
Submitted:
01 June 2023
Posted:
02 June 2023
You are already at the latest version
Abstract
Today, the most powerful approach to detect distant homologs of a protein is based on structure prediction and comparison. Yet this approach is still inapplicable to many viral proteins. Therefore, we developed a powerful sequence-based procedure to identify distant homologs of viral proteins. It relies on 3 main principles: 1) Traces of sequence similarity with a protein can persist beyond the significance cutoff of homology detection programs; 2) Candidate homologs can be identified among proteins with weak sequence similarity to the query, by using "contextual" information, e.g. taxonomy or type of host infected; 3) These candidate homologs can be validated using highly sensitive profile-profile comparison.As a test case, we applied our approach to a protein without known homologs, ORF4 of Lake Sinai virus (which infects bees). We discovered that ORF4 is composed of a domain that has homologs in proteins from >20 taxa of viruses infecting arthropods. We called it “Widespread, Intriguing, Versatile” (WIV) domain because it is found in proteins with a wide variety of domain organizations and functions. For example, WIV is encoded by the NSs protein of tospoviruses, a global threat to food security, which infect plants through arthropod vectors; by the protein encoded by RNA2 ORF1 of chronic bee paralysis virus, a widespread virus of bees; and by various proteins of cypoviruses, which infect the silkworm bombyx mori. WIV has a previously unknown structural fold, according to Alphafold predictions. In some viral species, WIV facilitates infection of arthropods, according to bibliographical evidence
Keywords:
insect viruses
; arthropod virus
; distant homology detection
; remote homology detection
; virulence factor
; tospovirus
; structure prediction
; cypovirus
; small protrusion domain
1. Introduction
Virus discovery is increasing at an exponential rate [1], and many newly sequenced viral genomes contain “orphan” proteins, i.e. proteins for which homologs could not be identified (e.g. [2,3]). Yet identifying homologs of a viral protein is crucial, as it may suggest a function, enable taxonomical classification, and illuminate viral evolution.
Unfortunately, two obstacles make the identification of homologs of viral proteins particularly challenging: 1) viral proteins diverge in sequence particularly fast, often beyond the reach of the most powerful automated sequence-based homology detection methods [4], such as HHblits [5]; 2) viral proteins are underrepresented in databases of sequence (such as PFAM), of 3D structures (such as the PDB), and of 3D models (at the time of writing, the Alphafold database of protein 3D models still does not include viral proteins [6], despite including models for most organisms). This underrepresentation means that many viral homologs are undetectable using even structure-based homology search, despite its tremendous progress in recent years [7,8,9].
To overcome these obstacles, we devised a recursive procedure that can identify extremely distant homologs of viral proteins based on their sequence. The procedure is based on the idea that it is extremely difficult to find distant homologs using sequence-based searches, but that given a candidate homolog, it is easy to confirm whether it is homologous to the query. This confirmation can be done by using a highly sensitive method, sequence profile comparison [10].
We present an application of our homology search procedure to a viral protein initially described as “orphan”, i.e. devoid of homologs, ORF4 from Lake Sinai virus. Lake Sinai virus is a recently discovered virus found worldwide, which infects honeybees [11,12], bumblebees [13] and ants [14]. Whether it contributes to bees’ colony collapse disorder [15] is still unknown.
We first identified distant homologs of Lake Sinai virus ORF4 by using our recursive, sequence-based procedure. We discovered that ORF4 is mainly composed of a domain that has homologs in proteins from a wide range of viruses that infect arthropods; these proteins have a large variety of functions and domain organizations. For this reason, we called this domain the “Widespread, Intriguing, Versatile” (WIV) domain. We predicted the 3D structure of the WIV domain using the highly reliable software Alphafold2 [8]. We validated particularly divergent candidate homologs using 3D structure comparison. Finally, we found bibliographical evidence that WIV is probably a virulence factor that facilitates viral infection of arthropods.
2. Results
Overview of our procedure to detect extremely distant homologs by using contextual information
To detect extremely distant homologs, we used a recursive procedure described in Figure 1. We had already used this procedure on several occasions (e.g. [4,16,17]), but had never formally presented it. It is based on the idea that it is extremely difficult to find distant homologs using sequence-based searches, but that is easy to confirm whether a candidate protein is homologous to the query. This idea can be further decomposed into 3 principles, described below.
Principle 1: Traces of sequence similarity can persist beyond the cutoff for statistical significance of programs for sequence homology detection.
Proteins detected by homology search programs such as Psiblast are considered homologous to the query if the statistical significance of their sequence similarity with the query (called “E-value”) is below a certain cutoff (typically E=10-3) [18]. However, these programs have the ability to return a long list of non-significant hits above the cutoff (up to an E-value of 1000 on the web-based version of Psiblast in the MPI toolkit [19]), which are often called “marginal” hits. Some of these hits might be homologs that have considerably diverged in sequence, which is why they are above the significance cutoff (the higher the E-value, the less significant the similarity is). The question is, how can we identify these divergent homologs? This can be done thanks to principles 2 and 3 below.
Principle 2: To identify candidate homologs in the list of non-significant hits, we can use "contextual" information, e.g. taxonomy or type of host infected.
"Contextual" information is the information associated with the primary sequence of a protein, such as gene location, gene order, taxonomy, protein size, domain co-occurrence, domain order, function, type of host infected, etc. Contextual information has been used since the beginning of bioinformatics to detect more distant homologs (e.g. [20,21,22]). Yet we noticed that it is particularly powerful in viruses for two reasons:
- -
- First, viruses tend to have very few genes (fewer than 10 for most RNA viruses, for example, compared to over 20,000 in humans). Thus, a weak similarity between two viral proteins is much more meaningful than, say, a weak similarity between two human proteins;
- -
- Second, some proteins are found primarily in viruses, or even restricted only to a certain type of viruses. For example, movement proteins of the 30K superfamily are restricted to plant-infecting viruses (as well as to certain plants) [23]. Therefore, a protein which has only weak similarity to a 30K movement protein, but which comes from a plant-infecting virus, might reasonably be considered a candidate homolog that would have considerably diverged in sequence.
In the present study, initial searches found that the WIV domain was only present in arthropod-infecting viruses (see below); we thus systematically considered weak hits from arthropod-infecting viruses as candidate homologs.
Principle 3: Candidate homologs can be validated using a highly sensitive method, pairwise profile-profile comparison.
Once a candidate homolog has been identified, it can be validated using a powerful method, HHpred pairwise comparison [10]. Briefly, in pairwise comparison mode, HHpred automatically performs 4 steps: a) it collects, in parallel, the sequences of homologs of the query protein and of the candidate homolog; b) it generates separate alignments of these sequences; c) it converts these alignments into representations called sequence "profiles"; and d) it compares these two profiles, as well as their predicted secondary structure. Comparing sequence profiles is much more powerful than comparing single sequences, because the profiles contain information about how the sequences of homologs can evolve [4,24]. The comparison can yield two results:
- -
- If HHpred detects a significant similarity, the two proteins are homologous;
- -
- If HHpred does not detect a significant similarity, either the two proteins are not homologous, or they are homologous but have diverged beyond recognition even by sequence profile methods. In such cases, only structure-based methods can confirm or infirm the homology (see below).
Based on these 3 principles, we designed a recursive procedure to identify distant homologs, described in Figure 1 (see also the Methods section). It is composed of 3 parts:
1) The first part (Figure 1, top) starts with a standard sequence-based homology search (step 1A) using highly sensitive software (Psi-blast [25] and HHblits [5]), using stringent significance cutoffs. Homologs detected in this step are aligned (step 1B) and are resubmitted to Psi-blast and HHblits until no new homolog is identified by this standard search. Then we proceed to part 2,
2) In the second part (Figure 1, middle) is an advanced sequence-based search, which takes advantage of contextual information. It starts with examining weak hits that only have marginal similarity to the query, among which we select candidates that have the appropriate contextual information (i.e. that come from certain taxa, or infect certain hosts) (step 2A). We filter out those candidates that are homologous to known domains (step 2B). Then we compare the sequence of each remaining candidate with the sequence alignment of the WIV domain (step 2C), using HHpred pairwise comparison. If HHpred detects significant similarity (which means that the candidate is homologous to WIV), we incorporate these validated candidates to the alignment of WIV domains (step 2D) and repeat the procedure from the start (part 1). Candidates for which HHpred detects no significant similarity might be divergent homologs of WIV. We examine them in part 3.
3) The third part (Figure 1, bottom) consists in a structure-based search. This step has only recently become possible thanks to the success of the software Alphafold2 [8] in reliably predicting 3D structures. First, using Alphafold2, we predict the structure of the divergent candidate homologs that could not be validated in part 2 (step 3A). Then, we compare their structure with that predicted for the WIV domain of Lake Sinai virus ORF4 (step 3B). If both structures are significantly similar, the candidate is homologous to WIV. Otherwise, the candidate is discarded.
This procedure continues until no new homolog is found. We will now describe its application to discovering homologs of the WIV domain.
The WIV domain is found in proteins from over 20 viral taxa, with a large variety of domain organizations
Lake Sinai virus ORF4 protein was initially classified as orphan (i.e. devoid of homologs) upon its discovery [12]. To identify distant homologs, we applied the procedure described above. We first present the results of parts 1 and 2 (respectively standard and advanced sequence-based homology search), and later the results of part 3 (structure-based homology search). In part 1 (Figure 1, top panel), we examined significant hits (E<10-3) returned by Psi-blast and HHblits, i.e. easily identifiable homologs, and noticed that they all came from viruses that infect arthropods. We thereby discovered that the Lake Sinai virus ORF4 protein is constituted by a standalone domain, which we will thereafter call it the WIV domain (see below). Therefore, in step 2, we considered as “candidate homologs” hits that both had weak sequence similarity to the WIV domain (10-3≤E<1000) and came from arthropod-infecting viruses. We verified these candidates using HHpred pairwise comparison (Figure 1, middle panel), and incorporated validated homologs in a new round of homology search (parts 1 and 2), until no new homolog was detected.
his procedure enabled us to detect homologs of Lake Sinai ORF4 in proteins from over 20 viral genera and unassigned taxa, corresponding to 11 viral orders (see Figure 2 and Table 1). Thus the WIV domain has an exceptionally wide taxonomic distribution. In addition, WIV occurs in proteins with a strikingly wide variety of domain organizations. We noted 4 main types of architectures:
- -
- As a standalone domain: WIV is found as a standalone domain in most positive-strand RNA viruses (Figure 2, panel A), except Picornavirales and an Amarillovirales; in a negative-strand RNA virus (Mononegavirales, panel B); and in some double-stranded DNA viruses (Pimascovirales, panel C). In some of these proteins, WIV is preceded by a signal peptide (e.g. in Lake Sinai virus ORF4).
- -
- Appended to a coiled-coil: In some species, WIV is appended to an N- or C-terminal coiled-coil, for example in Dougjudy virga-like virus RNA2 ORF1 (panel A), or in Wiseana iridescent virus gp049 (panel C);
- -
- Next to a double-stranded RNA-binding domain (dsRBD): WIV is wedged between a dsRBD domain and a capsid domain in some Picornavirales (panel A), and is located upstream of a dsRBD domain in a Ghabrivirales (panel D).
- -
- Downstream other types of domains: WIV is found at the very C-terminus of some proteins, such as Tospovirus NSs (panel B).
The SPD-like domain (panel D) has been discovered in this study, like the WIV domain (see main text). For cypoviruses (panel D), the segment that encodes each protein is indicated between brackets (e.g. S8 is segment 8).
The wide variety of domain contexts in which WIV occurs clearly shows that it is both structurally and functionally independent, justifying its name of "WIV", for “Widespread, Intruiguing, Versatile” domain.
