
Submitted:
30 May 2023
Posted:
02 June 2023
You are already at the latest version
Abstract
Ferroptosis is a newly discovered iron-dependent form of regulated cell death driven by phospholipid peroxidation, associated with processes including iron overload, lipid peroxidation and dysfunction of cellular antioxidant systems. Ferroptosis is found closely related to many diseases including cancer, at its every stage. Epithelial-mesenchymal transition (EMT) in malignant tumors that originate from epithelia promotes cancer cell migration, invasion and metastasis by disrupting cell-cell and cell-martrix junctions, cell polarity, etc. Recent studies have shown that ferroptosis appears to share multiple initiators and overlapping pathways with EMT in cancers and identify ferroptosis as a potential predictor of various cancer grade and prognosis. Cancer metastasis involves multiple steps including local invasion of cancer cells, intravasation, survival in circulation, arrest at a distant organ site, extravasation and adaptation to foreign tissue microenvironments, angiogenesis and the formation of “premetastatic niche”. Numerous studies have revealed that ferroptosis is closely associated with cancer metastasis. From the cellular perspective, ferroptosis has been implicated in the regulation of cancer metastasis. From the molecular perspective, the signaling pathways activated during the two events interweave. This review briefly introduces the mechanisms of ferroptosis, and discusses how ferroptosis is involved in cancer progression including EMT, cancer angiogenesis, invasion and metastasis.
Keywords:
ferroptosis
; cancer
; EMT
; angiogenesis
; metastasis
1. Ferroptosis
1.1. Brief Description of Iron Metabolism
Although only a trace element, iron plays an essential role in cellular metabolic processes. Iron is present in many forms in the diets and absorbed in the intestine. The most well-known way of absorption is in the form of non-heme iron [1]. Fe3+ in food is at first reduced to Fe2+ by duodenal cytochrome B and possibly other reducing enzymes, and then crosses the brush border of enterocytes via divalent metal-ion transporter 1 (DMIT1). Enterocyte iron is exported to the blood via ferroportin 1 (FPN1) on the basolateral membrane. Ferroxidase hephaestin on the basolateral membrane oxidizes Fe2+ to Fe3+ so that it binds to plasma transferrin (TF) and is distributed throughout the body in the form of transferrin-iron complexes. When iron-loaded TF binds to transferrin receptor 1 (TfR1) on cell plasma membrane, cells can take up iron by endocytosis. In addition, iron in diets can also be absorbed in the form of heme iron, mainly found in hemoglobin and myoglobin. However, the mechanism of heme iron absorption needs yet to be further investigated [2].
Iron uptake is essential for many key biological processes, such as oxygen transport, DNA replication, and mitochondrial function, etc. A large fraction of intracellular iron enters the mitochondria and is stored or used for the synthesis of heme and iron-sulfur clusters [3]. Heme is engaged in oxygen transport while iron-sulfur clusters are involved in electron transfer in the respiratory chain and act as cofactors for many other proteins required for critical cellular activities. Iron that is not utilized at once is stored in ferritin in ion state and can be mobilized by ferritin phagocytosis when needed. On the whole, the intracellular iron content is regulated by the Iron regulatory element-Iron regulatory protein (IRE-IRP) system, while the whole-body iron supply is primarily regulated by the hepcidin-ferroportin (FPN) axis in response to the body's iron requirements [4].
Iron is exerted from the body through sloughing of intestinal epithelial cells, exfoliation of skin cells, and physiologic blood loss due to menstruation or minor trauma to epithelial linings. These are passive pathways that are not regulated by iron levels or factors related to iron homeostasis [5].
1.2. Mechanisms of Ferroptosis
1.2.1. Iron Overload
The imbalance of iron metabolism homeostasis results in increased intracellular free iron, which is one of the important features of ferroptosis [6]. Although the specific mechanisms involved in ferroptosis are not yet clear, there is much evidence that ferrous iron plays the critical role in this process.
By using iron chelators, Dixon et al successfully blocked ferroptosis both in vivo and in vitro. They also found that exogenous iron supplementation increased the sensitivity of cells to ferroptosis inducers [7]. Hou et al observed an increase in cellular labile iron during the induction of ferroptosis [8]. Li et al found that excess iron, both heme and non-heme iron, can directly induce ferroptosis [9]. In addition, excess iron produces reactive oxygen species (ROS) through the Fenton reaction and the Haber-Weiss reaction, triggering oxidative stress and promoting lipid peroxidation through non-enzymatic reactions [10].
Studies about iron metabolism in ferroptosis suggest that key molecules in the process of iron metabolism might all be regulators of cellular sensitivity to ferroptosis [11]. Given that drug-resistant cancer cells are vulnerable to ferroptosis and that a number of organ injuries and degenerative pathologies are driven by ferroptosis, therapeutic ideas targeting ferroptosis might offer a new turnaround for many diseases.
1.2.2. Lipid Peroxidation
Polyunsaturated fatty acids (PUFAs) are susceptible to peroxidation during ferroptosis. The enzymes ACSL4 (Acyl-CoA synthetase long-chain family member 4) and LPCAT3 (lysophospholipid acyltransferase 3) are closely related to the synthesis of phospholipids containing polyunsaturated fatty acids (PUFA-PLs). ACSL4 can cause the enrichment of long PUFAs in cell membranes [12]. LPCAT3 promotes the combination of PUFAs with phospholipids to form PUFA-PLs, which are susceptible to free radicals-induced oxidation catalysed by lipoxygenase.
Fatty acid synthesis mediated by acetyl-CoA carboxylase (ACAC) as well as release mediated by lipophagy induce accumulation of intracellular free fatty acids, thereby accelerating ferroptosis. AMPK-mediated ACAC phosphorylation inhibits ferroptosis by preventing the synthesis of PUFAs [13].
1.2.3. Antioxidant Systems
The glutathione peroxidases (GPX) family is considered to be an important protective system in lipid peroxidation, among which GPX4 plays a key role in inhibiting ferroptosis by reducing phospholipid hydroperoxides to hydroxyphospholipid directly [16]. Drugs that inhibit GPX4 (Class Ⅱ ferroptosis-inducers) like RSL3, ML162, ML210, can induce ferroptosis [17]. The containing of selenocysteine confers GPX4 increased antiferroptotic activity [18], and GSH as an essential cofactor, helps GPX4 reduce lipid hydroperoxide to lipid alcohols [19].
