
Submitted:
01 June 2023
Posted:
02 June 2023
You are already at the latest version
Abstract
The objective of this study is to design and synthesize substituted η6-chromium tricarbonyl metal complexes carrying o-carborane units as potential boron neutron capture therapy (BNCT) agents. In this study, 1,2-Diphenyl-o-carborane units were used as starting materials to generate biologically active species. We investigated how the structural changes of 1,2-diphenyl-o-carborane substituted with chromium(0) tricarbonyl affect the biological properties; [(CO)3Cr]Ph2C2 (2) and [(CO)3Cr]2Ph2C2 (3) species were produced in moderate yields. The molecular structures of compounds 1–3 were identified and established by infrared (IR), 1H, 11B, and 13C nuclear magnetic resonance (NMR), and X-ray crystallography analyses. Crystal structures of o-carboranyl chromium complexes 1 [a = 10.859(1) Å, b = 24.953(3) Å, c = 13.938(2) Å, β = 111.854(2)º], 2 [a = 10.621(3) Å, b = 17.056(5) Å, c = 12.174(4) Å, β = 106.622(5)º], and 3 [a = 17.540(2) Å, b = 18.060(2) Å, c = 19.484(4) Å, α = 105.746(2)º, β = 110.226(2)º, γ = 91.256(2)º] were obtained. In vitro study using B16 and CT26 cancer cells containing the triphenyl-o-carboranyl chromium (0) complexes Ph3C2BCr2 and Ph3C2BCr3, which we have previously reported, the compounds 2 and 3 accumulated at higher levels than compounds Ph3C2BCr2 and Ph3C2BCr3. However, the phenylated o-carboranyl chromium(0) complexes have been found to be more cytotoxic than p-boronophenylalanine (BPA).
Keywords:
1
; 2-Diphenyl-o-carborane
; Chromium Metal Complexes
; Boron Neutron Capture Therapy
; Biological Evaluation
; Cytotoxicity
1. Introduction
Boron neutron capture therapy (BNCT) is a promising treatment for a variety of central nervous system (CNS) disorders, especially brain tumors. For example, BNCT is currently used as an adjunct to surgery for the treatment of glioblastoma multiforme [1]. The effect of BNCT is to suppress brain micrometastases that cannot be treated surgically or for which other treatment methods are not available [2]. BNCT destroys cancer cells with energy from targeted radiation bursts by reacting boron isotopes delivered to malignant cells with neutrons. Upon uptake into the cells, 10B is externally irradiated with neutrons and becomes unstable [11B]*, decaying to lithium (7Li3+) and releasing high-energy particles (4He2+). Another advantage of boron is that many 10B atoms form a polyhedral cluster, such as B10H102‒ and B12H122‒, enabling a high concentration of boron in the molecule, so that a satisfactory therapeutic effect can be expected. The high selectivity of neutron capture by the boron atoms and the effectiveness of treatment by it makes an acceptable alternative to other chemotherapy methods that destroy cancer cells by high toxicity.
o-Carborane, namely 1,2-dicarba-closo-dodecaborane, C2B10H12, is a boron-rich compound with an icosahedral structure and a diameter similar to that of a rotating benzene ring, nearly 1 nm, with high symmetry and remarkable stability under various conditions [3]. This cluster contains ten boron and two carbon atoms, making it well suited as a BNCT agent [4,5], and also has potential in other fields of drug discovery, molecular imaging, and targeted radionuclide therapy [6]. However, despite of these advantages, the high toxicity and low water solubility of boron agents remains a significant problem impairing the further clinical treatment [7].
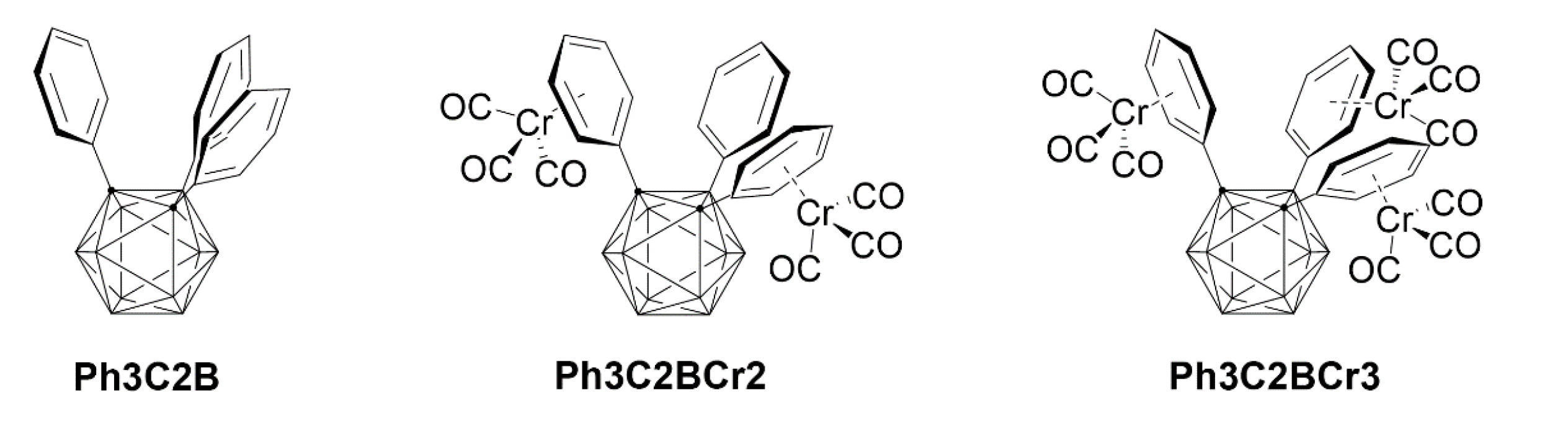
Recently, as shown in Chart 1, we reported the successful synthesis and structural characterization of 1,2,3-triphenyl-o-carboranes and the corresponding chromium complexes (Ph3C2B, Ph3C2BCr2, and Ph3C2BCr3) [8,9]. Among transition-metal π-complexes, η6-arene chromium(0) tricarbonyl complexes have been the subject of extensive development due to their significant applications in organic synthesis [10,11]. Moreover, the use of metal carbonyls in bioorganometallic chemistry is being increasingly developed to enhance the potential of chromium complexes [12‒15].
To the best of our knowledge, this is the first example of the application of η6-arene chromium(0) tricarbonyl-substituted o-carborane compounds in BNCT. Many studies have reported the introduction of various aryl groups to carbons and/or borons in o-carborane. However, no biological characteristics have yet been reported for them. This study is significant because it demonstrates the potential of phenylated o-carboranes with η6-chromium(0) tricarbonyl groups as potential BNCT agents.