The distribution of WIV suggests extensive horizontal transfer. In some cases, WIV is found only in a single species within an order, e.g. Hangzhou frankliniella intonsa flavivirus 1 (Figure 2A); Fushun monolepta xinmovirus (Figure 2B); and Hainan sediment Toti-like virus 9 [28] (Figure 2C). However, there are 5 large taxa in which all species encode a WIV domain. One is the genus orthotospovirus. The 4 other taxa are currently unclassified; their member species are presented in Table 2. These taxa are:

Table 2.
Unclassified viral taxa that contain at least 5 species, all of which encode a WIV domain.
| Taxon | Virus species in the taxon |
|---|---|
| Brandeis virus group | Brandeis virus, Beult virus, Muthill virus, Bofa virus, Marsac virus, Buckhurst virus, Hubei virga-like virus 18, Broome virga-like virus, Hubei virga-like virus 19, Zeugodacus cucurbitae negev-like virus, Erysiphe necator associated virga-like virus 2 |
| Dougjudy virga-like virus group | Dougjudy virga-like virus, Hangzhou merodon fulcratus virga-like virus 1, Leuven Virga-like virus 1, Virga-like virus 21, Atrato virga-like virus 6, Atrato virga-like virus 7, Hammarskog virga-like virus, Erysiphe necator associated virga-like virus 1 |
| Family Acypiviridae (proposed in [32]) | Acyrthosiphon pisum virus, Darwin bee virus 7, Hangzhou solinvi-like virus 2, Grapevine-associated RNA virus 1, Hubei picorna-like virus 55, Hubei picorna-like virus 56, Aphis citridus picorna-like virus, Rosy apple aphid virus, Changjiang crawfish virus 6, Lasius picorna-like virus 7, Electric ant virus 1 |
|
Solenopsis invicta virus 7-like group, closely related to the proposed family Acypiviridae (see Figure S1h in [34]). |
Solenopsis invicta virus 7, Apis-Picorna-like virus 5, PNG bee virus 9, Hangzhou sesamia inferens solinvi-like virus 1, YCA-associated virus-like sequence 8 [34], HVAC-associated RNA virus 1, Apis picorna-like virus 3, Bundaberg bee virus 8, Milolii virus, Lasius picorna-like virus 9 |
The genome of 9 viral species contains an unannotated coding sequence that has significant similarity with the WIV domain, detectable by using the software tblastn (see Methods). These species comprise Muthill virus, Bofa virus, Buckhurst virus and Marsac virus [35], Beult virus [29], Ceratitis capitata Negev-like virus 2 [36], Atractomorpha sinensis Negev-like virus 1 [37], Solenopsis invicta virus 8 [33], and Bat tymo-like virus (Genbank accession number NC_030844). We included the corresponding WIV domain of these viruses in the alignment presented in Supplementary File S1. These overlooked coding sequences are short (~300 nucleotides) and almost all are located at the very 3’ end of the genome, suggesting a bias of genome annotators against annotating short, 3’ coding sequences. Interestingly, in another species, Ek Balam virus [38], the 3’ end of the genome also contains an unannotated coding sequence with significant similarity to WIV, but it is interrupted by a stop codon.
Finally, a WIV domain is found in over a dozen proteins annotated as coming from arthropod viruses, in particular thrips. Their sequence is included in the alignment from Supplementary File S1.
The WIV domain is predicted to have a previously unknown fold composed of a 6-stranded β-sheet buttressed by 3 α-helices
We predicted the structure of the WIV domain (aa 29-129) of Lake Sinai virus ORF4 using Alphafold2 [8]. The structure is presented in Figure 3A. It is expected to be highly reliable (pLDDT = 0.95). WIV adopts a previously unknown fold, composed of a 6-stranded β-sheet buttressed by 3 α-helices (Figure 3A). Its topology is presented in Figure 3B: a long (~18 aas) helix (α1), followed by four β-strands (β1 to β4), by two helices (α2 and α3) and finally by two antiparallel β-strands (β5 and β6).
A) 3D structure of the WIV domain (aa 29-129) of Lake Sinai virus ORF4, predicted by Alphafold2. B) Topology of the WIV domain. C) Residues corresponding to positions conserved across WIV homologs (see text and multiple sequence alignment in Figure 4), visualized in a different orientation from those in panel A.
The WIV domain diverges considerably in sequence across viral taxa
Structure-based alignments are more reliable than sequence-based ones. Therefore, to generate an optimal alignment of the WIV domain, we first predicted the structures of two other representative WIV domains. We chose Tomato spot wilted virus NSs and Acyrthosiphon pisum virus polyprotein as representatives, because they have numerous close homologs in the database, a prerequisite for a good prediction by Alphafold2. Accordingly, Alphafold2 predicted their 3D structure with high expected reliability (plDDT = 92.8 and 91.3 respectively). The corresponding model coordinates are in supplementary files S2 and S3. Next, we generated a sequence alignment of the WIV domains based on the superposition of the WIV domain of the 3 representatives Lake Sinai virus ORF4, Tomato spot wilted virus NSs, and Acyrthosiphon pisum virus polyprotein, using Promals3D [39]. The resulting structure-based alignment is shown in Figure 4 (see Supplementary File S1 for the alignment in text format). Only the N-terminal two thirds of WIV have meaningful sequence conservation (aa 37-99 in Lake Sinai virus); in the last third of WIV, conserved positions in the alignment correspond mainly to conservation of hydrophobicity.
Four aa positions are well conserved across homologs of the WIV domain. They are boxed and indicated above the alignment in Figure 4:
1) Either a Q or a H (both large, polar aas) in most species, 9 aas after the start of the conserved region of WIV, in the middle of helix α1 (Q45 in Lake Sinai virus ORF4);
2) An A, or generally another tiny aa (G, S or C), 4 aas downstream of this conserved Q/H position (A49 in Lake Sinai virus ORF4);
3) An S, or generally a small aa, between strand β4 and helix α2 (S95 in Lake Sinai virus ORF4);
4) A Q, or generally a polar aa, 4 aas downstream of the conserved S position, in helix α2 (Q99 in Lake Sinai virus ORF4).
These residues are located towards the interior of the protein (Figure 3C), and therefore, their conservation is probably due to structural reasons. In the WIV domain of Lake Sinai virus, the aa corresponding to conserved position 1, Q45, is facing the aa corresponding to conserved position 3, S95 (Figure 3C). The aa corresponding to conserved position 4, Q99, is also located in the vicinity of these 2 aas (Figure 3C). Finally, the residue corresponding to the conserved position 2, A49, is not visible in the orientation depicted in Figure 3C, but is also located in the interior of the protein.
Structure-based alignment of representative WIV domain (see text). Protein accession numbers are in Table 1. For brevity, the term “virus” has been omitted in species names (e.g. “PNG bee 9” corresponds to “PNG bee virus 9”). Species names highlighted in bold are those for which experimental data regarding WIV are available. Aas substituted in the WIV domain of Tomato spotted wilt virus or of related tospoviruses are indicated in bold.
Proteins from several cypoviruses also encode WIV domain, located at the C-terminus
During step 2 of homology search (Figure 1, middle panel), we identified candidate homologs (i.e. marginal hits with an E-value 10-3<E<1000, from viruses infecting arthropods) in cypoviruses, both in Psiblast searches and in HHpred searches against the database of viral protein profiles Uniprot-SwissProt-viral70 (see Methods). Cypoviruses are double-stranded RNA viruses of the family Reoviridae, which infect arthropods. Their genome consists of 10 to 12 segments, and they have a non-enveloped, icosahedral capsid [40]. The cypoviral candidate proteins and the WIV domain had no significant sequence similarity (as seen using HHpred pairwise comparison), indicating that either 1) these candidates are not homologous to WIV, or 2) they are homologous but have diverged beyond identification by sequence-based methods.
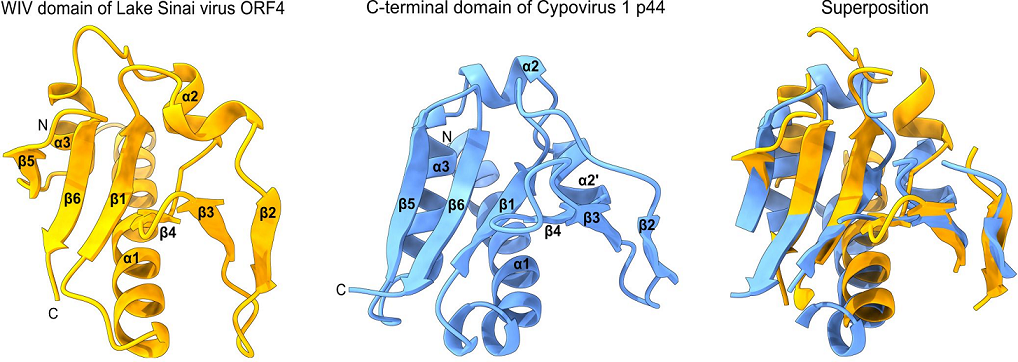
Such divergent homologs can be identified by structural comparison instead. Therefore, to determine whether the cypoviral protein candidates were genuine homologs of WIV, we conducted structure-based homology searches, i.e. part 3 of our procedure (Figure 1, bottom panel). We predicted the structure of the p44 protein of cypovirus 1, containing a candidate WIV domain. Alphafold2 returned a reliable model of p44 (plDDT = 86.8), predicting that it is composed of an N-terminal domain (aa 1-131), of a variable linker (aa 132-277), and of a C-terminal domain aa 278-389), corresponding to the candidate WIV domain. The predicted 3D structure of this C-terminal domain is shown in Figure 5 (middle panel). It has significant similarity with the structure of the WIV domain of Lake Sinai virus ORF4 (FATCAT E-value 3x10-5 with a RMSD of 3.29 Å), confirming that it homologous to WIV. Note in Figure 5 how the C-terminal domain of cypovirus 1 p44 has exactly the same arrangement of secondary structures as the WIV domain of Lake Sinai virus, in the same order. In conclusion, cypovirus 1 p44 contains a divergent C-terminal WIV domain.
A) WIV domain of Lake Sinai virus ORF4 (aa 37-127). B) WIV domain of cypovirus p44 (aa 278-389). C) Structural superposition, showing only common core regions (i.e. with no gaps, and aa distance < 4Å), identified with mTM-align [41].
We applied to the WIV domain of cypovirus 1 p44 the recursive homology search procedure described in Figure 1, and thereby also identified a WIV domain in cypoviruses 4, 5, 14, 18 and 23 (Table 3 and Figure 2D). Figure 6 presents a structure-based sequence alignment of cypoviral WIV domains (the alignment in text format is in Supplementary File S6). Cypoviral WIV domains are very distant from each other, and the alignment contains no well-conserved position; only general physio-chemical characters are conserved. The taxonomic distribution of WIV is mostly consistent with the phylogeny of cypoviruses [42], in which cypovirus 1, cypovirus 18 and cypovirus 14 are sister species, as are cypovirus 5 and Hubei lepidoptera virus 3. However, cypovirus 23 is not closely related to these species [42].
Structure-based alignment of the cypoviral WIV domains, based on the Alphafold models of cypovirus 1 and cypovirus 5 WIV. Conventions are the same as in Figure 4.
Some cypoviral proteins contain a domain upstream of WIV related to the SPD domain of cypoviral capsid protein VP1
We attempted to map the domain organization of cypoviral proteins that contain a WIV domain. First, we identified regions with meaningful sequence similarity with known domains, using HHpred (see Methods). HHpred only identified one domain, PARD (Poly ADP-ribose glycohydrolase), in cypovirus 5 P5, just upstream of WIV (Figure 2D).
