, M.; Stelitano, G.; Degiacomi, G.; Riccardi, G.; Chiarelli, L.R. Functional investigation of the antitubercular drug target Decaprenylphosphoryl-β-D-ribofuranose-2-epimerase DprE1/DprE2 complex. Biochemical and Biophysical Research Communications 2022, 607, 49-53. [CrossRef]
- Chhabra, S.; Kumar, S.; Parkesh, R. Chemical Space Exploration of DprE1 Inhibitors Using Chemoinformatics and Artificial Intelligence. ACS Omega 2021, 6, 14430-14441. [CrossRef]
- Iqbal, I.K.; Bajeli, S.; Akela, A.K.; Kumar, A. Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery. Pathogens 2018, 7, 1-30. [CrossRef]
- Wani, M.A.; Dhaked, D.K. Targeting the cytochrome bc1 complex for drug development in M. tuberculosis: review. Molecular Diversity 2022, 26, 2949-2965. [CrossRef]
- Hasenoehrl, E.J.; Wiggins, T.J.; Berney, M. Bioenergetic Inhibitors: Antibiotic Efficacy and Mechanisms of Action in Mycobacterium tuberculosis. Front Cell Infect Microbiol 2021, 10, 611683-611683. [CrossRef]
- Bajeli, S.; Baid, N.; Kaur, M.; Pawar, G.P.; Chaudhari, V.D.; Kumar, A. Terminal Respiratory Oxidases: A Targetables Vulnerability of Mycobacterial Bioenergetics? Front Cell Infect Microbiol 2020, 10, 1-22. [CrossRef]
- Foo, C.S.; Pethe, K.; Lupien, A. Oxidative Phosphorylation—an Update on a New, Essential Target Space for Drug Discovery in Mycobacterium tuberculosis. Applied Sciences 2020, 10. [CrossRef]
- Borisov, V.B.; Forte, E. Bioenergetics and Reactive Nitrogen Species in Bacteria. International Journal of Molecular Sciences 2022, 23. [CrossRef]
- Wiseman, B.; Nitharwal, R.G.; Fedotovskaya, O.; Schäfer, J.; Guo, H.; Kuang, Q.; Benlekbir, S.; Sjöstrand, D.; Ädelroth, P.; Rubinstein, J.L.; et al. Structure of a functional obligate complex III2IV2 respiratory supercomplex from Mycobacterium smegmatis. Nature Structural & Molecular Biology 2018, 25, 1128-1136. [CrossRef]
- Harrison Gregory, A.; Mayer Bridwell Anne, E.; Singh, M.; Jayaraman, K.; Weiss Leslie, A.; Kinsella Rachel, L.; Aneke Janessa, S.; Flentie, K.; Schene Miranda, E.; Gaggioli, M.; et al. Identification of 4-Amino-Thieno [2,3-d]Pyrimidines as QcrB Inhibitors in Mycobacterium tuberculosis. mSphere 2019, 4, e00606-00619. [CrossRef]
- Lupien, A.; Foo, C.S.-Y.; Savina, S.; Vocat, A.; Piton, J.; Monakhova, N.; Benjak, A.; Lamprecht, D.A.; Steyn, A.J.C.; Pethe, K.; et al. New 2-Ethylthio-4-methylaminoquinazoline derivatives inhibiting two subunits of cytochrome bc1 in Mycobacterium tuberculosis. PLOS Pathogens 2020, 16, e1008270. [CrossRef]
- Moraski, G.C.; Deboosère, N.; Marshall, K.L.; Weaver, H.A.; Vandeputte, A.; Hastings, C.; Woolhiser, L.; Lenaerts, A.J.; Brodin, P.; Miller, M.J. Intracellular and in vivo evaluation of imidazo [2,1-b]thiazole-5-carboxamide anti-tuberculosis compounds. PLOS ONE 2020, 15, e0227224. [CrossRef]
- Roy, K.K.; Wani, M.A. Emerging opportunities of exploiting mycobacterial electron transport chain pathway for drug-resistant tuberculosis drug discovery. Expert Opinion on Drug Discovery 2020, 15, 231-241. [CrossRef]
- Thompson, A.M.; Denny, W.A. Chapter Four–Inhibitors of enzymes in the electron transport chain of Mycobacterium tuberculosis. In Annual Reports in Medicinal Chemistry, Chibale, K., Ed.; Academic Press: 2019; Volume 52, pp. 97-130.
- Lee, B.S.; Kalia, N.P.; Jin, X.E.F.; Hasenoehrl, E.J.; Berney, M.; Pethe, K. Inhibitors of energy metabolism interfere with antibiotic-induced death in mycobacteria. Journal of Biological Chemistry 2019, 294, 1936-1943. [CrossRef]
- Yu, W.; Chiwala, G.; Gao, Y.; Liu, Z.; Sapkota, S.; Lu, Z.; Guo, L.; Khan, S.A.; Zhong, N.; Zhang, T. TB47 and clofazimine form a highly synergistic sterilizing block in a second-line regimen for tuberculosis in mice. Biomedicine & Pharmacotherapy 2020, 131, 110782. [CrossRef]
- Yu, W.; Yusuf, B.; Wang, S.; Tian, X.; Hameed, H.M.A.; Lu, Z.; Chiwala, G.; Alam Md, S.; Cook Gregory, M.; Maslov Dmitry, A.; et al. Sterilizing Effects of Novel Regimens Containing TB47, Clofazimine, and Linezolid in a Murine Model of Tuberculosis. Antimicrobial Agents and Chemotherapy 2021, 65, e00706-00721. [CrossRef]
- Cai, Y.; Jaecklein, E.; Mackenzie, J.S.; Papavinasasundaram, K.; Olive, A.J.; Chen, X.; Steyn, A.J.C.; Sassetti, C.M. Host immunity increases Mycobacterium tuberculosis reliance on cytochrome bd oxidase. PLOS Pathogens 2021, 17, e1008911. [CrossRef]
- Harikishore, A.; Chong, S.S.M.; Ragunathan, P.; Bates, R.W.; Grüber, G. Targeting the menaquinol binding loop of mycobacterial cytochrome bd oxidase. Molecular Diversity 2021, 25, 517-524. [CrossRef]
- Friedrich, T.; Wohlwend, D.; Borisov, V.B. Recent Advances in Structural Studies of Cytochrome bd and Its Potential Application as a Drug Target. International Journal of Molecular Sciences 2022, 23. [CrossRef]
- Foo Caroline, S.; Lupien, A.; Kienle, M.; Vocat, A.; Benjak, A.; Sommer, R.; Lamprecht Dirk, A.; Steyn Adrie, J.C.; Pethe, K.; Piton, J.; et al. Arylvinylpiperazine Amides, a New Class of Potent Inhibitors Targeting QcrB of Mycobacterium tuberculosis. mBio 2018, 9, e01276-01218. [CrossRef]
- Moosa, A.; Lamprecht Dirk, A.; Arora, K.; Barry Clifton, E.; Boshoff Helena, I.M.; Ioerger Thomas, R.; Steyn Adrie, J.C.; Mizrahi, V.; Warner Digby, F. Susceptibility of Mycobacterium tuberculosis Cytochrome bd Oxidase Mutants to Compounds Targeting the Terminal Respiratory Oxidase, Cytochrome c. Antimicrobial Agents and Chemotherapy 2017, 61, e01338-01317. [CrossRef]
- Sarathy, J.P.; Gruber, G.; Dick, T. Re-Understanding the Mechanisms of Action of the Anti-Mycobacterial Drug Bedaquiline. Antibiotics 2019, 8. [CrossRef]
- Gupta, S.; Fatima, Z.; Kumawat, S. Study of the bioenergetics to identify the novel pathways as a drug target against Mycobacterium tuberculosis using Petri net. Biosystems 2021, 209, 104509. [CrossRef]
- Jeon, A.B.; Ackart, D.F.; Li, W.; Jackson, M.; Melander, R.J.; Melander, C.; Abramovitch, R.B.; Chicco, A.J.; Basaraba, R.J.; Obregón-Henao, A. 2-aminoimidazoles collapse mycobacterial proton motive force and block the electron transport chain. Scientific Reports 2019, 9, 1513. [CrossRef]
- Odingo, J.; Bailey, M.A.; Files, M.; Early, J.V.; Alling, T.; Dennison, D.; Bowman, J.; Dalai, S.; Kumar, N.; Cramer, J.; et al. In Vitro Evaluation of Novel Nitazoxanide Derivatives against Mycobacterium tuberculosis. ACS Omega 2017, 2, 5873-5890. [CrossRef]
- Chao, A.; Sieminski, P.J.; Owens, C.P.; Goulding, C.W. Iron Acquisition in Mycobacterium tuberculosis. Chemical Reviews 2019, 119, 1193-1220. [CrossRef]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. European Journal of Medicinal Chemistry 2018, 155, 754-763. [CrossRef]
- Chiarelli, L.R.; Mori, M.; Beretta, G.; Gelain, A.; Pini, E.; Sammartino, J.C.; Stelitano, G.; Barlocco, D.; Costantino, L.; Lapillo, M.; et al. New insight into structure-activity of furan-based salicylate synthase (MbtI) inhibitors as potential antitubercular agents. Journal of Enzyme Inhibition and Medicinal Chemistry 2019, 34, 823-828. [CrossRef]
- Rohilla, A.; Khare, G.; Tyagi, A.K. Virtual Screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Scientific Reports 2017, 7, 4653. [CrossRef]
- Yang, L.; Hu, X.; Chai, X.; Ye, Q.; Pang, J.; Li, D.; Hou, T. Opportunities for overcoming tuberculosis: Emerging targets and their inhibitors. Drug Discovery Today 2022, 27, 326-336. [CrossRef]
- Khisimuzi, M.; Zhenkun, M. Mycobacterium tuberculosis DNA Gyrase as a Target for Drug Discovery. Infectious Disorders–Drug Targets 2007, 7, 159-168. [CrossRef]
- Khan, T.; Sankhe, K.; Suvarna, V.; Sherje, A.; Patel, K.; Dravyakar, B. DNA gyrase inhibitors: Progress and synthesis of potent compounds as antibacterial agents. Biomedicine & Pharmacotherapy 2018, 103, 923-938. [CrossRef]
- Godbole Adwait, A.; Ahmed, W.; Bhat Rajeshwari, S.; Bradley Erin, K.; Ekins, S.; Nagaraja, V. Targeting Mycobacterium tuberculosis Topoisomerase I by Small-Molecule Inhibitors. Antimicrobial Agents and Chemotherapy 2015, 59, 1549-1557. [CrossRef]
- Dwivedi, N.; Dube, D.; Pandey, J.; Singh, B.; Kukshal, V.; Ramachandran, R.; Tripathi, R.P. NAD+-Dependent DNA Ligase: A novel target waiting for the right inhibitor. Medicinal Research Reviews 2008, 28, 545-568. [CrossRef]
- Srivastava, S.K.; Tripathi, R.P.; Ramachandran, R. NAD+-dependent DNA Ligase (Rv3014c) from Mycobacterium tuberculosis: CRYSTAL STRUCTURE OF THE ADENYLATION DOMAIN AND IDENTIFICATION OF NOVEL INHIBITORS*. Journal of Biological Chemistry 2005, 280, 30273-30281. [CrossRef]
- Jian, Y.; Risseeuw, M.D.P.; Froeyen, M.; Song, L.; Cappoen, D.; Cos, P.; Munier-Lehmann, H.; van Calenbergh, S. 1-(Piperidin-3-yl)thymine amides as inhibitors of M. tuberculosis thymidylate kinase. Journal of Enzyme Inhibition and Medicinal Chemistry 2019, 34, 1730-1739. [CrossRef]