2. Results and Discussion
2.1. Synthesis of 1,2-Diphenyl-o-carborane and Corresponding Chromium Metal Complexes
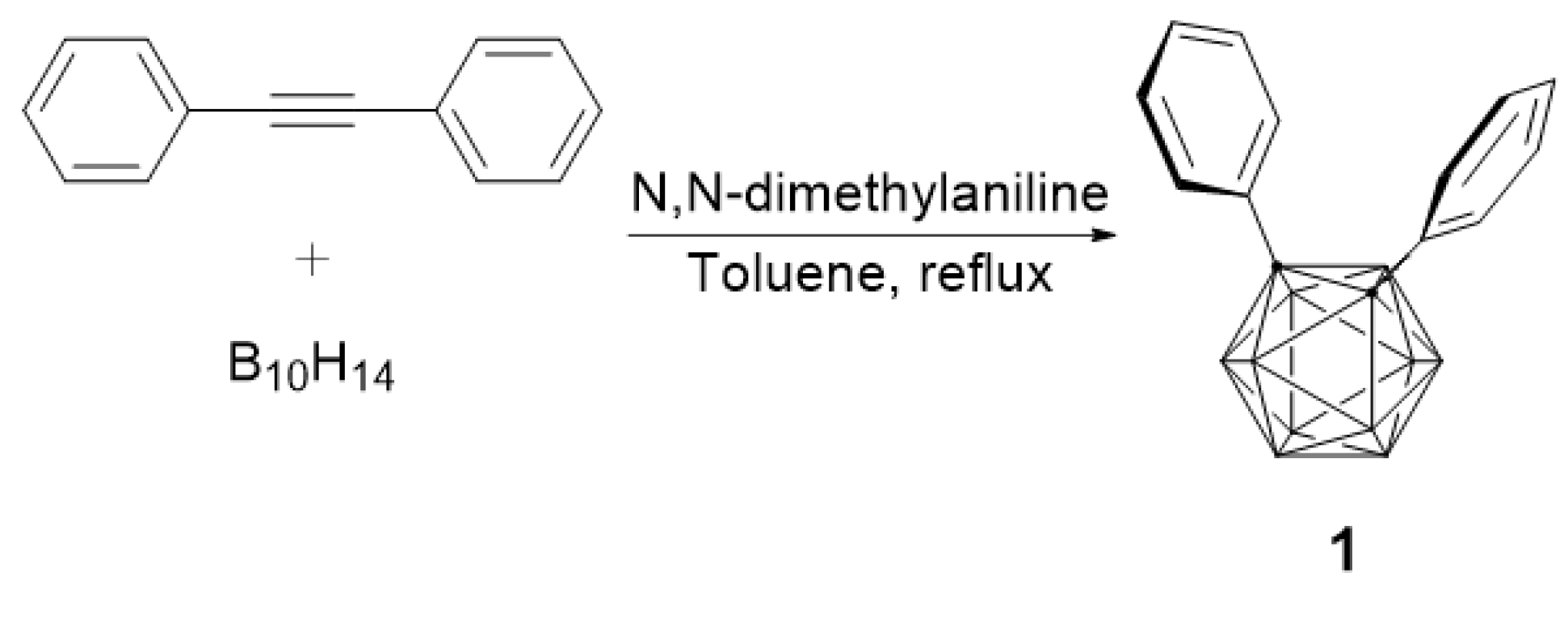
We previously reported the detailed synthesis of compounds Ph3C2B, Ph3C2BCr2, and Ph3C2BCr3 [8,9]. Compound 1 was synthesized by a modified procedure than previous results [18,19] as shown in Scheme 1. 1,2-Diphenyl-o-carborane (1) was prepared from decaborane (B10H14) and diphenylacetylene in moderate yield in toluene solvent using N,N-dimethylaniline as a base. As a result, we first synthesized icosahedral o-carborane with phenyl substituents on the two carbons (Scheme 1).
To compare with previous results, we tried to confirm the electron-withdrawing ability of o-carborane by reacting chromium(0) hexacarbonyl [Cr(CO)6] with two phenyl groups introduced into o-carborane [20]. The reaction of 1 with Cr(CO)6 produced η6-phenyl-coordinated mono- and bis-chromium complexes (2 and 3) in moderate yields through stoichiometric reactions, as shown in Scheme 2. Interestingly, these results showed that 1,2-diphenyl-o-carborane prefers stoichiometric reactions to 1,2,3-triphenyl-o-carborane.
2.2. IR and NMR Spectroscopy
The structure of 1,2-diphenyl-o-carborane (1) was proposed based on the assignments the 1H, 11B, and 13C NMR resonances before confirmation by X-ray diffraction (XRD). The 1H NMR spectrum of compound 1 showed resonances at around 7.35–7.31 ppm for the two phenyl rings. The 11B NMR spectrum showed resonances at around δ ‒11.18 and ‒2.52. In the 11B NMR, it can be seen that only two peaks appear due to high intramolecular symmetry. This pattern was confirmed to be consistent with the previously result [19]. The 13C NMR spectrum showed resonances at around δ 132.3–126.7. The infrared (IR) spectra of compounds 2 and 3 showed characteristic absorption bands of C≡O units at 1965,1890 (2), 1965, and 1892 cm‒1 (3). These values are lower than those of (C6H6)Cr(CO)3 [21,22,23,24,25]. The IR and NMR spectra clearly indicate Cr coordination to the phenyl groups of 2 and 3, with chemical shifts to the downfield region of δ 7.70–7.33 and 141.5–128.7 in the 1H and 13C NMR spectra, respectively.
2.3. X-ray Structural Studies of 1,2-Diphenyl-o-carborane and Corresponding Chromium Complexes
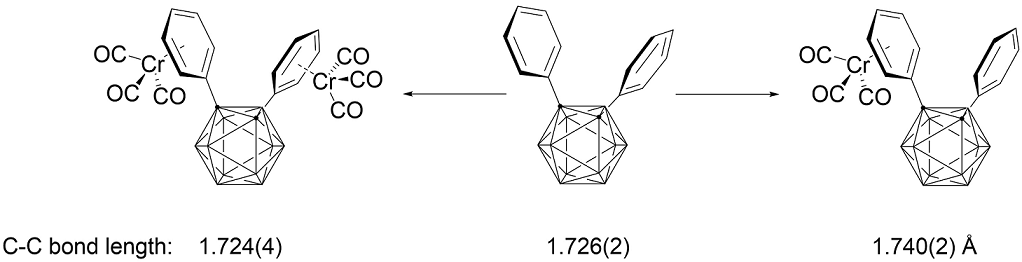
The selected crystallographic data and a summary of the intensity data collection parameters for 1, 2, and 3 are presented in Table 1 and Table 2, respectively. Detailed information on the structural determinations and structural features of compounds 1, 2, and 3 are provided in the Supplementary Materials and Appendix A. Additionally, the molecular structures and structural characteristics of Ph3C2B, Ph3C2BCr2, and Ph3C2BCr3 are also included (Figures S1‒S3 and Tables S10 and S11). Single-crystal X-ray structure determination revealed the structural authenticity of each compound and suggested alteration of the electronic structure based on the changes in the C‒C distance of carborane cage [26]. The C1‒C2 bond distance of 1 is 1.726(2) Å, which is significantly longer than the C1‒C2 bond distance of the unsubstituted o-carborane [1.629(6) and 1.630(6) Å]. Moreover, comparison of the C‒C distances in compounds 1 and Ph3C2B (Table S11) showed that the bonds in Ph3C2B are slightly longer than those in compound 1. Compound Ph3C2B contains further phenyl decorations at the boron atom while maintaining the active compound 1 platform [27,28].
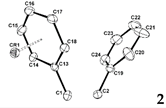
Single crystals of 1 and its chromium(0) tricarbonyl complexes, 2 and 3, suitable for X-ray crystallography, were obtained from dichloromethane solutions by slow evaporation. The general structural characteristics of compound 1 and its chromium complex are shown in Table 2. The molecular structures of compounds 1, 2, and 3 are shown in Figure 1, Figure 2 and Figure 3. Compound 1 was already reported in 1993 by Lewis and Welch [29]. Comparing the data, it can be seen that the bond lengths, angles and dihedral angles are nearly identical (Figure 1).
As shown in Figure 2, the chromium atom of compound 2 is coordinated to the phenyl ring of the carbon atom of o-carborane via a π-bond, similar to the previous results [8,9]. The chromium metal adopts the typical three-legged ‘piano stool’ geometry, with the expected geometrical parameters. The chromium metals are centered approximately over the phenyl rings, giving rise to Cr1‒C6H5 face (centroid) distances of 1.702 Å. The average C‒C bond length in the coordinated phenyl ring is 1.410 Å, which is 0.028 Å longer than the average bond length of 1.382 Å for C‒C bonds within the non-coordinated phenyl ring. The Cr‒CPh and Cr‒CO bonds had average values of 2.210 and 1.856 Å, respectively, which were within the normal range [30]. The average C‒O bond length in the chromium-coordinated carbonyl ligands is 1.143 Å, which is slightly shorter than that reported η6-arene chromium(0) tricarbonyl complexes [31‒35]. As expected, the C1‒C2 bond distance is 1.740(2) Å, a slightly increase over compound 1, and is essentially in the range of non-bonding character [36‒39]. Furthermore, as shown in Table 2 and S6, the torsion angles of the two phenyl rings of 1 and 2 changed upon coordination with the chromium atom.