Cystine/glutamate transporter (also known as system Xc-) transports cystine (Cys2) into cells, which is then oxidized to cysteine (Cys). Cysteine is used to synthesize GSH under the catalysis of glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS). System Xc- is consist of two subunits, SLC7A11 (solute carrier family 7A member 11) and SLC3A2 (solute carrier family 3A member 11). The expression and activity of SLC7A11 is positively regulated by the transcription factor nuclear factor E2-related factor 2 (NRF2) [20] and negatively regulated by p53 [21], which helps to control the level of GSH in ferroptosis. System Xc- inhibitors (class I ferroptosis-inducers) including erastin, sulfasalazine and sorafenib can thus induce lipid peroxidation and ferroptosis [17].
The NADPH-FSP1-CoQ10 pathway is another important antioxidant pathway independent from the system Xc-GSH-GPX4 axis. CoQ10 is mainly synthesized in mitochondria, and its reduced form, CoQ10H2, is a robust lipophilic antioxidant [22]. FSP1, ferroptosis inhibitor protein 1, is recruited to the plasma membrane to reduce CoQ10 to CoQ10H2, which effectively blocks the spread of lipid peroxides [23].
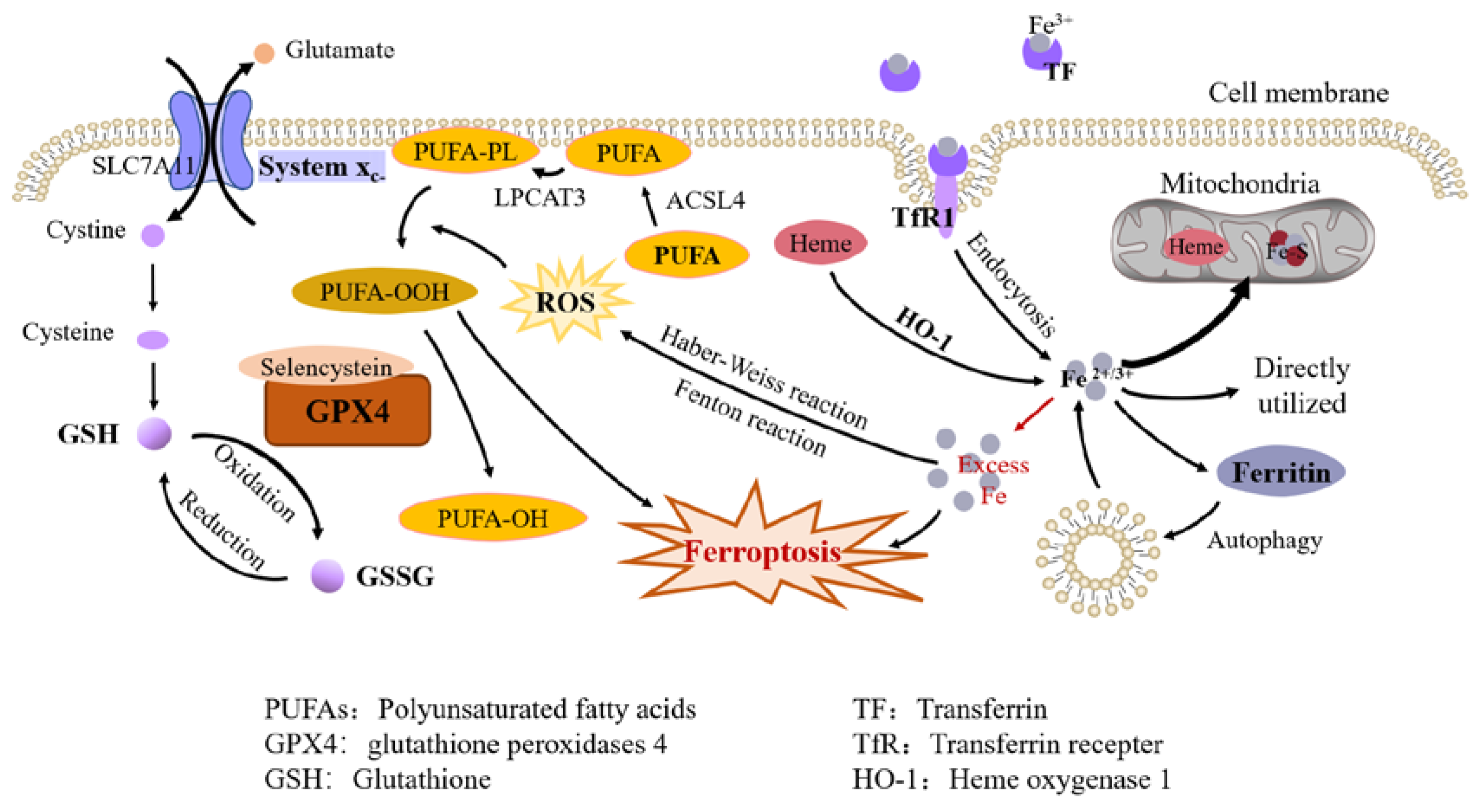
Figure 1.
Molecular mechanisms of ferroptosis.
2. Ferroptosis and EMT in Cancer
Epithelial-mesenchymal transition (EMT) is the process by which epithelial cells lose specific epithelial phenotypes and acquire mesenchymal phenotypes and behaviors. The concept of EMT was first proposed during embryonic development [24], and subsequently observed in other processes including tissue repair, organ fibrosis, and cancer metastasis [25].
2.1. Characteristics and Mechanisms of EMT
Epithelial cells are connected by cell junctions, including adhesion junctions, tight junctions, gap junctions and desmosomes. Cells are attached to the underlying basement membrane via hemidesmosomes, and present apical-basal polarity. During epithelial-mesenchymal transition, the cell junctions are substituted by cell-matrix junctions, and the apical-basal polarity is replaced by front-back polarity. The cytoskeleton is reprogrammed so that the cells turn into a spindle-shaped mesenchymal morphology and are conferred the ability of migration and invasion to the basement membrane [26]. Furthermore, hallmarks of cells are also greatly altered during EMT, mainly presented in the loss of epithelial markers and acquisition of mesenchymal markers. E-cadherin, the epithelial marker, is downregulated, while N-cadherin and vimentin, the mesenchymal markers, are upregulated during EMT [25].