- Fang, H.; Peng, B.; Ong, S.Y.; Wu, Q.; Li, L.; Yao, S.Q. Recent advances in activity-based probes (ABPs) and affinity-based probes (AfBPs) for profiling of enzymes. Chemical Science 2021, 12, 8288-8310. [CrossRef]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365-370. [CrossRef]
- Vandal, O.H.; Pierini, L.M.; Schnappinger, D.; Nathan, C.F.; Ehrt, S. A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nature Medicine 2008, 14, 849-854. [CrossRef]
- Simon, G.M.; Cravatt, B.F. Activity-based Proteomics of Enzyme Superfamilies: Serine Hydrolases as a Case Study. Journal of Biological Chemistry 2010, 285, 11051-11055. [CrossRef]
- Ortega, C.; Anderson, Lindsey N.; Frando, A.; Sadler, Natalie C.; Brown, Robert W.; Smith, Richard D.; Wright, Aaron T.; Grundner, C. Systematic Survey of Serine Hydrolase Activity in Mycobacterium tuberculosis Defines Changes Associated with Persistence. Cell Chemical Biology 2016, 23, 290-298. [CrossRef]
- Bachovchin, D.A.; Ji, T.; Li, W.; Simon, G.M.; Blankman, J.L.; Adibekian, A.; Hoover, H.; Niessen, S.; Cravatt, B.F. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proceedings of the National Academy of Sciences 2010, 107, 20941-20946. [CrossRef]
- Lentz, C.S.; Ordonez, A.A.; Kasperkiewicz, P.; La Greca, F.; O’Donoghue, A.J.; Schulze, C.J.; Powers, J.C.; Craik, C.S.; Drag, M.; Jain, S.K.; et al. Design of Selective Substrates and Activity-Based Probes for Hydrolase Important for Pathogenesis 1 (HIP1) from Mycobacterium tuberculosis. ACS Infectious Diseases 2016, 2, 807-815. [CrossRef]
- Babin, B.M.; Keller, L.J.; Pinto, Y.; Li, V.L.; Eneim, A.S.; Vance, S.E.; Terrell, S.M.; Bhatt, A.S.; Long, J.Z.; Bogyo, M. Identification of covalent inhibitors that disrupt M. tuberculosis growth by targeting multiple serine hydrolases involved in lipid metabolism. Cell Chemical Biology 2022, 29, 897-909.e897. [CrossRef]
- Li, M.; Patel, H.V.; Cognetta, A.B., III; Smith, T.C., II; Mallick, I.; Cavalier, J.-F.; Previti, M.L.; Canaan, S.; Aldridge, B.B.; Cravatt, B.F.; et al. Identification of cell wall synthesis inhibitors active against Mycobacterium tuberculosis by competitive activity-based protein profiling. Cell Chemical Biology 2022, 29, 883-896.e885. [CrossRef]
- Gun, M.A.; Bozdogan, B.; Coban, A.Y. Tuberculosis and beta-lactam antibiotics. Future Microbiology 2020, 15, 937-944. [CrossRef]
- Turner, J.; Muraoka, A.; Bedenbaugh, M.; Childress, B.; Pernot, L.; Wiencek, M.; Peterson, Y.K. The Chemical Relationship Among Beta-Lactam Antibiotics and Potential Impacts on Reactivity and Decomposition. Frontiers in Microbiology 2022, 13. [CrossRef]
- Lopez Quezada, L.; Smith, R.; Lupoli, T.J.; Edoo, Z.; Li, X.; Gold, B.; Roberts, J.; Ling, Y.; Park, S.W.; Nguyen, Q.; et al. Activity-Based Protein Profiling Reveals That Cephalosporins Selectively Active on Non-replicating Mycobacterium tuberculosis Bind Multiple Protein Families and Spare Peptidoglycan Transpeptidases. Frontiers in Microbiology 2020, 11. [CrossRef]
- de Munnik, M.; Lohans, C.T.; Langley, G.W.; Bon, C.; Brem, J.; Schofield, C.J. A Fluorescence-Based Assay for Screening β-Lactams Targeting the Mycobacterium tuberculosis Transpeptidase LdtMt2. ChemBioChem 2020, 21, 368-372. [CrossRef]
- Levine, S.R.; Beatty, K.E. Investigating β-Lactam Drug Targets in Mycobacterium tuberculosis Using Chemical Probes. ACS Infectious Diseases 2021, 7, 461-470. [CrossRef]
- Lehmann, J.; Cheng, T.-Y.; Aggarwal, A.; Park, A.S.; Zeiler, E.; Raju, R.M.; Akopian, T.; Kandror, O.; Sacchettini, J.C.; Moody, D.B.; et al. An Antibacterial β-Lactone Kills Mycobacterium tuberculosis by Disrupting Mycolic Acid Biosynthesis. Angewandte Chemie International Edition 2018, 57, 348-353. [CrossRef]
- Ravindran, M.S.; Rao, S.P.S.; Cheng, X.; Shukla, A.; Cazenave-Gassiot, A.; Yao, S.Q.; Wenk, M.R. Targeting Lipid Esterases in Mycobacteria Grown Under Different Physiological Conditions Using Activity-based Profiling with Tetrahydrolipstatin (THL). Molecular & Cellular Proteomics 2014, 13, 435-448. [CrossRef]
- Tallman, K.R.; Beatty, K.E. Far-red fluorogenic probes for esterase and lipase detection. Chembiochem : a European journal of chemical biology 2015, 16, 70-75. [CrossRef]
- Tallman, K.R.; Levine, S.R.; Beatty, K.E. Small-Molecule Probes Reveal Esterases with Persistent Activity in Dormant and Reactivating Mycobacterium tuberculosis. ACS Infectious Diseases 2016, 2, 936-944. [CrossRef]
- Tallman, K.R.; Levine, S.R.; Beatty, K.E. Profiling Esterases in Mycobacterium tuberculosis Using Far-Red Fluorogenic Substrates. ACS Chemical Biology 2016, 11, 1810-1815. [CrossRef]
- Touchette Megan, H.; Holsclaw Cynthia, M.; Previti Mary, L.; Solomon Viven, C.; Leary Julie, A.; Bertozzi Carolyn, R.; Seeliger Jessica, C. The rv1184c Locus Encodes Chp2, an Acyltransferase in Mycobacterium tuberculosis Polyacyltrehalose Lipid Biosynthesis. Journal of Bacteriology 2015, 197, 201-210. [CrossRef]
- Belardinelli, J.M.; Larrouy-Maumus, G.; Jones, V.; Sorio de Carvalho, L.P.; McNeil, M.R.; Jackson, M. Biosynthesis and Translocation of Unsulfated Acyltrehaloses in Mycobacterium tuberculosis. Journal of Biological Chemistry 2014, 289, 27952-27965. [CrossRef]
- Seeliger, J.C.; Holsclaw, C.M.; Schelle, M.W.; Botyanszki, Z.; Gilmore, S.A.; Tully, S.E.; Niederweis, M.; Cravatt, B.F.; Leary, J.A.; Bertozzi, C.R. Elucidation and Chemical Modulation of Sulfolipid-1 Biosynthesis in Mycobacterium tuberculosis*. Journal of Biological Chemistry 2012, 287, 7990-8000. [CrossRef]
- Ishikawa, F.; Tanabe, G.; Kakeya, H. Activity-Based Protein Profiling of Non-ribosomal Peptide Synthetases. In Activity-Based Protein Profiling, Cravatt, B.F., Hsu, K.-L., Weerapana, E., Eds.; Springer International Publishing: Cham, 2019; pp. 321-349.
- Li, W.; Deng, G.; Li, M.; Liu, X.; Wang, Y. Roles of Mucosal Immunity against Mycobacterium tuberculosis Infection. Tuberculosis Research and Treatment 2012, 2012, 791728. [CrossRef]
- Derrien, M.; van Passel, M.W.J.; van de Bovenkamp, J.H.B.; Schipper, R.; de Vos, W.; Dekker, J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 2010, 1, 254-268. [CrossRef]
- Mougous, J.D.; Leavell, M.D.; Senaratne, R.H.; Leigh, C.D.; Williams, S.J.; Riley, L.W.; Leary, J.A.; Bertozzi, C.R. Discovery of sulfated metabolites in mycobacteria with a genetic and mass spectrometric approach. Proceedings of the National Academy of Sciences 2002, 99, 17037-17042. [CrossRef]
- Mougous, J.D.; Lee, D.H.; Hubbard, S.C.; Schelle, M.W.; Vocadlo, D.J.; Berger, J.M.; Bertozzi, C.R. Molecular Basis for G Protein Control of the Prokaryotic ATP Sulfurylase. Molecular Cell 2006, 21, 109-122. [CrossRef]
- Yoon, H.Y.; Kim, H.J.; Jang, S.; Hong, J.-I. Detection of bacterial sulfatase activity through liquid- and solid-phase colony-based assays. AMB Express 2017, 7, 150. [CrossRef]
- Duckworth, B.P.; Wilson, D.J.; Nelson, K.M.; Boshoff, H.I.; Barry, C.E., III; Aldrich, C.C. Development of a Selective Activity-Based Probe for Adenylating Enzymes: Profiling MbtA Involved in Siderophore Biosynthesis from Mycobacterium tuberculosis. ACS Chemical Biology 2012, 7, 1653-1658. [CrossRef]
- Wolfe, L.M.; Veeraraghavan, U.; Idicula-Thomas, S.; Schürer, S.; Wennerberg, K.; Reynolds, R.; Besra, G.S.; Dobos, K.M. A chemical proteomics approach to profiling the ATP-binding proteome of Mycobacterium tuberculosis. Mol Cell Proteomics 2013, 12, 1644-1660. [CrossRef]
- Batt, S.M.; Minnikin, D.E.; Besra, G.S. The thick waxy coat of mycobacteria, a protective layer against antibiotics and the host’s immune system. Biochemical Journal 2020, 477, 1983-2006. [CrossRef]
- Vilchèze, C. Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs. Applied Sciences 2020, 10. [CrossRef]
- Grzegorzewicz, A.E.; Eynard, N.; Quémard, A.; North, E.J.; Margolis, A.; Lindenberger, J.J.; Jones, V.; Korduláková, J.; Brennan, P.J.; Lee, R.E.; et al. Covalent Modification of the Mycobacterium tuberculosis FAS-II Dehydratase by Isoxyl and Thiacetazone. ACS Infectious Diseases 2015, 1, 91-97. [CrossRef]
- Singh, B.K.; Singha, M.; Basak, S.; Biswas, R.; Das, A.K.; Basak, A. Fluorescently labelled thioacetazone for detecting the interaction with Mycobacterium dehydratases HadAB and HadBC. Organic & Biomolecular Chemistry 2022, 20, 1444-1452. [CrossRef]
- Rahlwes, K.C.; Dias, B.R.S.; Campos, P.C.; Alvarez-Arguedas, S.; Shiloh, M.U. Pathogenicity and virulence of Mycobacterium tuberculosis. Virulence 2023, 14, 2150449. [CrossRef]
- Khan, A.A.; Kamena, F.; Timmer, M.S.M.; Stocker, B.L. Development of a benzophenone and alkyne functionalised trehalose probe to study trehalose dimycolate binding proteins. Organic & Biomolecular Chemistry 2013, 11, 881-885. [CrossRef]
Figure 3.