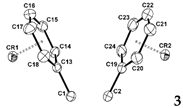
The X-ray crystal structure of 3 (Figure 3) reveals that the dichromium atoms adopt an η6-coordination with the two phenyl rings. The single crystal X-ray diffraction study of 3 revealed crystallization in the triclinic space group P‒1. Interestingly, even though it is a triclinic space group, the APEX3 program identified the Z value as 8, which made it difficult to solve. In the end, this was considered to be recognized by four molecules as one molecule, and the structure was interpreted by adjusting the Z value to 2. The geometrical parameters of complex 3 were within the expected ranges (Table 2). The average C‒C bond length in the chromium coordinated phenyl rings is 1.406 Å, which is 0.03 Å longer than the average bond length of 1.376 Å for the C‒C bonds in 1. The Cr‒CPh and Cr‒CO bond lengths were within the normal range [30], with average values of 2.212 and 1.851 Å, respectively. The average C‒O bond length in the chromium-coordinated carbonyl ligands is 1.138 Å, which is significantly shorter than those of reported η6-arene chromium(0) tricarbonyl complexes [31‒35]. As shown in Table 2, the chromium metals are centered approximately over the phenyl rings, giving rise to Cr1/Cr2‒C6H5 face (centroid) distances of 1.687 and 1.705 Å, respectively. Interestingly, the C1‒C2 bond length of 3 is shorter than that of 1 and 2, it was shown that contraction of this bond is due to favorable phenyl ring π* and carboranyl σ* orbital interactions do not occur in 3, as previous results [8,9]. As shown in Table 2 and S9, the torsion angles of the phenyl rings of 1 and 3 or 2 and 3 changed upon coordination with the chromium atoms.
2.4. Determination of IC50 and Incorporation of Boron into B16 and CT26 Cells
B16 mouse melanoma and CT26 colon carcinoma cells were treated with compounds 2, 3, Ph3C2BCr2, and Ph3C2BCr3 for 3 d, after which the cell viability was determined using the MTT [30-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. Compounds 2, 3, Ph3C2BCr2, and Ph3C2BCr3 showed higher cytotoxicity than BPA (Table 3), with IC50 (the half maximal inhibitory concentration) values in the range of 0.091–0.736 μM. Interestingly, the 1,2,3-triphenyl-o-carboranyl chromium tricarbonyl complexes (Ph3C2BCr2 and Ph3C2BCr3) showed higher cytotoxicity to the B16 and CT26 cells than complexes 2 and 3. The higher cytotoxicity of compounds Ph3C2BCr2 and Ph3C2BCr3 to B16 cells may be a result of the difference in the mechanism by which the phenyl groups and chromium(0) tricarbonyl moieties induce cell toxicity. Compounds 2, 3, Ph3C2BCr2, and Ph3C2BCr3 exhibited similar activities in the CT26 and B16 cells, with IC50 values in the range of 0.089–0.833 μM.
We next examined the level of intracellular accumulation of compounds 2, 3, Ph3C2BCr2, and Ph3C2BCr3 by determining their boron concentrations using ICP-OES. The intracellular boron uptake of compounds 2, 3, Ph3C2BCr2, and Ph3C2BCr3 in B16 and CT26 cells was higher than that of BPA (Table 3). Boron uptake from both bis- and tris-chromium tricarbonyl-substituted compounds, which included the 1,2,3-triphenyl-o-carboranyl chromium tricarbonyl complexes (i.e., Ph3C2BCr2 and Ph3C2BCr3), was lower. These results show that the introduction of phenyl groups or chromium metals into the carborane backbone increases cytotoxicity rather than an increase in boron accumulation in cancer cells.
3. Materials and Methods
3.1. General Procedure
All manipulations were performed under dry nitrogen or argon atmosphere using standard Schlenk techniques. Tetrahydrofuran (THF) was distilled from sodium and benzophenone under nitrogen atmosphere. Elemental analyses were performed using a Carlo Erba Instruments CHNS-O EA 1108 analyzer. High-resolution tandem mass spectrometry (JMS-HX 110/110A, Jeol Ltd.) data were acquired at the Korean Basic Science Institute. 1H, 11B, and 13C NMR spectra were recorded using a Bruker 600 spectrometer operating at 600.1, 150.9, and 192.6 MHz, respectively. All 11B chemical shifts were referenced to BF3·O(C2H5)2 (0.0 ppm), with a negative sign indicating an upfield shift. All proton and carbon chemical shifts were measured relative to the internal residual CHCl3 in the lock solvent (99.9% CDCl3). Decaborane was purchased from Katchem. N,N-dimethylaniline, 1,2-diphenylacetylene, n-BuLi (2.5 M in hexane), and chromium hexacarbonyl [Cr(CO)6] were purchased from Aldrich Chemicals.
3.2. Crystal Structure Determination
Crystals of 1, 2, and 3 were obtained from toluene or CH2Cl2, sealed in glass capillaries under argon atmosphere, and mounted on a diffractometer. Preliminary examination and data collection were performed using a Bruker SMART CCD detector system single crystal X-ray diffractometer equipped with a sealed-tube X-ray source (40 kV × 50 mA), using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The preliminary unit cell constants were determined using a set of forty-five narrow-frame (0.3° in ω) scans. Double-pass scanning was used to exclude noise. The collected frames were integrated using an orientation matrix determined from the narrow-frame scans. The SMART software package was used for data collection and SAINT was used for frame integration [16]. The final cell constants were determined by global refinement of the xyz centroids of the reflections harvested from the entire dataset. Structure solution and refinement were performed using the SHELXTL-PLUS software package [17].
3.3. Cell Viability Assay (MTT Assay)
The boron compounds were dissolved in dimethylsulfoxide (DMSO), and the resulting solution was diluted with Dulbecco’s modified Eagle’s medium (DMEM) (10% FCS), or p-boronophenylalanine (BPA) was directly dissolved in the same medium. In a 96-well culture plate (Falcon 3072), B16 mouse melanoma and CT26 colon carcinoma cells (5 × 103 cells/well) were cultured in five wells with medium containing the boron compounds at various concentrations, followed by incubation at for 72 h at 37 °C in a CO2 incubator. DMSO is non-toxic at concentrations less than 0.5%, and control experiments confirmed the non-toxicity of DMSO at the concentrations used in the present experiments. After incubation, the medium was removed, the cells were washed three times with phosphate-buffered saline [PBS (–)], and the CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay (MTT) was used to count the cells on a microplate reader. The concentration that resulted in a cell culture with 50% of the number of cells in the corresponding untreated group (IC50) is summarized in Table 3.
3.4. In Vitro Boron Incorporation into B16 and CT26 Cancer Cells
B16 and CT26 cancer cells were cultured in Falcon 3025 dishes (150 mm diameter). When the cell population increased to fill the dish (5 × 105 cells/dish), the boron compounds and BPA (10 μM) were added to the dishes. The cells were then incubated for 3 h at 37 °C in DMEM (20 mL of 10% FBS). The cells were washed three times with Ca/Mg-free PBS (–), collected with a rubber policeman, digested with a mixture of 60% HClO4–30% H2O2 (1:2) solution (2 mL), and finally decomposed for 1 h at 75 °C. After filtration through a membrane filter (Millipore, 0.22 mm), the boron concentration was determined using an inductively coupled plasma optical emission spectroscopy (ICP-OES) instrument [Avio 220 Max, PerkinElmer, Massachusetts, U.S.A)]. Each experiment was performed in triplicate.