EMT transcription factors (EMT-TFs) play a central role in the process of EMT. The major EMT-TFs include SNAI1/2, TWIST1/2, ZEB1/2, which are involved in the repression of epithelial associated genes and promotion of mesenchymal associated genes. SNAI1/2 and ZBE1/2 can directly bind to the E-box of the promoter of CDH1, which encodes E-cadherin, to repress its expression, while TWIST represses it indirectly [27]. SNAI1 and ZEB can downregulate the expression of genes that related the formation of tight junctions and apical-basal polarity. TWIST and ZEB1/2 can increase the expression of vimentin and N-cadherin, and SNAI1 can act as a transcriptional activator to directly prompt mesenchymal gene expression. Alterations in the expression of these genes can affect cell-cell adhesion, cell polarity and motility, resulting in epithelial-mesenchymal transition of cells [28].
EMT-TFs could all be regulated by multiple miRNAs and post-translational modifications, including phosphorylation, ubiquitination, SUMOylation, acetylation, and histone acetytransferases(HATs) [28,29].
Many other factors have been reported participated in EMT, including various growth factors, Wnt pathway, Notch pathway etc. For example, TGF-β interacts with the transcription factors, SNAI, ZEB and TWIST, through the Smad pathway to affect the transcription of epithelial and mesenchymal-associated genes [30].
2.2. Ferroptosis and EMT in Cancer
EMT is divided into three distinct subtypes, which are involved in embryonic development [25], wound healing and tissue regeneration [31], and cancer metastasis respectively. Type-Ⅲ EMT is involved the progression of cancer. As mentioned above, losing of cell-cell junctions and altering of cell polarity during EMT endow the cancer cells the ability to migrate, invade into adjacent tissues, intravasate into vascular or lymphatic vessels and then enter the systemic circulation [26].
Some studies suggest that ferroptosis and EMT in cancer promote each other, forming a positive feedback loop. E-cadherin inhibits EMT by affecting cell-cell contacts while protects cells from ferroptosis in experiments in vitro [32]. Enhancing the expression of EMT transcription factors such as SNAI, TWIST and ZEB reverses the ferroptosis sensitivity of cells. Overexpression of ZEB1 increases cellular susceptibility to ferroptosis, while silencing ZEB1 exhibits the opposite effect. Epigenetic reprogramming of EMT-TF, such as the elimination of CDH1 hypermethylation, promote the expression of E-cadherin, decreased ferroptosis susceptibility [33]. Studies on human adrenal cortical carcinoma lines have found that sensitivity to ferroptosis was augmented due to the increased iron accumulation, ROS production and decreased antioxidant genes in mesenchymal-like cells [34]. A strong correlation between ferroptosis and EMT was shown in RNA-seq and analysis of TCGA dataset, with the high tendency of ferroptosis and EMT suggesting poor prognosis of bladder cancer and lung adenocarcinoma [35,36]. Ferroptosis-related genes are used in the risk prognostic model of bladder cancer and colon adenocarcinoma [37]. While there is also research found that the EMT transcription factor Slug, also known as SNAI2, protects glioblastoma cell from ferroptosis by reversing the protein levels of SLC7A11, a subunit of system Xc-, which might contribute to the treatment of glioblastoma [38].
Ferroptosis is considered a regulator of EMT in the studies of various types of cancers. Inhibition of ferroptosis promotes epithelial-mesenchymal transition of cancer cells, leading to cancer migration, metastasis and poor prognosis. Both ferroptosis inhibition and EMT induction occurr in IRF2 overexpressed glioma cells and ARNTL2 overexpressed lung adenocarcinoma cells, which also suggest poor prognosis of both kinds of cancer [39]. Mechanistically, Some processes related with ferroptosis like ferritinophagy and ROS production were found to be involved in drug-induced EMT inhibition in hepatocellular carcinoma [40] and gastric cancer cell lines [41], and the inhibition of EMT was reversed with the Fer-1, a ferroptosis inhibitor. CST1 inhibits ferroptosis by increasing the stability of GPX4, a key protein of ferroptosis, and reducing intracellular reactive oxygen species, thereby promotes migration and invasion via EMT in gastric cancer cells [42]. In turn, the induction of ferroptosis prevents cancer cells from EMT, and inhibits cancer progression. NRF2 is an essential regulator of the cellular antioxidant system. Normally located in the cytoplasm, NRF2 is translocated to the nucleus and functions upon oxidative stress [8]. NRF2 expression is controlled mainly by Keap1 through proteasomal degradation [43]. The activation of the Keap1/NRF2 pathway facilitates the inhibition of EMT, a process that may act through the induction of ferroptosis [41,44]. Inhibition of PI3K/AKT signaling pathway induces ferroptosis in KLF2 overexpressed colorectal cancer(CRC) cells, which also results in EMT suppression of CRC cells and ultimately inhibits progression and metastasis of CRC [45].
In summary, iron plays an important regulatory role both in EMT and ferroptosis, and there is a complicated regulatory network between ferroptosis and EMT in cancer, which exhibits different interactions in specific contexts.
3. Ferroptosis and Cancer Invasion and Metastasis
3.1. Cancer Angiogenesis, Invasion and Metastasis
3.1.1. Cancer Angiogenesis
Angiogenesis is the process of growing new blood vessels from existing ones and includes degradation of the vascular basement membrane, activation, proliferation and migration of endothelial cells (ECs), and generation of new blood vessels. Angiogenesis is one of the characteristics of cancer development [46]. Under normal physiological conditions, the vascular system remains quiescent for a long time after maturation of the organism, with transient angiogenesis occurring only when the organism is damaged. Angiogenesis is essential for cancer growth and metastasis, because by constant angiogenesis, the oxygen and nutrient demands of tumor cells are met [47].