Schematic representation of the electron transport chain of Mycobacterium tuberculosis.
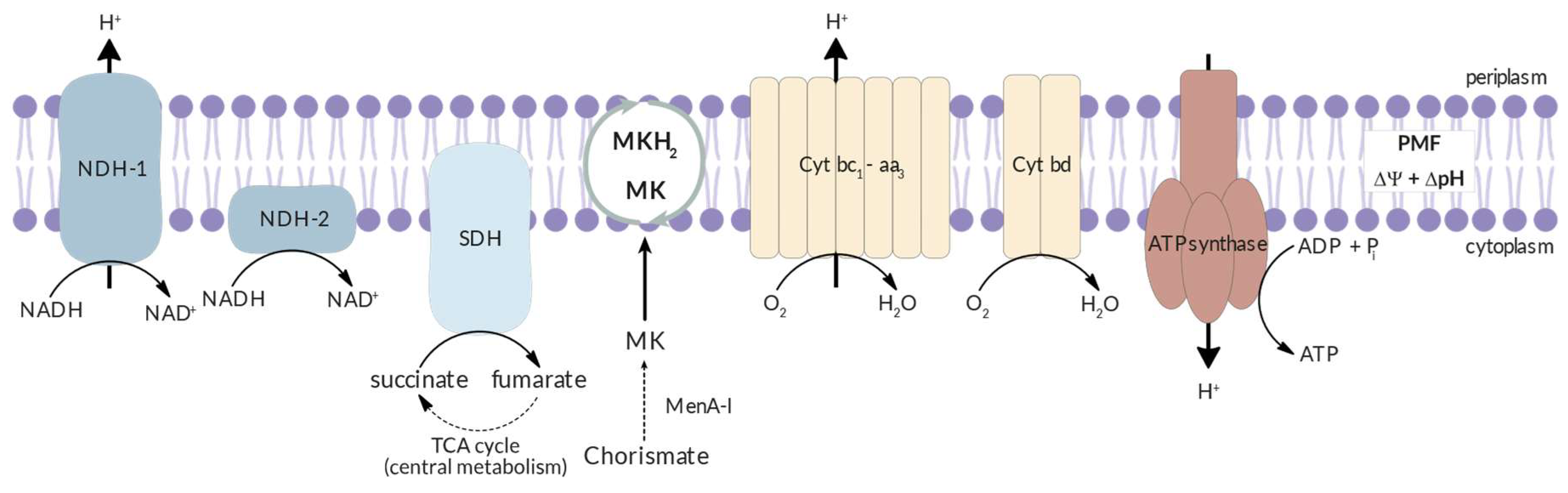
Figure 6.
Example of iron uptake inhibitors.
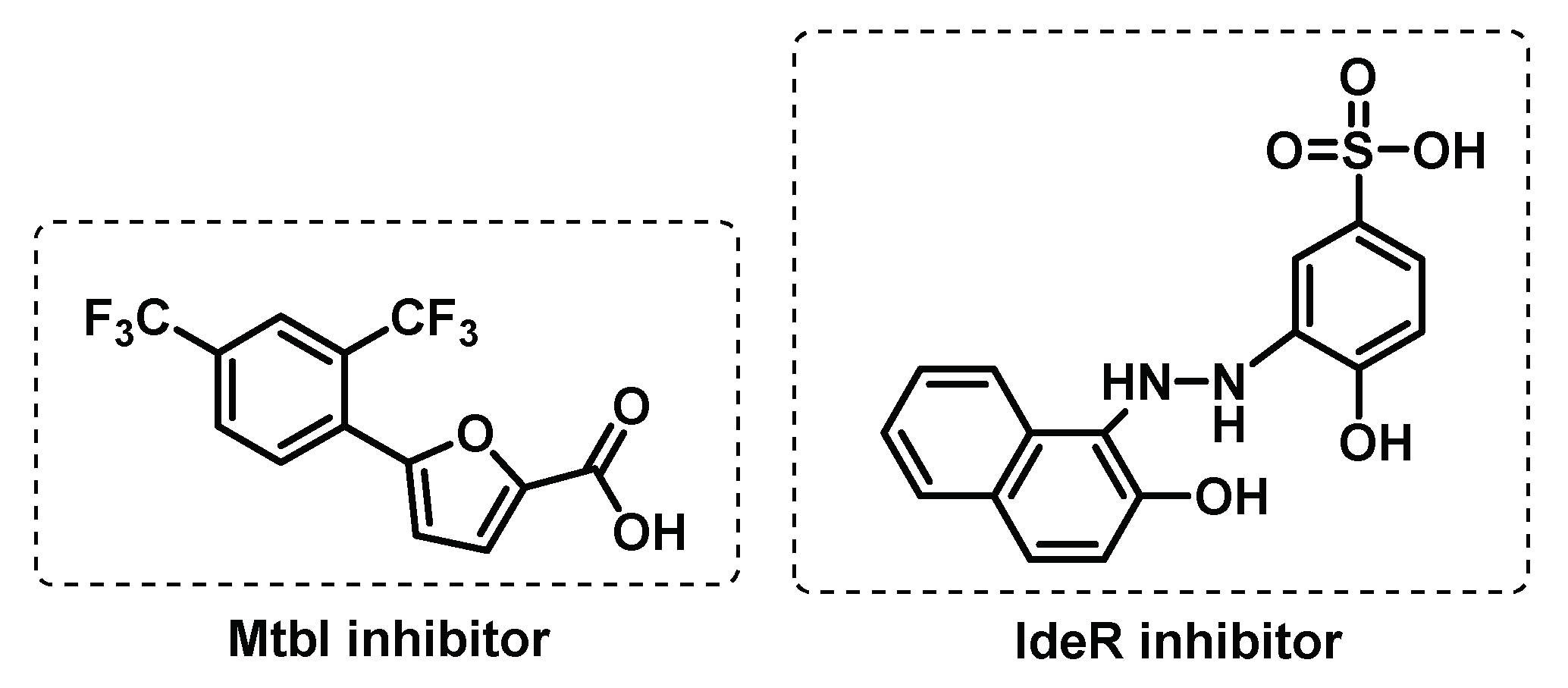
Figure 8.
(A) Structure of Activity-based probes and Affinity-based probes. Resume of common structures used as reporters, bioorthogonal handles and photo-crosslinkers. (B) Workflow for protein profiling using activity-based probes and affinity-based probes.
Figure 8.
(A) Structure of Activity-based probes and Affinity-based probes. Resume of common structures used as reporters, bioorthogonal handles and photo-crosslinkers. (B) Workflow for protein profiling using activity-based probes and affinity-based probes.
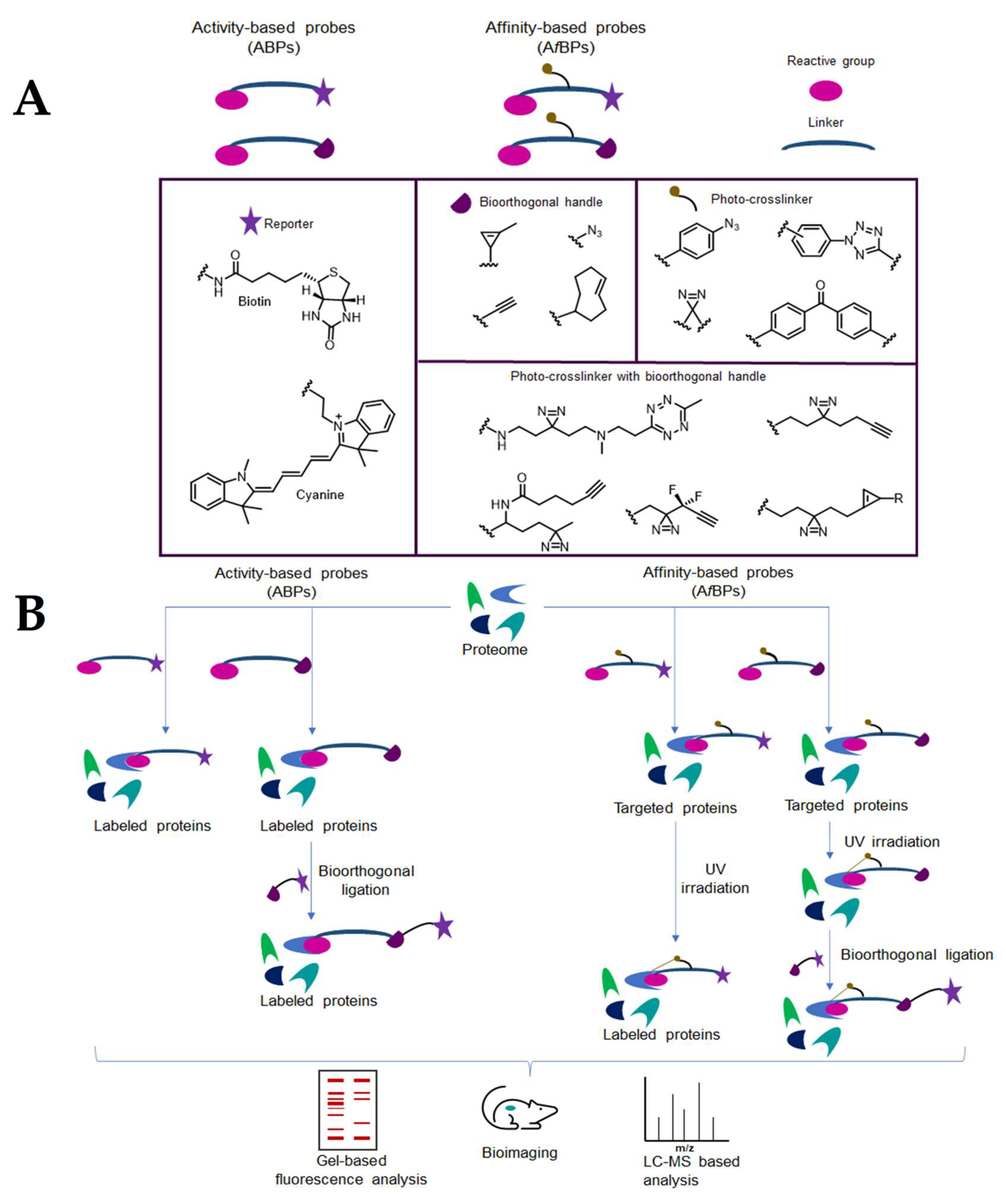
Figure 9.
Fluorophosphonate (FP) Activity-Based Probe.