Second, we attempted to predict the structure of the remaining regions, using Alphafold2. It returned a reliable prediction (plDDT=0.89) for the domain located upstream of WIV in cypovirus 1 p44 (aa 1-131). Figure 7 presents its structure, a helices/sheet/helices sandwich (its coordinates are in Supplementary File S7). It has significant similarity (DALI Z-score 12.5) to the structure of the SPD domain ("Small Protrusion Domain") of cypovirus 1 VP1, the major viral capsid protein [45] (aa 828-859 of VP1, shown in Figure 7B; PDB accession code 3IZX_C). The SPD domain of cypovirus VP1 stabilizes the assembly of the viral capsid [45], and accordingly mutations in it destabilize the capsid [46]. The SPD domain has only been observed so far in the capsid of cypoviruses, which is composed of a single shell, unlike the capsid of other Reovirales.
Owing to its structural similarity to the SPD domain, we called aa 1-131 of cypovirus 1 p44 an “SDP-like” domain. We could find no information regarding the function of the SPD-like domain, but in cypovirus 1 p44, it contains two experimentally verified glycosylation sites (aa N48 and N69), while a third one is located immediately downstream (aa N138), in the linker that separates the SPD-like domain from the WIV domains [47].
Figure 7C shows the good superposition between the predicted structure of the SPD-like domain of cypovirus 1 p44 and the SPD domain of cypovirus 1 VP1 (FATCAT P-value 2.8x10-6, RMSD 3.21Å over their whole length). The P8 protein of the closely related cypovirus 18 also contains an SPD-like domain, as indicated by a Psi-blast search.
Alphafold could also reliably predict the 3D structure of the N-terminal domain of cypovirus 5 P5 (pLDDT =0.92; structure not shown, see Supplementary File S8 for its coordinates), which also has significant structural similarity to the SPD domain of cypovirus 1 VP1.The P5 protein of the closely related Hubei lepidoptera virus 3 also contains an SPD-like domain, identifiable by Psi-blast. In contrast, Alphafold could not reliably predict the structure of the domain upstream of WIV in other cypoviral proteins, for lack of related sequences. Therefore, we could not determine whether they contain an SPD-like domain. In conclusion, the following proteins contain an SPD-like domain, located at the N-terminus: cypovirus 1 p44, cypovirus 18 P8, cypovirus 5 P5, and Hubei lepidoptera virus 3 P5.
Both the SPD and SPD-like domain are highly variable in sequence, even among species belonging to the same genus. For example, there is no detectable sequence identity between the SPD-like domain of cypovirus 1 p44 and that of cypovirus 5 P5. Likewise, we could only identify a domain with detectable similarity to the SPD domain of cypovirus 1 VP1 in VP1 of the closely related cypovirus 14. Thus, the fold of the SPD and SPD-like domains probably places few constraints on its sequence, allowing it to diverge very fast. Consequently an SPD-like domain may be present in many more proteins than those in which we have detected it.
A) SPD domain (“Small Protrusion Domain”) of cypovirus 1 VP1 capsid protein (PDB accession code 3izx-C). B) SPD-like domain (aa 1-131) of cypovirus 1 p44, located upstream of the WIV domain. C) Superposition of both domains.
Altogether, cypovirus WIV is found at the C-terminus of a wide variety of structurally and functionally unrelated proteins (Figure 2D), downstream of:
- -
- an SPD-like domain in the p44 protein of cypovirus 1 and the related protein P8 of cypovirus 18;
- -
- an SPD-like domain followed by a Poly ADP-ribose glycohydrolase (PARG) domain in the P5 protein of cypovirus 5 and Hubei lepidoptera virus 3;
- -
- an unknown domain (s) in the P8 protein of cypovirus 4, 14 and 23.
WIV is probably a virulence factor that facilitates infections of arthropods, according to bibliographical information
Bibliographical information about function or gene expression is available only for 4 proteins containing a WIV domain: 1) p15, in Bombyx mori macula-like virus; 2) NSs, in tospoviruses; 3) 206R, in invertebrate iridescent virus 6; and 4) p44, in cypovirus 1. We present this information below.
The p15 protein of Bombyx mori macula-like virus (BMLV, previously called bombyx mori latent virus) is essentially a standalone WIV domain (Figure 2A). BMLV was discovered in cell lines derived from the silk moth Bombyx mori, which it persistently infects, accumulating at extremely high levels [48]. It belongs to the family Tymoviridae (proposed genus: inmaculavirus [49]). BMLV produces a protein, p15, which has homologs in the other members of the proposed genus inmaculavirus (bee macula-like virus 2 and Nasturtium officinale macula-like virus 1), but not in the related genus maculavirus. The p15 mRNA is highly expressed in BMLV-infected silkworm cells (much more than the capsid protein mRNA) [50]. BMLV p15 is mostly located in the cytoplasm of infected Bombyx mori BmN cell lines [51] and is required to establish BmMLV infections in silkworm cells [50]. BMLV p15 had no RNA silencing suppressor activity in an Agrobacterium-mediated transient coexpression assay [50].
Some functional information is also available for tospovirus NSs. It contains a C-terminal WIV domain (aa 335-429 in tomato spotted wilt virus, see Figure 2B), preceded by a long (~300 aa) N-terminal domain. Among viruses encoding a WIV domain, tospoviruses are the only ones known to actively replicate both in arthropods and in plants, to which they are transmitted via thrips [52,53]. NSs is required for persistent infection and transmission by the thrips Frankliniella occidentalis [54]. NSs enhances baculovirus expression in various arthropod cell lines [55] and increased baculovirus virulence in a caterpillar [56]. In addition, NSs impairs RNA silencing in tick cells [57] and in thrips [58]. The NSs protein accumulated slower than the nucleocapsid (N) protein in primary cell cultures of thrips [59]. NSs is found in the cytoplasm, as is BMLV p15 (see above), where it is uniformly scattered, both in thrips cell cultures and in thrips infected with tomato spotted wilt virus, the type species of tospoviruses [59,60].
Despite this wealth of information on tospovirus NSs, to our knowledge, there is no data indicating whether it is the WIV domain and/or other regions of NSs that are responsible for impairing RNA silencing in arthropods, or for enabling infection and transmission by arthropods. The reason for this lack of data is that studies of the function of NSs by targeted mutations have been carried out exclusively in plants. We will present these studies only briefly (for a review, see [61]), since here we are mainly concerned with the role of WIV in arthropods.
On the one hand, NSs can enhance infection of plants (by impairing RNA silencing), and on the other hand, it can trigger resistance to infection in tomatoes [62]. Most substitutions that abolished the RNA silencing suppressor activity of NSs in plants or its ability to trigger resistance were found in the N-terminal third, i.e. aa 1-133 [63]. Yet several substitutions in the WIV domain also abolished the silencing suppressor activity of NSs, such as N355A/N356A [63], or L413A [64], in bold in Figure 4. Likewise, several substitutions in the WIV domain abolished its ability to trigger resistance in plants, such as L396A/S397A [63], in bold in Figure 4, which includes a substitution to the conserved S position (boxed in Figure 4). Both activities of NSs can be uncoupled: for example, another double substitution, S411A/Y412A (in bold in Figure 4), preserved the RNA silencing suppressor activity of NSs but abolished its ability to elicit resistance in plants [63]. Since the effect of these substitutions was only tested in plants, and not in arthropods, presenting them in further detail is beyond the scope of this study.
Besides the function of NSs, some mutational studies have also investigated its stability and multimerization. Substitutions within helix α1 of the WIV domain of watermelon silver mottle virus NSs abolished self-interaction of NSs but not its RNA silencing suppression activity [65]. In another study, the stability of NSs was decreased by substitution by an alanine of aa Y398 of watermelon silver mottle virus (corresponding to Y394 in NSs of tomato spotted wilt virus (strain Br20) - in bold in Figure 4), located in strand β4 [66]. Thus, WIV may be required for multimerization and stability of NSs.
Gene expression data are available for invertebrate iridescent virus 6 (also called "chilo iridescent virus"; genus iridovirus), in which the protein 206R is essentially a standalone WIV domain (Figure 2C). 206R belongs to the “immediate-early” class [67], i.e. is among the earliest viral genes expressed. Interestingly, the gene 206R is among the ones most targeted by small interfering RNAs [68]. This would be coherent as a counter-defense mechanism to prevent expression of 206R and of its WIV domain. The protein 206R has not been detected in virions [69].
Some information is also available for two cypoviral proteins containing a WIV domain. Cypovirus 1 p44 (also called NSP2) is a non-structural protein that plays a central role, since it interacts with most other viral proteins and drives the formation of viroplasms (i.e. cytoplasmic structures thought to be one of the main sites of viral replication) during infection [43]. P44 is localized close to intracellular membranes and to the endoplasmic reticulum [43]. Whether the C-terminal WIV domain of p44 contributes to viroplasm formation or binding to other viral proteins is unknown. In silkworms infected by cypovirus 1, the expression level of the mRNA encoding p44, i.e. segment S8, was lower than that of the main capsid protein VP1, encoded by segment S1 [70]. This is in contrast to the p15 mRNA of bombyx mori macula-like virus, whose level of expression was much higher than that of the capsid (see above).
Finally, Cypovirus 5 p61, which also contains a WIV domain, is a structural protein, i.e. it can be detected in virions [44].
We note that WIV is located near a known or putative RNA-binding domain in proteins with 3 types of organization, which may suggest a functional association with RNA binding:
1) in the polyprotein of various Picornavirales, in which WIV is located immediately downstream of a predicted double-stranded RNA-binding domain (dsRBD, see Figure 2A);
2) in the polyprotein of an artivirus (family Totiviridae) in which WIV occurs instead immediately downstream of a dsRBD domain (Figure 2D);
3) in cypovirus 1 p44, in which the region encompassing aa 104-201 binds single-stranded, but not double-stranded RNA [71]. This region corresponds mainly to the variable linker (aa 132-277) upstream of WIV (Figure 2D), but also encompasses a short part of the SPD-like domain (aa 1-131). The RNA binding of cypovirus 1 p44 is not sequence-specific (it might be mediated by the negative charge of the the linker, highly enriched in glutamic acid).
However, WIV is is by no means systematically appended to an RNA-binding domain; it also found as a standalone domain in numerous species (Figure 2). In fact, since RNA-binding activity is relatively common in proteins, its association with WIV in some taxa might be coincidental.
3. Discussion
Altogether, our results show that a domain of ~90 aas, WIV, is found in proteins of over 20 taxa of viruses infecting arthropods. In particular, WIV is encoded by tospoviruses, which infect plants through arthropod vectors, and are a global threat to food security [72]; by chronic bee paralysis virus, a widespread virus of honey bees, [73,74]; and by cypoviruses [40], which infect the silkworm bombyx mori, of economic importance for the production of silk.
According to bibliographical evidence, WIV is most probably a virulence factor, which enables infection of arthropods. There are no obvious common points between the proteins that encode a WIV domain for which experimental information is available (see last paragraphs above, before the Discussion). For example, in silkworms infected by cypovirus 1, the expression level of the mRNA encoding p44 was lower than that of the main capsid protein VP1 [70]. In contrast, the level of expression of the p15 mRNA of bombyx mori macula-like virus expression was much higher than that of the capsid [50]. However, the proteins p15 of Bombyx mori macula-like virus [51], tospovirus NSs [59,60], and cypovirus 1 p44 [43] have at least one point in common: they are found in the cytoplasm during infection.