The relationship between tumor growth and angiogenesis was investigated by Folkman, who found that when the blood supply to the tumor is cut off, the tumor shrinks or regresses due to ischemia [48]. Later, Dhar et al found significantly higher levels of angiogenesis in differentiated thyroid cancer [49], and Mohammed et al found that high levels of angiogenesis were closely associated with metastasis and recurrence as well as high mortality in colorectal cancer [50]. In addition, several independent research groups examined angiogenesis levels in various tumor samples using immunohistochemical techniques and found that angiogenesis was significantly upregulated in several common malignancies.
As research has progressed, the understanding of the mechanisms of cancer angiogenesis-related signaling pathways and their effects on cancer growth, metastasis, and invasion has increased. Two types of signaling pathways have been extensively studied, namely signaling pathways that regulate vascular permeability and those involved in vascular remodeling and vascular maturation. Among the signaling pathways that regulate vascular permeability, the most important is the VEGF/VEGFR signaling pathway. Among the regulatory pathways involved in vascular remodeling and maturation, the angiopoietin/Tie-2 signaling pathway is the most important. In addition, several other signaling pathways associated with angiogenesis are also involved in the regulation of vascular permeability and/or maturation. These signaling pathways are interrelated and interacted with each other to promote the proliferation of vascular ECs and induce tumor angiogenesis, thus providing favorable conditions for malignant tumor proliferation, invasion and metastasis [51].
3.1.2. Process of Cancer Invasion and Metastasis
Cancer metastasis is a multistep process that involves local invasion of cancer cells, intravasation, survival in circulation, arrest at a distant organ site, extravasation, and adaptation to foreign tissue microenvironments [52].
Mutations in oncogenes may be one of the initial leading factors in this process [53]. Active protein hydrolysis by the action of MMPs (matrix metalloproteinases) can alter the tumor microenvironment and promote the absence of the basement membrane barrier, providing suitable conditions for cancer cell invasion. After crossing the basement membrane, cancer cells enter the stroma and interact with tumor-associated stromal cells [54]. The stimulation of cancer cells makes stroma "reactive", while cytokines and growth factors, such as interleukin-6 (IL-6) and EGF (epidermal growth factor), secreted by stromal cells enhance the malignant traits of cancer cells [55,56].
Molecular changes allow cancer cells to cross the pericytes and endothelial cells barriers, promoting intravasation [57]. As mentioned above, cancer cells stimulate the formation of new blood vessels within their microenvironment. These new blood vessels are more tortuous and prone to leakiness compared to normal blood vessels, which creates favorable conditions for tumor cells to enter the circulation [58].
In order to reach distant organ sites, circulating tumor cells (CTCs) must cope with a certain amount of survival stresses [57]. Studies have shown that platelets and their activation play a crucial role in the spread of tumor cells. Platelets in circulation are cross-linked by adhesion molecules and bind fibrinogen to form an additional immune escape layer that can protect tumor cells from killing by NK cells [59,60].
Tumor cells that survive successfully in circulation can theoretically disseminate throughout the body, but in practice, certain types of tumors tend to metastasize to specific organs. Filder et al explain the distant metastasis of tumor cells with the "seed and soil" hypothesis [61]. They believe that successful metastasis depends on the interaction between metastatic cells and local homeostatic mechanisms. Another hypothesis suggests that CTCs themselves have a certain preference. Some cancer cells are able to adhere to specific tissues [62].
After being arrested by a distant organ, CTCs may begin to proliferate intraluminally and form a microcolony that eventually breaks the surrounding vessel wall [63]. In addition, cancer cells may penetrate the endothelial and pericyte layers, entering the tissue parenchyma directly. This process is known as extravasation. Primary tumors express proteins such as secreted protein angiopoietin-like-4 (Angptl4) and pleiotropically acting factors COX-2, and MMP, to disrupt the microenvironment of distant organ site and increase permeability [64,65].
After reaching the target organ site, tumor cells need to adapt to the new microenvironment and colonize there. Some researchers believe that "premetastatic niche" formed at target site by invading bone marrow-derived cells (BMDCs) are involved in this progress [66]. Erler et al demonstrated that lysyl oxidase (LOX) secreted by hypoxic breast tumor cells is able to accumulate at the target site and cross-link collagen IV, thereby recruiting CD11b+ cells. CD11b+ cells produce matrix metalloproteinase-2 (MMP2), enhancing the invasion and recruitment of BMDCs and promoting cancer metastasis [67].
3.2. Relationship between Ferroptosis and Cancer Angiogenesis, Invasion and Metastasis
Since ferroptosis was formally defined in 2012 [71], studies regarding ferroptosis and tumor biological behaviors have attracted a lot of interest. Studies have shown that there exist crosstalks between ferroptosis and tumor-associated signaling pathways, including RAS, TP53, NFE2L2, HIF pathways and so on, which serves as a basis for investigating new targets for tumor therapy [72].
Under certain conditions, hypoxia induces angiogenesis, which is controlled by a family of hypoxia inducible factors (HIFs), including HIF-1α and HIF-2α [73]. HIF-1α instability in non-small cell lung cancer cells has been reported to reduce the susceptibility to ferroptosis [74]. In addition, HIF-1α knockdown or HIF-2α ablation in renal clear cell carcinoma cells alone was found to reduce the susceptibility of tumor cells to ferroptosis [75]. Thus, the HIF pathway may be a major driver of ferroptosis susceptibility in several tumors and is essential for promoting ferroptosis in tumor cells.
Most of the factors that promote tumor angiogenesis play a role in inhibiting tumor ferroptosis. Exosomes accelerate tumor growth by participating in neovascularization. In RSL3-induced ferroptosis in breast cancer cells, exosome secretion is a protective mechanism against ferroptosis [76]. Exosomal miR-522 secreted by CAFs also inhibits ferroptosis in gastric cancer [77]. Integrins not only promote tumor metastasis, but also contribute to tumor angiogenesis. Integrin α6β4 protects breast cancer cell-adherent epithelial cells and cancer cells from erastin-induced ferroptosis [78], and integrins have an inhibitory role in driving the ferroptosis pathway in breast cancer cells.