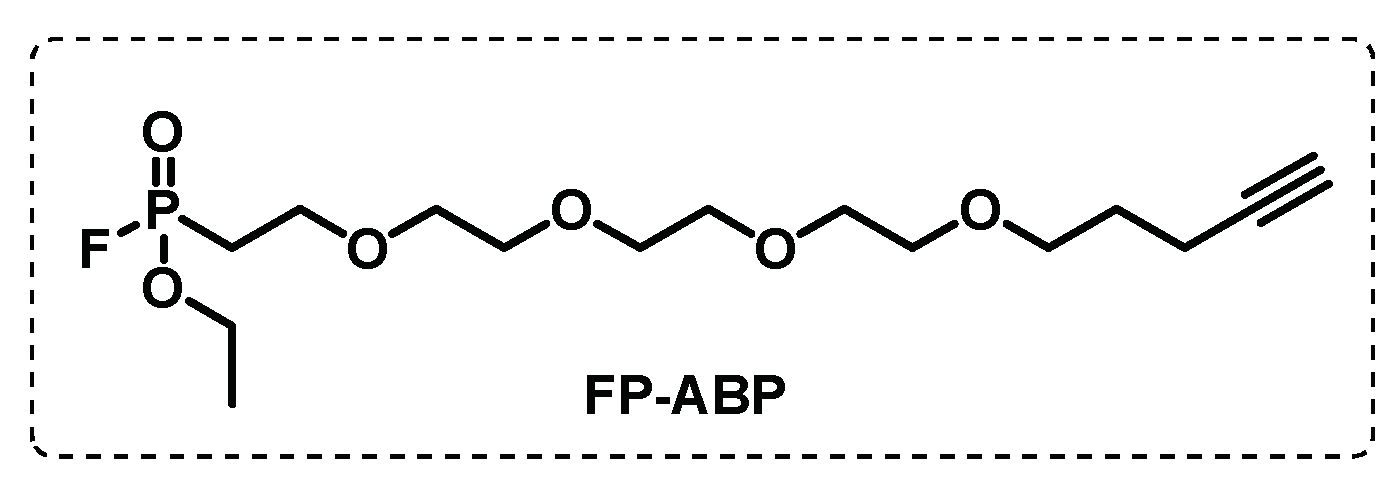
Figure 10.
(A) Isocoumarin based probe synthesised based on the inhibitor in purple, (B) Potent inhibitor design with nanomolar activity.
Figure 10.
(A) Isocoumarin based probe synthesised based on the inhibitor in purple, (B) Potent inhibitor design with nanomolar activity.
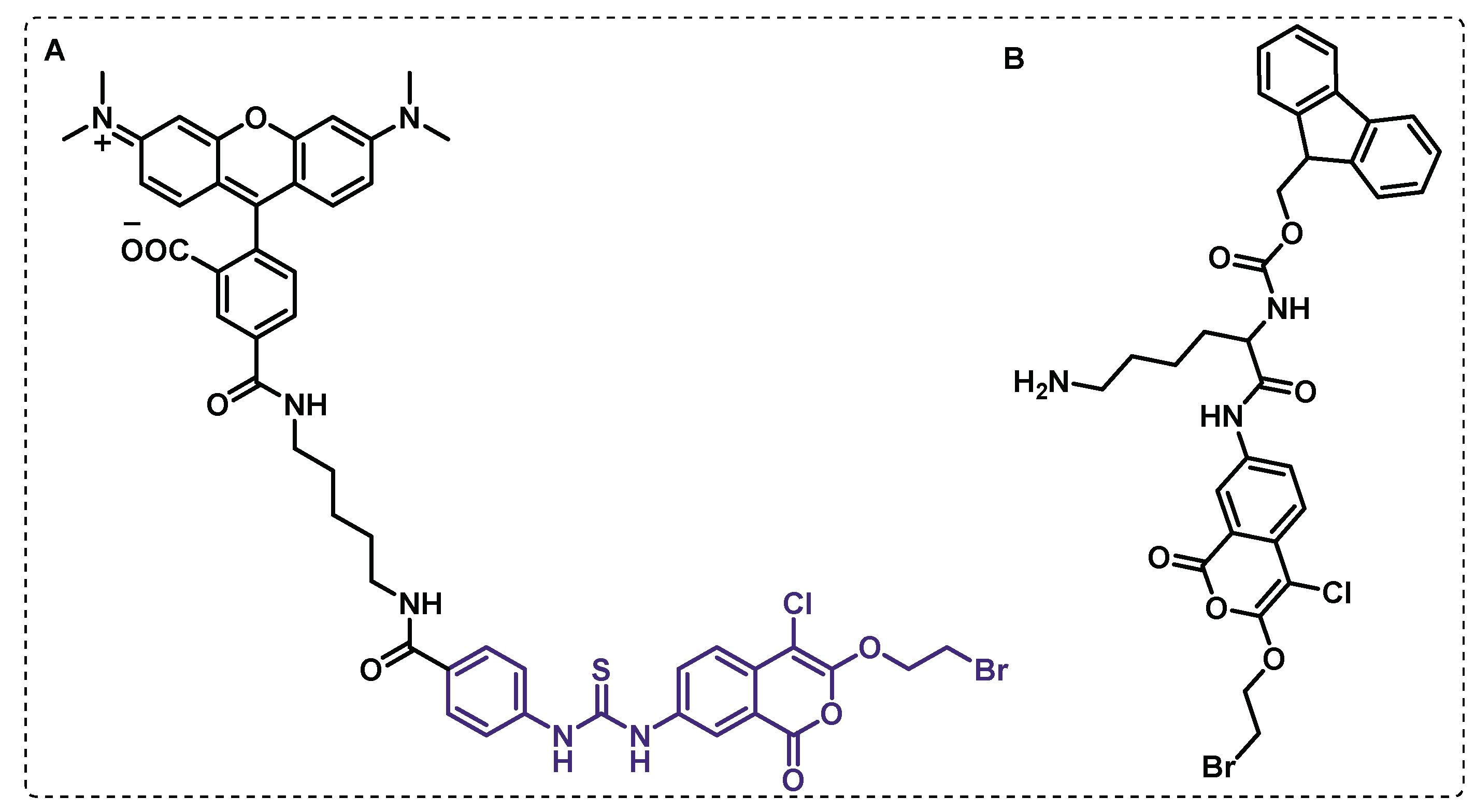
Figure 11.
(A) BMB034 ABP design based on JCP276 serine inhibitor. (B) Fluorophosfonate tetramethylrhodamine ABP.
Figure 11.
(A) BMB034 ABP design based on JCP276 serine inhibitor. (B) Fluorophosfonate tetramethylrhodamine ABP.
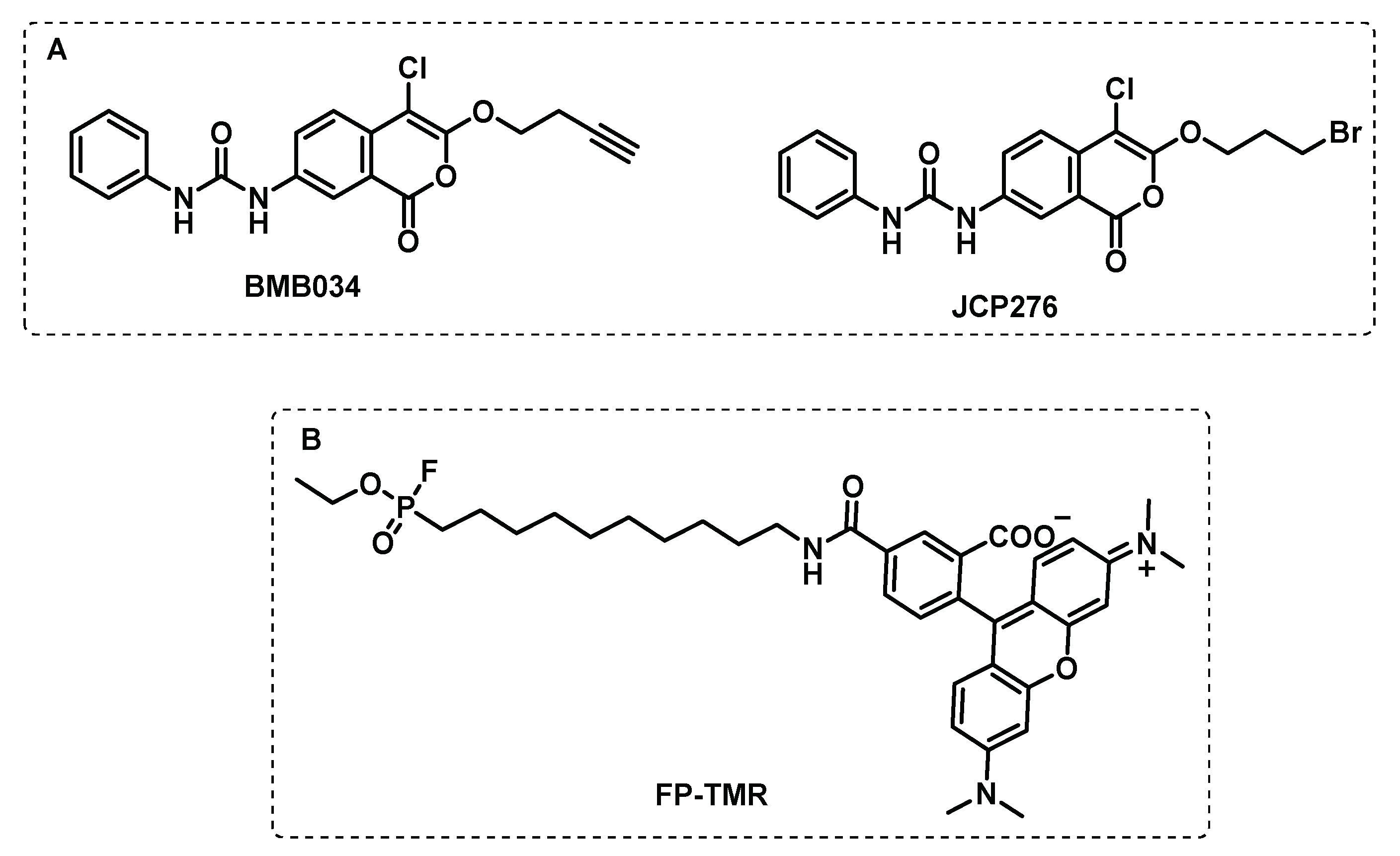
Figure 12.
Fluorophosfonate biotin probe.
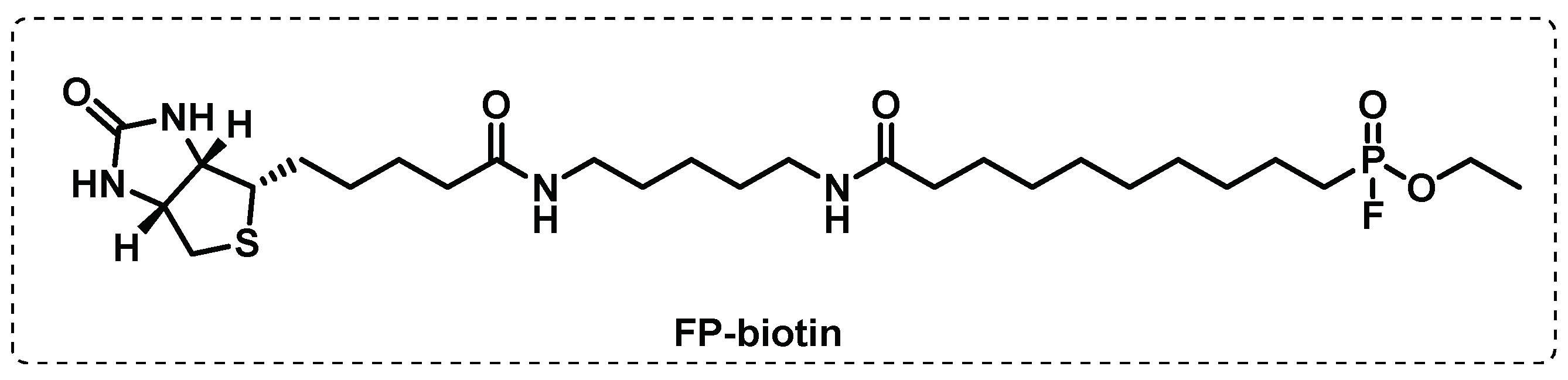
Figure 13.