The domain organization of proteins containing WIV provides support to our predictions
WIV is extremely divergent in sequence across taxa, and its 3D structure is only a model. Nevertheless, two arguments provide strong support to our predictions:
- -
- First, the reliability estimate (plDDT) provided by Alphafold2 has been proven to be accurate: a predicted structure with a plDDT≥0.90 is expected to be competitive with an experimentally determined structure [8]. The Alphafold2 structure for the WIV domain of Lake Sinai virus ORF4 has a plDDT of 0.95, and should therefore be close to the actual structure;
- -
- Second, the boundaries of the WIV domain frequently correspond exactly to an unassigned protein region between two known domains (or between a known domain and the extremity of a protein). For example, in the ORF1 of the virus wpk049shi07 [75] (Genbank accession QKE55054.1), related to Sinhaliviridae, the WIV domain, located between aa 1-113, is immediately followed by a 2A “StopGo” sequence (aa 127-139) (our observations; see Figure 2A, top). Such sequences (also called “Stop-Carryon”) mediate ribosome skipping during translation, which separates two proteins, akin to a cleavage, but without requiring a protease [76]. Their core motif is DxExNPGP, and the proteins are separated between the penultimate G and the final P (respectively G134 and P135 in the sequence of ORF1). Therefore, in this virus, the WIV domain should be found essentially as a standalone domain (with a short C-terminal extension, aa 114-134), providing strong biological support to our prediction.
Contextual information holds enormous untapped power for homology search in viruses
We presented here a procedure (Figure 1) to identify extremely distant homologs by harnessing contextual information (namely taxonomy and infected host). This procedure enabled us to identify a previously overlooked domain, WIV, present in nearly a hundred species of arthropod viruses (the full list is in Supplementary File S1). We had already used this procedure on several occasions to detect distant homologs [4,16,17] but had never formally presented it. It is based on the idea that it is extremely difficult to find distant homologs using sequence-based searches, but that given a candidate homolog, it is easy to confirm whether it is homologous to the query. This confirmation can be done by comparing the sequence profiles of close homologs of the query and of the candidate, using HHpred pairwise comparison [10].
An example, beyond WIV, will illustrate the power of our procedure: it has enabled us to identify homologs for all 4 proteins of chronic bee paralysis virus initially annotated as "orphan" (i.e. devoid of recognizable homologs) upon sequencing of the viral genome [77]. We had previously identified homologs for 3 of these orphan proteins [4]. First, we had discovered that the protein encoded by RNA1 ORF1, closely related to the N-terminal domain of the nodavirus replicase, was homologous to the alphavirus methyltransferase. This homology has since been experimentally confirmed by structural studies [78]. Second, we had discovered that the protein SP24, encoded by RNA2 ORF3, had homologs in a large group of viruses infecting plants and/or insects, related to Virgaviridae and Kitaviridae. Third, we had discovered that the predicted glycoprotein encoded by RNA2 ORF2 had homologs in the same group of viruses. Here, we discovered that the 4th orphan protein of chronic bee paralysis virus, encoded by RNA2 ORF1, is essentially a standalone WIV domain (Figure 2A). Thus, our procedure enabled us to identify homologs for all 4 orphan proteins of a phylogenetically isolated virus.
Methods that rely on contextual information to identify homologs have been presented elsewhere (e.g. [20,21,22]), but our approach combines two original elements which account for its power:
1) We focused on viruses, in which contextual information is particularly powerful. Indeed, viruses tend to encode much fewer proteins than other organisms, making a weak similarity between two viral proteins much more meaningful than for other organisms. In addition, some proteins are frequently found exclusively in certain types of viruses, making a weak similarity between two proteins belong to this type of viruses particularly meaningful (see Introduction).
2) We looked for candidate homologs among marginal hits up to extremely high E-values (E=1000), way beyond the cutoff of statistical significance (E=10-3) of Psi-blast and HHblits. By comparison, E=10 is the traditional cutoff below which marginal hits are presented on the web-based version of most homology detection software; and the highest E-value that we could find in the literature up to which marginal hits were examined by a homology detection method is E=100 (for the method MorFeus [79]). Using instead a relaxed E-value E=1000 enabled the detection of divergent WIV domains that otherwise could not have been detected, such as that of Feksystermes virus [80], an isolated Picornavirales.
Using contextual information to complement homology searches is necessary in viruses, despite the progress of structure prediction
It seems paradoxical that sequence-based homology searches should still be necessary despite the tremendous progress of structure prediction [7,8,81] and of structure-based comparison [9]. Yet at least in the medium term, many viral homologs will remain inaccessible to structure-based homology search, for 5 main reasons:
1) Viral proteins tend to have a more loosely packed structure, with less secondary structure elements, and more structural disorder, than their homologs in cellular organisms [82]. Thus, structural predictions are expected to be poorer for viral proteins;
2) There are few viral proteins with a solved 3D structure compared to bacterial or mammalian ones, perhaps because viral proteins are notoriously harder to produce and purify. As a consequence, the training datasets of Alphafold2 and of other methods proportionally comprise less viral protein structures (although more would be required, in view of their different characteristics, as outlined above). This most probably decreases these methods’ performance on viral proteins (although this has not yet been tested);
3) Viral proteins diverge extremely fast in sequence compared to proteins from other organisms, and thus for viral proteins, fewer close homologs are generally available (with the exception of well-sampled taxa such as betacoronavirus, the genus of SARS2-CoV). Yet close homologs are required both to enable prediction by methods that rely on sequence alignments (such as Alphafold2 [8]), and to train methods that do not rely on alignments (such as ESMfold [83]). As a consequence, structural predictions are expected to be less good for proteins from poorly sampled viruses than for other organisms (although again, this has not yet been tested);
4) Alphafold2 intrinsically fails to predict the structure of a number of proteins that have a stable 3D fold [84]; for these proteins, only sequence-based methods have the potential to identify distant homologs (this is true for all organisms, and not only for viruses);
5) Finally, at the time of writing, the Alphafold database does not include viral proteins (although it includes proteins from almost all other organisms) [6], making it impossible to identify protein homologs in viruses only by using structural comparisons.
Clearly, it would be necessary to automate the procedure presented here, since it is too time-consuming to be applied to the enormous number of orphan viral proteins being discovered. Automatically incorporating contextual information into sequence-based homology search might require scoring the contribution of each piece of contextual information (taxonomy, type of host infected, etc). Automated approaches will also need to be adapted to viral polyproteins, which raise special challenges, as they are often composed of many domains, which decrease the accuracy of homology searches. Such approaches are being developed (e.g. LAMPA [85]), and will need to be combined with an exploitation of contextual information for maximum efficiency.
4. Conclusion
In conclusion, we see 3 main implications to our findings. First, they imply that the NSs protein of tospoviruses is composed of two domains: an N-terminal one and a C-terminal WIV domain. This discovery will greatly facilitate functional and structural studies of NSs, an RNA silencing suppressor whose 3D structure and mechanism of action are still unknown despite two decades of studies [61]. Second, it will help us understand how arthropod viruses take control of their hosts. Finally, proteins containing a WIV domain might have biotechnological applications, by enhancing protein expression from insect virus platforms [86].
5. Materials and Methods
4.1. Sequence Alignment and Sequence-BASED Homology Search
We used Psi-Coffee [87] for standard multiple sequence alignment and Promals3D [39] for structure-based sequence alignment. Alignments are presented using Jalview [88] with the ClustalX colouring scheme [89]. To identify homologs, we used Psi-blast [25] (8 iterations against the databases nr30 or nr70) and HHblits [5], both with an E-value significance cutoff of 10−3, and both ran from the user-friendly, web-based version of the MPI toolkit [19]. To validate candidate homologs, HHpred [10] was run in pairwise comparison mode (accessible on the MPI toolkit by clicking “Align two sequences/MSAs”).
4.2. Prediction of Domain Organization
To predict domain organization, HHpred [10] was run against 3 databases: PFAM version 35 [90]; PDB [91] (January 2023 release); and Uniprot-SwissProt-viral70_3Nov_2021 (an unpublished database of sequence profiles of viral proteins, available on the web-based implementation of HHPred https://toolkit.tuebingen.mpg.de/tools/hhpred) on the MPI toolkit site [19]. We used an E-value significance cutoff of 10−6.
4.3. D structure Prediction
We predicted the 3D structure of proteins using Alphafold2-MMseqs2 ran from the Colabfold web server [92], a user-friendly implementation of Alphafold2 [8]. We set the number of recycles to 3 for initial explorations and 48 for high-quality predictions of individual domains. Alphafold2 outputs a measure of reliability of the 3D structure for each aa, pLDDT. pLDDT ≥ 0.70 corresponds to a reliable prediction, and pLDDT ≥ 0.90 to a highly reliable prediction (expected to be competitive with an experimentally solved 3D structure).