A research conducted by Chen et al regarding the anti-cancer mechanism of erianin revealed that erianin could inhibit cell proliferation, promote G2/M-phase arrest, trigger ferroptosis, and suppress migration in lung cancer cells [79]. Ferroptotic events, including ROS accumulation, GSH depletion, and lipid peroxidation, were significantly triggered following treatment with erianin. In addition, pretreatment with the ferroptosis inhibitor Fer-1, Lip-1, or DFO reduced erianin-induced cell death and suppressed cell migration. They hypothesized that erianin targets calmodulin (CaM) and activates the Ca2+/CaM signaling pathway, leading to elevated Ca2+ and Fe2+ levels that triggers ferroptosis.
You et al constructed a scoring system based on ferroptosis-related genes to investigate the relationship between ferroptosis and clinical features of ovarian cancer [80]. The results showed that cluster with higher ferroptosis-resistant-related genes has shorter median survival times. Critically, this cluster also exhibited aggressive growth patterns, including blood infiltration and lymphatic infiltration, suggesting that ferroptosis may affect ovarian cancer progression by regulating invasion ability.
Nassar et al investigated that DECR1, encoding the rate-limiting enzyme for oxidation of PUFAs, is highly expressed in prostate cancer (PCa) tissues [81]. DECR1 knockdown selectively inhibits β-oxidation of PUFAs, enhances oxidative stress and lipid peroxidation in mitochondria [82], and ultimately leads to ferroptosis. Nassar et al also suggested that the consistently increased expression of DECR1 in PCa tissue might contribute to PCa cell viability and invasive behaviour [81].
Ubellacker et al conducted a research on melanoma metastasis [83]. The results showed that melanoma cells in lymph experience less oxidative stress and form more metastasis than those in blood. Differences in lymph and plasma, such as higher levels of glutathione and oleic acid and lower levels of free iron, help reduce ferroptosis in melanoma cells in lymph. They also tentatively demonstrated that the function of oleic acid in protecting melanoma cells from ferroptosis is associated with acyl-CoA synthetase long-chain family member 3 (ACSL3).
Lu et al found that ACADSB, an acyl-CoA dehydrogenase localized in mitochondria and nucleus, was downregulated in colorectal cancer tissues. They demonstrated that the overexpression of ACADSB inhibits CRC cell migration, invasion, and proliferation, while promotes ferroptosis [84]. In other words, ACADSB may exert suppressive functions against colorectal cancer, which is consistent with the previously identified role of ACADSB in poor-differentiated hepatocellular carcinoma and renal clear cell carcinoma [85,86]. The mechanism of this function might be that ACADSB can promote the lipid metabolism via catalyzing the dehydrogenation of acyl-CoA derivatives [87].
Zhang et al discovered that circRHOT1 contributed to invasion and metastasis and attenuated ferroptosis in breast cancer by regulating the miR-106a-5p/STAT3 (signal transducer and activator of transcription 3) axis [88]. Cao et al showed that glucose-6-phosphate dehydrogenase (G6PD) can promote proliferation, migration and invasion of hepatocellular carcinoma (HCC) and inhibit ferroptosis by downregulating POR (cytochrome P450 oxidoreductase) [89]. Xu et al confirmed that SLC7A11 could promote the proliferation, migration, and invasion of renal cancer cells (RCCs) by enhancing GPX4 output, which in turn inhibits ferroptosis [90].
ACSL4, an important molecule in ferroptosis, is shown to be associated with migration, proliferation and invasion of 17β-estradiol-induced cancers [91]. The importance of hypoxia-inducible factor-1 (HIF-1) in tumor development has long been demonstrated [92]. Studies in recent years have shown that HIF is involved in regulating the expression of genes related to lipid metabolism, such as SCD1 and fatty-acid desaturase 2 (FADS2), thus affecting ferroptosis [74]. In addition, HIF-2 activates the expression of hypoxiainducible, lipid droplet-associated protein (HILPDA), which induces a ferroptosis-susceptible cell state [75]. While p53-mediated cell-cycle arrest, senescence, and apoptosis are critical barriers to cancer development, P53 also plays an important role in cellular metabolism, oxidative responses and ferroptosis [93]. P53 can both enhance ferroptosis by promoting the accumulation of lipid hydroperoxides and suppress ferroptosis by decreasing the accumulation of lethal lipid peroxides (LPO) [93,94,95].
A recent study conducted by Wang et al showed that RB1 loss/E2F activation sensitized cancer cells to ferroptosis by upregulating expression of ACSL4 and enriching ACSL4-dependent arachidonic acid–containing phospholipids [96]. Upon RB1 loss, E2F transcription factors induce ACSL4 expression, resulting in PUFA-PL accumulation and a high ferroptotic potential, which is kept in check by GPX4. This is consistent with the fact that GPX4 is abundantly expressed in most cancer cells. However, this strong ferroptotic potential is unblocked when RB1-deficient cells are treated with GPX4 inhibitors, resulting in massive ferroptosis [97].
Several studies have suggested an inextricable link between ferroptosis and cancer invasion and metastasis, but further studies are necessary to clarify the specific and comprehensive mechanisms.
On the other hand, research in this field can also provide new ideas for cancer treatment [79,98]. Small molecules-induced ferroptosis has been found to have a strong inhibition on tumor growth and enhances the sensitivity of chemotherapeutic drugs, especially in the condition of drug resistance [99].