Cephalosporine alkyne analogue probes.
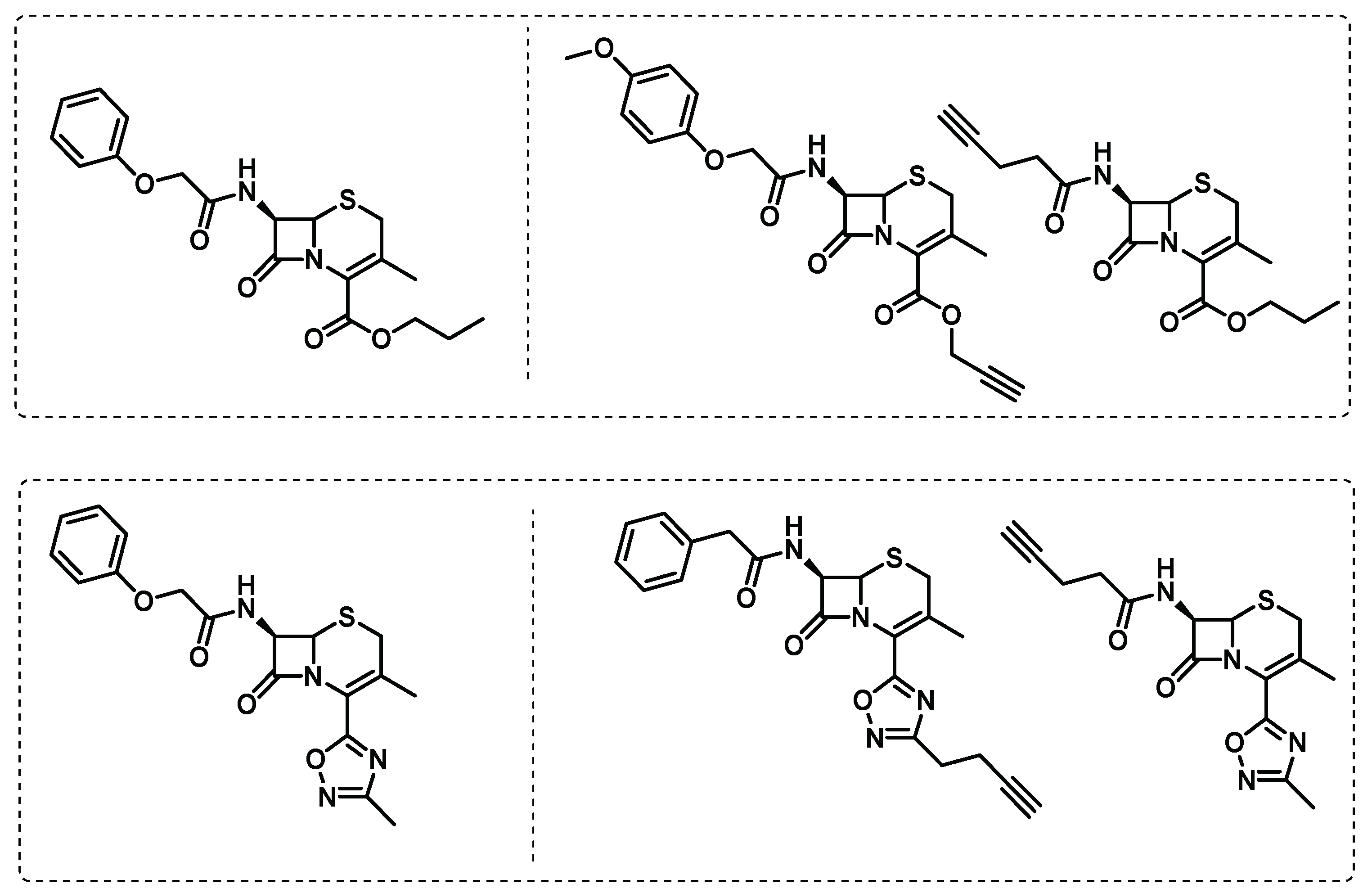
Figure 14.
Sulfone fluorogenic probes, benzoxadiazole (left) and fluorescein (right).
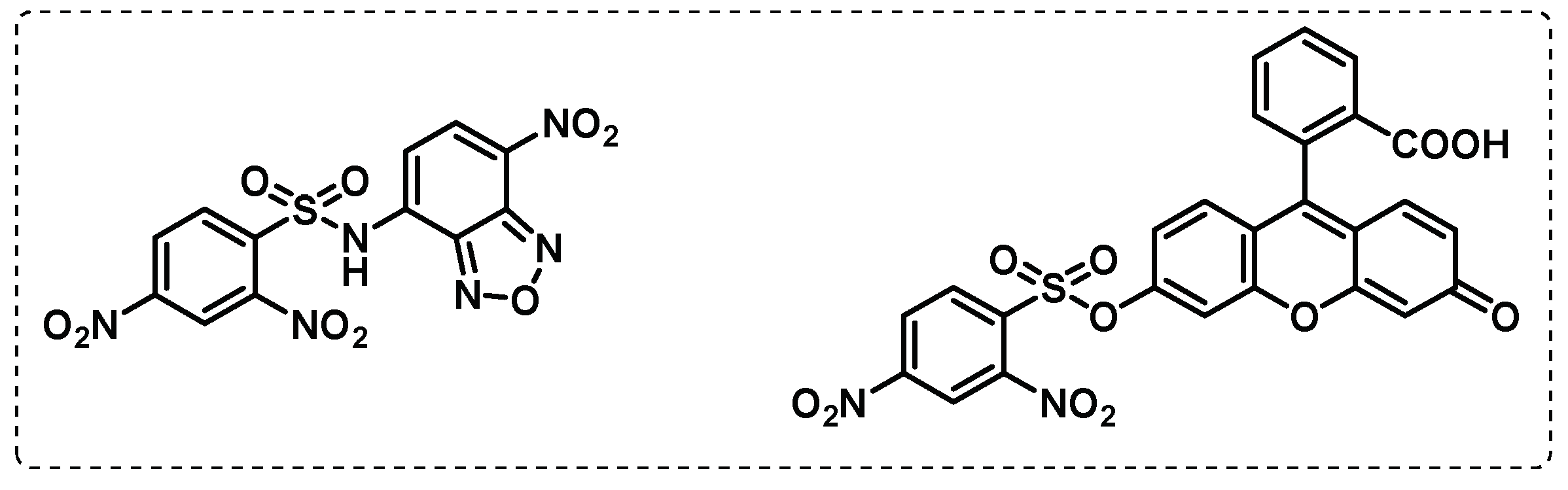
Figure 15.
Meropenem based ABP.
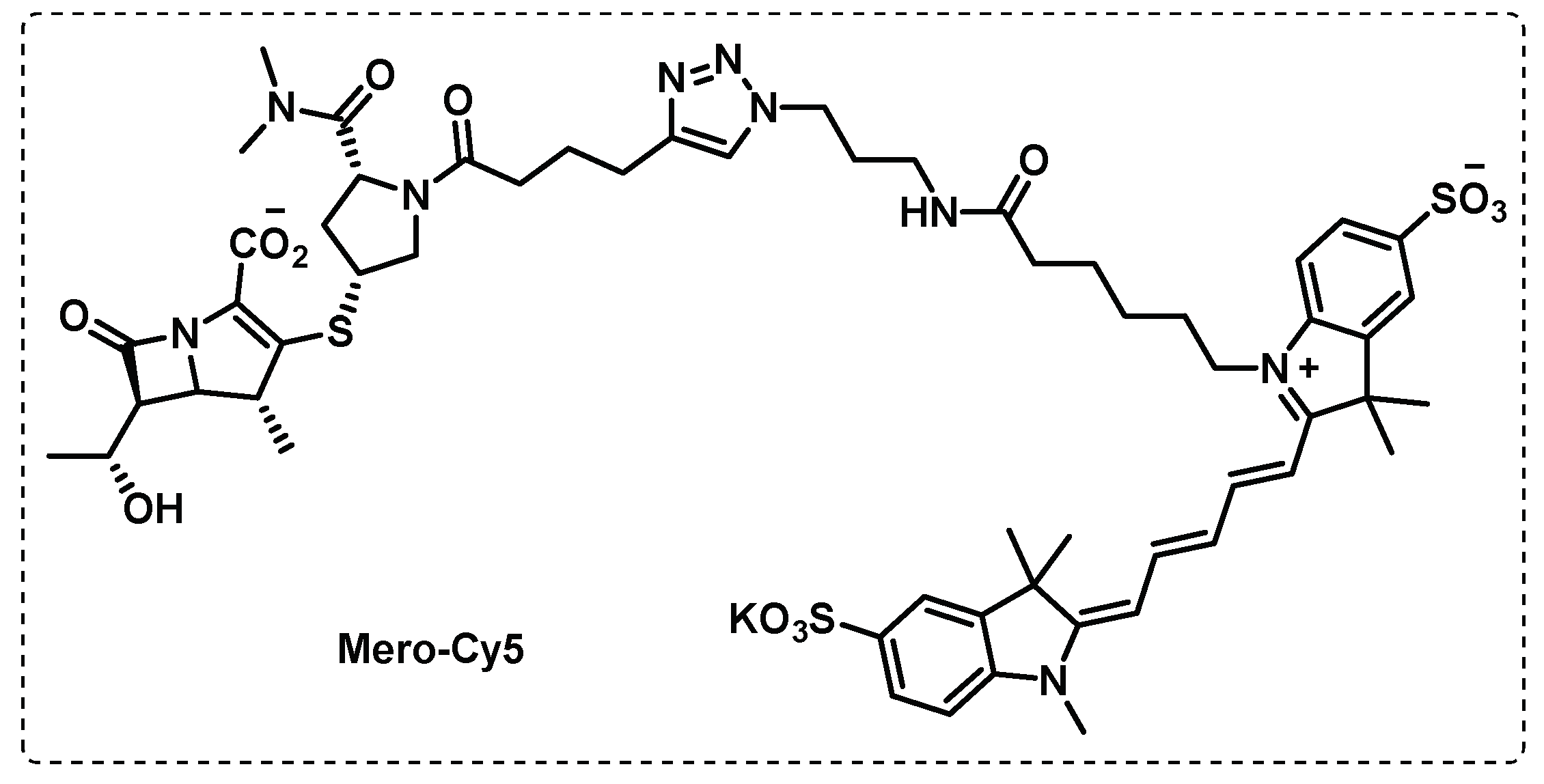
Figure 16.
β-lactone based ABP.