4.4. Structural Visualization and Alignment
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
We thank F Ferron for feedback on the structural analyses and comments on the manuscript; R Kormelink for feedback on Tospovirus NSs and comments on the manuscript; M Jamin and S von Bargen for feedback on the manuscript; U Neri and Y Wolf for generously providing guidance on analyzing viral metatranscriptomes (not included in the final study)
References
- Koonin EV, Krupovic M, Dolja VV. The global virome: How much diversity and how many independent origins? Environmental Microbiology. 2023;25: 40–44. [CrossRef]
- Zayed AA, Wainaina JM, Dominguez-Huerta G, Pelletier E, Guo J, Mohssen M, et al. Cryptic and abundant marine viruses at the evolutionary origins of Earth’s RNA virome. Science. 2022;376: 156–162. [CrossRef]
- Neri U, Wolf YI, Roux S, Camargo AP, Lee B, Kazlauskas D, et al. Expansion of the global RNA virome reveals diverse clades of bacteriophages. Cell. 2022;185: 4023-4037.e18. [CrossRef]
- Kuchibhatla DB, Sherman WA, Chung BYW, Cook S, Schneider G, Eisenhaber B, et al. Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J Virol. 2014;88: 10–20. [CrossRef]
- Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2012;9: 173–175. [CrossRef]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50: D439–D444. [CrossRef]
- Marx V. Method of the Year: protein structure prediction. Nat Methods. 2022;19: 5–10. [CrossRef]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596: 583–589. [CrossRef]
- van Kempen M, Kim SS, Tumescheit C, Mirdita M, Söding J, Steinegger M. Foldseek: fast and accurate protein structure search. Bioinformatics; 2022 Feb. [CrossRef]
- Hildebrand A, Remmert M, Biegert A, Söding J. Fast and accurate automatic structure prediction with HHpred: Structure Prediction with HHpred. Proteins. 2009;77: 128–132. [CrossRef]
- Daughenbaugh K, Martin M, Brutscher L, Cavigli I, Garcia E, Lavin M, et al. Honey Bee Infecting Lake Sinai Viruses. Viruses. 2015;7: 3285–3309. [CrossRef]
- Runckel C, Flenniken ML, Engel JC, Ruby JG, Ganem D, Andino R, et al. Temporal Analysis of the Honey Bee Microbiome Reveals Four Novel Viruses and Seasonal Prevalence of Known Viruses, Nosema, and Crithidia. Moritz RFA, editor. PLoS ONE. 2011;6: e20656. [CrossRef]
- Parmentier L, Smagghe G, de Graaf DC, Meeus I. Varroa destructor Macula-like virus, Lake Sinai virus and other new RNA viruses in wild bumblebee hosts ( Bombus pascuorum , Bombus lapidarius and Bombus pratorum ). Journal of Invertebrate Pathology. 2016;134: 6–11. [CrossRef]
- Bigot D, Dalmon A, Roy B, Hou C, Germain M, Romary M, et al. The discovery of Halictivirus resolves the Sinaivirus phylogeny. Journal of General Virology. 2017;98: 2864–2875. [CrossRef]
- McMenamin AJ, Flenniken ML. Recently identified bee viruses and their impact on bee pollinators. Current Opinion in Insect Science. 2018;26: 120–129. [CrossRef]
- Ahola T, Karlin DG. Sequence analysis reveals a conserved extension in the capping enzyme of the alphavirus supergroup, and a homologous domain in nodaviruses. Biol Direct. 2015;10: 16. [CrossRef]
- Rehanek M, Karlin DG, Bandte M, Al Kubrusli R, Nourinejhad Zarghani S, Candresse T, et al. The Complex World of Emaraviruses—Challenges, Insights, and Prospects. Forests. 2022;13: 1868. [CrossRef]
- Hu G, Kurgan L. Sequence Similarity Searching. Current Protocols in Protein Science. 2019;95: e71. [CrossRef]
- Zimmermann L, Stephens A, Nam S-Z, Rau D, Kübler J, Lozajic M, et al. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J Mol Biol. 2018;430: 2237–2243. [CrossRef]
- Coin L, Bateman A, Durbin R. Enhanced protein domain discovery using taxonomy. BMC Bioinformatics. 2004;5: 56. [CrossRef]
- Aravind L. Guilt by Association: Contextual Information in Genome Analysis: Figure 1. Genome Res. 2000;10: 1074–1077. [CrossRef]
- Boekhorst J, Snel B. Identification of homologs in insignificant blast hits by exploiting extrinsic gene properties. BMC Bioinformatics. 2007;8: 356. [CrossRef]
- Mushegian AR, Elena SF. Evolution of plant virus movement proteins from the 30K superfamily and of their homologs integrated in plant genomes. Virology. 2015;476: 304–315. [CrossRef]
- Dunbrack RL. Sequence comparison and protein structure prediction. Current Opinion in Structural Biology. 2006;16: 374–384. [CrossRef]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25: 3389–3402. [CrossRef]
- Paraskevopoulou S, Käfer S, Zirkel F, Donath A, Petersen M, Liu S, et al. Viromics of extant insect orders unveil the evolution of the flavi-like superfamily. Virus Evol. 2021;7: veab030. [CrossRef]
- Liu S, Sappington TW, Coates BS, Bonning BC. Sequences Encoding a Novel Toursvirus Identified from Southern and Northern Corn Rootworms (Coleoptera: Chrysomelidae). Viruses. 2022;14: 397. [CrossRef]
- Chen Y-M, Sadiq S, Tian J-H, Chen X, Lin X-D, Shen J-J, et al. RNA viromes from terrestrial sites across China expand environmental viral diversity. Nat Microbiol. 2022;7: 1312–1323. [CrossRef]
- Medd NC, Fellous S, Waldron FM, Xuéreb A, Nakai M, Cross JV, et al. The virome of Drosophila suzukii, an invasive pest of soft fruit. Virus Evol. 2018;4: vey009. [CrossRef]
- Kondo H, Chiba S, Maruyama K, Andika IB, Suzuki N. A novel insect-infecting virga/nege-like virus group and its pervasive endogenization into insect genomes. Virus Research. 2019;262: 37–47. [CrossRef]
- Mahar JE, Shi M, Hall RN, Strive T, Holmes EC. Comparative Analysis of RNA Virome Composition in Rabbits and Associated Ectoparasites. Pfeiffer JK, editor. J Virol. 2020;94: e02119-19. [CrossRef]
- Valles SM, Oi DH, Becnel JJ, Wetterer JK, LaPolla JS, Firth AE. Isolation and characterization of Nylanderia fulva virus 1, a positive-sense, single-stranded RNA virus infecting the tawny crazy ant, Nylanderia fulva. Virology. 2016;496: 244–254. [CrossRef]
- Valles SM, Rivers AR. Nine new RNA viruses associated with the fire ant Solenopsis invicta from its native range. Virus Genes. 2019;55: 368–380. [CrossRef]
- Lee C-C, Hsu H-W, Lin C-Y, Gustafson N, Matsuura K, Lee C-Y, et al. First Polycipivirus and Unmapped RNA Virus Diversity in the Yellow Crazy Ant, Anoplolepis gracilipes. Viruses. 2022;14: 2161. [CrossRef]
- Webster CL, Longdon B, Lewis SH, Obbard DJ. Twenty-Five New Viruses Associated with the Drosophilidae (Diptera). Evol Bioinform Online. 2016;12s2: EBO.S39454. [CrossRef]
- Hernández-Pelegrín L, Llopis-Giménez Á, Crava CM, Ortego F, Hernández-Crespo P, Ros VID, et al. Expanding the Medfly Virome: Viral Diversity, Prevalence, and sRNA Profiling in Mass-Reared and Field-Derived Medflies. Viruses. 2022;14: 623. [CrossRef]
- He Y-J, Ye Z-X, Zhang C-X, Li J-M, Chen J-P, Lu G. An RNA Virome Analysis of the Pink-Winged Grasshopper Atractomorpha sinensis. Insects. 2022;14: 9. [CrossRef]
- Charles J, Tangudu CS, Hurt SL, Tumescheit C, Firth AE, Garcia-Rejon JE, et al. Discovery of a novel Tymoviridae-like virus in mosquitoes from Mexico. Arch Virol. 2019;164: 649–652. [CrossRef]
- Pei J, Tang M, Grishin NV. PROMALS3D web server for accurate multiple protein sequence and structure alignments. Nucleic Acids Research. 2008;36: W30–W34. [CrossRef]
- Krell PJ. Reoviruses of Invertebrates (Reoviridae). Encyclopedia of Virology. Elsevier; 2021. pp. 867–882. [CrossRef]
- Dong R, Pan S, Peng Z, Zhang Y, Yang J. mTM-align: a server for fast protein structure database search and multiple protein structure alignment. Nucleic Acids Research. 2018 [cited 13 May 2022]. [CrossRef]
- Takatsuka J. A new cypovirus from the Japanese peppered moth, Biston robustus. Journal of Invertebrate Pathology. 2020;174: 107417. [CrossRef]
- Xu C, Wang J, Yang J, Lei C, Hu J, Sun X. NSP2 forms viroplasms during Dendrolimus punctatus cypovirus infection. Virology. 2019;533: 68–76. [CrossRef]
- Chavali VRM, Ghosh AK. Molecular cloning, sequence analysis and expression of genome segment 7 (S7) of Antheraea mylitta cypovirus (AmCPV) that encodes a viral structural protein. Virus Genes. 2007;35: 433–441. [CrossRef]
- Yu X, Ge P, Jiang J, Atanasov I, Zhou ZH. Atomic Model of CPV Reveals the Mechanism Used by This Single-Shelled Virus to Economically Carry Out Functions Conserved in Multishelled Reoviruses. Structure. 2011;19: 652–661. [CrossRef]
- Ren F, Swevers L, Lu Q, Zhao Y, Yan J, Li H, et al. Effect of mutations in capsid shell protein on the assembly of BmCPV virus-like particles. Journal of General Virology. 2021;102. [CrossRef]
- Zhu F, Li D, Song D, Huo S, Ma S, Lü P, et al. Glycoproteome in silkworm Bombyx mori and alteration by BmCPV infection. Journal of Proteomics. 2020;222: 103802. [CrossRef]
- Katsuma S, Tanaka S, Omuro N, Takabuchi L, Daimon T, Imanishi S, et al. Novel macula-like virus identified in Bombyx mori cultured cells. J Virol. 2005;79: 5577–5584. [CrossRef]
- Bejerman N, Debat H. Exploring the tymovirales landscape through metatranscriptomics data. Arch Virol. 2022;167: 1785–1803. [CrossRef]
- Katsuma S, Kawamoto M, Shoji K, Aizawa T, Kiuchi T, Izumi N, et al. Transcriptome profiling reveals infection strategy of an insect maculavirus. DNA Research. 2018;25: 277–286. [CrossRef]
- Feng Y, Zhang X, Kumar D, Kuang S, Liu B, Hu X, et al. Transient propagation of BmLV and dysregulation of gene expression in nontarget cells following BmLV infection. Journal of Asia-Pacific Entomology. 2021;24: 893–902. [CrossRef]