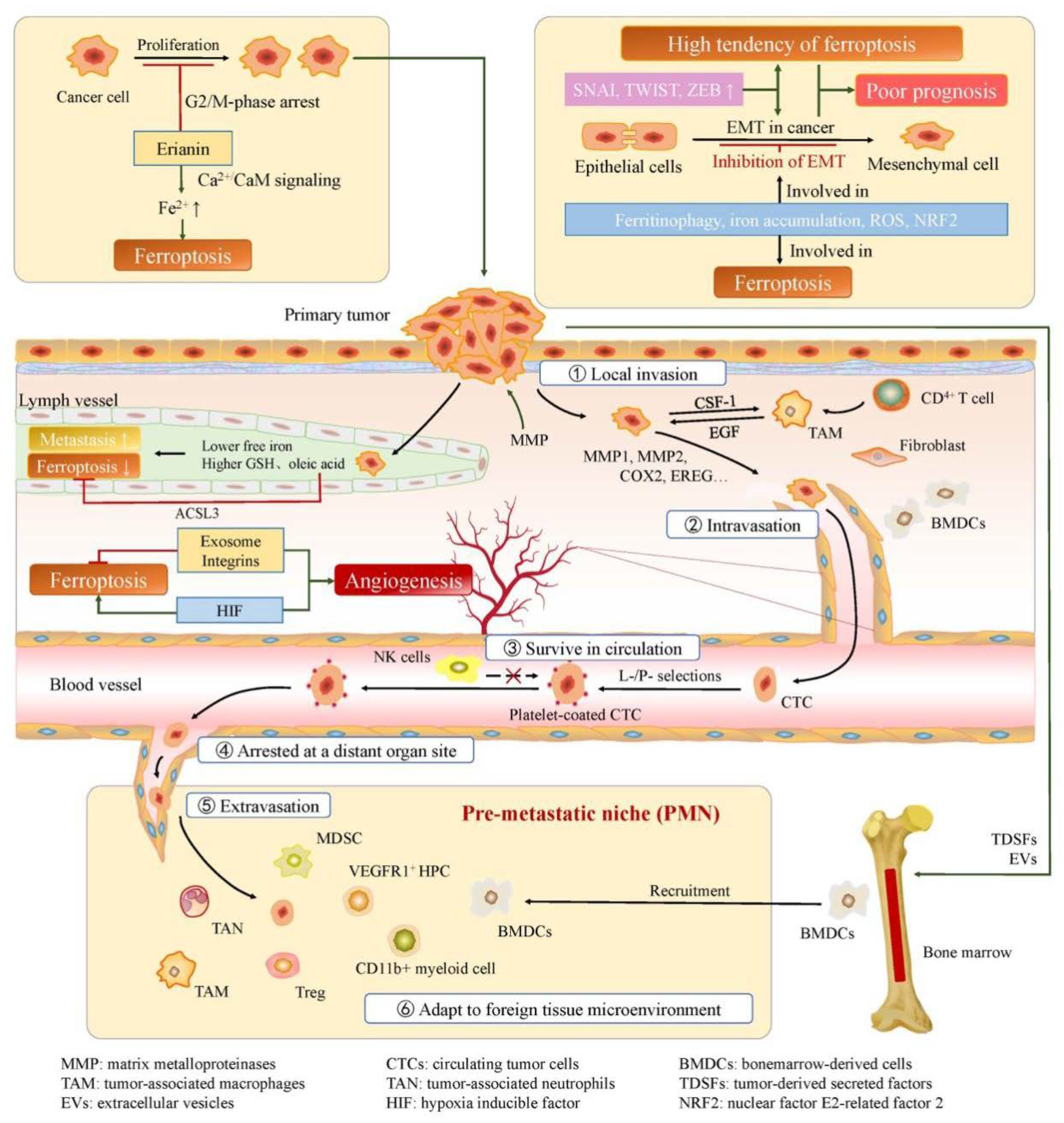
Figure 2.
Ferroptosis in cancer progression.
4. Conclusions
Since the discovery of ferroptosis as a novel regulated cell death, extensive attention has been drawn to investigate it, resulting in a more comprehensive understanding of iron metabolic processes and ferroptosis mechanisms. Recent studies have revealed close links between ferroptosis and tumor malignancy, which propose that the expression of ferroptosis-associated genes could serve as prognostic indicators for cancer patients, and that targeting ferroptosis may be a potential strategy to prevent cancer metastasis and drug resistance. Additional studies have demonstrate that ferroptosis inducers may be able to impede tumor infiltration and metastasis. Thus, the application of tumor therapy based on the molecular regulation mechanism of ferroptosis has a great research prospect and development potential.
Author Contributions
R.Z., J.C., S. W., and R.C. figured out the idea of writing this review. R.Z., J.C., S. W., and W.Z summarized the published results and drafted the manuscript. Q.Z. and R.C. revised the manuscript.
Funding
The review was funded by the Innovative Research Team of High-level Local Universities in Shanghai, and supported by National Natural Science Foundation of China 81872230, 82173352 to R.C.).
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fuqua, B.K.; Vulpe, C.D.; Anderson, G.J. Intestinal iron absorption. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2012, 26, 115–119. [Google Scholar] [CrossRef]
- West, A.-R.; Oates, P.-S. Mechanisms of heme iron absorption: Current questions and controversies. World J. Gastroenterol. 2008, 14, 4101–4110. [Google Scholar] [CrossRef]
- Dutt, S.; Hamza, I.; Bartnikas, T.B. Molecular Mechanisms of Iron and Heme Metabolism. Annu. Rev. Nutr. 2022, 42, 311–335. [Google Scholar] [CrossRef]
- Mleczko-Sanecka, K.; Silvestri, L. Cell-type-specific insights into iron regulatory processes. Am. J. Hematol. 2021, 96, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Koleini, N.; Shapiro, J.S.; Geier, J.; Ardehali, H. Ironing out mechanisms of iron homeostasis and disorders of iron deficiency. J. Clin. Investig. 2021, 131, e148671. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhang, Y.; Guo, H.; Hai, Y.; Luo, Y.; Yue, T. Mechanism and intervention measures of iron side effects on the intestine. Crit. Rev. Food Sci. Nutr. 2020, 60, 2113–2125. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Reviews. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol 2020, 22, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 2016, 113, E4966–4975. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat Chem Biol 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e421. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol Cell 2017, 68, 224–232.e224. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Stoker, M.; Perryman, M. An epithelial scatter factor released by embryo fibroblasts. J Cell Sci 1985, 77, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8. [Google Scholar] [CrossRef]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10. [Google Scholar] [CrossRef]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.-N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2–YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef]
- Lee, J.; You, J.H.; Kim, M.S.; Roh, J.L. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol 2020, 37, 101697. [Google Scholar] [CrossRef]
- Oliveira, T.; Hermann, E.; Lin, D.; Chowanadisai, W.; Hull, E.; Montgomery, M. HDAC inhibition induces EMT and alterations in cellular iron homeostasis to augment ferroptosis sensitivity in SW13 cells. Redox Biol 2021, 47, 102149. [Google Scholar] [CrossRef]
- Yao, J.; Zhang, Y.; Li, M.; Sun, Z.; Liu, T.; Zhao, M.; Li, Z. Single-Cell RNA-Seq Reveals the Promoting Role of Ferroptosis Tendency During Lung Adenocarcinoma EMT Progression. Front Cell Dev Biol 2021, 9, 822315. [Google Scholar] [CrossRef]
- Du, Y.; Miao, W.; Jiang, X.; Cao, J.; Wang, B.; Wang, Y.; Yu, J.; Wang, X.; Liu, H. The Epithelial to Mesenchymal Transition Related Gene Calumenin Is an Adverse Prognostic Factor of Bladder Cancer Correlated With Tumor Microenvironment Remodeling, Gene Mutation, and Ferroptosis. Front. Oncol. 2021, 11, 683951. [Google Scholar] [CrossRef]
- Shi, C.; Xie, Y.; Li, X.; Li, G.; Liu, W.; Pei, W.; Liu, J.; Yu, X.; Liu, T. Identification of Ferroptosis-Related Genes Signature Predicting the Efficiency of Invasion and Metastasis Ability in Colon Adenocarcinoma. Front Cell Dev Biol 2021, 9, 815104. [Google Scholar] [CrossRef]