3.5. Synthesis of 1,2-Diphenyl-o-carborane (1)
1,2-Diphenylethyne (2.2 g, 12.0 mmol) and B10H14 (decaborane) (1.22 g, 10.0 mmol) were dissolved in dry toluene (100 mL) at room temperature under argon atmosphere. N,N-dimethylaniline (2.78 mL, 24.0 mmol) was added to the reaction mixture, and the mixture was stirred at 110 °C for 24 h. After cooling, the solid residue was filtered off and the solvent was evaporated to dryness. The crude mixture was purified using silica gel column chromatography (hexane as the eluent). Recrystallization from CH2Cl2 provided 1 as colorless crystals (2.31 g, 78%). HRMS: Calcd. for [12C141H2011B10]+ 296.2568. Found: 296.2571. IR spectrum (KBr pellet, cm‒1): ν(CAr‒H) 3024, ν(B‒H) 2589, ν(C=CAr) 1601, 1500. 1H NMR (CDCl3, 600 MHz) δ 7.35 (m, 5H, Ph-H), 7.31 (m, 5H, Ph-H). 11B NMR (CDCl3, 192.6 MHz) δ ‒2.52, ‒11.18. 13C NMR (CDCl3, 150.9 MHz) δ 132.3, 130.1, 129.8, 129.2, 126.7 (Ph), 84.0 (Ccab).
3.6. Synthesis of 1-(Phenyl-η6-chromium(0) tricarbonyl)-2-phenyl-o-carborane (2)
Compound 1 (0.3 g, 1.0 mmol) and 1.2 equiv of [Cr(CO)6] (0.26 g, 1.2 mmol) were dissolved in a mixture of THF (5 mL) and di-n-butyl ether (50 mL). The mixture was refluxed for 72 h. The resulting dark reddish solution was cooled to room temperature and filtered through Celite. The solvent was evaporated under reduced pressure. After evaporation, the crude reaction mixture was purified by column chromatography [CH2Cl2:hexane (1:1) as the eluent] to give the chromium complex 2, which was then recrystallized from toluene to obtain red crystals. Yield: 88% (0.38 g, 0.88 mmol). HRMS: Calcd. for [12C171H2011B1052Cr116O3]+ 432.1821. Found: 432.1824. IR spectrum (KBr pellet, cm‒1): ν(CAr‒H) 3023, 3020, ν(B‒H) 2588, ν(C≡O) 1965,1890. 1H NMR (CDCl3, 600.1 MHz) δ 7.62 (m, 5H, Ph-H), 7.33 (m, 5H, Ph-H). 11B NMR (CDCl3, 192.6 MHz) δ ‒2.28, ‒4.11, ‒9.51, ‒11.45. 13C NMR (CDCl3, 150.9 MHz) δ 231.4 (Cr‒CO), 141.5, 137.9, 134.8, 132.7,131.3,130.8,128.7 (Ph), 85.6 (Ph‒Ccab).
3.7. Synthesis of 1,2-bis(phenyl-η6-chromium(0) tricarbonyl)-o-carborane (3)
A procedure analogous to the preparation of 2 was used to obtain red crystals. Yield: 57% (0.32 g, 0.57 mmol). HRMS: Calcd. for [12C201H2011B1052Cr216O6]+ 568.1073. Found: 568.1077. IR spectrum (KBr pellet, cm‒1): ν(CAr‒H) 3021, ν(B‒H) 2583, 2589; ν(C≡O) 1965, 1892. 1H NMR (CDCl3, 600.1 MHz) δ 7.70 (m, 4H, Ph-H), 7.48 (m, 3H, Ph-H), 7.39 (m, 3H, Ph-H). 11B NMR (CDCl3, 192.6 MHz) δ ‒2.65, ‒11.48. 13C NMR (CDCl3, 150.9 MHz) δ 233.1 (Cr-CO), 141.5, 140.3, 138.5, 135.1, 133.8, 131.1 (Ph), 86.7 (Ph-Ccab).
4. Conclusions
In conclusion, we described the synthesis, X-ray structures, and biological activities of a series of 1,2-diphenyl- and 1,2,3-triphenyl-o-carborane and the corresponding chromium(0) tricarbonyl transition metal complexes, which can be easily stoichiometrically substituted with transition metals to produce highly active biological molecules for BNCT. We presented a general and versatile method for the sequential introduction of phenyl groups into o-carborane and stoichiometrically-coordinated transition metal complexes using chromium(0) hexacarbonyl. Diphenyl- or triphenyl-o-carborane and its chromium complexes show higher cytotoxicity than p-boronophenylalanine in B16 and CT26 cancer cell lines, however, the boron accumulation is higher than that of p-boronophenylalanine.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1‒S3: Molecular structures of Ph3C2B, Ph3C2BCr2, and Ph3C2BCr3; Table S1‒S9: Bond lengths (Å), angles (º), and torsion angles (º) of compounds 1, 2, and 3; Table S10 and S11: Crystal data and structure refinement and comparison of selected bond lengths (Å), angles (º), and torsion angles (º) for 1‒3 and Ph3C2B, Ph3C2BCr2, and Ph3C2BCr3.
Author Contributions
Conceptualization, J.D.L.; Chemical experiments, T.J.H., D.K.I. and S.M.K.; Designed and performed biological tests, D.K.I. and J.D.L..; validation, J.D.L.; formal analysis, T.J.H., D.K.I., and S.M.K.; investigation, T.J.H., D.K.I. and S.M.K.; data curation, T.J.H. and J.D.L.; writing—original draft preparation, T.J.H. and S.M.K.; writing—review and editing, J.D.L.; supervision, J.D.L.; project administration, J.D.L.; funding acquisition, J.D.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Supplementary Materials are available online. Figures S1–S6: X-ray structures of compounds 1, 2, 3, Ph3C2B, Ph3C2BCr2 and Ph3C2BCr3, Tables S1–S6. Detailed information on the structural determinations and structural features of compounds 1, 2, 3, Ph3C2B, Ph3C2BCr2 and Ph3C2BCr3 are provided in the Supplementary Materials.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2022R1F1A1074095). We thank Prof. Kun-Ho Lee (Department of Biomedical Science, Chosun University) for assistance with the biological evaluation and discussion.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
CCDC 2262119, 2262120, and 2262121 contain supplementary crystallographic data for compounds 1, 2, and 3, respectively. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; or deposit@ccdc.cam.ac.uk). Additional Supporting Information can be found online in the Supporting Information tab.
References
- Yamamoto, T.; Nakai, K.; Matsumura, A. Boron neutron capture therapy for glioblastoma. Cancer Lett. 2008, 262, 143–152. [Google Scholar] [CrossRef]
- Pisarev, M.A.; Dagrosa, M.A.; Juvenal, G.J. Boron neutron capture therapy in cancer: past, present and future. Arq Bras Endocrinol Metab. 2007, 51, 852–856. [Google Scholar] [CrossRef]
- Lesnikowski, Z.J. Collect. Czech. Boron units as pharmacophores – New applications and opportunities of boron cluster chemistry. Collect. Czech. Chem. Commun. 2007, 72, 1646–1658. [Google Scholar] [CrossRef]
- Tjarks, W.; Tiwari, R.; Byun, Y.; Narayanasamy, S.; Barth, R.F. Carboranyl thymidine analogues for neutron capture therapy. Chem. Commun. 2007, 4978–4991. [Google Scholar] [CrossRef] [PubMed]
- Bregadze, V.I.; Sivaev, I.B. Polyhedral Boron Compounds for BNCT in Boron Science: New Technologies and Applications, CRC Press, 2011, Chapter 9, pp. 187‒207.