- Gupta R, Kwon S-Y, Kim ST. An insight into the tomato spotted wilt virus (TSWV), tomato and thrips interaction. Plant Biotechnol Rep. 2018;12: 157–163. [CrossRef]
- Whitfield AE, Falk BW, Rotenberg D. Insect vector-mediated transmission of plant viruses. Virology. 2015;479–480: 278–289. [CrossRef]
- Margaria P, Bosco L, Vallino M, Ciuffo M, Mautino GC, Tavella L, et al. The NSs Protein of Tomato spotted wilt virus Is Required for Persistent Infection and Transmission by Frankliniella occidentalis. Simon A, editor. J Virol. 2014;88: 5788–5802. [CrossRef]
- Oliveira VC, Bartasson L, de Castro MEB, Corrêa JR, Ribeiro BM, Resende RO. A silencing suppressor protein (NSs) of a tospovirus enhances baculovirus replication in permissive and semipermissive insect cell lines. Virus Research. 2011;155: 259–267. [CrossRef]
- de Oliveira VC, da Silva Morgado F, Ardisson-Araújo DMP, Resende RO, Ribeiro BM. The silencing suppressor (NSs) protein of the plant virus Tomato spotted wilt virus enhances heterologous protein expression and baculovirus pathogenicity in cells and lepidopteran insects. Arch Virol. 2015;160: 2873–2879. [CrossRef]
- Garcia S, Billecocq A, Crance J-M, Prins M, Garin D, Bouloy M. Viral suppressors of RNA interference impair RNA silencing induced by a Semliki Forest virus replicon in tick cells. Journal of General Virology. 2006;87: 1985–1989. [CrossRef]
- Kim C, Kim Y. In vivo transient expression of a viral silencing suppressor, NSs, derived from tomato spotted wilt virus decreases insect RNAi efficiencies. Arch Insect Biochem Physiol. 2022 [cited 12 Jan 2023]. [CrossRef]
- Nagata T, Storms MM, Goldbach R, Peters D. Multiplication of tomato spotted wilt virus in primary cell cultures derived from two thrips species. Virus Res. 1997;49: 59–66. [CrossRef]
- Wijkamp I, van Lent J, Kormelink R, Goldbach R, Peters D. Multiplication of tomato spotted wilt virus in its insect vector, Frankliniella occidentalis. J Gen Virol. 1993;74 ( Pt 3): 341–349. [CrossRef]
- Zhu M, van Grinsven IL, Kormelink R, Tao X. Paving the Way to Tospovirus Infection: Multilined Interplays with Plant Innate Immunity. Annu Rev Phytopathol. 2019;57: 41–62. [CrossRef]
- Turina M, Kormelink R, Resende RO. Resistance to Tospoviruses in Vegetable Crops: Epidemiological and Molecular Aspects. Annu Rev Phytopathol. 2016;54: 347–371. [CrossRef]
- de Ronde D, Pasquier A, Ying S, Butterbach P, Lohuis D, Kormelink R. Analysis of Tomato spotted wilt virus NSs protein indicates the importance of the N-terminal domain for avirulence and RNA silencing suppression: Mapping TSWV NSs Avr and RSS activity. Molecular Plant Pathology. 2014;15: 185–195. [CrossRef]
- Zhai Y, Bag S, Mitter N, Turina M, Pappu HR. Mutational analysis of two highly conserved motifs in the silencing suppressor encoded by tomato spotted wilt virus (genus Tospovirus, family Bunyaviridae). Arch Virol. 2014;159: 1499–1504. [CrossRef]
- Huang C-H, Foo M-H, Raja JAJ, Tan Y-R, Lin T-T, Lin S-S, et al. A Conserved Helix in the C-Terminal Region of Watermelon Silver Mottle Virus Nonstructural Protein S Is Imperative For Protein Stability Affecting Self-Interaction, RNA Silencing Suppression, and Pathogenicity. MPMI. 2020;33: 637–652. [CrossRef]
- Huang C-H, Hsiao W-R, Huang C-W, Chen K-C, Lin S-S, Chen T-C, et al. Two Novel Motifs of Watermelon Silver Mottle Virus NSs Protein Are Responsible for RNA Silencing Suppression and Pathogenicity. Ikegami T, editor. PLoS ONE. 2015;10: e0126161. [CrossRef]
- Yesilyurt A, Demirbag Z, van Oers MM, Nalcacioglu R. Conserved motifs in the invertebrate iridescent virus 6 (IIV6) genome regulate virus transcription. Journal of Invertebrate Pathology. 2020;177: 107496. [CrossRef]
- de Faria IJS, Aguiar ERGR, Olmo RP, Alves da Silva J, Daeffler L, Carthew RW, et al. Invading viral DNA triggers dsRNA synthesis by RNA polymerase II to activate antiviral RNA interference in Drosophila. Cell Reports. 2022;39: 110976. [CrossRef]
- İnce İA, Boeren SA, van Oers MM, Vervoort JJM, Vlak JM. Proteomic analysis of Chilo iridescent virus. Virology. 2010;405: 253–258. [CrossRef]
- Jiang L, Peng Z, Guo Y, Cheng T, Guo H, Sun Q, et al. Transcriptome analysis of interactions between silkworm and cytoplasmic polyhedrosis virus. Sci Rep. 2016;6: 24894. [CrossRef]
- Zhao SL, Liang CY, Zhang WJ, Tang XC, Peng HY. Characterization of the RNA-binding domain in the Dendrolimus punctatus cytoplasmic polyhedrosis virus nonstructural protein p44. Virus Research. 2005;114: 80–88. [CrossRef]
- Oliver JE, Whitfield AE. The Genus Tospovirus: Emerging Bunyaviruses that Threaten Food Security. Annu Rev Virol. 2016;3: 101–124. [CrossRef]
- Ribière M, Olivier V, Blanchard P. Chronic bee paralysis: A disease and a virus like no other? Journal of Invertebrate Pathology. 2010;103: S120–S131. [CrossRef]
- Beaurepaire A, Piot N, Doublet V, Antunez K, Campbell E, Chantawannakul P, et al. Diversity and Global Distribution of Viruses of the Western Honey Bee, Apis mellifera. Insects. 2020;11: 239. [CrossRef]
- Shan T, Yang S, Wang H, Wang H, Zhang J, Gong G, et al. Virome in the cloaca of wild and breeding birds revealed a diversity of significant viruses. Microbiome. 2022;10: 60. [CrossRef]
- de Lima JGS, Lanza DCF. 2A and 2A-like Sequences: Distribution in Different Virus Species and Applications in Biotechnology. Viruses. 2021;13: 2160. [CrossRef]
- Olivier V, Blanchard P, Chaouch S, Lallemand P, Schurr F, Celle O, et al. Molecular characterisation and phylogenetic analysis of Chronic bee paralysis virus, a honey bee virus. Virus Res. 2008;132: 59–68. [CrossRef]
- Zhan H, Unchwaniwala N, Rebolledo-Viveros A, Pennington J, Horswill M, Broadberry R, et al. Nodavirus RNA Replication Crown Architecture Reveals Proto-Crown Precursor and Viral Protein A Conformational Switching. Microbiology; 2022 Dec. [CrossRef]
- Wagner I, Volkmer M, Sharan M, Villaveces JM, Oswald F, Surendranath V, et al. morFeus: a web-based program to detect remotely conserved orthologs using symmetrical best hits and orthology network scoring. BMC Bioinformatics. 2014;15: 263. [CrossRef]
- Le Lay C, Shi M, Buček A, Bourguignon T, Lo N, Holmes E. Unmapped RNA Virus Diversity in Termites and Their Symbionts. Viruses. 2020;12: 1145. [CrossRef]
- Perrakis A, Sixma TK. AI revolutions in biology: The joys and perils of AlphaFold. EMBO Reports. 2021;22. [CrossRef]
- Tokuriki N, Oldfield CJ, Uversky VN, Berezovsky IN, Tawfik DS. Do viral proteins possess unique biophysical features? Trends in Biochemical Sciences. 2009;34: 53–59. [CrossRef]
- Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, et al. Evolutionary-scale prediction of atomic level protein structure with a language model. Synthetic Biology; 2022 Jul. [CrossRef]
- Bruley A, Mornon J-P, Duprat E, Callebaut I. Digging into the 3D Structure Predictions of AlphaFold2 with Low Confidence: Disorder and Beyond. Biomolecules. 2022;12: 1467. [CrossRef]
- Gulyaeva AA, Sigorskih AI, Ocheredko ES, Samborskiy DV, Gorbalenya AE. LAMPA, LArge Multidomain Protein Annotator, and its application to RNA virus polyproteins. Ponty Y, editor. Bioinformatics. 2020;36: 2731–2739. [CrossRef]
- Zhao Y, Sun J, Labropoulou V, Swevers L. Beyond Baculoviruses: Additional Biotechnological Platforms Based on Insect RNA Viruses. Advances in Insect Physiology. Elsevier; 2018. pp. 123–162. [CrossRef]
- Floden EW, Tommaso PD, Chatzou M, Magis C, Notredame C, Chang J-M. PSI/TM-Coffee: a web server for fast and accurate multiple sequence alignments of regular and transmembrane proteins using homology extension on reduced databases. Nucleic Acids Res. 2016;44: W339-343. [CrossRef]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25: 1189–1191. [CrossRef]
- Procter JB, Thompson J, Letunic I, Creevey C, Jossinet F, Barton GJ. Visualization of multiple alignments, phylogenies and gene family evolution. Nat Methods. 2010;7: S16-25. [CrossRef]
- Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 2021;49: D412–D419. [CrossRef]
- Burley SK, Bhikadiya C, Bi C, Bittrich S, Chen L, Crichlow GV, et al. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Research. 2021;49: D437–D451. [CrossRef]
- Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. 2022;19: 679–682. [CrossRef]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis: UCSF ChimeraX Visualization System. Protein Science. 2018;27: 14–25. [CrossRef]
- Li Z, Jaroszewski L, Iyer M, Sedova M, Godzik A. FATCAT 2.0: towards a better understanding of the structural diversity of proteins. Nucleic Acids Research. 2020;48: W60–W64. [CrossRef]
- Holm L. Dali server: structural unification of protein families. Nucleic Acids Research. 2022;50: W210–W215. [CrossRef]
Figure 1.
Our procedure to detect and verify distant homologs, using contextual information. NB: the cutoff used for statistically significant similarity varies depending on the software.
Figure 1.
Our procedure to detect and verify distant homologs, using contextual information. NB: the cutoff used for statistically significant similarity varies depending on the software.
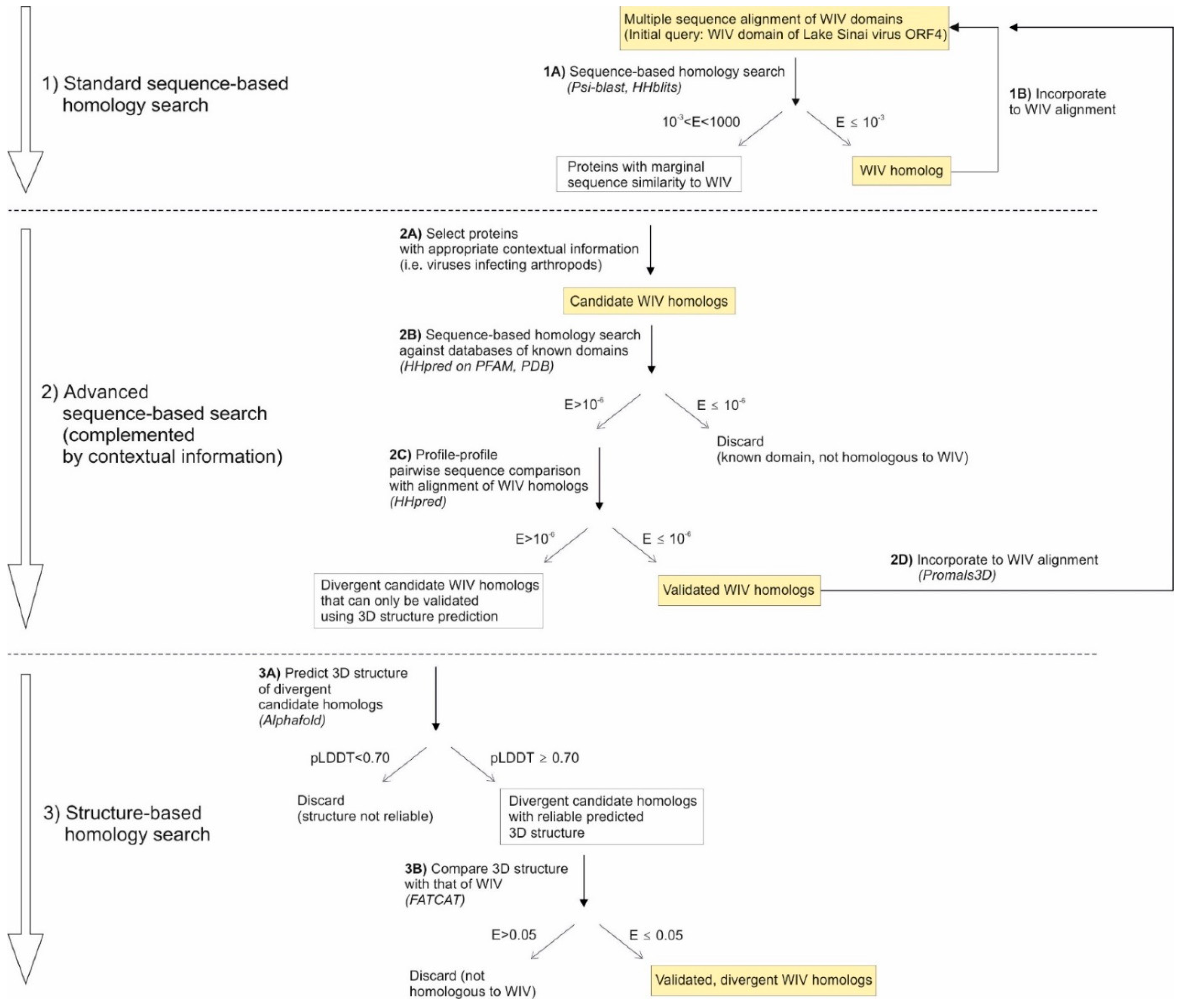
Figure 2.
The WIV domain is found in proteins from over 20 viral taxa, with a large variety of domain organizations.
Figure 2.
The WIV domain is found in proteins from over 20 viral taxa, with a large variety of domain organizations.

Figure 3.
The WIV domain has a previously unknown fold, according to predictions.