- Zhou, Y.; Qian, W.; Li, X.; Wei, W. NF-κB Inhibitor Myrislignan Induces Ferroptosis of Glioblastoma Cells via Regulating Epithelial-Mesenchymal Transformation in a Slug-Dependent Manner. Oxid Med Cell Longev 2023, 2023, 7098313. [Google Scholar] [CrossRef]
- Zhang, H.; Shan, G.; Jin, X.; Yu, X.; Bi, G.; Feng, M.; Wang, H.; Lin, M.; Zhan, C.; Wang, Q.; et al. ARNTL2 is an indicator of poor prognosis, promotes epithelial-to-mesenchymal transition and inhibits ferroptosis in lung adenocarcinoma. Transl Oncol 2022, 26, 101562. [Google Scholar] [CrossRef]
- Li, H.; Zhou, W.; Wei, H.; Li, L.; Wang, X.; Li, Y.; Li, S.; Li, C. Ferritinophagic Flux Was a Driving Force in Determination of Status of EMT, Ferroptosis, and NDRG1 Activation in Action of Mechanism of 2-Pyridylhydrazone Dithiocarbamate S-Acetic Acid. J Oncol 2021, 2021, 3015710. [Google Scholar] [CrossRef]
- Guan, D.; Zhou, W.; Wei, H.; Wang, T.; Zheng, K.; Yang, C.; Feng, R.; Xu, R.; Fu, Y.; Li, C.; et al. Ferritinophagy-Mediated Ferroptosis and Activation of Keap1/Nrf2/HO-1 Pathway Were Conducive to EMT Inhibition of Gastric Cancer Cells in Action of 2,2′-Di-pyridineketone Hydrazone Dithiocarbamate Butyric Acid Ester. Oxidative Med. Cell. Longev. 2022, 2022, 1–15. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Dong, C.; Chen, T.; Dong, A.; Ren, J.; Li, W.; Shu, G.; Yang, J.; Shen, W.; et al. CST1 inhibits ferroptosis and promotes gastric cancer metastasis by regulating GPX4 protein stability via OTUB1. Oncogene 2023, 42, 83–98. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, Y.; Liu, S.; Yin, H.; Duan, J.; Fan, L.; Zhao, X.; Jiang, B. Implications of Withaferin A for the metastatic potential and drug resistance in hepatocellular carcinoma cells via Nrf2-mediated EMT and ferroptosis. Toxicol Mech Methods 2023, 33, 47–55. [Google Scholar] [CrossRef]
- Li, J.; Jiang, J.L.; Chen, Y.M.; Lu, W.Q. KLF2 inhibits colorectal cancer progression and metastasis by inducing ferroptosis via the PI3K/AKT signaling pathway. J Pathol Clin Res 2023. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N Engl J Med 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Dhar, D.K.; Kubota, H.; Kotoh, T.; Tabara, H.; Watanabe, R.; Tachibana, M.; Kohno, H.; Nagasue, N. Tumor vascularity predicts recurrence in differentiated thyroid carcinoma. Am J Surg 1998, 176, 442–447. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Arif, S.H.; Pity, I.S. P53 expression and micro-vessel density in relation with 5-year survival in patients with colorectal cancer. Ann Med Surg (Lond) 2020, 57, 311–314. [Google Scholar] [CrossRef]
- Al-Ostoot, F.H.; Salah, S.; Khamees, H.A.; Khanum, S.A. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat Res Commun 2021, 28, 100422. [Google Scholar] [CrossRef]
- Zeeshan, R.; Mutahir, Z. Cancer metastasis - tricks of the trade. Bosn. J. Basic Med. Sci. 2017, 17, 172–182. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Sci. (New York N.Y.) 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Reviews. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat Rev Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirouskova, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef]
- Fidler, I.J. The pathogenesis of cancer metastasis: The 'seed and soil' hypothesis revisited. Nat. Reviews. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef]
- Brown, D.M.; Ruoslahti, E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell 2004, 5, 365–374. [Google Scholar] [CrossRef]
- Al-Mehdi, A.B.; Tozawa, K.; Fisher, A.B.; Shientag, L.; Lee, A.; Muschel, R.J. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: A new model for metastasis. Nat. Med. 2000, 6, 100–102. [Google Scholar] [CrossRef]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massagué, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef]
- Padua, D.; Zhang, X.H.F.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massagué, J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Reviews. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.-T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef]
- Khan, I.; Steeg, P.S. Endocytosis: A pivotal pathway for regulating metastasis. Br J Cancer 2021, 124, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Dang, H.X.; Lim, D.A.; Feng, F.Y.; Maher, C.A. Long noncoding RNAs in cancer metastasis. Nat Rev Cancer 2021, 21, 446–460. [Google Scholar] [CrossRef]
- Wang, B.; Guo, H.; Yu, H.; Chen, Y.; Xu, H.; Zhao, G. The Role of the Transcription Factor EGR1 in Cancer. Front Oncol 2021, 11, 642547. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Reviews. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol Life Sci 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Jiang, Y.; Mao, C.; Yang, R.; Yan, B.; Shi, Y.; Liu, X.; Lai, W.; Liu, Y.; Wang, X.; Xiao, D.; et al. EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics 2017, 7, 3293–3305. [Google Scholar] [CrossRef]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 2019, 10, 1617. [Google Scholar] [CrossRef]