- Armstrong, A.F.; Valliant, J.F. The bioinorganic and medicinal chemistry of carboranes: from new drug discovery to molecular imaging and therapy. Dalton Trans. 2007, 4240–4251. [Google Scholar] [CrossRef]
- Korbe, S.; Schreiber, P.J.; Michl, J. Chemistry of the Carba-closo-dodecaborate(−) Anion, CB11H12‒. Chem. Rev. 2006, 106, 5208–5249. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.F.; Hwang, J.-H.; Lee, J.-D.; Wee, K.-R.; Suh, I.-. H, Kang, S.O. A three-dimensional π-electron acceptor, tri-phenyl-o-carborane, bearing a rigid conformation with end-on phenyl units. Chem. Commun. 2013, 49, 9398–9400. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Ma, S.-Y.; Kang, S.O.; Lee, J.-D. B-phenylated o-carboranes and its chromium derivatives: Synthesis, electrochemical properties, and X-ray structural studies. J. Organomet. Chem. 2018, 865, 100–108. [Google Scholar] [CrossRef]
- Semmelhack, M.F. in: Abel, E.W.; Stone, F.G.A., Ed.; Wilkinson, G. (Eds), Transition Metal Arene Complexes: Nucleophilic Addition, Comprehensive Organometallic Chemistry II, vol. 12, Pergamon Press, Oxford, 1995, pp. 979‒1013. [Google Scholar]
- Rose-Munch, F.; Rose, E. in: Astruc, D. (Ed.), Arenetricarbonylchromium Complexes: Ipso, Cine, Tele Nucleophilic Aromatic Substitutions, Modern Arene Chemistry, Wiley-VCH, 2002, Chapter 11, pp. 368‒397.
- Jonson, T.R.; Mann, B.E.; Clark, J.E.; Foresti, R.; Green, C.J.; Motterlini, R. Metal Carbonyls: A New Class of Pharmaceuticals? Angew. Chem. Int. Ed. 2003, 42, 3722–3729. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Butler, I. Bioorganometallic Chemistry: A Future Direction for Transition Metal Organometallic Chemistry? Acc. Chem. Res. 1993, 26, 361–369. [Google Scholar] [CrossRef]
- Hess, A.; Metzler-Nolte, N. Transition metal labels on peptide nucleic acid (PNA) monomers. Chem. Commun. 1999, 885–886. [Google Scholar] [CrossRef]
- Baldoli, C.; Maiorana, S.; Licandro, E.; Zinzalla, G.; Perdicchia, D. Synthesis of Chiral Chromium Tricarbonyl Labeled Thymine PNA Monomers via the Ugi Reaction. Org. Lett. 2002, 4, 4341–4344. [Google Scholar] [CrossRef] [PubMed]
- Bruker Analytical X-Ray Division, SMART and SAINT, Madison, WI, 2002.
- Sheldrick, G.M. Bruker Analytical X-Ray Division, SHELXTL-PLUS Software Package, Madison, WI, 2002.
- Ohta, K.; Goto, T.; Endo, Y. 1,2-Dicarba-closo-dodecaboran-1-yl Naphthalene Derivatives. Inorg. Chem. 2005, 44, 8569–8573. [Google Scholar] [CrossRef] [PubMed]
- Kokado, K.; Nagai, A.; Chujo, Y. Poly(γ-glutamic acid) Hydrogels with Water-Sensitive Luminescence Derived from Aggregation-Induced Emission of o-Carborane. Macromolecules 2010, 43, 6463–6468. [Google Scholar] [CrossRef]
- Henly, T.J.; Knobler, C.B.; Hawthorne, M.F. Reactions of Anionic Carborane Nucleophiles with Chromium-Coordinated Haloarenes. Organometallics 1992, 11, 2313–2316. [Google Scholar] [CrossRef]
- Mahaffy, C.A.L.; Pauson, P.L. (η6-Arene)tricarbonylchromium Complexes, Inorganic Syntheses, vol. 19, 1979, pp. 154‒158.
- Fischer, R.D. IR-spektroskopische Untersuchungen der ν-CO-Banden an Metallcarbonylkomplexen rnit zentrisch-x-gebundenen organischen Ringsystemen. Chem. Ber. 1960, 93, 165–175. [Google Scholar] [CrossRef]
- Brown, D.A.; Sloan, H. Molecular-orbital Theory of Organometallic Compounds. Part I V.l Substitution Reactions of Tricarbonylbenzenechromium. J. Chem. Soc. 1963, 4389–4394. [Google Scholar] [CrossRef]
- Brown, D.A.; Raju, J.R. Infrared and Proton Magnetic Resonance Spectra of π-Complexes of Substituted Condensed Hydrocarbons. J. Chem. Soc. A. 1966, 1617–1620. [Google Scholar] [CrossRef]
- Adams, D.M.; Squire, A. Vibrational Spectra of Some Monosubstituted-π-arene Tricarbonylchromium Complexes and of Methyl Benzoate. J. Chem. Soc., Dalton Trans. 1974, 558‒565.
- Fox, M.A.; Nervi, C.; Crivello, A.; Low, P.J. Carborane radical anions: spectroscopic and electronic properties of a carborane radical anion with a 2n + 3 skeletal electron count. Chem. Commun. 2007, 2372–2374. [Google Scholar] [CrossRef]
- Boyd, L.A.; Clegg, W.; Copley, R.C.B.; Davidson, M.G. Fox, M.A.; Hibbert, T.G.; Howard, J.A.K.; Mackinnon, A.; Peace, R.J.; Wade, K. Exo-π-bonding to an ortho-carborane hypercarbon atom: systematic icosahedral cage distortions reflected in the structures of the fluoro-, hydroxy- and amino-carboranes, 1-X-2-Ph-1,2-C2B10H10 (X = F, OH or NH2) and related anions. Dalton Trans. 2004, 2786–2799. [Google Scholar]
- Fox, M.A.; Peace, R.J.; Clegg, W.; Elsegood, M.R.J.; Wade, K. Trends in ortho-carboranes 1-X-2-R-1,2-C2B10H10 (R = Ph, Me) bearing an exo-CN-bonded substituent group (X = NO, N=NRʹ or NHRʺ). Polyhedron 2009, 28, 2359–2370. [Google Scholar] [CrossRef]
- Lewis, Z. G.; Welch, A. J. Structure of 1,2-Diphenylcarbaborane, 1,2-Ph2-1,2-closo-C2B10H10. Acta Crystallogr., Sect. C. 1993, 49, 705–710. [Google Scholar] [CrossRef]
- Calhorda, M.J.; Frazão, C.F.; Martinho-Simões, J.A. Metal-Carbon “Bond Strengths” in Cr(CO)6, Cr(η-C6H6)2, and Cr(CO)3(η-C6H6). J. Organomet. Chem. 1984, 262, 305–314. [Google Scholar] [CrossRef]
- Rees, B.; Coppens, P. Electronic Structure of Benzene Chromium Tricarbonyl by X-ray and Neutron Diffraction at 78 ºK. Acta Crystallogr., Sect. B 1973, 29, 2516–2528. [Google Scholar] [CrossRef]
- Bailey, M.F.; Dahl, L.F. Three-Dimensional Crystal Structure of Benzenechromium Tricarbonyl with Further Comments on the Dibenzenechromium Structure. Inorg. Chem. 1965, 4, 1314–1319. [Google Scholar] [CrossRef]
- Wang, Y.; Angermund, K.; Goddard, R.; Kruger, C. Redetermination of the Experimental Electron Deformation Density of Benzenetricarbonylchromium. J. Am. Chem. Soc. 1987, 109, 587–589. [Google Scholar] [CrossRef]
- Czerwinski, C.J.; Guzei, I.A.; Riggle, K.M.; Schroeder, J.R.; Spencer, L.C. Haptotropic rearrangement in tricarbonylchromium complexes of 2-aminobiphenyl and 4-aminobiphenyl. Dalton Trans. 2011, 40, 9439–9446. [Google Scholar] [CrossRef]
- Guzei, I.A.; Spencer, L.C.; Buechel, S.C. Kaufmann, L.B. Czerwinski, C.J. Intricacies of ligand coordination in tricarbonylchromium(0) complexes with ortho- and para-fluorobiphenyls. Acta Crystallogr. Sect. C 2017, 73, 638–644. [Google Scholar] [CrossRef]
- Davidson, M. G.; Hibbert, T. G.; Howard, J. A. K.; Mackinnon, A.; Wade, K. Definitive crystal structures of ortho-, meta- and para-carboranes: supramolecular structures directed solely by C‒H···O hydrogen bonding to hmpa (hmpa = hexamethylphosporamide). Chem. Commun. 1996, 2285–2286. [Google Scholar] [CrossRef]
- Llop, J.; Viñas, C.; Oliva, J. M.; Teixidor, F.; Flores, M. A.; Kivekäs, R.; Sillanpää, R. Modulation of the C‒C distance in disubstituted 1,2-R2-o-carboranes. Crystal structure of closo 1,2-(SPh)2-1,2-C2B10H10. J. Organomet. Chem. 2002, 657, 232–238. [Google Scholar] [CrossRef]
- Oliva, J. M.; Allan, N. L.; Schleyer, P. v. R.; Viñas, C.; Teixidor, F. Strikingly Long C···C Distances in 1,2-Disubstituted ortho-Carboranes and Their Dianions. J. Am. Chem. Soc. 2005, 127, 13538–13547. [Google Scholar] [CrossRef] [PubMed]
- Hutton, B. W.; Maclntosh, F.; Ellis, D.; Herisse, F.; Macgregor, S. A.; McKay, D.; Petrie-Armstrong, V.; Rosair, G. M.; Perekalin, D. S.; Tricas, H.; Welch, A. J. Unprecedented steric deformation of ortho-carborane. Chem. Commun. 2008, 5345–5347. [Google Scholar] [CrossRef] [PubMed]
Chart 1.