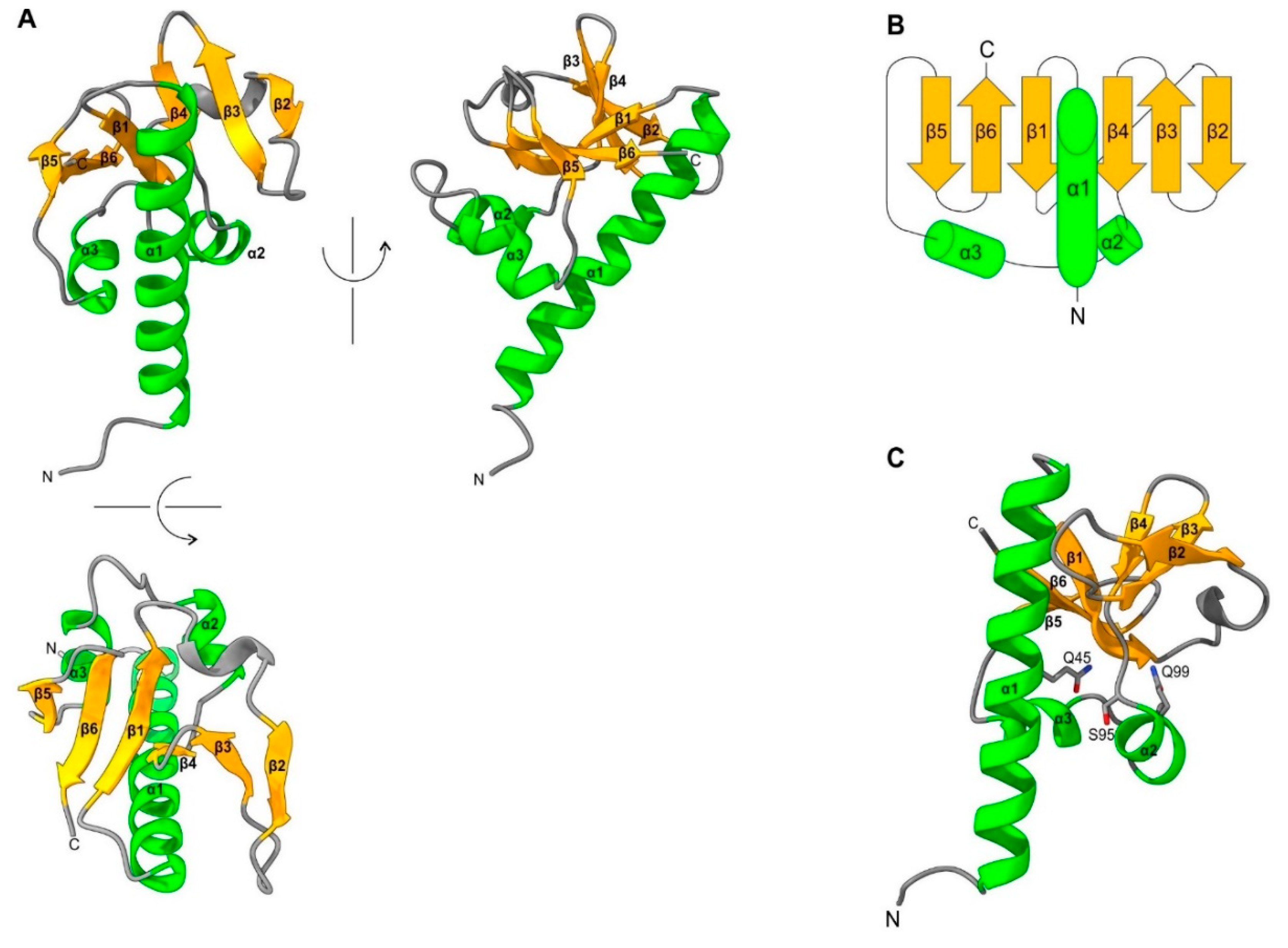
Figure 4.
Multiple sequence alignment of representative WIV domains.
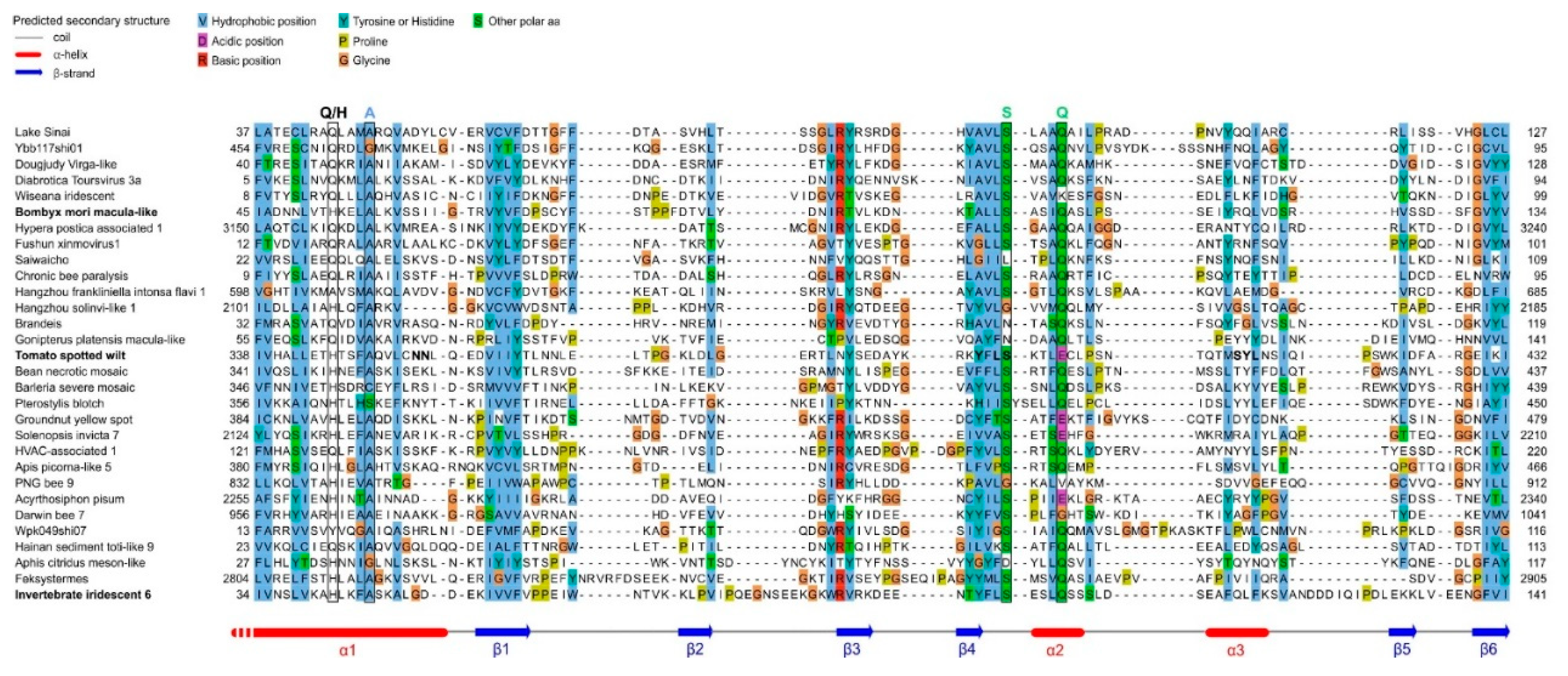
Figure 5.
The C-terminal domain of cypovirus p44 is homologous to the WIV domain of Lake Sinai virus ORF4.
Figure 5.
The C-terminal domain of cypovirus p44 is homologous to the WIV domain of Lake Sinai virus ORF4.
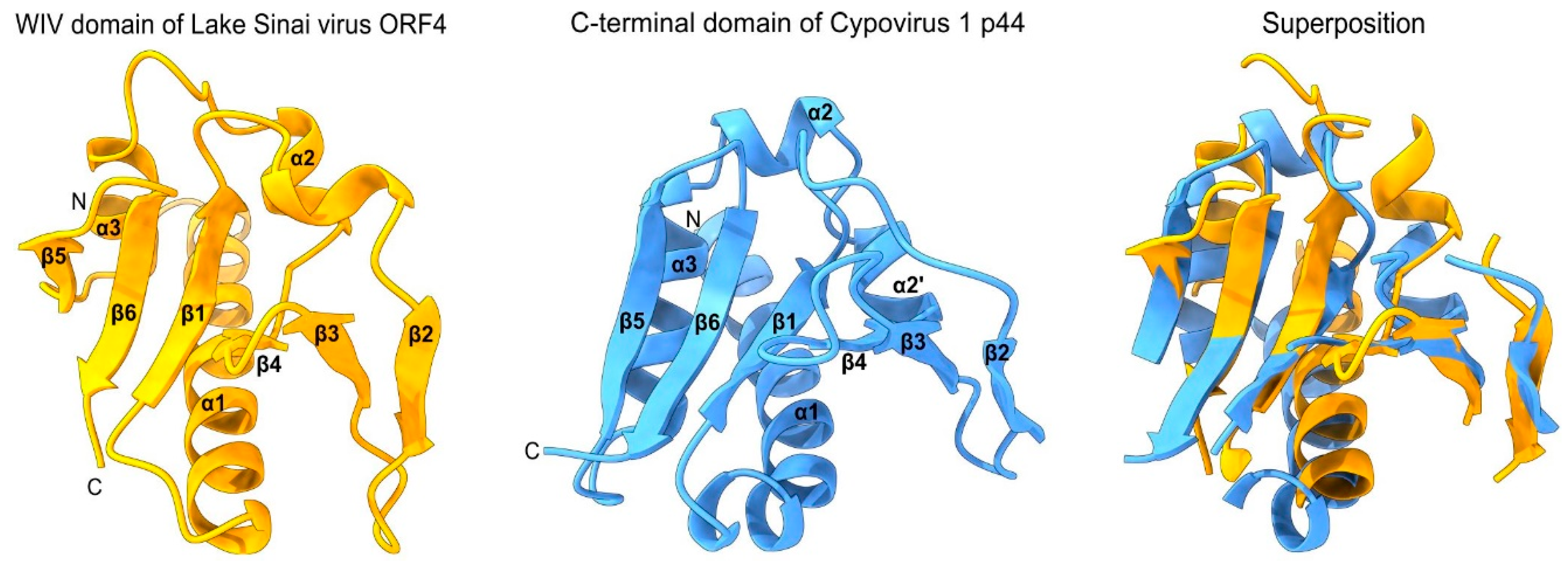
Figure 6.
Multiple sequence alignment of cypoviral WIV domains.
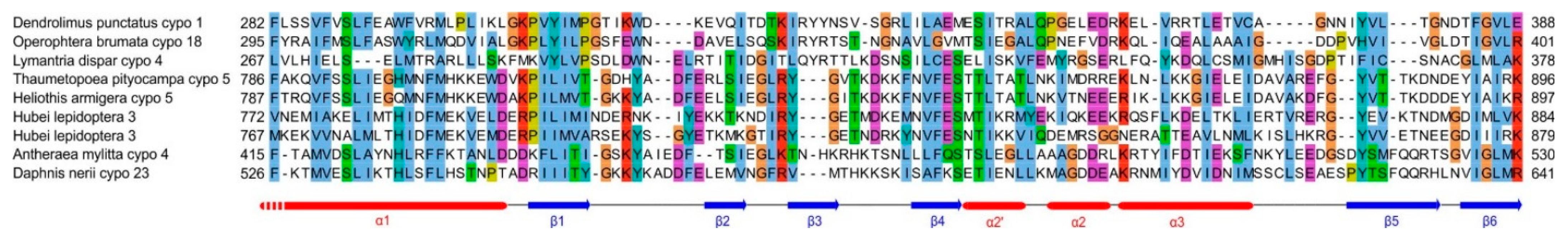
Figure 7.
The N-terminal domain of Cypovirus 1 p44 is structurally similar to the SPD domain of cypovirus 1 capsid protein VP1.
Figure 7.
The N-terminal domain of Cypovirus 1 p44 is structurally similar to the SPD domain of cypovirus 1 capsid protein VP1.