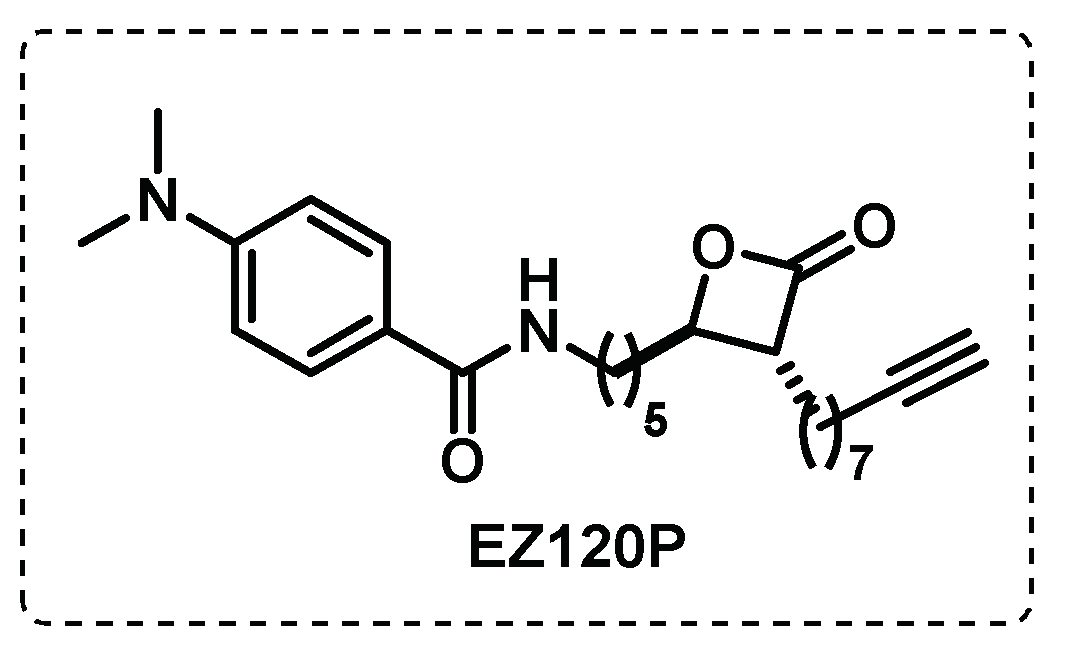
Figure 17.
Tetrahydrolipstatin based ABP.
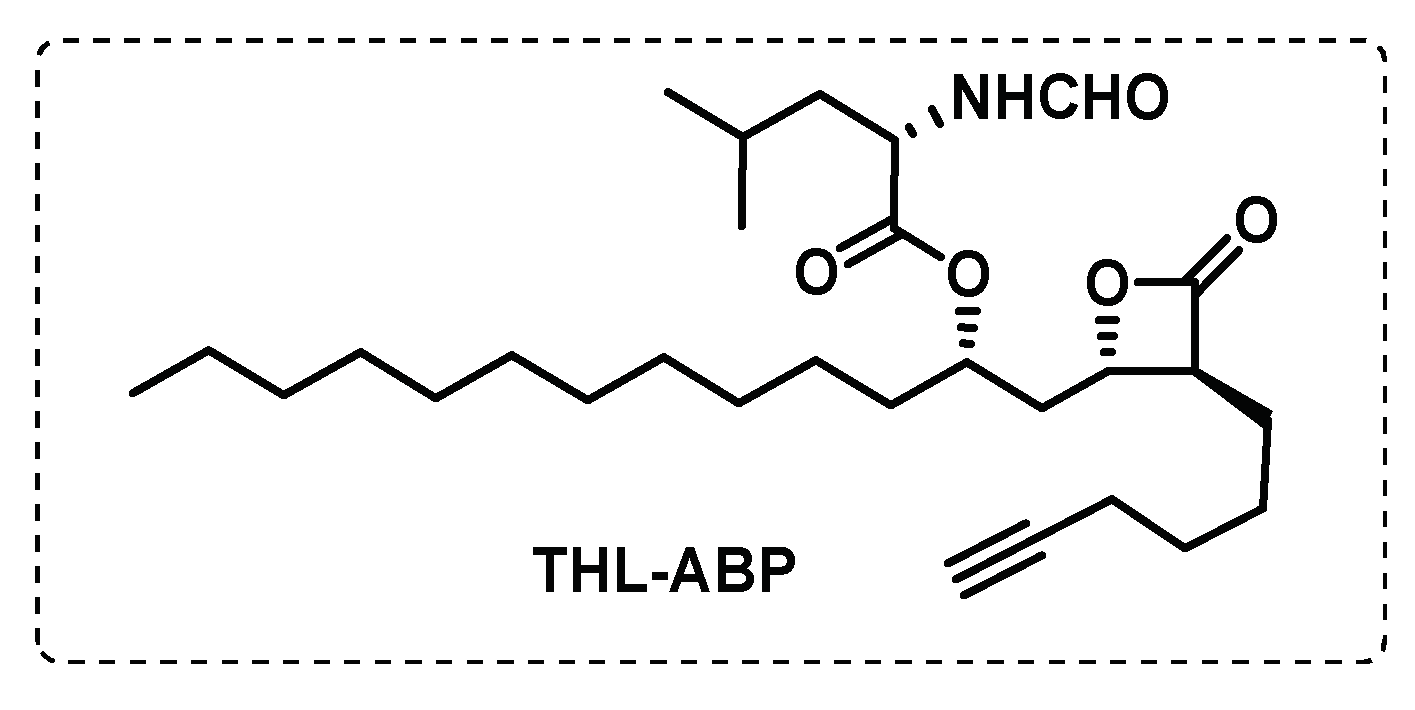
Figure 18.
Biotin probe (ActivX Desthiobiotin-FP) and fluorogenic probes (ActivX TAMRA-FP, DCF-AME) used to identify esterases.
Figure 18.
Biotin probe (ActivX Desthiobiotin-FP) and fluorogenic probes (ActivX TAMRA-FP, DCF-AME) used to identify esterases.
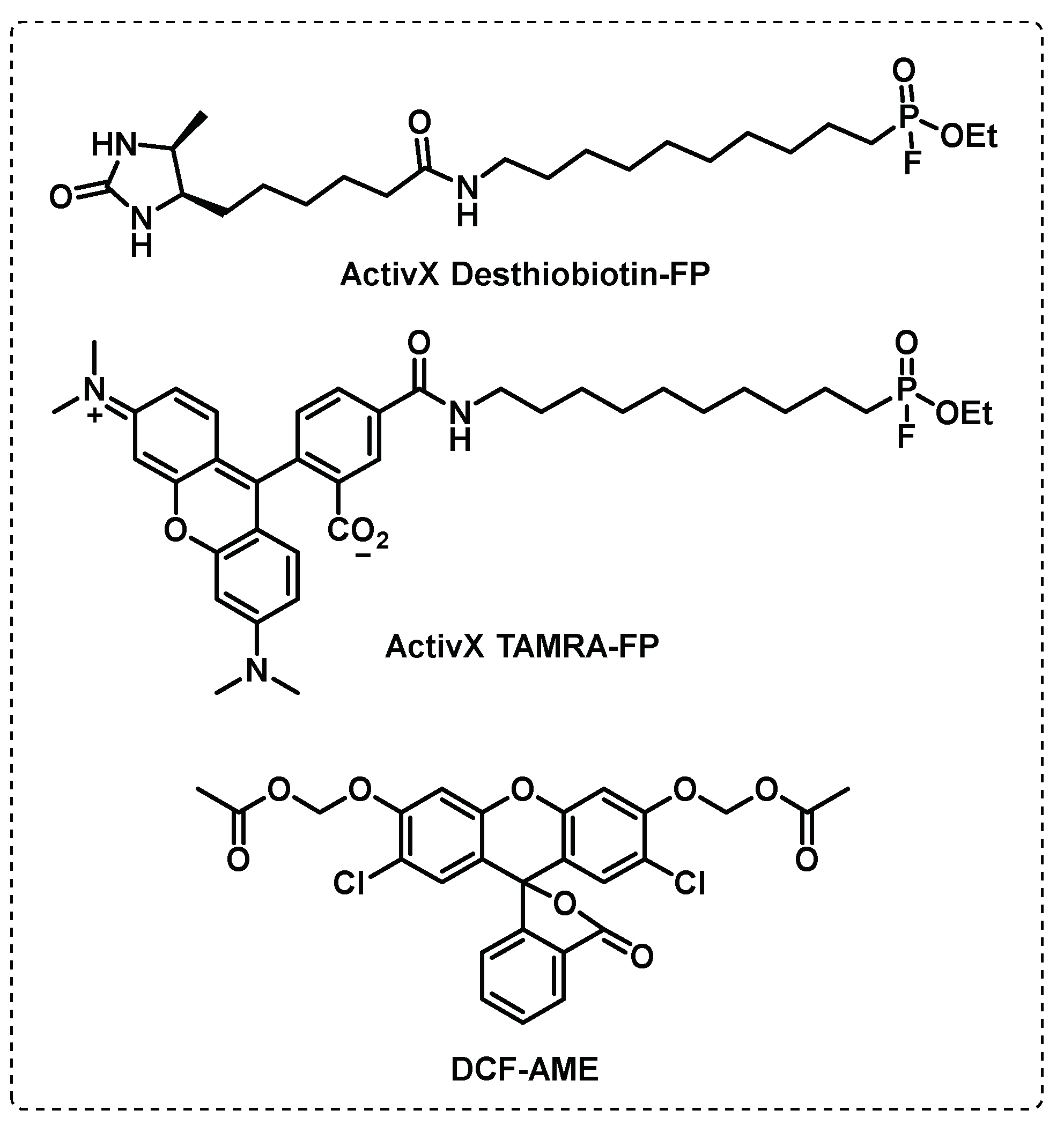
Figure 19.
3-decynoyl-NAC based ABPs.