Molecular structure of 1,2,3-triphenyl-o-carborane and its chromium(0) tricarbonyl complexes Ph3C2B, Ph3C2BCr2, and Ph3C2BCr3.
Chart 1.
Molecular structure of 1,2,3-triphenyl-o-carborane and its chromium(0) tricarbonyl complexes Ph3C2B, Ph3C2BCr2, and Ph3C2BCr3.
Scheme 1.
Preparation of 1, 2-diphenyl-o-carborane (1).
Scheme 2.
Preparation of mono- and bis- chromium complexes (2 and 3).
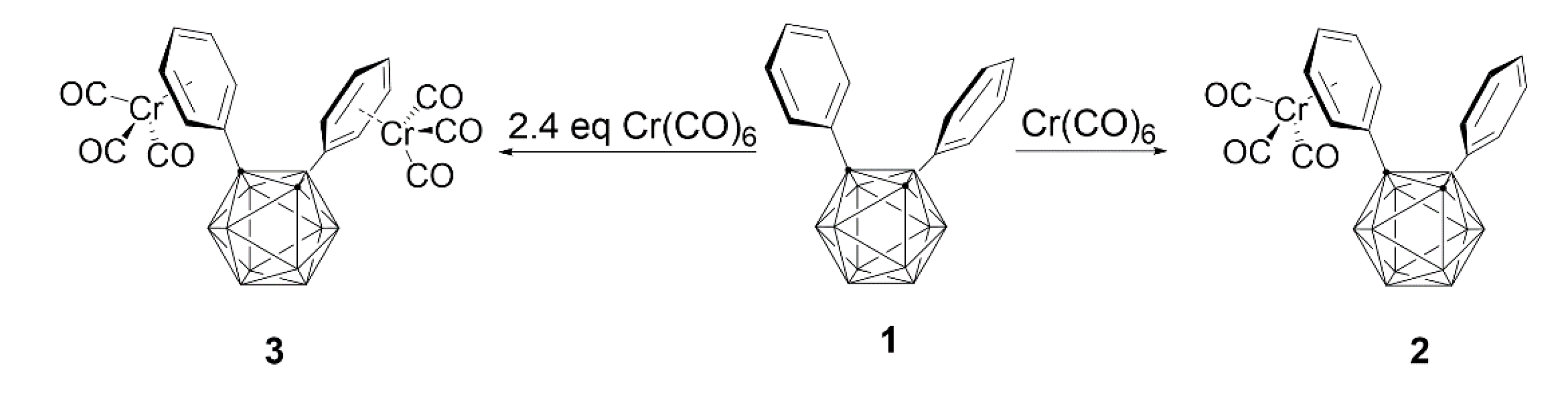
Figure 1.
ORTEP drawing (30% probability for thermal ellipsoids) of 1 (the hydrogen atoms are omitted for clarity).
Figure 1.
ORTEP drawing (30% probability for thermal ellipsoids) of 1 (the hydrogen atoms are omitted for clarity).
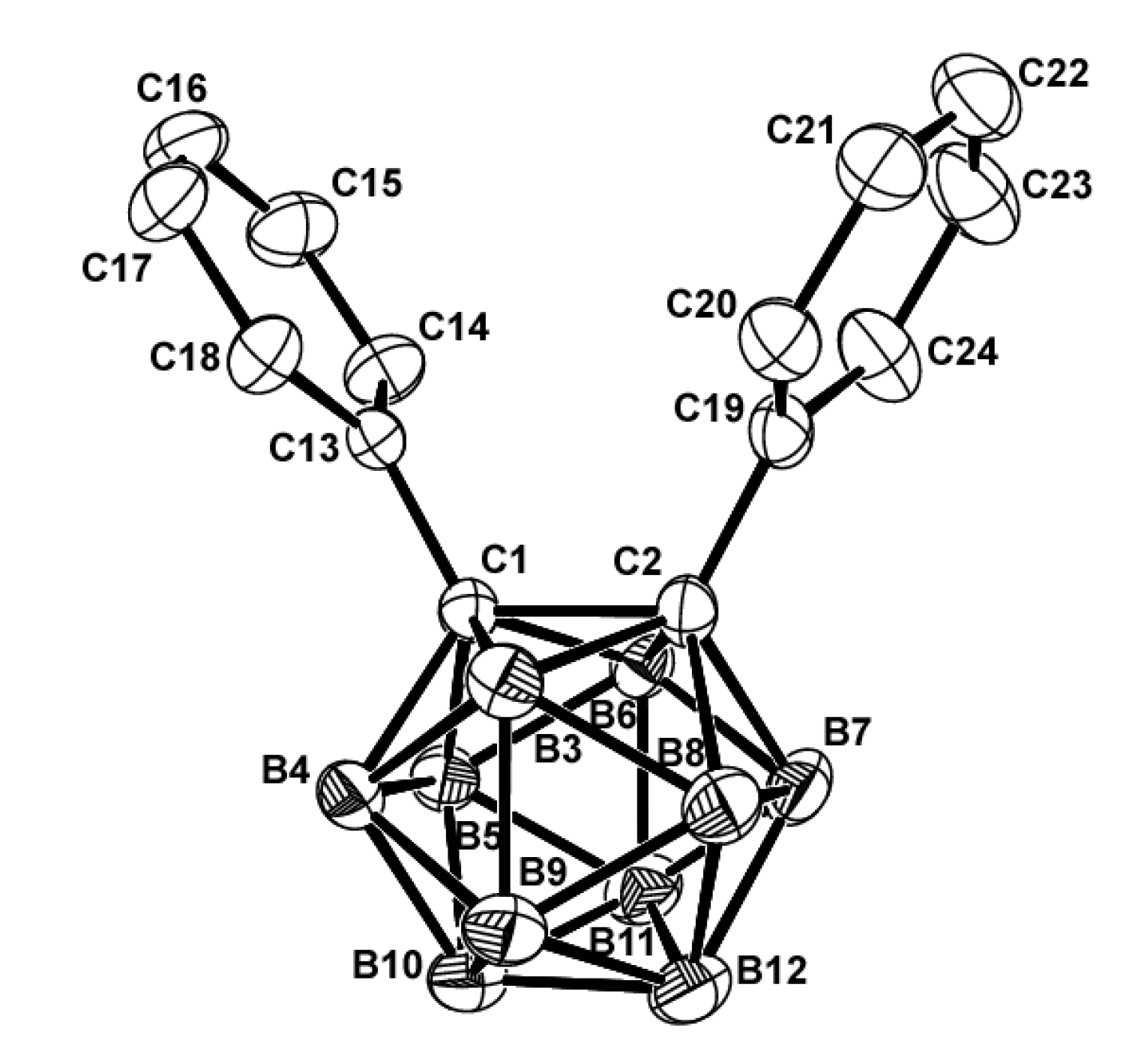
Figure 2.