- Strzyz, P. Iron expulsion by exosomes drives ferroptosis resistance. Nat Rev Mol Cell Biol 2020, 21, 4–5. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer 2020, 19, 43. [Google Scholar] [CrossRef]
- Brown, C.W.; Amante, J.J.; Goel, H.L.; Mercurio, A.M. The α6β4 integrin promotes resistance to ferroptosis. J Cell Biol 2017, 216, 4287–4297. [Google Scholar] [CrossRef]
- Chen, P.; Wu, Q.; Feng, J.; Yan, L.; Sun, Y.; Liu, S.; Xiang, Y.; Zhang, M.; Pan, T.; Chen, X.; et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 51. [Google Scholar] [CrossRef]
- You, Y.; Fan, Q.; Huang, J.; Wu, Y.; Lin, H.; Zhang, Q. Ferroptosis-Related Gene Signature Promotes Ovarian Cancer by Influencing Immune Infiltration and Invasion. J. Oncol. 2021, 2021, 1–16. [Google Scholar] [CrossRef]
- Zd, N.; Cy, M.; J, D.; Ij, B.; S, I.; Mm, C.; M, H.; Rk, S.; M, M.; As, D.; et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. eLife 2020, 9. [Google Scholar] [CrossRef]
- Blomme, A.; Ford, C.A.; Mui, E.; Patel, R.; Ntala, C.; Jamieson, L.E.; Planque, M.; McGregor, G.H.; Peixoto, P.; Hervouet, E.; et al. 2,4-dienoyl-CoA reductase regulates lipid homeostasis in treatment-resistant prostate cancer. Nat. Commun. 2020, 11, 2508. [Google Scholar] [CrossRef] [PubMed]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef]
- Lu, D.; Yang, Z.; Xia, Q.; Gao, S.; Sun, S.; Luo, X.; Li, Z.; Zhang, X.; Li, X. ACADSB regulates ferroptosis and affects the migration, invasion, and proliferation of colorectal cancer cells. Cell Biol. Int. 2020, 44, 2334–2343. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Battello, N.; Rothley, M.; Piorońska, W.; Sitek, B.; Ebert, M.P.; Hofmann, U.; Sleeman, J.; Wölfl, S.; Meyer, C.; et al. Liver cancer cell lines distinctly mimic the metabolic gene expression pattern of the corresponding human tumours. J. Exp. Clin. Cancer Res. : CR 2018, 37, 211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wu, Q.; Wang, Z.; Xu, R.; Hu, X.; Sun, Y.; Wang, Q.; Ju, F.; Ren, S.; Zhang, C.; et al. The promising novel biomarkers and candidate small molecule drugs in kidney renal clear cell carcinoma: Evidence from bioinformatics analysis of high-throughput data. Mol. Genet. Genom. Med. 2019, 7, e607. [Google Scholar] [CrossRef] [PubMed]
- Arden, K.C.; Viars, C.S.; Fu, K.; Rozen, R. Localization of short/branched chain acyl-CoA dehydrogenase (ACADSB) to human chromosome 10. Genomics 1995, 25, 743–745. [Google Scholar] [CrossRef]
- Zhang, H.; Ge, Z.; Wang, Z.; Gao, Y.; Wang, Y.; Qu, X. Circular RNA RHOT1 promotes progression and inhibits ferroptosis via mir-106a-5p/STAT3 axis in breast cancer. Aging 2021, 13, 8115–8126. [Google Scholar] [CrossRef]
- Cao, F.; Luo, A.; Yang, C. G6PD inhibits ferroptosis in hepatocellular carcinoma by targeting cytochrome P450 oxidoreductase. Cell. Signal. 2021, 87, 110098. [Google Scholar] [CrossRef]
- Xu, F.; Guan, Y.; Xue, L.; Zhang, P.; Li, M.; Gao, M.; Chong, T. The roles of ferroptosis regulatory gene SLC7A11 in renal cell carcinoma: A multi-omics study. Cancer Med. 2021, 10, 9078–9096. [Google Scholar] [CrossRef]
- Belkaid, A.; Ouellette, R.J.; Surette, M.E. 17β-estradiol-induced ACSL4 protein expression promotes an invasive phenotype in estrogen receptor positive mammary carcinoma cells. Carcinogenesis 2017, 38, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Q.; Russo, J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim Biophys Acta 2012, 1826, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A 2016, 113, E6806–e6812. [Google Scholar] [CrossRef] [PubMed]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.E.; Chen, J.; Lu, Y.; Bawcom, A.R.; Wu, J.; Ou, J.; Asara, J.M.; Armstrong, A.J.; Wang, Q.; Li, L.; et al. RB1-deficient prostate tumor growth and metastasis are vulnerable to ferroptosis induction via the E2F/ACSL4 axis. J Clin Invest 2023, 133. [Google Scholar] [CrossRef]
- Xie, W.; Agarwal, S.; Yu, J. Ferroptosis: The vulnerability within a cancer monster. J Clin Invest 2023, 133. [Google Scholar] [CrossRef]
- Wang, C.-x.; Chen, L.-h.; Zhuang, H.-b.; Shi, Z.-s.; Chen, Z.c.; Pan, J.-p.; Hong, Z.-s. Auriculasin enhances ROS generation to regulate colorectal cancer cell apoptosis, ferroptosis, oxeiptosis, invasion and colony formation. Biochem. Biophys. Res. Commun. 2022, 587, 99–106. [Google Scholar] [CrossRef]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front Pharmacol 2017, 8, 992. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