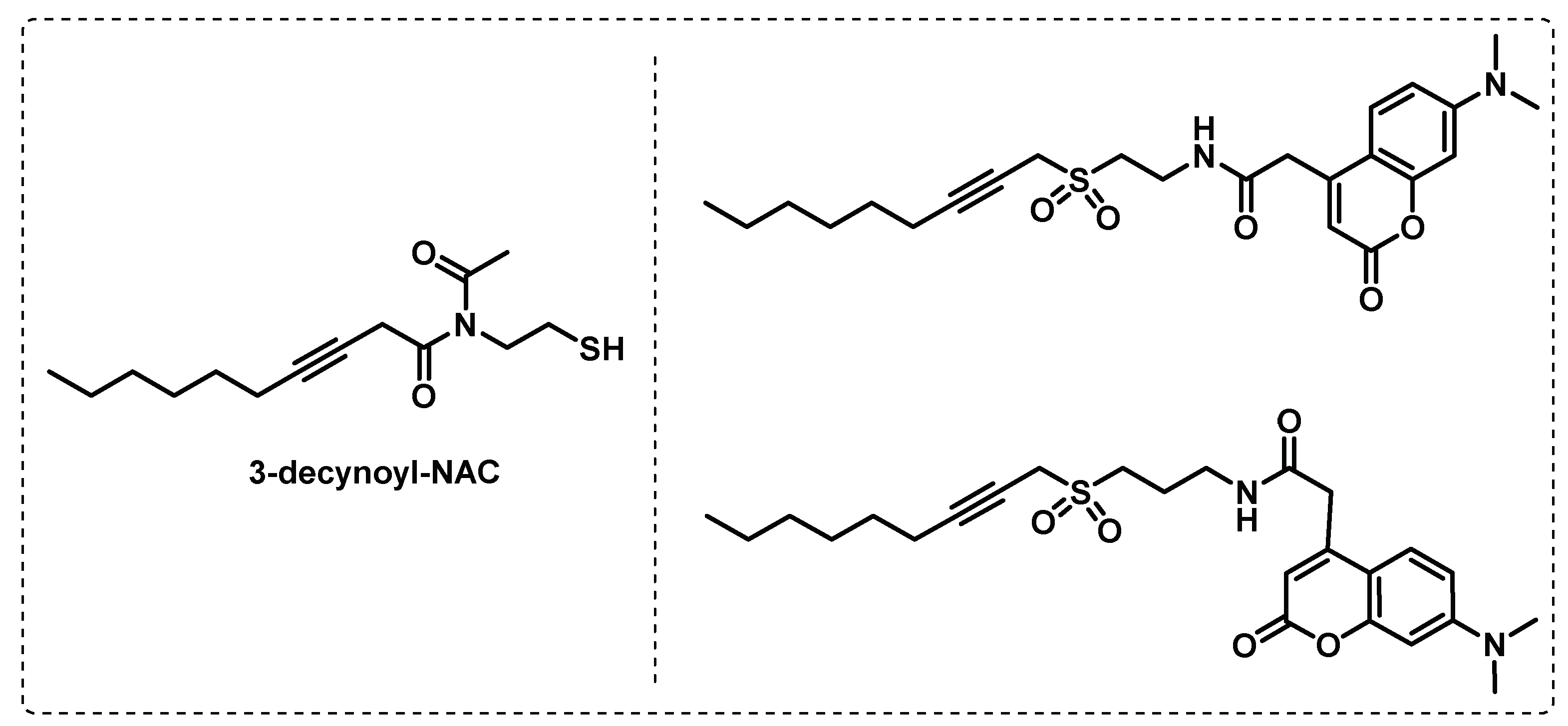
Figure 20.
Probe used by Yoon et al. to detect sulfatase activity.
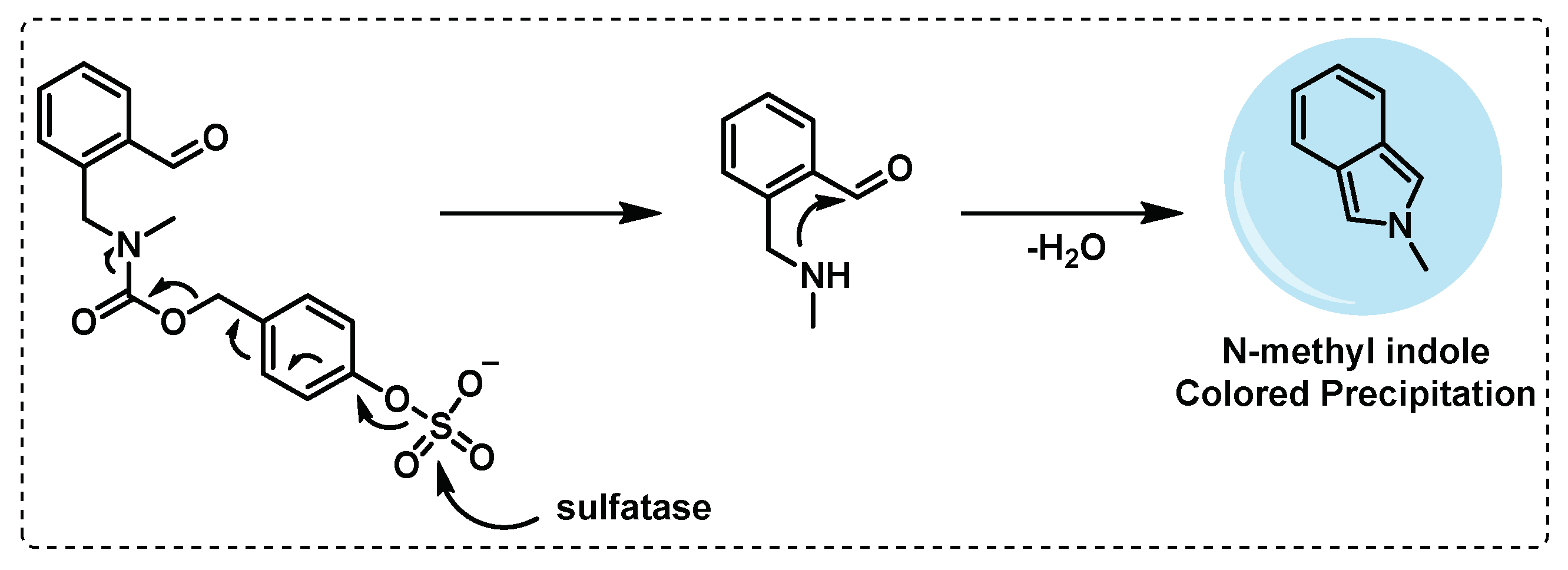
Figure 21.
Sal-AMs ABP.
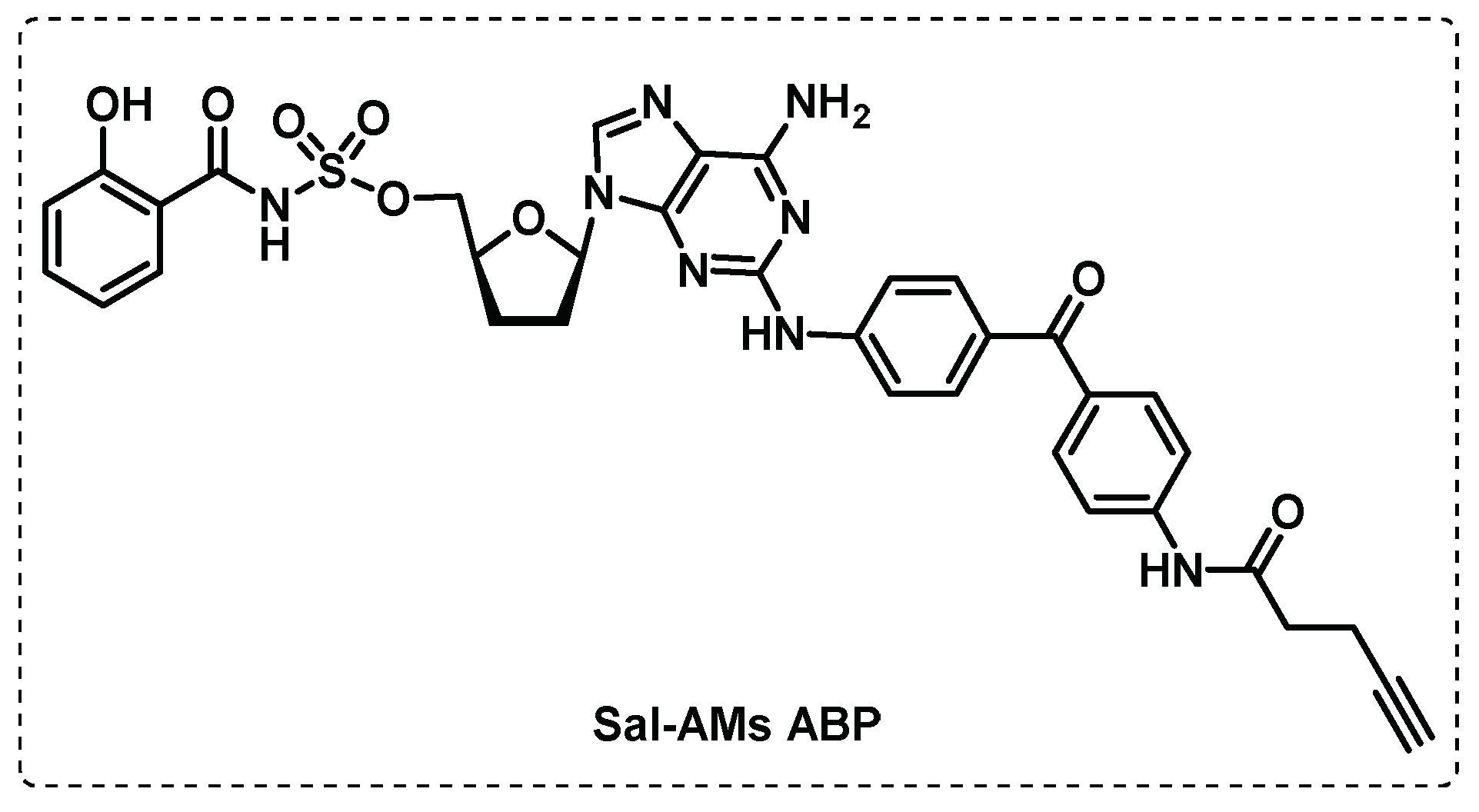
Figure 22.
Desthiobiotin-ATP probe.
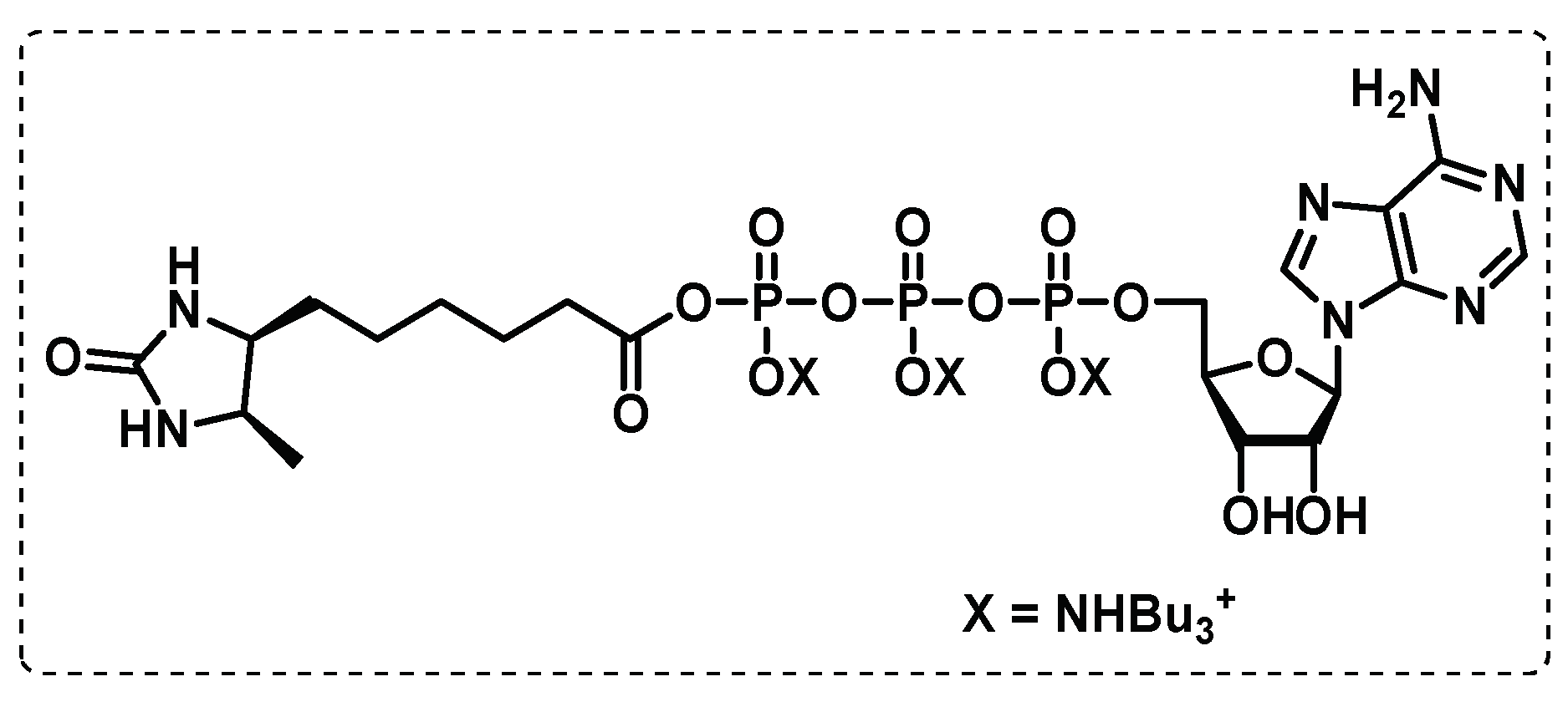
Figure 23.