ORTEP drawing (30% probability for thermal ellipsoids) of 2 (the hydrogen atoms are omitted for clarity).
Figure 2.
ORTEP drawing (30% probability for thermal ellipsoids) of 2 (the hydrogen atoms are omitted for clarity).
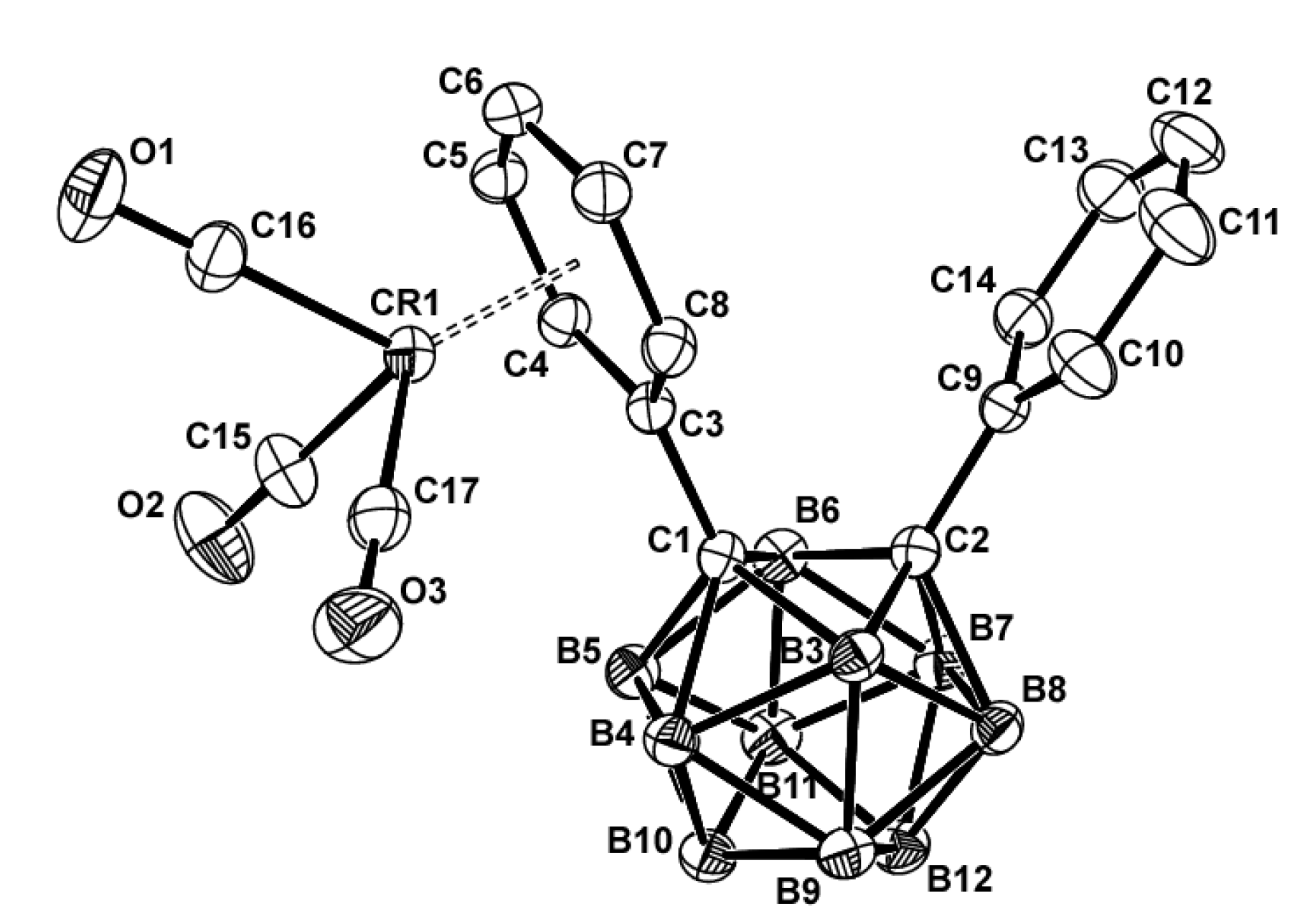
Figure 3.
ORTEP drawing (30% probability for thermal ellipsoids) of 3 (the hydrogen atoms are omitted for clarity).
Figure 3.
ORTEP drawing (30% probability for thermal ellipsoids) of 3 (the hydrogen atoms are omitted for clarity).
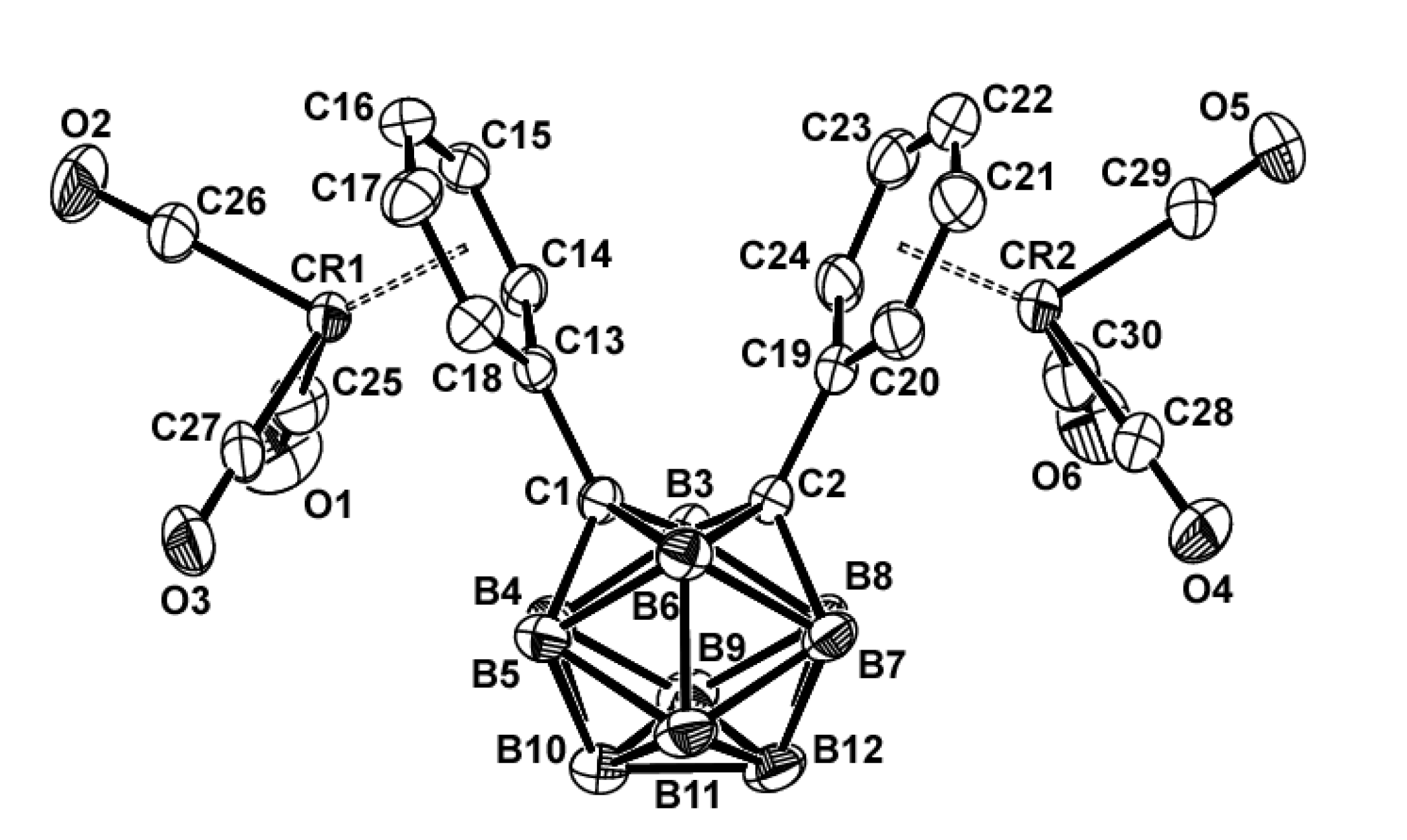

Table 1.