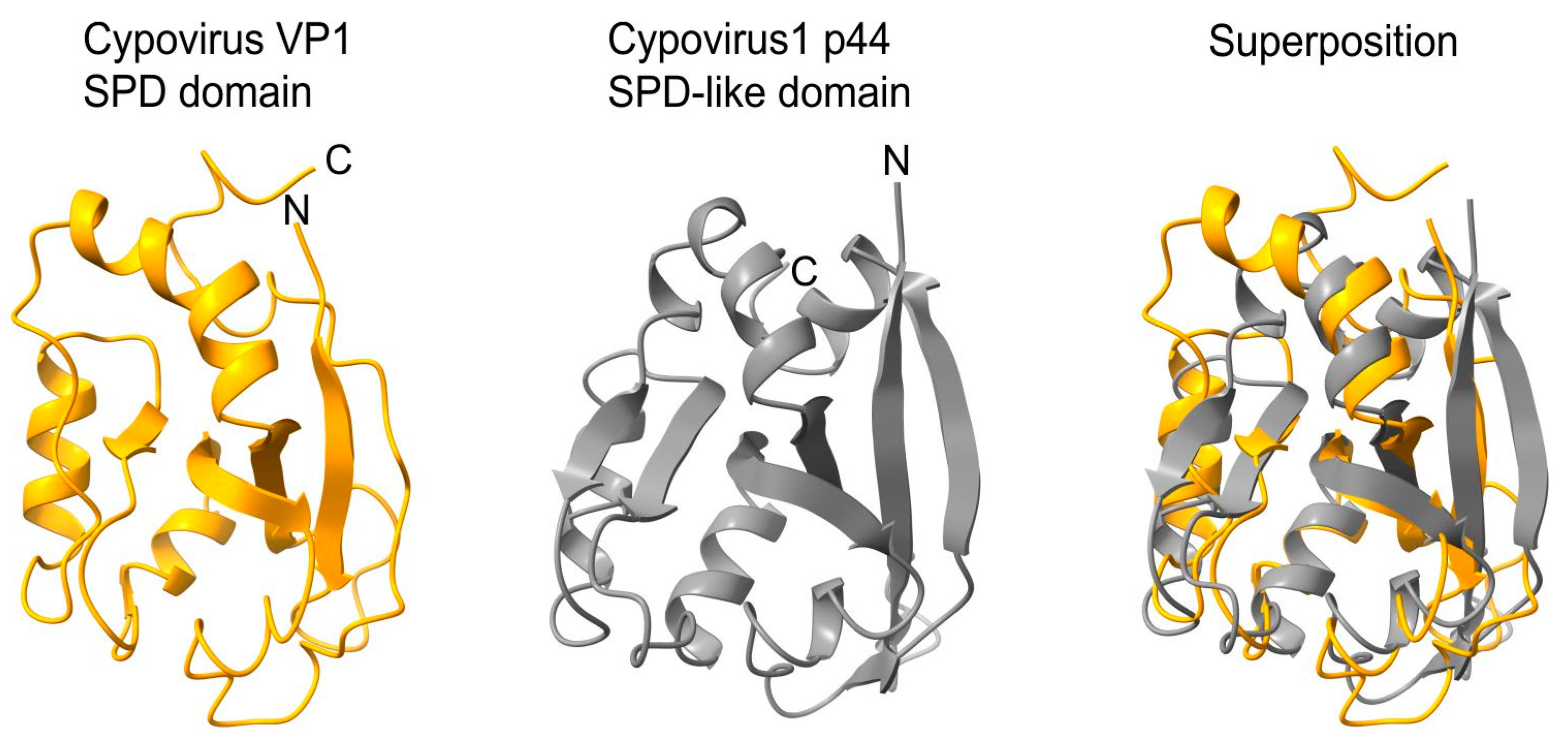
Table 1.
Proteins containing a WIV domain, from representative viral taxa.
| Species | Protein name | Genbank accession number | Genomic material | Phylum | Order | Family | Genus |
|---|---|---|---|---|---|---|---|
| Acyrthosiphon pisum virus | P1 | QAA78863.1 | +ssRNA | Pisuviricota | Picornavirales | Acypiviridae(1) | Acypivirus (proposed) |
| Aphis citricidus meson-like virus | ORF2a | QPD01783.1 | +ssRNA | Pisuviricota | Nidovirales | Mesoniviridae | Unclassified |
| Barleria severe mosaic virus | NSs | QVY47427.1 | -ssRNA | Negarnaviricota | Bunyavirales | Tospoviridae | Orthotospovirus |
| Bombyx mori Macula-like virus | p15 | YP_004464932 | +ssRNA | Kitrinoviricota | Tymovirales | Tymoviridae | Inmaculavirus (proposed) |
| Bean necrotic mosaic virus | NSs | YP_006468899.1 | -ssRNA | Negarnaviricota | Bunyavirales | Tospoviridae | Orthotospovirus |
| Brandeis virus | ORF5 | AVZ66287.1 | +ssRNA | Kitrinoviricata | Martellivirales | Brandeis virus group(1) | Unclassified |
| Chronic bee paralysis virus | ORF1 from RNA2 | YP_001911139.1 | +ssRNA | Kitrinoviricata | Nodamuvirales | Unclassified | Chroparavirus |
| Darwin bee virus 7 | Nonstructural polyprotein | AWK77849.1 | +ssRNA | Pisuviricota | Picornavirales | Acypiviridae-like(1) | Unclassified |
| Diabrotica toursvirus 3a | F4ORF11 | UOX61048.1 | dsDNA | Nucleocytoviricota | Pimascovirales | Ascoviridae(2) | Toursvirus |
| Dougjudy virga-like virus | ORF4 | QIJ70140.1 | +ssRNA | Kitrinoviricata | Martellivirales | Dougjudy virga-like virus group (1) | Virga-like?? |
| Feksystermes virus | Polyprotein | QRW42904.1 | +ssRNA | Pisuviricota | Picornavirales | Unclassified | Unclassified |
| Fushun monolepta lauta xinmovirus(1) | ORF3 | UHM27673.1 | -ssRNA | Negarnaviricota | Mononegavirales | Xinmoviridae | Unclassified |
| Gonipterus platensis Macula-Like virus | ORF3 | QWX94186.1 | +ssRNA | Kitrinoviricata | Tymovirales | Tymoviridae | Unclassified |
| Groundnut yellow Spot virus | NSs | YP_009665191.1 | -ssRNA | Negarnaviricota | Bunyavirales | Tospoviridae | Orthotospovirus |
| Hangzhou frankliniella intonsa flavivirus(1) | RNA-dependent DNA polymerase | UHK03321.1 | +ssRNA | Kitrinoviricata | Amarillovirales | Flaviviridae-like | Unclassified, “Large-genome flaviviruses” [26] |
| Hangzhou sesamia inferens solinvi-like virus(1) | RNA helicase | UHR49866 | +ssRNA | Pisuviricota | Picornavirales | Acypiviridae-like(1) | Unclassified, similar to Solenopsis invicta virus 7 |
| Hainan sediment toti-like virus 9 | Putative coat protein | UHS72513.1 | dsRNA | Duplornaviricota | Ghabrivirales | Totiviridae | Artivirus |
| Hubei picorna-like virus 36 | ORF2 | KX883970.1 (coding sequence: nt 9360-9647) | +ssRNA | Pisuviricota | Picornavirales | Iflaviridae | Iflavirus |
| Hypera postica associated virus(1) | Hypothetical protein | QUS52866.1 | +ssRNA | Pisuviricota | Picornavirales | Solinvirididae-like | Unclassified |
| HVAC-associated RNA virus(1) | Polyprotein | AVD69112 | +ssRNA | Pisuviricota | Picornavirales | Acypiviridae-like(1) | Unclassified, similar to Solenopsis invicta virus 7 |
| Invertebrate iridescent virus 6 | 206R | Q91FW4 | dsDNA | Nucleocytoviricota | Pimascovirales | Iridoviridae | Iridovirus |
| Lake Sinai virus | ORF4 | YP_009333196.1 | +ssRNA | Kitrinoviricata | Nodamuvirales | Sinhaliviridae | Sinaivirus |
| PNG bee virus 9 | Polyprotein | QKW94212.1 | +ssRNA | Pisuviricota | Picornavirales | Acypiviridae-like(1) | Unclassified, similar to Solenopsis invicta virus 7 |
| Pterostylis blotch virus | NSs | ULN99190.1 | -ssRNA | Negarnaviricota | Bunyavirales | Tospoviridae | Orthotospovirus |
| Saiwaicho virus | ORF4 | AWA82266.1 | +ssRNA | Kitrinoviricota | Martellivirales | Unclassified | Unclassified, Nelorpivirus-like |
| Solenopsis invicta virus 7 | Polyprotein | QBL75890.1 | +ssRNA | Pisuviricota | Picornavirales | Acypiviridae-like(1) | Unclassified |
| Solenopsis invicta virus 8 | ORF6 | MH727525.2 (coding sequence: nt 4189-4476) | +ssRNA | Pisuviricota | Picornavirales | Polycipiviridae | Sopolycivirus |
| Tomato spotted wilt virus (strain Br20) | NSs | ABI94070.1 | -ssRNA | Negarnaviricota | Bunyavirales | Tospoviridae | Orthotospovirus |
| Isolate H4_Bulk_46_scaffold_6139 | Hypothetical protein | MN034786 (coding sequence: nt 3-1841)(3) | +ssRNA | Pisuviricota | Picornavirales | Acypiviridae-like | Unclassified |
| Wiseana Iridescent virus | gp049 | YP_004732832 | dsDNA | Nucleocytoviricota | Pimascovirales | Iridoviridae | Chloriridovirus |
| Wpk049shi07 isolate(4) | ORF1 | QKE55054.1 | +ssRNA | Kitrinoviricata | Nodamuvirales | Sinhaliviridae-like | Sinaivirus-like |
| Ybb117shi01 isolate | ORF2 (hypothetical protein) | QJI53477 | +ssRNA | Pisuviricota | Picornavirales | Acypiviridae-like | Unclassified |
(1) Proposed family or taxon. See Table 2. (2) A new family, Toursviridae, has been suggested for this genus and for related species [27]. (3) The corresponding protein sequence lacks the C-terminus. (4) This species is mistakenly called “Picornaviridae sp.” but is in fact related to Sinhaliviridae.
Table 3.
Cypovirus proteins containing a WIV domain.
| Virus species | Protein name | Genomic RNA Segment | Genbank accession number | Bibliographical information |
|---|---|---|---|---|
|
Dendrolimus punctatus cypovirus 1 (a strain of Bombyx mori cypovirus 1) |
p44 (also called nsp2 or NS2 or NS) | S8 | NP_149153.1 | p44 is a non-structural protein that forms viroplasms during infection by Dendrolimus punctatus cypovirus [43]. |
| Hubei lepidoptera virus 3 (proposed as Lymantria dispar cypovirus 3) | P5 (also called VP5) | S5 | YP_009330260.1 | |
| Hubei lepidoptera virus 3 (isolate LdCPV3) | P5 (also called VP5) | S5 | QJB76100.1 | |
| Antheraea mylitta cypovirus 4 | p61 | S7 | ABF83587.1 | p61 is a structural protein, i.e. found in virions [44]. |
| Thaumetopoea pityocampa cypovirus 5 | P5 | S5 | AJC97792.1 | |
| Heliothis armigera cypovirus 5 | P5 | S5 | YP_001883319.1 | |
|
Lymantria dispar cypovirus 14 |
P8 | S8 | NP_149142.1 | |
| Operophtera brumata cypovirus 18 | P8 | S8 | ABB17218 | |
| Daphnis nerii cypovirus 23 | P5 | S5 | YP_009551580 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