Thioacetazone is activated by monooxygenase EthA and then binds covalently to HadA subunid of dehydratase complex HadAB.
Figure 23.
Thioacetazone is activated by monooxygenase EthA and then binds covalently to HadA subunid of dehydratase complex HadAB.
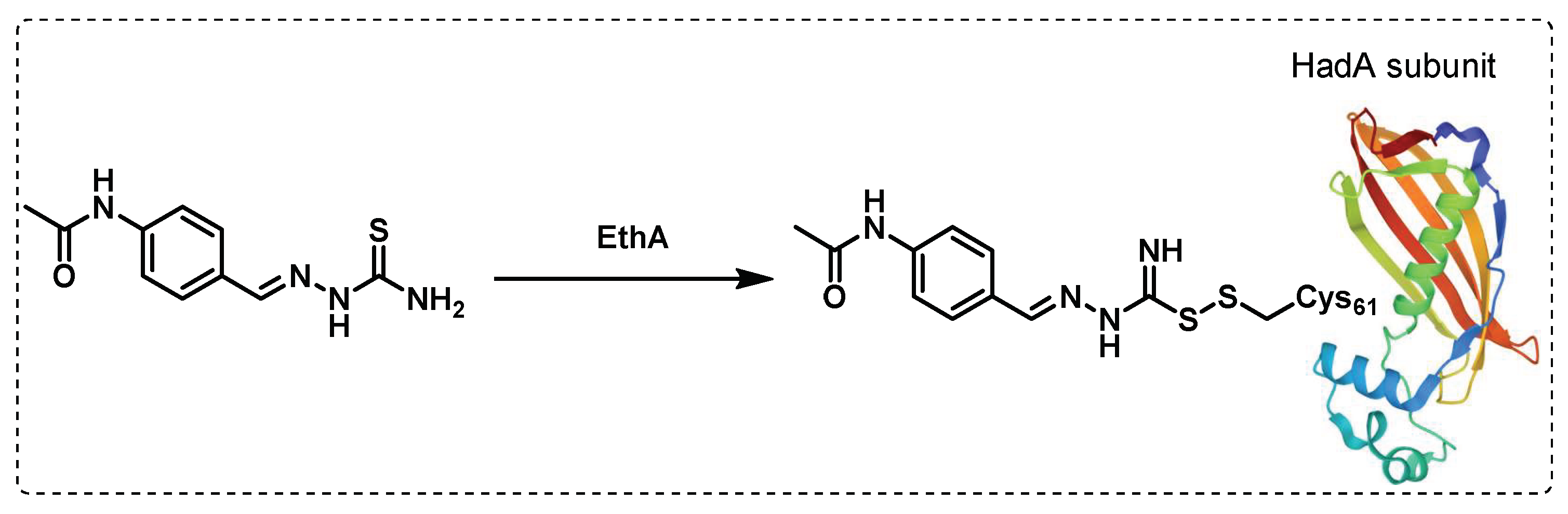
Figure 24.
TAC-affinity based probe.
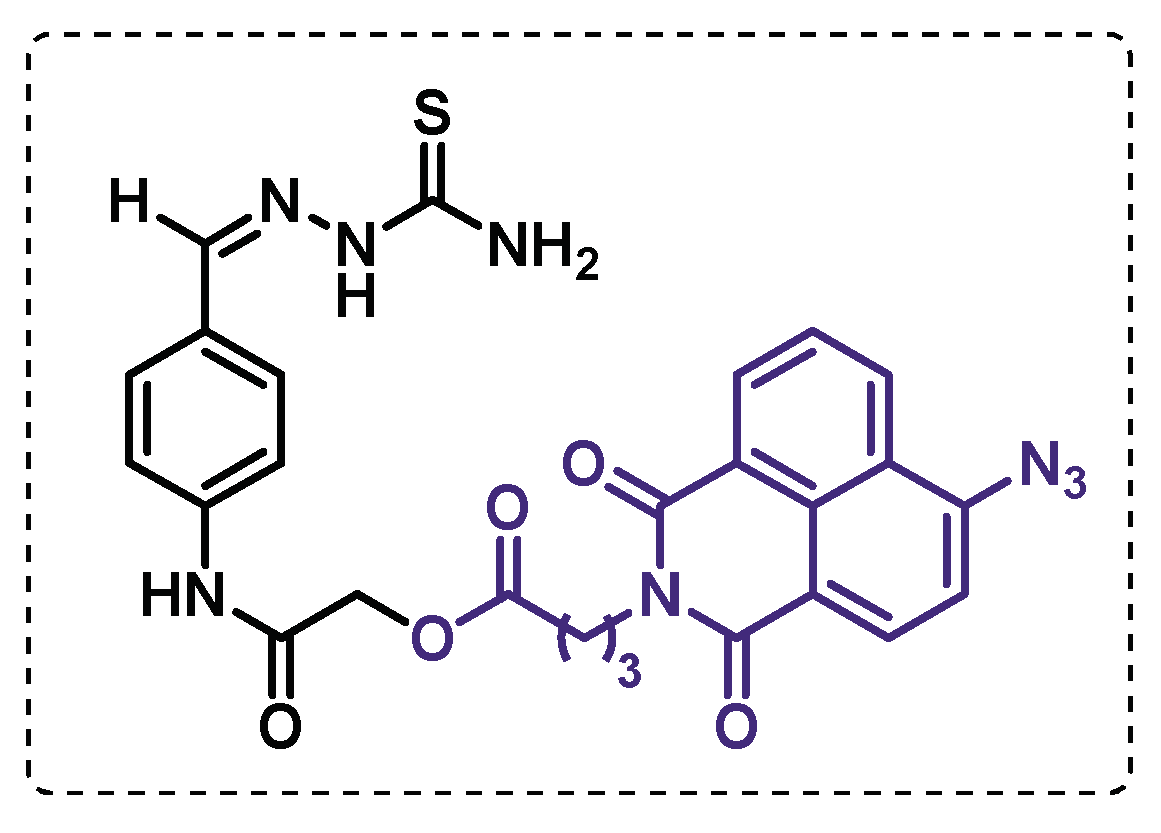
Figure 25.
AfBPP-TDM probe.
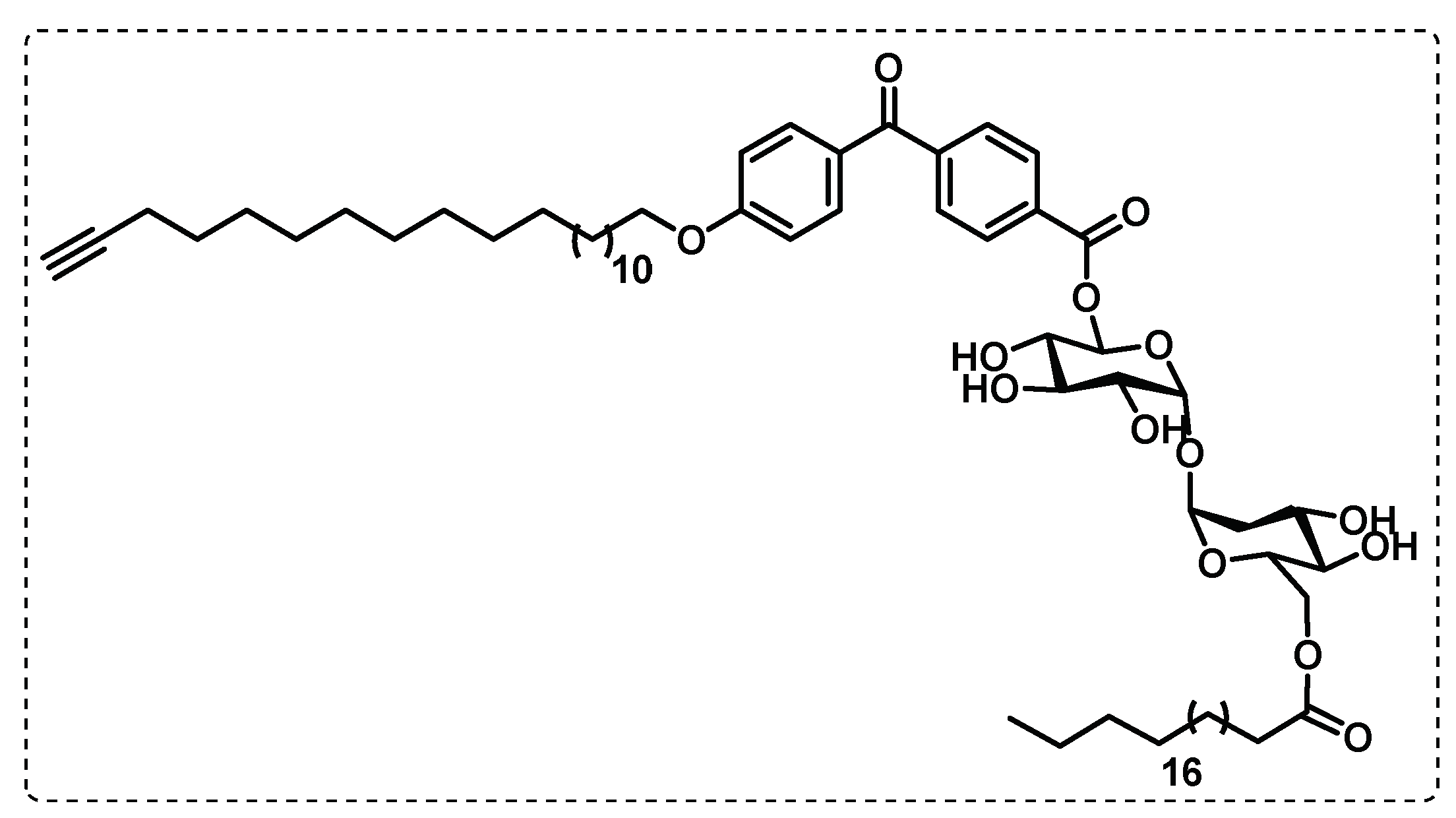
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