Crystal data and structure refinement of 1 ‒ 3.
| 1 | 2 | 3 | |
|---|---|---|---|
| Identification code Empirical formula Formula weight Temperature Wavelength Crystal system, space group Unit cell dimensions Volume Z, Dcalc F(000) Crystal size θ range for data collection Limiting indices Reflections collected/unique Completeness to θ = 28.38 Refinement method Data/restraints/parameters Goodness-of-fit on F2 Final R indices [I > 2θ(I)] R indices (all data) Largest diff. peak and hole |
K120504 C14H20B10 296.40 293(2) K 0.71073 Å Monoclinic, P21/n a = 10.859(1) Å b = 24.953(3) Å, β = 111.854(2)º c = 13.938(2) Å 3505.3(8) 8, 1.123 1232.0 0.15, 0.13, 0.12 1.63 to 28.37 ‒14 ≤ h ≤ 14, ‒33 ≤ k ≤ 32, ‒18 ≤ l ≤ 18 35826/8726 [R(int) = 0.0421] 99.3% Full-matrix least-squares on F2 8726/0/581 1.008 aR1 = 0.0616, bwR2 = 0.1545 aR1 = 0.1037, bwR2 = 0.1897 0.220 and ‒0.355 e.Å‒3 |
K130805 C17H20B10Cr1O3 432.43 293(2) K 0.71073 Å Monoclinic, P21/n a = 10.621(3) Å b = 17.056(5) Å, β = 106.622(5)º c = 12.174(4) Å 2113.2(1) 4, 1.359 880.0 0.17, 0.15, 0.13 2.12 to 28.46 ‒14 ≤ h ≤ 14, ‒22 ≤ k ≤ 22, ‒16 ≤ l ≤ 16 28490/5305 [R(int) = 0.0238] 99.2% Full-matrix least-squares on F2 5305/0/360 1.053 aR1 = 0.0332, bwR2 = 0.0958 aR1 = 0.0388, bwR2 = 0.1018 0.364 and ‒0.242 e.Å‒3 |
K131105 C20H20B10Cr2O6 568.46 293(2) K 0.71073 Å Triclinic, Pī a = 17.540(2) Å, α = 105.746(2)º b = 18.060(2) Å, β = 110.226(2)º c = 19.484(3) Å, γ = 91.256(2)º 5528.0(1) 2, 0.342 572 0.20, 0.20, 0.15 1.17 to 28.38 ‒23 ≤ h ≤ 23, ‒24 ≤ k ≤ 24, ‒26 ≤ l ≤ 25 44259/18014 [R(int) = 0.0398] 65.0% Full-matrix least-squares on F2 18014/0/1409 0.902 aR1 = 0.0563, bwR2 = 0.1505 aR1 = 0.0877, bwR2 = 0.1612 0.687 and ‒0.375 e.Å‒3 |
aR1 = ∑||Fo|-|Fc|| (based on reflections with Fo2>2σF 2), bwR2 = [∑[w(Fo2-Fc2)2]/∑[w(Fo2)2]]1/2; w = 1/[σ2(Fo2)+(0.095P)2]; , P = [max(Fo2, 0)+2Fc2]/3(also with Fo2>2σF 2).
Table 2.
Comparison of selected bond lengths (Å), angles (º), and torsion angles (º) for 1 ‒ 3.
 |
 |
 |
|
|---|---|---|---|
| PhC−C (av) | 1.376 | 1.400 | 1.406 |
| PhC−C (av)−Cr | 2.210 | 2.212 | |
| CabC−C | 1.726(2) | 1.740(2) | 1.724(4) |
| Cent−Cr | 1.702 | 1.696(av) | |
| Cr‒CO | 1.856(av) | 1.851(av) | |
| C1‒C13 | 1.507(2) | 1.499(2) | 1.502(4) |
| C2‒C19 | 1.501(2) | 1.500(2) | 1.510(4) |
| C13‒C1‒C2 | 118.3(1) | 116.6(1) | 116.4(2) |
| C19‒C2‒C1 | 119.0(1) | 119.6(1) | 116.1(2) |
| C1-C2-C19-C20 | 84.1(2) | 86.1(2) | 105.8(3) |
| C2-C1-C13-C14 | 81.7(2) | 102.3(1) | 112.4(3) |
Table 3.
Cytotoxicity (IC50) and Boron accumulation of B16 melanoma and CT26 colon carcinoma cells.
| Compds | B16 | CT26 | ||
|---|---|---|---|---|
| Cytotoxicity IC50 (M)a | Boron Accumulation (ppm)b | Cytotoxicity IC50 (M)a | Boron Accumulation (ppm)b | |
| 2 | 0.736 × 10‒6 (± 0.01) | 0.825 ± 0.003 | 0.833 × 10‒6 (± 0.03) | 0.755 ± 0.009 |
| 3 | 0.681 × 10‒6 (± 0.04) | 0.620 ± 0.002 | 0.314 × 10‒6 (± 0.07) | 0.694 ± 0.002 |
| 5 | 0.411 × 10‒6 (± 0.06) | 0.384 ± 0.006 | 0.164 × 10‒6 (± 0.05) | 0.402 ± 0.002 |
| 6 | 0.091 × 10‒6 (± 0.03) | 0.221 ± 0.001 | 0.089 × 10‒6 (± 0.08) | 0.247 ± 0.001 |
| BPA | 4.871 × 10‒5 (± 0.03) | 0.103 ± 0.002 | 3.862 × 10‒3 (± 0.04) | 0.514 ± 0.001 |
aB16 melanoma and CT26 colon carcinoma cancer cells (5 ×103 cells) were incubated for 72 h in the presence of compounds 2, 3, 4, and 6, and then the percentages of viable cells were determined by MTT assay. The drug concentrations required to inhibit cell viability by 50% (IC50) were determined from semi-logarithmic concentration-response plots, and the results represent the means ± s.d. of triplicate samples. bB16 and CT26 cells (5 ×105 cells) were incubated for 3 h in the presence of compounds 2, 3, 4, and 6 or BPA (10.8 ppm). After three times washes, the accumulated boron concentrations were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES). The values are the means ± s.d. from three samples.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



